

Abstract
Pathogenic bacteria introduce effector proteins directly into the cytosol of eukaryotic cells to promote invasion and colonization. OspG, a Shigella spp. effector kinase, plays a role in this process by helping to suppress the host inflammatory response. OspG has been reported to bind host E2 ubiquitin-conjugating enzymes activated with ubiquitin (E2∼Ub), a key enzyme complex in ubiquitin transfer pathways. A co-crystal structure of the OspG/UbcH5c∼Ub complex reveals that complex formation has important ramifications for the activity of both OspG and the UbcH5c∼Ub conjugate. OspG is a minimal kinase domain containing only essential elements required for catalysis. UbcH5c∼Ub binding stabilizes an active conformation of the kinase, greatly enhancing OspG kinase activity. In contrast, interaction with OspG stabilizes an extended, less reactive form of UbcH5c∼Ub. Recognizing conserved E2 features, OspG can interact with at least ten distinct human E2s∼Ub. Mouse oral infection studies indicate that E2∼Ub conjugates act as novel regulators of OspG effector kinase function in eukaryotic host cells.
Keywords: bacterial effector, E2 Ub-conjugating enzyme, kinase, Shigella, ubiquitin
See also: MF de Jong & NM Alto (January 2014)
Introduction
The human immune system orchestrates a multifaceted defense against invading enteric pathogens. In response, pathogenic bacteria have evolved a number of strategies to evade host defenses. Gram-negative pathogens like Shigella spp., Yersinia spp., and pathogenic Escherichia coli utilize a Type III secretion apparatus to introduce a suite of bacterial effector proteins that can target host cell signaling pathways and function to facilitate bacterial entry, survival, and replication (Phalipon & Sansonetti, 2007). Many of these effectors work to prevent host innate immune responses, including activation of nuclear factor-κB (NFκB) and mitogen-activated protein kinase (MAPK), signaling pathways that govern expression of immune-response genes (Ogawa et al, 2008). Typical of many eukaryotic signaling cascades, activation and regulation of these pathways rely heavily upon post-translational modification of proteins involving both phosphorylation and ubiquitination.
Bacterial effector kinases, phosphatases, and acetyl transferases have been identified that can manipulate or disrupt key host signaling pathways involving protein phosphorylation [e.g. SteC from Salmonella (Poh et al, 2008), and YpkA (Juris et al, 2000), YopH, and YopJ from Yersinia (Guan & Dixon, 1990; Bliska et al, 1991)]. Likewise, numerous effectors that engage host ubiquitination machinery and target ubiquitin (Ub) signaling pathways have also been described. In eukaryotes, targeted modification of proteins by Ub requires three sequential enzyme activities; an E1 Ub-activating enzyme, an E2 Ub-conjugating enzyme, and an E3 Ub-ligase. Pathogens such as Shigella and Salmonella exploit this signaling pathway by encoding multiple effector proteins that can function as E3 ubiquitin ligases and deubiquitinases [e.g. the IpaH, SspH, and SseL enzymes (Rohde et al, 2007; Rytkönen et al, 2007)]. These bacterial enzymes must recognize and repurpose host Ub-transfer machinery and/or substrate proteins to disrupt Ub signaling. To function and compete in a eukaryotic environment, bacterial effectors are thought to be highly active enzymes (Levin et al, 2010), though certain effectors exhibit weak enzyme activity when isolated and require interaction with eukaryotic factors to stimulate activity. For instance, the effector phospholipase ExoU from Pseudomonas aeruginosa requires interaction with host Ub for enhanced enzymatic activity (Anderson et al, 2011).
OspG, an effector first identified in Shigella spp. but also found in strains of Yersinia and enterohemorrhagic E. coli, appears to exploit both eukaryotic ubiquitin and phosphorylation signaling pathways during pathogenesis. Kim et al (2005) showed that OspG (i) exhibits weak kinase activity in vitro, (ii) acts to slow proteasomal degradation of the NFκB inhibitor IκBα in cells during Shigella infection, and (iii) interacts directly with E2∼Ub conjugates. Recently, Zhou et al (2013) demonstrated that binding to Ub could enhance OspG kinase activity, but the significance of OspG/E2∼Ub interactions remained unclear. To investigate the role of OspG during pathogenesis, its function as a kinase, and the significance of its interactions with E2∼Ub conjugating enzymes, we determined a crystal structure of an OspG/E2∼Ub complex. The structure shows that OspG is a minimal kinase domain that retains key catalytic elements also found in eukaryotic kinases. While isolated OspG appears to be highly dynamic and weakly active, complex formation stabilizes a highly active conformation via interaction with both subunits of the E2∼Ub conjugate. Mutations that selectively target the OspG/E2∼Ub interaction show reduced kinase activity in vitro and reduced ability to attenuate the immune response in a mouse model for Shigella infections, confirming a key role for the interaction in a host cell environment. Our results provide structural and functional insights into a class of effector kinases from medically important enteric pathogens and illustrate a mechanism by which bacteria exploit host proteins as regulatory cofactors to aid in pathogenesis.
Results
Crystal structure of the OspG/UbcH5c-O∼Ub complex
To define the molecular interactions that govern assembly, we determined the crystal structure of the OspG/UbcH5c-O∼Ub complex to 2.70 Å (Fig 1A and Table 1). The covalent oxyester linkage between the UbcH5c active site (mutated from cysteine to serine) and the Ub C-terminus is maintained in the complex (Supplementary Fig S1A) and binding to OspG does not significantly alter the conformations of either UbcH5c or Ub. Pairwise RMSDs of backbone atoms are <0.4 Å for both the UbcH5c and Ub subunits (compared to PDB IDs 1X23 and 1UBQ, respectively) with minor structural variation observed in the Ub K48-loop. Both the UbcH5c and Ub subunits contact OspG, but bind to distinct surfaces, placing the Ub and UbcH5c subunits in an extended conformation with respect to each other (Fig 1A and B). The OspG binding surface of UbcH5c (encompassing ∼547 Å2 of solvent-accessible surface) is comprised of residues located in Helix 1 (K4, R5, and K8), Loop 4 (D59, F62, K63), and Loop 7 (S91, Q92, W93, P95, and A96). Ub binds to OspG using an extensive surface (∼907 Å2) that is centered about the L8-I44-V70 hydrophobic patch, but also involves the loop containing Ub residues A46, G47, and K48, and residues S57, D58, Y59, and N60 located in a short helical segment (Fig 1A and B and Supplementary Fig S1B).
Figure 1.
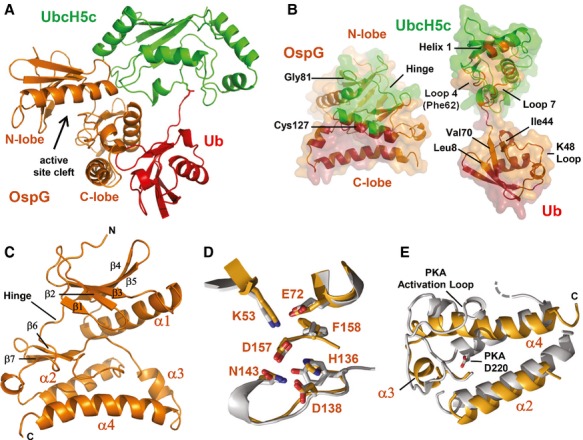
- Ribbon representation of the OspG/UbcH5c-O˜Ub crystal structure determined at 2.70 Å. Ub (red) is covalently linked to UbcH5c (green) and associates in an extended conformation with OspG (orange). The OspG active site formed by a cleft between the N-and C-lobes is marked and faces away from the UbcH5c˜Ub conjugate.
- ‘Open book’ representation of the OspG/UbcH5c-O˜Ub interfaces. Surfaces are colored according to their interacting protein and landmark features are labeled.
- Ribbon representation of the OspG kinase structure looking into the active site cleft. Secondary structure elements are labeled.
- Global superposition (Supplementary Fig S1D) of OspG and PKA (PDB ID 1ATP; Zheng et al, 1993) overlay important structural and catalytic residues. OspG residues are labeled and the corresponding PKA residues are shown in Supplementary Fig S1C).
- Superposition of OspG and PKA highlights differences in the arrangement of helices in the C-terminal lobe of each kinase. OspG helices are labeled. The structure of OspG is shown through the C-terminus whereas PKA has an additional 115 residues that are hidden. All PKA residue numbering used here and in the text matches PDB entry 1ATP.
Table 1.
Data collection and refinement statistics
| OspG/UbcH5c-O∼Ub | |
|---|---|
| Data collection | |
| Space group | P6322 |
| Cell dimensions | |
| a,b,c (Å) | 183.87, 183.87, 63.97 |
| α, β, γ (°) | 90, 90, 120 |
| Resolution (Å) | 50–2.70 (2.80–2.70)a |
| Rsym | 0.202 (1.0) |
| I/σI | 16.39 (2.58) |
| Completeness (%) | 99.97 (99.83) |
| Redundancy | 19.1 (11.2) |
| Refinement | |
| Resolution (Å) | 44.16–2.70 |
| No. reflections | 17947 |
| Rwork/Rfree | 0.244/0.291 |
| No. atoms | 3226 |
| Protein | 3165 |
| Ligand/ion | 0 |
| Water | 61 |
| B-factors | |
| Protein | 22.20 |
| Water | 14.10 |
| R.m.s. deviations | |
| Bond lengths (Å) | 0.005 |
| Bond angles (°) | 0.92 |
| Ramanchandran statistics | |
| No. of non-glycine/proline residues | 344 |
| Residues in most favored regions | 91.3% |
| Residues in additionally allowed regions | 8.4% |
| Residues in generously allowed regions | 0.0% |
| Residues in disallowed regions | 0.3% |
Values in parentheses are for highest-resolution shell.
OspG represents a minimal kinase domain that lacks added structural features typically found in eukaryotic kinases. The structure of OspG in the complex is well defined by the electron density with the exception of a 10x-His tag and 25 native residues at the N-terminus that are disordered and may encode a sequence necessary for recognition by the Type III secretion apparatus. Like other kinases, OspG has an N-terminal lobe composed of a long α-helix and 5-stranded β-sheet that is connected to a predominantly α-helical C-terminal lobe by a short hinge segment (Fig 1C). OspG contains only four α-helical segments and lacks a helix corresponding to cAMP-dependent protein kinase (PKA) αD (Supplementary Fig S1C and D). To our knowledge, all other kinase structures solved to date have additional C-terminal α-helical elements (αG, αH and αI in PKA; Supplementary Fig S1C and D) that interact with both the αE and αF helices. Instead, the position of these helices is occupied by the Ub subunit within the OspG/UbcH5c∼Ub complex (Fig 1A). Finally, though many of the hydrophobic residues that make up the catalytic-and regulatory-spines described for PKA and other eukaryotic kinases (Taylor et al, 2012) have structural analogues in OspG, comparisons of protein contact maps reveal that OspG has significantly fewer direct contacts between its N-terminal and C-terminal lobes overall (Supplementary Fig S1E).
Complex formation with UbcH5c∼Ub appears to compensate for the minimal number of intra-protein contacts observed between the N-and C-terminal lobes of the OspG kinase. UbcH5c interactions occur primarily with OspG residues in the N-terminal lobe (OspG residues D47, A78, F79, Y80, G81, E83) and the hinge segment (OspG residues L99, R100, P102) (Fig 1B). Some additional UbcH5c contacts are observed for OspG residues in the loop connecting the short β-strands β6 and β7 in the C-terminal lobe located directly behind the ATP binding pocket (OspG residues E149, S150, F154). The Ub subunit binds exclusively to the OspG C-terminal lobe, docking the Ub K48-loop into a pocket formed by the triangular arrangement of OspG α2, α3, and α4 helices (Fig 1B and C). Ub makes extensive contacts with OspG that include α2-helix residues E119, S120, L123, Q124, I126, C127, N130, E131, and I135, α3-helix residues Y162, Y165, and Y166, and α4-helix residues K170, K173, D177, L180, Q181, and Y188 (Supplementary Fig S1B). Thus, simultaneous contacts to both subunits of the E2∼Ub conjugate via two different surfaces align the positions of the OspG N-and C-terminal lobes.
Comparisons to structurally similar proteins using the Dali server (Holm & Rosenström, 2010) reveal that OspG adopts a structure very similar to kinases that crystallized in active conformations. Critical structural and catalytic features that define eukaryotic protein kinases are preserved in OspG. A Glu-Lys salt bridge located between the αC helix and β-strand 3, a structural hallmark of active kinases (Taylor et al, 2012), is formed by OspG residues E72 and K53 (structurally analogous to PKA residues E91 and K72, respectively; Fig 1D, Supplementary Fig S1C). The DFG motif, which contains both structural and catalytic residues, consists of only DF in OspG. However the conformations of OspG D157 and F158 are nearly identical to those observed for analogous residues in PKA (D184 and F185 in PKA; Fig 1D). Likewise, the positions of OspG H136, D138, and N143 (corresponding to PKA residues Y164, D166, and N171 that define the HxDxxxxN catalytic motif) also overlay with the corresponding PKA residues. The overall structure of the OspG ATP-binding pocket is very similar to kinase structures determined in the presence of bound ligands. Undefined electron density reminiscent of ATP and Mg2+ was observed in the OspG ATP binding pocket (Fig 2A). Notably, ATP and Mn2+ from the PKA structure can be docked directly into the OspG active site by aligning the kinase domains (Fig 2B). Thus, the above observations are consistent with OspG adopting an active kinase conformation in the context of the OspG/UbcH5c∼Ub complex.
Figure 2.
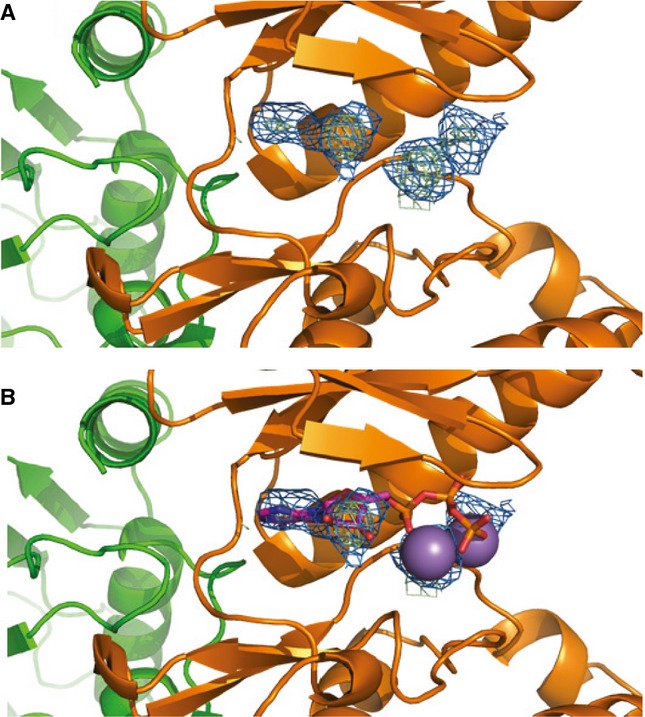
- 2Fo-Fc composite map (blue) and Fo-Fc difference map (green) plotted at 2σ showing unknown density in the OspG active site.
- Comparison with structurally aligned PKA (PDB ID 1ATP) suggests the density could be bound ATP/Mn2+.
Where the structure of OspG differs markedly from eukaryotic kinases is the conformation of helices in the C-terminal lobe. In eukaryotic kinases the conformation of the peptide backbone that positions the catalytic Asp in the HxDxxxN-motif is dependent upon a key hydrogen bond to a highly conserved Asp (PKA D220 in helix αF; Fig 1E). OspG does not have a structurally analogous Asp residue and the position of the N-terminal segment of the OspG α2 helix differs markedly from the PKA αF helix. Instead, the middle residue of the HxD motif, OspG Y137, interacts with I176 and R179 in the α4 helix helping to stabilize an unusual triangular arrangement of the OspG C-terminal lobe helices (α2, α3, and α4 helices in Fig 1C). Given the extensive number of observed OspG and Ub intermolecular contacts, Ub binding likely stabilizes the arrangement of OspG C-lobe helices and, therefore, critical catalytic residues.
OspG forms complexes with various human E2∼Ub conjugates
Interactions of E2∼Ub conjugates with proteins not directly involved in Ub transfer reactions have rarely been reported. Using a two-hybrid approach, Kim et al (2005) found that OspG could interact with several E2s, including members of the Ube2D, Ube2E, and Ube2L families. With approximately 40 E2s encoded in the human genome, defining which E2s form complexes with OspG may provide additional functional insight. To augment the previous two-hybrid studies, we determined which E2s co-purify with OspG from transiently transfected HeLa cells and from extracts generated from Caco-2 intestinal epithelial cells (Table 2).
Table 2.
OspG Interactions with E2∼Ub
| Total Peptides |
|||
|---|---|---|---|
| E2 | HeLa transfection-IP | Caco-2 extract-Pulldown | Binding in vitro |
| UBE2D1 | Not Observed | Not Observed | YES |
| UBE2D2 | 2 | 2 | YES |
| UBE2D3 | 3 | 1 | YES |
| UBE2E1 | 1 | 4 | YES |
| UBE2E2 | Not Observed | 2 | YES |
| UBE2E3 | Not Observed | Not Observed | YES |
| UBE2K | Not Observed | 14 | YES |
| UBE2N | Not Observed | Not Observed | YES |
| UBE2L3 | 23 | 28 | YES |
| UBE2L6 | 11 | Not Observed | YES |
| UBE2S | Not Observed | Not Observed | NO |
| F62A-UBE2D3 | N/A | N/A | NO |
YFP-tagged OspG was transiently transfected into HeLa cells and fluorescence microscopy showed that OspG was diffusely distributed throughout transfected cells (Supplementary Fig S2A). Mass spectrometry analysis of YFP-immunoprecipitates identified UbcH5b (Ube2D2), UbcH5c (Ube2D3), Ube2E1, UbcH7 (Ube2L3), and UbcH8 (Ube2L6) as OspG-associated E2s that were not present in the YFP-only control (Table 2). In a second approach, extracts were prepared from Caco-2 intestinal epithelium cells and incubated with recombinant GST-tagged OspG. OspG complexes were affinity purified on glutathione resin, washed repeatedly, and eluted. Mass spectrometry analysis identified the E2s Ube2D2, Ube2D3, Ube2E1, Ube2E2, Ube2K, and Ube2L3 as present only in the GST-OspG sample. These E2s are the same as those identified above with the addition of Ube2E2, Ube2K and Ube2L6 (Table 2).
The ability of the E2s identified above to bind to OspG as E2∼Ub conjugates was confirmed in vitro (Table 2, Supplementary Fig S2B). GST-OspG was incubated with a panel of purified E2s representing the above families. UbcH5a (Ube2D1), Ube2E3, and Ubc13 (Ube2N) were also included in these experiments. Prior to mixing with OspG, each E2 was pre-incubated with E1, Ub, and MgCl2 in the presence or absence of ATP to compare OspG interactions with E2∼Ub conjugates relative to the free components. OspG-containing complexes were isolated on GST-affinity resin, washed, and released by boiling in SDS-sample buffer under both reducing and non-reducing conditions. SDS-PAGE followed by Coomassie staining shows that OspG binds each E2∼Ub conjugate with enough affinity to be observed by this procedure (Supplementary Fig S2B and S5B, Fig 3B), whereas individual E2 and Ub subunits that were not covalently tethered via a thioester linkage were not observed.
Figure 3.

- UbcH5c F62 (green) binds to a shallow pocket formed by OspG residues Y80, L99, P102, and F154.
- Input: non-reducing samples of reaction mixtures prior to the addition of GST-OspG showing generation of E2˜Ub conjugates from wild-type UbcH5c, F62A-UbcH5c, and Ube2S. Pulldown: reducing SDS-PAGE shows GST-OspG only forms a high-affinity complex with wild-type UbcH5c˜Ub.
Source data are available online for this figure.
Comparison of the various human E2∼Ub conjugates that form complexes with OspG suggest they share common structural features. The UbcH5c residues described above that contact OspG are the same as those commonly involved in E2/E3 interactions and are often conserved (Supplementary Fig S3A and B). The most extensive side chain interactions involve UbcH5c residues F62 and P95, which contact a shallow hydrophobic pocket formed by OspG residues F79, Y80, L99, P102 and F154 (Fig 3A). UbcH5c P95 is highly conserved among human E2s and all the E2s found to interact with OspG have an aromatic or large hydrophobic residues at the position structurally analogous to UbcH5c F62. Mutation of UbcH5c F62 to alanine abolishes the observed interaction between UbcH5c∼Ub and OspG in in vitro pulldown assays (Fig 3B). We also failed to observe an interaction with Ube2S∼Ub, which has an alanine at this position (Fig 3B). Therefore, OspG can potentially bind an array of E2∼Ub conjugates within the host cell, with the only commonalities being the Ub moiety and conservation in the OspG/E2 interaction interface (Supplementary Fig S3).
OspG binds isolated UbcH5c and Ub with low affinity
To understand the contributions each protein subunit makes toward complex formation, we examined pairwise interactions using NMR spectroscopy. Addition of unlabeled OspG to an equimolar mixture of 15N-labeled UbcH5c and Ub exhibited different binding characteristics as compared to interaction with 15N-UbcH5c-O∼Ub (compare Figs 4A and B). Severe linewidth broadening was observed upon addition of OspG to 15N-labeled UbcH5c-O∼Ub (Fig 4B, compare black and red spectra), consistent with the formation of a high affinity, high molecular weight complex. High quality NMR spectra of the complex can be obtained if the protein subunits are perdeuterated (which often allows NMR data collection on higher molecular weight proteins and complexes; Supplementary Fig S4A), showing that the observed reduction in resonance intensity is not simply due to non-specific self-association. By comparison, addition of OspG to 15N-labeled UbcH5c and Ub resulted in much more moderate perturbations, indicative of weaker interactions. The Ub and UbcH5 interaction surfaces, identified by the largest observed resonance perturbations in NMR titration experiments, map to the same surfaces identified in the crystal structure. The sensitivity of NMR titrations for weak interactions confirms that while the individual components interact with low affinity, covalent linkage of UbcH5c and Ub to form the E2∼Ub conjugate allows both subunits to bind simultaneously for a high affinity interaction.
Figure 4.
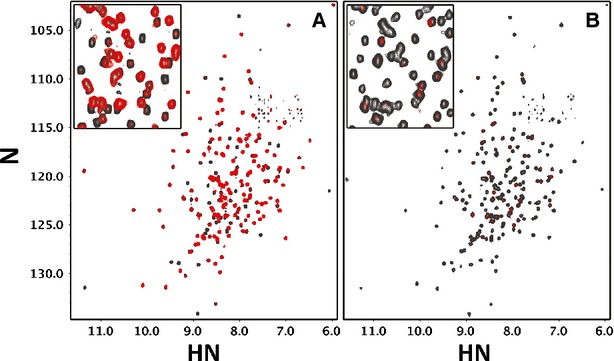
OspG binds free UbcH5c and Ub with weak affinity.
A, B 1H,15N-TROSY spectra of (A) free UbcH5c and Ub and (B) the UbcH5c-O˜Ub conjugate before and after addition of 1 molar equivalent of GST-OspG (grey and red, respectively). OspG shows weak interactions with free UbcH5c and Ub (A). The perturbed UbcH5c and Ub resonances map to the E3-binding interface of UbcH5c and the region surrounding the I44 hydrophobic surface of Ub as shown in the crystal structure. An enlarged region of the center (inset) shows linewidth broadening of the UbcH5c-O˜Ub spectrum after addition of OspG (B), illustrating a substantial increase in affinity attained via conjugation of UbcH5c and Ub subunits.
We also note that HSQC spectra of free OspG show a high degree of heterogeneity and resonance broadening, consistent with the existence of significant conformational exchange and/or protein self-association (Supplementary Fig S4B). As our structure would indicate, the N-and C-terminal lobes most likely make very few inter-domain contacts and would thus exhibit conformational exchange with respect to each other. Therefore, the crystal structure and NMR experiments indicate that OspG/UbcH5c∼Ub complex formation promotes a stable conformation of OspG.
OspG kinase activity is enhanced upon binding UbcH5c∼Ub
To assess how complex formation alters OspG function, we used isothermal titration calorimetry (ITC) to compare binding of nonhydrolyzable ATP-γ-S to either free OspG or the OspG/UbcH5c∼Ub complex. Very little heat associated with binding was observed during ITC experiments upon addition of concentrated ATP-γ-S to free OspG, though the measurements were clearly above the level of mixing observed in control experiments (Fig 5A, compare left and center). In contrast, addition of ATP-γ-S to the OspG/UbcH5c∼Ub complex showed substantially higher heats of binding (Fig 5A, compare center and right). The interaction was not strong enough to obtain a complete titration curve to calculate a Kd for ATP binding to the OspG/UbcH5c∼Ub complex. These results suggest that there is a marked increase in the affinity of OspG/UbcH5c∼Ub for ATP-γ-S compared to OspG alone. Another interpretation is that binding of ATP-γ-S to free OspG is entropically driven whereas there is a large enthalpic contribution observed on binding to the OspG/UbcH5c∼Ub complex. This scenario suggests a change in mechanism for ATP binding. In either case, complex formation has a significant affect on OspG/ligand interactions.
Figure 5.
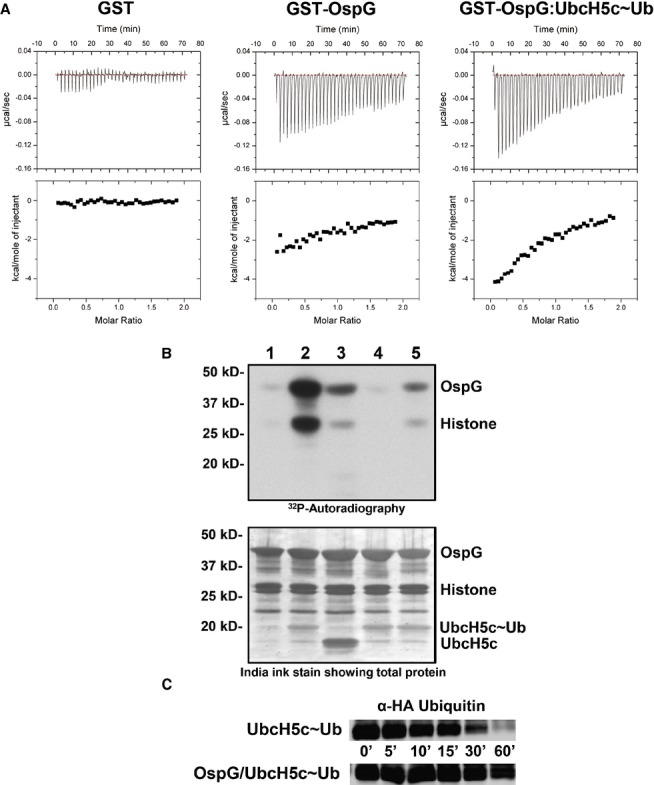
- Raw heat outputs and integrated isotherms for titration of 1 mM ATP-γ-S into 50 μM GST (left), GST-OspG (center), or GST-OspG/UbcH5c-O˜Ub complex (right).
- (1-5) γ32P-ATP kinase assay following phosphorylation of GST-OspG and Histone H1. 1) OspG alone, 2) premixed OspG + UbcH5c-O˜Ub, 3) premixed OspG + UbcH5c + Ub (unconjugated), 4) premixed K53M-OspG + UbcH5c-O˜Ub, 5) premixed C127R-OspG + UbcH5c-O˜Ub. The panel below depicts the proteins present in each reaction mixture.
- OspG stabilizes the UbcH5c˜Ub conjugate. 5 μM samples of purified UbcH5c˜Ub or OspG/UbcH5c˜Ub were reacted with 50 mM Lysine over a 60-min time course. Non-reducing gel samples were separated by SDS-PAGE, transferred to nitrocellulose, and visualized by Western analysis for the N-terminal HA epitope on Ub. Decay of the UbcH5c˜Ub species is shown.
Source data are available online for this figure.
OspG has been shown to have some auto-phosphorylation activity (Kim et al, 2005) which appears to target a serine residue located in the linker between the GST and OspG subunit in fusion constructs (Zhou et al, 2013). We found that Histone H1 can also be used as a proxy substrate in kinase activity assays (Fig 5B). Free OspG exhibits very weak kinase activity (Fig 5B, lane 1), while the OspG/UbcH5c∼Ub complex exhibits substantially greater activity in phosphorylating both GST-OspG and Histone H1 (Fig 5B, lane 2). Kinase activity depends on the covalent linkage between UbcH5c and Ub, as measured activity is significantly lower in the presence of an equimolar mixture of free UbcH5c and Ub (Fig 5B, lane 3). As expected, disruption of the OspG Lys53-Glu72 salt bridge by mutation of Lys53 to Met abolishes OspG kinase activity (Fig 5B, lane 4). Finally, the integrity of the interaction between E2∼Ub and OspG is critical as introduction of a point mutation at the Ub-binding interface, C127R-OspG, greatly diminished kinase activity in the presence of the UbcH5c∼Ub conjugate (Fig 5B, compare lanes 2 and 5; Fig 1B). Thus, both the ATP-binding and kinase activity assays are consistent with a model in which the OspG kinase domain is activated upon binding to an intact UbcH5c∼Ub conjugate.
OspG-binding stabilizes the UbcH5c∼Ub conjugate
In the complex, OspG holds the E2∼Ub conjugate in an extended conformation with the E2 active site fully accessible to solvent. Our previous studies of E2∼Ub conjugates in solution showed that the E2 and Ub subunits are highly dynamic with respect to each other (Pruneda et al, 2011). Interaction with a RING E3 Ub-ligase promotes closed conformations of the E2∼Ub that are associated with increased reactivity toward nucleophiles, as demonstrated by reactivity toward the free amino acid Lysine (Wenzel et al, 2011; Dou et al, 2012; Plechanovová et al, 2012; Pruneda et al, 2012). When the same assay is used to measure the reactivity of the UbcH5c∼Ub conjugate in the context of the OspG complex, the stability of the E2∼Ub is substantially increased compared to isolated UbcH5c∼Ub (Fig 5C). This contrasting result nicely demonstrates that, while the closed E2∼Ub conformations induced by RING E3 ligases are highly reactive, the extended conformation maintained by OspG is highly stable.
Comparison of OspG to related effector kinases
A number of bacterial effector kinases have been identified in certain strains of Yersinia (e.g. YspK) and enterohemorrhagic E. coli (Nobe et al, 2009) that show high sequence homology to Shigella OspG. The closely related effectors share 90% or higher sequence identity with OspG and at least 95% sequence similarity. Side-chain contacts to UbcH5c and Ub are 100% conserved in these effectors and strongly suggest they similarly require activation by E2∼Ub conjugates (Supplementary Fig S5A). A second group of effectors that includes NleH1 and NleH2 (also found in enterohemorrhagic E. coli) are often functionally compared to OspG. However, NleH1 and NleH2 kinases share only approximately 25% primary sequence identity with approximately 50% similarity in the kinase domain (Supplementary Fig S5A). These effectors have additional N-terminal regions, but intriguingly, sequence alignment show that NleH1 and NleH2 have small kinase domain C-terminal lobes like that of OspG (Supplementary Fig S5A), implying that they may also bind other proteins in this region. We note that only 13% of residues are conserved between OspG and NleH1 in the potential E2∼Ub contact surfaces (Supplementary Fig S5A), suggesting that NleH1 and NleH2 may not bind E2∼Ub conjugates. Consistent with this notion, we were unable to observe an interaction between GST-tagged NleH1 and NleH2 from O157:H7 Escherichia coli and UbcH5c∼Ub in pulldown assays (Supplementary Fig S5B). In contrast to OspG, NleH1 and NleH2 show robust kinase activity when purified from bacteria (Gao et al, 2009 and Supplementary Fig S5C) and we did not detect additional catalytic enhancement in the presence of Ub or the Ub-like proteins NEDD8, SUMO1, or ISG15 (Supplementary Fig S5C). Thus, NleH1 and NleH2 do not require interaction with Ub or these Ub-related molecules for activity, though we cannot rule out that the C-terminal lobe in these kinases may potentially interact with other eukaryotic proteins.
A murine model of shigellosis demonstrates OspG/E2∼Ub binding is critical for OspG function
To examine the importance of OspG/E2∼Ub complex formation during Shigella pathogenesis, we applied a recently developed oral murine model of Shigella infection that displays many characteristics of shigellosis (Martino et al, 2005). Infection of BALB/c mice with wild-type streptomycin-resistant Shigella resulted in weight loss and colonization of the gut (Fig 6). Shigella was observed in the feces of mice after 1 day of infection and continued throughout the course of the experiment. Consistent with the results of Bernardini and coworkers, infection with wild-type Shigella did not result in mortality of mice. In contrast, infection with Shigella deleted for ospG (Shigella ΔospG) resulted in a reliable 30% mortality after 3 days of infection (Fig 6A). Mice that had not succumbed by the third day of infection became more active and began to gain weight. No mice mortality was observed after 3 days of infection. The most severely sick (by clinical score) ΔospG infected mice often displayed colons that were discolored and devoid of fecal pellets (Fig 6B). Mice infected with ΔospG did not display diarrhea but liquid was regularly observed on the fur around the anus of approximately 30% of the mice during days two and three post infection. These gross pathologies were not observed in mice infected with wild-type Shigella. Infection with wild-type Shigella or Shigella ΔospG, however, resulted in similar bacterial burdens in the feces and from homogenates of the cecum or liver (Fig 6C, Supplementary Fig S6A and B). Additionally, there were no obvious histological differences comparing the wild-type and Shigella ΔospG infected mouse colons (Fig 6D). Infected mice show some infrequent patchy mucosal and submucosal infiltration by mononuclear cells though the crypt architecture is mostly left intact. We conclude that OspG is required to down-regulate acute disease, although the precise mode of action awaits further investigation.
Figure 6.
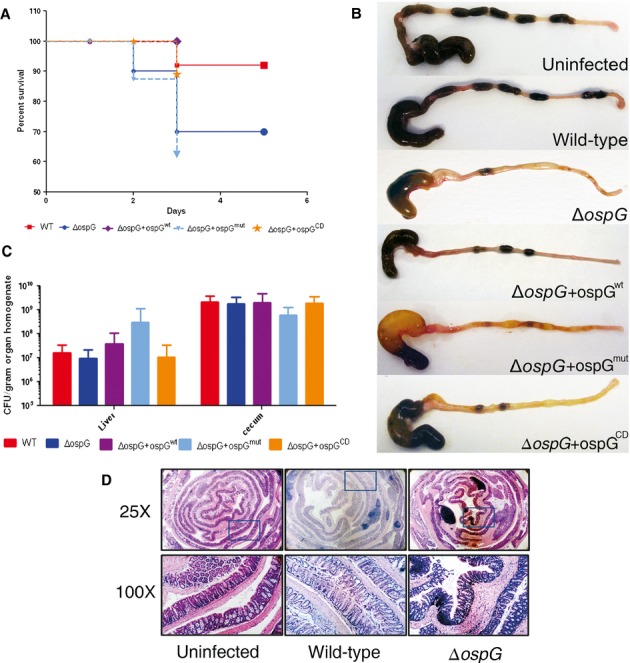
OspG/E2∼Ub binding is important for OspG functions during murine model shigellosis.
A–C Mortality (A) (n = 10–25 per group), P = 0.03, gross morphology of cecum and colon (B), and CFU counts (C) in homogenized liver (n = 5–15 per group, P > 0.05) and cecum content (n = 5–15 per group, P > 0.05), at day 3 post oral infection of BALB/c mice with WT, ΔospG, ΔospG+ospGwt, ΔospG+ospGmut, and ΔospG+ospGcd Shigella flexneri (1 × 108 CFU oral gavage).
D Representative mouse colon sections showing a low and high magnification (area encompassed by the blue box is shown in 100X panels). The low power image shows the proximal regions on the outside of the roll. The high power images were chosen to show more than one run of crypts. Infected mice show some infrequent patchy mucosal and submucosal infiltration by mononuclear cells though the crypt architecture is mostly left intact. An example where the architecture is lost can be seen in the 100X image of the WT infected animal.
We used the mouse infection model to directly test the role of the OspG/E2∼Ub complex during Shigella pathogenesis. We complemented the ΔospG strain by re-introducing wild-type or variant ospG directly into the virulence plasmid. Similar to the wild-type Shigella strain, infection of mice with the ΔospG+ospGwt strain did not result in a lethal infection, demonstrating the efficacy of our complementation strategy (Fig 6A). Furthermore, both the ΔospG and ΔospG+ospGwt strains showed similar effector secretion profiles to the wild-type strain in vitro (Supplementary Fig S7). Infection with the ΔospG+ospGmut strain bearing the G81R and C127R mutations at the E2∼Ub interface (Fig 1B), however, resulted in a 30% mortality, identical to the strain lacking OspG entirely (Fig 6A). In addition, the colons of the severely sick mice infected with the Shigella ΔospG+ospGmut strain were devoid of fecal pellets and discolored, similar to those of mice infected with the ΔospG strain (Fig 6B). Colons from mice infected with Shigella ΔospG+ospGwt had colons more similar to those infected with wild-type Shigella (Fig 6B). Infection with a catalytically deficient ospG (ΔospG+ospGCD) strain bearing the catalytically-dead K53M mutation at the OspG active site resulted in an intermediate phenotype. There was no statistical difference in mortality between mice infected with wild-type and the ΔospG+ospGCD; however, the gross histology of the colons from the ΔospG+ospGCD more closely resembles those from the ΔospG mutant than the wild-type (Fig 6A and B). We conclude that OspG is required for decreasing the severity of Shigella infection in mice and that this activity depends upon interaction with host E2∼Ub conjugates. In addition, our data support E2∼Ub-mediated OspG kinase regulation in vivo.
Discussion
Bacterial pathogens have developed numerous strategies to inhibit or evade host immune responses. In most cases, successful colonization requires bacterial effector proteins that target key signaling pathways to be introduced directly into the cytosol of host cells. Many effectors have been found to be highly active enzymes that can out-compete their eukaryotic counterparts (Levin et al, 2010). However, not all effectors exhibit high levels of enzyme activity on their own, but rather require interaction with additional eukaryotic factors for their function. The Yersinia virulence factor YpkA is dependent on interaction with a eukaryotic-derived host factor, actin, for activation of its kinase activity (Juris et al, 2000). The activity of the phospholipase ExoU, a key virulence factor in P. aeruginosa pathogenesis, has been shown to be stimulated by interaction with Ub, poly-Ub chains, or Ub-modified proteins (Anderson et al, 2011). Since bacterial cells do not contain Ub, this strategy ensures that the effector will not be active until it is introduced into the eukaryotic host. Here, we present the structure of the Shigella effector protein OspG and show that it requires interaction with host E2∼Ub conjugates to stimulate its kinase activity in vitro and its virulence function in a mouse infection model.
Complex formation is essential for maximal activity and OspG mutations that target the E2∼Ub binding interfaces impact kinase activity in vitro and in mouse infection models. Though these mutations are far removed from the OspG active site and kinase regions associated with substrate binding, mice infected with Shigella harboring OspG mutations in the E2∼Ub interface show the same mortality rates as mice infected with Shigella lacking OspG entirely. The mortality observed in these experiments likely arises from the severity of the host immune response which ΔospG Shigella strains have difficulty attenuating. Although a recent report suggests that Ub is sufficient to activate the kinase activity of OspG (Zhou et al, 2013), we find the more relevant species in activating OspG kinase activity is the E2∼Ub conjugate (Fig 5B). An important consequence of the large and extended contact surface in the complex is proper orientation of the N-and C-terminal lobes of the kinase—a result that Ub binding alone cannot recapitulate. Comparisons of protein contact maps derived from structurally similar kinases (i.e. RIO1, RIO2, CDK-like kinase, and IL1 receptor-associated kinase; PDB IDs 1ZTH, 1ZAR, 4AGU, and 2NRU, respectively) reveal that the kinase domain of OspG has fewer of the inter-lobe contacts that typically stabilize an active conformation (Supplementary Fig S1D). To offset this deficiency, OspG co-opts E2∼Ub conjugates, which are likely available in abundance in the host cell (Haas & Rose, 1982). Though both the E2 and Ub subunits bind simultaneously, the complex is not likely to be completely rigid. The flexibility of the E2∼Ub thioester linkage could accommodate any OspG inter-lobe motions required during ligand binding and catalysis.
Enhancement of OspG kinase activity could be the primary affect of E2∼Ub/OspG complex formation. In this case, it is expected that Shigella harboring a kinase dead mutant will have the same phenotype as OspG null or OspG mutants that prevent E2∼Ub binding. The intermediate levels of disease/pathology observed in our mouse models relative to OspG null or E2∼Ub binding mutants (Fig 6) suggest that E2∼Ub conjugates may play an additional role in OspG function. We note that OspG can form complexes with at least 10 distinct human E2∼Ub conjugates (Table 2, Supplementary Fig S2). Peptides from Ube2L3 and Ube2L6 were the most highly represented E2 class after mass analysis of OspG interacting E2s derived from HeLa cells and Caco-2 cell extracts. It is certainly possible that OspG has greater selectivity for these E2s in vivo, and Ube2L3 and Ube2L6 could play an important role in OspG localization and/or substrate recognition. Alternatively, we note that OspG recognizes E2 structural elements in Helix 1, Loop 4, and Loop 7 (Fig 1B) that are highly conserved among a large number of human E2s (Supplementary Fig S3). A second possibility is that complex formation with any of a number of human E2∼Ub conjugates is sufficient to enhance kinase activity and the common OspG/Ub subunits of the complex are sufficient for full OspG function. Thus, OspG may not have to rely on complex formation with a specific E2∼Ub conjugate for function during pathogenesis.
OspG represents a minimal kinase domain, having no activation loop and only three C-terminal lobe helices. The lack of an OspG activation loop and deviation from the HxD-motif is similar to the so-called non-RD kinases, many of which are constitutively active. Overall, OspG displays the highest structural similarity to ATP-bound conformations of the RIO-family of atypical protein kinases present in all kingdoms of life (Supplementary Fig S8). Positions of αC helices, catalytic loops, and metal-binding loops in RIO kinases are very similar to those observed in the structure of OspG. OspG and the RIO-family kinases also lack an activation loop that is found in many eukaryotic kinases. This suggests that once OspG forms a complex with an E2∼Ub conjugate, the kinase will then be constitutively active. However, in contrast to OspG, the RIO kinase domains have a fourth C-terminal helix and the key Asp residue that orients the HRD-catalytic loop, features commonly found in other eukaryotic kinases.
Interestingly, OspG binds an extended, less reactive conformation of the UbcH5c∼Ub conjugate. This conformation draws an interesting distinction with RING-E3 dependent processes that promote closed E2∼Ub conformations to facilitate Ub transfer (Supplementary Fig S9A and B). Structural and biochemical studies of the E2 active site have described a model in which a conserved asparagine (UbcH5c N77) interacts with the carbonyl of the thioester to stabilize an oxyanion intermediate via hydrogen bonding (Wu et al, 2003). The same side chain also plays an important structural role in stabilizing the position of nearby active site residues in Loop 8 of the E2 (Berndsen et al, 2013). In addition, a conserved aspartate (UbcH5c D117) located adjacent to the E2 active site is proposed to suppress the pKa of the incoming lysine thereby enhancing its reactivity (Yunus & Lima, 2006; Plechanovová et al, 2012). Although all these features are conserved in the OspG/UbcH5c∼Ub complex, assays that measure the intrinsic reactivity of the E2∼Ub thioester linkage reveal the E2∼Ub is much less reactive toward lysine in the context of the OspG/E2∼Ub complex as compared to free E2∼Ub in solution (Fig 5C). The UbcH5c∼Ub conformation is much more similar to an E2∼Ub conjugate poised for trans-thiolation as proposed for a HECT-type E3 ligase reaction (Supplementary Fig S9C). Taken together, the observations suggest that the conformation of E2 active site residues in the complex is not optimal for Ub-transfer activity to lysine. Thus, formation of the OspG/UbcH5c∼Ub complex enhances kinase activity while reducing reactivity of the E2∼Ub conjugate, a reciprocal effect that may serve to increase the lifetime of an active kinase complex.
In conclusion, the OspG/UbcH5c∼Ub complex provides the first atomic-level view of a bacterial effector kinase domain. The requirement for OspG to interact with E2∼Ub conjugates to regulate catalytic activity defines a new function for E2∼Ubs. Sequence analyses identify a number of other pathogenic bacteria encoding minimal effector kinases that may function via a similar mode of regulation. The structure and activation mechanism described herein establish a basis for devising strategies aimed at identifying the cellular target(s) of OspG and, more broadly, the role of the ubiquitin system in host-pathogen response.
Materials and Methods
Plasmids, protein expression, and purification
Plasmid constructs, expression, and purification of E1, E2s, and Ub were as previously described (Christensen et al, 2007). For crystallography, ospG was cloned into a pET16 expression vector. Cells containing the ospG plasmid were grown for 16 h at 16°C after induction at OD 0.6–0.8 with 0.2 mM IPTG. Inclusion bodies containing OspG were solubilized with 8M urea, 25 mM Tris (pH 7.6), 200 mM NaCl, 10 mM NH4Cl, 2 mM DTT and refolded through serial two-fold dilutions of the urea with intermittent centrifugation to remove aggregated protein. The final 1M-0M urea dilution was performed by dialyisis. The protein was then concentrated and purified further by SDX75 size-exclusion chromatography into 25 mM sodium phosphate (pH 7.0), 150 mM NaCl. For all other experiments, OspG was expressed as a GST-fusion protein from the pGEX-4T-2 vector and purified using GSH resin according to the manufacturer's protocol and SDX75 size exclusion chromatography into 25 mM sodium phosphate (pH 7.0), 150 mM NaCl.
Crystallization and data collection
UbcH5c-O∼Ub was prepared as previously described using the active site C85S mutation in UbcH5c to improve conjugate stability and the S22R mutation to prevent self-assembly of the conjugated species (Levin et al, 2010). The OspG/UbcH5c-O∼Ub complex was purified over SDX75 size exclusion chromatography into 50 mM HEPES (pH 7.5), 100 mM NaCl and concentrated to 200 μM. Crystals were grown by sitting drop vapor diffusion at 4°C in a 2 μl drop (1 μl protein, 1 μl reservoir solution). The optimal condition for crystal growth was 0.1 M Tris (pH 8.5), 20% EtOH from the Hampton Research Crystal Screen 2 kit. Crystals were cryoprotected in paraffin oil and shipped to the Stanford Synchrotron Radiation Lightsource for data collection on beamline 9–2. 180 frames at 1° intervals were collected at 100 K using a MAR325 detector with a crystal-detector distance of 350 mm. The X-ray wavelength was 0.9795 Å. The datasets were processed using DENZO and SCALEPACK (Otwinowski & Minor, 1997).
Structure solution and refinement
Phases were calculated by molecular replacement using the PHASER (McCoy et al, 2007) program within the CCP4 software suite (Winn et al, 2011) and the UbcH5c structure (PDB ID 1X23) followed by residues 1–72 of the Ub structure (1UBQ). A single solution was found in spacegroup P6322 with one OspG/UbcH5c-O∼Ub complex in the asymmetric unit. Parrot (Cowtan, 2010) and Buccaneer (Cowtan, 2006) were used for density modification and automated model building of OspG. The model was subjected to iterative refinement with REFMAC5 (Murshudov et al, 2011) and manipulation in COOT (Emsley et al, 2010). Molprobity (Chen et al, 2010) and TLSMD (Painter & Merritt, 2006) were used during and after refinement. All structure figures were prepared using PyMOL (The PyMOL Molecular Graphics System). The atomic coordinates and structure factors have been deposited into the Protein Data Bank (code 4BVU).
Cell culture and Immunoprecipitation
Caco-2 or HeLa cells were grown in DMEM (Gibco) supplemented with 10% FBS (or 5% NCS for HeLa) and penicillin/streptomycin. HeLa cells were transfected with Lipofectamine 2000 (Invitrogen) for 4 h before growth medium was changed and cells incubated 24–36 h. HeLa cells in 100-mm dishes were transfected with DNA encoding N-terminally YFP-tagged OspG in the pDEST6.2 vector. After approximately 48 h, cells were harvested in 20 mM HEPES (pH 7.4), 150 mM NaCl, 1 mM EDTA, 1% Triton X-100 containing complete-Mini protease inhibitor (Roche). Lysates were centrifuged at 13,000 × g for 20 min at 4°C. The clarified lysates were incubated with GFP-Trap-M magnetic beads (ChromoTek) for 1 h at 4°C with rocking. Beads were washed 4 × 1 ml lysis buffer using a handheld magnet (Invitrogen) for bead capture. Protein was eluted by incubation of beads with 0.2 M glycine (pH 2.5) for 10 min. Supernatant was removed and neutralized with 2M Tris (pH 8.8), then further processed for mass analysis.
Cell staining
HeLa cells were grown on fibronectin coated coverslips (BD Biosciences) and transfected using Lipofectamine 2000 for 4 h before the growth medium was changed and cells incubated 24–36 h. Cells were fixed with 4% (v/v) paraformaldehyde in PBS for 20 min at room temperature, permeabilized in PBS supplemented with 0.2% BSA and 0.1% Triton X-100 for 10 min, and stained with the appropriate primary antibodies for 4 h at 4°C. Cells were washed three times in PBS and incubated with Alexa Fluor-conjugated secondary antibodies (1:500) for 2 h at room temperature. Cells were washed again and mounted on glass slides using ProLong antifade media (Invitrogen). Actin was labeled by incubation with Texas Red-phalloidin. Nuclei were stained with DAPI (4′,6′-diamidino-2-phenylindole) in the final step before mounting. Cells were imaged with a Zeiss LSM 510 META confocal microscope.
OspG pulldowns from cell extract
Caco-2 cells were grown to confluency and lysed in 50 mM Tris (pH 7.6), 150 mM NaCl, 1 mM EDTA, and 1% NP-40 with protease (Sigma) and phosphatase (Roche) inhibitor cocktails. 30 μl of GSH resin (Invitrogen) was pre-loaded with 30 μl of 50 μM protein (GST-OspG, GST-OspG/UbcH5c-O∼Ub, or GST alone) and washed three times with 500 μl cold 50 mM Tris (pH 7.6) 150 mM NaCl buffer. Pre-loaded resin was incubated with 100 μl of 1 mg/ml cell extract on ice for 30 min, then washed seven times with 500 μl cold Tris/NaCl buffer. Resin was eluted with 50 μl of Tris/NaCl + 10 mM GSH and processed for mass spectrometry analysis.
Mass spectrometry
Samples for mass analysis were digested with trypsin and peptides were separated by nanoflow reversed-phase chromatography and directly eluted into a Velos Orbitrap mass spectrometer (Thermo Scientific). Injected peptides were fragmented via collision-activated dissociation and detected in the linear ion trap of the mass spectrometer. Resulting mass spectra were searched against the human IPI database, to which the OspG protein sequence was added. Identified peptides were filtered to a 1% false-discovery rate. Only those proteins containing peptides found exclusively in the OspG containing samples (i.e. not the GST or YFP tag control) were considered to be binding partners of OspG for follow-up in vitro studies.
In vitro pulldown of OspG/E2∼Ub complexes
Prior to addition of GST-OspG, 35 μl of 10 μM E2, 1 μM human E1, 20 μM Ub, and 5 mM MgCl2 were incubated at 37°C for 15 min with or without the addition of 2.5 mM ATP. After a 10 μl gel sample was collected (input), 10 μl of GSH resin loaded with 12.5 μl of 25 μM GST-tagged protein was added and the mixture was incubated on ice for 30 min. After washing two times with 250 μl cold 25 mM sodium phosphate (pH 7.0) with 150 mM NaCl, the resin was eluted with addition of 35 μl SDS-PAGE load dye. For samples that were pre-incubated with ATP, non-reducing load dye was used to preserve the E2∼Ub thioester.
NMR spectroscopy
NMR spectra were recorded on a 500 MHz Bruker Avance II (University of Washington) or a 600 MHz Varian INOVA spectrometer (Pacific Northwest National Laboratories, Richland, WA). Spectra were recorded at 25°C in 25 mM sodium phosphate (pH 7.0) 150 mM NaCl, 10% D2O. 1H,15N-HSQC-TROSY experiments were collected on 200 μM labeled protein with or without the addition of 1 molar equivalent of unlabeled GST-OspG. Datasets were processed using NMRPipe/NMRDraw (Delaglio et al, 1994), and visualized with NMRView (Johnson & Blevins, 1994).
Isothermal calorimetry
Measurements were made on a MicroCal ITC-200 calorimeter with freshly prepared proteins at 25°C. Protein and ATP-γ-S were made up in 25 mM HEPES (pH 7.0), 150 mM NaCl, 5 mM MgCl2, quantified by absorbance (with an ε260 of 15.4 mM/cm for ATP), and degassed under vacuum before use. A range of concentrations and conditions were tested, but the most binding information was obtained with 1.5 μl additions of 1 mM ATP-γ-S at 3-min intervals into 50 μM protein (GST, GST-OspG, or GST-OspG/UbcH5c-O∼Ub). Reverse titrations of 100 μM protein (GST-OspG or GST-OspG/UbcH5c-O∼Ub) into 1 mM ATP-γ-S were also performed to obtain an approximate measure of the total enthalpy of binding.
Kinase assays
Kinase assays were performed with purified proteins and Histone H1 (Sigma) as a proxy substrate. OspG alone, OspG mutants or OspG-conjugate complexes (∼6.7 μM final) were incubated in 25 mM Tris-HCl (pH 7.5), 10 mM MgCl2, 100 μM ATP with 5 μg histone and 0.5 μCi γ32P-ATP for 20 min at 30°C with shaking. Reactions were stopped by addition of 5X Laemlli sample buffer and boiling for 5 min. Kinase reactions were separated by SDS-PAGE on 4–12% gradient gels (Invitrogen). After transfer to nitrocellulose, membranes were washed three times for 5 min in PBS + 0.3% Tween-20 and stained with 0.2% India ink in the same buffer. Membranes were dried and exposed to autoradiographic film.
Bacterial strains and cloning
Streptomycin-resistant strain Shigella flexneri serotype 5a (M90T-Sm) was used (Onodera et al, 2012). Shigella was routinely cultured with appropriate antibiotics in or on Trypticase Soy Broth (TSB), with or without 20 mg/ml agar and 0.01% Congo red. To construct the ospG::tetRA mutant, ospG was deleted in M90T using lambda red recombination in a manner similar to that described in (Baba et al, 2006). For complementation studies, ospGwt, ospGmut (G81R, C127R), or ospGcd (K53M) was introduced into the M90T virulence plasmid under control of the constitutive lac promoter using the pJQ200 suicide integration vector (Quandt & Hynes, 1993). Oligonucleotides used in construction of the ospG mutant and for the complementation strategy are listed in Supplementary Table S1, and the detailed methods are provided in the Supplementary Data S1.
Mouse infection
Five-to eight-week-old female BALB/c mice were obtained from Charles River (St Constant, QC, Canada). A minimum of 10 mice of age 6-8 weeks were used for each experiment. Experiments were undertaken in the IWK Health Centre under the approval of the University Committee on Laboratory Animals, Dalhousie University. Mouse infections were performed as described by Martino (Martino et al, 2005 and Supplementary Data S1). The mice were monitored for either 3 or 5 days with daily assessments on severity of clinical illness and collection of fecal samples to determine bacterial burden. Clinical illness was scored based on appearance (grooming, urine staining, and coat quality), behavior (response to stimulus, posture, lethargy), appetite, hydration, temperature, and weight loss. Mice that had lost >20% of their initial body weight or those that did not respond to stimulus were euthanized. At the culmination of the experiment, blood samples were collected and organs were harvested for histology and bacterial burden measurements (see Supplementary Data S1).
Data analysis
A non-parametric Kruskal–Wallis test was used for statistical analysis of bacterial counts in feces and organs with a P-value < 0.05 for significance, followed by a Dunn's multiple comparison (Sellge et al, 2010). Survival curves were analyzed using a Log-rank (Mantel–Cox) test with significance defined as a P-value < 0.05.
Acknowledgments
We thank V. Vittal and S. Libby for helpful discussions and critical reading of the manuscript and C. Lesser (Harvard) for an OspG plasmid. Portions of this research were carried out at the Stanford Synchrotron Radiation Lightsource, a Directorate of SLAC National Accelerator Laboratory and an Office of Science User Facility operated for the U.S. Department of Energy Office of Science by Stanford University. The SSRL Structural Molecular Biology Program is supported by the DOE Office of Biological and Environmental Research, and by the National Institutes of Health, National Institute of General Medical Sciences (including P41GM103393). The contents of this publication are solely the responsibility of the authors and do not necessarily represent the official views of NIGMS, NCRR or NIH. A portion of this research was performed using NMR facilities at EMSL, a national user facility sponsored by the Department of Energy's Office of Biological and Environmental Research and located at PNNL. This work was supported by National Institutes of Health Grants R01 GM098503 (P.S.B) and R01 GM088055 (R.E.K.), a Career Development Award from the Northwest Regional Center of Excellence (U54 AI057141, PSB), and a U.S. Public Health Service Grant NRSA 2T32 GM007270 from the National Institute of General Medical Sciences (J.N.P.). JRR is supported by Canadian Institute of Health Research (MOP-102594).
Author contributions
JNP determined the OspG crystal structure with assistance from ILT and RES. Biochemical assays were designed, performed, and analyzed by JNP, FDS, JDS, REK, and PSB. Mass spectrometry data was collected and analyzed by DLS and JV. Mouse model experiments were designed, performed, and interpreted by AD, AWS, and JRR. JNP and PSB prepared the manuscript with contributions from FDS, AD, JRR, and REK.
Conflict of interest
The authors declare that they have no conflict of interest.
Supplementary information
for this article is available online: http://emboj.embopress.org
References
- Anderson DM, Schmalzer KM, Sato H, Casey M, Terhune SS, Haas AL, Feix JB, Frank DW. Ubiquitin and ubiquitin-modified proteins activate the Pseudomonas aeruginosa T3SS cytotoxin, ExoU. Mol Microbiol. 2011;82:1454–1467. doi: 10.1111/j.1365-2958.2011.07904.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baba T, Ara T, Hasegawa M, Takai Y, Okumura Y, Baba M, Datsenko KA, Tomita M, Wannter BL, Mori H. Construction of Escherichia coli K-12 in-frame, single-gene knockout mutants: the Keio collection. Mol Sys Biol. 2006;2:1–11. doi: 10.1038/msb4100050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berndsen CE, Wiener R, Ian WY, Ringel AE, Wolberger C. A conserved asparagine has a structural role in ubiquitin-conjugating enzymes. Nat Chem Biol. 2013;9:154–156. doi: 10.1038/nchembio.1159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bliska JB, Guan KL, Dixon JE, Falkow S. Tyrosine phosphate hydrolysis of host proteins by an essential Yersinia virulence determinant. Proc Natl Acad Sci USA. 1991;88:1187–1191. doi: 10.1073/pnas.88.4.1187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen VB, Arendall WB, Headd JJ, Keedy DA, Immormino RM, Kapral GJ, Murray LW, Richardson JS, Richardson DC. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr D Biol Crystallogr. 2010;66:12–21. doi: 10.1107/S0907444909042073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christensen DE, Brzovic PS, Klevit RE. E2-BRCA1 RING interactions dictate synthesis of mono-or specific polyubiquitin chain linkages. Nat Struct Mol Biol. 2007;14:941–948. doi: 10.1038/nsmb1295. [DOI] [PubMed] [Google Scholar]
- Cowtan K. The Buccaneer software for automated model building. 1. Tracing protein chains. Acta Crystallogr D Biol Crystallogr. 2006;62:1002–1011. doi: 10.1107/S0907444906022116. [DOI] [PubMed] [Google Scholar]
- Cowtan K. Recent developments in classical density modification. Acta Crystallogr D Biol Crystallogr. 2010;66:470–478. doi: 10.1107/S090744490903947X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delaglio F, Grzesiek S, Vuister GW, Zhu G, Pfeifer J, Bax A. NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J Biomol NMR. 1994;6:277–293. doi: 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
- Dou H, Buetow L, Sibbet GJ, Cameron K, Huang DT. BIRC7–E2 ubiquitin conjugate structure reveals the mechanism of ubiquitin transfer by a RING dimer. Nat Struct Mol Biol. 2012;19:876–883. doi: 10.1038/nsmb.2379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of Coot. Acta Crystallogr D Biol Crystallogr. 2010;66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao X, Wan E, Mateo K, Callegari E, Wang D, Deng W, Puente J, Li F, Chaussee MS, Finlay BB, Lenardo MJ, Harwidge PR. Bacterial effector binding to ribosomal protein s3 subverts NF-κB function. PLoS Pathog. 2009;5:e1000708. doi: 10.1371/journal.ppat.1000708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guan KL, Dixon JE. Protein tyrosine phosphatase activity of an essential virulence determinant in Yersinia. Science. 1990;249:553–556. doi: 10.1126/science.2166336. [DOI] [PubMed] [Google Scholar]
- Haas AL, Rose IA. The mechanism of ubiquitin activating enzyme. A kinetic and equilibrium analysis. J Biol Chem. 1982;257:10329–10337. [PubMed] [Google Scholar]
- Holm L, Rosenström P. Dali server: conservation mapping in 3D. Nucleic Acids Res. 2010;38:W545–W549. doi: 10.1093/nar/gkq366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson BA, Blevins RA. NMRView: a computer program for the visualization and analysis of NMR data. J Biomol NMR. 1994;4:603–614. doi: 10.1007/BF00404272. [DOI] [PubMed] [Google Scholar]
- Juris SJ, Rudolph AE, Huddler D, Orth K, Dixon JE. A distinctive role for the Yersinia protein kinase: actin binding, kinase activation, and cytoskeleton disruption. Proc Natl Acad Sci USA. 2000;97:9431–9436. doi: 10.1073/pnas.170281997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim DW, Lenzen G, Page AL, Legrain P, Sansonetti PJ, Parsot C. The Shigella flexneri effector OspG interferes with innate immune responses by targeting ubiquitin-conjugating enzymes. Proc Natl Acad Sci USA. 2005;102:14046–14051. doi: 10.1073/pnas.0504466102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levin I, Eakin C, Blanc MP, Klevit RE, Miller SI, Brzovic PS. Identification of an unconventional E3 binding surface on the UbcH5∼Ub conjugate recognized by a pathogenic bacterial E3 ligase. Proc Natl Acad Sci USA. 2010;107:2848–2853. doi: 10.1073/pnas.0914821107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martino MC, Rossi G, Martini I, Tattoli I, Chiavolini D, Phalipon A, Sansonetti PJ, Bernardini ML. Mucosal lymphoid infiltrate dominates colonic pathological changes in murine experimental shigellosis. J Infect Dis. 2005;192:136–148. doi: 10.1086/430740. [DOI] [PubMed] [Google Scholar]
- McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. Phaser crystallographic software. J Appl Crystallogr. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murshudov GN, Skubák P, Lebedev AA, Pannu NS, Steiner RA, Nicholls RA, Winn MD, Long F, Vagin AA. REFMAC5 for the refinement of macromolecular crystal structure. Acta Crystallogr D Biol Crystallogr. 2011;67:355–367. doi: 10.1107/S0907444911001314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nobe R, Nougayrède JP, Taieb F, Bardiau M, Cassart D, Navarro-Garcia F, Mainil J, Hayashi T, Oswald E. Enterohaemorrhagic Escherichia coli serogroup O111 inhibits NF-κB-dependent innate responses in a manner independent of a type III secreted OspG orthologue. Microbiology. 2009;155:3214–3225. doi: 10.1099/mic.0.030759-0. [DOI] [PubMed] [Google Scholar]
- Ogawa M, Handa Y, Ashida H, Suzuki M, Sasakawa C. The versatility of Shigella effectors. Nat Rev Microbiol. 2008;6:11–16. doi: 10.1038/nrmicro1814. [DOI] [PubMed] [Google Scholar]
- Onodera NT, Ryu J, Durbic T, Nislow C, Archibald JM, Rohde JR. Genome sequence of Shigella flexneri serotype 5a strain M90T Sm. J Bacteriol. 2012;194:3022–3022. doi: 10.1128/JB.00393-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otwinowski Z, Minor W. Denzo and Scalepack. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- Painter J, Merritt EA. Optimal description of a protein structure in terms of multiple groups undergoing TLS motion. Acta Crystallogr D Biol Crystallogr. 2006;62:439–450. doi: 10.1107/S0907444906005270. [DOI] [PubMed] [Google Scholar]
- Phalipon A, Sansonetti PJ. Shigella's ways of manipulating the host intestinal innate and adaptive immune system: a tool box for survival? Immunol Cell Biol. 2007;85:119–129. doi: 10.1038/sj.icb7100025. [DOI] [PubMed] [Google Scholar]
- Plechanovová A, Jaffray EG, Tatham MH, Naismith JH, Hay RT. Structure of a RING E3 ligase and ubiquitin-loaded E2 primed for catalysis. Nature. 2012;489:115–120. doi: 10.1038/nature11376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poh J, Odendall C, Spanos A, Boyle C, Liu M, Freemont P, Holden DW. SteC is a Salmonella kinase required for SPI-2-dependent F-actin remodelling. Cell Microbiol. 2008;10:20–30. doi: 10.1111/j.1462-5822.2007.01010.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pruneda JN, Littlefield PJ, Soss SE, Nordquist KA, Chazin WJ, Brzovic PS, Klevit RE. Structure of an E3:E2∼Ub complex reveals an allosteric mechanism shared among RING/U-box ligases. Mol Cell. 2012;47:933–942. doi: 10.1016/j.molcel.2012.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pruneda JN, Stoll KE, Bolton LJ, Brzovic PS, Klevit RE. Ubiquitin in motion: structural studies of the ubiquitin-conjugating enzyme∼ubiquitin conjugate. Biochemistry. 2011;50:1624–1633. doi: 10.1021/bi101913m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quandt J, Hynes MF. Versatile suicide vectors which allow direct selection for gene replacement in gram-negative bacteria. Gene. 1993;127:15–21. doi: 10.1016/0378-1119(93)90611-6. [DOI] [PubMed] [Google Scholar]
- Rohde JR, Breitkreutz A, Chenal A, Sansonetti PJ, Parsot C. Type III secretion effectors of the IpaH family are E3 ubiquitin ligases. Cell Host Microbe. 2007;1:77–83. doi: 10.1016/j.chom.2007.02.002. [DOI] [PubMed] [Google Scholar]
- Rytkönen A, Poh J, Garmendia J, Boyle C, Thompson A, Liu M, Freemont P, Hinton JCD, Holden DW. SseL, a Salmonella deubiquitinase required for macrophage killing and virulence. Proc Natl Acad Sci USA. 2007;104:3502–3507. doi: 10.1073/pnas.0610095104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sellge G, Magalhaes JG, Konradt C, Fritz JH, Salgado-Pabon W, Eberl G, Bandeira A, Di Santo JP, Sansonetti PJ, Phalipon A. Th17 cells are the dominant T cell subtype primed by Shigella flexneri mediating protective immunity. J Immunol. 2010;184:2076–2085. doi: 10.4049/jimmunol.0900978. [DOI] [PubMed] [Google Scholar]
- Taylor SS, Ilouz R, Zhang P, Kornev AP. Assembly of allosteric macromolecular switches: lessons from PKA. Nat Rev Mol Cell Biol. 2012;13:646–658. doi: 10.1038/nrm3432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The PyMOL Molecular Graphics System, Version 1.5.0.4 Schrödinger, LLC.
- Wenzel DM, Lissounov A, Brzovic PS, Klevit RE. UBCH7 reactivity profile reveals parkin and HHARI to be RING/HECT hybrids. Nature. 2011;474:105–108. doi: 10.1038/nature09966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winn MD, Ballard CC, Cowtan KD, Dodson EJ, Emsley P, Evans PR, Keegan RM, Krissinel EB, Leslie AGW, McCoy A, McNicholas SJ, Murshudov GN, Pannu NS, Potterton EA, Powell HR, Read RJ, Vagin A, Wilson KS. Overview of the CCP4 suite and current developments. Acta Crystallogr D Biol Crystallogr. 2011;67:235–242. doi: 10.1107/S0907444910045749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu PY, Hanlon M, Eddins M, Tsui C, Rogers RS, Jensen JP, Matunis MJ, Weissman AM, Wolberger CP, Pickart CM. A conserved catalytic residue in the ubiquitin-conjugating enzyme family. EMBO J. 2003;22:5241–5250. doi: 10.1093/emboj/cdg501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yunus AA, Lima CD. Lysine activation and functional analysis of E2-mediated conjugation in the SUMO pathway. Nat Struct Mol Biol. 2006;13:491–499. doi: 10.1038/nsmb1104. [DOI] [PubMed] [Google Scholar]
- Zheng J, Trafny EA, Knighton DR, Xuong NH, Taylor SS, Ten Eyck LF, Sowadski JM. 2.2 Å refined crystal structure of the catalytic subunit of cAMP-dependent protein kinase complexed with MnATP and a peptide inhibitor. Acta Crystallogr D Biol Crystallogr. 1993;49:362–365. doi: 10.1107/S0907444993000423. [DOI] [PubMed] [Google Scholar]
- Zhou Y, Dong N, Hu L, Shao F. The Shigella type three secretion system effector OspG directly and specifically binds to host ubiquitin for activation. PLoS ONE. 2013;8:e57558. doi: 10.1371/journal.pone.0057558. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.