

Abstract
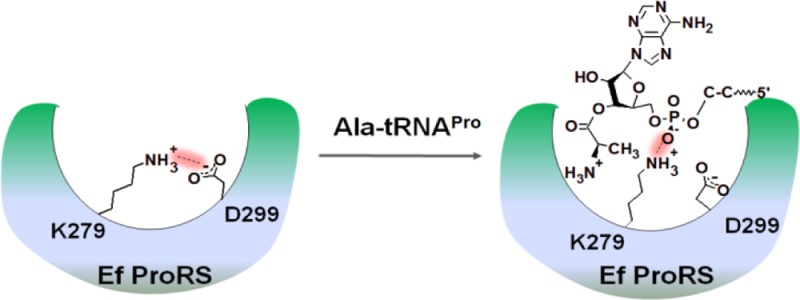
To ensure high fidelity in translation, many aminoacyl-tRNA synthetases, enzymes responsible for attaching specific amino acids to cognate tRNAs, require proof-reading mechanisms. Most bacterial prolyl-tRNA synthetases (ProRSs) misactivate alanine and employ a post-transfer editing mechanism to hydrolyze Ala-tRNAPro. This reaction occurs in a second catalytic site (INS) that is distinct from the synthetic active site. The 2′-OH of misacylated tRNAPro and several conserved residues in the Escherichia coli ProRS INS domain are directly involved in Ala-tRNAPro deacylation. Although mutation of the strictly conserved lysine 279 (K279) results in nearly complete loss of post-transfer editing activity, this residue does not directly participate in Ala-tRNAPro hydrolysis. We hypothesized that the role of K279 is to bind the phosphate backbone of the acceptor stem of misacylated tRNAPro and position it in the editing active site. To test this hypothesis, we carried out pKa, charge neutralization, and free-energy of binding calculations. Site-directed mutagenesis and kinetic studies were performed to verify the computational results. The calculations revealed a considerably higher pKa of K279 compared to an isolated lysine and showed that the protonated state of K279 is stabilized by the neighboring acidic residue. However, substitution of this acidic residue with a positively charged residue leads to a significant increase in Ala-tRNAPro hydrolysis, suggesting that enhancement in positive charge density in the vicinity of K279 favors tRNA binding. A charge-swapping experiment and free energy of binding calculations support the conclusion that the positive charge at position 279 is absolutely necessary for tRNA binding in the editing active site.
Aminoacyl-tRNA synthetases (AARSs) play an important role in the translation of the genetic code. AARSs catalyze the covalent attachment of amino acids to the 3′-end of their cognate tRNAs and are responsible for maintaining the fidelity of the aminoacylation process. The similar sizes and/or chemical structures of many amino acid side chains make their accurate discrimination by AARSs challenging. As a result, misactivation of noncognate amino acids and misacylation of tRNAs sometimes occur. Accuracy in the coupling of amino acids with their cognate tRNAs is achieved not only through substrate specificity but also through product editing. Approximately half of the 20 AARSs employ editing mechanisms for hydrolyzing misactivated amino acids (pretransfer editing) and misacylated tRNAs (post-transfer editing).1
Prolyl-tRNA synthetases (ProRSs) are class II synthetases that catalyze covalent attachment of proline to the 3′-end of tRNAPro. ProRSs from all three kingdoms of life are found to misactivate noncognate alanine and cysteine and form Ala-tRNAPro and Cys-tRNAPro.2,3 To prevent mistranslation of the proline codon, bacterial ProRSs have evolved pre- and/or post-transfer editing mechanisms. Previous studies of Escherichia coli (Ec) ProRS have demonstrated that the aminoacylation domain is the site of pretransfer editing,4 whereas an insertion domain located between the class II synthetase conserved motifs 2 and 3 (INS, ∼180 amino acids) is the post-transfer editing active site that hydrolyzes Ala-tRNAPro (Figure 1).5,6 In contrast, Cys-tRNAPro is deacylated in trans by a free-standing editing protein known as YbaK.7−9
Figure 1.
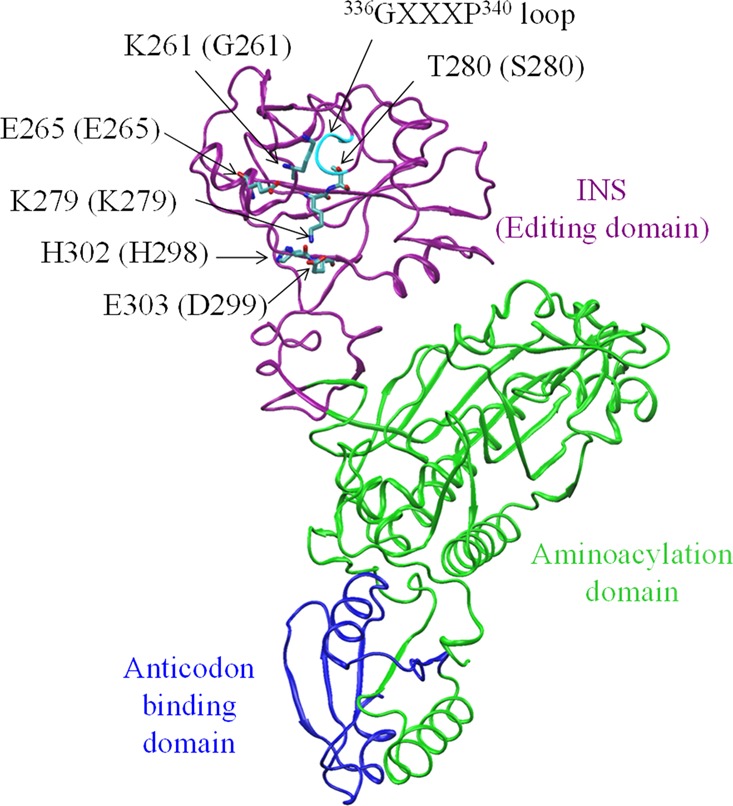
Ribbon representation of the three-dimensional structural model of the monomeric form of Ec ProRS. The homology model was derived from the X-ray crystal structure of Ef ProRS (PDB code: 2J3L).16 The catalytically important residues and the GXXXP motif of Ec ProRS are shown by licorice representation; the Ef ProRS residues are shown in parentheses.
Recent efforts have focused on the molecular mechanism of the post-transfer editing reaction catalyzed by INS homologues.10−12 Despite several studies, the role of lysine 279 (K279) of the INS domain, which is strictly conserved in bacterial ProRSs,5 has remained poorly understood. This residue is also strongly conserved among most of the INS-like superfamily of editing domains.12,13 Earlier experimental studies revealed that K279 is crucial for the post-transfer editing reaction.5 The substitution of K279 to alanine resulted in a post-transfer editing defective enzyme; only ∼5% activity was retained compared to the wild-type (WT) enzyme, suggesting a role in substrate binding and/or catalysis. The YbaK protein, which is a homologue of the INS domain, possesses a strictly conserved lysine that is analogous to K279 at position 46. The conservative change of K46R results in a ∼44-fold reduced Cys-tRNAPro deacylation activity as compared to WT YbaK.12 This observation suggested that the conserved lysine of INS and its homologues may play a role in catalysis.7 However, subsequent studies did not support the direct involvement of K279 of ProRS10 or K46 of YbaK14 in deacylation of aminoacyl-tRNAPro. Through extensive computational and experimental analysis, it was demonstrated that in the case of INS, the 2′-OH of the A76 ribose of tRNA activates a catalytic water molecule required for the hydrolysis of Ala-tRNAPro. The proton transfer from the catalytic water to O3′ of the substrate is facilitated by the backbone carbonyl of G261. The tetrahedral intermediate formed during hydrolytic cleavage is stabilized by the backbone amides of the conserved 331GXXXP335 loop.10
YbaK exploits the chemistry of its substrate cysteine sulfhydryl in a unique cyclization mechanism12 but shares some catalytic features with INS. In particular, YbaK catalysis also involves hydrogen-bonding interactions between the 2′-OH of A76 and a conserved GXXXP loop to stabilize reaction intermediates and products, and protein backbone atoms for proton transfer steps.14 Therefore, the reduced catalytic activity observed for the K46R YbaK protein is likely to originate from surface/electrostatic incompatibility due to delocalization of the positive charge over a larger area, which in turn affects the binding of misacylated Cys-tRNAPro. Although these studies rule out a direct catalytic involvement of K279 in the Ala-tRNAPro hydrolysis, the precise role of K279 in Ala-tRNAPro deacylation remains unanswered.
On the basis of previous experimental, substrate docking, and MD simulation results,10,14 we hypothesized that the role of the conserved lysine may be to anchor the phosphate backbone of the acceptor stem of misacylated tRNAPro and position it into the editing active site. However, the quantitative contribution of K279 to binding versus catalysis has been difficult to address experimentally. It is technically not feasible to prepare mischarged tRNAPro at high enough concentrations to derive KM by varying the misacylated tRNA concentrations. Moreover, the main tRNA binding determinant of Ec ProRS is the anticodon interaction, and therefore any contribution of K279 to binding/positioning of the 5′-CCA-Ala end of the tRNA is difficult to measure and quantify. Therefore, in this work, we have attempted to address the elusive role of K279 of bacterial ProRSs using computational approaches. In particular, the binding versus catalytic role of K279 was investigated by calculating its pKa. As previously observed by Sekimoto et al.,15 if a lysine is involved in catalysis as a general base that polarizes a catalytic water molecule, its pKa is expected to be lower than that of a free lysine (∼10.5). In contrast, a higher pKa value would stabilize the protonated state of this residue, which may facilitate substrate binding through electrostatic interaction. In addition to pKa calculations, charge-neutralization calculations were performed to evaluate the impact of neighboring polar residues on the protonation state of K279. We also probed the role of K279 in substrate binding by computing the Gibbs free energies for the WT and mutant ProRSs. Complementary experimental studies were also performed. Our results support the hypothesis that an electrostatic interaction between K279 and the tRNA phosphate backbone is critical for hydrolytic editing of misacylated tRNAPro.
Materials and Methods
E. coli and Enterococcus faecalis (Ef) ProRSs possess high sequence identity (48%). In particular, the active site residues within 6 Å of K279 are highly similar (93% similar and 50% identical) between these two enzymes. Because the X-ray crystallographic structure of Ec ProRS is not currently available and the accuracy of the computational methods used in this study depends on the accuracy of the structure, computational studies were performed with the X-ray crystallographic structure of Ef ProRS as a starting structure (PDB code: 2J3L).16 All experimental studies were performed with purified WT and mutant Ec ProRS (see below).
Computational Methods
The pKa of K279 (both in WT and D299A Ef ProRS, Figure 1) and K306 (as a control, Figure S1, Supporting Information) were computed using a single subunit of Ef ProRS (PDB code: 2J3L, chain B) in a substrate-free state. Separately, the binding affinity of the substrate analogue 5′-CCA-Ala was determined theoretically using the same subunit of Ef ProRS. A truncated INS editing domain was first generated that included all residues within 30 Å of K279. Free energy of binding was calculated using a previously reported model, where the substrate analogue 5′-CCA-Ala was docked into the isolated INS domain (residues 242–388) of Ef ProRS.10 The double mutant (K279D/D299K) was prepared by swapping the position of the two residues, K279 and D299, located in the INS domain. Combined quantum mechanical and molecular mechanical (QM/MM) calculations were used for the pKa calculation,17 while the binding free energies were computed using pure molecular mechanical potentials. Visualizations, manipulations, and analyses of the protein structure were carried out using the Visual Molecular Dynamics (VMD) program.18 All free energy calculations were performed with explicit solvent molecules using the CHARMM program suite.19 For all simulations, the all-atom CHARMM22 force field was used.20 The density functional theory (DFT) calculations were performed using the B3LYP functional and 6-31+g(d,p) basis set.21,22 All computations were carried out on an in-house server at the University of Wisconsin-Eau Claire Chemistry Department containing 11 nodes, each with 8 Intel Xeon E5430 processors.
Stochastic Boundary Setup of Molecular Systems
The centers of the simulated systems for the calculations of pKa and binding free energy were taken as the geometric average of the atoms of K279 and 5′-CCA-Ala, respectively. In both simulations, the protein segment of interest was submerged into a water box, and all water molecules beyond 30 Å from the center were deleted. This spherical, solvated active site was simulated using the stochastic boundary condition,23 the details of which have been described earlier.24,25 Briefly, atoms within 24 Å were treated with Newtonian dynamics, while Langevin’s dynamics was applied to the region 24–30 Å, where the frictional forces between the atoms were gradually increased toward the boundary. Water molecules were treated by the three-point-charge TIP3P model.26 All nonbonded interactions were truncated using a switching function between 12 and 14 Å, and the dielectric constant was kept at unity. The SHAKE algorithm27 was used to constrain the bond lengths and bond angles of the hydrogen atoms. A time step of 1 fs was used in the leapfrog Verlet algorithm for integration.28,29 For conformation sampling, MD simulations were performed using a constant number of particles, volume, and temperature (NVT) conditions producing a canonical ensemble. The Helmholtz free energy obtained using the NVT ensemble has been approximated to be equal to the Gibbs free energy in the present study. Finally, to keep the reaction zone intact, a deformable boundary potential corresponding to a 30 Å solvent sphere30 was applied to all solvent atoms in the system.
Calculation of pKa
The atoms of K279 (or K306, Figure S1, Supporting Information), located in the center of the solvated enzyme, were treated quantum mechanically using self-consistent charge density-functional tight-binding (SCC-DFTB) theory.31,32 All other atoms belonging to the enzyme and the solvent were simulated with molecular mechanics. The boundary between the QM and MM regions was treated with the link atom method,33 where one link atom was placed between the Cβ and the Cα atoms of the lysine.
The calculation of proton affinity in the condensed phase was performed using the thermodynamic diagram shown in Scheme 1. In the first step, the electrostatic interaction between the proton and the ε-amino nitrogen atom of the lysine was gradually reduced by converting the proton to an uncharged dummy atom (D) following the standard procedure of Kirkwood’s thermodynamic integration (TI) method, details of which have been described earlier. This provides the change in Gibbs free energy for the process (LysNH3+ → LysNH2···D) in Scheme 1. The second step is the conversion of the dummy atom to a gas-phase proton, and the change in Gibbs free energy of this process can be equated to the gas-phase standard Gibbs free energy of an isolated proton, ΔG°(g, H+) with a value of −6.28 kcal/mol.35 In the third step, the gas-phase proton is solvated and the Gibbs free energy is equal to the solvation free energy of a free proton, ΔGsolv°(H+), which is −264.0 kcal/mol.36
Scheme 1. Scheme for the pKa Calculation of Lys279 using Kirkwood’s Thermodynamic Integration Method.

The procedures for carrying out the first step of Scheme 1, that is, the computation of ΔGTI°(LysNH3+ → LysNH2···D), have been discussed earlier.25 Briefly, the proton is converted over sequential steps to a dummy atom with an effective charge reduction from +1 to 0. To study the response of the active site to the nonintegral charge on the proton, the enzyme system was simulated with a hybrid energy function (Uhybrid) derived by linearly combining the potential energies of the protonated (ULysNH3+) and the deprotonated (ULysNH2···D) states of the enzyme-bound lysine at different proportions. Mathematically, this is carried out using the coupling parameter, λ:
| 1 |
A total of 11 separate windows were used. In each window, an ensemble of conformations was generated by carrying out 500-ps MD simulations, each time perturbing the charge of the proton by a small amount. The partial derivative of the Gibbs free energy with respect to the coupling parameter (∂G/∂λ) was calculated from the ensemble-averaged potential energy difference in each window as described by Rauschnot et al.25 Finally, the free energy change, ΔGTI°(LysNH3+ → LysNH2···D), was computed by integrating (∂G/∂λ) over the coupling parameter (λ) from 0 to 1:
| 2 |
As illustrated in Scheme 1, the aqueous free energy of deprotonation of K279, ΔG°(aq), was obtained as the sum of the three quantities as in eq 3:
| 3 |
The above free energy change was corrected by including three correction terms in eq 4:
| 4 |
The first correction term represents Born’s correction to include the solvation effects beyond 30 Å (ΔGBorn).37 The second term accounts for the zero-point energy difference (ΔZPE) of the lysine for its protonated and deprotonated states. The last term is the high-level correction (HLC), which accounts for the difference between DFT and SCC-DFTB-computed energies for the gas-phase proton addition reaction of an isolated lysine molecule.
The pKa was calculated from the corrected aqueous free energy change, ΔGcorr°(aq), using the following equation:
| 5 |
where R and T are the universal gas constant (1.987 cal mol–1 K–1) and the temperature (298 K), respectively.
Charge-Neutralization Calculations
The impact of neighboring evolutionarily conserved polar residues on the protonation state of K279 was studied by examining their effects on the proton transfer equilibrium. To quantify the electrostatic impact of specific charged residues on the protonated state of the lysine, the change in the electrostatic component of the QM/MM interaction energy was calculated using eq 6:
| 6 |
where Eelecδ and Eelec are the interaction energies (between K279 and a specific charged residue) before and after the charge on the specific residue is abolished. Therefore, ΔEelec contains the net effect of the partial charges (indicated by δ) of all atoms of the specific charged residue on the protonated state of K279.
Relative Binding Free Energy Calculation
The relative binding free energy calculations were carried out using the molecular mechanics-based free energy perturbation (FEP) method.38,39 The solvated substrate-bound INS domain was equilibrated for 500 ps, after which 500 ps of further simulation was carried out. The binding free energy of a substrate, ΔGbind° (aq), can be computed by following the thermodynamic diagram39 in Scheme 2. The substrate is annihilated by converting the molecule to a dummy molecule, wherein atoms have no electrostatic, hydrogen bonding, or van der Waals interactions. Following Scheme 2, the free energy of binding can be written as
| 7 |
Scheme 2. A Thermodynamic Diagram for the Calculation of Gibbs Free Energy of Binding of the 5′-CCA-Ala Substrate to the INS Domain.

The first term, ΔGannih°(S) is the free energy change due to the annihilation of the substrate and was not calculated as it cancels out in the calculation of the relative binding free energy between the WT and the mutant ProRS (represented as MUT, hereafter). The second term is computed by employing Zwanzig’s FEP formulation38
| 8 |
where the UE...S and UE...D denote the potential energies for enzyme in the substrate-bound and substrate-annihilated states, respectively, kB is Boltzmann’s constant, and the quantity in ⟨ ⟩ indicates the ensemble-average computed on the enzyme bound state (E···S). The relative binding free energy of the WT and the mutant ProRS was computed from the difference between ΔGbind°(MUT, aq) and ΔGbind(WT, aq) using eq 9:
| 9 |
Experimental Methods
Materials
Proline and alanine (>99% pure) were obtained from Sigma. [γ-32P] ATP and [32P]PPi were from Perkin-Elmer, Shelton, CT. Primers for site-directed mutagenesis and PCR were purchased from Integrated DNA Technologies, Coralville, IA.
Enzyme Preparation
Overexpression and purification of WT and mutant Ec ProRS, tagged with six histidine residues (His6) at the N-terminus, were performed as described previously.40,41 Plasmids encoding E303A, E303D, E303K, K279E, and K279E/E303K mutant variants of Ec ProRS were generated by site-directed mutagenesis of pCS-M1S41 using primers listed in Table S1 (Supporting Information). Results of the mutagenesis were confirmed by DNA sequencing (University of Wisconsin, Biotechnology Center-Madison). Protein expression was induced in Ec SG13009 (pREP4) competent cells with 0.1 mM isopropyl β-d-thiogalactoside for 4 h at 37 °C. Histidine-tagged proteins were purified using a Talon cobalt affinity resin, and the desired protein was eluted with 100 mM imidazole. Protein concentrations were determined initially by the Bio-Rad Protein Assay (Bio-Rad Laboratories) followed by active-site titration.42
RNA Preparation
E. coli tRNAPro was transcribed using T7 RNA polymerase from BstN1-linearized plasmids as described43 and purified by denaturing 12% polyacrylamide gel electrophoresis.
ATP-PPi Exchange Assays
The ATP-PPi exchange assay was performed at 37 °C according to the published method.44 The concentration of proline was varied between 0.025 and 50 mM, and 10 nM WT and 20 nM mutant enzyme concentrations were used for proline activation. Kinetic parameters were determined from Lineweaver–Burk plots and represent the average of at least three determinations.
ATP Hydrolysis Assays
ATP hydrolysis reactions to monitor pretransfer editing were carried out as described previously.4 The alanine concentration used was 500 mM and the reactions were initiated with a final ProRS concentration of 0.5 μM.
Aminoacylation Assays
Aminoacylation assays were performed under standard conditions45−47 with 0.5 μM tRNAPro, 3 μM [3H] proline, 20 μM proline, and 100 nM ProRS.
Aminoacylated tRNA
Aminoacylated tRNA for use in deacylation assays was prepared at room temperature according to published conditions.2E. coli alanyl-tRNA synthetase (8 μM) was used to acylate G1:C72/U70 tRNAPro (8 μM) in the presence of [14C]Ala (∼250 μM) in buffer containing 50 mM HEPES (pH 7.5), 4 mM ATP, 25 mM MgCl2, 20 mM β-mercaptoethanol, 20 mM KCl, and 0.1 mg/mL bovine serum albumin.
Deacylation Assays
Deacylation assays were carried out at room temperature according to published conditions2 with slight modifications. Reactions contained about 1 μM G1:C72/U70 [14C]-Ala-tRNAPro, 50 mM HEPES (pH 7.5), 5 mM MgCl2, and were initiated with 0.5 μM ProRS. Background deacylation observed in a reaction lacking ProRS was subtracted from each reaction.
Results
Computational Results
Theoretical pKa Calculation for Lysine
The plot of Gibbs free energy change for the K279 of WT Ef ProRS as a function of the coupling parameter λ is shown in Figure 2. The high correlation coefficient (>0.99) is consistent with the assumption of linear variation of energy upon gradual perturbation of the site due to deprotonation. The Gibbs free energy of the process of converting the lysine proton to a dummy atom, ΔGTI°(LysNH3+ → LysNH2···D), was computed using integration of the linear function from λ = 0 to 1 (eq 2). The corrected Gibbs free energy of deprotonation, ΔGcorr(aq), is obtained using eqs 3 and 4. The computed pKa (eq 5) of the ε-amino group of K279 is 14.2 (Table 1), which is ∼3 units higher than the pKa of an isolated lysine (10.53). Thus, the proton on the ε-amino group of K279 in INS is more difficult to deprotonate compared to that of an isolated lysine. We hypothesize that the elevated pKa of K279 originates from salt-bridge formation with the adjacent acidic residue, D299. This is supported by the computed pKa of the same lysine in a D299A mutant, which was found to be 11.0; this value is close to the experimental value of 10.53 for a free lysine. To validate the computational protocol, the pKa of a lysine located on the surface (K306, Figure S1) was also calculated and was found to be 11.3, which is only slightly above the pKa of a free lysine (Table 1).
Figure 2.

A plot of the partial derivative of Gibbs free energy with respect to the coupling parameter λ, (∂G/∂λ), as a function of the coupling parameter, λ.
Table 1. Decomposition of the Components of the Gibbs Free Energy of Deprotonation of K279 and K306 for Various Ef ProRS Systemsa.
| free energy (kcal/mol) |
|||
|---|---|---|---|
| components | K279 (WT) | K279 (D299A) | K306 (WT) |
| ΔGTI°(LysNH3+ → LysNH2···D)b | 291.1 | 286.8 | 287.2 |
| ΔG°(g, H+)c | –6.28 | –6.28 | –6.28 |
| ΔGsolv°(H+)d | –264.0 | –264.0 | –264.0 |
| ΔGBorne | –5.5 | –5.5 | –5.5 |
| ΔZPEf | –9.4 | –9.4 | –9.4 |
| HLCg | 13.3 | 13.3 | 13.3 |
| ΔGcorr°(aq) | 19.3 | 15.0 | 15.4 |
| pKa | 14.2 | 11.0 | 11.3 |
ΔGTI°(LysNH3+ → LysNH2···D) is the Gibbs free energy computed using SCC-DFTB following the formalism of thermodynamic integration.34
ΔG°(g,H+) is the reported value of the standard Gibbs free energy of a proton the in gas phase.35
ΔGsolv°(H+) is the standard Gibbs free energy of solvation of a proton obtained from the theoretical study by Kelley et al.36
ΔGBorn is Born’s correction in the Gibbs free energy to account for the solvation effect beyond 30 Å, which is the limit of our spherical boundary.
ΔZPE is the difference in the zero-point vibrational energies of the protonated and deprotonated state of the lysine calculated using DFT.
HLC is equal to the difference between SCC-DFTB and DFT computed Born–Oppenheimer energies for the gas-phase proton addition reaction of an isolated lysine molecule.
The Gibbs free energy of deprotonation was decomposed into its various components, and the values are given in Table 1. It appears that the zero-point correction for the protonated and deprotonated states accounts for ∼9 kcal/mol of energy. Also, the high-level correction indicates that the SCC-DFTB-computed gas-phase Born–Oppenheimer energies for the deprotonation reaction of the lysine molecule is underestimated by a significant amount (13 kcal/mol) compared to those obtained using DFT.
The charge neutralization calculations were performed to determine the effect of neighboring residues on the protonation state of K279. The results suggest that the polar residues H298 and D299 of Ef ProRS (Figure 3a), which correspond to H302 and E303 of Ec ProRS (Figure 3b), stabilize the protonated state of K279. D299 forms a salt bridge with K279 (Figure 3a), and as indicated in Figure 4 neutralization of this negative charge has the most significant impact of all the residues tested on the interaction energy (ΔEelec) between K279 and D299. The 15 kcal/mol higher value of ΔEelec indicates a significant destabilizing effect of charge neutralization.
Figure 3.

(a) The highly conserved K279 and the surrounding polar residues are shown in the editing active site of Ef ProRS. (b) Sequence alignment of the relevant portion of INS domain from Ec and Ef ProRS.
Figure 4.

Change in the QM/MM interaction energy due to charge removal of neighboring polar residues of K279, averaged over 100 conformations.
Relative Binding Free Energy Calculation
To examine if binding of the backbone of the tRNA acceptor stem to INS is favored by the lysine at position 279, the free energy of binding of 5′-CCA-Ala to the editing domain was compared between WT INS and the ProRS mutants. For the charge-swapped double mutant, K279D/D299K, the relative binding free energy, ΔΔGbind(aq), was computed to be 12.8 kcal/mol (Table 2). The positive ΔΔGbind(aq) value indicates less favorable binding of 5′-CCA-Ala to the editing domain of the double mutant. A severe reduction in binding affinity was also observed for the K279E variant (ΔΔGbind(aq) = 33.7 kcal/mol). In contrast, an enhanced binding affinity for the 5′-CCA-Ala substrate was observed for the D299K variant (ΔΔGbind(aq) = −39.6 kcal/mol). This observed trend indicates that the presence of lysine at position 279 is essential for the binding of misacylated tRNA to the INS. Visualization of the substrate-bound structures (Figure 5) revealed that the 5′-CCA-Ala substrate is positioned farther away from the lysine in the charge-swapped double mutant. In particular, the nonbridging oxygen of the phosphate anion of the terminal A76 of 5′-CCA-Ala is located ∼10.5 Å from the ε-amino group of K299 in the double mutant, whereas the distance between the same nonbridging oxygen of the phosphate anion and the ε-amino group of K279 is ∼5.0 Å in the case of WT INS. In contrast, a much shorter distance was observed between the nonbridging phosphate and ε-amino nitrogen of K279 in the case of D299K variant (2.7 Å, figure not shown). The presence of an additional positive charge at position 299 caused an increased electrostatic interaction between the phosphate group of the terminal A76 and K279.
Table 2. Relative Gibbs Free Energy of Binding of 5′-CCA-Ala to the INS Domain of WT and Mutant Ef ProRS, Obtained Using FEP Calculations.
| energy (kcal/mol) |
||
|---|---|---|
| Ef ProRS | ΔGFEP°(WT···D → WT···S) | ΔΔGbind° (aq) |
| WT | –415.4 | |
| K279D/D299K | –402.6 | 12.8 |
| K279E | –381.7 | 33.7 |
| D299K | –455.0 | –39.6 |
Figure 5.
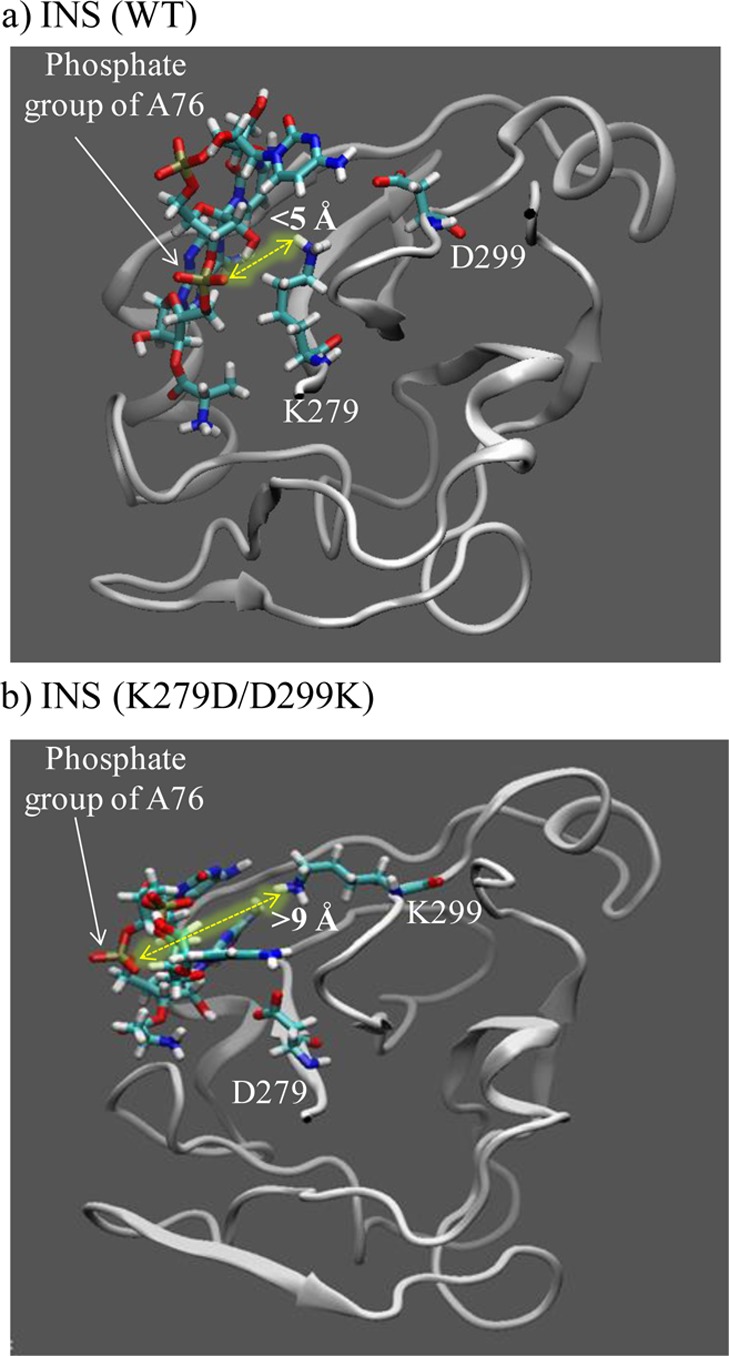
Altered conformation of the 5′-CCA-Ala bound to the INS double mutant (b) compared to WT Ef ProRS (a). The protein segment is shown in new cartoon representation, whereas the 5′-CCA-Ala substrate is represented in licorice.
Experimental Results
To evaluate the computational findings, we have purified mutants of Ec ProRS proteins wherein K279 and E303 were substituted with other polar or neutral residues. E303 is the equivalent of D299 in Ef ProRS. Kinetic studies were performed to analyze the enzymatic activities of the WT enzyme and all ProRS variants (vide infra).
Amino Acid Activation
To examine the impact of mutation of E303 on the function of Ec ProRS, this residue was substituted with alanine, aspartic acid, and lysine. Using the ATP-PPi exchange reaction, we found that substitution of residue 303 with neutral, negatively charged, or positively charged residues result in only a minor 2–4-fold decrease in proline activation (Table 3). Similarly, only ∼2-fold reduced activity was observed for the K279E variant and the K279E/E303K double mutant.
Table 3. Kinetic Parameters for Proline Activation by WT and Mutant Ec ProRSa.
| Ec ProRS | kcat (s–1) | KM (mM) | kcat/KM (mM–1 s–1) | relative kcat/KM | fold decrease |
|---|---|---|---|---|---|
| WTb | 12.6 ± 4.9 | 0.18 ± 0.03 | 71 | 1.0 | |
| E303A | 5.2 ± 0.6 | 0.216 ± 0.004 | 24 | 0.34 | 3.0 |
| E303D | 4.6 ± 0.5 | 0.199 ± 0.005 | 23 | 0.32 | 3.1 |
| E303K | 5.1 ± 1.5 | 0.30 ± 0.03 | 17 | 0.24 | 4.2 |
| K279E | 5.7 ± 0.9 | 0.22 ± 0.04 | 26 | 0.37 | 2.7 |
| K279E/E303K | 5.9 ± 0.9 | 0.19 ± 0.01 | 31 | 0.44 | 2.3 |
Results are the average of three trials with the standard deviation indicated.
Data is from ref (47).
Aminoacylation Activity
We next determined the effect of mutation of E303 and K279 on aminoacylation activity using conditions where the initial rate of the reaction was proportional to kcat/KM. A small decrease in the aminoacylation efficiency for the E303 variants was observed compared to WT ProRS (Figure 6a). The aminoacylation activity of the K279E variant and the K279E/E303K double mutant was essentially the same as WT ProRS (Figure 6b).
Figure 6.

Initial rates of aminoacylation of tRNAPro with proline by WT and the five variants of Ec ProRS. For clarity, the results are presented in two panels, (a) and (b). The assays were performed at room temperature with 0.5 μM tRNAPro and 100 nM Ec ProRS. Linear fits of the data are shown.
Pretransfer Editing Activity
We also investigated the pretransfer editing activity for all variants by monitoring the hydrolysis of the misacylated alanyl-adenylate (Ala-AMP). For the five variants (K279E, E303A, E303D, E303K, and K279E/E303K) of Ec ProRS, an approximately 20–40% reduction in pretransfer editing activity was observed relative to WT ProRS (Figure 7).
Figure 7.

Relative pretransfer editing activity of WT and mutant Ec ProRS. The assay was performed at 37 °C using 4 μM ProRS and 500 mM alanine. Results are reported as percent activity relative to WT, which was set to 100%.
Post-Transfer Editing Activity
The impact of mutation of K279 and E303 on post-transfer editing was investigated by monitoring hydrolysis of mischarged Ala-tRNAPro. A severe impact on the post-transfer editing activity (∼25-fold) was observed for the K279E mutant (Figure 8). In addition, the deacylation activity of the charge-swapped K279E/E303K double mutant was undetectable (>50-fold decrease). Thus, even though they interact via a salt bridge, swapping the positions of the two residues does not restore activity. These results are consistent with the computational results showing a significant decrease in the free energy of binding of 5′-CCA-Ala to the editing domain of the K279D/D299K variant compared to the WT enzyme (Table 2). Interestingly, the E303K variant was found to be significantly more active in hydrolyzing Ala-tRNAPro compared to the WT enzyme. Slightly enhanced post-transfer editing activity was also observed for E303A ProRS. On the other hand, no significant change in activity was observed for the conservative E303D substitution variant (Figure 8).
Figure 8.

Deacylation of Ala-tRNAPro by WT and mutant Ec ProRS. The assays were performed at room temperature with 1 μM G1:C72/U70 [14C]Ala-tRNAPro and 0.5 μM Ec ProRS.
Discussion
Lysine 279 plays a crucial role in post-transfer editing activity by the INS domain of Ec ProRS, and analogous lysine residues are found in the majority of INS-like editing domain homologues.13 Recent experimental and computational studies have revealed no direct involvement of this residue in catalysis.10,11,14 The docked structure of 5′-CCA-Ala bound to INS indicates that the role of K279 may be limited to substrate binding.11 Indeed, experiments performed with high concentrations of K279A ProRS (5–50 μM) showed significant levels of deacylation of Ala-tRNAPro, which supports a role for K279 in binding (data not shown). However, there are technical challenges in quantifying the binding versus catalytic role of K279. Binding studies of WT versus K279E/E303K ProRS were carried out using a fluorescently labeled semisynthetic two-piece tRNA, but very similar affinities were measured in each case (data not shown). This is most likely due to the major role of the anticodon interaction in tRNAPro binding.16,48,49 To overcome these experimental obstructions, in the present study computational approaches were employed to address the binding versus catalytic role of the highly conserved K279.
Protein electrostatics is well-known to play an important role in protein structure and function, and many functionally important amino acid residues exhibit an altered pKa relative to that of the free amino acid. For example, pKa perturbations have been observed for amino acid residues that contribute to ligand binding, as well as to protein–protein interactions.50,51 To explore the precise role of the highly conserved and functionally critical K279 residue present in the INS domain of bacterial ProRS and free-standing editing domain homologues, we computed its pKa in various environments. The high pKa value of 14 calculated for the ε-amino group of K279 in WT ProRS is due to the presence of neighboring polar residues, especially due to the salt-bridge with D299. This is evident from the three-unit decrease in the pKa of K279 in the presence of the secondary D299A mutation (Table 1). Aspartate 299 is moderately conserved in bacterial ProRSs. Analysis of the multiple sequence alignment of 492 ProRS sequences showed 65% conservation of D/E at that position. Earlier NMR studies have revealed that the pKa of a carboxylate side chain of aspartate/glutamate decreases because of salt bridge interactions involving basic residues such as arginine and lysine.52,53 Similarly, the pKa of lysine is expected to increase in the presence of a neighboring acidic residue because the positive charge on the ε-amino nitrogen atom is stabilized by the salt bridge interaction. A computational charge neutralization study indeed demonstrated that the protonated state of K279 in Ef ProRS is significantly stabilized by D299; destabilization of the system was evident upon neutralization of this closely located (within 4 Å) acidic residue and to a lesser extent other surrounding polar residues (Figure 4). As discussed in Sekimoto et al.,15 the high pKa of K279 suggested its role in substrate binding by favoring a strong interaction with a phosphate oxygen of the tRNA backbone.
As expected, kinetic studies show that site-directed substitution of E303 and K279 has little impact on enzyme functions that occur in the aminoacylation active site pocket, that is, amino acid activation, aminoacylation, and pretransfer editing reactions. In contrast, a more significant impact was observed in the post-transfer editing reaction. Earlier, it was shown that K279A ProRS is severely defective in hydrolyzing Ala-tRNAPro.5 In addition, the analogous lysine residue in the free-standing YbaK editing domain (K46) is critical for deacylation of Cys-tRNAPro.12 Of all the YbaK variants examined, K46A was the most deleterious but did not result in a complete loss in activity (∼66-fold). The data were consistent with a role in positioning of the misaminoacylated tRNAPro substrate in the active site.12 In the present study, we observed that the K279E Ec ProRS variant is also severely defective in post-transfer editing. The fact that the double mutation K279E/E303K, which is expected to restore a salt bridge between positions 279 and 303, failed to restore hydrolysis activity supports our claim that the presence of a positively charged residue at position 279 is essential for Ala-tRNAPro deacylation. A computational study of the free energy of binding of 5′-CCA-Ala revealed tighter binding of the substrate to the WT INS domain relative to the double mutant. Thus, the combined computational and experimental results support the conclusion that K279 is involved in binding and positioning the misacylated tRNAPro into the post-transfer editing active site. Although a K279R ProRS variant was not tested, a previous study of Haemophilus influenzae YbaK showed that even a conservative K46R change resulted in a large decrease in editing activity (∼44-fold).12
Although the charge neutralization calculations with Ef ProRS revealed stabilization of K279 by the closely positioned D299 (E303 of Ec ProRS), enhancement in post-transfer editing efficiency was observed for both E303K and E303A variants relative to the WT enzyme. This finding suggests that the presence of a positively charged or neutral residue at position 303 of Ec ProRS enhances the positive charge density at position 279, which is likely to increase the binding affinity of K279 for the backbone of the tRNA acceptor stem. These experimental and computational findings suggest that the role of the neighboring glutamate is not only limited to stabilizing the protonated K279 residue. E303 also appears to modulate the positive charge density of the active site that is used to anchor the phosphate backbone of the substrate during catalysis. The salt bridge between K279 and E303 helps to maintain an optimum interaction between the mischarged tRNA and K279. The increase in positive charge density due to the substitution of E303 with a neutral or basic residue, which likely results in tighter binding of the misacylated tRNA, may slow product release.
Conclusions
Previous mutational studies suggested that K279 is important for Ala-tRNAPro deacylation by the bacterial ProRS INS domain, and the analogous lysine is critical for Cys-tRNAPro deacylation by the homologous single-domain YbaK protein. Recent computational studies of ProRS and YbaK did not reveal any active participation of this residue in catalysis. On the basis of both experimental and computational results, it has been speculated that the role of this strictly conserved basic residue in prokaryotic-like ProRSs and related editing domains is to bind the misacylated tRNA substrate and position it in the editing active site. In the present study, pKa and free energy of binding calculations, as well as charge-swapping experiments, are consistent with a role for K279 in binding the phosphate group of misacylated tRNAPro. A general role for lysine in substrate binding through electrostatic interactions has been observed in many enzymes. For example, the conserved histidine residues in the active site of bovine pancreatic ribonuclease are actively involved in catalytic function, while the active site lysine serves only to bind the phosphate anion.54 Taken together, the combined computational and experimental studies reported here strongly support the role of K279 in substrate binding but not in catalysis. Interestingly, one member of the INS superfamily of editing domains, ProXp-z, does not have a conserved lysine in the analogous position to K279 but instead has a conserved asparagine residue.13 Future studies will address the activity of this editing domain homologue and the role of the asparagine residue in catalytic function and substrate binding.
Acknowledgments
We gratefully acknowledge the computational support from the Learning and Technology Services, University of Wisconsin-Eau Claire. We would also like to thank Ms. Irene C. Mueller and Mr. Mathew J. Tschudy for their contribution in purifying some of the Ec ProRS mutant proteins.
Glossary
Abbreviations Used
- AARS
aminoacyl-tRNA synthetase
- Ec
Escherichia coli
- Ef
Enterococcus faecium
- DFT
density functional theory
- FEP
free energy perturbation
- HLC
high-level correction
- INS
insertion domain
- MD
molecular dynamics
- NVT
constant particle, volume and temperature
- ProRS
prolyl-tRNA synthetase
- QM/MM
quantum mechanical/molecular mechanical
- SCC-DFTB
self-consistent charge density-functional tight-binding
- TI
thermodynamic integration
- WT
wild-type
- ZPE
zero-point energy
Supporting Information Available
A table containing the list of primers used in this study and a figure showing the three-dimensional structure of Ef ProRS (PDB code: 2J3L)16 are presented as supporting materials. Figure S1 shows the two lysine residues (K279 and K306) used in the pKa calculations. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
This work was supported in part by National Institute of Health [Grant Numbers GM049928 (K.M.-F.) and GM085779 (S.H.)] and by the Office of Research and Sponsored Programs of the University of Wisconsin-Eau Claire, Eau Claire, WI.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Mascarenhas A., Martinis S. A., An S., Rosen A. E., and Musier-Forsyth K. (2009) Fidelity Mechanisms of the Aminoacyl-tRNA Synthetases, in Protein Engineering (L R. U., and C K., Eds.) pp 153–200, Springer Verlag, New York. [Google Scholar]
- Beuning P. J.; Musier-Forsyth K. (2000) Hydrolytic editing by a class II aminoacyl-tRNA synthetase. Proc. Natl. Acad. Sci. U. S. A. 97, 8916–8920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahel I.; Stathopoulos C.; Ambrogelly A.; Sauerwald A.; Toogood H.; Hartsch T.; Soll D. (2002) Cysteine activation is an inherent in vitro property of prolyl-tRNA synthetases. J. Biol. Chem. 277, 34743–34748. [DOI] [PubMed] [Google Scholar]
- Hati S.; Ziervogel B.; Sternjohn J.; Wong F. C.; Nagan M. C.; Rosen A. E.; Siliciano P. G.; Chihade J. W.; Musier-Forsyth K. (2006) Pre-transfer editing by class II prolyl-tRNA synthetase: role of aminoacylation active site in “selective release” of noncognate amino acids. J. Biol. Chem. 281, 27862–27872. [DOI] [PubMed] [Google Scholar]
- Wong F. C.; Beuning P. J.; Nagan M.; Shiba K.; Musier-Forsyth K. (2002) Functional role of the prokaryotic proline-tRNA synthetase insertion domain in amino acid editing. Biochemistry 41, 7108–7115. [DOI] [PubMed] [Google Scholar]
- Wong F. C.; Beuning P. J.; Silvers C.; Musier-Forsyth K. (2003) An isolated class II aminoacyl-tRNA synthetase insertion domain is functional in amino acid editing. J. Biol. Chem. 278, 52857–52864. [DOI] [PubMed] [Google Scholar]
- An S.; Musier-Forsyth K. (2004) Trans-editing of Cys-tRNAPro by Haemophilus influenzae YbaK protein. J. Biol. Chem. 279, 42359–42362. [DOI] [PubMed] [Google Scholar]
- An S.; Musier-Forsyth K. (2005) Cys-tRNA(Pro) editing by Haemophilus influenzae YbaK via a novel synthetase.YbaK.tRNA ternary complex. J. Biol. Chem. 280, 34465–34472. [DOI] [PubMed] [Google Scholar]
- Ruan B.; Soll D. (2005) The bacterial YbaK protein is a Cys-tRNAPro and Cys-tRNA Cys deacylase. J. Biol. Chem. 280, 25887–25891. [DOI] [PubMed] [Google Scholar]
- Kumar S.; Das M.; Hadad C. M.; Musier-Forsyth K. (2012) Substrate and enzyme functional groups contribute to translational quality control by bacterial prolyl-tRNA synthetase. J. Phys. Chem. B 116, 6991–6999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar S.; Das M.; Hadad C. M.; Musier-Forsyth K. (2012) Substrate specificity of bacterial prolyl-tRNA synthetase editing domain is controlled by a tunable hydrophobic pocket. J. Biol. Chem. 287, 3175–3184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- So B. R.; An S.; Kumar S.; Das M.; Turner D. A.; Hadad C. M.; Musier-Forsyth K. (2011) Substrate-mediated fidelity mechanism ensures accurate decoding of proline codons. J. Biol. Chem. 286, 31810–31820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vargas-Rodriguez O.; Musier-Forsyth K. (2013) Exclusive use of trans-editing domains prevents proline mistranslation. J. Biol. Chem. 288, 14391–14399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar S.; Das M.; Hadad C. M.; Musier-Forsyth K. (2013) Aminoacyl-tRNA substrate and enzyme backbone atoms contribute to translational quality control by YbaK. J. Phys. Chem. B 117, 4521–4527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sekimoto T.; Matsuyama T.; Fukui T.; Tanizawa K. (1993) Evidence for lysine 80 as general base catalyst of leucine dehydrogenase. J. Biol. Chem. 268, 27039–27045. [PubMed] [Google Scholar]
- Crepin T.; Yaremchuk A.; Tukalo M.; Cusack S. (2006) Structures of two bacterial prolyl-tRNA synthetases with and without a cis-editing domain. Structure 14, 1511–1525. [DOI] [PubMed] [Google Scholar]
- Bhattacharyya S.; Stankovich M. T.; Truhlar D. G.; Gao J. (2007) Combined quantum mechanical and molecular mechanical simulations of one- and two-electron reduction potentials of flavin cofactor in water, medium-chain acyl-CoA dehydrogenase, and cholesterol oxidase. J. Phys. Chem. A 111, 5729–5742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humphrey W.; Dalke A.; Schulten K. (1996) VMD: visual molecular dynamics. J. Mol. Graph. 14(33–38), 27–38. [DOI] [PubMed] [Google Scholar]
- Brooks B. R.; Bruccoleri R. E.; Olafson B. D.; States D. J.; Swaminathan S. J. (1983) CHARMM: A program for macromolecular energy, minimization, and dynamics calculations. J. Comput. Chem. 4, 187. [Google Scholar]
- MacKerell A. D. J.; Bashford D.; Bellott M.; Dunbrack R. L. J.; Evanseck J. D.; Field M. J.; Fischer S.; Gao J.; Gou J.; Ha S.; Joseph-McCarthy D.; Kuchnir L.; Kuczera K.; Lau F. T. K.; Mattos C.; Michnick S.; Ngo T.; Nguyen D. T.; Prodhom B.; Reiher W. E. I.; Roux B.; Schelenkrich M.; Smith J. C.; Stote R.; Straub J.; Watanbe M.; Wiórkiewicz-Kuczera J.; Yin D.; Karplus M. (1998) Allatom empirical potential for molecular modeling and dynamics studies of proteins. J. Phys. Chem. B 102, 3586. [DOI] [PubMed] [Google Scholar]
- Becke A. D. (1988) Density-functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. A 38, 3098. [DOI] [PubMed] [Google Scholar]
- Lee C.; Yang W.; Parr R. G. (1988) Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 37, 785. [DOI] [PubMed] [Google Scholar]
- Brooks C. L. III; Brunger A.; Karplus M. (1985) Active site dynamics in protein molecules: a stochastic boundary molecular-dynamics approach. Biopolymers 24, 843–865. [DOI] [PubMed] [Google Scholar]
- Mueller R. M.; North M. A.; Yang C.; Hati S.; Bhattacharyya S. (2011) Interplay of flavin’s redox states and protein dynamics: an insight from QM/MM simulations of dihydronicotinamide riboside quinone oxidoreductase 2. J. Phys. Chem. B 115, 3632–3641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rauschnot J. C.; Yang C.; Yang V.; Bhattacharyya S. (2009) Theoretical determination of the redox potentials of NRH:quinone oxidoreductase 2 using quantum mechanical/molecular mechanical simulations. J. Phys. Chem. B 113, 8149–8157. [DOI] [PubMed] [Google Scholar]
- Jorgensen W. L.; Chandrasekhar J.; Madura J. D.; Impey R. W.; Klein M. L. (1983) Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 79, 926. [Google Scholar]
- Ryckaert J. P.; Ciotti G.; Berensden H. J. C. (1977) Numerical integration of the Cartesian equations of motion of a system with constraints: molecular dynamics of n-alkanes. J. Comput. Phys. 23, 327–341. [Google Scholar]
- Hockney R. W. (1970) The potential calculation and some applications. Methods Comput. Phys. 9, 136–211. [Google Scholar]
- Verlet L. (1967) Computer “experiments” on classic fluids. I. Thermodynamical properties of Lennard-Jones molecules. Phys. Rev. 159, 98–103. [Google Scholar]
- Brooks C. L. I.; Karplus M. (1983) Deformable stochastic boundaries in molecular dynamics. J. Chem. Phys. 79, 6312. [Google Scholar]
- Cui Q.; Elstner M.; Kaxiras E.; Frauenheim T.; Karplus M. (2001) A QM/MM Implementation of the Self-Consistent Charge Density Functional Tight Binding (SCC-DFTB) Method. J. Phys. Chem. B 105, 569–585. [Google Scholar]
- Elstner M.; Cui Q.; Munih P.; Kaxiras E.; Frauenheim T.; Karplus M. (2003) Modeling zinc in biomolecules with the self consistent charge-density functional tight binding (SCC-DFTB) method: applications to structural and energetic analysis. J. Comput. Chem. 24, 565–581. [DOI] [PubMed] [Google Scholar]
- Singh U. C.; Kollman P. A. (1986) A combined ab initio quantum mechanical and molecular mechanical method for carrying out simulations on complex molecular systems: Applications to the CH3Cl + Cl– exchange reaction and gas phase protonation of polyethers. J. Comput. Chem. 7, 718–730. [Google Scholar]
- Kirkwood J. G. (1935) Statistical mechanics of fluid mixtures. J. Chem. Phys. 3, 300–313. [Google Scholar]
- Range K.; Riccardi D.; Cui Q.; Elstner M.; York D. M. (2005) Benchmark calculations of proton affinities and gas phase basicities of molecules important in the study of biological phosphoryl transfer. Phys. Chem. Chem. Phys. 7, 3070. [DOI] [PubMed] [Google Scholar]
- Kelly C. P.; Cramer C. J.; Truhlar D. G. (2006) Aqueous solvation free energies of ions and ion-water clusters based on an accurate value for the absolute aqueous solvation free energy of the proton. J. Phys. Chem. B 110, 16066–16081. [DOI] [PubMed] [Google Scholar]
- Born M. (1920) Volumen and Hydratationswarne der Ionen. Z. Phys. 1, 45–48. [Google Scholar]
- Zwanzig R. W. (1954) High-temperature equation of state by a perturbation method. I. J. Phys. Chem. 22, 1420–1426. [Google Scholar]
- Miyamoto S.; Kollman P. A. (1993) Absolute and relative binding free energy calculations of the interaction of biotin and its analogs with streptavidin using molecular dynamics/free energy perturbation approaches. Proteins 16, 226–245. [DOI] [PubMed] [Google Scholar]
- Burke B.; Lipman R. S.; Shiba K.; Musier-Forsyth K.; Hou Y. M. (2001) Divergent adaptation of tRNA recognition by Methanococcus jannaschii prolyl-tRNA synthetase. J. Biol. Chem. 276, 20286–20291. [DOI] [PubMed] [Google Scholar]
- Stehlin C.; Heacock D. H. 2nd; Liu H.; Musier-Forsyth K. (1997) Chemical modification and site-directed mutagenesis of the single cysteine in motif 3 of class II Escherichia coli prolyl-tRNA synthetase. Biochemistry 36, 2932–2938. [DOI] [PubMed] [Google Scholar]
- Fersht A. R.; Ashford J. S.; Bruton C. J.; Jakes R.; Koch G. L.; Hartley B. S. (1975) Active site titration and aminoacyl adenylate binding stoichiometry of aminoacyl-tRNA synthetases. Biochemistry 14, 1–4. [DOI] [PubMed] [Google Scholar]
- Liu H.; Musier-Forsyth K. (1994) Escherichia coli proline tRNA synthetase is sensitive to changes in the core region of tRNA(Pro). Biochemistry 33, 12708–12714. [DOI] [PubMed] [Google Scholar]
- Heacock D.; Forsyth C. J.; Shiba K.; Musier-Forsyth K. (1996) Synthesis and aminoacyl-tRNA synthetase inhibitory activity of prolyl adenylate analogs. Bioorg. Chem. 24, 273–289. [Google Scholar]
- Musier-Forsyth K.; Scaringe S.; Usman N.; Schimmel P. (1991) Enzymatic aminoacylation of single-stranded RNA with an RNA cofactor. Proc. Natl. Acad. Sci. U. S. A. 88, 209–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanford B.; Cao B. V.; Johnson J. M.; Zimmerman K.; Strom A. M.; Mueller R. M.; Bhattacharyya S.; Musier-Forsyth K.; Hati S. (2012) Role of coupled-dynamics in the catalytic activity of prokaryotic-like prolyl-tRNA synthetases. Biochemistry 51, 2146–2156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson J. M.; Sanford B. L.; Strom A. M.; Tadayon S. N.; Lehman B. P.; Zirbes A. M.; Bhattacharyya S.; Musier-Forsyth K.; Hati S. (2013) Multiple pathways promote dynamical coupling between catalytic domains in Escherichia coli prolyl-tRNA synthetase. Biochemistry 52, 4399–4412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beuning P. J.; Musier-Forsyth K. (1999) Transfer RNA recognition by aminoacyl-tRNA synthetases. Biopolymers 52, 1–28. [DOI] [PubMed] [Google Scholar]
- Das M.; Vargas-Rodriguez O.; Goto Y.; Suga H.; Musier-Forsyth K. (2013) Distinct tRNA recognition strategies used by a homologous family of editing domains prevent mistranslation. Nucleic Acid Res. 10.1093/nar/gkt1332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kundrotas P. J.; Alexov E. (2006) Electrostatic properties of protein-protein complexes. Biophys. J. 91, 1724–1736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frick D. N.; Rypma R. S.; Lam A. M.; Frenz C. M. (2004) Electrostatic analysis of the hepatitis C virus NS3 helicase reveals both active and allosteric site locations. Nucleic Acids Res. 32, 5519–5528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomlinson J. H.; Ullah S.; Hansen P. E.; Williamson M. P. (2009) Characterization of salt bridges to lysines in the protein G B1 domain. J. Am. Chem. Soc. 131, 4674–4684. [DOI] [PubMed] [Google Scholar]
- Sundd M.; Iverson N.; Ibarra-Molero B.; Sanchez-Ruiz J. M.; Robertson A. D. (2002) Electrostatic interactions in ubiquitin: stabilization of carboxylates by lysine amino groups. Biochemistry 41, 7586–7596. [DOI] [PubMed] [Google Scholar]
- Roberts G. C.; Dennis E. A.; Meadows D. H.; Cohen J. S.; Jardetzky O. (1969) The mechanism of action of ribonuclease. Proc. Natl. Acad. Sci. U. S. A. 62, 1151–1158. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.