

Abstract
Superoxide dismutase 1 (SOD1) proteins harboring mutations linked to familial Amyotrophic Lateral Sclerosis (FALS) uniformly show heightened potential to form high molecular weight structures. Here, we examine the domains of SOD1 that are involved in forming these structures (aggregates) and study the role of intra and intermolecular disulfide bonds. An analysis of disease mutations identified to date reveals a non-random distribution with predominant occurrence at residues within highly conserved β-strands or at highly conserved residues in loop domains. Using a cell transfection assay for aggregation, we determined that no single domain in SOD1 is indispensable in the formation of sedimentable aggregates, suggesting multiple potential motifs in the protein mediate non-native interactions. By a cell-free aggregation assay, analysis of transgenic mouse tissues, and mutagenesis approaches, we found evidence that redox conditions may modulate SOD1 aggregation; reduction of the native intra-molecular disulfide bonds may predispose SOD1 to unfolding and aggregation, whereas non-native inter-molecular disulfide linkages may help stabilize aggregates in vivo. The results suggest a possible mechanism for diversity in the structures formed by different SOD1mutants; and define a potential contribution of redox conditions to SOD1 aggregation.
Introduction
Cu/Zn superoxide dismutase (SOD1), a principle intracellular antioxidant enzyme, metabolizes superoxide radicals to molecular oxygen and hydrogen peroxide (McCord and Fridovich 1969; Fridovich 1974). Since the first mutation in SOD1 was linked to Familial Amyotrophic Lateral Sclerosis (FALS) (Rosen et al. 1993), a large number of SOD1 variants have been identified (http://alsod.iop.kcl.ac.uk/Index.aspx). Collectively SOD1 mutations account for about 20% of the familial cases of the disease, which is characterized by selective degeneration of motor neurons. Primarily a cytosolic protein (Crapo et al. 1992), the active SOD1 enzyme consists of a homodimer of 2 subunits (for reviews see (Fridovich 1974; Fridovich 1986)). Each subunit has a Greek key β barrel structure consisting of six β strands and a large loop domain near the middle of the protein (Parge et al. 1992). Each subunit also contains four cysteine residues, 6, 57, 111, and 146. One intra-molecular disulfide bond between C57 and C146 of each subunit facilitates its correct folding, and thus stabilizes the active homodimeric structure (Arnesano et al. 2004). To date, more than 100 mutations at over 70 residues in the 153 amino acid SOD1 protein have been found to cause ALS (http://alsod.iop.kcl.ac.uk/Index.aspx). Most mutations are point substitutions, with a few causing early termination or frameshift near the carboxyl terminus. Most or all FALS mutations render SOD1 more susceptible to the disulfide reduction (Tiwari and Hayward 2003), most accelerate the rate of protein turnover (Borchelt et al. 1994; Ratovitski et al. 1999), some eliminate superoxide scavenging activity (Borchelt et al. 1994; Potter and Valentine 2003; Valentine and Hart 2003), and some reduce the ability of enzyme to bind one (Zn usually Cu sometimes) or both of its metal cofactors (Crow et al. 1997a; Potter and Valentine 2003).
The challenge has been to understand how the specific disease phenotype arises through a “gain-of-property” mechanism from all these mutations scattered throughout the protein. Under the premise that different SOD1 mutations cause FALS through a common mechanism, a limited understanding of the disease mechanism has been gained by examining the chemistry of multiple mutants. This approach has established that disease develops independently of the native enzymatic activity of SOD1; the toxicity of the mutant enzymes results from an acquired property that is absent or much lower in the wild-type protein (Borchelt et al. 1994; Gurney et al. 1994; Borchelt et al. 1995; Reaume et al. 1996). We have reported that mutations that eliminate histidines that coordinate the binding of Cu to SOD1 do not diminish toxicity (Wang et al. 2003); suggesting that potentially toxic catalytic activities of mutant SOD1, such as production of hydroxyl-like radicals (Yim et al. 1996; Wiedau-Pazos et al. 1996; Yim et al. 1997) or covalent nitration of tyrosine residues (Crow et al. 1997a; Crow et al. 1997b; Estévez et al. 1999) – chemistries attributed to enzyme with Cu correctly bound in the active site, may not be required for toxicity. Reducing Cu loading to mutant enzymes that normally bind Cu very efficiently, by eliminating the Cu chaperone for SOD1 (CCS), similarly does not diminish the toxicity of mutants (Subramaniam et al. 2002). Most relevant to the present study, work by our group and others have collectively examined at least 8 different FALS mutants in mice and 13 mutants in cell models, having shown all to possess a significantly heightened potential to form sedimentable structures with poor detergent solubility (Johnston et al. 2000; Shinder et al. 2001; Wang et al. 2002a; Wang et al. 2002b; Wang et al. 2003; Jonsson et al. 2004; Watanabe et al. 2005; Wang et al. 2005).
In the present study, we sought to understand the molecular basis of SOD1 aggregation by identifying features in the protein sequence that are critical for aggregate formation in vitro and in vivo. We analyzed the distribution of disease mutations documented to date to determine whether mutation positions coincide with structural features. Then using a cell transfection assay for aggregation, we asked whether there is a critical domain required for the assembly of high molecular weight aggregates. Last, we investigated the potential roles of intra- and inter-molecular disulfide bonds in the process of SOD1 aggregation. Our data suggest that the redox state of the cell may play a role in destabilizing native SOD structure and/or stabilizing non-native structures. We also find that unlike many other aggregation-prone proteins that have been examined, we find no critical domain in SOD1 that catalyzes assembly. Multiple domains of the protein appear to inherently posses the ability to mediate interactions that result in detergent-insoluble, sedimentable, structures. In this context, mutations at multiple sites in the protein may expose a domain that can mediate self-self interactions to form non-native oligomeric structures.
Materials and Methods
Tissue culture transfection and transgenic mice
All SOD1 variants were generated in the cDNA of the human gene by PCR strategies and expressed by the pEF-BOS vector (Mizushima and Nagata 1990; Wang et al. 2003). Confluent (90%) HEK 293 cells, cultured by standard procedures in 60 mm wells, were transfected using Lipofectamine 2000 (Invitrogen, Carlsbad, CA) and harvested in 24 hours for analyses of SOD1 aggregation.
All lines of SOD1 transgenic animals in this study have been previously characterized; the G93A variant [B6SJL-TgN(SOD1-G93A)1Gur; Jackson Laboratory, Bar Harbor, ME], the G85R variant (line 164) (Bruijn et al. 1997), the G37R variant (line 29) (Wong et al. 1995), the H46R/H48Q variant (lines 139) (Wang et al. 2002a; Wang et al. 2002b), the H46R/H48Q/H63G/H120G (or Quad, line 87 or 125) (Wang et al. 2003), the L126Z mice (line 171) (Wang et al. 2005), and wild-type SOD1 over-expressing mice (line 76) (Wong et al. 1995). Mouse lines were maintained by crossing transgenic males to non-transgenic [C57BL/6J X C3/HeJ F1] females (Jackson laboratories, Bar Harbor, ME). All studies involving mice were approved by the JHU Institutional Animal Care and Use Committee.
SOD1 aggregation assay by differential extraction
The assay for SOD1 aggregation by differential detergent extraction and centrifugation has been previously described (Wang et al. 2003). Briefly, cultured cell pellets or spinal cords were homogenized with a probe sonicator in 200 μl per 60 mm well, or 1:10 W:V, respectively, of extraction buffer 1 (10mM Tris-HCl pH 8.0, 1mM EDTA pH 8.0, 100mM NaCl, 0.5% Nonidet P40, and proteinase inhibitor). 200μl of the extract was centrifuged at > 100,000 g for 10 minutes in a Beckman AirFuge to separate the non-ionic detergent-insoluble pellet (P2) from the supernatant (S1). The P2 fraction was either resuspended in 75 μl of buffer 3 (see below) for analysis, or re-extracted in 200 μl of buffer 2 (10mM Tris-HCl pH 8.0, 1mM EDTA pH 8.0, 100mM NaCl, 0.5% Nonidet P40, 0.5% deoxycholic acid, and 0.25% SDS) and re-pelleted by centrifugation at > 100,000 g for 10 minutes in the AirFuge to yield the final pellet (P3), which was resuspended in 75 μl of buffer 3 (10mM Tris-HCl pH 8.0, 1mM EDTA pH 8.0, 100mM NaCl, 0.5% Nonidet P40, 0.5% deoxycholic acid, and 2% SDS). Protein concentration were then measured in all fractions (BCA method, Pierce, Rockford, IL), and analyzed by immunoblotting. Cell transfection, detergent extraction, and differential centrifugation were independently carried out at least 3 times for each construct.
Immunoblotting
Standard SDS-PAGE was performed in 18% Tris-HCL Criterion gels (Bio-Rad, Hercules, CA); samples were boiled for 10 minutes in Laemmli sample buffer (Laemmli 1970) with or without 2.5% β-mercaptoethanol (βME). Immunoblots were probed with one of two polyclonal antibodies as noted in Figure Legends: the hSOD1 antibody (hAb), a peptide antiserum binding to amino acids 24-36 [not conserved between mouse and human SOD1]; and the m/hSOD1 antibody (m/hAb), a peptide antiserum recognizing amino acids 124-136 [completely conserved between mouse and human protein].
Cell-free protein aggregation assay
Brains were homogenized using a Dounce homogenizer in phosphate buffered saline (PBS, pH 7.4) with proteinase inhibitors. The homogenate was centrifuged at 150,000 g for 50 minutes at 4 °C, and the supernatant was adjusted to 2 mg/ml of protein with PBS. To induce aggregation, 50 μl of supernatant were added to a 200 μl tube, and shaken on a titer plate shaker (750 rpm) at 37 °C for 12 hrs. The resulting samples were centrifuged in an AirFuge (> 100,000 g) for 10 minutes, and the pellets are washed twice with PBS. The resulting pellets were considered aggregated proteins, and resuspended in PBS with 2% SDS before mixing with Laemmli sample buffer (with or without βME as noted in the Legends) and further analyses by SDS-PAGE. In vitro assays involved the addition of dithiothreitol/DTT (Invitrogen, Carlsbad, CA) as described in Results.
Results
ALS mutations occur primarily within conserved β-strand domains
Comparative sequence analysis of gene sequence across phyla is a means to identify amino acids that are critical to either enzymatic activity or structure. To determine how the locations of point mutations align with conserved sequences among SOD1 genes derived from divergent species, we searched the NIH Entrez databases for SOD1 sequences across eukaryotic phyla, from mammals to fish, birds, plants, and fungi. A comparison of the protein sequences from these species revealed conservation of coding sequence during evolution (Fig. 1 and Supplemental Information). Of the 153 total amino acids in SOD1, 112 are conserved in mammals (capital letters), with 70 of them absolutely conserved across eukaryotic phyla (bold capitals larger font). We observed that ALS mutations identified to date occur in a non-random pattern (Fig. 1, Table 1). Of the 67 positions (red font) where disease-causing mutations are documented (http://alsod.iop.kcl.ac.uk/Index.aspx), 60 occur at residues conserved in mammals. The frequency of mutation at conserved residues is over three times higher than that at residues not conserved in mammals (the probability of random occurrence, p < 10−4). Forty-nine mutations occur at positions that are extremely conserved across all eukaryotic phyla; the frequency is again over three times higher than that at residues not conserved in eukaryotes (p < 10−8). Approximately 50% of the protein participates in the formation of the core β-barrel structure and mutations in β-strand domains occur at about twice the rate as loop domains of the protein (43 mutated positions out of 75 β-strand residues, p < 0.005).
Fig 1. Non-random distribution of ALS mutations in SOD1 protein sequence.
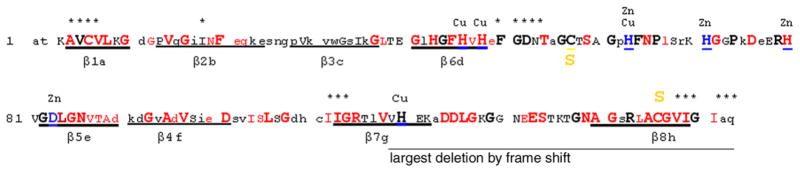
A comparative sequence analysis of SOD1 genes from various eukaryotic species is summarized in the diagram (sequence information available in Supplemental Information). Capitalized letter codes denote residues that are absolutely conserved in mammals; bold capitals in a larger font denote residues that are highly conserved across all phyla of eukaryotes; and red letter codes denote positions mutated in SOD1-linked FALS. Eight β-strands are underlined. Cu or Zn binding residues are labeled and blue-underlined. Cys 57 and 146, which form the native disulfide, are labeled. Asterisks mark residues that are part of the dimer interface.
Table 1.
Non-random distribution of ALS mutations in SOD1.
| N-terminus | Middle loop | C-terminus | Full length | |
|---|---|---|---|---|
| Residues in the segment | 1–37 | 38–90 | 91–153 | 1–153 |
| Overall mutation rate at the segment | 13/37 | 22/53 | 32/63 | 67/153 |
| Mutation rate at residues conserved in eukaryotes, Re | 9/10 | 14/29 | 26/31 | 49/70 |
| Mutation rate at residues NOT conserved in eukaryotes, Rne | 4/27 | 8/24 | 6/32 | 18/83 |
| p (Re = Rne) | <10−8 | |||
| Mutation rate at residues conserved in mammals, Rm | 10/19 | 20/46 | 30/47 | 60/112 |
| Mutation rate at residues NOT conserved in mammals, Rnm | 3/18 | 2/7 | 2/16 | 7/41 |
| p (Rm = Rnm) | <10−4 | |||
| Mutation rate at β strand residues, Rb | 12/29 | 13/17 | 18/29 | 43/75 |
| Mutation rate at non-β strand residues, Rnb | 1/8 | 9/36 | 14/34 | 24/78 |
| p (Rb = Rnb) | <0.005 |
Note: p is the 2-tail probability using the Fisher’s Exact Test for the difference between mutation rates of the dichotomous groups of residues.
SOD1 can readily be divided into three segments that participate in different structural features of the protein (Fig. 1, Table 1). In the N-terminal 37 residues, which are almost entirely β-strand domain (encompass the first 3 β-strand domains), mutations occur at 12 of 29 positions in these three β-strands. The large Zn loop segment encompasses residues 38 to 90 and includes two β-strand domains outside the beta-barrel and a large hydrophilic domain. Within the hydrophilic domain, mutations occur at only 8 positions; whereas, in the two β-strand domains of the Zn loop, mutations occur at 13 of 17 positions. The C-terminal residues 91 to 153, which encompass the last three β-strand domains and the structurally (and catalytically) important electrostatic loop, are frequently mutated; 32 positions overall with 18 occurring in β-strand domains. Overall, mutations clearly occur in β-strand residues much more frequently.
Mapping aggregation prone domains in SOD1
The higher frequency of ALS mutations occurring at conserved or β-strand residues suggested that destabilization of the compact structure of SOD1 protein may be important for the toxicity. Destabilized, β-strand rich proteins often demonstrate increased aggregation propensity (Valentine and Hart 2003), but it is unclear how SOD1 forms aggregates and whether there is a critical domain mediating the process. Large deletions of the C-terminal region of the protein have been associated with the disease. The largest simple deletion occurs by the mutation of L126 to stop. Several other deletions result from frameshift mutations, with the largest occurring from a frameshift immediately after Val118 (Val118insAAAACstop at 150) (http://alsod.iop.kcl.ac.uk/Index.aspx). In a previous study, we noted that spinal cords of diseased mice accumulated truncated forms of SOD1 that lack portions of either the N- or C-terminus. Moreover, we generated experimental deletions of N- and C-terminal domains of wild-type SOD1 creating variants that were prone to aggregate (Wang et al. 2003).
To further map domains in SOD1 that may be critical for the formation of self-associating structures, we constructed additional deletion constructs to compare to our previously generated N1-75 and C49-153 constructs (Fig. 2) (Wang et al. 2003). The series of deletion constructs included the following; N1-75WT, N1-75(H46R/H48Q), Δ57-79WT, Δ57-79(H46R/H48Q), Δ49-56WT, Δ49-56(A4V), Δ49-56(FS126), and C49-153 (Fig. 2). In a cell transfection assay for SOD1 aggregation, all constructs tested, as compared to wild-type SOD1, produced significantly greater amounts of protein that wes detergent insoluble. For some mutants, such as the N1-75WT and N1-75(H46R/H48Q) constructs which are particularly unstable, all detectable forms of the protein were in the detergent-insoluble fraction [Fig. 2 – a very faint band for N1-75(H46R/H48Q) is visible adjacent to the lane containing N1-75WT]. Similarly, the majority of the C49-153WT protein was also detergent-insoluble. The Δ57-79WT protein, which lacks amino acids contained in exon 3 including most of the hydrophilic domain of the Zn loop, also produced detergent-insoluble species; adding the double histidine FALS mutations did not significantly alter the aggregation potential of this deletion mutant.
Fig 2. No single domain in SOD1 mediates aggregation.
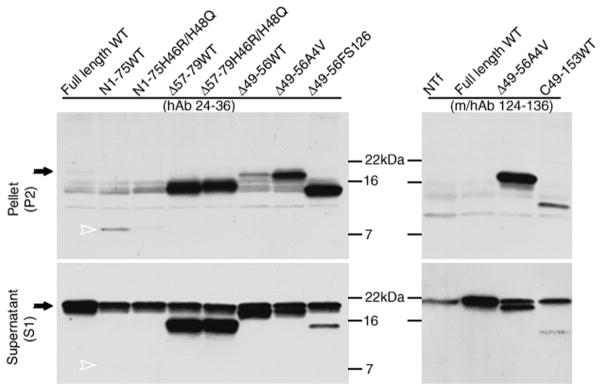
Using the cell-transfection aggregation-assay described in Materials and Methods, SOD1 in the detergent-soluble or insoluble fractions was detected by immunoblotting. Two peptide antibodies were used to detect different fragments; the hSOD1 antiserum (hAb, specific for the human protein) recognizes residues 24-36, a sequence divergent in mouse and human SOD1. The m/hSOD1 antiserum (m/hAb) recognizes residues 124-136, conserved between mouse and human SOD1. Although highly expressed, wild-type SOD1 was barely detectable in the insoluble fractions while abundant in soluble fractions. In contrast, all tested mutants accumulated, to varying degrees, in insoluble fractions (P2). Some deletion mutants were detectable only in the insoluble fractions. The solid arrows point to the position of wild-type full-length SOD1; the open arrowheads point to the position of the most unstable N(1–75) fragment. The images shown are representative of 3 independent repetitions of the experiment.
From this series of constructs, it is apparent that domains in the first 57 amino acids of SOD1 can mediate aggregation; N1-75 aggregates as does SOD1Δ57-79. However, as previously reported (Wang et al. 2003) and shown here, C-terminal fragments, such as C49-153, also form aggregates (Fig. 2). From the foregoing series, the only sequence common to all aggregating mutants was between residues 49 and 56. Unlike the other large deletions, mutants deleted of these residues (Δ49-56) produced little aggregated protein (Figure 2B). However, when FALS mutations (A4V or FS126) were introduced into the SOD1Δ49-56 backbone, detergent-insoluble species were readily generated. Collectively, these studies indicate that multiple domains in SOD1 posses the potential to form detergent-insoluble structures. Whether all domains in SOD1 are quantitatively identical in aggregation potential is uncertain and best determined by studies of purified protein in vitro. The main point we note is that no single domain in SOD1 must be present for self-association to occur.
Contribution of intra- and inter-molecular disulfide bonds to SOD1 aggregation
Full-length SOD1 contains 4 cysteine residues located at codons 6, 57, 111, and 146; Cys 57 and 146 normally form an intra-molecular disulfide, which would be lost in the various naturally occurring C-terminal truncation mutants. In this next series of experiments, we investigated the role of the normal disulfide bond and the potential role of aberrant inter-molecular disulfide bonds in the formation of mutant SOD1 aggregates. We developed an in vitro cell-free system that is based on high speed supernatant fractions from the brains of transgenic mice expressing WT, G37R, G85R, G93A, H46R/H48Q, H46R/H48Q/H63G/H120G (or Quad), and L126Z variants of SOD1. In contrast to WT SOD1 transgenic mice, all mutant transgenic mice studied here develop motor neuron disease between the age of 3 months and 10 months (see Materials and Methods for reference list). The expression levels of SOD1 protein in these mice have been described previously (see Materials and Methods). With the exception of SOD1-L126Z, which is very unstable and hence almost undetectable in its soluble form (Fig. 3A), the high speed supernatant fractions of brain from these mice contained readily detectable soluble mutant protein. Brain supernatants from transgenic mice, free of sedimentable insoluble aggregates, were incubated at 37°C with shaking for 12 hrs to induce aggregation. With subsequent high-speed centrifugation, a new pellet fraction formed, and insoluble SOD1 was assayed by immunoblotting. This cell-free aggregation assay was performed with or without fresh DTT in the solution, and then insoluble fractions were electrophoresed in gels with or without β-mercaptoethanol (βME). The most accurate assessments of levels of newly insoluble protein were gauged in gels electrophoresed in the presence of βME (Fig. 3B lower panel) because here the protein runs as a single band on the gel. As opposed to wild-type mice, brain fractions from the mutant mice generated considerable insoluble protein (Fig. 3B all lanes contain pellet fractions). However, the effect of reducing agent on the generation of insoluble material varied with mutation. With the addition of 20 mM DTT, the G37R and G93A variants, which are more wild-type-like (Potter and Valentine 2003; Valentine and Hart 2003), produced much more insoluble material (Fig. 3B). By contrast, the same concentration of DTT had no effect on wild-type SOD1 (Fig. 3B). However, when 20 mM reduced glutathione (G6013, Sigma, St. Louis, MO), a more natural reducing agent, was added to the homogenates from wild-type mice, then wild-type SOD1 could be induced to form insoluble material (not shown). Paradoxically, the addition of DTT slowed the generation of insoluble material in brain homogenates from mice that express metal-binding-region mutants [G85R, H46R/H48Q, Quad, L126Z] (Fig. 3B). The contrast between the wild-type-like mutants [G37R and G93A] and the medal-binding-region mutants [G85R, H46R/H48Q, Quad, and L126Z] is shown by assessing the relative change in cell-free aggregation (Fig. 3C). These comparisons indicated that for mutants that achieve a more native-like structure, destabilization of the normal intra-molecular disulfide bond may contribute to the formation of aberrant structures.
Fig 3. Contributions of intra- and inter-molecular disulfide bonds to SOD1 aggregation in a cell-free assay.

A cell-free assay for SOD1 aggregation was developed by fractionating brain extracts of young SOD1 transgenic mice [non-transgenic (NTg), WT, G37R, G93A, G85R, H46R/H48Q, H46R/H48Q/H63G/H120G (Quad), and L126Z; all 2 months old], in non-ionic detergent and utilizing the high-speed supernatant fraction (the soluble fraction) as starting material (see Methods). (a) At the starting time point, all the SOD1 proteins are soluble, and their relative steady-state levels are shown by loading equal amounts of total proteins on SDS-PAGE followed by immunoblotting with the hSOD1 antiserum. (b) A 50 μl solution of supernatant protein (2 μg/μl) was incubated at 37°C to induce aggregation in the presence or absence of 20mM DTT as described in Methods. At 12 hrs, the resulting suspensions were centrifuged to harvest aggregated proteins, which were analyzed by SDS-PAGE in the absence (top panel) or presence (lower panel) of 2.5% βME before immunoblotting with hSOD1 antiserum. (c) Quantitative comparison of the effects of 20mM DTT on the aggregation rates of two different types of SOD1 mutants is shown. The sensitivity of aggregation to DTT, the Y axis, was measured as a common logarithm of the ratio of insoluble SOD1 produced in the presence of DTT to that without DTT. The relative levels of aggregates were measured by densitometry of immunoblot signals, on the reducing SDS-PAGE gels, from 3 independent assays (error bars represent standard deviations). The samples were divided into two groups; the wild-type-like mutants G37R and G93A, and the metal-binding-region mutants G85R, H46R/H48Q, Quad, and L126Z.
In comparing the gels containing equal amounts of sample treated with βME versus samples there were not reduced (upper panel of Fig. 3B), we note that all of the mutants produce aggregates that run as high-molecular-weight smears in the gel when βME is omitted from the sample buffer (compare the intensity of the monomeric size SOD1 band in the upper and lower panels of Fig. 3B). These data indicate that oxidation of cysteine residues and/or the formation of intermolecular disulfide bonds is contributing to the formation of sedimentable structures.
Next, we prepared P3 fractions (material that sediments in SDS – See Materials and Methods) from homogenates of spinal cord from mice expressing different FALS variants that were symptomatic (ages vary according to model). In the presence of β-mercaptoethanol (βME), most of the protein in detergent-insoluble fractions of spinal cord was dissociated into monomeric species (~17 kDa) (Fig. 4A). When the insoluble fractions were analyzed by non-reducing SDS-PAGE, insoluble SOD1 migrating at the top of the gel or as a higher-molecular-weight smear was obviously increased with a proportional decrease in the amount of protein migrating at the monomeric size (Fig. 4 compare A and B; a higher exposure of L126Z shown in Fig. 5). The increase in high-molecular-weight SOD1 under the non-reducing condition is specific to the insoluble SOD1, as SOD1 in the soluble fraction at comparable concentrations, with boiling, showed no signs of oxidation under the same conditions (Fig. 5). These data indicate that in vivo, oxidation of cysteine residues, which presumably form intermolecular disulfide bridges, participates in the generation of detergent-insoluble structures.
Fig 4. Inter-molecular disulfide bonds contribute to formation of high-molecular-weight species of SOD1 in spinal cords from symptomatic transgenic mice.

From symptomatic SOD1 transgenic mice expressing G37R, G93A, G85R, H46R/H48Q, H46R/H48Q/H63G/H120G (Quad), or L126Z, and age-matched controls of SOD1-WT over-expressing or non-transgenic (NTg) mice, spinal cord tissues were homogenized and fractionated in detergent as described in Methods. The ionic-detergent-insoluble pellet fractions (P3) were boiled in the SDS sample buffer, and then electrophoresed in the presence (a) or absence (b) of reducing agent (2.5% βME) followed by immunoblotting with the hSOD1 antiserum. All lanes contain 5μg of protein from the insoluble pellet. The images shown are representative of 3 independent repetitions of the experiment.
Fig 5. Inter-molecular disulfide bonds are specific to the insoluble forms of SOD1 from symptomatic transgenic mice.
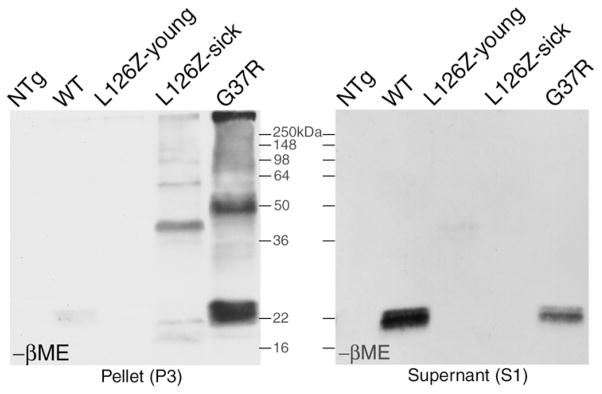
From symptomatic SOD1 transgenic mice expressing stable mutant G37R, or unstable mutant L126Z, and controls [young L126Z mice, old SOD1-WT over-expressing, and non-transgenic (NTg) mice], spinal cord tissues were homogenized and fractionated into insoluble and soluble fractions as described in Methods. (a) The insoluble pellet fractions (P3) were boiled in the SDS sample buffer, and then electrophoresed in the non-reducing SDS-PAGE followed by immunoblotting with the hSOD1 antiserum. (b) The soluble fraction (S1), with a much higher SOD1 concentration than in insoluble fractions, were diluted to have SOD1 concentrations comparable to insoluble fractions, and underwent the same procedures of boiling, SDS-PAGE without βME, and immunoblotting. Note the absence of high-molecular-weight oxidized SOD1 forms.
To further clarify the role of intermolecular disulfide bonding in mutant SOD1 oligomerization we constructed a series of mutants with point mutations in cysteine residues and examined their potentials to aggregate when expressed at high levels in cultured cells (Fig. 6). The cysteine mutants were compared to SOD1-G37R, which has one of the lowest aggregation potentials in the cell transfection assay and represents the minimum achievable level of aggregation in these assays (Wang et al. 2003). Cysteine 6 is highly conserved and mutations to Gly or Phe have been associated with FALS (http://alsod.iop.kcl.ac.uk/Index.aspx). Cysteine 111 is not conserved and not a known site of FALS mutations. Neither cysteine is involved in normal intra-molecular disulfide bonding of subunits (Parge et al. 1992). A variant of SOD1 encoding mutations of both Cys 6 and 111 to serine was found to remain detergent-soluble (Fig. 6). Two additional variants were constructed in which all 4 Cys residues were mutated to serine, with or without the A4V FALS mutation; SOD1-A4V shows a very high aggregation potential even with a relatively short half life in cell transfection assays (Wang et al. 2003). However, again, neither of these two variants spontaneously formed detergent-insoluble species in the cell transfection assay (Fig. 6).
Fig 6. Cysteine mutations diminish the ability of SOD1 to form aggregates in cultured cells.
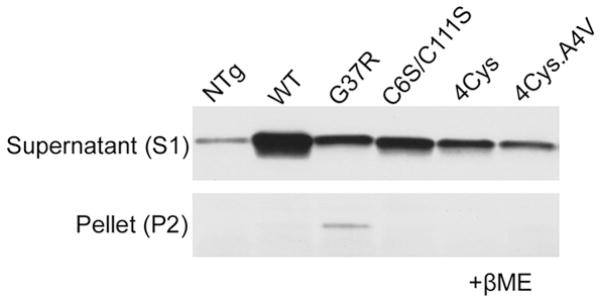
HEK 293 cells were transfected with SOD1 variants encoding mutations at cysteine residues, mutations linked to FALS, or a combination of both. Aggregation potentials are analyzed by comparing the ratios of detergent-insoluble to soluble protein, which are fractionated as described in Methods. By immunoblotting, SOD1 levels in the supernatant S1 fraction, which contains the majority of expressed SOD1 protein, reflect the shortened half-lives and thus lower steady-state levels after introduction of mutations. When sedimentable insoluble SOD1, or aggregated forms, were analyzed, only FALS mutant G37R showed a clearly detectable accumulation of detergent-insoluble protein. Mutating cysteine 6 and 111 that do not form the native disulfide, or all four cysteines did not inherently heighten the protein’s aggregation potential. A4V, previously shown to have a higher aggregation potential than G37R (Wang et al. 2003), shows a dramatically reduced aggregation potential when all 4 Cys residues are mutated (last lane).
These data are consistent with the view that redox environment may influence the aggregation of mutant SOD1. Reduction of intra-molecular disulfide bonds may help destabilize normal structure of some mutants, but the formation of inter-molecular disulfide bonds, between SOD1 monomers, or among other proteins, may be a mechanism to stabilize high-molecular-weight forms of mutant SOD1.
Discussion
In the present study, we identified features of SOD1 that modulate its capacity to aggregate. First, we note that FALS mutations in SOD1 are not entirely random. Mutations occur at a much higher frequency in residues of the β-strands of the protein, which are critical elements of normal structure. Second, we note that for wild-type-like mutants, which have thermal stabilities most similar to normal protein (Potter and Valentine 2003), reducing the native disulfide bond facilitates the formation of detergent-insoluble structures. Third, we find that inter-molecular disulfide may work as “glue” that stabilizes aggregated SOD1 structure. Lastly, and somewhat to our surprise, we find that multiple domains in the protein possess the ability to spontaneously aggregate, which suggests low specificity and the possibility for diverse conformations in oligomer assembly.
In analyzing the distribution of FALS mutations in SOD1 in the context of known structural elements and sequences across all phyla, we noted a predominant (~2:1) localization of disease-causing mutations in β-strand domains of the protein. Those mutations that occur outside of these domains almost invariably fall in highly conserved residues. The less conserved hydrophilic domain of the Zn loop is relatively devoid of ALS mutations. It is generally accepted that the most conserved residues of a protein are most likely to be critical for either functional structure or to compose the active site of the protein (enzyme). In the case of SOD1, the active site consists of the coordinated binding of the Cu cofactor, and positively charged residues of the electrostatic loop (Parge et al. 1992). Hence, we can infer that most of the highly conserved amino acids play a profound role in maintenance of structure. One consequence of ALS mutations in these conserved residues is to diminish the structural stability of the protein. Indeed, the half-lives for most tested ALS mutants are decreased (Borchelt et al. 1995; Borchelt et al. 1994; Ratovitski et al. 1999). The thermal stability of many ALS variants is also decreased (Deng et al. 1993), particularly when metal cofactors are removed from the enzyme (Rodriguez et al. 2002). Studies of crystal structure have been reported for a limited number of mutants, noting decreased order in the structure of certain domains in the protein (Lindberg et al. 2002; DiDonato et al. 2003; Elam et al. 2003). Our deletion analysis study indicates that both N- and C-terminal β-strand domains of SOD1, which are where the vast majority of FALS mutations occur, were independently capable of assembling into detergent-insoluble structures. The less conserved hydrophilic domain of the Zn loop also does not contain elements required for aggregation. Notably, in the larger deletion mutants we produced, FALS mutations had little impact on aggregation potential; possibly because native structure was already destroyed. In the less dramatic deletion mutation that removed residues 49-56, the presence of FALS mutations dramatically enhanced aggregation potential. These data are consistent with the notion that destabilization of the structure is the immediate consequence of these mutations. If structure is already abrogated by some other perturbation, then FALS mutations lose potency.
Assuming that the process by which mutant SOD1 aggregates involves specific self-association of the protein, there are a number of examples in which a specific domain in the protein mediates assembly of oligomeric structures. For example, the NAC domain of α-synuclein is necessary for fibril assembly (Bodles et al. 2001). Similarly, the prion protein also contains a specific aggregation-prone domain, which if deleted abrogates the formation of oligomeric structure (Norstrom and Mastrianni 2005). In amyloid B-peptide (Aβ), the addition of 2 amino acids to the C-terminus of Aβ40 to Aβ42, produced via differential proteolytic of the amyloid precursor protein, results in peptides with vastly different potentials to aggregate in vivo (McGowan et al. 2005). In SOD1, Elam and colleagues demonstrated, in crystal structures of two metal-deficient mutants, a specific non-native interaction between the electrostatic loops of dimeric mutant enzyme, suggesting that some type of gain of interaction could be adopted by this specific domain of the enzyme (Elam et al. 2003). Our data, however, indicate that there is no one particular domain in SOD1 that is the dominant proponent of aggregate formation.
An alternative mechanism by which SOD1 oligomers might form is that aggregation may not require specific interactions among aggregation-prone domains but that multiple domains can mediate non-native intermolecular interactions. For example, one mechanism we have suggested involves domain swapping interactions (Wang et al. 2003) where evolutionarily conserved intra-molecular interactions between β-strand domains are replaced by intermolecular interactions that mimic the natural interaction (Bennett et al. 1994; Bennett et al. 1995). Other mechanisms include the propensity of beta-turn structures to stack into helical fibril structures. Adjacent β-strand domains of the core β-barrel structure will be prone to form hairpin folds to create two extended β-strands separated by a turn. The stacking of such structures in helical fibrils has been a suggested mechanism of aggregation for both β-amyloid peptide and polyglutamine proteins (Guo et al. 2004; Thakur and Wetzel 2002; Williams et al. 2004). Such a mechanism could explain the ability of both N- and C-terminal domains of SOD1 to form detergent-insoluble structures. Given the wide distribution of ALS mutations, the second scenario in which the core structures of oligomers could utilize different β-strand sequences seems to be more likely. By such a mechanism, there is no need for a uniform unfolding pathway, but rather local changes induced by mutation could expose nearby β-strand domains, facilitating intermolecular interactions. Whether the final structure achieved by all FALS mutants is identical is unknown. It remains possible that the most thermodynamically stable oligomers may share similar features. However, we noted that mice expressing the SOD1-L126Z mutant fail, as compared to all other available mouse models, to develop thioflavin-S reactive structures (which are presumed to be composed of aggregated SOD1) (Wang et al. 2005). Hence it is possible that mutant SOD1 oligomers are structurally diverse.
Our data suggest that the reducing environment of the cell may influence the aggregation rate of SOD1 variants. In the cell-free assays of aggregation, the G37R and G93A mutants, which are wild-type-like in their properties (Potter and Valentine 2003), were slow to form detergent-insoluble structures. However, reducing agents dramatically stimulated aggregation, suggesting that a second hit may influence the rate at which these proteins oligomerize. These observations are consistent with a recent study by Furukawa and O’Halloran who showed that reduction of disulfide bonds stimulated purified SOD1 proteins to aggregate in vitro (Furukawa and O’Halloran 2005). Paradoxically, those mutants that are inherently more compromised structurally, which include the metal-binding-region mutants of H46R/H48Q and SOD1-Quad that we have analyzed, appear to aggregate more slowly in the presence of reducing agents, in vitro. In all mice we have examined, a portion of the mutant SOD1 that accumulates in the detergent-insoluble fraction migrates as βME sensitive high-molecular-weight species. Indeed, for SOD1-L126Z the disulfide-linked forms account for the majority of the insoluble species. Only the SOD1 that partitions into the detergent insoluble fraction possesses these non-native intermolecular disulfide linkages. SOD1 in the soluble fraction experiences all the same treatments and yet contains no such oxidized species. Therefore, it appears that intermolecular disulfide bonds occur prior to isolation and we suggest that these disulfide bonds may act to stabilize some types of multimeric structures.
Although we focus discussion on intermolecular disulfide bonds, it is important to note that non-native intramolecular bonds could also occur, that is bonds involving Cys residues other than the native bond between C57 and C146, and that such bonds could stabilize non-native conformations. Hence, non-native disulfide bonding could play a role in both misfolding of the monomer and stabilization of multimers.
Our cell culture studies with experimental mutations of Cys residues in SOD1 produced results consistent with the idea that intermolecular disulfide bonds may play a role in the formation of detergent insoluble, sedimentable, SOD1 species. Mutation of all 4 Cys residues in SOD1 dramatically attenuated the ability of the A4V mutation to induce aggregation. Although other conformational consequences of Cys mutations are possible, this result suggests an important role for disulfide bonds in the formation of SOD1 aggregates in cytosol. However, we concede that other data regarding the location of FALS mutations to Cys residues present the possibility that intermolecular disulfide bonding is not critical. Mutations at Cys 6 to Gly or Phe have been described in FALS (http://alsod.iop.kcl.ac.uk/Index.aspx) as have mutations at Cys 57 and Cys 146 (see Supplemental Information). In cell transfection assays of aggregation, mutation of Cys residues 6 and 111 to serine (Ser is more common at 111 than Cys in most species, whereas Cys 6 is more highly conserved – see Supplemental Information) did not induce the formation of detergent-insoluble species. The Cys 6 to Ser substitution we utilized is more conservative with regard to charge than the FALS mutations at this residue. Nevertheless, this result indicates that mutation of conserved residues per se is not sufficient to induce SOD1 to aggregate spontaneously. It is possible that the combined mutation of Cys 111 with Cys 6, and the loss of 2 Cys residues, suppressed aggregation by reducing the ability to form intermolecular disulfide bonds; whereas in the case of FALS mutants, the substitution of any one Cys residue does not significantly reduce the ability to form intermolecular disulfide bonds. Interestingly, of the three Cys residues, Cys 111 is the least conserved but is also the only one that is not a target of FALS mutation. Clearly, additional study of both experimental and disease-associated mutations of Cys residues in SOD1, in both cell and mouse models, is required to better understand the role of intermolecular disulfide bonding in SOD1 aggregation in vivo.
One aspect of the data that is entirely consistent is that destabilization of normal disulfide bonds can induce SOD1 to aggregate. Almost all of the C-terminal truncation mutants of SOD1 that are associated with FALS would eliminate Cys 146 and thus break the normal intramolecular disulfide bond. Further as noted above, disease causing substitution mutations have also been described at Cys 57 and Cys 146. The in vitro aggregation of FALS mutants that are most like wild-type (possess intact disulfide linkage) was greatly stimulated by reducing agents. As noted above, Furukawa and O’Halloran have demonstrated reduction of purified SOD1 induces aggregation. We have observed that wild-type SOD1 in cell-free brain extracts can be induced to form detergent-insoluble species in the presence of reduced glutathione (J. Wang, personal observation). Thus it is possible that the level of cellular reductants may influence the rate at which mutant SOD1 aggregates. Whether changes in redox potential of the cell are stimulatory or inhibitory may well depend upon the stage of the disease. Early in disease, a highly reducing environment may stimulate the unfolding of some mutants, allowing misfolding. However, late in the disease a loss of reducing potential may facilitate the formation of high-molecular-weight species.
Although we can be relatively convinced that aggregation of SOD1 is a common property and could well be linked to the “toxic gain of property”, how aggregates (or oligomers) of SOD1 injure motor neurons remains unsolved. Among the processes that could play a role are; entrapment of critical cellular factors, such as the anti-apoptotic activity bcl-2, (Pasinelli et al. 2004); disruption of mitochondrial metabolism (Liu et al. 2004); formation of pore-forming structures (Caughey and Lansbury 2003; Ray et al. 2004); and inhibition of the ubiquitin proteasome system (Johnston et al. 2000). We also do not fully understand why SOD1 aggregates are relatively restricted to regions of the CNS that contain motor neurons, despite widespread expression in CNS and all organs (Wang et al. 2003; Wang et al. 2005). In a recent study we demonstrated that the aggregation of SOD1-L126Z is attenuated by αB-crystallin (a small heat shock protein with chaperone activities) and that this protein was most abundantly expressed in spinal cord cells that did not accumulate aggregated forms of this mutant (Wang et al. 2005). Whether other chaperone activities modulate tissue or cell specificity is uncertain. The rate at which different tissues turnover mutant SOD1 could also be a modifier, as very unstable C-terminal truncation mutants appear to accumulate only in motor neurons (Wang et al. 2005; Jonsson et al. 2004). In the present study, we find that redox potential of the cell could be a modifying factor. Collectively, there may be several factors which together leave motor neurons selectively vulnerable. Still, if aggregates, or oligomers, are the culprit, then how do they kill? Perhaps in the case of SOD1, the mechanism is not particularly specific. Over-expression of neurofilament (mutant only) (Lee et al. 1994), tau (both mutant and wild-type)(Ishihara et al. 1999; Lewis et al. 2000; Götz et al. 2001), α-synuclein (mutant only) (Giasson et al. 2002; Lee et al. 2002), and peripherin (wild-type) (Beaulieu et al. 1999), by pan-neuronal promoters, have all been shown to induce motor neuron disease with protein aggregate pathology. Perhaps, specificity is governed at the level of aggregate formation, with motor neurons being unable to tolerate the cytosolic accumulation of misfolded protein as a consequence of one, or many, toxic mechanisms.
Supplementary Material
Acknowledgments
We thank Ms Jodi McBride and Dr. Valeria Culotta for providing C6S/C111S SOD1 mutant. This study was supported by grants from the National Institutes of Neurologic Disease and Stroke (R01 NS 37225 and R01 NS 047225), the Muscular Dystrophy Association, the ALS Association, and by the Robert Packard Center for ALS Research at The Johns Hopkins University.
References
- Arnesano F, Banci L, Bertini I, Martinelli M, Furukawa Y, O’Halloran TV. The unusually stable quaternary structure of human Cu,Zn-superoxide dismutase 1 is controlled by both metal occupancy and disulfide status. J Biol Chem. 2004;279:47998–48003. doi: 10.1074/jbc.M406021200. [DOI] [PubMed] [Google Scholar]
- Beaulieu JM, Nguyen MD, Julien JP. Late onset death of motor neurons in mice overexpressing wild-type peripherin. J Cell Biol. 1999;147:531–544. doi: 10.1083/jcb.147.3.531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett MJ, Choe S, Eisenberg D. Domain swapping: entangling alliances between proteins. Proc Natl Acad Sci U S A. 1994;91:3127–3131. doi: 10.1073/pnas.91.8.3127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett MJ, Schlunegger MP, Eisenberg D. 3D domain swapping: a mechanism for oligomer assembly. Protein Sci. 1995;4:2455–2468. doi: 10.1002/pro.5560041202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bodles AM, Guthrie DJ, Greer B, Irvine GB. Identification of the region of non-Abeta component (NAC) of Alzheimer’s disease amyloid responsible for its aggregation and toxicity. J Neurochem. 2001;78:384–395. doi: 10.1046/j.1471-4159.2001.00408.x. [DOI] [PubMed] [Google Scholar]
- Borchelt DR, Guarnieri M, Wong PC, Lee MK, Slunt HS, Xu ZS, Sisodia SS, Price DL, Cleveland DW. Superoxide dismutase 1 subunits with mutations linked to familial amyotrophic lateral sclerosis do not affect wild-type subunit function. J Biol Chem. 1995;270:3234–3238. doi: 10.1074/jbc.270.7.3234. [DOI] [PubMed] [Google Scholar]
- Borchelt DR, Lee MK, Slunt HH, Guarnieri M, Xu ZS, Wong PC, Brown RH, Jr, Price DL, Sisodia SS, Cleveland DW. Superoxide dismutase 1 with mutations linked to familial amyotrophic lateral sclerosis possesses significant activity. Proc Natl Acad Sci USA. 1994;91:8292–8296. doi: 10.1073/pnas.91.17.8292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruijn LI, Becher MW, Lee MK, Anderson KL, Jenkins NA, Copeland NG, Sisodia SS, Rothstein JD, Borchelt DR, Price DL, Cleveland DW. ALS-linked SOD1 mutant G85R mediates damage to astrocytes and promotes rapidly progressive disease with SOD1-containing inclusions. Neuron. 1997;18:327–338. doi: 10.1016/s0896-6273(00)80272-x. [DOI] [PubMed] [Google Scholar]
- Caughey B, Lansbury PT. Protofibrils, pores, fibrils, and neurodegeneration: separating the responsible protein aggregates from the innocent bystanders. Annu Rev Neurosci. 2003;26:267–298. doi: 10.1146/annurev.neuro.26.010302.081142. [DOI] [PubMed] [Google Scholar]
- Crapo JD, Oury T, Rabouille C, Slot JW, Chang LY. Copper, zinc superoxide dismutase is primarily a cytosolic protein in human cells. Proc Natl Acad Sci USA. 1992;89:10405–10409. doi: 10.1073/pnas.89.21.10405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crow JP, Sampson JB, Zhuang Y, Thompson JA, Beckman JS. Decreased zinc affinity of amyotrophic lateral sclerosis-associated superoxide dismutase mutants leads to enhanced catalysis of tyrosine nitration by peroxynitrite. J Neurochem. 1997a;69:1936–1944. doi: 10.1046/j.1471-4159.1997.69051936.x. [DOI] [PubMed] [Google Scholar]
- Crow JP, Ye YZ, Strong M, Kirk M, Barnes S, Beckman JS. Superoxide dismutase catalyzes nitration of tyrosines by peroxynitrite in the rod and head domains of neurofilament-L. J Neurochem. 1997b;69:1945–1953. doi: 10.1046/j.1471-4159.1997.69051945.x. [DOI] [PubMed] [Google Scholar]
- Deng HX, Hentati A, Tainer JA, Iqbal Z, Cayabyab A, Hung WY, Getzoff ED, Hu P, Herzfeldt B, Roos RP, Warner C, Deng G, Soriano E, Smyth C, Parge HE, Ahmed A, Roses AD, Hallewell RA, Pericak-Vance MA, Siddique T. Amyotrophic lateral sclerosis and structural defects in Cu,Zn superoxide dismutase. Science. 1993;261:1047–1051. doi: 10.1126/science.8351519. [DOI] [PubMed] [Google Scholar]
- DiDonato M, Craig L, Huff ME, Thayer MM, Cardoso RM, Kassmann CJ, Lo TP, Bruns CK, Powers ET, Kelly JW, Getzoff ED, Tainer JA. ALS mutants of human superoxide dismutase form fibrous aggregates via framework destabilization. J Mol Biol. 2003;332:601–615. doi: 10.1016/s0022-2836(03)00889-1. [DOI] [PubMed] [Google Scholar]
- Elam JS, Taylor AB, Strange R, Antonyuk S, Doucette PA, Rodriguez JA, Hasnain SS, Hayward LJ, Valentine JS, Yeates TO, Hart PJ. Amyloid-like filaments and water-filled nanotubes formed by SOD1 mutant proteins linked to familial ALS. Nat Struct Biol. 2003;10:461–467. doi: 10.1038/nsb935. [DOI] [PubMed] [Google Scholar]
- Estévez AG, Crow JP, Sampson JB, Reiter C, Zhuang Y, Richardson GJ, Tarpey MM, Barbeito L, Beckman JS. Induction of nitric oxide-dependent apoptosis in motor neurons by zinc-deficient superoxide dismutase. Science. 1999;286:2498–2500. doi: 10.1126/science.286.5449.2498. [DOI] [PubMed] [Google Scholar]
- Fridovich I. Superoxide dismutases. Adv Enzymol Relat Areas Mol Biol. 1974;41:35–97. doi: 10.1002/9780470122860.ch2. [DOI] [PubMed] [Google Scholar]
- Fridovich I. Superoxide dismutases. Adv Enzymol Relat Areas Mol Biol. 1986;58:61–97. doi: 10.1002/9780470123041.ch2. [DOI] [PubMed] [Google Scholar]
- Furukawa Y, O’Halloran TV. Amyotrophic lateral sclerosis mutations have the greatest destabilizing effect on the apo- and reduced form of SOD1, leading to unfolding and oxidative aggregation. J Biol Chem. 2005;280:17266–17274. doi: 10.1074/jbc.M500482200. [DOI] [PubMed] [Google Scholar]
- Giasson BI, Duda JE, Quinn SM, Zhang B, Trojanowski JQ, Lee VMY. Neuronal α–synucleinopathy with severe movement disorder in mice expressing A53T human α-synuclein. Neuron. 2002;34:521–533. doi: 10.1016/s0896-6273(02)00682-7. [DOI] [PubMed] [Google Scholar]
- Götz J, Chen F, Barmettler R, Nitsch RM. Tau filament formation in transgenic mice expressing P301L tau. J Biol Chem. 2001;276:529–534. doi: 10.1074/jbc.M006531200. [DOI] [PubMed] [Google Scholar]
- Guo JT, Wetzel R, Xu Y. Molecular modeling of the core of Abeta amyloid fibrils. Proteins. 2004;57:357–364. doi: 10.1002/prot.20222. [DOI] [PubMed] [Google Scholar]
- Gurney ME, Pu H, Chiu AY, Dal Canto MC, Polchow CY, Alexander DD, Caliendo J, Hentati A, Kwon YW, Deng HX, Chen W, Zhai P, Sufit RL, Siddique T. Motor neuron degeneration in mice that express a human Cu,Zn superoxide dismutase mutation. Science. 1994;264:1772–1775. doi: 10.1126/science.8209258. [DOI] [PubMed] [Google Scholar]
- Ishihara T, Hong M, Zhang B, Nakagawa Y, Lee MK, Trojanowski JQ, Lee VM. Age-dependent emergence and progression of a tauopathy in transgenic mice overexpressing the shortest human tau isoform. Neuron. 1999;24:751–762. doi: 10.1016/s0896-6273(00)81127-7. [DOI] [PubMed] [Google Scholar]
- Johnston JA, Dalton MJ, Gurney ME, Kopito RR. Formation of high molecular weight complexes of mutant Cu, Zn-superoxide dismutase in a mouse model for familial amyotrophic lateral sclerosis. Proc Natl Acad Sci USA. 2000;97:12571–12576. doi: 10.1073/pnas.220417997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jonsson PA, Ernhill K, Andersen PM, Bergemalm D, Brannstrom T, Gredal O, Nilsson P, Marklund SL. Minute quantities of misfolded mutant superoxide dismutase-1 cause amyotrophic lateral sclerosis. Brain. 2004;127:73–88. doi: 10.1093/brain/awh005. [DOI] [PubMed] [Google Scholar]
- Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature (London) 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- Lee MK, Marszalek JR, Cleveland DW. A mutant neurofilament subunit causes massive, selective motor neuron death: implications for the pathogenesis of human motor neuron disease. Neuron. 1994;13:975–988. doi: 10.1016/0896-6273(94)90263-1. [DOI] [PubMed] [Google Scholar]
- Lee MK, Stirling W, Xu Y, Xu X, Qui D, Mandir AS, Dawson TM, Copeland NG, Jenkins NA, Price DL. Human α-synuclein-harboring familial Parkinson’s disease-linked Ala-53-Thr mutation causes neurodegenerative disease with α-synuclein aggregation in transgenic mice. Proc Natl Acad Sci U S A. 2002;99:8968–8973. doi: 10.1073/pnas.132197599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis J, McGowan E, Rockwood J, Melrose H, Nacharaju P, Van Slegtenhorst M, Gwinn-Hardy K, Paul MM, Baker M, Yu X, Duff K, Hardy J, Corral A, Lin WL, Yen SH, Dickson DW, Davies P, Hutton M. Neurofibrillary tangles, amyotrophy and progressive disturbance in mice expressing mutant (P301L) tau protein. Nat Genet. 2000;25:402–405. doi: 10.1038/78078. [DOI] [PubMed] [Google Scholar]
- Lindberg MJ, Tibell L, Oliveberg M. Common denominator of Cu/Zn superoxide dismutase mutants associated with amyotrophic lateral sclerosis: Decreased stability of the apo state. Proc Natl Acad Sci USA. 2002;99:16607–16612. doi: 10.1073/pnas.262527099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Lillo C, Jonsson PA, Vande VC, Ward CM, Miller TM, Subramaniam JR, Rothstein JD, Marklund S, Andersen PM, Brannstrom T, Gredal O, Wong PC, Williams DS, Cleveland DW. Toxicity of familial ALS-linked SOD1 mutants from selective recruitment to spinal mitochondria. Neuron. 2004;43:5–17. doi: 10.1016/j.neuron.2004.06.016. [DOI] [PubMed] [Google Scholar]
- McCord JM, Fridovich I. Superoxide dismutase. An enzymic function for erythrocuprein (hemocuprein) J Biol Chem. 1969;244:6049–6055. [PubMed] [Google Scholar]
- McGowan E, Pickford F, Kim J, Onstead L, Eriksen J, Yu C, Skipper L, Murphy MP, Beard J, Das P, Jansen K, Delucia M, Lin WL, Dolios G, Wang R, Eckman CB, Dickson DW, Hutton M, Hardy J, Golde T. Abeta42 is essential for parenchymal and vascular amyloid deposition in mice. Neuron. 2005;47:191–199. doi: 10.1016/j.neuron.2005.06.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizushima S, Nagata S. pEF-BOS, a powerful mammalian expression vector. Nucleic Acids Res. 1990;18:5322. doi: 10.1093/nar/18.17.5322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norstrom EM, Mastrianni JA. The AGAAAAGA palindrome in PrP is required to generate a productive PrPSc-PrPC complex that leads to prion propagation. J Biol Chem. 2005;280:27236–27243. doi: 10.1074/jbc.M413441200. [DOI] [PubMed] [Google Scholar]
- Parge HE, Hallewell RA, Tainer JA. Atomic structures of wild-type and thermostable mutant recombinant human Cu,Zn superoxide dismutase. Proc Natl Acad Sci USA. 1992;89:6109–6113. doi: 10.1073/pnas.89.13.6109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasinelli P, Belford ME, Lennon N, Bacskai BJ, Hyman BT, Trotti D, Brown RH., Jr Amyotrophic lateral sclerosis-associated SOD1 mutant proteins bind and aggregate with Bcl-2 in spinal cord mitochondria. Neuron. 2004;43:19–30. doi: 10.1016/j.neuron.2004.06.021. [DOI] [PubMed] [Google Scholar]
- Potter SZ, Valentine JS. The perplexing role of copper-zinc superoxide dismutase in amyotrophic lateral sclerosis (Lou Gehrig’s disease) J Biol Inorg Chem. 2003;8:373–380. doi: 10.1007/s00775-003-0447-6. [DOI] [PubMed] [Google Scholar]
- Ratovitski T, Corson LB, Strain J, Wong P, Cleveland DW, Culotta VC, Borchelt DR. Variation in the biochemical/biophysical properties of mutant superoxide dismutase 1 enzymes and the rate of disease progression in familial amyotrophic lateral sclerosis kindreds. Hum Mol Genet. 1999;8:1451–1460. doi: 10.1093/hmg/8.8.1451. [DOI] [PubMed] [Google Scholar]
- Ray SS, Nowak RJ, Strokovich K, Brown RH, Jr, Walz T, Lansbury PT., Jr An intersubunit disulfide bond prevents in vitro aggregation of a superoxide dismutase-1 mutant linked to familial amytrophic lateral sclerosis. Biochemistry. 2004;43:4899–4905. doi: 10.1021/bi030246r. [DOI] [PubMed] [Google Scholar]
- Reaume AG, Elliott JL, Hoffman EK, Kowall NW, Ferrante RJ, Siwek DF, Wilcox HM, Flood DG, Beal MF, Brown RH, Jr, Scott RW, Snider WD. Motor neurons in Cu/Zn superoxide dismutase-deficient mice develop normally but exhibit enhanced cell death after axonal injury. Nature Genetics. 1996;13:43–47. doi: 10.1038/ng0596-43. [DOI] [PubMed] [Google Scholar]
- Rodriguez JA, Valentine JS, Eggers DK, Roe JA, Tiwari A, Brown RH, Jr, Hayward LJ. Familial Amyotrophic Lateral Sclerosis-associated Mutations Decrease the Thermal Stability of Distinctly Metallated Species of Human Copper/Zinc Superoxide Dismutase. J Biol Chem. 2002;277:15932–15937. doi: 10.1074/jbc.M112088200. [DOI] [PubMed] [Google Scholar]
- Rosen DR, Siddique T, Patterson D, Figlewicz DA, Sapp P, Hentati A, Donaldson D, Goto J, O’Regan JP, Deng HX, Rahmani Z, Krizus A, McKenna-Yasek D, Cayabyab A, Gaston SM, Berger R, Tanzi RE, Halperin JJ, Herzfeldt B, Van den Bergh R, Hung WY, Bird T, Deng G, Mulder DW, Smyth C, Laing NG, Soriano E, Pericak-Vance MA, Haines J, Rouleau GA, Gusella JS, Horvitz HR, Brown RH., Jr Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature. 1993;362:59–62. doi: 10.1038/362059a0. [DOI] [PubMed] [Google Scholar]
- Shinder GA, Lacourse MC, Minotti S, Durham HD. Mutant Cu/Zn-Superoxide Dismutase Proteins Have Altered Solubility and Interact with Heat Shock/Stress Proteins in Models of Amyotrophic Lateral Sclerosis. J Biol Chem. 2001;276:12791–12796. doi: 10.1074/jbc.M010759200. [DOI] [PubMed] [Google Scholar]
- Subramaniam JR, Lyons WE, Liu J, Bartnikas TB, Rothstein J, Price DL, Cleveland DW, Gitlin JD, Wong PC. Mutant SOD1 causes motor neuron disease independent of copper chaperone-mediated copper loading. Nat Neurosci. 2002;5:301–307. doi: 10.1038/nn823. [DOI] [PubMed] [Google Scholar]
- Thakur AK, Wetzel R. Mutational analysis of the structural organization of polyglutamine aggregates. Proc Natl Acad Sci U S A. 2002;99:17014–17019. doi: 10.1073/pnas.252523899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tiwari A, Hayward LJ. Familial Amyotrophic Lateral Sclerosis Mutants of Copper/Zinc Superoxide Dismutase Are Susceptible to Disulfide Reduction. J Biol Chem. 2003;278:5984–5992. doi: 10.1074/jbc.M210419200. [DOI] [PubMed] [Google Scholar]
- Valentine JS, Hart PJ. Misfolded CuZnSOD and amyotrophic lateral sclerosis. Proc Natl Acad Sci U S A. 2003;100:3617–3622. doi: 10.1073/pnas.0730423100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Slunt H, Gonzales V, Fromholt D, Coonfield M, Copeland NG, Jenkins NA, Borchelt DR. Copper-binding-site-null SOD1 causes ALS in transgenic mice: aggregates of non-native SOD1 delineate a common feature. Hum Mol Genet. 2003;12:2753–2764. doi: 10.1093/hmg/ddg312. [DOI] [PubMed] [Google Scholar]
- Wang J, Xu G, Borchelt DR. High molecular weight complexes of mutant superoxide dismutase 1: age-dependent and tissue-specific accumulation. Neurobiol Dis. 2002a;9:139–148. doi: 10.1006/nbdi.2001.0471. [DOI] [PubMed] [Google Scholar]
- Wang J, Xu G, Gonzales V, Coonfield M, Fromholt D, Copeland NG, Jenkins NA, Borchelt DR. Fibrillar inclusions and motor neuron degeneration in transgenic mice expressing superoxide dismutase 1 with a disrupted copper-binding site. Neurobiol Dis. 2002b;10:128–138. doi: 10.1006/nbdi.2002.0498. [DOI] [PubMed] [Google Scholar]
- Wang J, Xu G, Li H, Gonzales V, Fromholt D, Karch C, Copeland NG, Jenkins NA, Borchelt DR. Somatodendritic accumulation of misfolded SOD1-L126Z in motor neurons mediates degeneration: {alpha}B-crystallin modulates aggregation. Hum Mol Genet. 2005 doi: 10.1093/hmg/ddi236. [DOI] [PubMed] [Google Scholar]
- Watanabe Y, Yasui K, Nakano T, Doi K, Fukada Y, Kitayama M, Ishimoto M, Kurihara S, Kawashima M, Fukuda H, Adachi Y, Inoue T, Nakashima K. Mouse motor neuron disease caused by truncated SOD1 with or without C-terminal modification. Brain Res Mol Brain Res. 2005;135:12–20. doi: 10.1016/j.molbrainres.2004.11.019. [DOI] [PubMed] [Google Scholar]
- Wiedau-Pazos M, Goto JJ, Rabizadeh S, Gralla EB, Roe JA, Lee MK, Valentine JS, Bredesen DE. Altered reactivity of superoxide dismutase in familial amyotrophic lateral sclerosis. Science. 1996;271:515–518. doi: 10.1126/science.271.5248.515. [DOI] [PubMed] [Google Scholar]
- Williams AD, Portelius E, Kheterpal I, Guo JT, Cook KD, Xu Y, Wetzel R. Mapping abeta amyloid fibril secondary structure using scanning proline mutagenesis. J Mol Biol. 2004;335:833–842. doi: 10.1016/j.jmb.2003.11.008. [DOI] [PubMed] [Google Scholar]
- Wong PC, Pardo CA, Borchelt DR, Lee MK, Copeland NG, Jenkins NA, Sisodia SS, Cleveland DW, Price DL. An adverse property of a familial ALS-linked SOD1 mutation causes motor neuron disease characterized by vacuolar degeneration of mitochondria. Neuron. 1995;14:1105–1116. doi: 10.1016/0896-6273(95)90259-7. [DOI] [PubMed] [Google Scholar]
- Yim HS, Kang JH, Chock PB, Stadtman ER, Yim MB. A familial amyotrophic lateral sclerosis-associated A4V Cu, Zn-superoxide dismutase mutant has a lower Km for hydrogen peroxide. Correlation between clinical severity and the Km value. J Biol Chem. 1997;272:8861–8863. doi: 10.1074/jbc.272.14.8861. [DOI] [PubMed] [Google Scholar]
- Yim MB, Kang JH, Yim HS, Kwak HS, Chock PB, Stadtman ER. A gain-of-function of an amyotrophic lateral sclerosis-associated Cu,Zn-superoxide dismutase mutant: An enhancement of free radical formation due to a decrease in Km for hydrogen peroxide. Proc Natl Acad Sci USA. 1996;93:5709–5714. doi: 10.1073/pnas.93.12.5709. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.