

Abstract
Protein kinase Cζ (PKCζ) is phosphorylated at the activation loop and the turn motif (TM). However, the TM kinase and functional relevance of TM phosphorylation remain largely unknown. We demonstrate that PKCζ TM is phosphorylated directly by the mTORC2 complex, and this phosphorylation is required for maintaining PKCζ kinase activity and stability. Functionally, mTORC2 regulates the activity of Rho family of GTPases, and therefore the organization of the actin cytoskeleton, through the control of PKCζ activity. Taken together, our findings identify PKCζ as a novel substrate and downstream effector of mTORC2 signaling.
Keywords: actin cytoskeleton, mTOR, phosphorylation, PKCζ, protein stability
Introduction
PKCζ, an atypical member of the PKC family, has been widely implicated in the regulation of cell polarity, tumorigenesis, and immune response 1, 2. However, due to its unique domain composition and relatively low sequence similarity with conventional and novel PKCs, the molecular mechanism underlying the regulation of PKCζ is less understood. The N-terminal regulatory domain in PKCζ consists of a PB1 and a single cysteine-rich zinc-finger domain; as a consequence, PKCζ is insensitive to diacylglycerol- and Ca2+-mediated activation due to lack of functional C1 and C2 domains 3. In addition, PKCζ differs in the phosphorylation sequence that are conserved among AGC (protein kinase A/G/C) kinase family members. The conventional and novel PKCs are known to be phosphorylated at three conserved phosphorylation sites, namely the activation loop (A-loop), the turn motif (TM), and the hydrophobic motif (HM) 4. In the case of PKCζ, only the A-loop and the TM are available for phosphorylation, while the HM is replaced by Glu, a phospho-mimetic residue 3. PDK-1 has been identified as the A-loop kinase for all PKC isozymes including PKCζ 4, 5.
mTOR is a serine/threonine kinase which exists in two distinct functional complexes named mTOR complex 1 (mTORC1) and 2 (mTORC2) 6. Both mTOR complexes contain multiple protein components with raptor and rictor functioning as the key nucleating members in mTORC1 and mTORC2, respectively. More recently, it has been shown that mTORC2 components are required for the phosphorylation of both the TM and the HM in conventional PKC isozymes and Akt, and the TM phosphorylation allows the maturation and stablization of the kinases 7, 8, 9, 10. However, the nature of the TM kinase and the functional importance of TM phosphorylation in PKCζ remain unknown.
In this study, we report the identification of mTOR in the mTORC2 complex as the upstream kinase that phosphorylates the TM of PKCζ in vitro and in cells. We show that mTOR-mediated phosphorylation prevents the degradation of PKCζ and promotes its kinase activity. Furthermore, PKCζ functions downstream of mTORC2 to control actin cytoskeleton orgnization.
Results and Discussion
The TM phosphorylation of PKCζ requires mTORC2 components
In our effort to study the regulation of PKCζ, we found that the phosphorylation of PKCζ at T560 (the TM) was completely lost in rictor knockout (Ric−/−) MEF cells, whereas T410 (the A-loop) of PKCζ was phosphorylated to a similar extent as in WT MEF cells (Fig 1A, lanes 1–4). As a control, insulin-induced phosphorylation of Akt at S473, a known substrate of mTORC2, was absent in Ric−/− MEF cells as well. To confirm the specificity of rictor in regulating the TM phosphorylation of PKCζ, a Myc-tagged rictor was re-introduced into Ric−/− MEF cells. As a consequence, the TM phosphorylation of PKCζ, as well as insulin-induced phosphorylation of Akt at S473, was restored (Fig 1A, lanes 5–6). The level of T560 phosphorylation was lower in Myc-Rictor transfected Ric−/− MEF cells compared to WT MEF cells. This is likely due to the fact that only ∼30% of cells were transfected after transient transfection. In addition, we found that the TM phosphorylation of PKCζ was absent in Ric−/− MEF cells regardless of serum or EGF stimulation, whereas the A-loop of PKCζ was constitutively phosphorylated in both WT and Ric−/− MEF cells (supplementary Fig S1A and B). As a control, the phosphorylation of Akt at S473 was rapidly induced upon both serum and EGF treatment in WT MEF cells but completely absent in Ric−/− MEF cells.
Figure 1.
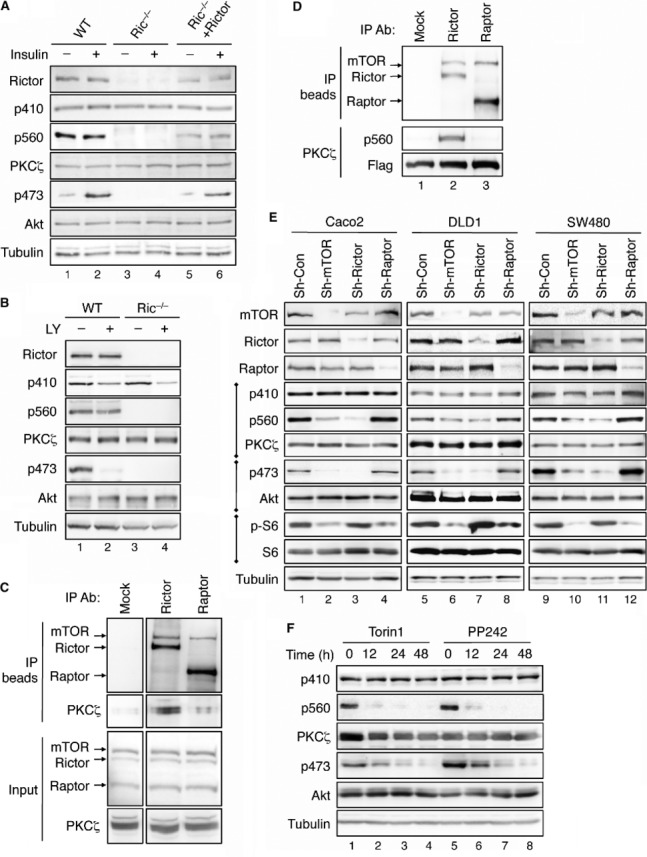
- The TM phosphorylation of PKCζ requires the expression of rictor. The following cells, including WT, rictor knockout (Ric−/−), and Ric−/− MEF cells transfected with Myc-Rictor, were serum-starved for 4 h and subsequently treated with insulin (0.1 μM) for 15 min. Cells lysates were analyzed by immunoblotting using indicated antibodies.
- The TM phosphorylation of PKCζ is insensitive to the inhibition of PI3K activity. WT and Ric−/−MEF cells were treated with LY294002 (30 μM) in serum-free medium for 1 h.
- PKCζ is associated with the mTORC2 complex. 293T cell lysates were immunoprecipitated with either protein A/G beads alone (Mock, lane 1) or antibodies to rictor or raptor coupled to protein A/G beads (lanes 2–3, respectively). The immunoprecipitates and 5% of the input were analyzed by immunoblotting with indicated antibodies.
- PKCζ is phosphorylated by the mTORC2 complex in vitro. 293T cell lysates were immunoprecipitated with either protein A/G beads alone (Mock) or antibodies to rictor or raptor coupled to protein A/G beads. In vitro phosphorylation reactions were carried out by incubating the immunoprecipitates with partially purified PKCζ as the substrate.
- Knockdown of rictor but not raptor results in a decrease in TM phosphorylation of PKCζ. Stable raptor, rictor, or mTOR knockdown colon cancer cells were analyzed for the phosphorylation of PKCζ, Akt, and rpS6 using phospho-specific antibodies.
- The TM of PKCζ is dephosphorylated in cells treated with mTOR kinase inhibitors. 293T cells were treated with Torin1 (100 nM, lanes 1–4) or PP242 (1 μM, lanes 5–8) for the indicated time.
Source data are available online for this figure.
Interestingly, PKCζ remained phosphorylated after serum starvation for 4 h and growth factor treatment had no obvious effect on stimulating the phosphorylation at either the A-loop or the TM of PKCζ in WT MEF cells. To further determine if alteration of PI3K activity affects the phosphorylation of PKCζ, MEF cells were treated with LY294002 for 1 h. As expected, the phosphorylation of Akt was abolished upon inhibition of PI3K (Fig 1B). However, the TM phosphorylation of PKCζ remained largely unchanged whereas the A-loop phosphorylation was decreased (Fig 1B). These data suggest that the A-loop phosphorylation of PKCζ is sensitive to PI3K as previously reported 5, 11, however, to a less extent compared to Akt. More importantly, the TM phosphorylation of PKCζ is insensitive to PI3K but requires the expression of rictor. This constitutive nature of TM phosphorylation may suggest a co-translational mechanism that has been reported for mTORC2-mediated TM phosphorylation of Akt 12.
Identification of PKCζ as a novel substrate of mTOR in the mTORC2 complex
Since rictor is a key component in the mTORC2 complex, we tested the hypothesis that PKCζ TM is phosphorylated by mTOR in the mTORC2 complex. Co-immunoprecipitation experiments showed that PKCζ interacted with rictor and mTOR in mTORC2, but not raptor in mTORC1 (Fig 1C, lanes 2–3). Moreover, PKCζ was phosphorylated in vitro at T560 by mTOR in the complex with rictor (Fig 1D, lane 2), whereas the raptor-mTOR complex was unable to phosphorylate PKCζ (Fig 1D, lane 3), suggesting that the T560 site is a specific substrate of mTORC2. We next tested whether silencing mTORC2 components affects the TM phosphorylation of PKCζ in three different colon cancer cell lines. Indeed, PKCζ phosphorylation at T560, as well as Akt phosphorylation at S473, was largely abolished in rictor and mTOR knockdown cells (Fig 1E). In contrast, the TM phosphorylation was not affected by silencing raptor, thus confirming the specificity of mTORC2. As a control, the phosphorylation of S6, a substrate of S6 kinase, was decreased in raptor and mTOR knockdown cells (Fig 1E). Furthermore, the A-loop phosphorylation of PKCζ remained unchanged in raptor, rictor, or mTOR knockdown cells, suggesting that the A-loop of PKCζ is not regulated by either mTOR complex.
Finally, we examined the phosphorylation of PKCζ in cells treated with mTOR kinase inhibitors. Inhibition of mTOR activity led to complete dephosphorylation of T560 in PKCζ, while had no effect on T410 phosphorylation. The phosphorylation of Akt at S473 was dephosphorylated in cells treated with mTOR inhibitors as well (Fig 1F). Since PKCι, another member of the atypical PKC family, is highly homologous to PKCζ, we also determined if PKCι TM is phosphorylated by mTORC2. Similar to PKCζ, the TM phosphorylation of PKCι was completely lost in mTOR inhibitor-treated 293T cells whereas the A-loop was not affected (supplementary Fig S1C). Thus, PKCι TM (T564 in human PKCι) is also a substrate of mTORC2. However, we did not detect PKCι expression (the mouse homologue is also known as PKCλ) in WT or Ric−/− MEF cells using the PKCι/λ-specific antibody. Results shown in all subsequent experiments performed in MEF cells are considered PKCζ-specific. Although earlier studies have suggested that PKCζ TM is phosphorylated via autophosphorylation 13, our findings provide direct evidence that mTORC2 is responsible for phosphorylating the TM of atypical PKCs, including PKCζ and PKCι.
The TM phosphorylation is a key determinant of PKCζ kinase activity
We next determined whether the TM phosphorylation is required for PKCζ activity. Although PKCζ is basally phosphorylated at both the A-loop and the TM (Fig 1), mutating either site to Ala (T410A or T560A) largely had not effect on the phosphorylation at the other site (Fig 2A), thus confirming that the A-loop and the TM phosphorylation are independently mediated by two kinases. The activity of PKCζ expressed in Ric−/− MEF cells was markedly decreased as the result of loss of TM phosphorylation (Fig 2B). Moreover, similar to the A-loop mutant T410A, the TM phosphorylation-deficient mutant (T560A) was inactive, whereas the phospho-mimetic mutants T410E and T560D were equally active in vitro (Fig 2C). There was a modest increase in WT PKCζ activity upon insulin treatment (Fig 2B and C). However, insulin had no effect on stimulating the activity of phosphorylation-deficient mutants, and the activity of phospho-mimetic mutants was similar to that of WT PKCζ after insulin treatment (Fig 2C). Collectively, our data show that loss of phosphorylation at the TM has a similar detrimental effect on the kinase activity of PKCζ as losing phosphorylation at the A-loop. Thus, phosphorylation at both sites is required for PKCζ activation.
Figure 2.

- The A-loop and TM of PKCζ are independently phosphorylated. 293T cells transfected with Flag-PKCζ, Flag-PKCζ/T410A, or Flag-PKCζ/T560A were serum-starved overnight and subsequently treated with insulin for 15 min.
- The kinase activity of PKCζ is decreased in Ric−/− MEF cells. WT and Ric−/− MEF cells were serum-starved for 4 h and treated with insulin for 15 min. Cell lysates were immunoprecipitated with the PKCζ antibody and the kinase activity was assessed using MBP as the substrate. The level of MBP phosphorylation catalyzed by WT PKCζ under control condition was set to 1 and all other conditions were normalized accordingly. Total MBP in each reaction was visualized on the membrane by Ponceau S staining.
- Phosphorylation of PKCζ at both T410 and T560 sites are required for its kinase activity. 293T cells transfected with vector, Flag-PKCζ, Flag-PKCζ/T410A, Flag-PKCζ/T410E, Flag-PKCζ/T560A, or Flag-PKCζ/T560D were serum starved overnight and subsequently treated with insulin for 15 min. Cell lysates were immunoprecipitated with Flag beads and the kinase activity was determined using MBP as the substrate.
Source data are available online for this figure.
The TM phosphorylation controls the protein stability and ubiquitination of PKCζ
The phosphorylation of Akt and conventional PKC isozymes has been implicated in maintaining the protein stability 7. It remains unknown whether the phosphorylation of PKCζ controls its stability in cells. Since inhibition of HSP90 disrupts proper folding of multiple protein kinases including PKC isozymes 7, we first examined the rate of PKCζ degradation in the presence of HSP90 inhibitor, 17-AAG. The WT PKCζ was a very stable protein even in cells treated with 17-AAG (Fig 3A). However, loss of phosphorylation at the TM drastically reduced the half-life of PKCζ, whereas the phosphorylation-deficient mutation at the A-loop had no effect on PKCζ stability (Fig 3A and B). Consistent with our finding that PKCζ was basally phosphorylated at both the TM and the A-loop in cells, the phospho-mimetic mutants were equally stable as WT PKCζ (Fig 3C). In addition, the degradation of endogenous PKCζ in Ric−/− MEF cells was significantly faster compared to that of WT MEF cells (Fig 3D and E). A similar decrease in Akt stability was observed in Ric−/− MEF cells (Fig 3D) 7. Furthermore, loss of TM phosphorylation in PKCζ drastically increased the level of ubiquitination suggesting faster turnover of the protein (Fig 3F, lanes 3–4). The ubiquitination of T560D mutant was markedly lower than that of T560A but slightly higher compared to WT PKCζ (Fig 3F, lanes 5–6). This is likely because the phospho-mimetic residues are usually not perfect substitutions for phosphate groups. More importantly, the ubiquitination of PKCζ expressed in Ric−/− MEF cells was largely increased as well (Fig 3G). Taken together, our data demonstrate that mTORC2-mediated TM phosphorylation plays a critical role in stabilizing PKCζ and lack of TM phosphorylation leads to increased ubiquitination and degradation of PKCζ.
Figure 3.
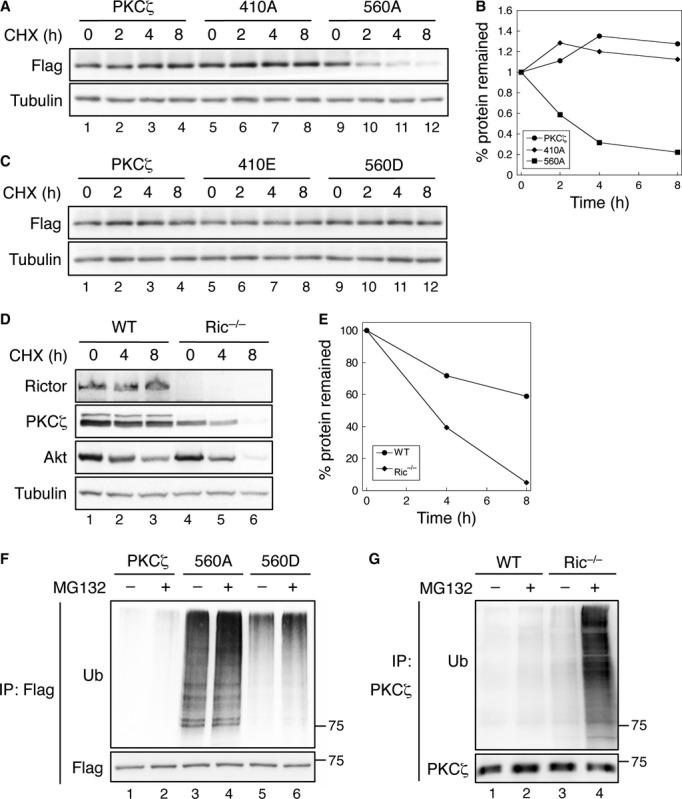
- Inhibition of Hsp90 activity destabilizes PKCζ/T560A mutant. 293T cells transiently transfected with Flag-PKCζ, Flag-PKCζ/T410A, or Flag-PKCζ/T560A were pre-treated with Hsp90 inhibitor 17-AAG for overnight and subsequently treated with cycloheximide (CHX). Cell lysates were analyzed for PKCζ expression.
- Graph showing the relative amount of WT and mutant PKCζ remained in cells following CHX treatment. Western blots shown in (A) were quantified by normalizing the amount of total PKCζ to that of tubulin.
- The expression of phospho-mimetic mutant PKCζ is not affected by Hsp90 inhibitor. 293T cells transfected with Flag-PKCζ, Flag-PKCζ/T410E, or Flag-PKCζ/T560D were treated and analyzed as described in (A).
- Inhibition of Hsp90 activity destabilizes PKCζ in Ric−/− MEF cells. WT and Ric−/− MEF cells were treated and analyzed as described in (A).
- Graph showing the relative amount of PKCζ remained in WT and Ric−/− MEF cells following CHX treatment.
- The TM phosphorylation protects PKCζ from ubiquitination-mediated degradation. 293T cells transfected with Flag-PKCζ, Flag-PKCζ/T560A, or Flag-PKCζ/T560D were immunoprecipitated with Flag beads.
- Ubiqutination of PKCζ is enhanced in Ric−/− MEF cells. WT and Ric−/− MEF cell lysates were immunoprecipitated with the PKCζ antibody. The levels of ubiquitination of PKCζ were detected using the ubiquitin antibody.
PKCζ functions downstream of mTORC2 to control actin cytoskeleton organization
In mammalian cells, actin cytoskeleton reorganization is a dynamic and complex process that is orchestrated by activation of Rho family small GTPases, particularly Rho, Rac, and Cdc42. Previous studies have shown that mTORC2 modulates actin cytoskeleton 14. However, the underlying molecular mechanism is less clear. We found that the morphology and the organization of actin cytoskeleton in WT and Ric−/− MEF cells showed striking differences. Specifically, when adhered to fibronectin-coated coverglasses, the formation of lamellipodia was readily observed in WT MEF cells which coincided with cortical organization of actin (Fig 4A). Ric−/− MEF cells generally showed elongated and more spindle-like morphology associated with abundant actin stress fiber formation, however, apparently lacking lamellipodia (Fig 4B). More examples of WT and Ric−/− MEF cells are shown in supplementary Fig S2. The percentage of cells with lamellipodia and cortical actin organization was 73.6% and 18.4% in WT and Ric−/− MEF cells, respectively, whereas stress fibers were observed in other cells (Fig 4C). Since actin cytoskeleton organization is directly controlled by Rho family small GTPases 15, we next determined if the activation of these GTPases is affected by rictor and PKCζ. As shown in Fig 4D, the amount of activated Rac1 and Cdc42 was decreased by 90% and 50%, respectively, in Ric−/− MEF cells compared to WT cells. A similar decrease in Rac1 and Cdc42 activation was observed when Pkcζ was knocked down in WT MEF cells. Consistent with the notion that RhoA activation is associated with stress fiber formation 15, the amount of activated RhoA was markedly increased in Ric−/− and Pkcζ knockdown MEF cells (Fig 4D).
Figure 4.
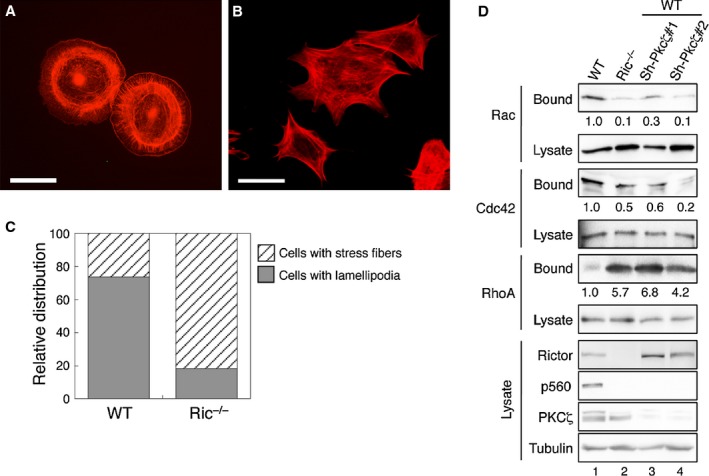
Ritor regulates actin cytoskeleton organization by controlling Rho family small GTPases.
A–B Actin cytoskeleton rearrangement is disrupted in rictor null MEF cells. WT (A) and Ric−/− (B) MEF cells seeded on fibronectin-coated coverslips were treated with EGF for 15 min. Actin was stained with Alexa594-phalloidin. Scale bar, 50 μm.
C The percentages of WT and Ric−/− cells with lamellipodia or stress fibers as indicated by actin staining were quantified and expressed graphically. WT, 73.6% with lamellipodia and 26.4% with stress fibers (n = 205); Ric−/−, 18.4% with lamellipodia and 81.6% with stress fibers (n = 136, P < 0.0001 by chi-square test comparing between the WT and Ric−/− groups).
D Loss of rictor or Pkcζ expression alters the activation of Rac1, Cdc42, and RhoA. WT, Ric−/− and two different Pkcζ knockdown MEF cells were subjected to Rac1/Cdc42 and RhoA activity assays. The relative activation of small GTPases was quantified by normalizing the amount of bound Rac1, Cdc42, or RhoA to that of total lysates.
To determine if decreased PKCζ activity in Ric−/− MEF cells contributes to altered actin cytoskeleton organization, we overexpressed EGFP-T560D in these cells. Indeed, the cortical organization of actin and lamellipodia formation were largely restored in cells expressing T560D PKCζ whereas stress fibers remained in untransfected cells (Fig 5A and B). As a control, overexpression of EGFP-T560A mutant in Ric−/− MEF cells had little effect on promoting lamellipodia formation (Fig 5C-D). More examples of T560D and T560A expressing Ric−/− MEF cells are shown in supplementary Fig S3. The percentage of cells with lamellipodia was determined to be 63.8% and 30.4% in T560D and T560A expressing Ric−/− MEF cells, respectively (Fig 5E). The patterns of actin organization remained unchanged in untransfected cells. Furthermore, overexpression of T560D PKCζ in Ric−/− MEF cells rescued the defect in Rac1 and Cdc42 activation and normalized the activity of RhoA (Fig 5F and G). In marked contrast, overexpression of T560A mutant had no effect on the activation of Rac1, Cdc42, or RhoA in Ric−/− MEF cells (supplementary Fig S4). Collectively, these results demonstrate that PKCζ is likely the downstream effector of rictor in controlling actin cytoskeleton rearrangement by modulating the activity of Rho family small GTPases.
Figure 5.
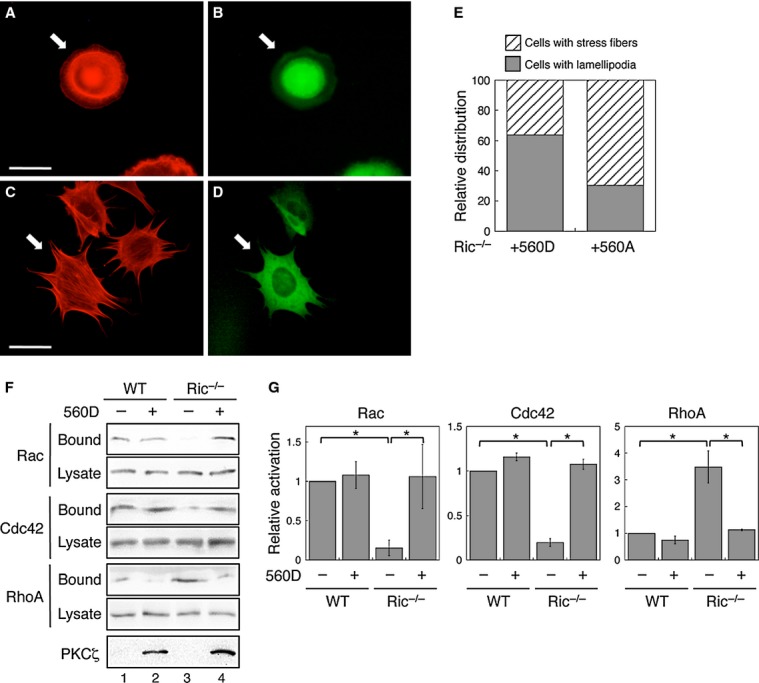
Overexpression of PKCζ/T560D rescues the actin cytoskeleton organization in Rictor null MEF cells.
A–D Ric−/− MEF cells transfected with EGFP-PKCζ/T560D (A and B) or EGFP-PKCζ/T560A (C and D) were seeded on fibronectin-coated coverslips and treated with EGF. Actin was visualized using Alexa594-phalloidin (A and C), whereas the transfected cells were visualized by EGFP (B and D). Arrows mark the transfected cells. Scale bar, 50 μm.
E The percentages of Ric−/− cells transfected with EGFP-PKCζ/T560D or EGFP-PKCζ/T560A with lamellipodia or stress fibers were quantified and expressed graphically. Note that only the GFP-positive cells (transfected cells) were included in the quantification. EGFP-PKCζ/T560D, 63.8% with lamellipodia and 36.2% with stress fibers (n = 72); EGFP-PKCζ/T560A, 30.4% with lamellipodia and 69.6% with stress fibers (n = 79, P < 0.0001 by chi-square test comparing between the two groups).
F Overexpression of PKCζ/T560D rescues the activation of Rac1, Cdc42, and RhoA. WT and Ric−/− MEF cells transfected with vector or Flag-PKCζ/T560D were subjected to Rac1/Cdc42 and RhoA activity assays.
G The relative activation of each small GTPase was quantified by normalizing the amount of bound to that of total cellular proteins. Data shown in graphs represent the mean ± SEM (n = 3, * indicates P < 0.01 by Student's t-test).
Recent studies have placed rictor and mTORC2 at the forefront of tumor initiation and progression. Identification of PKCζ as a substrate of mTORC2 to control lamellipodia formation and cytoskeleton organization suggests that PKCζ may play a role in regulating tumor cell migration and metastasis downstream of mTOR. Indeed, PKCζ has been linked to rictor-dependent breast cancer metastasis 16. A recent study has identified lethal giant larvae 2 (LLGL2) as a substrate of atypical PKCs to regulate cell migration 17. By elucidating the role of TM phosphorylation, our study demonstrates that the constitutive phosphorylation of PKCζ at the TM not only protects it from proteasome-mediated degradation but also ensures the full activation of the kinase.
Materials and Methods
Cell culture, antibodies, and expression constructs
Cell culture conditions and sources of antibodies as well as all plasmids used are detailed in the Supplementary Information.
Immunoprecipitation and in vitro phosphorylation assays
For all immunoprecipitation experiments cells were lysed in the lysis buffer containing 1% Triton (50 mM Na2HPO4, 1 mM sodium pyrophosphate, 20 mM NaF, 2 mM EDTA, 2 mM EGTA, 1% Triton X-100, 1 mM DTT, 200 μM benzamidine, 40 μg ml−1 leupeptin, 200 μM PMSF) except where maintaining the integrity of mTOR complexes is required, in which case the lysis buffer containing 0.3% CHAPS instead of 1% Triton X-100 was used 10. For in vitro phosphorylation of PKCζ, endogenous mTORC1 and mTORC2 complexes were immunoprecipitated from the detergent-solubilized 293T cell lysates using the raptor or rictor antibody coupled to protein A/G beads. To generate unphosphorylated PKCζ, 293T cells transfected with Flag-PKCζ were lysed and immunoprecipitated with anti-Flag agarose, the Flag beads were treated with λ-PPase in the phosphatase reaction buffer (50 mM Tris-HCl, pH 7.5, 100 mM NaCl, 2 mM DTT, 0.1 mM EGTA, and 0.01% Brij 35) at 30°C for 1 h to remove phosphates. Dephosphorylated Flag-PKCζ was eluted from the beads with the Flag peptide, dialyzed, and subsequently used as the substrate. In vitro phosphorylation reactions were initiated by adding the raptor and rictor immunoprecipitates to the dephosphorylated and partially purified Flag-PKCζ in the kinase buffer (25 mM Tris-HCl, pH7.4, 10 mM MgCl2, 5 mM β-glycerophosphate, and 200 μM ATP). Reactions were carried out at 30°C for 30 min and stopped by adding SDS sample buffer.
To assay the kinase activity PKCζ, 293T cells transfected with WT and mutant Flag-PKCζ were lysed and incubated with anti-Flag agarose. For endogenous PKCζ, WT and Ric−/− MEF cell lysates were immunoprecipitated using the anti-PKCζ mAb coupled to protein A/G beads. The beads were washed and used as the kinase to phosphorylate myelin basic protein (MBP) as substrate. The kinase reactions were carried out as described above in the kinase buffer containing 5 μg MBP and 10 Ci [γ-32P]ATP. MBP phosphorylation was detected by exposing the membrane to X-ray film and the amount of PKCζ was analyzed using immunoblotting.
Ubiquitination and degradation of PKCζ
To examine the protein stability of PKCζ, cells were pre-treated with HSP90 inhibitor 17-AAG (1 μM) for overnight and subsequently treated with cycloheximide (CHX, 20 μg/ml) or MG-132 (20 μM) as indicated. Specific experimental procedures are described in the Supplementary information.
RhoA/Rac1/Cdc42 activity assays
RhoA and Rac1/Cdc42 activity was assessed using GST-tagged Rho-binding domain of Rhotekin (RBD) and GST-tagged p21 binding domain of PAK1 (GST-PBD), respectively, as described previously 18. Briefly, cells grown to ∼70% confluency in regular growth medium were collected in RhoA or Rac1/Cdc42 lysis buffer and cleared extracts were incubated for 30 min at 4°C with glutathione beads coupled with GST-RBD or GST-PBD to pull down activated RhoA or Rac1/Cdc42, respectively. The amount of total and active RhoA, Rac1, and Cdc42 was determined by Western blotting.
Acknowledgments
We thank Dr. David Sabatini (MIT) for providing wild-type and Ric−/− MEF cells, and Drs. Nathanael Gray and David Sabatini for providing Torin1. This work was supported by NIH R01CA133429 (T.G.) and American Cancer Society RSG0822001TBE (T.G.).
Conflict of Interest
The authors declare that they have no conflict of interest.
Supplementary information for this article is available online: http://embor.embopress.org
References
- Diaz-Meco MT, Moscat J. The atypical PKCs in inflammation: NF-kappaB and beyond. Immunol Rev. 2012;246:154–167. doi: 10.1111/j.1600-065X.2012.01093.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wodarz A, Nathke I. Cell polarity in development and cancer. Nat Cell Biol. 2007;9:1016–1024. doi: 10.1038/ncb433. [DOI] [PubMed] [Google Scholar]
- Suzuki A, Akimoto K, Ohno S. Protein kinase C lambda/iota (PKClambda/iota): a PKC isotype essential for the development of multicellular organisms. J Biochem. 2003;133:9–16. doi: 10.1093/jb/mvg018. [DOI] [PubMed] [Google Scholar]
- Newton AC. Regulation of the ABC kinases by phosphorylation: protein kinase C as a paradigm. Biochem J. 2003;370:361–371. doi: 10.1042/BJ20021626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou MM, Hou W, Johnson J, Graham LK, Lee MH, Chen CS, Newton AC, Schaffhausen BS, Toker A. Regulation of protein kinase C zeta by PI 3-kinase and PDK-1. Curr Biol. 1998;8:1069–1077. doi: 10.1016/s0960-9822(98)70444-0. [DOI] [PubMed] [Google Scholar]
- Laplante M, Sabatini DM. mTOR signaling in growth control and disease. Cell. 2012;149:274–293. doi: 10.1016/j.cell.2012.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Facchinetti V, Ouyang W, Wei H, Soto N, Lazorchak A, Gould C, Lowry C, Newton AC, Mao Y, Miao RQ, Sessa WC, Qin J, Zhang P, Su B, Jacinto E. The mammalian target of rapamycin complex 2 controls folding and stability of Akt and protein kinase C. EMBO J. 2008;27:1932–1943. doi: 10.1038/emboj.2008.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikenoue T, Inoki K, Yang Q, Zhou X, Guan KL. Essential function of TORC2 in PKC and Akt turn motif phosphorylation, maturation and signalling. EMBO J. 2008;27:1919–1931. doi: 10.1038/emboj.2008.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacinto E, Facchinetti V, Liu D, Soto N, Wei S, Jung SY, Huang Q, Qin J, Su B. SIN1/MIP1 maintains rictor-mTOR complex integrity and regulates Akt phosphorylation and substrate specificity. Cell. 2006;127:125–137. doi: 10.1016/j.cell.2006.08.033. [DOI] [PubMed] [Google Scholar]
- Sarbassov DD, Guertin DA, Ali SM, Sabatini DM. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science. 2005;307:1098–1101. doi: 10.1126/science.1106148. [DOI] [PubMed] [Google Scholar]
- Le Good JA, Ziegler WH, Parekh DB, Alessi DR, Cohen P, Parker PJ. Protein kinase C isotypes controlled by phosphoinositide 3-kinase through the protein kinase PDK1. Science. 1998;281:2042–2045. doi: 10.1126/science.281.5385.2042. [DOI] [PubMed] [Google Scholar]
- Oh WJ, Wu CC, Kim SJ, Facchinetti V, Julien LA, Finlan M, Roux PP, Su B, Jacinto E. mTORC2 can associate with ribosomes to promote cotranslational phosphorylation and stability of nascent Akt polypeptide. EMBO J. 2010;29:3939–3951. doi: 10.1038/emboj.2010.271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Standaert ML, Bandyopadhyay G, Kanoh Y, Sajan MP, Farese RV. Insulin and PIP3 activate PKC-zeta by mechanisms that are both dependent and independent of phosphorylation of activation loop (T410) and autophosphorylation (T560) sites. Biochemistry. 2001;40:249–255. doi: 10.1021/bi0018234. [DOI] [PubMed] [Google Scholar]
- Sarbassov DD, Ali SM, Kim DH, Guertin DA, Latek RR, Erdjument-Bromage H, Tempst P, Sabatini DM. Rictor, a novel binding partner of mTOR, defines a rapamycin-insensitive and raptor-independent pathway that regulates the cytoskeleton. Curr Biol. 2004;14:1296–1302. doi: 10.1016/j.cub.2004.06.054. [DOI] [PubMed] [Google Scholar]
- Nobes CD, Hall A. Rho, rac, and cdc42 GTPases regulate the assembly of multimolecular focal complexes associated with actin stress fibers, lamellipodia, and filopodia. Cell. 1995;81:53–62. doi: 10.1016/0092-8674(95)90370-4. [DOI] [PubMed] [Google Scholar]
- Zhang F, Zhang X, Li M, Chen P, Zhang B, Guo H, Cao W, Wei X, Cao X, Hao X, Zhang N. mTOR complex component Rictor interacts with PKCzeta and regulates cancer cell metastasis. Cancer Res. 2010;70:9360–9370. doi: 10.1158/0008-5472.CAN-10-0207. [DOI] [PubMed] [Google Scholar]
- Kjaer S, Linch M, Purkiss A, Kostelecky B, Knowles PP, Rosse C, Riou P, Soudy C, Kaye S, Patel B, Soriano E, Murray-Rust J, Barton C, Dillon C, Roffey J, Parker PJ, McDonald NQ. Adenosine-binding motif mimicry and cellular effects of a thieno[2,3-d]pyrimidine-based chemical inhibitor of atypical protein kinase C isoenzymes. Biochem J. 2013;451:329–342. doi: 10.1042/BJ20121871. [DOI] [PubMed] [Google Scholar]
- Gulhati P, Bowen KA, Liu J, Stevens PD, Rychahou PG, Chen M, Lee EY, Weiss HL, O'Connor KL, Gao T, Evers BM. mTORC1 and mTORC2 regulate EMT, motility, and metastasis of colorectal cancer via RhoA and Rac1 signaling pathways. Cancer Res. 2011;71:3246–3256. doi: 10.1158/0008-5472.CAN-10-4058. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.