

Abstract
Many transmembrane helices contain serine and/or threonine residues whose side chains form intrahelical H-bonds with upstream carbonyl oxygens. Here, we investigated the impact of threonine side-chain/main-chain backbonding on the backbone dynamics of the amyloid precursor protein transmembrane helix. This helix consists of a N-terminal dimerization region and a C-terminal cleavage region, which is processed by γ-secretase to a series of products. Threonine mutations within this transmembrane helix are known to alter the cleavage pattern, which can lead to early-onset Alzheimer’s disease. Circular dichroism spectroscopy and amide exchange experiments of synthetic transmembrane domain peptides reveal that mutating threonine enhances the flexibility of this helix. Molecular dynamics simulations show that the mutations reduce intrahelical amide H-bonding and H-bond lifetimes. In addition, the removal of side-chain/main-chain backbonding distorts the helix, which alters bending and rotation at a diglycine hinge connecting the dimerization and cleavage regions. We propose that the backbone dynamics of the substrate profoundly affects the way by which the substrate is presented to the catalytic site within the enzyme. Changing this conformational flexibility may thus change the pattern of proteolytic processing.
Introduction
The transmembrane domains (TMDs) of integral membrane proteins are mostly composed of hydrophobic amino acids. Many of them also contain polar amino acids that contribute to helix-helix interactions, cofactor binding, etc. (1). That polar amino acids serve structural roles supporting protein function is indicated by the fact that mutations of Ser, Thr, and Cys within TMDs frequently cause hereditary diseases (2,3). Ser and Thr are the most abundant of the polar amino acids as each represents ∼5% of TMD residues (4). The majority of Ser and Thr at position i of a helix form intrahelical H-bonds with main chain carbonyl oxygens at i-3 and/or i-4 positions, which was originally revealed by analyzing crystallized soluble proteins (5,6). More recently, similar observations have been made by investigating the growing database of crystallized membrane proteins and by molecular dynamics (MD) simulation of model helices (7–11). As a result of side-chain/main-chain backbonding, Ser and Thr can slightly increase helix bending and induce local helix opening (7,12). The impact of Ser and Thr backbonds on helix bending can be more pronounced in the presence of Pro suggesting its dependence on sequence context (13).
The TMD of the amyloid precursor protein (APP, for sequence see Fig. 1 A) is cleaved by the intramembrane protease presenilin, the catalytic subunit of the γ-secretase complex. Cleavage at ε-, ζ-, and γ-sites of the APP TMD produces a series of amyloid beta (Aβ) peptides that form toxic oligomers and amyloid plaques, one of the hallmarks of Alzheimer’s disease (reviewed in (14–17)). The dynamics of the APP TMD helix has attracted considerable interest because proteolysis of helical substrates by proteases requires at least its local and transient unwinding (18). Solid-state NMR experiments in micelles showed that the TMD is essentially helical at the γ-sites but unfolded downstream of the ε-sites (19). NMR studies in the lipid-embedded state reported the region around the γ-sites to be fully helical in DOPG, whereas helical and nonhelical conformations were found in neuronal lipids (20). NMR studies also detected a flexible hinge at the diglycine motif G37G38 (C99 numbering, Fig. 1 A, (21)). Most recently, we systematically studied APP TMD helix dynamics by amide hydrogen/deuterium exchange (DHX) experiments and MD simulations (22,23). The results revealed helix fraying at both termini and corroborated the existence of the hinge where a helical dimerization domain (24) within the N-terminal half, denoted TM-N, connects to the TM-C helix that harbors the cleavage sites (22). This hinge has been suggested to play a role in positioning the substrate within the enzyme (21). A peptide harboring the cleavage region forms a helix whose flexibility does not exceed that of most tested TMDs whose amino acid composition is close to average (23). This was surprising given that a bioinformatic analysis had previously suggested low helix propensities around the cleavage sites due to an abundance of helix-destabilizing amino acids (25). Interestingly, the cleavage region contains two Thr residues and mutating them can cause aberrant substrate processing leading to familial early-onset Alzheimer’s disease (FAD) (17). The side chains of both Thr form H-bonds with main-chain carbonyls of the preceding helical turn as detected by NMR spectroscopy (21,26) and MD simulation (22).
Figure 1.
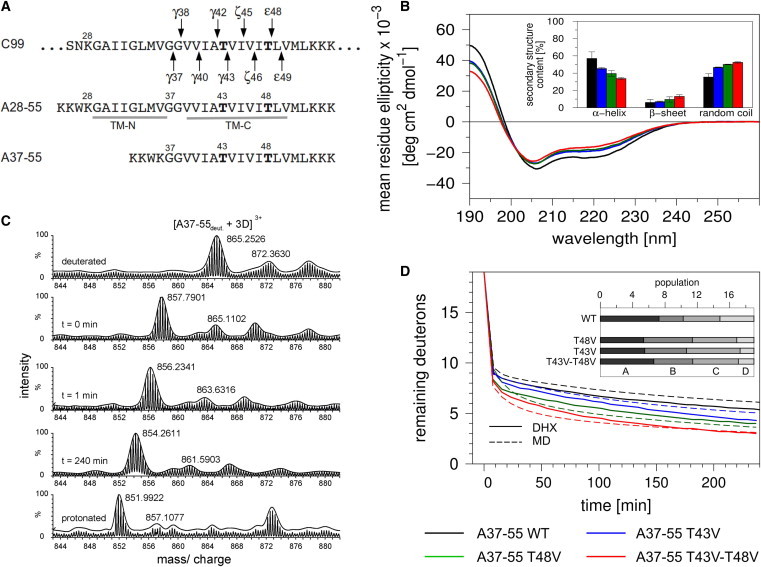
Experimental examination of APP TM-C secondary structure and backbone dynamics. (A) Sequences of the predicted APP TMD (C99 numbering, arrows indicate cleavage sites) and the model peptides used here. Mutated Thr at positions 43 and 48 are in bold. All peptides contain an additional N-terminal KKWK sequence for better solubility and photometric quantification. (B) Averaged CD spectra and calculated secondary structure contents (inset, n = 3–6, means ± SD) of A37-55 WT and mutant peptides. (C) Representative mass spectra of the triply charged WT A37-55 ion from different time points of a DHX experiment. (D) DHX kinetics (n = 3, SD ≤ 0.8 D for t < 5 min, and ≤ 0.25 D for the remainders of the kinetics; error bars not shown). The kinetics were fit with a maximum entropy method using D(t = 0 min) = 19 as the maximum number of hydrogen-bonded amide deuterons. The inset shows the sizes of the four kinetic subclasses of deuteron populations. The individual classes exhibit the following apparent rate constants: A (kDX > 60 h−1), B (kDX ≈ 12 h−1), C (kDX ≈ 0.3 h−1), and D (kDX < 0.1 h−1). Broken lines represent kinetics calculated from the corresponding MD trajectories with good correspondence to the experimental curves (WT, χ2 = 0.38; T43V, χ2 = 0.29; T48V, χ2 = 0.22; T43V/T48V, χ2 = 0.16). All experiments were carried out in TFE solution containing 20% water at pH = 5. To see this figure in color, go online.
Here, we examined the role of Thr residues in regulating the helix dynamics of the APP TMD. The comparison of wild-type (WT) and mutant TMD peptides by experimental work and MD simulation shows that side-chain/main-chain backbonding of Thr has dramatic impacts on the stability and the local dynamics of this helix. In addition, the mutations strongly affect the relative movements of TM-N and TM-C subhelices. Conceivably, this could alter presentation of the different cleavage sites at the catalytic site of presenilin.
Materials and Methods
Peptide synthesis
Peptides were synthesized by Fmoc chemistry by PSL, Heidelberg, Germany, and were >90% pure as judged by mass spectrometry.
Circular dichroism (CD) spectroscopy
Peptides were dissolved in 80% (v/v) 2,2,2-trifluoroethanol (TFE) and 10 mM NH4Ac, pH 5 at 30 μM. For each sample, 10 accumulated CD spectra from 185–260 nm were obtained using a Jasco J-710 CD spectrometer with a 0.1 data pitch, 1 s response, 100 nm/min scan velocity, 100 mdeg/cm sensitivity, and a path length of either 0.5 or 1 mm (27). Mean molar residue ellipticities were calculated based on peptide concentrations as determined by ultraviolet spectroscopy using an extinction coefficient 5,600 M-1cm−1. Secondary structure contents were estimated by deconvoluting the CD spectra using the program CDNN/PEPFIT that is based on peptide-derived reference spectra (28).
DHX experiments of synthetic peptides, electrospray ionization mass spectrometry (ESI-MS), and data evaluation
Solutions of deuterated peptide (100 μM in 80% (v/v) deuterated (d) TFE in 10 mM ND4Ac) were diluted 1:20 with protonated solvent (80% (v/v) TFE in 10 mM NH4Ac) to 5 μM final peptide concentration at 20°C. pH reading was corrected by adding 0.4 units to obtain pD = 5.0. Aliquots were taken after 0, 10 s, 20 s, and 30 s and quenched on ice and by adding 0.5% (v/v) formic acid, which lowered the pH to ∼2.6. After 40 s, mass/charge ratios were recorded in continuous mode by injecting reaction mixtures into the ion source of the mass spectrometer for 4 h (29). Mass spectra were acquired in positive-ion mode using a Waters Q-ToF Ultima with one scan/s and evaluated as described (27). For continuous measurements, five scans were accumulated and smoothed, and centered mass/charge ratios were obtained for intervals of 10 s. The triply charged isotope patterns were smoothed with the Savitzky-Golay algorithm with 25 measuring points and a data pitch of five. The numbers of remaining deuterons was calculated as described (27). The distributions of DHX rate constants were analyzed using the maximum entropy method (MEM) implemented in the program MemExp, version 4.0 (30,31) as described previously (23).
MD simulations, TrajectoryAnalysis, and calculation of DHX rates
The simulations were performed in 80% (v/v) TFE/water as described (23) using the CHARMM force field (32). Briefly, the monomer with initial ideal α-helical backbone conformation and charged termini was solvated in a rectangular solvent box (10.3 nm × 6.0 nm × 6.0 nm) containing ≈ 2340 water and ≈2340 TFE molecules and six neutralizing chloride ions. Peptide and solvent were equilibrated for 12 ns with gradual release of constraints on backbone atoms (1 ns with a force constant k = 5 kcal/(mol Å2), 1 ns with k = 2 kcal/(mol Å2), 10 ns without restraints) followed by 200 ns of free dynamics at constant temperature and pressure (T = 293 K, p = 0.1 MPa). The last 150 ns were used for analysis. All simulations were performed with the program NAMD (33). Structures were saved every ps for analysis. Analysis of H-bond populations, average lifetimes, autocorrelation functions, and global conformational parameters was carried out with homebuilt programs using routines provided with the CHARMM software version 35 (34) for extraction of the respective time series. Local conformational helix parameters (e.g., rise per residue) were calculated using the program HAXIS (35). An H-bond was counted as closed if the H…O distance is below 0.26 nm and the donor-H to acceptor- O angle is within 180° ± 60°. Average lifetimes of the intrahelical H-bonds are calculated from i), the number of frames where either the α-helical or the 310-helical H-bond is formed, and ii), the number of transitions from the closed to the open state (note that a switch between α and 310 H-bond was not counted as H-bond breaking).
To assess the convergence of the simulations, correlation times of the α-helical H-bonds were determined (see Fig. S1 in the Supporting Material). Correlation times in the range from 100 ps to 10 ns indicate sufficient sampling (36) within our 150 ns simulations. Statistical uncertainties were calculated by dividing the trajectories into nonoverlapping windows of 30 ns length and evaluating the SD of the mean values (66% confidence interval).
The calculation of DHX rates from MD simulations is based on i), the probability by which an amide H-bond opens to a distance larger than 0.3 nm, and ii), the local concentration of the exchange catalyst [OH−], which was calculated from the local water concentration around the amides, and iii), the chemical exchange rates of unfolded model peptides as described (23).
Three collective coordinates define the global helix dynamics: the bending angle Θ between the N-terminal and C-terminal part of the TMD, the rotation Φ of the TM-C with respect to TM-N, and the interhelical distance d measured as distance between the Cα atoms of residues 33 and 48. The geometrical definition of these coordinates is given in Fig. S2. To collect frames with similar global backbone conformation, clustering of the time series of these coordinates (cluster radius 1.6) was carried out using the ART-2′ algorithm (37) as implemented in CHARMM version 35 (34). To check for stable clustering, cluster sizes and cluster centers were calculated from time series with a randomized order of frames. Because the three patterns used for clustering have different ranges of values and different units, they were normalized to unit variance before clustering.
Results
We examined the contributions of side-chain/main-chain backbonding of the Thr residues at positions 43 and 48 of the APP cleavage region (Fig. 1 A) to the dynamics of the TMD helix. To this end, we compared peptides representing the complete TMD (A28-55) or the TM-C cleavage region (A37-55) to mutants in terms of stability and backbone dynamics. In the mutants, Thr was exchanged for Val, which largely maintains side-chain volume. All experiments and simulations were done in 80% (v/v) TFE in aqueous buffer to mimick the water-filled catalytic cleft of presenilin as done previously (22).
Stability and flexibility of the cleavage domain are restricted by threonine backbonding
Here, we investigated the cleavage region by experimental approaches. For this purpose, we preferred studying A37-55 over the complete TMD whose highly dynamic dimerization domain and hinge region (22) would mask subtle variations of the flexibility of the cleavage domain.
CD spectroscopy showed that the helicity of WT A37-55 is strongly reduced by T43V, T48V, and the T43V/T48V double mutation as indicated by the line shapes of the spectra and their quantitative evaluation. Decreased helicity is mainly compensated for by increased random coil content suggesting that our peptides exist in helix/coil equilibrium (Fig. 1 B). The flexibility of the helices was investigated by recording DHX kinetics, which is a powerful way to analyze the conformational equilibria along a protein helix because the extent and kinetics of successive exchange reports transient local amide H-bond opening (27,38,39). DHX of exhaustively (>95%) deuterated peptides was continuously monitored in 80% (v/v) TFE (pH = 5.0) by determining the molecular masses of the triply charged peptide ions using ESI-MS. The isotope envelopes gradually shifted toward lower mass/charge values with incubation time (Fig. 1 C). A gradual mass shift is diagnostic of uncorrelated exchange (40). Depending on the peptide, recording DHX kinetics for 4 h exchanged ∼13–16 deuterons of a total of 19 amides.
Interestingly, both single mutants, and in particular the T43V/T48V double mutant, exchange considerably faster than the WT (Fig. 1 D, full lines). The different local folding/unfolding equilibria along a helix give rise to deuterons exchanging with different rates. The apparent rates and the numbers of respective deuterons were recovered from the kinetics using a MEM (23,31). For all peptides, four kinetic classes can be distinguished, denoted A, B, C, and D in the order of decreasing rates. The flexibility of a helix is mainly reflected by the numbers of deuterons within these classes (that vary with the type of peptide much more than their apparent rate constants given in the legend of Fig. 1) (23). Accordingly, the faster amide exchange displayed by the mutant helices is mainly reflected by larger numbers of class B and C deuterons at the expense of the slower class C, i.e., amide deuterons are redistributed from slower to faster classes (Fig. 1 D, inset).
Taken together, these experiments clearly demonstrate a stabilizing effect of both Thr residues because mutating them results in lower helicity and faster amide exchange. We also assessed whether the mutations would increase the flexibility of the A37-55 helix beyond that of peptides representing natural TMDs (23). Fig. S3 shows that even unleashing the TM-C backbone dynamics by the T43V/T48V double mutation does not increase it above that of these natural TMDs.
Removing Thr backbonding distorts the TMD and alters bending motions
Here, we performed atomistic MD simulations of the WT and mutant helices to provide detailed insights into the impact of side-chain/main-chain backbonding on their structure and dynamics. Simulations were performed in 80% (v/v) TFE in water, i.e., a solvent matching our experimental conditions.
First, we studied the experimentally characterized isolated TM-C cleavage domain represented by A37-55. The MD simulations were validated by computing DHX kinetics from the fraction of open amide H-bonds and the local water concentration around the amides (23). A statistical comparison between MD-derived (Fig. 1 D, broken lines) and experimental kinetics (Fig. 1 D, full lines) shows close agreement between the backbone dynamics observed by the simulations and by the DHX experiment (χ2 <0.4).
In the case of the WT helix, calculation of block-averaged H-bond occupancies revealed that H-bonds are simultaneously donated by the Thr backbone amide and its side-chain hydroxyls to the i-4 carbonyl oxygens of the main chain in 94% (T43) or 97% (T48) of all frames, respectively. WT A37-55 forms ≥80% α-helical structure (i, i-4 bonding) from G38 to L49 (Fig. 2 A, broken lines). H-bonding that is characteristic of 310-helix formation (i, i-3 bonding) can substitute for reduced α-helical bonding and accounts for up to ∼15% of all H-bonds. The H-bond occupancies reveal a significant impact of the mutations because T43V and T48V strongly reduce amide H-bond occupancies at positions N-terminal from the mutated sites and the double mutant reduces α-helicity to <70% over an extended stretch from G37 to T43.
Figure 2.
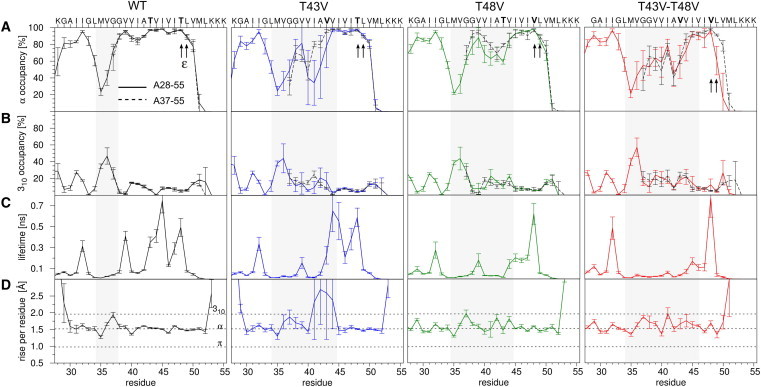
Local backbone dynamics of the TMD model peptides probed by MD simulations. (A) α-Helical amide H-bond occupancy as defined by the fraction of frames where the carbonyl oxygen at position i forms an intrahelical H-bond to the amide at position i+4 of A28-55 (continuous lines), or A37-55 (broken lines) peptide variants. (B) 310- Helical H-bond occupancy where H-bonds extend from O(i) to HN(i+3). (C) Average lifetimes of intrahelical H-bonds calculated from the numbers of frames where either the α- or the 310-helical H-bond is closed, and the numbers of closed-to-open switches during the simulation time. Note that a transition between α- and 310-bonding is not counted as H-bond breaking. (D) Rise-per-residue counted from Cα at i to Cα at i-1. Error bars correspond to the SD from block averaging using nonoverlapping time windows of 30 ns length. The shaded area indicates core regions, which deviate from ideal α-helical structure as defined by the rise-per-residue parameter. The ε-cleavage sites are marked with arrows. To see this figure in color, go online.
Second, we examined the complete A28-55 helix to investigate the movements of the TM-N helix relative to TM-C at the G37G38 hinge where 310-H-bonding predominates (Fig. 2, A and B). In a comparison of WT A28-55 (Fig. 2 A, solid lines) and A37-55 (broken lines) helices, the extent of Thr backbonding and amide H-bonding are similar for most parts of the helices as noted previously (23). A significant difference in α-helical occupancies between the long and short forms is only seen for T43V from V40 to T43. Overall, the helix-destabilizing impact of the Thr mutations is preserved with the full TMD. Specifically, all mutations decrease the occupancy of α-H-bonds near the hinge. The simulation of the A28-55 helix was previously validated by comparison with DHX kinetics (22).
The dynamics along the helix backbones relates to the average H-bond lifetimes. The amide H-bond lifetimes exhibit strong site-specificity and range from ∼20 to ∼800 ps with the WT helix. The T43V mutation reduces the lifetime of the V39-to-T43 amide H-bond by ∼60% (Fig. 2 C). Similarly, T48V reduces the V44-to-T48 amide H-bond lifetime by ∼80%. Open-times are much shorter (in the range from 5 to 50 ps) and vary much less than the lifetimes of the closed H-bonds along the core region of WT and mutant helices. Only H-bonds in the highly distorted turn around position 43 in the T43V mutant open for >10 ns (data not shown).
To evaluate the impact of the changes in H-bond strength on helix geometry, we compared the rise-per-residue, i.e., the distance between neighboring Cα atoms, that distinguishes α- (1.5 Å), 310- (2.0 Å), and π-helices (1.0 Å) (41). Furthermore, helix distortions (like kinks and twists) can be detected and quantified by their typical signatures (35). The shaded areas in Fig. 2 indicate non-α-helical regions thus defined. The rise-per-residue signature of the 34–38 regions reflects the G37G38 hinge, which is present in the WT helix and all mutants (Fig. 2 D). Compared to WT, the mutations induce additional distortions in the case of T48V and even lead to complete unfolding of the helix around position 43 in T43V. Remarkably, the helix distortions observed for the single mutants are not additive in the double mutant. The broadly increased helix flexibility seen for the double mutant (see Fig. 2, A and C), appears to counteract the local unfolding induced by T43V and results in more subtle conformational distortions along the whole TM-C region.
Local distortions can affect the global conformations and dynamics of helices. Therefore, we compared WT and mutant A28-55 in terms of three fundamental collective modes that define the movement of TM-C relative to TM-N: the bending angle Θ, the rotation angle Φ, and an arbitrarily chosen interhelical distance d measured between Cα atoms at positions 33 and 48 (see Fig. S2 for definitions). Clustering the trajectories according to these three collective coordinates (see Methods) leads to 8–11 clusters using a cluster radius of 1.6. Fig. 3 A shows representatives of the clusters, defined as those frames with the shortest distance to the respective cluster centers. The three most highly populated clusters collectively represent ∼50% of the frames for each type of TMD and are shown in color. The distributions of the collective modes are shown by the polar plots and boxes given in Fig. 3 B (note that the Cα atom of G33 chosen as the point of reference is located on the positive x axis and thus has the rotation angle 0°; the Cα atoms of G37 at the diglycine hinge are located at rotation angles of 320° ± 20°). Comparing these global helix parameters reveals dramatic impacts of the mutations on helix conformation. The WT helix conformers exhibit mostly moderate bending angles; the rotation angles locate mainly in the fourth quadrant, which indicates that bending is anisotropic and mainly over the hinge with G37G38 being located on the concave side of the bend. The T43V mutation strongly amplifies the bending motions that are mainly directed into the second quadrant (G37G38 on the convex side). More pronounced TM-C versus TM-N fluctuations of T43V relative to WT is also reflected by the broader Cα33–Cα48 distance distribution. A very different behavior is observed for T48V where the rotation angles indicate strong bending into the third quadrant. The double mutation induces over the hinge bending reminiscent of WT but with larger bending angles as well as a broader distribution of rotation angles and interhelical distances.
Figure 3.
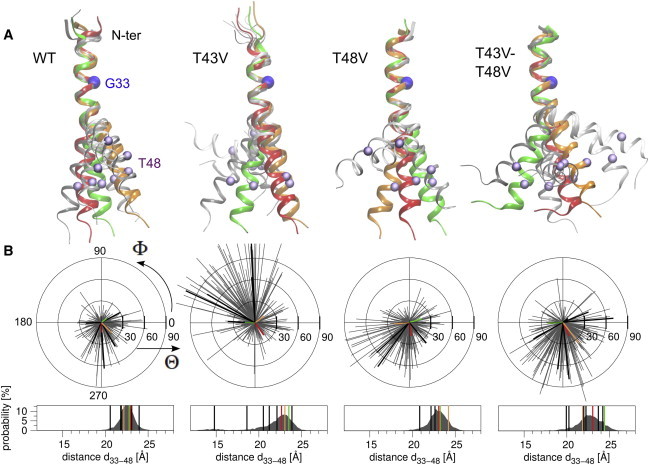
Global backbone dynamics of A28-55 helices. The movement of TM-C relative to TM-N is characterized by three collective coordinates: bending angle Θ, rotation angle Φ, and interhelical distance d (see Fig. S2 for definitions). To collect structures with similar conformations, the time series of these coordinates were clustered. (A) The overview shows representative helix conformations taken as the frame with the shortest distance to the centers of the respective cluster. The representatives of the three clusters with the highest populations are shown in red, orange, and green. These three clusters cover 50% of the backbone conformations sampled for each mutant. Cα atoms at position 33 (blue) and 48 (violet) are indicated as spheres. (B) Quantification of collective dynamics. The length of the gray lines in the polar plots represent bending angles scaled from 0° to 120°, the rotation is indicated as a polar angle from 0° to 360°. A rotation of 0° corresponds to bending in the direction of the Cα atom of G33. Shown are values for every 200th time step. The normalized distributions of the Cα33 to Cα48 distances d are shown in the boxes. The collective coordinates of the representatives of the three most highly populated clusters are indicated by colored lines, the values for the other clusters are shown as black lines. To see this figure in color, go online.
Discussion
Our results reveal a dramatic impact of side-chain/main-chain backbonding on the local and the global structure and dynamics of the APP TMD helix. This has implications for the general understanding of the impact of backbonding on the architecture of TMDs and for its role in intramembrane proteolysis in the case of the APP TMD.
Implications for understanding the impact of backbonding on TMD helix architecture
Mutating the Thr residues decreases the average helicity and accelerates DHX kinetics of peptides representing the TM-C cleavage region. In agreement with this, MD simulations show that these mutations reduce amide H-bonding within regions N-terminal of the mutated sites. The reductions in H-bonding correlate to reduced lifetimes of the closed state of these bonds, rather than to increased open state lifetimes. This indicates that removing Thr backbonding destabilizes the helix by facilitating its local unfolding. In addition to these changes in amide H-bonding, the removal of side-chain/main-chain backbonds increases the repertoire of potential movements of the TM-C cleavage region relative to the TM-N dimerization region, as indicated by increased bending angles and different directions of bending at the G37G38 hinge. The impacts of both mutations are very different: T43V causes local unfolding at the turn around the site of mutation, while only extended bending is seen with T48V. The observation that the double mutation reverses the effect of T43V suggests that the subtle distortions induced by T48V prevent local unfolding by T43V. It is apparent, therefore, that changes in local helix dynamics, as induced by removal of side-chain/main-chain backbonding, can translate into altered global dynamics suggesting a hierarchy of interconnected local and global fluctuations. Furthermore, our results support the notion that noncanonical conformations within TMD helices can be encoded intrahelically, rather than resulting from interhelical interactions (42). Extensive side-chain/main-chain backbonding within membrane-spanning helices has been noted before in crystallized polytopic membrane proteins (7,9) and simulated model helices (8,10–12). The drastic effects of removing the backbonds seen in this study may relate to the presence of the preexisting G37G38 hinge. It has been noted before that Ser and Thr backbonding can either increase or decrease a Pro-induced bend, depending on the precise sequence of the motif and the resulting local geometry (13). Furthermore, the previous simulations were done in hydrophobic environments that stabilize a helix (22). In contrast to that, our present work was done in a solvent of medium polarity, which includes 20% water due to reasons discussed in the next section.
Implications of backbonding for the cleavage of the APP TMD by presenilin
How could the dynamics of the APP TMD helix affect its intramembrane proteolysis? Upon recognition by presenilin or another subunit of γ-secretase, a substrate TMD must gain access to the active site. This may be associated with the loss of noncovalent bonds formed during initial encounter where the associated enthalpy loss may be compensated for by the entropy gain due to partial substrate unfolding during its repositioning within the enzyme (43). As a result, highly dynamic substrate TMDs may form a productive substrate/enzyme complex much faster than rigid ones. Cleavage of the APP TMD is believed to be initiated at alternative sites, termed ε48and ε49. This results in two distinct product lines along which subsequent proteolysis at alternate ζ- and γ-sites produces tri- and tetrapeptides until the most abundant Aβ40 as well as the less abundant Aβ42 and Aβ38 peptides are released (44). This cleavage pattern suggests a helical substrate, and it is believed that the preference of initial cleavage at ε48 over ε49 determines the Aβ40/Aβ42 ratio (15,16). Successive cleavage is likely to require the helix to slide past the catalytic aspartates in a process that may be facilitated by its flexibility. The cleavage reaction proper requires helix unfolding to allow formation of the tetrahedral intermediate between the scissile bond and catalytic residues (18). Because the catalytic cleft contains water (45), which can increase the dynamics of the bound substrate helix, we chose solvent-containing water to mimick this environment in our work; including TFE prevents peptide aggregation in our experimental assays (22).
Is there evidence that side-chain/main-chain backbonding affects substrate processing? It has been known for quite some time that mutations at T43 and T48 can change the Aβ42/Aβ40 ratio. For example, exchanging T43 to Ala, Phe, or Pro strongly increased the Aβ42/Aβ40 ratio, whereas a T43S mutation had only minor effects. Similarly, a T48F mutation favored Aβ42 production (46,47). This is consistent with the idea that abolishing Thr backbonding has a significant impact on the efficiency of proteolysis, whereas Ser backbonding maintains it. In FAD mutants, T43 is mutated by Ala or Ile and T48 is exchanged to Pro (reviewed in (17)), which results in increased Aβ42/Aβ40 ratios (48,49). In the case of T43I, the increased Aβ42/Aβ40 ratio has been related to a favored entry into the Aβ42 product line by preferential initial cleavage at ε48 at an overall reduced efficiency of ε-cleavage (50).
Our present results show that the Thr mutations tested do not significantly change the local dynamics at the ε-sites. Furthermore, ε-site dynamics does not exceed the dynamics at equivalent sites of some simulated nonsubstrate TMDs (23). We thus consider it unlikely that local helix dynamics controls ε-cleavage. In an alternative model, we propose that the bending and rotation of the enzyme-bound substrate TMD around the G37G38 hinge region could regulate the exposure of both ε-sites to the catalytic Asp residues. This, in turn, may define the efficiency and specificity of ε-cleavage that is seen to change after mutating Thr (48,49). In other words, removal of Thr backbonding could translate into altered accessibility of the initial cleavage sites to the active site of the enzyme. In addition, helix dynamics may affect subsequent proteolysis at ζ- and γ-sites as well as the kinetics of substrate release by affecting the kinetics of substrate movement within the enzyme after the initial ε-cut has been made. Therefore, side-chain/main-chain backbonding could influence the pattern of resulting cleavage products in multiple ways. Although we have chosen Thr-to-Val exchanges here to minimize the change in side-chain volume, determining the impact of FAD mutations on substrate helix dynamics will be an interesting subject of future investigations. It is clear that the substrate helix embedded within presenilin is likely to exhibit different structural properties compared to the free helix and that the conformations detected here for A28-55 and its mutants may not explore the full conformational space of these extremely dynamic helices. On the other hand, it has been noted before that the intrinsic dynamics of a protein explores the types of conformational changes that may occur upon binding to other proteins (51). Apart from affecting presentation of the scissile bond after binding to the enzyme, a recent MD simulation suggests that FAD mutations may also affect the vertical orientation, helix bending, and backbone hydration of the APP TMD, which could affect its recognition by γ-secretase (52).
Most recently, the rate of intramembrane proteolysis performed by rhomboids have been shown to be independent on substrate affinity and the kinetics of the actual cleavage reaction. Rather, the catalytic efficiency of rhomboids was proposed to depend on the exposure of the scissile bond (53). Conceivably, the catalytic efficiency of both types of intramembrane protease might be governed by the kinetics of substrate positioning, which depends on the global dynamics around the cleavage sites.
Acknowledgments
This work was supported by the Deutsche Forschungsgemeinschaft (LA699/14-1), the Bundesministerium für Forschung und Technologie (grant 01GI0724), the State of Bavaria, and the Center of Integrative Protein Science Munich (CIPSM). We also thank the Leibniz Supercomputing Centre, Garching, for computing time at the SuperMUC (project pr42ri).
Footnotes
Oxana Pester’s present address is Roche Diagnostics GmbH, Nonnenwald 2, 82377 Penzberg, Germany.
Daniel Hornburg’s present address is Max Planck Institute of Biochemistry, Am Klopferspitz 18, 82152 Martinsried, Germany.
Supporting Material
References
- 1.Curran A.R., Engelman D.M. Sequence motifs, polar interactions and conformational changes in helical membrane proteins. Curr. Opin. Struct. Biol. 2003;13:412–417. doi: 10.1016/s0959-440x(03)00102-7. [DOI] [PubMed] [Google Scholar]
- 2.Partridge A.W., Therien A.G., Deber C.M. Missense mutations in transmembrane domains of proteins: phenotypic propensity of polar residues for human disease. Proteins. 2004;54:648–656. doi: 10.1002/prot.10611. [DOI] [PubMed] [Google Scholar]
- 3.Joh N.H.J., Min A., Bowie J.U. Modest stabilization by most hydrogen-bonded side-chain interactions in membrane proteins. Nature. 2008;453:1266–1270. doi: 10.1038/nature06977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Senes A., Gerstein M., Engelman D.M. Statistical analysis of amino acid patterns in transmembrane helices: the GxxxG motif occurs frequently and in association with beta-branched residues at neighboring positions. J. Mol. Biol. 2000;296:921–936. doi: 10.1006/jmbi.1999.3488. [DOI] [PubMed] [Google Scholar]
- 5.Gray T.M., Matthews B.W. Intrahelical hydrogen bonding of serine, threonine and cysteine residues within alpha-helices and its relevance to membrane-bound proteins. J. Mol. Biol. 1984;175:75–81. doi: 10.1016/0022-2836(84)90446-7. [DOI] [PubMed] [Google Scholar]
- 6.McGregor M.J., Islam S.A., Sternberg M.J. Analysis of the relationship between side-chain conformation and secondary structure in globular proteins. J. Mol. Biol. 1987;198:295–310. doi: 10.1016/0022-2836(87)90314-7. [DOI] [PubMed] [Google Scholar]
- 7.Ballesteros J.A., Deupi X., Pardo L. Serine and threonine residues bend alpha-helices in the chi(1) = g(-) conformation. Biophys. J. 2000;79:2754–2760. doi: 10.1016/S0006-3495(00)76514-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ash W.L., Stockner T., Tieleman D.P. Computer modeling of polyleucine-based coiled coil dimers in a realistic membrane environment: insight into helix-helix interactions in membrane proteins. Biochemistry. 2004;43:9050–9060. doi: 10.1021/bi0494572. [DOI] [PubMed] [Google Scholar]
- 9.Chamberlain A.K., Lee Y., Bowie J.U. Snorkeling preferences foster an amino acid composition bias in transmembrane helices. J. Mol. Biol. 2004;339:471–479. doi: 10.1016/j.jmb.2004.03.072. [DOI] [PubMed] [Google Scholar]
- 10.Johansson A.C., Lindahl E. Amino-acid solvation structure in transmembrane helices from molecular dynamics simulations. Biophys. J. 2006;91:4450–4463. doi: 10.1529/biophysj.106.092767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Del Val C., White S.H., Bondar A.N. Ser/Thr motifs in transmembrane proteins: conservation patterns and effects on local protein structure and dynamics. J. Membr. Biol. 2012;245:717–730. doi: 10.1007/s00232-012-9452-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Deupi X., Olivella M., Pardo L. Influence of the g- conformation of Ser and Thr on the structure of transmembrane helices. J. Struct. Biol. 2010;169:116–123. doi: 10.1016/j.jsb.2009.09.009. [DOI] [PubMed] [Google Scholar]
- 13.Deupi X., Olivella M., Pardo L. Ser and Thr residues modulate the conformation of pro-kinked transmembrane alpha-helices. Biophys. J. 2004;86:105–115. doi: 10.1016/S0006-3495(04)74088-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Haass C., Selkoe D.J. Soluble protein oligomers in neurodegeneration: lessons from the Alzheimer’s amyloid beta-peptide. Nat. Rev. Mol. Cell Biol. 2007;8:101–112. doi: 10.1038/nrm2101. [DOI] [PubMed] [Google Scholar]
- 15.Lichtenthaler S.F., Haass C., Steiner H. Regulated intramembrane proteolysis—lessons from amyloid precursor protein processing. J. Neurochem. 2011;117:779–796. doi: 10.1111/j.1471-4159.2011.07248.x. [DOI] [PubMed] [Google Scholar]
- 16.Kaden D., Munter L.M., Multhaup G. The amyloid precursor protein and its homologues: structural and functional aspects of native and pathogenic oligomerization. Eur. J. Cell Biol. 2012;91:234–239. doi: 10.1016/j.ejcb.2011.01.017. [DOI] [PubMed] [Google Scholar]
- 17.Weggen S., Beher D. Molecular consequences of amyloid precursor protein and presenilin mutations causing autosomal-dominant Alzheimer’s disease. Alzheimer’s Res. Ther. 2012;4:9. doi: 10.1186/alzrt107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li M., Phylip L.H., Gustchina A. The aspartic proteinase from Saccharomyces cerevisiae folds its own inhibitor into a helix. Nat. Struct. Biol. 2000;7:113–117. doi: 10.1038/72378. [DOI] [PubMed] [Google Scholar]
- 19.Sato T., Tang T.C., Smith S.O. A helix-to-coil transition at the epsilon-cut site in the transmembrane dimer of the amyloid precursor protein is required for proteolysis. Proc. Natl. Acad. Sci. USA. 2009;106:1421–1426. doi: 10.1073/pnas.0812261106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lu J.X., Yau W.M., Tycko R. Evidence from solid-state NMR for nonhelical conformations in the transmembrane domain of the amyloid precursor protein. Biophys. J. 2011;100:711–719. doi: 10.1016/j.bpj.2010.12.3696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Barrett P.J., Song Y., Sanders C.R. The amyloid precursor protein has a flexible transmembrane domain and binds cholesterol. Science. 2012;336:1168–1171. doi: 10.1126/science.1219988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pester O., Barrett P.J., Langosch D. The backbone dynamics of the amyloid precursor protein transmembrane helix provides a rationale for the sequential cleavage mechanism of γ-secretase. J. Am. Chem. Soc. 2013;135:1317–1329. doi: 10.1021/ja3112093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pester O., Götz A., Langosch D. The cleavage domain of the amyloid precursor protein transmembrane helix does not exhibit above-average backbone dynamics. ChemBioChem. 2013;14:1943–1948. doi: 10.1002/cbic.201300322. [DOI] [PubMed] [Google Scholar]
- 24.Munter L.M., Voigt P., Multhaup G. GxxxG motifs within the amyloid precursor protein transmembrane sequence are critical for the etiology of Abeta42. EMBO J. 2007;26:1702–1712. doi: 10.1038/sj.emboj.7601616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Beel A.J., Sanders C.R. Substrate specificity of gamma-secretase and other intramembrane proteases. Cell. Mol. Life Sci. 2008;65:1311–1334. doi: 10.1007/s00018-008-7462-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nadezhdin K.D., Bocharova O.V., Arseniev A.S. Dimeric structure of transmembrane domain of amyloid precursor protein in micellar environment. FEBS Lett. 2012;586:1687–1692. doi: 10.1016/j.febslet.2012.04.062. [DOI] [PubMed] [Google Scholar]
- 27.Stelzer W., Poschner B.C., Langosch D. Sequence-specific conformational flexibility of SNARE transmembrane helices probed by hydrogen/deuterium exchange. Biophys. J. 2008;95:1326–1335. doi: 10.1529/biophysj.108.132928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Poschner B.C., Reed J., Hofmann M.W. An automated application for deconvolution of circular dichroism spectra of small peptides. Anal. Biochem. 2007;363:306–308. doi: 10.1016/j.ab.2007.01.021. [DOI] [PubMed] [Google Scholar]
- 29.Poschner B.C., Quint S., Langosch D. Sequence-specific conformational dynamics of model transmembrane domains determines their membrane fusogenic function. J. Mol. Biol. 2009;386:733–741. doi: 10.1016/j.jmb.2008.12.077. [DOI] [PubMed] [Google Scholar]
- 30.Steinbach P.J. Inferring lifetime distributions from kinetics by maximizing entropy using a bootstrapped model. J. Chem. Inf. Comput. Sci. 2002;42:1476–1478. doi: 10.1021/ci025551i. [DOI] [PubMed] [Google Scholar]
- 31.Steinbach P.J. Filtering artifacts from lifetime distributions when maximizing entropy using a bootstrapped model. Anal. Biochem. 2012;427:102–105. doi: 10.1016/j.ab.2012.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.MacKerell A.D., Bashford D., Karplus M. All-atom empirical potential for molecular modeling and dynamics studies of proteins. J. Phys. Chem. B. 1998;102:3586–3616. doi: 10.1021/jp973084f. [DOI] [PubMed] [Google Scholar]
- 33.Phillips J.C., Braun R., Schulten K. Scalable molecular dynamics with NAMD. J. Comp. Chem. 2005;26:1781–1802. doi: 10.1002/jcc.20289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Brooks B.R., Brooks C.L., 3rd, Karplus M. CHARMM: the biomolecular simulation program. J. Comput. Chem. 2009;30:1545–1614. doi: 10.1002/jcc.21287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Guo Z., Kraka E., Cremer D. Description of local and global shape properties of protein helices. J. Mol. Model. 2013;19:2901–2911. doi: 10.1007/s00894-013-1819-7. [DOI] [PubMed] [Google Scholar]
- 36.Allen M.P., Tildesley D.J. Oxford University Press; 1987. Computer Simulations of Liquids. [Google Scholar]
- 37.Karpen M.E., Tobias D.J., Brooks C.L., 3rd Statistical clustering techniques for the analysis of long molecular dynamics trajectories: analysis of 2.2-ns trajectories of YPGDV. Biochemistry. 1993;32:412–420. doi: 10.1021/bi00053a005. [DOI] [PubMed] [Google Scholar]
- 38.Bahar I., Wallqvist A., Jernigan R.L. Correlation between native-state hydrogen exchange and cooperative residue fluctuations from a simple model. Biochemistry. 1998;37:1067–1075. doi: 10.1021/bi9720641. [DOI] [PubMed] [Google Scholar]
- 39.Demmers J.A., Haverkamp J., Killian J.A. Electrospray ionization mass spectrometry as a tool to analyze hydrogen/deuterium exchange kinetics of transmembrane peptides in lipid bilayers. Proc. Natl. Acad. Sci. USA. 2000;97:3189–3194. doi: 10.1073/pnas.050444797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Xiao H., Hoerner J.K., Kaltashov I.A. Mapping protein energy landscapes with amide hydrogen exchange and mass spectrometry: I. A generalized model for a two-state protein and comparison with experiment. Protein Sci. 2005;14:543–557. doi: 10.1110/ps.041001705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Riek R.P., Rigoutsos I., Graham R.M. Non-alpha-helical elements modulate polytopic membrane protein architecture. J. Mol. Biol. 2001;306:349–362. doi: 10.1006/jmbi.2000.4402. [DOI] [PubMed] [Google Scholar]
- 42.Rigoutsos I., Riek P., Novotny J. Structural details (kinks and non-alpha conformations) in transmembrane helices are intrahelically determined and can be predicted by sequence pattern descriptors. Nucleic Acids Res. 2003;31:4625–4631. doi: 10.1093/nar/gkg639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Whitford P.C., Sanbonmatsu K.Y., Onuchic J.N. Biomolecular dynamics: order-disorder transitions and energy landscapes. Rep. Prog. Phys. 2012;75:076601. doi: 10.1088/0034-4885/75/7/076601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Fukumori A., Fluhrer R., Haass C. Three-amino acid spacing of presenilin endoproteolysis suggests a general stepwise cleavage of gamma-secretase-mediated intramembrane proteolysis. J. Neurosci. 2010;30:7853–7862. doi: 10.1523/JNEUROSCI.1443-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lazarov V.K., Fraering P.C., Li H. Electron microscopic structure of purified, active gamma-secretase reveals an aqueous intramembrane chamber and two pores. Proc. Natl. Acad. Sci. USA. 2006;103:6889–6894. doi: 10.1073/pnas.0602321103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lichtenthaler S.F., Ida N., Beyreuther K. Mutations in the transmembrane domain of APP altering gamma-secretase specificity. Biochemistry. 1997;36:15396–15403. doi: 10.1021/bi971071m. [DOI] [PubMed] [Google Scholar]
- 47.Lichtenthaler S.F., Wang R., Beyreuther K. Mechanism of the cleavage specificity of Alzheimer’s disease gamma-secretase identified by phenylalanine-scanning mutagenesis of the transmembrane domain of the amyloid precursor protein. Proc. Natl. Acad. Sci. USA. 1999;96:3053–3058. doi: 10.1073/pnas.96.6.3053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Page R.M., Gutsmiedl A., Steiner H. Beta-amyloid precursor protein mutants respond to gamma-secretase modulators. J. Biol. Chem. 2010;285:17798–17810. doi: 10.1074/jbc.M110.103283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Munter L.M., Botev A., Multhaup G. Aberrant amyloid precursor protein (APP) processing in hereditary forms of Alzheimer disease caused by APP familial Alzheimer disease mutations can be rescued by mutations in the APP GxxxG motif. J. Biol. Chem. 2010;285:21636–21643. doi: 10.1074/jbc.M109.088005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chávez-Gutiérrez L., Bammens L., De Strooper B. The mechanism of γ-secretase dysfunction in familial Alzheimer disease. EMBO J. 2012;31:2261–2274. doi: 10.1038/emboj.2012.79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tobi D., Bahar I. Structural changes involved in protein binding correlate with intrinsic motions of proteins in the unbound state. Proc. Natl. Acad. Sci. USA. 2005;102:18908–18913. doi: 10.1073/pnas.0507603102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Dimitrov M., Alattia J.-R., Fraering P.C. Alzheimer’s disease mutations in APP but not gamma-secretase modulators affect epsilon-cleavage-dependent AICD production. Nat. Commun. 2013;4:2246. doi: 10.1038/ncomms3246. [DOI] [PubMed] [Google Scholar]
- 53.Dickey S.W., Baker R.P., Urban S. Proteolysis inside the membrane is a rate-governed reaction not driven by substrate affinity. Cell. 2013;155:1270–1281. doi: 10.1016/j.cell.2013.10.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.