

1. Introduction
Redox reactions play important roles in almost all biological processes, including photosynthesis and respiration, which are two essential energy processes that sustain all life on earth. It is thus not surprising that biology employs redox-active metal ions in these processes. It is largely the redox activity that makes metal ions uniquely qualified as biological cofactors and makes bioinorganic enzymology both fun to explore and challenging to study.
Even though most metal ions are redox active, biology employs a surprisingly limited number of them for electron transfer (ET) processes. Prominent members of redox centers involved in ET processes include cytochromes, iron–sulfur clusters, and cupredoxins. Together these centers cover the whole range of reduction potentials in biology (Figure 1). Because of their importance, general reviews about redox centers1−77 and specific reviews about cytochromes,8,24,78−90 iron–sulfur proteins,91−93 and cupredoxins94−104 have appeared in the literature. In this review, we provide both classification and description of each member of the above redox centers, including both native and designed proteins, as well as those proteins that contain a combination of these redox centers. Through this review, we examine structural features responsible for their redox properties, including knowledge gained from recent progress in fine-tuning the redox centers. Computational studies such as DFT calculations become more and more important in understanding the structure–function relationship and facilitating the fine-tuning of the ET properties and reduction potentials of metallocofactors in proteins. Since this aspect has been reviewed extensively before,105−110 and by other reviews in this thematic issue,2000,2001,2002 it will not be covered here.
Figure 1.

Reduction potential range of redox centers in electron transfer processes.
2. Cytochromes in Electron Transfer Processes
2.1. Introduction to Cytochromes
Cytochromes are a major class of heme-containing ET proteins found ubiquitously in biology. They were first described in 1884 as respiratory pigments (called myohematin or histohematin) to explain colored substances in cells.81,111 These colored substances were later rediscovered in 1920 and named “cytochromes”, or cellular pigments.112 The intense red color combined with relatively high thermodynamic stability makes cytochromes easy to observe and to purify. As of today, more than 70 000 cytochromes have been discovered.78 In addition, due to their small size, high solubility, and well-folded helical structure and the presence of the heme chromophore, cytochromes are one of the most extensively studied classes of proteins spanning several decades.79
Cytochromes are present mostly in the inner mitochondrial membrane of eukaryotic organisms and are also found in a wide variety of both Gram-positive and Gram-negative bacteria.113,114 Cytochromes play crucial roles in a number of biological ET processes associated with many different energy metabolisms. Additionally, cytochromes are involved in apoptosis in mammalian cells.115 Further description of the latter role of cytochromes is beyond the scope of this review, which is solely focused on the role of cytochromes in ET. For a similar reason, another family of cytochromes, the cyts P450 (CYP), which catalyze the oxidation of various organic substrates such as metabolites (lipids, hormones, etc.) and xenobiotic substances (drugs, toxic chemicals, etc.), will not be discussed in this review either.
A number of books and reviews have appeared in the literature describing the role of cytochromes as ET proteins.8,24,78−90 Here we summarize studies on both native and designed cytochromes and their roles in biological ET processes.
2.2. Classification of Cytochromes
Cytochromes are classified on the basis of the electronic absorption maxima of the heme macrocycle, such as a, b, c, d, f, and o types of heme. More specifically, these letter names represent characteristic absorbance maxima in the UV–vis electronic absorption spectrum when the heme iron is coordinated with pyridine in its reduced (ferrous) state, designated as the “pyridine hemochrome” spectrum (Figure 2).
Figure 2.

Representative pyridine hemochromogen spectra of hemin cofactors: (A) heme b, (B) heme a, and (C) heme d1. The spectrum of pyridine ferrohemochrome c is similar to that of heme b. Reprinted with permission from ref (116). Copyright 1992 Springer-Verlag.
Table 1 shows the maximum peak positions and their corresponding extinction coefficients of the pyridine hemochrome spectra of various classes of cytochromes. These differences arise from different substituents at the β-pyrrole positions on the periphery of the heme.
Table 1. UV–Vis Spectral Parameters of Pyridine Hemochrome Spectra of Various Types of Cytochromesa.
| pyridine hemochromogen |
|||||
|---|---|---|---|---|---|
| heme | position of α peak (nm) | εmM (at α peak) | α peak (nm) of reduced protein | example | ref |
| protoheme IX (b) | 557 | 34.4 | 557–563 | cyt b6f complex | (117) |
| heme c | 550 | 29.1 | 549–561 | cyt c | (118) |
| heme a | 587 | 26 | 587–611 | cyt aa3 oxidase | (117) |
| heme d | 613 | 630–635 | cyt bd oxidase | (116) | |
| heme d1 | 620 | 24 | 625 | cyt cd1 nitrite reductase | (116) |
| heme o | 553 | 560 | cyt bo3 oxidase | (119) | |
Adapted with permission from ref (116). Copyright 1992 Springer-Verlag.
The word “heme” specifically describes the ferrous complex of the tetrapyrrole macrocyclic ligand called protoporphyrin IX (Figure 3).81 It is the precursor to various types of cytochromes through different peripheral substitutions. Figure 3 shows a schematic of these various types of hemes.
Figure 3.

Different types of heme found in cytochromes.
The b-type cytochromes have four methyl substitutions at positions 1, 3, 5, and 8, two vinyl groups in positions 2 and 4, and two propionate groups at positions 6 and 7, resulting in a 22-π-electron porphyrin. Hemes a and c are biosynthesized as derivatives of heme b. In heme a, the vinyl group at position 2 of the porphyrin ring of heme b is replaced by a hydroxyethylfarnesyl side chain while the methyl group at position 8 is oxidized to a formyl group. These substituents make heme a more hydrophobic as well as more electron-withdrawing than heme b due to the presence of farnesyl and formyl groups, respectively. Covalent cross-linking of the vinyl groups at β-pyrrole positions 2 and 4 of heme b with Cys residues from the protein yields heme c, where the vinyl groups of heme b are replaced by thioether bonds.
The covalent cross-linking of the two Cys residues from the protein to the porphyrin ring occurs at the highly conserved -Cys-Xxx-Xxx-Cys-His- sequences (Xxx=any amino acid). This cross-linking covalently attaches heme c to the protein. The histidine residue in the conserved sequence serves as an axial ligand to the heme iron. In heme d, two cis-hydroxyl groups are inserted at positions 5 and 6 on the β-pyrrole, which renders heme d as a 20-π-electron chlorin. Heme d1 contains two ketone groups in place of the vinyl groups at positions 2 and 4, while two acetate groups are added to positions 1 and 3 of the tetrapyrrole macrocycle, resulting in 18-π-electron isobacteriochlorins. The hemes f is similar to heme c, with the difference in the ligands that coordinate to the heme iron at the axial position (called axial ligands) make hemes c and f spectroscopically distinct.
Common axial ligands found in cytochromes are shown in Figure 4. With the exception of cytochromes c′ (cyts c′), all cytochromes with ET function contain 6-coordinate low-spin (6cLS) hemes axially ligated to amino acids such as His or an N-terminal amine group. Two axial His residues act as ligands to the heme iron in b-type cytochromes. The only example of bis-Met axial coordination to heme b is observed in the iron storage protein bacterioferritin.120,121 A common axial His ligand is found in all cyts c, where the axial His is a part of the conserved -Cys-Xxx-Xxx-Cys-His- sequence, through which the heme is covalently attached to the protein. The most commonly encountered second axial ligand in c-type cytochromes is Met with the exception of multiheme c-type cytochromes, which generally display bis-His axial ligation of the heme iron (section 2.3.6).80 In most cases, the His ligands are coordinated to the heme iron by their Nε atom. However, an example of Nδ coordination has been reported.122 The f-type cytochromes contain the same type of heme with one axial His ligand, as in cyts c; the only exception is in the nature of the second axial ligation in that the second axial ligand is the NH2 group of an N-terminal tyrosine instead of the most commonly found Met or His as the second axial ligand.123 Not surprisingly, the variation in the axial ligation makes each heme type electronically unique, resulting in different out-of-plane distortions of the heme iron from the heme plane (Figure 4) as well as different spectroscopic features (Table 1).
Figure 4.
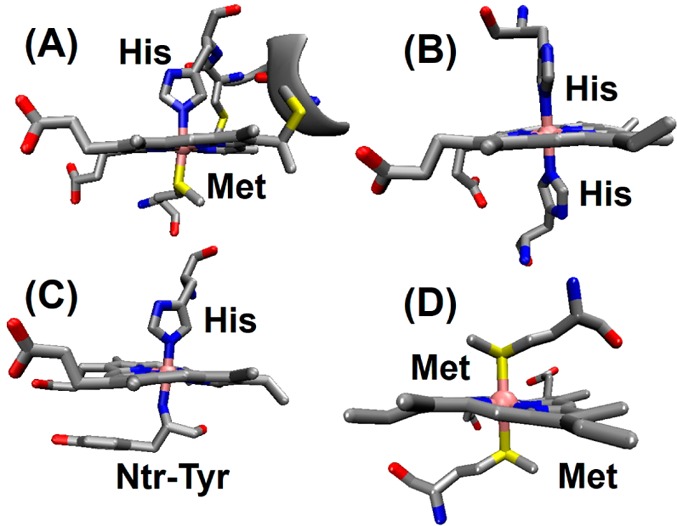
Commonly found heme axial ligands in various cytochromes. (A) Class I cyts c (PDB ID 3CYT) uses His/Met axial ligation. (B) Cyts b and multiheme cyts c contain bis-His ligation (bovine liver cyt b5, PDB ID 1CYO). (C) An unusual His/amine ligation is found only in cyt f (PDB ID 1HCZ). (D) Bis-Met ligation is encountered in bacterioferritin (PDB ID 1BCF). For c-type cytochromes the conserved -Cys-Xxx-Xxx-Cys-His- ligation and its covalent linkage to the heme via Cys residues are shown.
2.3. Native Cytochromes c
2.3.1. Functions of Cytochromes c
Cytochromes c are involved in biological ET processes in both aerobic and anaerobic respiratory chains. In aerobic respiration, they are involved in the mitochondrial respiratory chain to produce the energy currency ATP by transferring electrons from the transmembrane bc1 complex to cyt c oxidase.85,86 In addition, cyts c have also been recently discovered to play a crucial role in programmed cell death (apoptosis), where they activate the protease involved in cell death, caspase 3.124−126 Other examples where c-type cytochromes are involved in ET include the reduction of sulfate to hydrogen sulfide, conversion of nitrogen to ammonia in nitrogen fixation, reduction of nitrate to dinitrogen in denitrification, in phototrophs that use light energy to carry out various cellular processes, and in methylotrophs that use methane or methanol as the carbon source for their growth. Detailed descriptions of the roles of cyts c in these cases will be discussed in the following sections.
As cyts c are involved in numerous crucial biological processes, they have been used extensively as a hallmark system to study biological ET by site-directed mutagenesis, which have elucidated the regions of the protein that are critical for their ET properties as well as fine-tuning the reduction potentials.87,127−131 In addition, various inorganic redox couples have been covalently appended to surface sites of cyts c to study intraprotein ET pathways.24,132,133 Various complexes of cyts c with other protein partners have also been prepared to study interprotein ET pathways.134−149
2.3.2. Classifications of Cytochromes c
Cytochromes c generally contain ∼100–120 amino acids. Biosynthesis of cyts c involves the formation of two thioether bonds between two Cys residues and the two vinyl groups of heme b by post-translational modification.150,151 Primary amino acid sequence alignment shows that the residue identity of cyts c is 45–100% among eukaryotes. The electronic spectra of cyts c are dominated by the allowed porphyrin π → π* transitions that are mixed together with interelectronic repulsions that give rise to an intense band at ∼410 nm (called the Soret or γ band) and two weaker signals in the 500–600 nm range (the α and β bands). The reduced form of the protein shows a Soret band at 413 nm and sharp α and β bands at 550 nm (ε = 29.1 mM–1 cm–1) and 521 nm (ε = 15.5 mM–1 cm–1), respectively, with a ratio of α to β bands of 1.87 (Table 1). The electronic spectra of cyts c from other sources are very similar to that of horse heart cyt c. Originally classified by Ambler,89,152 cyts c have been divided into four major classes on the basis of the number of hemes, position and identity of the axial iron ligands, and reduction potentials (Table 2).
Table 2. Axial Ligand Types and Reduction Potentials of Various Cytochromesa.
| cytochrome | axial ligand | heme type | E (mV)b | mutant | E (mV) | ref |
|---|---|---|---|---|---|---|
| Nitrosomonas europaea diheme cyt c peroxidase | His/Met | class I | 450 | (153, 154) | ||
| Rhodocyclus tenuis THRC cyt c | class IV | 420 | (155) | |||
| HP1 | His/Met | 420 | ||||
| HP2 | His/Met | 110 | ||||
| LP1 | bis-His | 60 | ||||
| LP2 | His/Met | |||||
| Rhodopseudomonas viridis THRC cyt c | class IV | 380 | (156,157) | |||
| H1 (c559) | His/Met | 330 | ||||
| H3 (c556) | His/Met | 20 | ||||
| H2 (c552) | bis-His | –60 | ||||
| H4 (c554) | His/Met | |||||
| Rhodobacter capsulatas cyt c2 | His/Met | class I | 373 | Gly29Ser | 330 | (158−160) |
| Pro30Ala | 258 | |||||
| Tyr67Cys | 348 | |||||
| Tyr67Phe | 308 | |||||
| Chlamydomonas reinhardtii cyt f | His/Ntr-Tyr | cyt f | 370 | Tyr1Phe | 369 | (161) |
| Tyr1Ser | 313 | |||||
| Val3Phe | 373 | |||||
| Phe4Leu | 348 | |||||
| Phe4Trp | 336 | |||||
| Tyr1Phe/Phe4Tyr | 370 | |||||
| Tyr1Ser/Phe4Leu | 289 | |||||
| Val3Phe/Phe4Trp | 342 | |||||
| Rhodospirillum rubrum cyt c2 | His/Met | class I | 324 | (156) | ||
| Pseudomonas aeruginosa cyt c nitric oxide reductase | His/Met | class I | 310 | (162) | ||
| bis-His | cyt b | 345 | ||||
| Pseudomonas aeruginosa cyt c peroxidase | His/Met | class I | 320 | (163) | ||
| Arthrospira maxima cyt c6 | His/Met | class I | 314 | (164) | ||
| Saccharomyces cerevisiae iso-2-cyt c | His/Met | class I | 288 | Asn52Ile | 243 | (130) |
| Saccharomyces cerevisiae iso-1-cyt c | His/Met | class I | 272 | Arg38Lys | 249 | (131, 165−173) |
| 285 | Arg38His | 245 | ||||
| 290 | Arg38Gln | 242 | ||||
| Arg38Asn | 238 | |||||
| Arg38Leu | 231 | |||||
| Arg38Ala | 225 | |||||
| Asn52Ala | 257 | |||||
| Asn52Ile | 231 | |||||
| Tyr67Phe | 234 | |||||
| Phe82Leu | 286 | |||||
| Phe82Tyr | 280 | |||||
| Phe82Ile | 273 | |||||
| Phe82Trp | 266 | |||||
| Phe82Ala | 260 | |||||
| Phe82Ser | 255 | |||||
| Phe82Gly | 247 | |||||
| Pseudomonas aeruginosa cyt c551 | His/Met | class I | 276 | (156) | ||
| horse cyt c | His/Met | class I | 262 | Met80Ala | 82 | (158, 174) |
| Met80His | 41 | |||||
| Met80Leu | –42 | |||||
| Met80Cys | –390 | |||||
| rat cyt c | His/Met | class I | 260 | Pro30Ala | 258 | |
| Pro30Val | 261 | |||||
| Tyr67Phe | 224 | |||||
| Rhodopseudomonas palustris cyt c556 | His/Met | class II | 230 | (80) | ||
| Escherichia coli cyt b562 | His/Met | cyt b (class II) | 168 | Phe61Gly | 90 | (175, 176) |
| Phe65Val | 173 | |||||
| Phe61Ile/Phe65Tyr | 68 | |||||
| His102Met | 240 | |||||
| Arg98Cys/His102Met | 440 | |||||
| Alicycliphilus denitrificans cyt c′ | His/Met | class II | 132 | (80) | ||
| Rhodopseudomonas palustris cyt c′ | His/Met | class II | 102 | (80) | ||
| cytochrome b5 | bis-His | cyt b | form A | 80 | (177) | |
| form B | –26 | |||||
| Desulfovibrio vulgaris cyt c553 | His/Met | class I | 37 | Met23Cys | 29 | (156, 178) |
| 20 ± 5 | Gly51Cys | 28 | ||||
| Met23Cys/Met23Cys | 88 | |||||
| Gly51Cys/Gly51Cys | 105 | |||||
| bovine liver microsomal cyt b5 | bis-His | cyt b | 3 | protoheme IX dimethyl ester | 70 | (179) |
| Saccharomyces cerevisiae cyt b2 | bis-His | cyt b | –3 | (156) | ||
| Chromatium vinosum cyt c′ | His | class II | –5 | (80) | ||
| rat liver microsomal cyt b5 | bis-His | cyt b | –7 ± 1 | (129, 180) | ||
| Rhodospirillum rubrum cyt c′ | His/Met | class I | –8 | (80) | ||
| tryptic bovine hepatic cyt b5 | His/Met | class I | –10 ± 3 | Val61Lys | 17 | (181) |
| Val61His | 11 | |||||
| Val61Glu | –25 | |||||
| Val61Tyr | –33 | |||||
| Allochromatium vinosum triheme cyt c | bis-His | class III | –20 | (182) | ||
| His/Met | –200 | |||||
| His-Cys/Met | –220 | |||||
| Rhodobacter sphaeroides cyt c′ | His/Asn | cyt c | –22 | (183) | ||
| cyt b6f complex | bis-His | cyt b | –45 | (184) | ||
| –150 | ||||||
| Thermosynechococcus elongates PS cyt c550 | His/Met | class I | –80 | in the absence of mediators | 200 | (185) |
| MamP magnetochrome | His/Met | class I | –76 | (186) | ||
| rat liver OM cyt b5 | bis-His | cyt b | –102 | His63Met | 110 | (187, 188) |
| Val45Leu/Val61Leu | –148 | |||||
| protoheme IX dimethyl ester | –36 | |||||
| Desulfovibrio desulfuricans Norway cyt c3 | bis-His | class III | –132 | (78) | ||
| bis-His | –255 | |||||
| bis-His | –320 | |||||
| bis-His | –360 | |||||
| Chlorella nitrate reductase cyt b557 | bis-His | cyt b | –164 | (189, 190) | ||
| Ectothiorhodospira shaposhnikovii cyt b558 | bis-His | cyt b | –210 | (191) | ||
| Azotobacter vinelandii bacterioferritin | bis-His | cyt b | –225 | (192) | ||
| (in the presence of a nonheme iron core) | –475 | |||||
| Desulfovibrio vulgaris Hildenborough cyt c3 | bis-His | class III | –280 | (192, 193) | ||
| bis-His | –320 | |||||
| bis-His | –350 | |||||
| bis-His | –380 | |||||
| Synechocystis sp. cyt c549 | bis-His | –250 | (78) | |||
| Arthrospira maxima cyt c549 | His/Met | –260 | (164) |
Adapted with permission from ref (78). Copyright 2004 Elsevier.
All reduction potentials listed in this review are versus standard hydrogen electrode (SHE) or normal hydrogen electrode (NHE).
The class I cyts c include small (8–120 kDa) soluble proteins containing a single 6cLS heme moiety and display a range of reduction potentials from −390 to +450 mV versus standard hydrogen electrode (SHE) (Table 2).78 On the basis of sequence and structural alignments, class I cyts c have further been partitioned into 16 different subclasses.88 The majority of the subclasses include mitochondrial cyts c and purple bacterial cyts c. Examples of other subclasses represent a wide variety of different sources, including cyts c551, cyts c4, cyts c5, and cyts c6 from Pseudomonas, Chlorobium cyt c555, Desulfovibrio (Dv.) cyts c553, c550 from cyanobacteria and algae, Ectothiorhodospira cyts c551, flavocytochromes c, methanol dehydrogenase-associated cyt c550 or cL, cyt cd1 nitrite reductase, the cyt subunit associated with alcohol dehydrogenase, nitrite reductase-associated cyt c from Pseudomonas, and cyt c oxidase subunit II from Bacillus.78
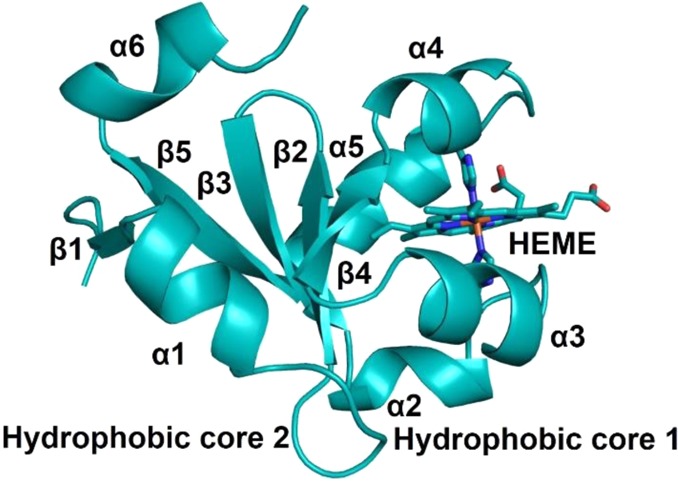
Class I cyt c domains are characterized by their signature cyt c fold and the presence of an N-terminal conserved -Cys-Xxx-Xxx-Cys-His- sequence containing cysteines for covalent cross-linking of the heme to the protein and the His, which acts as the axial ligand to the heme iron. The class I cyt c fold is recognized as having a total of five α-helices arranged in a unique tertiary structure. There are two helices, one each at the N- and C-termini, represented as α1 and α5, respectively. In between, there is a small helix, α3 (also called the 50s helix in mitochondrial cyts c), followed by two other helices, α4 and α5, which are known as the 60s helix and 70s helix, respectively, in mitochondrial cyts c. The 70s helix precedes a loop toward the C-terminus that contains the second axial ligand, Met, to the heme iron. There are examples where the second axial ligand is a residue other than Met, e.g., Asn or His, or is even absent.79 In many cases, this core cyt c domain can be found fused to other membrane proteins. General features of the class I cyt c fold are shown in Figure 5.
Figure 5.
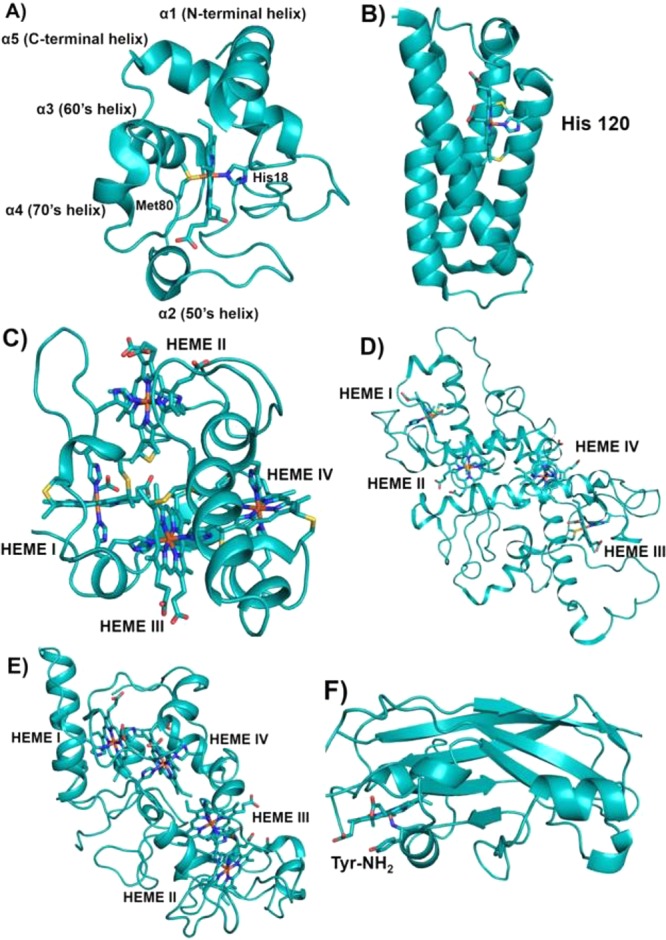
Schematic representations of various classes of cyts c. (A) Class I cyt c fold with His/Met heme axial ligands (PDB ID 3CYT). Mitochondrial designation of the helices is also shown. (B) Four-helix bundle cyt c′ belongs to class II cyt c having a 5c heme with His120 as the sole axial ligand (PDB ID 1E83). (C) Tetraheme cyt c3 belongs to class III cyt c with bis-His ligation to all four hemes (PDB ID 1UP9). Hemes I and III are attached to the protein via the highly conserved -Cys-Xxx-Xxx-Cys-His- sequence, whereas hemes II and IV are covalently bound to the protein by a -Cys-Xxx-Xxx-Xxx-Xxx-Cys-His- motif. In (A)–(C) the covalent attachment of the heme to the protein via Cys residues is shown. (D) Tetraheme cyt c from the photosynthetic reaction center (RC) belongs to class IV cyt c. Hemes I, II, and III have His/Met axial ligands, while heme IV has bis-His axial ligation to the heme iron (PDB ID 2JBL). (E) Cyt c554 from Nitrosomonas europaea belongs to a class of its own. Hemes I, III, and IV have bis-His-ligated heme iron, whereas heme II is 5c with His as the only axial ligand (PDB ID 1BVB). Heme numbering in (C)–(E) is according to their attachment occurring along the protein’s primary sequence. (F) Cyt f from chloroplast is unique from all other classes of cytochromes in that it mostly contains β-sheets and the heme is 6c with a His and N-terminal backbone NH2 group of a Tyr residue (PDB ID 1HCZ). It has been included as a subclass of cyt c because the heme is covalently bound to the protein via the highly conserved -Cys-Xxx-Xxx-Cys-His- signature motif for heme attachment ubiquitously found in c-type cytochromes.
The class II cyts c consist of a c-type heme covalently attached to the highly conserved C-terminal -Cys-Xxx-Xxx-Cys-His- sequence, as in class I cyts c, with the Cys residues and the His as one of the axial ligands.80 Four α-helices and a left-handed twisted overall structure represent this subclass of cyts c (Figure 5). The second axial ligand to the heme iron is variable.194,195 The subclass cyt c′ is axially coordinated to a single His imidazole ligand, lacks the second axial ligand, and has a relatively small range of reduction potentials ranging from approximately −200 to +200 mV.8,84,90 Members from this subclass represent a wide range of sources that include photosynthetic, denitrifying, nitrogen-fixing, methanotrophic, and sulfur oxidizing bacteria. This class has two subclasses based on the distinct spin states displayed by the heme. Subclass IIa of cyt c′ displays high-spin (HS) ferrous [Fe(II), S = 2] electronic configurations, while the ferric form shows either a HS S = 5/2 state or S = 3/2, S = 5/2 mixture of spin states.196−202 The subclass IIa proteins, isolated from Rhodopseudomonas palustris, Rhodobacter (Rb.) capsulatus, and Chromatium (Ch.) vinosum, display a large amount of the S = 3/2 ground state in the spin-state admixture, ranging from 40% to 57% as determined from electron paramagnetic resonance (EPR) simulations.196,201,203 The second subclass, IIb, includes cyt c556 from Rp. palustris,204Rb. sulfidophilus,205 and Agrobacterium tumefaciens(80) and cyt c554 from Rb. sphaeroides,206 which contain heme in the low-spin (LS) configuration. This subclass of proteins has a second axial ligand to the heme iron which is a Met residue located close to the N-terminus. Class II cyts display reduction potentials ranging from −5 to +230 mV (Table 2).
Class III cyts c include proteins containing multiple hemes with bis-His ligation and display reduction potentials in the range of −20 to −380 mV (Table 2).80,88,152,207−212 In some cases this class of cytochromes have up to 16 heme cofactors and display no structural similarity with other classes of cyts c. They are found as terminal electron donors in bacteria involved in sulfur metabolism.213 These bacteria utilize sulfur or oxidized sulfur compounds as terminal electron acceptors in their respiratory chain. One of the best studied proteins in this class is cyt c3 (∼13 kDa) (Figure 5) from Desulfovibrio, which acts as a natural electron acceptor and donor in hydrogenases and ferredoxins.214 The overall protein fold containing two β-sheets and three to five α-helices is conserved among the known structures of cyts c3 as well as the orientation of the four hemes which are located in close proximity to each other, with each of the heme planes being nearly perpendicular to the others.88 Each heme displays a distinct reduction potential spanning a range from −200 to −400 mV.215−219 Cyt c555.1, also known as cyt c7 (∼9 kDa, 70 amino acids), from Desulfuromonas acetoxidans is another class III cyt c that contains three hemes.220 These proteins have been proposed to be involved in ET to elemental sulfur as well as in the coupled oxidation of acetate and dissimilatory reduction of Fe(III) and Mn(IV) as an energy source in these bacteria.221 In cyt c7, two of the hemes have a reduction potential of −177 mV and the third heme has a reduction potential of −102 mV.222
Class IV cyts c fall into the category of large molar mass (∼35–40 kDa) cytochromes that contain other prosthetic groups in addition to c-type hemes such as flavocytochromes c and cyts cd.152 One example of class IV cyts c is revealed by the X-ray structure of the photosynthetic reaction center (RC) from Rhodpseudomonas viridis, where light energy is harvested and converted to chemically useful energy. The cyt c in the RC consists of four c-type heme moieties covalently bound to subunit C of the RC. Three of the hemes have His/Met axial ligation, while the fourth heme is bis-His-ligated. The four hemes are oriented in two types of pairs. The porphyrin planes of hemes I/III and II/IV are orientated parallel to each other, while the porphyrin planes of each pair of hemes are mutually perpendicular to each pair’s porphyrin planes (Figure 5).223
Cyt c554 is another tetraheme cytochrome that is involved in the ET pathway of the biological nitrogen cycle in the oxidation of ammonia in Nitrosomonas europaea.122,224 This family of cytochromes does not fall into either class III or class IV cytochromes and has been proposed to belong to a class of its own. A pair of electrons are passed from hydroxylamine oxidoreductase (HAO) to two molecules of cyt c554 upon oxidation of hydroxylamine to nitrite. One of the hemes is HS, and the other three are 6cLS with reduction potentials of +47, +47, −147, and −276 mV, respectively. Porphyrin planes of hemes III and IV are oriented almost perpendicular to each other, while the heme pairs I/III and II/IV have parallel orientation (Figure 5). The sets of parallel hemes overlap at an edge, and such heme orientation has been observed in HAO and cyt c nitrite reductase.
Cyt f is a high-potential (Table 2) electron acceptor of the chloroplast cyt b6f complex involved in oxygenic photosynthesis by passing electrons from photosystem II to photosystem I of the RC.123,225 Cyt f accepts electrons from a Rieske-type iron–sulfur cluster and passes electrons to the copper protein plastocyanin. Cyt f consists of two domains primarily of β-sheets and is anchored to the membrane by a transmembrane segment, while most of the protein is located on the lumen side of the thylakoid membrane. The heme is also located on the lumen side at the interface of the two domains and is covalently attached to the protein via the signature sequence of cyts c, -Cys-Xxx-Xxx-Cys-His-. The β-sheet fold has not been observed in any other families of cytochromes and is thus unique to cyts f. Intriguingly, this family of cytochromes also contains an unusual second axial ligation to the heme iron, an N-terminal −NH2 group of a Tyr residue (Figure 5).
Quite uniquely, the only exception to the bis-Cys covalent attachment of the c-type hemes via the conserved -Cys-Xxx-Xxx-Cys-His- motif in cyt c is found in eukaryotes from the phylum Euglenozoa, including trypanosome and Leishmania parasites. In the mitochondrial cyt c of these organisms, the heme is attached to the protein via a single Cys residue from the heme binding motif -Ala (Ala/Gly)-Gln-Cys-His-.226−228
2.3.3. Conformational Changes in Class I Cytochromes c Induced by Changes in the Heme Oxidation State
Many structural studies have been undertaken to determine whether there is any effect on the protein structure associated with different oxidation states of the heme iron. These studies include X-ray and NMR structures of oxidized and reduced cyts c from various sources,229−235 which indicate that the oxidation state of the heme iron has a minimal effect on the tertiary structures of the proteins (Figure 6). The major changes are observed in the conformation of some amino acid residues located close to the heme pocket. Among these residues, Asn52, Tyr67, Thr78, and a conserved water (wat166) molecule show maximal changes in conformations depending on the oxidation state of the heme iron. These conserved residues,236 along with the conserved water molecule, the axial ligand Met80, and heme propionate 7, form a hydrogen-bonding network around the heme site. The high-resolution X-ray structure of yeast iso-1-cytochrome c shows that in the reduced state the heme is significantly distorted from planarity into a saddle shape. The degree of heme distortion in the oxidized state is even more pronounced compared to that of the reduced state, suggesting that the planarity of the heme group is dependent on the oxidation state of the iron. The major change in the bond length of the heme iron ligands is observed in the case of axial Met80, which increases from 2.35 to 2.43 Å in going from the reduced to the oxidized state. On the contrary, the other axial ligand, His18, shows a minute change of 0.02 Å, from 1.99 to 2.01 Å.230
Figure 6.
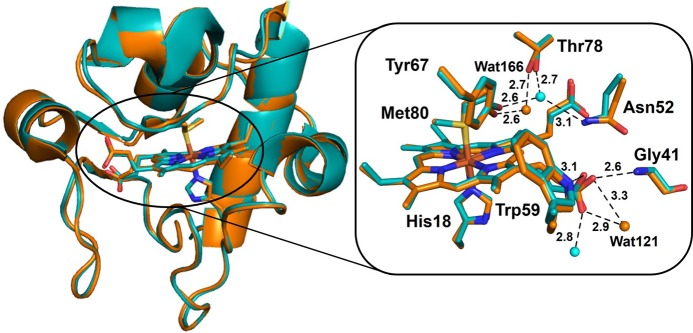
Overall structural overlay of the reduced (cyan, PDB ID 1YCC) and oxidized (orange, PDB ID 2YCC) iso-1-cyt c (left). A close look at the heme site and the nearby residues is shown on the right along with some hydrogen bond interactions.
In the reduced state of iso-1-cytochrome c, the conserved water molecule is hydrogen bonded to Asn52, Tyr67, and Thr78 (Figure 6). Upon oxidation wat166 undergoes a 1.7 Å displacement toward the heme, which results in the loss of the hydrogen bond to Asn52, but interactions with Tyr67 and Thr78 are retained. Figure 6 shows an overlay of the residues near the heme pocket between the reduced and oxidized states of iso-1-cytochrome c.87
Further analysis suggested that wat166 plays a key role in stabilizing both oxidation states of the heme iron by reorienting the dipole moment, by changing the heme iron–wat166 distance, and by variations in the nearby H-bonding network. Another noticeable change is observed in the H-bonding between a conserved water, wat121, and heme propionate 7. In the reduced state, wat121 and Trp59 are hydrogen-bonded to O1A and the O2A oxygen of propionate 7, respectively. In the oxidized state, interaction between Trp59 and O2A of the heme propionate weakens, while that of O2A and the conserved Gly41 increases. Additionally, wat121 moves by 0.5 Å and causes a bifurcated hydrogen bond between both O1A and O2A of the propionate.230 Thus, it appears that there are three major regions that show significant changes in conformation between the two oxidation states: heme propionate 7, wat166, and Met80. A conserved region that does not show mobility between oxidation states is the region encompassing residues 73–80 in iso-1-cytochrome c, which is linked to the three major regions of conformation change through Thr78. On the basis of this observation, it has been suggested that region 73–80 acts as a contact point with redox partners and triggers the necessary conformational changes in other parts of the protein that are required to stabilize both oxidation states of cyt c.230 A contrasting observation from NMR studies is that wat166 moves 3.7 Å away from the heme iron when going from the reduced to the oxidized state, rather than moving toward the heme iron.237,238
Similar to the changes of heme propionate observed in eukaryotes, cyts c2(160,239−242) and c6(220,243,244) from some prokaryotes also display conformational changes in the heme propionate between the reduced and oxidized states of the protein. In the cases of cyt cH (reduces methanol oxidase in methylotropic bacteria) from Methylobacterium extorquens and cyt c552(245−247) (electron donor to a ba3–cytochrome c oxidase) from T. thermophilus, there is no conserved water molecule in the heme pocket, suggesting that the water-mediated H-bonding network is not a critical requirement for ET.248−250
2.3.4. Cytochromes c as Redox Partners to Other Enzymes
In the following sections we summarize some specific examples of native enzymes that use cyts c as the native electron donor for performing various biochemical processes.
2.3.4.1. Cytochrome c as a Redox Partner to Cytochrome c Peroxidases
Cytochrome c peroxidases (CcPs) are a family of enzymes that catalyze the conversion of H2O2 to water and are found in both eukaryotes and prokaryotes. Eukaryotic CcPs are located in the inner mitochondrial membrane and contain a single heme cofactor, heme b, while prokaryotic CcPs are located in the periplasmic space and contain two covalently bound c-type hemes,251,252 one of which is a low-potential (lp) heme and the other is a high-potential (hp) heme. In general, the physiological electron donors to bacterial CcPs are monoheme cyts c, although other donors such as azurin (Az) or pseudoazurin have also been found in some bacteria.253 The hp heme is located at the C-terminal domain and has a more positive reduction potential than cyt c as it accepts electrons from cyt c. The reduction potential for the hp heme varies depending on the organism; e.g., the Ps. aeruginosa CcP hp site has a reduction potential of +320 mV,163 the Rb. capsulatus CcP hp site a reduction potential of +270 mV,254 and the N. europaea CcP hp site a reduction potential of +130 mV.154 The electrons are then transferred from the hp heme to the lp heme of CcP. In some organisms, e.g., Ps. aeruginosa and Rb. capsulatus, the hp heme should be in the ferrous state for the enzyme to be active,254,255 whereas in other cases the enzyme is fully functional even with the ferric state of the hp heme, e.g., in N. europaea.154 The axial ligands for the hp heme are a His and a Met, similar to most c-type cytochromes. The lp heme is the site for H2O2 reduction. It is located at the N-terminal domain and has two His residues as axial ligands. The lp heme also displays a wide range of reduction potentials from as low as −330 mV in Ps. aeruginosa(163) to as high as +70 mV in N. europaea CcP.154 Electron transfer between the hp and lp hemes, which are 10 Å apart, is thought to occur through tunneling.255
Cyts c interact with CcP at a small surface patch of the enzyme which has a hydrophobic center and a charged periphery.256 The small size of the surface patch suggests that the interaction of the enzyme with the electron donor is transient, but at the same time is highly specific, which ensures complex formation due to desolvation of the surface waters and binding of cyt c. The charged periphery has been shown to be important to guide the donor toward the surface site, but it does not increase the specificity of the interactions or the ET rate.257 Mutagenesis studies in Rb. capsulatus CcP have shown that the interface at which the enzyme interacts with its electron donor cyt c2 involves nonspecific salt bridge interactions, as the extent of the interaction is dependent on the ionic strength of the solution.258 In contrast, in Ps. nautica CcP, the interaction surface between the enzyme and the electron donor cyt c is highly hydrophobic on the basis of studies which showed that the enzyme was active across a wide range of ionic strength of the solution.259 Studies from Pa. denitrificans CcP have shown that two molecules of horse heart cyt c are able to bind to the enzyme surface.260 Binding of an “active” and “waiting” cyt c in a ternary complex with the enzyme has been proposed to improve the ET rate. Structural studies of Pa. denitrificans CcP with the monoheme cyt c has shown that the heme of the donor binds above the hp heme of CcP, while the two molecules of horse heart cyts c bind between the two hemes of the enzyme.261
2.3.4.2. Cytochrome c as a Redox Partner to Denitrifying Enzymes: Nitrite, Nitric Oxide, and Nitrous Oxide Reductases
Denitrification is a stepwise process in the biological nitrogen cycle where nitrogen oxides act as electron acceptors and are sequentially reduced from nitrate to nitrite, nitrite to nitric oxide, nitric oxide to nitrous oxide, and finally nitrous oxide to nitrogen. These four steps of the nitrogen cycle are catalyzed by a diverse family of enzymes, viz., nitrate reductase, nitrite reductase, nitric oxide reductase, and nitrous oxide reductase, all of which are induced under anoxic conditions.262−264 Various cyt c domains act as electron donors in the denitrification process. Reduction of nitrite to nitric oxide is catalyzed by one of the two structurally diverse enzymes that also have different catalytic sites: (a) cytochrome cd1 nitrite reductase (cyt cd1 NiR)265,266 and (b) multicopper nitrite reductase (CuNiR).267,268 Cyt cd1 NiRs are periplasmic, soluble heterodimeric enzymes containing an ET cyt c domain and a catalytic cyt d1 domain in each subunit, while multicopper nitrite reductases are homotrimeric enzymes containing T1Cu as an ET site and T2Cu as a catalytic site. Cyts c552 are the putative electron donors of cyt cd1.269 Multicopper nitrite reductases have cupredoxin-like folds and use azurins and pseudoazurins as their biological redox partner, and as such are not expected to have cyt c domains. Contrary to this expectation, two instances have been found where a fusion of multicopper nitrite reductase and cyt c domains was discovered in the genomes of Chromobacterium violaceum and Bdellovibrio bacteriovorus, where in both cases the cytochrome c domain is present at the end of a ∼500-residue-long sequence.79 These cyt c sequences are similar to those of the caa3 oxidase sequences.
Nitric oxide reductases (NORs) are integral membrane proteins that catalyze the two-electron reduction of nitric oxide to nitrous reductase.270,271 A recent X-ray structure of the Gram-negative bacterium Ps. aeruginosa cyt c-dependent NOR (cNOR) (Figure 7) shows that the enzyme consists of two subunits.272 The NorB subunit is the transmembrane subunit and contains the binuclear active site consisting of an HS heme b3 and a nonheme iron (FeB) site. It also houses an LS ET cofactor heme b. NorC is a membrane-anchored cyt c and contains a c-type heme. Electrons are received from cyt c552 or azurin to the heme c, which then passes the electrons to LS heme b and then to HS heme b3 of the catalytic binuclear active site. The reduction potentials are +310, +345, +60, and +320 mV for heme c, heme b, heme b3, and the FeB sites, respectively.162
Figure 7.
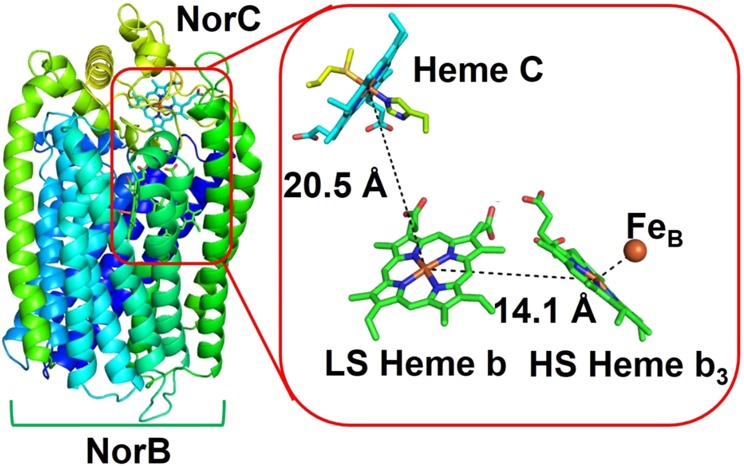
X-ray structure of cyt c-dependent NOR (cNOR) (PDB ID 3O0R) from Ps. aeruginosa.
2.3.4.3. Cytochromes c as Redox Partners to Molybdenum-Containing Enzymes
Mononuclear molybdenum-containing enzymes constitute a group of enzymes that catalyze a diverse set of reactions and are found in both eukaryotes and prokaryotes.273,274 The general function of these groups of enzymes is to catalytically transfer an oxygen atom to and from a biological donor or acceptor molecule, and these enzymes are thus referred to as molybdenum oxotransferases. These enzymes possess a Mo=O unit at their active site and an unusual pterin cofactor which coordinates to the metal via its dithiolene ligand moiety. These Mo-containing enzymes are generally classified into three families depending on their structures and the reactions that they catalyze. The first one is xanthine oxidase from cow’s milk, which has an LMoVIOS(OH) (L = pterin) catalytic core and generally catalyzes the hydroxylation of carbon centers. The second family includes sulfite oxidase from avian or mammalian liver with a core coordination consisting of an LMoVIO2(S–Cys) moiety that catalyzes the transfer of an oxygen atom to or from the substrate’s lone pair of electrons. The third family of oxotransferases shows diversity in both structure and function and uses two pterin ligands instead of only one used by the first two classes. The reaction occurs at the active site core containing L2MoVIO(X), where X could be Ser as in DMSO reductase or Cys as in assimilatory nitrate reductase.
Xanthine oxidases have been reported to be coexpressed with three cyt c domains in Bradyrhizobium japonicum, Bordetella bronchiseptica, Ps. aeruginosa, and Ps. putida; however, the exact cause of this association is not well understood as these enzymes use flavins as their redox partners.79 Sulfite oxidase catalyzes the oxidation of sulfite to sulfate using 2 equiv of oxidized cyt c as physiological oxidizing substrates (Scheme 1).273 The molybdenum is reduced from the VI to the IV oxidation state, and the reducing equivalents are then transferred sequentially to the cyt c in the oxidative half-reaction. The assimilatory nitrate reductases (NRs) are found in algae, bacteria, and higher plants which uptake and utilize nitrate.273 These enzymes contain a cyt b557 and flavin adenine dinucleotide (FAD) in addition to the Mo center. Electrons flow from FAD to cyt b557 to the Mo center under physiological conditions. The midpoint reduction potentials for FAD and cyt b557 from Chlorella NR have been determined to be −288 and −164 mV, respectively.189,190,275 The Mo center displays reduction potentials of +15 mV for the MoVI/V couple and −25 mV for the MoV/IV couple. These reduction potentials indicate that the physiological direction of electron flow is thermodynamically favorable. The cyt b557 domain of NR is homologous to the mammalian cyt b5, yeast flavo-cyt b2, and cyt b domain of sulfite oxidase.276
Scheme 1. Scheme Showing the Oxidation of Sulfite to Sulfate by Cyt c in Sulfite Oxidase.
Reprinted from ref (273). Copyright 1996 American Chemical Society.
The DMSO reductase family consists of a number of enzymes from bacterial and archaeal sources that display remarkable sequence similarity. Respiratory DMSO reductases are periplasmic and use membrane-anchored multiheme cyts c as electron donors that transfer electrons from the quinine pool to the periplasmic space. These cytochromes are about 400 amino acids long and are encoded in the same operon as the enzyme. In some γ-proteobacteria, the tetraheme cyts c occur as a fusion to the C-terminal cyt c binding domain of the enzyme. On the other hand, in some ε-proteobacteria single-domain cyts c have been coexpressed with the DMSO reductase and act as electron donors to the enzyme. Nonetheless, the cyt c sequences from both types of proteobacteria are clustered together, suggesting that even though the mechanism of ET is different, they are functionally similar.79 Even though these ET proteins in DMSO reductases are referred to as cyts c because they contain c-type hemes, their structural folds do not fall into the uniquely defined category of cyt c folds as mentioned in section 2.3.2.
2.3.4.4. Cytochrome c as a Redox Partner to Alcohol Dehydrogenase
The type II quinohemoprotein alcohol dehydrogenases are periplasmic enzymes that catalyze the oxidation of alcohols to aldehydes and transfer electrons from substrate alcohols first to the pyrroloquinoline quinone (PQQ) cofactor, which subsequently transfers electrons to an internal heme group that is found in a cyt c domain.277 This cyt c domain of about 100 residues contains three α-helices in the core cytochrome domain and is similar to the cyt c domain in p-cresol methylhydroxylase (PCMH) from Ps. putida(278) and the cyt c551i from Pa. denitrificans.279
2.3.4.5. Involvement of Cytochromes c in Photosynthetic Systems
Photosynthesis involves the conversion of light energy to useful chemical forms of energy, which is accomplished by two large membrane protein complexes, photosystem I (PSI) and photosystem II (PSII).280 The catalytic cores of the two PSs are referred to as the reaction centers, which have [4Fe−4S] clusters and quinines as terminal electron acceptors for PSI and PSII, respectively. Like algae and higher plants, cyanobacteria also use PSI and PSII to convert light energy to chemical forms by producing oxygen from water oxidation. Even though cyanobacteria have a bis-His-coordinated PS-C550 cyt subunit in their PSII, apparently there is no redox role of this cytochrome.281,282 Being located at the lumenal surface of the enzyme, PS-C550 cytochrome acts as an insulator of the catalytic core from reductive attack and contributes to structural stabilization of the complex.283,284 The low midpoint reduction potentials of the soluble protein from −250 to −314 mV exclude any redox role of this class of cytochromes.285−288 When complexed with PSII, more positive values of reduction potentials have been determined.288,289 A reduction potential of +200 mV in PS-C550 cytochrome from Thermosynechococcus elongates has recently been reported,185 which suggests a possible role of this cytochrome in ET in PSII, despite a long distance (∼22 Å) between the PS-C550 cytochrome and its nearest redox center, the Mn4Ca cluster.290
In cyanobacteria, cyt c6 is known to act interchangeably with the copper protein plastocyanin as an electron donor to PSI, depending on the availability of copper,291−293 while in higher plants plastocyanin is the exclusive electron donor. On the basis of this observation, it has been proposed that cyt c6 is the older ancestor, which has been replaced by plastocyanin during evolution due to the shortage of iron in the environment.294
Another cytochrome, cyt cM, is found exclusively in cyanobacteria, but its role is ambiguous. It has been shown to be expressed under stress-induced conditions such as intense light or cold temperatures where the expression of both cyt c6 and plastocyanin is suppressed.295 Thus, it would be tempting to believe that cyt cM is a third electron donor to PSI in cyanobacteria under stress conditions, but experimental evidence goes against this hypothesis.296
2.3.4.6. Cytochrome c as a Single-Domain Oxygen Binding Protein
Sphaeroides heme protein (SHP) is an unusual c-type cytochrome which was discovered in Rb. sphaeroides.183 SHP (∼12 kDa) has a single HS heme with a reduction potential of −22 mV and an unusual His/Asn axial heme coordination in the oxidized form. SHP is spectroscopically distinct from cyts c′, which also have a HS heme. SHP was shown to bind oxygen transiently during slow auto-oxidation of the heme. The Asn axial ligand was shown to swing away upon reduction of the heme or binding of small molecules such as cyanide or nitric oxide. The distal pocket of SHP shows marked resemblance to other heme proteins that bind gaseous molecules.297 It has been suggested that SHP could be involved as a terminal electron acceptor in an ET pathway to reduce small ligands such as peroxide or hydroxylamine.297
2.3.5. Cytochrome c Domains in Magnetotactic Bacteria
Magnetotactic bacteria consist of a group of taxonomically and physiologically diverse bacteria that can align themselves with the geomagnetic field.298 The unique property of these bacteria is due to the presence of iron-rich crystals inside their lipid vesicles forming an organelle, referred to as the magnetosome. From sequence analysis, three proteins, MamE, MamP, and MamT, in the Gram-negative bacterium Magnetospirillum magneticum AMB-1 that contribute to the formation of the magnetosome have been discovered to contain a double -Cys-Xxx-Xxx-Cys-His- motif, characteristic of cyts c.186 All three proteins were expressed and purified in E. coli. Subsequent characterization of these proteins confirmed that MamE, MamP, and MamT indeed belong to c-type cytochromes, and they have been designated as “magnetochromes”. Midpoint reduction potentials were determined to be −76 and −32 mV for MamP and MamE, respectively. The presence of cyts c proteins in magnetotactic bacteria is intriguing and suggests that these proteins take part in ET, although the exact nature of their ET partners is not known. It has been hypothesized that the magnetochromes can either donate electrons to Fe(III) and participate in magnetite [mixture of Fe(III) and Fe(II)] formation or accept electrons from magnetite to maintain a redox balance, or they can act as redox buffers to maintain a proper ratio of maghemeite (all ferric irons) and magnetite.
2.3.6. Multiheme Cytochromes c
Multiheme cyts c occur as both soluble and membrane-anchored ET proteins in many enzymes across diverse functionalities.79,299 Triheme cyts c7 from Geobacter sulfurreducens and Dm. acetoxidans are involved in ET for Fe(III) respiration,207,300−303 although their exact roles are not known. These proteins have conserved secondary structural elements consisting of double-stranded β-sheet at the N-terminus followed by several α-helices. The protein displays a miniaturized version of the cyt c3 fold where heme II and the surrounding protein environment are missing (Figure 8). The arrangement of hemes is conserved in cyts c7 in terms of the distances between heme iron atoms and the angles between heme planes. Hemes I and IV are almost parallel to each other and are mutually perpendicular to heme III, which is in close contact with hemes I and IV. NMR and docking experiments suggest that heme IV is the region of interaction with similar physiological partners, while the other interacting partner would most likely interact through the region near hemes I and III. Such differences in interaction surfaces might play a role in choosing the right redox partners to perform different physiological functions.
Figure 8.
(A) X-ray structure of triheme cyt c7 (PDB ID 1HH5). All the hemes are bis-His-ligated. Cyt c7 is a minimized version of cyt c3 where heme II is missing. (B) Spatial arrangement of the four hemes in flavocytochrome c3 fumarate reductase (PDB ID IQO8). The heme irons of the heme pair II and III are in close proximity at 9 Å from each other, and the heme edges are 4 Å away.
An unusual triheme cyt c is DsrJ from the purple sulfur bacterium Allochromatium vinosum that is a part of a complex involved in sulfur metabolism.182,304 Sequence analysis suggested the presence of three distinct c-type hemes containing bis-His, His/Met, and a very unusual His/Cys axial ligation, respectively. Subsequent cloning and expression of DsrJ in E. coli indeed confirmed the presence of three hemes, and EPR data showed the presence of partial His/Cys coordination to one of the hemes (His/Met is another possibility). From redox titrations, reduction potentials of the hemes were determined to be −20, −200, and −220 mV, respectively. Although the exact role of DsrJ is still unknown, its involvement in catalytic functions rather than in ET has been hypothesized.182
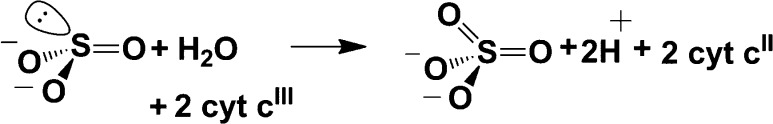
Other examples of multiheme cyts c include a tetraheme cyt c (NapC) involved in nitrate reductase from Pa. denitrificans,305 an Fe(III)-induced tetraheme flavocytochrome c3 (Ifc3)306 in fumarate reductase (Fcc3) from Sh. frigidimarina, an HAO containing eight heme groups for hydroxylamine oxidation in N. europaea,307 and a pentaheme nitrite reductase (NrfA) for nitrite reduction in Sulfurospirillum deleyianum.308,309 A periplasmic flavocytochrome c3 which is an isozyme of the soluble Fcc3 is also induced by Fe(III).310−312 The X-ray structure of this protein shows that the tetraheme arrangement in Fcc3 includes an intriguing heme pair where the two irons are only 9 Å from one another and the closest heme edges are within 4 Å (Figure 8).
The four hemes from Ifc3 and Fcc3 can be superimposed on four of the eight hemes in HAO.307 All four hemes of Ifc3 overlay on four of the hemes from the pentaheme NrfA,308 and all five hemes from NrfA overlay on five of the HAO hemes. Lastly, two hemes from Ifc3 overlay on two of the four hemes of cyt c554(122) from N. europaea, all four hemes of which overlay on four hemes from HAO. Despite such similarities in heme arrangement, there is no resemblance in the primary sequence of these enzymes. Nevertheless, such similar heme arrangements in these proteins suggest that they share a common ancestor, but have evolved divergently to perform four different reactions, viz., Fe(III) reduction, fumarate reduction, hydroxylamine reduction, and nitrite reduction.313 Some membrane-bound multiheme cytochromes, belonging to the NapC/NirT family, contain four heme binding sequences that have evolved due to gene duplication of diheme domains.314 In NapC and CymA all four hemes are 6cLS with bis-His axial ligation and display reduction potentials of +10 and −235 mV, respectively.305,313
Sh. oneidensis MR-1 is a facultative anaerobe that is capable of using many terminal electron acceptors such as DMSO or metal oxides such as ferrihydrite and manganese dioxide outside the outer cell membrane, accepting electrons from the quinol pool and the tetraheme protein CymA.317−325 Electron transfer in Sh. oneidensis MR-1 is facilitated by two periplasmic decaheme cyts c, DmsE, which supplies electrons to DMSO, and MtrA, which is involved in ET to metal oxides (Figure 9). Both of these decaheme proteins have been proposed to be involved in a long-range ET across a ∼300 Å “gap” 326 (∼230 Å periplasmic gap and ∼40–70 Å thick outer membrane). Using protein film voltammetry, a potential window between −90 and −360 mV and an ET rate of ∼122 mV s–1 were measured for DmsE at pH 6.315 The measured reduction potential window for DmsE is shifted ∼100 mV lower than what was observed in MtrA,327−329 although the rate of ET is similar in both proteins. Although the MtrA and DmsE families of decaheme proteins facilitate long-range ET in Sh. oneidensis, it is not clear how ET is feasible across a 300 Å gap, especially given the fact that MtrA spans only 105 Å in length.330 Clearly, the arrangement of hemes must play a crucial role; however, the exact mechanism of this ET process is yet to be determined. A recent NMR study proposes the presence of two independent redox pathways by which the ET occurs from the cytoplasm to electron acceptors on the cell surface across the periplasmic gap in MtrA,331 one involving small tetraheme cyt c (STC) and the other involving FccA (flavocytochrome c). Both of these proteins interact with their redox partners CymA (donor) and MtrA (acceptor) through a single heme and show a large dissociation constant for protein–protein complex formation. Together, these facts suggest that a stable multiprotein redox complex spanning the periplasmic space does not exist. Instead, ET across the periplasmic gap is facilitated through the formation of transient protein–protein redox complexes.
Figure 9.

(A) Schematic model for DMSO reduction by DmsEFAB and iron reduction by MtrABC(DEF). Flows of electrons are shown with arrows. DmsE and MtrA(D) are proposed to accept electrons from the menaquinone pool via CymA. Multiheme groups in CymA, MtrACDF, and DmsE are shown. IM = inner membrane, and OM = outer membrane. (B) “Staggered-cross” orientation of the hemes in outer membrane decaheme MtrF (PDB ID 3PMQ). Heme numbering is shown as Roman numerals, heme–iron distances are shown in orange, and distances between heme edges are shown in blue. (A) Reprinted with permission from ref (315). Copyright 2012 Biochemical Society. (B) Adapted from ref (316) Copyright 2011 National Academy of Sciences.
MtrF is a decaheme c-type cytochrome found in the outer membrane of Sh. oneidensis MR-1 (Figure 9) which has been proposed to transfer electrons to solid substrates through the outer membrane, like its homologue MtrC, with the help of periplasmic MtrA and a membrane barrel protein, MtrE, that facilitates ET by forming contact between MtrA and MtrF.332,333 A recent crystal structure of MtrF shows that the protein consists of four domains, domains I and III containing β-sheets and domains II and IV being α-helices.316 The arrangement of the 10 bis-His-ligated hemes is like a “staggered cross” where four hemes (I, II, VI, VII) are almost coplanar with each other and are almost perpendicular to a group of three hemes (III, IV, V and VIII, IX, X) that are parallel to each other (Figure 9).
The reduction potentials of the hemes in MtrF lie in the range of 0 to −312 mV as determined by both solvated and protein film voltammetry. Unfortunately, reduction potentials of individual hemes have not been possible to assign due to their similar chemical nature. Molecular dynamics simulations show an almost symmetrical free energy profile for ET. Additionally, the computed reorganization energy range from 0.75 to 1.1 eV is consistent for partially solvent exposed heme cofactors capable of overcoming the energy barrier for ET.334,335 Further molecular details of ET in MtrF are unknown.
Multiheme cyts c also act as ET agents in the Fe(III)-respiring genus Shewanella.299 However, due to the fact that Fe(III) is soluble only at pH < 2, these organisms face the problem of moving electrons from the cytoplasm across two cell membranes to the extracellular space to reduce the insoluble extracellular species. It has been proposed that these organisms circumvent this problem by employing a number of tetraheme and decaheme cyts c which act as “wires” to transfer electrons between the inner and outer membranes.313,336
For tetraheme cyts c3, hemes I and III are covalently attached to the protein segment by a conserved -Cys-Xxx-Xxx-Cys-His- sequence, while hemes II and IV are linked to the protein with the two Cys residues occurring in the sequence -Cys-Xxx-Xxx-Xxx-Xxx-Cys-His-.337,338 Although the overall orientation of hemes is conserved, the order of heme oxidation varies from source to source.217,339,340 The hemes in cyts c3 display redox cooperativity, such that the reduction potential of one heme is dependent on the oxidation state of the other hemes. The reduction potentials of the hemes in cyts c3 are also dependent on the pH, called the redox-Bohr effect,340−342 due to the interactions of the heme propionates in the H-bonding network and/or electrostatic interactions with the residues in the vicinity.341,343−345
Type I cyts c3 are soluble, periplasmic proteins and contain a patch of positively charged residues close to heme IV which have been proposed to interact with its partners.346 This class of cyts c3 mediate ET between periplasmic hydrogenases and transmembrane ET complexes where the electron acceptor is thought to be type II cyts c3. Type II cyts c3 are structurally similar to those of type I, but lack the lysine patch.347 It was proposed that type I cyts c3 receive electrons from hydrogenase and deliver them to type II cyts c3. Recent experimental evidence shows that these two types of cyts c3 form a complex with each other and are indeed physiological partners, but type I cyts c3 transfer only one electron to type II cyts c3 in solution.348,349
2.3.7. Cytochromes b5
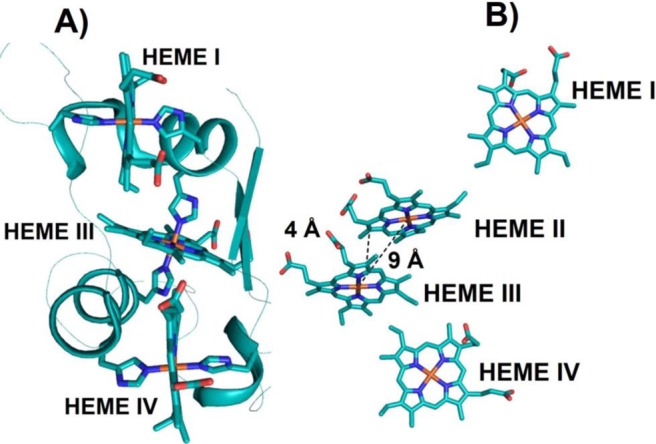
Cyts b5 are ET hemoproteins containing bis-His-ligated b-type hemes and are found ubiquitously in bacteria, fungi, plants, and animals. Cyts b5 display reduction potentials that span a range of ∼400 mV.350−353 Mitochondrial and microsomal cyts b5 are membrane-bound, while those from bacteria and erythrocytes are soluble. In addition, there are various cyt b5-like proteins that act as redox partners in various enzymes such as flavocytochrome b2 (l-lactate dehydrogenase), sulfite oxidase, assimilatory nitrate reductase, and cyt b5/acyl lipid desaturase fusion proteins. The structures of cyts b5 from various sources reveal that there are two hydrophobic cores on each side of a β-sheet that belong to the α + β class (Figure 10).350 The larger hydrophobic core contains the heme binding crevice, while the smaller hydrophobic core is proposed to have only a structural role. About 3% of deoxyhemoglobin in adults is oxidized to inactive methemoglobin.354 Soluble cyts b5 in erythrocytes reduce methemoglobin to the functionally reduced deoxy form that binds oxygen. For this reaction electrons are transferred from NADH to methemoglobin via NADH cyt b5 reductase and cyt b5.355 Microsomal cyts b5 are found in the membranes of the endoplasmic reticulum anchored to the membrane by a stretch of 22 hydrophobic residues.353 Microsomal cyts b5 are known to function by transferring electrons in fatty acid desaturation, cholesterol biosynthesis, and hydroxylation reactions involving cyts P450.356
Figure 10.
Schematic representation of the X-ray structure of bovine cyt b5 that belongs to the α + β class (PDB ID 1CYO). Two hydrophobic core domains, six α-helices, five β-strands, and 6c bis-His-ligated heme are shown. Adapted from ref (357). Copyright 2011 American Chemical Society.
Two different forms of cyt b5 have been detected in rat hepatocyte; one is associated with the membrane of the endoplasmic reticulum (microsomal, or Mc, cyt b5), while the other is anchored to the outer membrane of liver mitochondria (OM cyt b5).358−362 These two types of cyt b5 display a reduction potential difference of 100 mV (−107 mV for OM cyt b5,187,363 −7 mV for Mc cyt b5).180 The rat OM cyt b5 is involved in the reduction of cytosolic ascorbate radical using NADH as the electron source.364,365 The mammalian OM cyt b5 and Mc cyt b5 have three different domains, an N-terminal hydrophilic domain that binds the heme, an intermediate hydrophobic domain, and a C-terminal hydrophilic domain. The N-terminal heme binding domains for both types of cyts b5 have very similar structural folds consisting of six α-helices and four β-strands. The heme is bound in a pocket formed by four α-helices and a β-sheet formed by two of the β-strands.141,366 Studies relating to the complex formation and ET rates between cyts b5 and its redox partners suggest that the nature of interactions between two proteins is primarily electrostatic and the heme edges of cyts b5 make contacts with electron donors and acceptors.350 Within this general area, there are multiple overlapping sites with which cyts b5 interact with its various partners.
A gene encoding a cyt b5-type heme from the protozoan intestinal parasite Giardia lamblia was recently cloned into E. coli as a soluble protein.367 The spectroscopic properties of this cloned cyt b5 are similar to those of the microsomal cyts b5, and homology modeling suggests the presence of a bis-His-ligated heme. Residues near the heme binding core from Giardia cyt b5 are comprised of charged amino acids and differ from those of other families of cyt b5. The reduction potential of the heme was determined to be −165 mV.
2.3.7.1. Heme Orientation Isomers in Cytochromes b5
Solution NMR studies of the soluble fragment of cyt b5 suggested the coexistence of two different species that contained two orientation isomers (forms A and B, Figure 11) of heme that are related by a 180° rotation about an axis through the heme α,γ-meso-carbon atoms.368−372
Figure 11.

Two orientation isomers (A and B forms) of heme observed in solution studies of the soluble fragment of cyt b5. The two isomers are related by a 180° rotation around the α,γ-meso-carbon atoms.
The relative population of the two isoforms A and B varies from species to species. In bovine and rabbit, the A/B ratio is ∼10/1,177,368,370,373 20/1 in chicken cyt b5,374 6/4 in rat Mc cyt b5,374 and 1/1 in the OM cyt b5.375 Even though reconstitution of apo cyt b5 with heme resulted in the initial formation of a 1/1 ratio of species A and B, they converted back to the proportion found in the thermodynamically stable native state after some time.370,373 Reduction potentials of +0.8 and −26.2 mV were calculated for isoforms A and B, respectively, from spectroelectrochemical titrations.177 Interaction between the 2-vinyl group and side chains of residues 23 and 25 was initially thought to be the driving factor that dictated the heme orientation isomers.368,374,376 This theory was disputed in later studies.375 It is now generally accepted that the heme itself can adapt to the surrounding environment by a rotation of the porphyrin plane around an axis perpendicular to the iron, which is proposed to be the determining factor that caused the different heme orientation in species A and B.376−378 Several studies have indicated that residue His39 is the major determining factor of the electronic state that orients the molecular orbitals for easy ET through the exposed pyrrole ring III and meso-carbon heme edge.370,379,380
2.3.8. Cytochrome b562
Cyt b562 is a 106-residue monomeric heme protein of unknown function found in the periplasm of E. coli. It is a four-helix bundle protein where the helices are oriented antiparallel to each other (Figure 12).381,382
Figure 12.
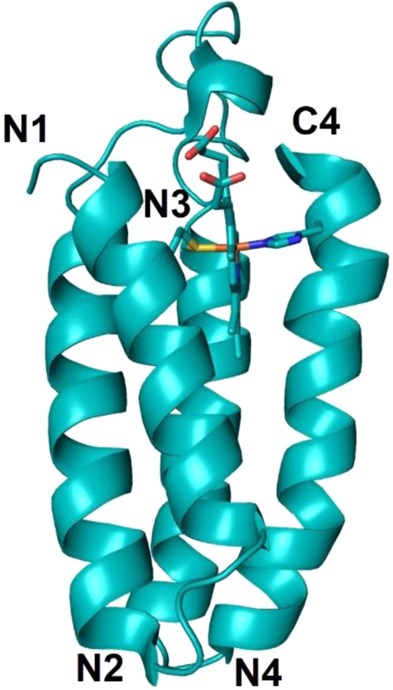
NMR structure of the antiparallel four-helix bundle cyt b562 (PDB ID 1QPU). His/Met axial coordination to the heme iron is shown.
The protein has a noncovalently bound 6cLS heme with His102 and Met7 axial ligands, even though this protein is structurally homologous to cyt c′ that contains a covalently bound 5cHS c-type heme. In the oxidized unfolded state, the heme of cyt b562 is converted to 5cHS with His102 as the only axial ligand.383 The folding properties of this protein are highly dependent on the pH. At pH 7 the reduction potential of the heme in the folded state is 189 mV, while that of the unfolded state is −150 mV, suggesting that the reduced state has a greater driving force for folding than the oxidized state.176,384−387 Unfolding of the oxidized state of the protein occurs reversibly with a midpoint GuHCl concentration of 1.8 M, while the reduced state shows irreversible unfolding at >5 M GuHCl due to heme dissociation. Folding of the reduced state has been shown to be triggered by photoinduced ET to the oxidized form of the protein under 2–3 M GuHCl concentrations. A folding rate of 5 μs was extrapolated in the absence of denaturant, which is similar to the intrachain diffusion time scale of the polypeptide.388
2.4. Designed Cytochromes
In addition to studying native systems by a top-down approach, in recent decades, many groups have adopted a bottom-up approach of building minimal functional proteins that mimic natural ones. The theoretical simplicity and ubiquity of cytochromes has made them appealing targets for design, and a number of artificial cytochrome-mimicking proteins have been engineered, with varying levels of sophistication. In this issue of Chemical Reviews, Pecoraro and co-workers give a thorough review of protein design strategies and successes, including designed heme ET proteins.3000 Here, we give a brief account focusing on the redox properties of designed 6-coordinate heme proteins mimicking ET cytochromes.
2.4.1. Designed Cytochromes in de Novo Designed Protein Scaffolds
Two de novo heme proteins called VAVH25(S–S) and retro(S–S)389 were designed to bind heme in a bis-His coordination, by strategically engineering His residues into the de novo cystine-cross-linked, homodimeric four-helix bundle called α2.390−392 Both sequences yielded artificial cytochromes with dissociation constants for heme in the submicromolar range, and spectroscopic properties of these proteins were consistent with low-spin bisimidazole-ligated heme, with reduction potentials of −170 and −220 mV for each of the proteins. Although these potentials are nearly unchanged from the potentials of bisimidazole heme in aqueous solution, the success of incorporation demonstrated the power of rational de novo design and set the stage for rapid development of more complex and nativelike structures. Using an alternative tetrameric protein scaffold, consisting of two pairs of disulfide linked α-helices, a series of proteins mimicking the heme b domain of cytochrome bc1 were also designed by strategic placement of histidine residues. The designed proteins incorporated either two or four hemes per bundle,393 with potentials of the individual sites reported to range from −230 to −80 mV in the tetraheme construct. More impressively, the sites showed cooperative redox properties, with the presence of a second ferric heme site proposed to raise the potential of the first by ∼115 mV through electrostatic interactions (vide infra).393,394 In a systematic study of the electronic properties of this scaffold, varying the heme, pH, and local charge could achieve a potential range of 435 mV (−265 to +170 mV),395 over half the 800 mV range covered by native cytochromes. Interestingly, investigation of the more natural mutation of one of the His ligands with a Met resulted in only a 30 mV increase in reduction potential, and substitution of heme b with heme c gave no significant change.396 Rational mutagenesis of several core residues, as well as incorporation of helix–turn–helix and asymmetric disulfide bonds, further improved the structural rigidity and uniqueness of the designed scaffolds.397,398 Subsequently, this maquette system was extended in a variety of ways to achieve coupling to electrode surfaces,399 incorporation of non-natural amino acid ligands,400 and binding of two different hemes—which mimics the structure of ba3 oxidases.401 Particularly exciting is the demonstration of coupling of ET and protonation of carboxylate residues on the protein,402−404 which is relevant for understanding and engineering proton pumping.
On the basis of recent developments in structural understanding of cytochrome bc1 and improvements in computational modeling, Ghirlanda et al. investigated designing a more structurally unique mimic of the bc1 complex. The structure of the heme b binding portion of bc1 was modeled as a coiled coil, and secondary coordination sphere interactions to the coordinating histidines, such as conserved Gly, Thr, and Ala residues, were added to stabilize the orientation of the His ligand and tune its electronic properties (Figure 13A).405 The potentials were measured by cyclic voltammetry (CV) as −76 and −124 mV in the oxidative and reductive directions, respectively, at pH 8, significantly higher than the potential of aqueous bisimidazole heme and earlier bis-His-ligated designed proteins. The hysteresis in the potentials is attributed to conformational reorganization of the ligating His residues between the oxidized and reduced forms. The model was further improved by linking and expression as a single chain for more efficient structure determination studies,408 as well as incorporation into a membrane.409
Figure 13.

Structural models of designed cytochrome models in de novo scaffolds. (A) A design model for a homodimeric four-helix tetraheme binding protein inspired by cyt bc1. Remade from coordinates courtesy of G. Ghirlanda and W. F. DeGrado.405 (B) Schematic representation of monomeric four-α-helix maquettes used to mimic ET cytochromes. Reprinted with permission from ref (406). Copyright 2013 Macmillan Publishers Ltd. (C) Crystal structure of Co(II) mimichrome IV (PDB 1PYZ).407
Most recently, Dutton and co-workers have reported the design and thorough characterization of a monomeric, single-chain four-α-helix bundle maquette protein, which can bind up to two hemes (Figure 13B). It is particularly noteworthy for the subject of this review that the redox properties of this scaffold as a function of charge distribution were systematically analyzed. By raising the total charge uniformly from −16 to +11, the reduction potential of both hemes changed from −290 to −150 mV, as expected. Furthermore, the potentials of the hemes could be changed individually by only increasing the charge at one end of the protein; the potentials of the individual hemes were −240 and −150 mV. Finally, it was demonstrated that the reduced negatively charged protein could transfer an electron to native cytochrome c with rate constants approaching those of native photosynthetic and respiratory electron transport chains. Such a single-chain four-helix bundle was also used to build an artificial oxygen binding cytochrome c with an intramolecular B-type ET heme with a 60 mV lower reduction potential, mimicking a natural ET chain.410
More rational computational protein design algorithms have also been brought to bear on the de novo design of artificial cytochromes. Xu and Farid used the algorithm named CORE411 to design a nativelike four (27 amino acid)-helix bundle that binds two to four hemes in a bis-His fashion.412 The α-helical character was confirmed by circular dichroism (CD), and the binding affinity for the first 2 equiv was determined to be in the micromolar range, while, due to negative cooperativity, the remaining sites had Kd > 3 mM. The measured potentials for the diheme and tetraheme protein were −133 to −91 and −190 to −0110 mV, respectively.
While the rationally guided design strategies described above have been very successful, the lack of a priori knowledge about the necessary structural features for design of functional metalloproteins limits the scope of sequence and structure space that is probed by the strategy. As a complementary approach, Hecht and co-workers have utilized a semirational “binary code” library generation method to produce 15 74-residue sequences that formed helical bundles and bound heme,413 one with submicromolar affinity. Extending this scaffold further produced five 102-residue sequences with higher stabilities and more “nativelike” structures.414 Analysis of a handful of these proteins revealed spectroscopic features typical of low-spin heme proteins and reduction potentials ranging from −112 to −176 mV.415 Furthermore, it was demonstrated that at least one construct was electrically competent on an electrode.416 A similar semirational combinatorial approach was utilized by Haehnel and co-workers, who combined it with template-assisted synthetic protein (TASP) methods, in which two sets of antiparallel helices are templated onto a polypeptide ring, to design and screen an impressive library of 399 cytochrome b mimicking four-helix bundles.417,418 Using a colorimetric screen, the potentials were estimated to range from −170 to −90 mV. It was also demonstrated that the proteins could be incorporated onto electrodes419,420 and achieved estimated ET rate constants comparable to those of native cytochromes.
A number of smaller, water-soluble peptide-based cytochrome mimics have also been developed, utilizing one or two short α-helical peptides. Two groups independently developed heme compounds with covalently attached, short α-helix-forming peptides, with His ligands. In one case, peptide-sandwiched mesoheme (PSM) compounds were prepared by covalently attaching a 12-mer peptide to each of the two propionate groups of the heme via amide bonds with lysine groups on the peptide.421 Although the helicity of the free peptide was low, upon ligatation of the heme, the helicity was seen by CD to increase to ∼50%, and the electronic spectra were consistent with bis-His heme ligation, similar to b-type cytochromes.421,422 Further work suggested that aromatic side chain interaction with the heme, such as Phe and Trp, improves helix stability and heme binding,423 and covalent linkage of the peptide termini via disulfide bonds resulted in further stabilization.424 Studies of the redox properties of a PSM and a mutant with an Ala to Trp mutation, (called PSMW), highlight the importance of stability in determining reduction potential, with more stable helix binding in PSMW lowering the reduction potential by 56 mV (−281 to −337 mV), due to the increased ability of the His ligands to stabilize the Fe(III) state.425 The authors propose that this effect may also explain the difference in potential between mitochondrial and microsomal cyts b5.
Similarly, short α-helical peptides, based on the heme binding peptide fragment of myoglobin, have been covalently attached to deuterohem by a similar amide-bond attachment strategy, yielding compounds known as mimochromes.426 It is noteworthy that the peptides retained their α-helical character even in the absence of heme binding.426,427 The stability of the model was further improved in later revisions by enhancing the intramolecular interpeptide interactions through extending the peptide (mimochrome II)428 or rational mutagenesis (mimochrome IV).429 A crystal structure of the Co(II) derivative of mimochrome IV has been obtained and substantiates the designed structure (Figure 13C).407 The reduction potential of Fe mimochrome (IV) at pH 7 is −80 mV, though it exhibits strong pH dependence over the range of pH from 2 to 10 (∼+30 to −170 mV).429 The low-pH dependence is attributed to the His ligands unbinding from the heme, while the high-pH transition is proposed to be caused by deprotonation of a nearby arginine; however, this is surprising due to the 4 orders of magnitude higher apparent acidity and requires further investigation to be proven. Still, it is exciting that this simple mimic is well folded enough to be crystallized and has a potential in the range of those of native cytochromes.
Intermediate between these covalently attached heme–peptide models and full polyhelical bundles described above, heme protein complexes consisting of heme ligated by designed short peptides that are not covalently attached have also been developed.430−434 Studies on the binding of a variety 15-mer peptides showed a strong correlation between peptide–heme affinity and reduction potential (−304 to −218 mV), with lower potentials for more stable complexes, consistent with the results of studies on PSMs.425,431 The overall low potential was attributed to the inability of the small peptides to reduce the strong dielectric constant of the solvent, as native proteins do (vide infra). To further improve the stability, two peptides were covalently linked at both ends by disulfide ligands, resulting in a series of cyclic dipeptide heme binding motifs, with reduction potentials ranging from −215 to −252 mV.433
Interestingly, in a step away from the helix bundle paradigm, Isogai and co-workers were able to rationally design a series of de novo proteins that would fold into a globin fold, but with only ∼25% sequence identity to sperm whale myoglobin.435,436 Although the proteins were designed for a 5-coordinate myoglobin-like heme binding site, the resulting proteins were consistent with 6-coordinate bis-His-ligated heme. In these scaffolds, the reduction potential was in the range of −170 to −200 mV, similar to that of aqueous bis-Im heme, which was attributed to higher solvent access to the heme due to the molten-globular state of the proteins. This was further supported by the re-engineering of a nonheme globin protein, phycocyanin, into a heme binding protein (vide infra), which had a more unique, hydrophobic, and nativelike core structure and 50 mV higher reduction potential.437
2.4.2. Designed Cytochromes in Natural Scaffolds
In addition to designing scaffolds for cytochromes de novo, an appealing alternative strategy is to make use of the diversity of natural proteins as scaffolds. One of the most straightforward approaches is to convert a non-cytochrome heme protein into a cytochrome by site-directed mutagenesis. Along these lines, various myoglobins have also been redesigned into bis-His cytochrome-like proteins, similar to b5, by mutating the valine near the heme at position E11 to histidine (Figure 14A).438−440 The spectroscopic features of reduced and oxidized forms of these mutants are consistent with low-spin bis-His-ligated heme, and the crystal structure confirms the ligation.440 The mutations result in a 170 mV decrease in the reduction potential of myoglobin, from ∼60 to ∼−110 mV.
Figure 14.
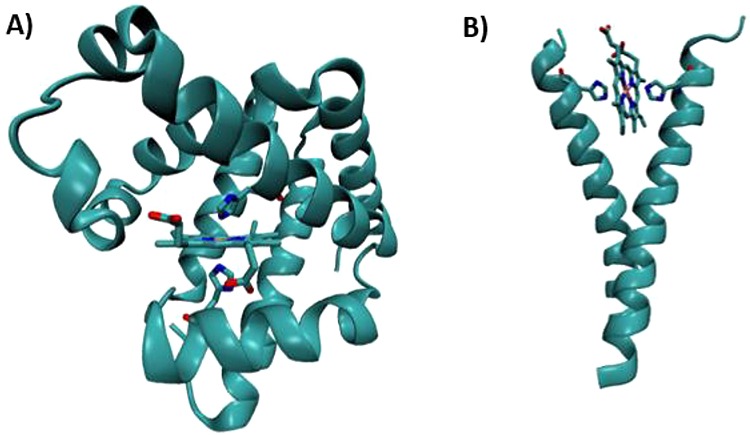
Structural models of designed cytochrome models in native scaffolds. (A) X-ray crystallographic model of a pig myoglobin designed to have cytochrome-like bis-His ligation (PDB ID 1MNI).440 (B) Molecular dynamics model of a histidine mutant of the membrane protein, glycophorin A, designed to bind heme in a cytochrome-like manner.441 Coordinates provided by courtesy of G. Ghirlanda.
Similarly, natural nonheme proteins can also be designed to bind heme in a manner consistent with the cytochrome binding motif. As briefly mentioned above, Isogai and co-workers introduced two histidines into the natural nonheme plant globin phycocyanin437 to generate a heme binding site. Although the protein was designed as a myoglobin mimic, the spectral features were consistent with low-spin bis-His coordination, similar to that of cytochromes b, with a one-electron reduction potential of −120 mV.
Heme binding sites have also similarly been designed into native α-helical bundle proteins that do not have native heme binding sites. Starting with the DNA binding protein rop, a specific bis-His heme binding protein was designed by removing surface histidines and introducing two internal histidine residues.442 An alternative His/Met binding mode was also investigated.443 Both proteins displayed electronic spectra characteristic of low-spin heme, with reduction potentials of −155 and −88 mV, respectively. A cytochrome-like heme binding site was also designed into the transmembrane protein glycophorin A (Figure 14B).441,444 Each of the proteins bound heme with submicromolar affinity, and the presence of aromatic phenylalanine residues near the heme lowered the reduction potential from −128 to −172 mV.
2.4.3. Conversion of One Cytochrome Type to Another
In addition to designing cytochrome sites in non-cytochrome proteins, several groups have investigated the conversion of one type of cytochrome into another.445−449 Conversion of c-type to b-type cytochrome has been achieved in cytochrome c552 by removing the Cys residues in the -Cys-Xxx-Xxx-Cys-His- heme binding motif with the Cys11Ala/Cys14Ala double mutation.447 CD and NMR spectra confirmed that the structure of the protein and heme site was maintained.447,450 However, it was found that the removal of the c-type heme binding motif destabilized the protein toward chemical and thermal denaturation. While the electron-withdrawing potential of the vinyl groups of heme b relative to the thioether groups of heme c would be expected to raise the potential,80 the resulting protein had a reduction potential of 170 mV, 75 mV lower than that of the wild type, suggesting that the electronic structure of the porphyrin is not the major determinant of the reduction potential difference between cytochromes c and b (discussed in section 2.5).
Conversion from cyt b562 to c-type heme has been achieved by introducing the conserved -Cys-Xxx-Xxx-Cys-His- motif into the wild-type protein by means of two mutations (Arg98Cys and Tyr101Cys).449,451 The resulting c-type cytochrome displayed enhanced stability toward chemical denaturants, maintaining the same protein fold and axial His ligation. c-type heme attachment has also been achieved in cytochrome b5 by introducing a surface cysteine residue with the Asn57Cys mutation.448 The resulting holoprotein was isolated in four forms, with distinct forms of heme, one of which contained covalently attached heme and a hemochrome α-band at 553 nm, intermediate between those of b-type (556 nm) and c-type (551 nm) heme, suggesting the presence of a single c-type thioether linkage. NMR further confirmed the stereochemical nature of this linkage, and the protein displayed a reduction potential of −19 mV, 23 mV lower than that of the wild-type b5.
2.5. Structural Features Controlling the Redox Chemistry of Cytochromes
Being involved in distinct ET pathways, each cytochrome has evolved its ET properties to match those of its redox partners. Therefore, reduction potentials of cytochromes span a range of almost 1 V, from −475 mV in bacterioferritin from Azotobacter vinelandii(192,452) to +450 mV in the heme c of diheme cytochrome c peroxidase of N. europaea(153,154) vs the SHE.453 Through a variety of studies, many properties have been found to be important in determining the redox properties of heme proteins. As expected, the molecules in the first coordination sphere of the iron, namely, the four pyrrole groups of the porphyrin and the axially coordinating residues, are important in determining the baseline reduction potential, as they interact directly with the iron center. These interactions are also fine-tuned by the secondary coordination sphere—chemical moieties that interact with the primary coordination sphere ligands and adjust their properties. Secondary coordination sphere interactions, such as H-bonding, can cause strengthening or weakening of ligand–metal interactions. The overall charge as well as the electrostatic environment of the metal center, which is determined by the surrounding charge, dipole distribution, and solvent accessibility, also critically modulates the redox properties.
2.5.1. Role of the Heme Type
It is known that c-type hemes tend to be found in cytochromes with more extreme potentials (much lower or much higher) relative to b-type hemes; however, it is unclear whether a direct causative relationship exists. One way to probe the role of the heme type in a way that is less dependent on other factors is to replace the heme in one protein with another. In studies of the de novo designed four-helix bundles, the strongest effect on reduction potential was attributed to the nature of the heme,395 though unnatural hemes were used in the study. In the more natural protein cases, several groups have interconverted b- and c-type hemes.445−449 It has been found, however, that this interconversion shows little inherent effect on the reduction potential447,448 with no clear trend. For instance, it was found that converting the c-type heme in cyt c552 into a b-type heme by mutating away the conserved Cys residues lowered the reduction potential by 75 mV.447 On the other hand, introducing a thioether bond between heme in cytochrome b5 and the protein, and therefore converting the b-type heme into a c-type heme, also lowered the potential by 23 mV.448 It is clear that the choice of heme c over heme b has little effect on the reduction potential, and other effects, such as structural changes or solvent accessibility, may play a bigger role.
If the choice of heme c or heme b does not play a significant role in determining the reduction potentials of cytochromes, one may wonder why organisms invest in the energetically expensive process of synthesizing c-type linkages. Though the exact reason that Nature has chosen c-type hemes in certain proteins remains to be fully understood, several hypotheses have been proposed.454−456 It is suggested that multiheme cytochromes, such as c3, with largely exposed hemes in close proximity may utilize heme anchoring as a strategy to ensure stable heme binding in the absence of well-defined hydrophobic interactions.457 Similarly, the high-potential cyts c, with His/Met coordination, may use covalent anchoring as a strategy to prevent heme dissociation due to the relatively weaker binding of methionine to ferric heme.457 Alternatively, it is proposed that covalent heme attachment may help in protein folding and stability454,456 or may strengthen the Fe–His bond and help maintain a low-spin state.456 Regardless, the choice of heme c over heme b likely does not itself directly tune the reduction potential in a significant or consistent way, but may allow the protein greater flexibility in achieving other functionality and tuning the potential by other means, such as solvent accessibility.
In addition to hemes b and c, heme a is a unique heme used for ET in enzymes such as heme copper oxidases (HCOs). The heme incorporates two unique peripheral structural features, namely, a hydroxyethylfarnesyl group and a formyl group, and these functional groups have been suggested to play a role in tuning the reduction potential of the heme. While heme a has been replaced with other hemes in a native system,458 detailed studies of how this substitution affects the redox chemistry of the protein have not been reported. Using their de novo designed scaffold (vide supra), Gibney and co-workers459 have studied the redox properties of hemes a and b, as well as diacetyl heme, and found that the electron-withdrawing acyl groups increased the potential by ∼160 mV. This effect can be fully accounted for by the 200-fold lower affinity of the ligands for the oxidized form over the reduced form of the heme, and it is proposed that the hydrophobic farnesyl group serves to anchor the heme stably in the protein460 to compensate for the lower affinity of the ferric state.
2.5.2. Role of Ligands
In addition to the heme type, the identity of the axial ligands sets the baseline for the reduction potentials of cytochromes.457 Between the two most common ligands (His and Met), it has been found that the Met ligation generally raises the potential of the heme by ∼100–150 mV, relative to the His ligation.461−463 However, contrary to this theory, early work by Sligar and co-workers found that redesigning bis-His cyt b5 into a His/Met cyt lowered the reduction potential by ∼240 mV. This opposite change in the reduction potential was attributed to the change in the spin state of the heme, from low-spin bis-His to high-spin His/Met cyt.464 More consistent with the theory, it was demonstrated that conversion of bis-His to His/Met ligation in cyts c3 results in a reduction potential increase of 160–180 mV.192 Similarly, using a proteolytic fragment of cyt c, it was found that methionine ligation in cyts c contributes 130 mV to the energy.386 Conversely, a 105 mV drop in the reduction potential was observed when the methionine in cytochrome c551 was replaced with a histidine.463 Interestingly, Hay and Wydrzynski462 observed a 260 mV decrease in reduction potential when they substituted the native Met ligand in cyt b562 with His, yielding a typical bis-His cyt. This decrease is greater than ∼150 mV, and the authors attribute it to destabilization of the fold and increased solvent exposure, which is known to significantly lower the potential (vide infra). In contrast, an Arg98Cys and His102Met double mutant of the same protein, cyt b562, shows 6cLS bis-Met axial ligation at low pH, with a reduction potential of +440 mV, ∼180 mV higher than that of native His/Met cyt b562.465 The authors note that the effect of bis-Met ligation is likely to be slightly higher at ∼200 mV, as they expect the c-type thioether heme linkage to lower the potential. The stereochemical alignment of the axial methionine ligands results in an almost axial symmetry of the heme, caused by a 110° change in the torsion angle between the sulfur lone pairs.466 The reduction potential of this protein is 665 mV higher than that of the only other known bis-Met axially ligated heme system in bacterioferritin (−225 mV)176 in which the ground state of the oxidized form of the heme is highly rhombic in nature.120,121,467 Therefore, factors other than the differences in the ligand coordination are most likely to be involved to account for the reduction potential difference.78 In general, all else being equal, the preference of soft methionine thioether for the softer ferrous heme over the harder ferric heme contributes to a ∼100–200 mV increase in reduction potential over His ligation.
2.5.3. Role of the Protein Environment
2.5.3.1. Solvent Exposure
Consistently, one of the most important factors in raising the reduction potentials of the cytochromes is the extent of heme burial in the protein or, alternatively, the extent of solvent exposure of the heme.178,187,386,457,468−473 The basis for this effect lies in the lower dielectric constant of proteins relative to aqueous solution, which significantly destabilizes the charged ferric site over the neutral ferrous state of the heme. For instance, Tezkan et al. estimated that solvent exclusion accounts for ∼240 mV of the potential increase in cyt c.386 Similarly, in a thorough computational study of heme proteins spanning an 800 mV range of potentials, Zheng and Gunner identified that heme solvent exclusion accounts for ∼20% of the reduction potential difference between proteins.457 Interestingly, the same study found less correlation between the reduction potentials and the remaining individual factors or energy terms, yet the computation was able to faithfully reproduce and account for heme protein potentials over an 800 mV range. This study elegantly demonstrates that the reduction potential is determined by an intricate balance of numerous factors of comparable energy.
2.5.3.2. Secondary Coordination Sphere of the Ligand
Although the nature of the ligand itself determines primary interaction energies with the heme, and therefore is the primary determinant of the reduction potential, the electronic character of the ligand can be further modulated by secondary noncovalent interactions, such as hydrogen bonds. These so-called secondary coordination sphere effects have been shown to be influential in determining the potentials of a number of heme proteins, including cytochromes.230,472,474−477 For instance, in cyt c in particular, Bowman et al. demonstrated that strengthening the hydrogen bond between the proximal His ligand and a backbone carbonyl through peripheral mutations resulted in an almost 100 mV decrease in the reduction potential, attributable to increased imidazolate character.474 Similarly, Berguis et al. show in three different mutants of yeast iso-1-cyt c that a disruption of the hydrogen bond from tyrosine 67 to the methionine ligand consistently decreases the potential by 56 mV, due to an increase in electron density on the Met sulfur, stabilizing the ferric form of the heme,230,476 and Ye et al., found that the presence of hydrogen bonds between Gln64 and the axial Met ligand in Ps. aeruginosa and Hydrogenobacter thermophilus cyt c lowered the potential by 15–30 mV.477 In addition, aromatic interactions with the axial ligand have also been implicated in tuning the heme reduction potentials. For instance, it was shown that Tyr43, which interacts with the π system of His 34, contributed a ∼35–45 mV decrease in reduction potential.478 Therefore, although the identity of the ligand is a primary determinant of the reduction potential of the heme, the secondary coordination sphere interactions with it also play a role of similar magnitude in determining the reduction potential.
2.5.3.3. Local Charges and Electrostatics
Another important means by which cytochromes have been found to modulate their reduction potentials is through the judicious use of charge and electrostatic interactions. For instance, by comparison and selective mutagenesis of the structurally homologous cyts c6 and c6A, it was demonstrated that the interaction of the positive dipole of the amide group of a carefully positioned glutamine (residues 52 in c6 and 51 in c6A) with the heme is a strategy used by Nature to raise the reduction potential by ∼100 mV.479 Similarly, Lett et al. observed an increase in the reduction potential of cytochrome c by 117 mV through the Tyr48Lys mutation.480 Tyr48 is involved in a H-bonding interaction with a heme propionate, and it is likely that introduction of lysine at this position stabilizes the propionate negative charge and destabilizes the ferric heme state. It has also been shown that replacement of a neutral residue in contact with the heme in myoglobin with a polar or negatively charged residue can reduce the potential by up to 200 mV.481 Furthermore, a library screen of cytochrome b562 mutants at four residues near the heme binding site identified mutations that could gradually tune the potential over a 160 mV range.482 Even relatively distant surface electrostatic interactions have been shown to control the redox function of cytochromes.483 These reports demonstrate the critical role of local charge in determining the reduction potential of the heme. In general, negative local charges stabilize the ferric state and lower the reduction potential, and the magnitude of this effect can be comparable to that of ligand substitution or ligand secondary coordination sphere effects.
In addition to charge interactions, more subtle effects such as electrostatic interactions can also play an important role in determining redox properties. As discussed in section 5.2.2 below, a conserved aromatic residue in cyt b6f is found to be in contact with the heme f at position 4, and the identity of the aromatic residue differs between cyanobacteria and algae. Interconversion between Phe and Trp at this position accounts for about half of the 70 mV difference between these proteins.161 The origin of this effect is attributed to differential interaction of the side chain electrostatic potentials with the porphyrin π system and the Fe orbitals. A similar effect has also been reported in cyt c3, where a phenylalanine in contact with heme I is proposed to maintain its low potential by a π–π interaction with the porphyrin π system.484
Since many charged residues around the heme, such as Glu, Asp, Lys, and Arg, as well as the heme propionate group itself, can be protonated or deprotonated depending on the pKa values of the residues and pH of the solution, protonation states of these groups will affect the reduction potential of the heme by preferentially stabilizing one redox state over the other. Therefore, the pH of the solution can have significant effects on the reduction potentials in various cytochromes.342,485−490 For example, protonation of a heme propionate in cyt c contributed an increase of 65 mV to the reduction potential.485 Similar effects of 60 and 75 mV have been reported in cyt c551(491,492) and in cyt b559,490 respectively. In cyt c2, pH-dependent reduction potentials covered a range of ∼150 mV, between pH 4 and pH 10.493 In their de novo designed maquette, Dutton and co-workers observed a 210 mV range of reduction potentials over a pH range of 3.5–10, and such a change was attributed to the involvement of Glu residues near the heme site.494 Furthermore, the role of the propionate charge has been investigated specifically by studies in which the carboxylate groups have been neutralized to their ester form. An increase of reduction potential by ∼60 mV was reported,495,496 consistent with those obtained from the studies described above.
A special case of the effect of local charges on reduction potential is the cooperativity between nearby hemes in multiheme cytochromes.497 It is known that the presence of multiple hemes in various oxidation states greatly affects the macroscopic or observable reduction potentials of the hemes. For instance, it has been demonstrated in multiheme cyt c3 that the interaction energy between hemes can shift the reduction potential by 50–60 mV.498−500 It is suggested that this effect may be mediated by electrostatic interactions also involving local aromatic groups.484 The cooperativity between hemes in multiheme cytochromes is proposed to be a major factor in their reduction potential regulation.
In cyt c3, the redox-Bohr effect can result in pKa differences of up to 2.8 pH units, and the coupling between protonation has been linked to cooperativity between the hemes, resulting in concerted two-ET steps.340,501,502 On the other hand, the pH-dependent reduction potential difference, over a range of 10 pH units, can be ∼200 mV.503 Such property is crucial for proper charge separation to generate a promotive force that drives ATP synthesis.343,504 Similarly, this coupling of proton and ET plays a key role in the proton pumping mechanism of cytochrome c oxidase. Although there are several proposed mechanisms, they share the common theme that proton uptake to the heme sites and release into the P-side of the membrane are driven by charge compensation during ET events from the low-spin to high-spin heme.505−507 It is clear that local electrostatic interactions at heme redox centers are of immense physiological importance.
2.5.3.4. Heme Distortion/Ruffling
Another significant contributor to heme redox properties is the plasticity of the heme. It is now well-known that heme distortion or ruffling plays an important role in the electronic sturcture of the porphyrins,508,509 due to decreased delocalization of the π electrons.510−516 While the phenomenon has been described in many heme proteins, including cytochromes,512,513,515,517,518 thorough investigation of how it affects redox properties is limited. Recently, Marletta and co-workers demonstrated that protein-induced heme distortion can account for up to a 170 mV increase in potential in the heme nitric oxide/oxygen binding protein.513 Furthermore, a basic computational model was implemented by Senge and co-workers, and it was estimated that porphyrin distortion can account for 54 mV of the difference between hemes in a bacterial tetraheme cytochrome.519 Further investigation is needed to gain a more detailed understanding of the role of heme distortion in the redox properties of typical cytochromes.
3. Fe–S Redox Centers in Electron Transfer Processes
3.1. Introduction to Fe–S Redox Centers
Fe–S proteins are among the oldest metalloproteins on earth. The early atmosphere, under which both sulfur and iron were abundant, enabled the spontaneous assembly of these two elements into clusters, mainly containing four iron and four sulfur atoms.91,520 Early life took advantage of the redox properties of these clusters and used them as redox centers. Despite the later shift to a more oxidizing environment on earth, the established Fe–S proteins continued to be used as electron carriers. Thus, these proteins are found ubiquitously throughout all kingdoms of life and play roles in crucial processes such as photosynthesis and respiration. The wide range of reduction potentials these proteins can accommodate and their diverse structural motifs allow them to interact with different redox partners, acting as electron carriers in a variety of biological processes.91−93
The Fe–S proteins were first discovered in the 1960s on the basis of their unique g = 1.9 EPR signal that appears upon reduction and was not observed before for any metalloproteins.9000−9002 This discovery was aided by the abundance of these proteins and their unique spectral features and often highly charged nature, which made them easier to purify and analyze. Studies of these proteins were further facilitated by advances in molecular biology and recombinant protein expression, allowing the use of site-directed mutagenesis to unravel important features of these proteins and their function.
While the Fe–S centers are well-known for their function as electron carriers, they are also known to be involved in the active sites of many enzymes, performing several functions521 such as reduction of disulfide bonds and initiation or stabilization of radical chain reactions,523,525,529 or serving as Lewis acids.524,527,543 In addition, the Fe–S centers can simply function as structural elements that stabilize the protein or another active site in the protein.523,525,527,529 Furthermore, the sensitivity of the Fe–S centers to an oxidative environment and their range of redox states make them good candidates for sensing oxidative and metal stress and balancing the oxidative homeostasis of the cells.93,525,526,527,530−533,543 Functions in DNA repair have also been reported for several Fe–S proteins.532,534 Recently, a function for Fe-S proteins has been proposed in formation of FemoCo cluster.522 Finally it has been shown that the Fe–S proteins can be used as a storage for sulfur or iron.529,532 This review focuses exclusively on the ET function of the Fe–S proteins.
3.2. Classification of Fe–S Redox Centers and Their General Features
The Fe–S clusters are often classified on the basis of the number of iron and sulfur atoms in the cluster, as suggested by the Nomenclature Committee of the International Union of Biochemistry (IUB) in 1989.535 In this convention, the elements of the core cluster (iron and inorganic sulfur atoms) are placed in brackets with the oxidized level of the core cluster shown as a superscript outside the brackets (e.g., [2Fe–2S]2+). A comma or a slash in the superscript can show multiple possible oxidation states. A more expanded notation can be used to show the ligands and the overall charge of the whole cluster, including those ligands. Another common classification of Fe–S clusters, which is used in this review, is based on the protein type. This scheme classifies the Fe–S centers on the basis of not only the number of iron and sulfur atoms but also certain structural motifs and spectroscopic and electrochemical properties. In this classification, the Fe–S proteins are divided into major groups as follows: rubredoxins (Rd’s; [1Fe–4S]), ferredoxins (low-potential [2Fe–2S], [4Fe–4S], [3Fe–4S], [3Fe–4S][4Fe–4S], and [4Fe–4S][4Fe–4S]), Rieske proteins (which are high-potential [2Fe–2S] proteins), and high-potential iron–sulfur proteins (HiPIPs, which are high-potential [4Fe–4S] proteins) (Table 3). In addition, we will also describe more complex Fe–S proteins that contain multiple Fe–S cofactors or Fe–S cofactors coupled with other cofactors, such as heme.92,93,523,526,529,536−540
Table 3. Classification of Fe–S Proteins.
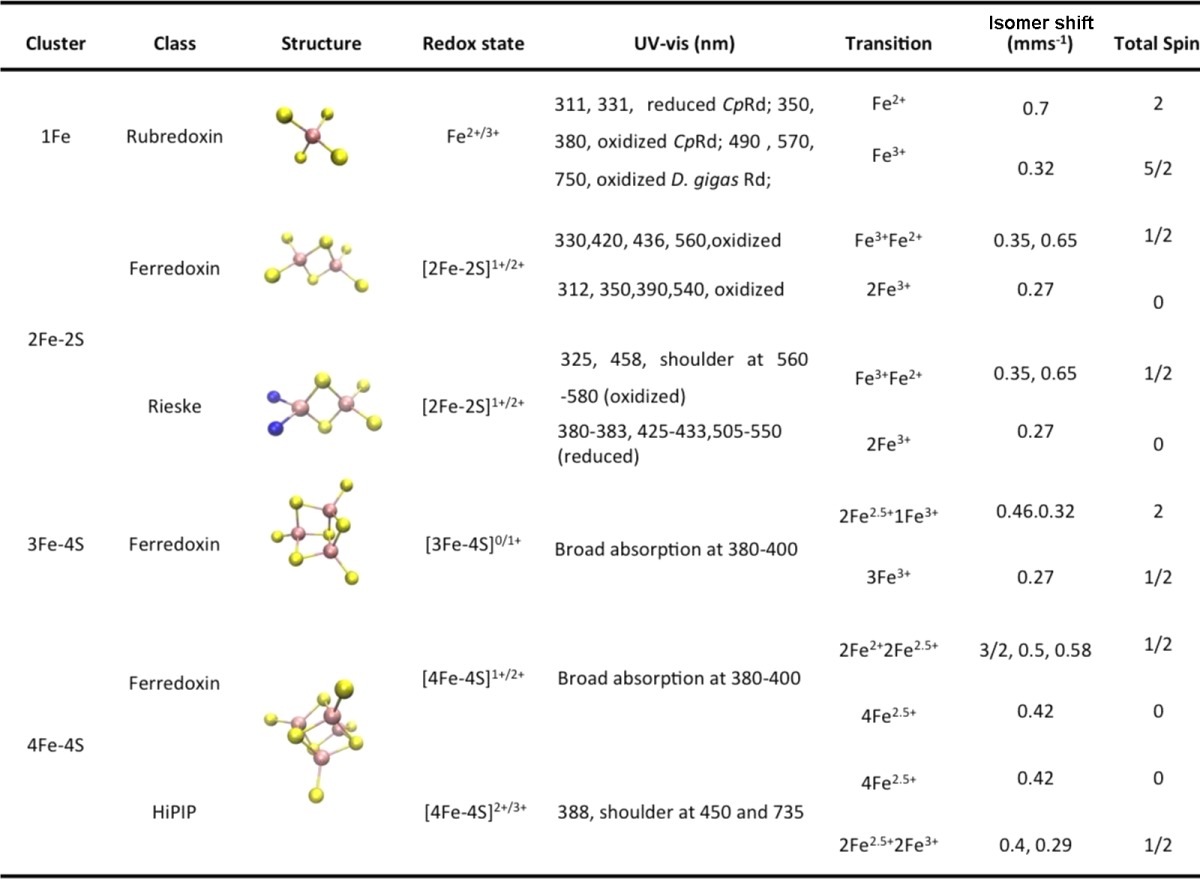
Though certain structural elements may differ between them, members of each class of Fe–S proteins usually consist of a common structural motif. Between classes the overall structure is distinct. Despite these overall structural differences, however, the geometries of the Fe–S clusters are quite similar, especially within each cluster class. The iron cofactor has a distorted tetrahedral geometry in almost all the Fe–S proteins. In the case of proteins with more than one iron, the S–S distance is usually 1.3 times longer than the Fe–Fe distance.523 Each iron atom is coordinated by a total of four ligands, typically cysteine or inorganic sulfurs, although other ligands have been observed. For instance, in Rieske proteins, two cysteine ligands have been replaced with histidines. In some [3Fe–4S] clusters, an aspartate serves as a ligand to iron. In certain enzymes such as aconitase, a hydroxyl group from the solvent is shown to be one of the ligands.541
While the geometry of Fe and its coordinating cysteine/sulfur ligands is very similar in all Fe–S proteins, the amino acid sequences and peptide motifs that accommodate these clusters differ significantly even in a given class, resulting in further categorization of each group. Interestingly, the ligands of the Fe–S proteins usually reside within loop regions. This structural flexibility is important in accommodating the geometric requirement of the Fe–S clusters and thus minimizing the reorganization energy required for rapid ET. The iron site has large spin-polarization effects, strong Fe–S covalency, and spin coupling through inorganic sulfurs.542 The strong covalency and the delocalization features of Fe–S proteins result in a low reorganization energy, mostly by lowering the inner sphere effects. Gas-phase DFT calculations give the following reorganization energies for different Fe–S proteins in vacuum: 0.41 eV (1Fe, Rd) < 0.45 eV (4Fe, HiPIP) < 0.64 eV (4Fe, Fd) < 0.83 eV (2Fe, Fd).542
The sulfur atom has several advantages over other ligands for coordinating Fe: it can occupy 3d orbitals of the iron, while the effects of its nuclear charge are not significant, and as a weak ligand, it can keep iron in a high-spin state.543 However, it imparts an intrinsic instability to the cluster, as sulfur is vulnerable to oxidation. Moreover, due to having a weak ligand, Fe in Fe-S clusters is in a high spin state.523 As a result, the Fe–S clusters are usually very sensitive to oxidation, hydroxylation, and other chemical modifications.523 In fact, one of the characteristic features of Fe–S clusters is their being “acid-labile”.1,538 The protein provides a protective, hydrophobic environment around the Fe–S clusters, excluding solvent and improving stability.523
The Fe–S proteins have long been the focus of bioinorganic studies due to their rich electronic structure and magnetism. The presence of iron as the core redox-active center provides researchers with a wealth of techniques to investigate this site which are not easily applicable to most other redox-active metals. A very intriguing feature of the Fe–S proteins is the presence of mixed-valence species, which have been the subjects of extensive investigations. All common bioinorganic methods have been applied to study Fe–S proteins, including EPR, electron–nuclear double resonance (ENDOR)/electron spin echo envelope modulation (ESEEM), 1D and 2D NMR, X-ray absorption spectroscopy (XAS) analysis, X-ray crystallography, Mössbauer, and CD/magnetic circular dichroism (MCD). Information can be deduced even with simple electronic absorption spectroscopy techniques.537,538,540
3.3. Biosynthesis of Fe–S Proteins
In vitro studies have shown that the Fe–S proteins can be reconstituted by addition of FeCl3 and Na2S in a reductive environment.539,544,545 The presence of iron and sulfur in the solution is sufficient for formation of a [4Fe–4S] cluster.543 Despite the straightforward in vitro assembly, the assembly of the Fe–S clusters in vivo is a more precise and complex process. Multiple experiments have been performed with the aim of elucidating the exact mechanism of assembly of different Fe–S clusters, and every year, new discoveries are made in this field. Nif, Isc, and Suf cluster-binding systems are the most common systems involved in in vivo assembly of Fe–S proteins.520 These systems are abundant in different organisms, and many organisms have more than one of them. Briefly, all of these systems require a cysteine desulfurase to produce sulfur from l-cysteine, a scaffold that plays the role of a carrier for the formation of the cluster, and a carrier to transfer the cluster to the final protein. The source of iron remains to be definitively elucidated. The Nif system is dedicated to maturation of nitrogenase and was first found in Azotobacter vinelandii. Isc and Suf systems, in contrast, are more general, and homologues of these systems are found in mitochondria and chloroplasts, respectively. The two systems are conserved between bacteria and eukaryotes. The Isc system utilizes five proteins: IscU that acts as a scaffold, IscS that generates sulfur from cysteine, HscA/B that facilitates the transfer of the cluster to the protein, and the ferredoxin. The Suf system is composed of two subcomplexes: One is SufBCD that can bind to and transfer the [4Fe–4S] cluster to proteins. In this subcomplex, SufB acts as a scaffold, SufD is important for iron entry, and SufC is an essential ATPase. The other is the SufSE subcomplex that acts as a heterodimer and donates sulfur to the cluster. SufS is the major component with cysteine desulfurase activity, and SufE enhances its activity. Several classes of proteins are important in transferring the cluster to the apoprotein, but the so-called A-type proteins are the most common. Recently, members of cytosolic iron-sulfur cluster assembly machinery have been found as main components of the Fe–S biogenesis in cytosol. The Fe–S biogenesis is tightly regulated and correlated to oxidative and metal stresses.520,547−554
3.4. Native Fe–S Proteins
3.4.1. Rubredoxin
3.4.1.1. Structural Aspects
Rd is the simplest among Fe–S proteins. It is a robust small protein usually composed of 45–54 amino acids with a molar mass of 6–7 kDa mainly found in bacteria, archaea, and anaerobes. It contains a monoiron center, coordinated by four cysteines from two Cys-(Xxx)2-Cys-Gly segments, with a distorted tetrahedral geometry (Figure 15).555,556 Sequence alignment reveals that the four cysteine residues are conserved in rubredoxins from different sources. Moreover, nearby glycine and proline residues, several aromatic residues such as tyrosine, tryptophan, and phenylalanine, and two charged lysine residues are conserved as well. However, a novel rubredoxin has been identified in several members of the Desulfovibrio genus, possessing an N-terminal Cys-(Xxx)4-Cys segment.557
Figure 15.
Crystal structure of CpRd (PDB ID 1IRO) at 1.1 Å resolution. (a) Overall fold of chain A of CpRd. The Fe(Cys)4 center is displayed as a ball-and-stick representation. (b) NH···S H-bond interactions around the Fe(Cys)4 center of CpRd. The side chains of C6, C39, V8, Y11, L41, and V44 are omitted for clarity. Color code: Fe, green; C, cyan; S, yellow, O, red; N, blue.
Rubredoxin from mesophilic Cl. pasteurianum (CpRd) is among the most well studied members of the family,556 and rubredoxin from hyperthermophilic archaeon Pyrococcus furiosus (PfRd) is one of the most thermally stable proteins with a melting temperature of 200 °C.558 The overall fold of rubredoxin is composed of a three-strand antiparallel β-sheet with a hydrophobic core and two loops containing the coordinating cysteines with pseudo-2-fold symmetry (Figure 15). The loop carrying ligands Cys6 and Cys39 (numbering of CpRd), buried inside the protein, is more constrained by the rigid aromatic core of the protein. In combination with a bulky aliphatic residue (Ile/Leu/Val33), these conserved aromatic residues contribute to the stabilization of the overall three-dimensional structure as well as exclusion of water from the metal center.559,560 Charged residues, mainly glutamate and aspartate, are distributed over the surface and result in high solubility and a very acidic isoelectric point of about 4. The metal binding site is close to the protein surface, between the two binding loops, and metal incorporation contributes to stabilization of the protein as well.
The two coordinating loops exhibit a pseudo-2-fold symmetry about the [Fe(Cys)4] center with six NH···S H-bonds in a range of 3.5–3.9 Å. The Fe–S bond distances to the buried Cys6 and Cys39 ligands are slightly longer (2.28–2.30 Å on the basis of three different rubredoxins) than those of Cys9 and Cys42, which are close to the surface (2.25–2.26 Å). This is possibly because Cys6 and Cys39 are involved in two H-bonds with the backbone amides of Thr7/Val8 and Pro40/Leu41, respectively, while Cys9 and Cys42 have only one H-bond donor each, from the backbone amides of Tyr11 and Val44, respectively (numbering of CpRd, Figure 15b).561,562 Nine sp3-hybridized C–H···S weak hypervalent interactions are identified by 13C NMR in CpRd, which contribute to stabilization of the protein as well.563,564 X-ray absorption near-edge spectral (XANES) fitting of the oxidized forms of recombinant CpRd at pH 8.0 gave a bond length of 2.27(1) Å for Fe(III)–S,562 comparable to the average bond length of 2.26(3) Å from crystal structures.556
3.4.1.2. Function
The electron-rich iron center of rubredoxin is redox-active, and its Fe(II)/Fe(III) couple is involved in a variety of biological ET processes.565 No significant structural changes are observed by NMR and crystallographic studies when the ferric center is reduced. Slight lengthening of the Fe–S bonds by an average of 0.096 Å (CpRd),566 0.033 Å (PfRd),555 or 0.012 Å (Leu41Ala CpRd),567 as well as shortening of the cysteine involved in H-bonds has been observed, consistent with the valence change of the metal center. DFT calculations reveal that the Fe–S center of rubredoxin from Dv. vulgaris has a low reorganization energy during oxidation due to high Fe–S bond covalency and large electronic relaxation, which makes it well suited for fast ET.568
Rubredoxin from Ps. oleovorans (PoRd) forms an ET complex with rubredoxin reductase in its physiological environment and provides a good system for studies of interprotein ET. PoRd transfers electrons from rubredoxin reductase to a membrane-bound ω-hydroxylase for aliphatic and aromatic hydrocarbon oxidation. The ET from NADH to Rd is gated by a rate-limiting adiabatic step preceding the ET step.569−572
Similarly, rubredoxin from Ps. aeruginosa is involved in alkane oxidation by transferring electrons from NAD(P)H via NAD(P)H:rubredoxin reductase to the terminal electron acceptor.573 FAD-dependent NAD(P)H:rubredoxin reductase has been cocrystallized with RubA2(PA5350), an AlkG2-type rubredoxin from Ps. aeruginosa closely related to PfRd,574 and diffracted to 2.45 Å. The shortest distance between redox centers has been determined to be 6.2 Å, which leads to an estimated maximum ET rate in the nanosecond range.575,576
Rubredoxin from Dv. gigas is important in the oxidative stress defense system in anaerobic organisms by functioning as the redox partner of NADH:rubredoxin oxidoreductase and rubredoxin:dioxygen oxidoreductase561,577−579 and transferring electrons from ferredoxin:NADP+ oxidoreductase to superoxide reductase (SOR) to reduce O2 or reactive oxygen species (ROS).580−582 It also donates electrons to rubrerythrin or diiron SORs (i.e., rubredoxin oxidoreductase or desulfoferrodoxin; see section 3.4.2.4) to reduce hydrogen peroxide or superoxide, respectively, in Dv. vulgaris.583
Rubredoxin is an electron acceptor of carbon monoxide dehydrogenase and pyruvate ferredoxin oxidoreductase in Chlorobium tepidum(584) and intracellular lactase dehydrogenase in Dv. vulgaris Miyazaki F.585 Furthermore, nucleomorph-encoded rubredoxin has been discovered to associate with PSII and proposed to branch electrons from PSII to plastid membrane-located pathways or replace some of the ET proteins in photosynthesis machinery under certain circumstances.586
Rubredoxin also exhibits high electron self-exchange rates (kese). For example, the kese of CpRd has been determined to be 3 × 105 M–1 s–1 at 30 °C in 50 mM potassium phosphate at pH 7.587 DFT calculations reveal that pathways through the two surface cysteines dominate in the electron self-exchange process and surface-accessible amides H-bonded to the cysteines play an important role as well.568
3.4.1.3. Important Structural Features
The reduction potential of the metal cofactor in a protein is generally determined by its ionization energy, electronic structure, reorganization energy, and solvent accessibility during the redox process.588 Specifically in the case of rubredoxin, the NH···S H-bonding interactions and water solvation of the active site are proposed to have a significant influence on the reduction potential of the iron center. The reduction potentials of rubredoxins vary in the range of −100 to +50 mV vs SHE (those of the model complexes are around −1 V vs SHE)92,588−590 and can be divided into two categories by the residue at position 44 (Table 4).590 Rubredoxins such as mesophilic CpRd with lower reduction potentials have a Val residue at position 44 followed by Gly 45, while those such as hyperthermophilic PfRd with higher reduction potentials (∼50 mV difference) have an Ala residue at position 44 followed by Pro 45. Mutating Ala44 of CpRd to Val increases the reduction potential, and changing Val44 of PfRd to Ala decreases the reduction potential (Table 4). The short peptide Ala44Pro45 has higher backbone stability, and consequently a higher probability of orienting the backbone dipole toward the redox center.591−595 No correlation between reduction potential and Fe–S bond covalency of CpRd and PfRd has been observed by sulfur K-edge XAS studies.596
Table 4. Reduction Potentials for Simple Rubredoxinsa.
| class | source | Emνb (mV) |
|---|---|---|
| I (V44) | Clostridium pasteurianum | –77, −53 |
| Chlorobium limicolac | –61 | |
| Butyribacterium methyltrophicum | –40 | |
| Heliobacillus mobiliz | –46 | |
| Pyrococcus furiosus A44V | –58 | |
| Cp Pf chimerasd | –46 to −67 | |
| II (A44) | Clostridium pasteurianum V44A | –24, +31 |
| Pyrococcus furiosus | 0 to +31 | |
| Desulfovibrio vulgarise H | 0 | |
| Desulfovibrio vulgarisf M | +5 | |
| Desulfovibrio gigas | +6 | |
| Megasphaera elsdenii | +23 | |
| Cp Pf chimerasd | +63 to +69 |
Reprinted with permission from ref (590). Copyright 2002 The Royal Society of Chemistry.
Versus SHE.
f. sp. thiosulfatophylum.
Constructions of fused domains from Clostridium pasteurianum and Pyrococcus furiosus.
Strain Hildenborough.
Strain Mivazaki.
The reduction potential of rubredoxin is pH-independent in the pH range of 5–10, but pressure- and temperature-dependent. The reduction potentials of CpRd and PfRd have been reported to linearly decrease with an increase of temperature (−1.6 and −1.8 mV/°C, respectively) and decrease of pressure (0.028 and 0.033 mV/atm, respectively).597 The phenomena could be rationalized by the dielectric constant change of a solvent such as water, which is lower at higher temperature and lower pressure, and consequently less efficient in protein solvation. Since the stability of a protein oxidation state is dependent on the solvent–solute interactions to neutralize the excess charge, the oxidized state of Rd with less net charge is more stable at high temperatures and low pressures.598
Replacement of one of the surface cysteines with serine in CpRd resulted in a significant decrease of the reduction potential by up to 200 mV, while for internal cysteines only a 100 mV decrease was observed (Table 5). Sulfur K-edge XAS studies of wild-type CpRd and the four Ser mutants revealed an increase in the pre-edge energy of the Cys for all four mutants compared to the wild type, indicating higher d orbital energy for the mutants, arising from the more electronegative olate serine ligand, which will lower the reduction potential as observed experimentally. Consistent with the pre-edge data, extended X-ray absorption fine structure (EXAFS) fitting shows longer average Fe–S bonds for the four mutants. DFT calculations also indicate that an alkoxide ligand stabilizes Fe(III) better than a thiolate ligand. Changes of solvent accessibility, H-bonding, and the electrostatic field around the site are other factors possibly involved.599,600 The Ser mutants display strong pH dependence, possibly arising from the protonation of coordinating oxygen of Ser following reduction at neutral or low pH.601−603
Table 5. Reduction Potentials for CpRdsa.
| protein | E°, mV | protein | E°, mV | protein | E°, mV |
|---|---|---|---|---|---|
| native | –76 | G43A | –93 | V44G | 0 |
| recombinant | –77 | G43V | –123 | V44A | –24 |
| C6S | –170 | G10V/G43A | –134 | V44I | –53 |
| C39S | ∼−190 | G10V/G43V | –163 | V8G/V44G | +39 |
| C9S | –284 | V8G | –7 | V8I/V44I | –13 |
| C42S | –273 | V8A | –44 | V44I/V44I | –55 |
| G10A | –104 | V8L | –82 | V44L | –87 |
| G10V | –119 | V8I | –81 |
Square wave voltammetry data, vs SHE.
Mutations of the secondary sphere residues have been conducted mainly on the conserved residues, and potential changes of 100 mV in both directions have been achieved (Table 5).604,605 In recombinant CpRd, Gly43Ala eliminates the Val44 NH···S Cys42 H-bonding interactions, and a Gly10Val mutation significantly perturbs the overall structure of Cys9 containing loop by increasing steric hindrance. Replacement by Val decreases the reduction potential more than Ala, and the mutations lower reduction potentials up to −86 mV.604,606,607 Side chain variation of surface residue 44 of CpRd also could influence the reduction potential of the metal center. Three mutants, Val44Ile, Val44Ala, and Val44Gly, increase the reduction potential to −53, −24, and 0 mV, respectively, from −77 mV of the wild type. The increase of E° is well correlated with a decrease of the NH···S H-bond distance determined by 15N NMR. A possible explanation of the trend is that the shortening of H-bonds might lead to increased capacity for electron delocalization or decreased electron donation from the sulfur ligands and finally to a higher reduction potential of the metal center.608,609 Similarly, quantum mechanical calculations reveal that shortening of H-bonds would decrease the energy of the reduced state faster than that of the oxidized state and result in increased reduction potential.610
Electrostatic effects of the charged residues make important contributions to the reduction potential of the iron center as well. Two neutral surface residues, Val8 and Leu41, of CpRd close to the iron center were replaced by positively charged Arg, and the resulting mutants displayed increased reduction potentials as expected. However, mutants Val8Asp and Leu41Asp, in which two negatively charged residues were incorporated, also displayed higher reduction potentials. The mutations might have also changed the solvent accessibility, and consequently the dielectric constant around the metal center, leading to complicated effects difficult to predict and explain simply by Coulomb’s law.611,612
A series of unnatural analogues of tyrosine have been incorporated into the Tyr10 position of PfRd close to the sulfur of Cys38 (3.95 Å at the closest point) by native chemical ligation methods, and the reduction potentials of the resulting proteins are linearly correlated with the Hammett σp of the para substituent of the phenyl ring. Electron-donating groups shift E° to more negative values (Tyr10 PfRd, −78.0 mV; Phe10 PfRd, −69.5 mV; 4-F-Phe10 PfRd, −61.5 mV vs SHE), and electron-withdrawing groups shift E° to more positive values (4-NO2-F10 PfRd, −49.5 mV; 4-CN-F10 PfRd, −43.5 mV vs SHE).613 The trend is not well correlated with the dipole movement of the side chain614 and is proposed to arise from either electrostatic interaction615,616 or modulation of the H-bond strength between the sulfur of Cys38 and residue 10.617−619
3.4.1.4. Spectroscopic Features
Ferrous rubredoxin is colorless, with weak absorptions centered at 311 and 331 nm. On the other hand, ferric rubredoxin displays strong absorption peaks at 350, 380, 490, and 570 nm from ligand to metal charge transfer (LMCT) of the σ orbital and a weak peak at 750 nm from the π orbital of the cysteinyl sulfur to the metal center (Figure 16a). Mutating one of the Cys residues to Ser still gives LMCT bands in ferric form, but with the peaks shifted to higher energy together with some changes of intensity, consistent with a decreased S to Fe(III) LMCT contribution.562 CD spectra of rubredoxins display minima at 202 and 226 nm from β-sheet structures in the protein.620−622
Figure 16.

Representative spectra of rubredoxins. (a) UV–vis spectra of ferric and ascorbate reduced ferrous (inset) CpRd. (b) Mössbauer spectra of dithionite reduced ferrous CpRd measured at 4.2 K under a magnetic field applied parallel to the γ-rays. Reprinted from ref (623). Copyright 2002 American Chemical Society. (c) EPR spectra of CpRd. Reprinted with permission from ref (611). Copyright 1996 Elsevier.
Mössbauer spectra of ferrous rubredoxin as purified give parameters of an S = 2 Hamiltonian with D = 5.7(3) cm–1, E/D = 0.25(2), δ = 0.70(3) mm/s, and ΔEQ = −3.25(2) mm/s (Figure 16b).623 Consistent with the Mössbauer studies, experiments using broad-band quasi-optical HF-EPR reveal a D value of 4.8 ± 0.2 cm–1 and E/D of 0.25 ± 0.01.624 The ferric form is high-spin as well, as determined by EPR spectroscopy, with a set of signals arising from an S = 5/2 spin state, including g = 4.3 from the middle Kramers doublet and g = 9.5 from the lowest Kramers doublet (Figure 16c). The Mössbauer spectrum of the oxidized form of CpRd shows δ = 0.24 ± 0.01 mm/s at 4.2 K.603,625
The Fe–S covalency has also been probed using single-molecule AFM by measuring the mechanical stabilities of Fe(III)–thiolate bonds. The rupture forces of interior Fe–S bonds of PfRd are greater than those of surface Fe–S bonds, consistent with other experimental observations.626 The mechanical stability of Fe–S bonds also shows good correlation with the NH···S H-bond strength reflected by the reduction potential.627
The dynamic properties of the redox iron center are important for the redox properties of a protein. 57Fe nuclear resonance vibrational spectroscopy (NRVS) of the oxidized form of PfRd, which is sensitive to all normal modes involving the Fe center, shows bands around 70, 150, and 364 cm–1. The 70 cm–1 signal is from collective motion of some or all of the coordinating cysteines with respect to the iron center. The ∼150 cm–1 signal mostly involves S–Fe–S bending motion composed of a doubly degenerate E mode (ν2) and a mixed T2 ν4 mode of Td symmetry. The strong signal between 355 and 375 cm–1 is mainly from an asymmetric Fe–S stretch mode, ν3, of Td symmetry, consistent with an average value of 362 cm–1 from Raman spectra of Dv. gigas (Dg) Rd. In the case of the reduced form, the asymmetric Fe–S stretching modes shift to 300–320 cm–1, bending modes shift slightly lower, and collective motion modes at ∼70 cm–1 do not change substantially. Derived force constants of both stretching and bending modes are higher in the oxidized form than in the reduced form.614,628
The resonance Raman spectra of oxidized Rd display the strongest band at ∼315 cm–1 from totally symmetric Fe–S4 breathing modes.614 The force constant of the ν3 frequency is lower than in synthetic models, probably because of the H-bonding to the S of the cysteines in the protein scaffold.589
1H NMR has been utilized to study the magnetic properties of ferrous rubredoxin. Broadening and shifting of signals are observed due to the presence of iron. To avoid the strong paramagnetism of iron, other metals such as Zn, Cd, and Hg were used as a surrogate of Fe(II) for structural studies. Paramagnetic contact shifts in 1H, 2H, 13C, and 15N nuclei of oxidized CpRd have been measured experimentally, and the data are consistent with high-level all-electron density functional calculations based on high-resolution crystal structures. Computational studies reveal that the experimental hyperfine shifts are mainly from Fermi contact interactions.629,630 NMR has also been applied in measuring the magnetic susceptibility anisotropies of both oxidized and reduced CpRd, demonstrating that pseudocontact has negligible contributions to hyperfine shifts.631
3.4.2. Rubredoxin-like Proteins
3.4.2.1. Flavorubredoxin
Flavorubredoxin is a type of protein containing a rubredoxin-like domain coupled to a flavodiiron protein and a flavodoxin domain binding one flavin mononucleotide.632,633 It has been isolated from E. coli and Moorella (M.) thermoacetica and discovered to be involved in ET pathways in reduction of nitric oxide and conversion of CO2 to acetate.634−637 The reduction potentials of flavorubredoxins from E. coli have been determined to be −140 ± 20 mV at pH 7.6635 and −120 ± 20 mV at pH 7.5.636 The reduction potential of flavorubredoxin from M. thermoacetica is −30 ± 10 mV at pH 7.0.638,639
3.4.2.2. Diiron Rubredoxins
Diiron rubredoxin is composed of two [FeCys4] domains connected by a 70–80 amino acid linker.570,640 It can be readily prepared from corresponding monoiron rubredoxin by precipitation and resolubilization and is proposed to be the physiological form of rubredoxin. Though less stable, it can transfer electrons from reduced spinach ferredoxin reductase to cytochrome c just as the monoiron form. The midpoint reduction potential of both of the two-electron reduction processes is −10 mV vs SHE at pH 7.0, similar to that of monoiron rubredoxins.641
3.4.2.3. Desulforedoxin
Desulforedoxin (Dx), isolated from sulfate reducing bacterium Dv. gigas, is an α2 dimer with 36 amino acids for each subunit. Each dimer contains a four-stranded antiparallel β-sheet and several turns and interchain short β-sheets. Each monomer has a high-spin rubredoxin-like [Fe(Cys)4] center. The iron center is near the protein surface, coordinated by four cysteine residues, Cys9-Xxx-Xxx-Cys12 and Cys28-Cys29. Unlike rubredoxin, two of the four coordinating cysteines are consecutive, making the tetrahedral coordination geometry distorted (Figure 17).642,643 In addition, Dx only has one aromatic residue, while Rd has up to six. The Fe–S bond lengths of Dx range from 2.25 to 2.36 Å, and the S–Fe–S angles vary from 102° to 119°.
Figure 17.
Crystal structure of desulforedoxin from Dv. gigas (PDB ID 1DXG). The [FeCys4] centers are displayed in ball-and-stick mode and denoted. The backbones of coordinating cysteines are omitted for clarity. Color code for the ball-and-stick mode: cyan, carbon; green, iron; yellow, sulfur.
Oxidized Dx displays three major UV–vis absorptions centered at 278, 370, and 507 nm. The 370 and 507 nm absorptions arise from the sulfur to iron charge transfer, and the extinction coefficient of the 507 nm absorption is 4580 M–1 cm–1 per monomer, falling in the normal range of Fe–S proteins.
Unlike the nearly rhombic EPR features of oxidized Rd (E/D = 0.28),644 the EPR spectrum of oxidized Dx displays an S = 5/2 site with near axial symmetry, with g = 4.1, 7.7, and 1.8 from the ground Kramers doublet and g = 5.7 from the middle Kramers doublet.645 This difference reflects different geometric and electronic structures of the two iron sites. D = 2.2 ± 0.3 cm–1, ΔEQ = −0.75 mm/s, and δ = 0.25 mm/s are obtained by Mössbauer studies of oxidized Dx. The parameters of reduced Dx from Mössbauer studies are D = −6 cm–1, E/D = 0.19, ΔEQ = 3.55 mm/s, and δ = 0.70 mm/s. The positive ΔEQ value of reduced Dx indicates that the ground-state orbital is mainly dx2–y2, while the ΔEQ value of reduced Rd is correlated to pure dz2 as the ground-state orbital.642
Insertion of a Gly residue or Pro-Val residues between Cys28 and Cys29 makes the ferric center of Dx nearly spectroscopically identical to that of Rd. However, both mutations are detrimental to the protein stability.646
Similar to Rd, Dx associates with other metal centers in biological systems. For example, desulfoferredoxin (Dfx) possesses a binding motif for the Dx-type [FeCys4] center associated with another nonheme monoiron center with N/O ligands647 (see section 3.4.2.4). Moreover, Dx in Dv. gigas is reported to transfer electrons to SOR more efficiently than Rd.648
3.4.2.4. Desulfoferrodoxin
Dfx is an α2 dimer with a molar mass of ∼28 kDa belonging to the diiron superoxide reductase family.649,650 Each monomer contains an [FeCys4] center (center I) and a nonheme iron center coordinated by a four-His–one-Cys motif (center II).651 The 1.9 Å resolution crystal structure reveals that center I is structurally similar to the metal center of Dx.652 The midpoint reduction potential of center I is around 0 mV, falling in the range of [FeCys4] centers in Dx and Rd.647,653−656
Replacement of Cys13 of Dfx from Dv. vulgaris Hildenborough with serine results in a [1Fe–3Cys–1Ser] center instead of the Rd/Dx-like center. Redox titration reveals no influence on the reduction potential of center II by such a mutation, indicating the independence of the two cofactors.657 On the other hand, reduction potentials of Dfx from hyperthermophilic archaeon Archaeoglobus fulgidus are +60 mV for center I and +370 mV for center II,649 while E° is +230 mV for monoiron SOR containing only the center II cofactor from the same genome.658 The difference in E° implies possible involvement of center I of Dfx in facilitating the reduction of center II.654
3.4.2.5. Rubrerythrins
Rubrerythrin (Rr), an α2 dimer, is a nonheme iron protein with peroxidase and in vitro ferroxidase activity.583,659 Each monomer contains a diiron–oxo site in the middle of a four-helix bundle, and an [FeCys4] center at the C-terminus.660,661 The [FeCys4] center is structurally very similar to that of Rd, yet the midpoint reduction potentials are estimated to be +230 mV at pH 8.6 and +281 mV at pH 7.0, much higher than the normal value of around 0 mV for Rd centers.662,663 The crystal structure reveals the dramatic potential increase and pH-dependent behavior might be due to the polar and solvent-exposed environment around the iron center created by nearby residues, including Asn160, His179, and Ala176, which are not conserved in Rd.660,664 Replacement of the iron in the Rd-like domain with zinc inhibits the peroxidase activity of the protein, indicating the essential role of the Rd domain in the ET process.665
Desulforubrerythrin, a unique member of the rubrerythrin family, has been isolated recently from Campylobacter jejuni. It is an α4 protein, and each 24 kDa monomer is composed of three domains: a Dx-like N-terminal domain, a four-helix bundle domain containing a μ-oxo-bridged diiron site, and an Rd-like C-terminal domain. The reduction potentials of the [FeCys4] centers in the N-terminal and C-terminal domains are +240 ± 30 and +185 ± 30 mV, respectively, at pH.7.0 vs SHE.666
Nigerythrin is an α2 dimer containing one diiron–oxo center and an [FeCys4] center, very similar to rubrerythrin. The reduction potential of the Rd-like center in nigerythrin from Dv. vulgaris is +280 mV vs SHE at pH 7.5, comparable to that of Rr as well.663,667,668
3.4.3. Ferredoxins
3.4.3.1. Introduction
The term ferredoxin refers to a wide range of small, low molar mass Fe–S proteins that function solely as electron carriers in different biological pathways including photosynthesis and respiration.669 Ferredoxins first were observed on the basis of their distinct rhombic EPR feature with g = 1.9. EPR studies with 57Fe later confirmed that the signal is from a nonheme iron.670 Evolution of H2S gas upon acid treatment was an indicator of the presence of inorganic sulfur in this protein.1,539,671 All ferredoxins share some common features: They are all low molar mass, highly acidic proteins that contain iron and inorganic or acid-labile sulfurs.1,669 The Fe–S cluster resides in a hydrophobic patch within the protein and gives the proteins a distinctive dark-brown color.672,673 All ferredoxins go through a partial decrease in absorbance upon reduction. Reduction can be achieved through chemical treatment by sodium hydrosulfite or enzymatic treatment with H2 gas and hydrogenase. The pattern of reduction is dependent on the method and extent of reduction. After reduction, a rhombic EPR signal appears with g < 2 (exact value depending on the cluster type).539,672 Ferredoxins usually have low reduction potentials with an average of −400 mV and spanning a range of 800 mV depending on the cluster type, protein structure, H-bonding network, water solubility of the cluster, and ligands to the iron. This wide range enables ferredoxins to serve as redox partners to a variety of molecules in a number of important biological reactions. Due to the high acidity, these proteins usually have high affinity for (diethylamino)ethanol (DEAE)-sepharose and can be easily purified by acetone precipitation and DEAE-facilitated separation. The purity can be monitored by the ratio of A390/A280.539,672,673 It has been shown that the proteins can usually be reconstituted by treatment with iron and Na2S under reducing conditions (in the presence of β-mercaptoethanol).539,672−674
All of the low reduction potential ferredoxins seem to have evolved from a common ancestral polypeptide.91 Despite different types, CD and optical rotatory dispersion (ORD) studies show that all ferredoxins have a very similar polar active site environment around the cluster in which the iron assumes tetrahedral coordination geometry. The similarity of extinction coefficients of their electronic absorption bands, mainly due to metal to ligand charge transfer, also indicates a similar electronic structure of the iron center.539 Despite somewhat surface-exposed iron, the reaction of the proteins with iron chelators is usually slow, unless denaturing conditions are applied.539 The ferredoxins are further divided into subcategories on the basis of the number of iron molecules present in the cluster.
3.4.3.2. [2Fe–2S] Clusters
3.4.3.2.1. Structural Aspects
As their name suggests, [2Fe–2S] clusters are a class of one-electron transport ferredoxins containing two iron atoms that are coordinated in a distorted tetrahedral geometry by two inorganic sulfurs and four cysteine thiolates from the protein. The [2Fe–2S] cluster is not completely planar, having a small tilt in the plane of the first and second irons. Three of the four cysteines come from one loop in the structure of the protein, with the other one being at the tip of a β-strand in a different loop (3 + 1 arrangement). The cluster is positioned close to the surface of the protein, surrounded by hydrophobic residues. Except for the vicinity of the cluster, the surface of [2Fe–2S] ferredoxins is highly acidic, covered with a large number of Asp and Glu residues. This acidic patch is used to interact with the basic surface of redox partners. After initial alignment through these electrostatic interactions, hydrophobic interactions between the two surfaces and water exclusion further facilitate the ET between the proteins.540,677 A role for the orientation of redox partners with regards to each other has been proposed in ET rates.678 Lack of complete complementarity between the two surfaces ensures the separation of oxidized ferredoxin and initiation of a new cycle.540 There are several NH···S H-bonds from backbone amides to the sulfurs of the cluster, with sulfur ligands of Fe1 (the iron closer to the surface) being involved in more H-bonds than those of Fe2.678,683 It appears that the Fe–Fe and Fe–Sγ bonds lengthen upon reduction while the H-bonds strengthen and shorten, consistent with increased negative charge on S.683,685
Despite these similar features, [2Fe–2S] ferredoxins can be further divided into three subcategories on the basis of differences in sequence and structural alignments and in the ligand Cys motifs (Figure 18). The details about each category are briefly explained below.677
Figure 18.
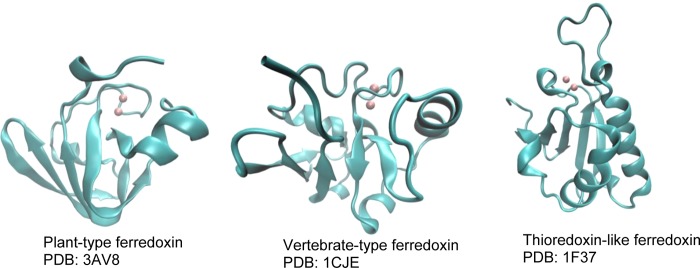
Structures of three classes of [2Fe–2S] ferredoxins. Notice that, in their physiological form, thioredoxin-like ferredoxins function as a dimer.
3.4.3.2.1.1. Plant-Type Clusters
The archetype of plant-type ferredoxins is chloroplast ferredoxin I. The members of this family share a common β-grasp structural motif, which consists of three to five β-strands, with one to three adjacent α-helices and some additional secondary structures and loops.91 Three of four coordinating Cys residues are in a loop with a conserved Cys-(Xxx)4-Cys-(Xxx)2-Cys motif, and the fourth Cys is 29 amino acids away. The cluster is usually buried at one end of the protein in a hydrophobic environment.677,682,683 Although plant-type ferredoxins have high sequence homology, there are multiple isoforms of them in each organism, which suggests different roles of the isoforms in different evolutionary and physiological conditions. Acidic residues are usually distributed in an asymmetric fashion, resulting in a dipole with its negative end near the Fe–S cluster. This dipole is shown to be important in docking of the ferredoxin into its redox partner.679−681 Several H-bonds anchor the cluster to the protein and are known to be important in fine-tuning the reduction potential of the protein. A water channel with five water molecules connects the solvent to the proximity of the cluster in the C-terminal region of the protein.677,683−686
3.4.3.2.1.2. Mammalian/Mitochondrial Cluster
Mostly known for their hydroxylating role, these clusters include mammalian [2Fe–2S] proteins as well as some bacterial [2Fe–2S] proteins. The archetypes of this class are adrenodoxin and bacterial putiredoxin. The overall fold and structure of this class are very similar to those of plant-type clusters with the exception that they have an additional interaction loop,91 a large hydrophobic domain that is used as an interacting domain with the redox partner. The conserved ligating motif of this class is Cys-(Xxx)5-Cys-(Xxx)2-Cys, with the fourth cysteine 35–37 residues away from the third ligand, further away than in plant-type structures. This group has a very flexible C-terminal which is very difficult to crystallize, but can be captured in the presence of its redox partner. It also has a compact α + β structure, characteristic of ferredoxins. Interestingly, the same fold has been observed in enzymes containing Fe–S clusters as well as some unrelated proteins that are void of Fe–S clusters. There has been evidence of structural changes upon reduction in some loops as well as the C-terminus. The solvent channel is shorter in mammalian-type ferredoxins compared to plant-type ferredoxins.677,681,682
3.4.3.2.1.3. Thioredoxin-like Clusters
These proteins are only reported in bacteria, mostly in proteobacteria and cyanobacteria.689 They were first discovered in Cl. pasteurianum(687) and Azotobacter vinelandii(670) due to their spectroscopic features, which are distinct from those of common [2Fe–2S] ferredoxins. Their sequence and positioning of the cysteine ligands differ significantly from those of other ferredoxins. These differences were further confirmed by analyzing vibrational bands in resonance Raman studies.688 Two features in the structure of this class are known to cause these differences: a distortion of the loop containing the Cys ligands and a H-bond between two cysteine residues. Proteins of this class function as a dimer, each monomer having a thioredoxin-like fold, despite low sequence homology (∼7%). Two regions are notably distinct between these proteins and thioredoxins: a protruding surface loop that has been shown to have no significant function and an α-helix in one subunit and a short helix in the other subunit that are important in interaction689 between the two subunits. The cluster lies within two loop regions in the periphery of subunits in a conserved motif of Cys-(Xxx)10−12-Cys-(Xxx)29−34-Cys-(Xxx)3-Cys.682,692 The fourth cysteine is placed in a protruding loop, which is absent in other ferredoxins. Several studies showed that the position of this Cys is flexible and that it can be moved to other positions in the loop.690,691 Some members of this class contain five cysteines instead of four. ESEEM studies and mutational analyses showed that loss of one of these cysteine residues can be compensated by the other four.9003 There are a small number of conserved residues in the family, including the four cysteine ligands and some cysteines in the dimer interface. The overall common structure has five β-strands, two long α-helices, and an additional short helix. The Cys ligands of the more buried iron are provided by the loop that is longer. The cluster itself shows some deviation from other ferredoxins with two irons. One difference is a more compressed angle between two sulfurs of Cys ligands and Fe2 (the more buried iron atom), and the other is a longer distance between one of the Cys residues and Fe2 than other Fe–S distances. The cluster is more surface-exposed in this class than the other two classes of [2Fe–2S] ferredoxins.91,689,692,693
3.4.3.2.2. Function
3.4.3.2.2.1. Plant-Type Ferredoxins
Plant-type ferredoxins can usually be found in the stroma of chloroplasts of higher plants and algae as well as the cytoplasm of cyanobacteria. Ferredoxins play a role as the first electron acceptor in the stromal side of chloroplasts and function mainly as electron distributors in photosynthesis. They are also involved in a variety of other functions such as sulfur and nitrogen assimilation, biosynthesis of several compounds such as chlorophyll, and redox homeostasis of the cell.538
The most important and well-studied function of these proteins is the transfer of two electrons in two consecutive steps from photoreduced PSI to ferredoxin:NADP reductase (FNR), which will result in final CO2 assimilation.538,683 The FNR forms a 1:1 complex with reduced ferredoxin and uses NADP+ to oxidize the ferredoxin. The NADP+ and ferredoxin have separate binding sites in FNR. It has been shown that binding of one of these substrates (ferredoxin or NADP+) weakens the binding of the other. Once oxidized, the ferredoxin has a lower binding affinity to FNR and dissociates from the complex, while a second reduced ferredoxin will replace it to complete the cycle.2003 In organs that produce NADPH by the pentose phosphate cycle, FNR acts in the reverse direction and reduces ferredoxin.677
Ferredoxin also distributes electrons from photoreduced PSI to ferredoxin-dependent enzymes such as nitrite reductase, glutamate synthase, and ferredoxin:thioredoxin reductase (FTR) for nitrogen and sulfur assimilation. Cyanobacteria have a vegetative ferredoxin that functions in photosynthesis and a heterocyst ferredoxin that transfers electrons to nitrogenase. Ferredoxin from halobacteria can function as an electron carrier in α-keto acid decarboxylation or in nitrite reduction.694
One of the most studied realms in the field of ferredoxins is their interaction patterns with their redox partners. These complexes have been studied using several techniques such as cross-linking, NMR, isothermal titration calorimetry (ITC), and site-directed mutagenesis; however, it is not completely understood whether ferredoxin uses the same surface, partially overlapping surfaces, or totally different surfaces for interacting with different redox partners. The most likely hypothesis is that ferredoxin acts as a mobile electron carrier between PSI and other redox partners.677
3.4.3.2.2.2. Interactions with Other Proteins
3.4.3.2.2.2.1. Interaction with Ferredoxin:NADP+ Reductase (FNR)
The most well-known partner of plant-type ferredoxins is FNR. It has been shown that ferredoxin and FNR have very tight binding with Kd in the range of 10–7–10–8 M.677 As discussed previously, several surface amino acid residues are conserved in ferredoxins, and mutation of these amino acids revealed important factors in interaction between these redox partners. Binding of ferredoxin to FNR cause a negative shift in Em of ferredoxin, which is suggested to be important in more efficient ET between the two proteins. Laser flash photolysis is one of the techniques that has been used to analyze the reactivity of several ferredoxin mutants from Anabaena. Among the conserved residues, Phe65 was the only one essential for tight binding between ferredoxin and FNR.684,697 Ser47, Glu94, and Phe65 were also shown to be important in the rapid ET between the two partners, though conservative mutations to other similar residues were tolerated.677,695 Interestingly, mutating residues adjacent to the above three residues had a much less effect on the activity.684 Mutational studies of Glu92 in spinach ferredoxin, which is analogous to Glu94 in Anabaena, resulted in decreased activity, but much less significant than that of the former. More interestingly, this mutation resulted in an increase in reduction potential and stimulation of NADPH–cytochrome c reductase activity catalyzed by FNR. These mutants were more efficient in transferring electrons in the direction opposite that of the physiological ET pathway.2004 Although several studies have shown significant correlation between ET and reduction potential, ET changes are thought to be more likely a result of changes in protein orientation and transient changes in configuration rather than a consequence of reduction potential changes. A thorough study of the mutants with laser flash photolysis showed very similar effects of Glu92/94 mutation in both spinach and Anabaena variants, hence suggesting a difference between these results and previous NAD+ photoreduction results.677 ITC studies suggested entropy as the main driving force of complex formation, meaning that hydrophobic interactions are the major forces governing the efficient interaction between the two partners. The proposed binding surfaces of many ferredoxins are covered with water, so the binding of the partners will release water molecules and favor the reaction entropically.694,698
Several models of complexes between ferredoxins and FNRs have been made on the basis of experimental evidence coming from chemical modification, cross-linking, partial proteolysis, and mutational studies, as well as homology models. These models predicted the binding site between ferredoxin and FNR to be a large hollow surface near the dimethylbenzyl ring edge of the flavin in FNR. The binding will bring the Fe–S cluster and the flavin close, so that they can transfer electrons. While ferredoxin has an excess of positive charge on the binding surface, FNR has a net negative charge on its binding surface. The specific orientations of dipoles in the two proteins have been shown to be important in recognition between the two partners. Another model proposes that electrostatic potential complementation plays an important role. The two models differ in the orientation of the ferredoxin molecule about the axis perpendicular to the protein–protein surface.677,679,680 Cross-linking experiments have been done to study the complex between ferredoxin and FNR (Figure 19). The cross-linked molecule showed oligomer states in the crystal structure that might be relevant to in vivo interactions.699
Figure 19.
Structure of ferredoxin (right) cross-linked to FNR (left), PDB ID 3W5U. As shown, red acidic patches of ferredoxin are positioned in contact with blue basic residues of FNR. A zoomed-in figure of the region containing the cofactors (Fe–S and FAD) is shown at the bottom.
3.4.3.2.2.2.2. Interaction with Ferredoxin:Thioredoxin Reductase, Nitrate, Nitrite and Sulfite Reductase, and Glutamate Synthase
Reduced ferredoxin donates electrons to FTR to reduce thioredoxin, which is involved in multiple steps of the Krebs carbon cycle. FTR is found only in oxygenic photosynthetic organisms. Chemical modification of acidic residues on the surface showed that the Glu92–94 acidic patch is important for the interaction between the two partners. A model has been proposed on the basis of the crystal structures of the two partners. In this model, ferredoxin docks into the opposite site of the flat, disklike structure of FTR in such a way as to position itself close to the [4Fe–4S] cluster and the redox-active disulfide bond in FTR.700 In this ternary complex, two successive one-ET reactions take place. The complex between ferredoxin and FTR has very high affinity, with both electrostatic and hydrophobic interactions being involved.
Site-directed mutagenesis and chemical modification studies suggest that the same site of ferredoxin is responsible for interacting with nitrite reductase, sulfite reductase, and glutamate synthase.677,701,702 The surface is formed in low ionic strength, indicating a role for electrostatic interactions in formation of the complex.694 Another site has also been proposed for sulfite reductase (SiR).694,703 While less is known for SiR, NMR analyses of the contact shifts between the presumed complex confirmed the important role of acidic surface residues on complex formation.694
Nitrate reductase is found in cyanobacteria and performs two-electron reduction of nitrate to nitrite. It has been shown that there is only one ferredoxin binding site in nitrate reductase, so the reduction proceeds in two separate consecutive steps.694
Nitrite reductase performs six-electron reduction of nitrite to ammonia. As with nitrate reductase, only one binding site exists for ferredoxin. A conserved Trp residue has been shown to play an important role in ET between the two partners.694
A loop close to the [3Fe–4S] cluster of glutamate synthase is responsible for binding of ferredoxin. CD analyses showed that neither of the two proteins underwent significant conformational changes upon binding.694
3.4.3.2.2.2.3. Interaction with Photosystem I
Photosystem I (PSI) is an essential part of the photosynthetic ET pathway in cyanobacteria and plants. This multisubunit complex is a membrane-bound system that harvests light and helps convert it into a chemical potential. The complex consists of multiple chlorophylls, carotenoids, phylloquinones, bound lipids, and [4Fe–4S] clusters. Three subunits at the stromal site of PSI are involved in docking and reducing ferredoxin I: PsaC (with [4Fe–4S] clusters FA and FB), PsaD, and PsaE. FA, FB, and FX are three low-potential [4Fe–4S] clusters that lie in the stromal side of the PSI complex. FA and FB are bound to PsaC, and FB functions as a terminal electron acceptor (Figure 20). FX is an interpolypeptide cluster, positioned between PsaA and PsaB, and has the most negative reduction potential reported so far for a [4Fe–4S] cluster (−705 mV).705
Figure 20.

Structure of PSI (PDB ID 1JB0). The top left figure shows the overall structure, and the bottom figure shows all the cofactors in the system. The top right figure shows the PsaC, PsaD, and PsaE sites with FA and FB. Ferredoxin binds in the interface between PsaC, PsaD, and PsaE.
In vitro studies and cross-linking experiments revealed PsaD as the main docking site for ferredoxin I. A binding site for PsaC has been also proposed on the basis of mutational studies. It has been shown that PsaD and FNR compete with each other in binding to ferredoxin, yet no ternary complex has been observed.705
3.4.3.2.2.3. Mammalian-Type and Thioredoxin-like Ferredoxins
The main function of mammalian-type ferredoxins is ET in the mitochondrial ET chain, ET to P450’s, and Fe–S biosynthesis. It has been shown that adrenodoxin has very tight binding to both adrenodoxin reductase and cytochrome P450 on the order of 10–7–10–8 M.677 As with ferredoxin, adrenodoxin interacts with its redox partners through an acidic surface,706 with Asp76 and Asp79 being essential for the binding. The overlapping interaction surface supports a mobile carrier hypothesis for the adrenodoxin. A model based on the crystal structures of the partners suggests that adrenodoxin binds in the cleft between two domains of adrenodoxin reductase, resulting in a distance of 16 Å between the Fe–S cluster and the isoalloxazine ring of the FAD in the reductase.707,708 A specific ET path between the two has also been proposed.708 Several studies on putiredoxin have shown the same overlapping surface for reductase and P450 interaction. The crystal structure of the complex between adrenodoxin and adrenodoxin reductase further confirmed the importance of charged Asp and Glu residues on the surface of ferredoxin in the formation of the complex (Figure 21).709
Figure 21.
Structure of adrenodoxin (right) in complex with adrenodoxin reductase (left) (PDB ID 1E6E). As shown, red acidic patches of adrenodoxin are positioned against blue basic residues of adrenodoxin reductase. A zoom-in region of the cofactors (Fe–S and FAD) is shown at the bottom.
No certain function has been determined for thioredoxin-like ferredoxins yet. However, their abundance in nitrogen-fixing bacteria suggests a role in nitrogen metabolism. Some molecular dynamics and docking studies have shown an interaction surface with this class of proteins and the MoFe protein of nitrogenase, suggesting a role as electron carriers to this complex.692,689,710
To analyze the ET activity of [2Fe–2S] ferredoxins, a simple spectroscopic assay can be performed using cytochrome c as the final electron acceptor.677 A wealth of mutational studies showed the importance of entropy as the main driving force in this interaction. While positive surface charges are important in bringing the two proteins into proximity, hydrophobic interactions are the major players in stabilizing the complex.694
3.4.3.2.3. Important Structural Features
The reduction potentials of ferredoxins from plants and mammals are between −460 and −300 mV.683,694 On average, mammalian ferredoxins have higher reduction potentials than plant-type ferredoxins,683 due to different patterns of electron delocalization, as observed by NMR.711 Interestingly, mammalian ferredoxins show an ionic strength- and pH-dependent redox behavior.712 The average reduction potential for the thioredoxin-like class is around −300 mV.689 Multiple methods have been used to measure reduction potentials of ferredoxins, including direct protein film voltammetry,714 and spectrochemical redox titration.714
Several factors have been reported to be important in fine-tuning the reduction potentials of ferredoxins. The overall protein fold and solvent accessibility of the cluster are known to be important in giving a low reduction potential range to ferredoxins compared to Rieske centers that also have a [2Fe–2S] cluster core. These factors are discussed in more detail in the section on Rieske centers (section 3.4.4).
Models of [2Fe–2S] proteins have been used to analyze the reduction potential properties. These analyses have shown the nature of the peptide to be important in reduction potential determination and behavior.717 Other factors such as the H-bonding network from backbone amides to sulfurs and overall charge of the protein are reported to play a role in determining the reduction potential value within [2Fe–2S] ferredoxin classes. In all the classes, there is a conserved H-bonding network, with sulfurs ligating the higher potential iron being involved in more H-bonds (Figure 22).677,683
Figure 22.
H-bonding network in plant-type ferredoxins.
It has been suggested that the charge and H-bonding pattern differences between Thioredoxin-like ferredoxins and plant-type ferredoxins is the cause of differences in their reduction potential. Indeed, point mutations near the active site that change the charge of thioredoxin-like ferredoxin resulted in a 100 mV change in reduction potential.689 Three kinds of mutations were found to influence the reduction potential in thioredoxin-like ferredoxins the most: replacing Cys ligands, swapping ligands or changing the loop containing them, and changing the charge in the vicinity of the cluster.689 Interestingly, changing the loop (either insertion or deletion) resulted in a reduction potential correlated with the sum of the charged residues left in the loop. Cys → Ser mutations caused a decrease in reduction potential.690,721 A 100 mV change in reduction potential was observed upon mutating one of the Cys residues in thioredoxin-like ferredoxins that have five Cys residues.690 Cys to ser mutants of Anabaena [2Fe−2S] cluster showed that the changes in reduction potential is dependent on the position of ligating Cys.678 Mutations of Glu94 and Ser47 of Anabaena ferredoxin showed a significant increase in the reduction potential of this protein mostly due to rearrangement of the H-bonding network as well as removal of a negative charge close to the cluster.9004
3.4.3.2.4. Spectroscopic Features
All [2Fe–2S] ferredoxins share very similar UV–vis spectra with a protein peak at 280, a near ultraviolate peak at 330 nm, and visible absorptions at 420 and 463 nm, and a shoulder at 560 nm in the oxidized form (Figure 23). The relative intensities of the 420 and 460 bands are inverted in thioredoxin-like ferredoxins compared with the other two groups. Depending on the hydrophobicity and H-bonding pattern around iron atoms, one of them, usually the one closest to the surface, is reduced more easily. After reduction, the spectral intensity decreases to about 50% of that of the oxidized form and the band positions are altered to a maximum at 540, with small peaks at 460, 390, 350, and 312 nm.538,539 These proteins show similar CD and ORD spectra. A red shift was observed in the spectra after selenium substitution. Strong positive bands between 420 and 460 nm in the oxidized form dominate the CD spectra. The reduced state has negative bands at 440 and 510 nm. From these CD analyses, bands from dz2 → dxz and dz2 → dyz have been assigned.539
Figure 23.

Representative spectra of [2Fe–2S] ferredoxins:728 (a) UV–vis spectra of reduced (thin line) and oxidized (thick line) forms of ferredoxin from Aquifex aeolicus; (b) X-band EPR of [2Fe–2S]+ ferredoxin from Aq. aeolicus at 20 K; (c) Mössbauer of the [2Fe–2S]2+ state of ferredoxin from Aq. aeolicus at 4.2 K in zero field (upper) and an 8.0 T applied field parallel to the observed γ radiation (lower). Reprinted from ref (728). Copyright 2002 American Chemical Society.
Ferredoxins were first identified through their unique EPR signal in the reduced state (Figure 23). The two irons in the Fe(III) state each have a spin of S = 5/2 and are antiferromagentically coupled, resulting in a final diamagnetic EPR-silent species. Upon reduction of one of the iron ions to Fe(II), the net spin changes to 1/2 and a rhombic EPR signal at g = 1.94 is observable at temperatures below 100 K. When the iron in the protein is replaced with 57Fe, the sample shows a broader or split EPR signature, proving that the signal is from iron.670 Studies with S33 showed that hyperfine splitting from S contributes to broadening of the signal at g = 1.94.539,722,723 ENDOR experiments were performed and provided information complementary to that of EPR that is required for computer simulation of Mössbauer data. These studies showed two nonequivalent iron sites in the reduced form, consistent with the Mössbauer results. The same studies also revealed some protons that are coupled to irons in the cluster.539 While all studies are consistent with a localized electronic structure of the irons in the reduced state, a Cys → Ser mutant of a thioredoxin-like ferredoxin showed a valence-delocalized S = 9/2 feature in EPR, which was further analyzed by variable temperature magnetic circular dichroism.724
Due to the centrosymmetric core of [2Fe–2S] ferredoxins (D2d, oxidized; C2v, reduced), the ungerade vibrations are Raman-inactive and the protein has fewer features than its counterpart Rieske centers. They show a characteristic Bt3u peak at around the 283–291 cm–1 region, which shifts to 263–273 in the reduced form. Other features are an Agt peak at 329–338 cm–1, a B1ut peak at 350–357 cm–1 (mostly Fe–St stretching mode), and an Agb peak at 387–400 cm–1 in the oxidized form. These peaks appear at 307–314, 319–328, and 370–385 cm–1 in the reduced form, respectively. Resonance Raman spectra of thioredoxin-like ferredoxins are substantially different from those of the other two categories due to different cluster environments. The main peaks are observed at 208, 290, 313, 335, 353, 366, 387, and 404 cm–1 in the oxidized form and at 267, 280, 310, 328, 370, and 390 cm–1 in the reduced form.725
It was first shown by Mössbauer that upon reduction one of the irons changes to Fe2+ (Figure 23). Mössbauer of the oxidized state shows a narrow quadruple doublet with δ = 0.27 mm/s relative to iron and a splitting of 0.6 mm/s. The doublet position is temperature-independent, and the splitting shows a slight decrease at temperatures higher than 200 K. The spectrum in the reduced form is temperature-dependent and more complex, primarily because of magnetic hyperfine interactions and quadruple interactions. The reduced state shows δ = 0.55–0.59 mm/s at 200 K. The A tensor of these proteins is more symmetric along the z axis. In the reduced state, Mössbauer of ferredoxins reveals two quadruple doublets, one at δ = 0.30 mm/s and the other at δ = 0.72 mm/s, indicating two localized irons.529,539,726
NMR studies show that, in the reduced state, the protein has a mixed-valence Fe2+/Fe3+ state, with the iron closer to the surface being in the Fe2+ form. Solvent exchange studies by NMR suggested that reduction of the cluster might increase accessibility of protons to the cluster. NMR studies were used to analyze the interaction of ferredoxins with their redox partners to find their contact points. Chemical shift changes upon reduction have been assigned. NMR has also been extensively used for structure assignment. NMR studies showed differences between plant-type and mammalian-type ferredoxins. While plant-type proteins show a downfield shift of Cys ligands in the reduced state, with the ligands of Fe3+ showing Curie-type behavior and Fe2+ ligands showing anti-Curie behavior, vertebrate-type proteins have both upfield and downfield signals of cysteine ligands in their reduced state, and all show Curie-type behavior.539,727
3.4.3.3. [3Fe–4S] and [4Fe–4S] Clusters
3.4.3.3.1. Structural Aspects
These clusters are mainly found in bacteria and usually consist of either one or two [3/4Fe–4S] clusters. [4Fe–4S] clusters are known to be the first clusters formed in the early earth environment and function as ubiquitous ET members in most anaerobic bacteria. The cluster takes the form of a distorted cube, with iron and sulfur atoms positioned alternatively in the apexes. Three inorganic sulfurs and one thiol from a cysteine in the protein coordinate each iron. The cysteine ligands are arranged in a C-(Xxx)2-C-(Xxx)2-C motif, the so-called classic [4Fe–4S] motif. The cluster resides in a common ferredoxin motif (βαββαβ) with four β-strands, two linking helices, and cluster binding loops. This fold is the most ancient ferredoxin fold and is very versatile, with lots of insertions and deletions observed in different proteins of the family.91,92,539
The 2[4Fe–4S] or eight iron clusters are hypothesized to emerge from a gene duplication of the ancestral [4Fe–4S] cluster.91 A clostridial 2[4Fe–4S] protein was the first ferredoxin discovered. Due to its high iron content, a large portion of the protein consists of inorganic materials in these proteins.91 The positions of cysteines in all [4Fe–4S] or 2[4Fe–4S] proteins are very similar. The proteins with two clusters can be divided into five subcategories on the basis of their sequence and evolutionary relationship, including the clostridial type, chromatium type from green and purple bacteria, azotobacter [3Fe–4S][4Fe–4S] type, archaebacteria type, and single [4Fe–4S] type.729 The essence of this characterization is sequence homology of 27 ferredoxins and their deviation from basal architecture, which is a two-subunit structure resulting from gene duplication with a three-linker connector and an (Xxx)7-CysI-(Xxx)2-CysII-(Xxx)2-CysIII-(Xxx)8-CysIV motif in each subunit (Figure 24).729
Figure 24.

Structures of the five classes of two-subunit ferredoxins.
Clostridial-type ferredoxins follow the basal architecture and have a conserved motif of Cys-(Xxx)2-Cys-Gly-(Xxx)-Cys-(Xxx)3-Cys-Pro. This motif usually contains no other cysteine except in the case of a small number of proteins, including PaFd, which contains a ninth cysteine in its 22 position. The proteins consist of two homologous halves that arrange in a pseudo 2-fold symmetry, with three of the cysteine ligands coming from one half and the fourth cysteine being provided by the second half, adjacent to a proline. In 2[4Fe–4S] clusters, the [4Fe–4S] clusters are surrounded by two antiparallel β-strands and two α-helices. In the final arrangement of the protein, two sets of antiparallel β-sheets with two strands lie beneath the clusters and two short helices are positioned on top of the cluster. An array of water molecules facilitates H-bonding between two halves of the protein. In clostridial ferredoxins, there is a conserved Pro after the last coordinating Cys. Although mutations of this Pro has shown that it is not necessary for the cluster arrangement, it provides an optimal environment for the next cluster by both providing hydrophobicity and supporting a specific turn mode for binding.91,674,730
In contrast, chromatium-type ferredoxins in most cases contain a ninth cysteine in positions 2–8, between the second and third cysteines in the clostridial core. They also have a C-terminal extension relative to the clostridial sequences. Further classifications within this class are possible on the basis of the position of their ninth cysteine and the length and arrangement of their extension, including photosynthetic ferredoxins, chromatium-type ferredoxins, and dimeric 2[4Fe–4S] ferredoxins. Chromatium-type ferredoxins have their ninth cysteine close to cluster I. In addition, they have an extended loop and a short α-helix next to cluster II. The presence of this loop results in a positive Fe–S–Cα–Cβ torsion angle, compared to the negative angle in clostridial-type ferredoxins. Moreover, the backbone orientation around this loop is changed so that this cluster I has one less NH···S H-bond.731 Lack of this H-bond results in a slightly shorter Fe–S bond. These clusters are unstable at room temperature, at pH values below 6.5, and in the presence of oxygen.674
The azotobacter-type ferredoxins have two residues inserted after CysII in their subunit 1, and the CysII is mutated to Ala. Their subunit 2 is intact, apart from a 48- or 49-residue extension of the C-terminus. While this extension is similar within members of the group, it differs substantially from that of other groups.729
The archaebacteria-type ferredoxins have a conserved central domain in each subunit, but further modifications are observed in regions before or after this domain, such as an extension of the N-terminus, or an insertion before the linker. CysII in this class is mutated to an Asp, resulting in a [3Fe–4S] cluster that can become a [4Fe–4S] cluster under certain conditions.729
The single [4Fe-4S] group has both domains, but the conserved motif in subunit II is disrupted due to replacement of two to four of the cysteines with other nonligating residues. Members of this group cannot be grouped further due to differences in their sequence and structure.729
Chemical modification studies showed that neither the N- nor C-terminal Fe–S binding motif can form a stable cluster in 2[4Fe–4S] proteins, but their combination will result in formation of a stable cluster.674 Using a protein maquette of [4Fe–4S] ferredoxins and step-by-step replacement and truncation of amino acids, several minimal essential features have been derived for formation of a [4Fe–4S] cluster, including the spacing between Cys residues, the importance of noncoordinating amino acids in assembling and stabilizing the cluster, preferable use of Cys ligands, the requirement of only three Cys ligands for formation of a single cluster, and the requirement of only a consensus core motif of CysIleAlaCysGlyAlaCys.732 Figure 25 shows consensus motifs in [3/4Fe−4S] ferredoxins.
Figure 25.

Consensus sequences in ferredoxins. Reprinted with permission from ref (740). Copyright 2007 University Science Books.
The [3Fe–4S] cluster can be thought of as a cubane [4Fe–4S] cluster missing one of the irons. This class is found exclusively in bacteria, mainly anaerobic bacteria, and is involved in anaerobic metabolism. The [3Fe–4S] clusters can emerge from oxidative damage of [4Fe–4S] clusters, as in the case of aconitase, or treatment of 4Fe clusters with potassium ferricyanide or can be found as intrinsic constituents of natural proteins, such as mitochondrial complex II and nitrate reductase. In all cases, the true reason for the presence of such clusters is not yet completely understood. It has been shown that [3Fe–4S] and [4Fe–4S] clusters can be interconverted under certain physiological conditions and the exchange between 3Fe and 4Fe can be used as a regulatory mechanism. The [3Fe-4S] clusters have the Cys-(Xxx)2-Cys-(Xxx)2-Cys motif similar to the [4Fe-4S] clusters but the middle Cys is replaced by an Asp in most of them.734 It has been shown that replacement of the Asp with Cys can change the cluster into a complete [4Fe–4S] type.734,735 Addition of two extra amino acids between the second and third cysteines can also change a [4Fe–4S] cluster into a [3Fe–4S] cluster.733
Another common motif, Cys-(Xxx)7-Cys, is found in [3Fe-4S] cluster of 7Fe-containing proteins, some of which are thermostable and air-stable. Another Cys following this motif serves as the third ligand to the cluster. The presence of seven irons in [3Fe–4S][4Fe–4S] clusters has been confirmed by a combination of techniques such as EPR, Mössbauer, and X-ray crystallography. There are examples of Asp residues and hydroxyl groups from the solvent as ligands. As with 2[4Fe–4S] clusters, the [3Fe–4S][4Fe–4S] clusters are capable of two-ET. The [3Fe–4S] cluster can be found in two states: [3Fe–4S]1+ and [3Fe–4S]0, with overall spins of 1/2 and 2, respectively. H-bonds play an important role in stabilizing the reduced state. The number of these bonds is related to the extent of solvent accessibility of iron, but there are on average six such interactions that direct protons to the site.737 The N-terminal structure of the 7Fe proteins is similar to that of 8Fe proteins, consisting of a central part with four β-strands that have the Fe–S cluster in the middle. Two short α-helices connect the loops in β-sheets. The structure has a partial 2-fold symmetry that is disrupted at the N-terminus by differences in Cys ligands to the [3Fe–4S] cluster. There are two nonligand Cys residues next to each cluster. Although the clusters are positioned close to the surface, the presence of hydrophobic and aromatic residues protects them from the solvent. The [3Fe–4S] cluster is very similar to the [4Fe–4S] cluster, with Fe–Fe distances shorter than S–S distances, and very similar Fe–S distances. However, the protein matrix distorts the [3Fe–4S] cluster, while the [4Fe–4S] cluster is more symmetric.737
Conserved hydrophobic residues are shown to be important for the stability of the protein but not for ET.738,1085 The thermostable ferredoxins have been shown to have extra salt bridges in their C-terminus as well as an extra flexible hydrophobic loop.739
3.4.3.3.2. Function
The [4Fe–4S] clusters are important in hydrogen evolution in anaerobic bacteria, in which the reduced form of ferredoxin transfers electrons to H+ as the final acceptor. In Clostridium, reduction of ferredoxin is coupled to pyruvate oxidation. The hydrogenase complex further oxidizes the reduced ferredoxin. Ferredoxins have been shown to be important in reactions that couple oxidation of the substrate with reduction of NAD(P)+, flavin mononucleotide (FMN), FAD, riboflavin, sulfite, and N2. They can bridge excitation of chlorophyll by light to reduction of NAD. Conversion of formate to CO2 is often ferredoxin-coupled.9005
The role of [3Fe–4S] clusters is less well-known. It has been reported that they can act in sulfite reduction. A role as iron storage has also been proposed. The [3Fe–4S] clusters have been observed in the monooxygenase system of Streptomyces griseolus.741
The 2[4Fe–4S] clusters are mainly found in anaerobic bacteria and Clostridium species. However, there are multiple reports of their occurrence in other organisms such as Micrococcus lactolyticus, Peptostreptococcus esldenii, Methanobacillus omelianski, certain photosynthetic bacteria such as Ch. vinosum, Chlorobium limicola, and Rb. capsulatus, and several extremophiles.674
There are several ways to test the activity of [3/4Fe–4S] ferredoxins. Clostridial-type ferredoxins are usually assayed using their ability to reduce NADP either in an NADP:ferredoxin reductase system or in a phosphoroclastic system. Coupling H2 oxidation to the reduction of an organic dye is another assay used to monitor the concentration and activity of ferredoxins.674
3.4.3.3.3. Important Structural Elements
The [3/4Fe–4S] clusters, like other Fe–S clusters, display very low reduction potentials. The reduction potential of [4Fe–4S] clusters usually ranges from −250 to −650 mV, with an average of −400.540,737 The common reduction potential for [3Fe–4S] clusters ranges from −50 to −450 mV, with an average of −100 to −150.540,737,887 Several methods have been used to monitor the reduction potential of the clusters, such as potentiometric CD titration, direct CV, and spectroscopic potentiometry.737,742 In the case of 7/8Fe proteins, the reduction potentials of the two sites can be similar (isopotential) or differ by values as high as 192 mV.743 The same factors that control the reduction potential of clusters affect the reduction potential of each cluster within a multiple-cluster protein. Usually the greater the difference between the reduction potentials of two clusters, the lower the ET rate between the two. Mutational analyses of conserved residues that are thought to be important in the intramolecular ET showed no significant decrease, but less stability. It was postulated that the geometry and relative orientation of the two clusters are the factors important in determining this rate. A role for amide dipoles has also been suggested.737 It has been shown that the number of these bonds and more importantly the overall dipole around the cluster play essential roles in the reduction potential.718,719
A major part of reduction potential analyses of these types of ferredoxins deal with roots of differences between them and HiPIPs. These types of studies are discussed in detail in the section on HiPIPs (section 3.4.5).
Peptide models of [4Fe–4S] proteins showed that the reduction potential of the center is dependent on the number of Cys residues in the oligomer and will stabilize higher oxidation states, hence decreasing the reduction potential, with increasing cysteines. These studies also showed the importance of NH···S in determining the reduction potential of 4Fe ferredoxins and their difference from HiPIPs.717
The reduction potential of the [3Fe–4S] cluster is pH-dependent. The pH dependence is related to proton transfer via the conserved Asp next to the cluster.745 Mutation of this Asp to Asn lowers the proton transfer and gates oxidation. Other studies show a less significant role for the conserved Asp, suggesting protonation of the cluster itself as the main cause of the pH-dependent behavior.746 Also, it has been shown that, in a protein film electrochemical setup, a hyper-reduced [3Fe–4S]2– cluster can be formed.716,747
The presence of a fifth Cys residue close to the cluster can lead to formation of a SH···S H-bond and tune the activity by lowering the reduction potential.9006 This effect is important in fine-tuning the reduction potential of proteins with two clusters. Moreover, there are around 15 partial positive charges in ferredoxins that result in an overall positive environment of the cluster, which is suggested to be a reason for the lower reduction potential of these ferredoxins compared to their higher reduction potential counterparts, HiPIPs.674
Introduction of a His near the cluster of a 7Fe protein caused a 100–200 mV increase in the reduction potential. The reduction potential of this variant was pH-dependent. At pH values where the His was protonated, this large increase in reduction potential was attributed to placement of a positive charge next to the cluster. A dipole moment directed toward the cluster was proposed as the main cause of increased reduction potential when the His was neutral.277
Mutations of conserved Pro in CpFd resulted in changes of the reduction potentials of the two clusters. NMR studies of these mutants showed that signals from the β-proton to cysteine sulfur were changed by these mutations.730 Mutational analysis of conserved Asp and Glu residues in the CpFd show negligible changes in the redox properties.748 Replacement of AvFdI amino acids with their counterparts in PaFd showed no change except for small changes in the case of a Phe → Ile mutation, casting doubt on the role of single amino acids in the reduction potential differences.749 A Cys→ Ala mutation resulted in a 100 mV lower reduction potential of the cluster, mainly due to changes in coordination geometry and accommodating a new Cys ligand.750
Resonance Raman studies on the cluster showed a very similar environment of the cluster in different proteins and suggested a role for the Fe–S–Cα–Cβ torsional angle in fine-tuning the reduction potential of the site.618
Solvent accessibility and cluster solvation also play important roles in determining the reduction potential of these clusters. More buried clusters have higher reduction potentials.92,749,752
The protein dipole Langevine dipole (PDLD) model was used to analyze the important features of the reduction potential. On the basis of these calculations, the number and orientation of amide dipoles, and not necessarily their involvement in H-bonding, are the most important factor sin defining the reduction potential. Addition of more amide dipoles by site-directed mutagenesis indeed resulted in a more positive reduction potential in cases where the backbone conformation did not change drastically.737 Another study suggested that not the absolute number of H-bonds, but the net dipole moment on the cluster is the determining factor in the reduction potential of the cluster.752
While factors important in determining reduction potentials of [3/4Fe–4S] clusters have been found, their effects are not conclusive. It seems that different factors have different degrees of importance in different proteins. While surface charges seem not to be important in CpFd, they showed significant effects on the reduction potential in other proteins.753 Studies on CvFd showed that the two clusters have different reduction potentials, with one being extremely low, ∼−600 mV. Although it seems that the cluster with classical geometry should be the one with a normal reduction potential, thorough mutational and electrochemical studies on this protein proved it to be the other way.753
3.4.3.3.4. Spectroscopic Features
Proteins with more than one cluster are usually brown in color, with a broad absorption in the 380–400 nm region. Usually an R(390 nm)/Z(280 nm) ratio of more than 0.7 is observed for these proteins.9005 CD and MCD analyses showed that the [3Fe–4S] cluster of 7Fe proteins is protonated at acidic pH.539,746
The [4Fe–4S] clusters go from a 2Fe3+–2Fe2+ EPR-silent state (S = 0) to an Fe3+–3Fe2+ (S = 1/2) state with an EPR signal of around 1.96, while [3Fe–4S] clusters have an EPR signal with a feature at 2.01, going from [3Fe–4S]1+ to [3Fe–4S]0 (Figures 26 and 27). Although the EPR signals are similar between this class of ferredoxins and [2Fe–2S] ferredoxins, the relaxation time of the [2Fe–2S] clusters differs from that of the [4Fe–4S] clusters, with a common trend of [2Fe–2S] < [3Fe–4S] < [4Fe–4S]3+ < ferredoxin-type [4Fe–4S]1+. Therefore, the temperature dependence of the EPR signal can be used as a guide to the cluster type. However, care should be taken in analysis of the signals, because spin–spin interactions between clusters can lead to an enhanced relaxation time.754
Figure 26.

Representative spectra of [4Fe–4S] proteins. (a, left) UV–vis of the oxidized form. Reprinted with permission from ref (760) Copyright 2005 Springer-Verlag. (b, middle) EPR of the [4Fe–4S]1+ state. Reprinted with permission from ref (761). Copyright 1999 Elsevier. (c, right) Mössbauer of the [4Fe−4S]2+ cluster of the E. coli FNR protein, T = 4.2 K (top), and the [4Fe−4S]1+ cluster of E. coli sulfite reductase, T = 110 K (bottom). Reprinted with permission from ref (529). Copyright 1997 American Association for the Advancement of Science.
Figure 27.

Representative spectra of the [3Fe–4S] cluster. (a, left top) UV–vis of the oxidized form and (b, right) temperature-dependent EPR of the [3Fe–4S]1+ cluster. Reprinted with permission from ref (762). Copyright 2002 Elsevier. (c, left bottom) Mössbauer of the [3Fe–4S]1+ (top) and [3Fe–4S]0 (bottom) clusters . Reprinted with permission from ref (529). Copyright 1997 American Association for the Advancement of Science.
The [3Fe–4S] clusters have a Mössbauer spectrum with one quadruple doublet at δ = 0.27 mm/s, showing three equivalent Fe3+ sites in the oxidized state (Figure 27). The reduced form shows two doublets with a 1/2 ratio in intensity. The minor doublet at δ = 0.32 mm/s is assigned to Fe3+, while the major doublet at δ = 0.46 mm/s is attributed to a delocalized mixed-valence Fe2.5+ state.529 The Mössbauer features of the [4Fe–4S]2+ cluster have been discussed in detail in the section dealing with the spectroscopic features of HiPIPs (section 3.4.5.5).
NMR is one of the tools that has been extensively used to analyze [3/4Fe–4S] clusters. A higher number of total hyperfine shifted resonances in NMR can indicate the presence of more than one cluster in a given protein. Nine or twelve contact shifts are usually observed for [3Fe–4S] or [4Fe–4S] clusters, respectively. The [4Fe–4S] clusters are identified by the presence of peaks with anti-Curie temperature dependence, while Curie-type behavior is indicative of a [3Fe–4S] cluster. Typical 7Fe ferredoxins show five downfield peaks, two with Curie-temperature-dependent behavior. There are, however, 7Fe proteins with quite different NMR spectra and more downfield peaks. These 7Fe proteins usually have a short symmetric motif. A peak at 30.0 ppm is characteristic of mononuclear 3Fe clusters.736 In NMR studies of the [3Fe–4S] clusters, it has been shown that the contact shifts of His close to the conserved Asp are pH-dependent and correlate with the pKa of the Asp residue. Also, the effects of disulfide bonds in the shifts were studied.755 NMR of [4Fe–4S] clusters showed very similar shifts for all Cys residues in the oxidized form. Upon reduction, a similar pattern is observed for all [4Fe–4S] proteins, with two Cys residues showing Curie-like behavior (Fe2.5+) and two showing anti-Curie behavior (Fe2+). This also suggests that there are two isoforms with an Fe2.5+ pair on the Cys I/III or Cys II/IV pair. The former is more preferred, and this preference is stronger when a disulfide bond is present, as shown by NMR studies.755 The effects of other ligating residues were also analyzed in terms of NMR contact shift. NMR was also used to analyze the self-exchange rate and hence reorganization energy in ferredoxins.756 NMR studies provided structures of several ferredoxins such as [4Fe–4S] ferredoxin from Tt. maritima.757
The resonance Raman spectra of [4Fe–4S] ferredoxins can be explained without considering coupling between Fe–S and δ(S–C–C) modes. For these proteins at least seven ν(Fe–Sβ) bands and three ν(Fe–St) bands are observable, with a band at 340 cm–1 being the most prominent due to total symmetry of the cubane structure. Resonance Raman was also used to study Se complexes of ferredoxins as well as the presence of [3Fe–3S] clusters. Resonance Raman studies revealed the solvent accessibility of H-bonds to the cluster, the distorted D2d symmetry of the cluster, and Fe–S–Cα–Cβ torsion angles.618,758 NRVS was also used to study the dynamics and the oxidized and reduced states of the [4Fe–4S] cluster.759
3.4.3.4. Ferredoxin-like Proteins
A class of so-called plant ferredoxin-like proteins (PLFPs) has been discovered in the past few years. These proteins are known to play a role in several cellular processes. The first PFLP was discovered in sweet pepper. The protein consists of three domains: a N-terminal signal peptide, a [2Fe–2S] domain, and a casein kinase II phosphorylation (CK2P) site at the C-terminus. Phosphorylation of this domain is postulated to be important in resistance to pathogens in Arabidopsis thaliana,763 and PLFPs are evolved in plant defense mechanism pathways.
[4Fe–4S] ferredoxin-like proteins are also common and are found in some bacteria with a modified Cys-(Xxx)2-Cys-(Xxx)2-Cys-(Xxx)3-Cys motif at the N-terminus or Cys-(Xxx)2-Cys-(Xxx)8-Cys-(Xxx)3-Cys-(Xxx)5-Cys at the C-terminus. The ferredoxin-like protein in Rhizobium meliloti is shown to be important in nitrogen fixation. The protein is located in an operon with nif genes. Mutational analyses and molecular modeling showed the importance of extra amino acids in positioning the loop in a way that it could incorporate the cluster efficiently.764,765
A PLFP has been discovered in Erwinia carotovora that is regulated by quorum sensing. This ferredoxin has similarity to plant ferredoxins with no significant similarity to bacterial ferredoxins.766,767 PFLP genes in Helicobacter pylori and its corresponding ferredoxin reductase have been shown to be important in imparting metronidazole resistance to the bacteria.768 PFLPs are known to be important in enhancing plant resistance to bacterial pathogens. Transgenic expression of PFLP from sweet pepper in calla lily resulted in more resistance to soft rot bacterial diseases.769 The same transformation in tobacco, orchid, and rice plants enhanced their resistance to Xanthomonas oryzae pv. oryzae.767
3.4.4. Rieske Centers
3.4.4.1. Introduction/History
Rieske proteins are [2Fe–2S] iron–sulfur proteins that are distinguished by their unique His2-Cys2 ligation motif. The first example of these proteins was discovered by Rieske in 1964, who observed an EPR signal with g = 1.90 in the cytochrome bc1 complex (complex III of the mitochondrial electron transport chain770). Similar EPR signals were later observed in the b6f complex of the photosynthetic chain, the membrane of bacteria with a hydroquinone oxidizing ET chain, and soluble bacterial dioxygenases. The coordination environment was first established by ENDOR and ESEEM spectroscopy and further proved by the crystal structure.773 There have been multiple reports of the presence of several isoforms of Rieske proteins in the genome of prokaryotes. The presence of these isoforms most likely aids the organism to adapt better to environmental changes.771
3.4.4.2. Structural Aspects
3.4.4.2.1. Primary Structure/Amino Acid Sequence
The first Rieske protein to be sequenced was the Rieske protein from the bc1 complex of Neurospora crassa.772 Subsequently, other gene sequences of multiple Rieske proteins from a wide range of organisms have been obtained. Sequence alignment and analysis revealed a Cys-Xxx-His-(Xxx)15–47-Cys-Xxx-Xxx-His motif as the conserved motif for [2Fe–2S] ligands.773 On the basis of this sequence analysis, the proteins can be divided into Rieske and Rieske-type subcategories.
Rieske proteins can be found in bc complexes such as the bc1 complex of mitochondria and bacteria, the b6f complex of chloroplast, and corresponding subunits in menaquinone oxidizing bacteria. Three residues other than Fe–S ligands are also conserved in this class of Rieske proteins, two of which are cysteine residues that form a disulfide bond important in the stability of the protein,774 and the other is a Gly in a conserved Cys-Xxx-His-Xxx-Gly-Cys-(Xxx)12–44-Cys-Xxx-Cys-His motif. Mutational analysis of this class confirmed the presence of two histidines and four cysteines essential for cluster formation.775,776 Rieske proteins that are not part of the bc complex also belong to this class. Some of these proteins are within complexes that are not well identified, and some belong to organisms that are devoid of the bc complex, such as TRP from T. aquaticus and SoxF and SoxL from Sl. acidocaldarius.773,777
Rieske-type proteins are typically part of water-soluble dioxygenases. This class of proteins can be further divided into four separate groups. Bacterial Rieske-type ferredoxins are water-soluble ET proteins with a [2Fe–2S] cluster that show no similarity to common ferredoxins and share a conserved Cys-Xxx-His-(Xxx)16–17-Cys-Xxx-Xxx-His motif. They have diverse sequences, but their three-dimensional structures are very similar to those of other Rieske proteins. Bacterial Rieske-type oxygenases have a Rieske center and a mononuclear nonheme iron in their active site. In addition to four Rieske ligands, four other residues are conserved in these proteins, including two glycine residues, one tryptophan, and one arginine. Naphthalene dioxygenase (NDO) is the archetype of this class. Eukaryotic homologues of bacterial Rieske-type oxygenases also have a ligand set for Rieske coordination and a site for mononuclear nonheme iron. Choline monooxygenase and CMP-N-acetylneuraminic acid hydroxylase are examples of this class. Lastly, there are proteins that have a putative Rieske binding site, with a common motif of Cys-Pro-His-(Xxx)16-Cys-Pro-Xxx-His, but the presence of a Rieske cluster has not been confirmed in them yet.773
3.4.4.2.2. Three-Dimensional Structure/Crystallographic Analysis
Crystal structures of several Rieske proteins from different categories have been solved. All Rieske proteins share the so-called “Rieske fold”. This fold consists of three antiparallel β-sheets that form a double β-sandwich (Figure 28). Sheet 1 consists of three conserved strands, 1, 10, and 9. Strands 2, 3, and 4 form sheet 2, and strands 5–8 are in sheet 3. Sheet 2 is longer and interacts with both sheets 1 and 3. The interactions between sheets 2 and 1 are mostly of hydrophobic nature. Most conserved residues are found in the loop regions connecting the β-strands, especially loops β1−β2, β2−β3, and β8−β9 (the so-called “Pro loop”).91,773
Figure 28.
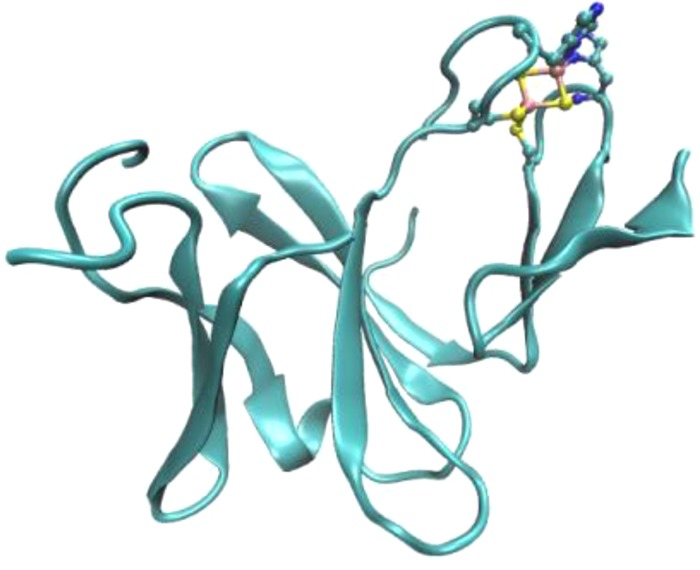
Minimal Rieske fold with three β-sheets and loops coordinating the [2Fe–2S] cluster with two His ligands and two Cys ligands (from PDB ID 1NDO).
The cluster binding subdomain is mainly located in sheet three and two of its adjacent loops (β4−β5 and β6−β7). Each loop provides one of the cysteine and histidine ligands, so the pattern is 2 + 2, in contrast to the 3 + 1 pattern observed in most ferredoxins. In mitochondrial and chloroplast Rieske proteins, there is a disulfide bridge that connects the loops in Rieske proteins. This disulfide bond is of prominent importance in maintaining structural integrity in these proteins because their loops are exposed to solvent. Rieske-type proteins do not have this conserved disulfide bridge. It has been argued that this difference is due to the fact that buried Rieske complexes are stable without the need to disulfide bond.773
Rieske proteins from bc1 or b6f complexes have an additional “Pro loop” with a highly conserved sequence of Gly-Pro-Ala-Gly that covers the cluster and has been shown to be critical for the stability.773,779 In most cases the Fe2+ is the one that is more surface-exposed, and it is this iron atom that has two exposed His ligands. In buried Rieske complexes such as NDO, the histidines are not solvent-exposed and usually form H-bonds with acidic side chains in the active site.780 The geometry of the Fe–S cluster is the same among all Rieske proteins, forming a distorted tetrahedral conformation. In contrast to the Cys ligands which impart a tetrahedral geometry, the His ligands accommodate a geometry that is closer to octahedral (Figure 29).773
Figure 29.

Structure of the bc1 complex from chicken (PDB ID 3H1J) and its Rieske protein and Rieske center (left) and structure of the b6f complex from Mastigocladus laminosus (PDB ID 1VF5) and its Rieske protein and Rieske center (right).
Multiple H-bonds constrain and stabilize the cysteine ligands, which are conserved between most bc1 and b6f Rieske proteins. They are three bonds with sulfur S1, two with sulfur S2, two with Sy of cysteine in loop 1, and 1 with Sy of loop 2. Usually there are H-bonds between the sulfurs of coordinating cysteines and the main chain nitrogen of residue i + 2. These H-bonds are known to stabilize type I turns. Two of these H-bonds are of the OH···S type, one from a conserved Ser to the bridging S1 and one from a conserved Tyr to the Cys in loop 1. Rieske proteins from menaquinol oxidizing organisms lack this Ser···Cys H-bond. Rieske-type proteins lack three of these conserved H-bonds due to a lack of the conserved Ser and Tyr. Multiple site-directed mutagenesis studies confirmed the importance of these two H-bonds in maintaining the high reduction potential of Rieske proteins.773,781
Despite the high degree of structural similarity between different Rieske and Rieske-type proteins, each category has its unique features. It seems that although the cluster-binding site and the minimal Rieske fold are highly conserved among all classes of Rieske and Rieske-type proteins, there are multiple insertions between elements of this minimal fold, mainly in loop regions. These significant differences make sequence alignments of Rieske proteins controversial, compared to their ribosomal RNA alignments.782 Rieske-type ferredoxins have the closest structure to the minimal fold. Rieske proteins from the b6f complex usually have a C-terminal extension that is known to be important in stabilizing the open conformation required for the activity. The same role was proposed for helix–loop insertion in mitochondrial Rieske proteins. Chloroplast Rieske proteins also show a distortion in the β-sheets, forming a β-barrel rather than a β-sandwich.773 Novel disulfide bonds have been reported at the C-terminus of a thermophilic Rieske protein from Acidianus ambivalence that are reported to be important in higher stability of the protein.783 A disulfide bond and extended C-terminal region insertion have been observed in archaeal Rieske proteins.784 Some acidophilic proteins have extended β-strands in their cluster binding domain. The peptide bond orientation differs in the Pro loop of bc1 and b6f complexes in regard to the cis or trans configuration.773 Some Rieske proteins have a very long loop in place of the Pro loop that is important for interacting with redox partners.785 Although the pattern of H-bonding and salt bridges is similar, it is not identical, and the residues that are involved are not conserved.773 Another difference between Rieske proteins lies in their surface charge distribution. These differences are required for interactions with different redox partners. Different charge distribution also reflects the variation of pH in which the proteins work, as exemplified by a net negative charge on the surface of some acidophilic proteins.786
The Rieske fold and the geometry of the cluster are unique to Rieske and Rieske-type proteins and differ significantly from those of the other class of [2Fe–2S] ferredoxins. The most similar geometries are those of rubredoxins and the zinc-ribbon domain, suggesting that the Rieske fold may have arisen from a mononuclear ancestral fold.91
3.4.4.3. Function
3.4.4.3.1. Rieske Clusters: Cytochrome bc Complexes
Mitochondrial bc1 complexes and chloroplast b6f complexes are multisubunit proteins with four redox centers organized in three subunits: two heme b centers in a transmembrane domain of cytochrome b, cytochrome c1/f, and the Rieske iron–sulfur protein. All of them oxidize hydroquinone (ubihydroquinone/plastohydroquinone) and transfer electrons to either cytochrome c or plastocyanin, generating a proton gradient across the membrane through the Q-cycle. For proper function of this cycle, the hydroquinone oxidation reaction is strictly coupled. The Rieske protein is responsible for hydroquinone oxidation and acts as the first electron acceptor. Electron transfer is accomplished by direct interaction between the exposed His ligand and the quinone substrate.787 Since the function of the Fe–S cluster in these protein complexes is tied to hemes, a more detailed explanation will be presented in section 5.
3.4.4.3.2. Rieske-Type Clusters: Dioxygenases
Rieske-type clusters are part of aromatic ring hydroxylating dioxygenase enzymes that catalyze the conversion of aromatic compounds to cis-arenediols, a key step in aerobic degradation of aromatic compounds.788 Dioxygenases contain a reductase, a terminal oxygenase, and often a [2Fe–2S] ferredoxin. The reductase part can be of two types: ferredoxin–NADP or glutathione. The oxygenase part contains a Rieske center and a mononuclear nonheme iron center (Figure 30). The Rieske center transfers an electron from ferredoxin or reductase to the iron center.788 Although these two centers are in different domains that are far apart in a single subunit (45 Å), the quaternary structure with 3-fold symmetry will bring them to a close distance within 12 Å. In most cases the His ligand of the Rieske center and one of the His ligands of iron are bridged by an Asp residue, ensuring the rapid ET between the two centers (Figure 31). The removal of this conserved Asp abolishes the activity without changing the metalation.789−791 In the case of 2-oxoquinoline monooxygenase, the Asp changes its position after reduction of the Rieske center to H-bond with a His ligand that is protonated upon reduction. This repositioning will cause a conformational change that results in generation of a 6-coordinated iron geometry which is more active.792 It has also been suggested that the H-bonds provided by this Asp can help the Rieske center and catalytic center to sense the redox state and ligand state of each other. Mutational studies have been implemented to discover sites that are important in specific interactions between these Rieske centers and their redox partners.793
Figure 30.
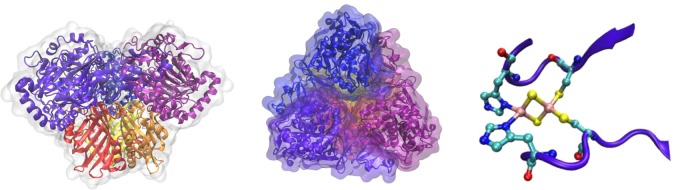
Structure of naphthalene 1,2-dioxygenase (PDB ID 1NDO), the archetype of Rieske-type proteins from two different views, and a close-up of the active site Rieske center.
Figure 31.
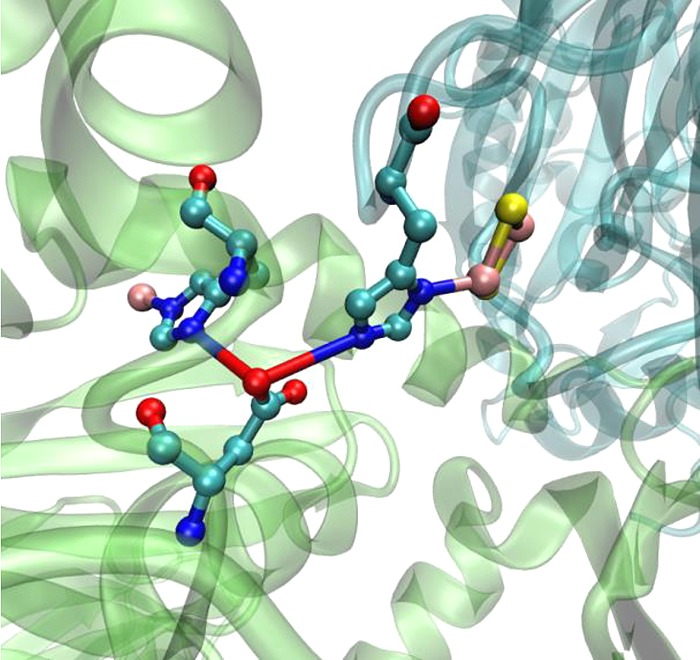
Interface between two monomers of naphthalene dioxygenase, NDO. Asp205 from the polypeptide chain on the left bridges two His residues that are ligands to the Fe–S cluster and catalytic nonheme iron center (PDB ID 1NDO).
3.4.4.4. Important Structural Elements
As with any other ET centers, the reduction potential of Rieske centers is one of the most important factors in determining its ET rate and conveying its activity.794 Any changes in the reduction potential of Rieske and Rieske-type proteins have been shown to affect their activity and the kinetics of the ET between these centers and their redox partners. Reduction potentials of Rieske centers vary in a wide range of −100 to +490 mV, which is significantly higher than the average reduction potentials of ferredoxins. In general, any factor that selectively stabilizes either the reduced or oxidized state of a Rieske center will influence its reduction potential.773 The difference between the overall charge of the cluster (0/–1 in the case of Rieske proteins vs −2/–3 in the case of ferredoxins) and electronegativity of the ligands (histidine vs cysteine) is the main reason for the higher reduction potential of Rieske proteins. Different H-bonds to bridging or terminal sulfurs and solvent exposure of the clusters are the main determinants of different reduction potentials within the Rieske family. The reduction potential range differs depending on the type of Rieske complex: 265–310 mV in the bc1 complex and around 320 mV in the b6f complex. The reduction potentials of menahydroquinone oxidizing complexes are 150 mV lower than that of the ubihydroquinone bc1 complex (the same difference that is observed between the two types of quinones).773 This lower reduction potential has been attributed to a lack of a H-bond donated from a conserved Ser, which is absent in the former class of Rieske proteins. Different methods of reduction potential measurement have been applied to Rieske proteins, such as chemical redox titration monitored by EPR795 or CD796 and direct cyclic voltammetry,797,798 which enables measurement of thermodynamic parameters.780 CV experiments also showed for the first time the second reduction step to a 2Fe2+ state at very low reduction potentials (∼−840 mV).797
Computational studies showed that the cluster distortions caused by the protein environment play a prominent role in tuning the reduction potential of the center. Accordingly, using active site structures determined from x-ray crystallography will result in calculations that agree much better with experimental values than idealized structures.800
An interesting feature of Rieske proteins is their pH-dependent reduction potential, which decreases with increasing pH and is attributed to deprotonation of a group in contact with the Rieske complex.773,801 This phenomenon can be observed in the oxidized state where the pKa values of one of the His ligands are near physiological pH (two pKa values of 7.8 and 9.6 vs one pKa of around 12.5 in the reduced state802). This pH dependence can be important in interactions and binding of Rieske proteins to their redox partners. Moreover, this redox-dependent ionization may be very important for their physiological function, as these proteins are part of proton-coupled ET systems. The biomimetic models of Rieske clusters prove the dependence of the reduction potential of the center on the protonation state of its His ligands.803 Shifts in the UV–vis absorption peaks and CD features upon pH titration are consistent with the two protonation states of the oxidized form.804 Several studies have shown that multiple inhibitors can bind to the sites close to the cluster and affect the reduction potential of the site.787,805,806
In a related study, diethyl pyrocarbonate (DEPC) was used to react with and trap deprotonated His. Addition of this ligand caused reduction of the cluster as well as an increase in the overall reduction potential, a phenomenon that was observed in the case of inhibitors such as stigmatellin, immobilizing it in the b conformation. Moreover, if the protein was reduced first, no addition would be observed, due to a lack of available deprotonated His.806,807 Analysis of some pH-independent low reduction potential Rieske proteins suggests that the coupling between the cluster oxidation state and the His protonation state also has a role in determining the reduction potential of the cluster.808
The reduction potentials of Rieske-type clusters are lower than those of Rieske clusters, with values around −150 to −100 mV.773,780,788 One reason for this difference is a lack of three out of eight conserved H-bonds of Rieske proteins in Rieske-type proteins (Figure 32).781 Reduction potential of Rieske-type proteins is pH-independent due to less solvent accessibility in comparison to Rieske proteins.809,810 There are examples of Rieske-type proteins that have very similar active site structure to Rieske centers, but different loop orientations cause disruption of the H-bonding network, resulting in proteins with reduction potentials around −150 mV.810 A Rieske-type ferredoxin has been found with a reduction potential around 170 mV. The higher reduction potential in this Rieske-type protein has been attributed to the presence of amino acid substitutions in positions around the metal center.795
Figure 32.

Differences in the H-bond pattern between the Rieske fragment of naphthalene dioxygenase, NDO (PDB ID 2NDO), the water-soluble Rieske fragment of the bc1 complex, ISF (PDB ID 1RIE), and the Rieske fragment from the b6f complex, RFS (PDB ID 1RFS). Reprinted with permission from ref (773). Copyright 1999 Elsevier.
The most important residues involved in the H-bonding network in Rieske proteins are a conserved serine and a conserved tyrosine. It has been suggested that this H-bond network stabilizes the reduced state by charge delocalization, thereby increasing the reduction potential.773,811 The electrostatic environment of the protein is another feature that can influence the reduction potential, meaning that the presence of charged residues on their own can increase the reduction potential of the center. In one study, removal of negatively charged residues in the vicinity of the Rieske center in Rieske ferredoxin from biphenyl dioxygenase of Burkholderia sp. resulted in a pKa of the His ligands similar to that of mitochondrial Rieske proteins.812
Mutational analyses have been extensively used to reveal features that are important in tuning the reduction potential. Gly143Asp, Pro146Leu, and Pro159Leu mutations in the Pro loop resulted in a shift of about 50–100 mV toward more negative reduction potentials, mostly due to distortion in the Fe–S environment and changes in the H-bond network around it.773 The cluster content was decreased to 32–70% in these mutants. Another study showed that mutations in the loop containing Fe-S ligands are the ones that alter reduction potential.779
Several site-directed mutations were made with the goal of understanding the role of H-bonds from conserved Ser and Tyr in different organisms.781,813,814,817 Mutations of Ser to Ala and Tyr to Phe both decreased the reduction potential.781,815 When both mutations were made, the effects on the reduction potential were observed to be additive. It was shown that these mutations do not influence the stability of the cluster or its interaction with quinone. However, the activity was decreased, demonstrating the importance of the reduction potential in hydroquinone oxidation activity.9007 These mutations also increased the pKa values of the His ligands. Different effects were observed when these two residues were mutated into other amino acids. Mutations of Tyr to nonphenolic amino acids targeted the Rieske protein to cytosolic proteolytic cleavage machinery. A Ser to Cys mutation resulted in expression of proteins that could no longer incorporate a Rieske cluster, and in cases where it could, a slight increase in the reduction potential was observed. A Ser to Thr mutation resulted in a protein with moderate changes in the midpoint potential.781
Mutations of a conserved Thr that packs tightly against the Pro loop resulted in a lower reduction potential and a significant decrease in the activity.774 Mutations of a conserved Leu residue that is supposed to protect the cluster from solvent were analyzed as well.816,817 Leu136Gly/Asp/Arg/His mutants were analyzed and showed low activity and altered reduction potential. Replacement of Leu with a neutral residue such as Ala caused a similar change in both reduction potential and pKa values of the His ligands, suggesting a causative effect of a change in water accessibility.816 Mutation to a negative residue such as Asp has marginal effects on the reduction potential, probably due to movement of the Asp side chain from His and its solvation. However, placing a positive charge here resulted in a significant increase in the reduction potential.817
Several mutations in a flexible linker distant from the cluster binding site have been shown to increase the reduction potential.773 Mutations in a hinge region were shown to increase the Em of the Rieske center of Rb. capsulatus. These mutations affect the reduction potential in two ways: by altering the interaction mode with quinone, which is known to affect the reduction potential, and by altering the positioning of the [2Fe−2S]-containing domain of the Rieske protein, which can impart changes in both the reduction potential and the EPR signal shape.818 Mutations in the residues involved in disulfide bridge formation also showed decreased reduction potential values. This lower reduction potential is mainly due to removal of polarizable Cys groups and disturbance of the loop conformation and pattern of H-bonds.817,819 Analyses of a protein with a reduced disulfide also showed a small decrease in the reduction potential that was attributed mainly to changes in the H-bonding pattern and enthalpic effects.820
Similar mutational studies of conserved residues close to the cluster binding domain of Rieske-type proteins have also been performed, showing different effects depending on the mutation type. Mutations of a conserved Asp residue in Rieske oxygenase resulted in a lower reduction potential mainly due to deprotonation of a His ligand caused by loss of a H-bond from Asp.822
Another important factor in determining the reduction potential is the condition in which the protein performs its function. Studies on extremophilic organisms revealed that Rieske centers from acidophilic organisms have more positive midpoint potentials than neutral centers whereas the potentials of acidophilic Rieske centers are significantly lower than the expected value. Interestingly, the pKa of the His ligand also shifted correspondingly in these extremophilic organisms.786,823
It should be noted that there are exceptions to these general statements. There are high reduction potential Rieske proteins, such as sulredoxin, which lacks the hydroxyl group responsible for redox modulation and shows a different pH-dependent redox response compared to other high reduction potential Rieske proteins.824
3.4.4.5. Spectroscopic Features of Rieske and Rieske-Type Proteins
As with other Fe–S proteins, Rieske proteins have broad absorption spectra resulting from overlapping bands from S → Fe3+ charge transfer (Figure 33). CD and MCD spectroscopic techniques were used to deconvolute some of these spectra. In their oxidized form, Rieske proteins have absorptions at 325 and 458 nm and a shoulder around 560–580 nm. Upon reduction, the position of the bands shifts to 380–383, 425–432, and 505–550 nm and the intensity of the bands drops by 50%. The CD spectrum of Rieske proteins has features that are unique among Fe–S proteins, showing two positive bands between 310 and 350 nm, a negative band at 375–380 nm, and a set of positive bands between 400 and 500 nm in the oxidized form. In the reduced form, the CD spectrum shows a positive band at 314 nm, a negative band at 384–390 nm, a negative band at 500 nm, and a band at 760 nm.773 These bands are attributed to d–d transitions of Fe2+ from the lowest lying d orbital into t2g sets. The strong negative band at 500 nm in the reduced state is an indicator of the redox state even in the presence of other cofactors such as heme.809 The CD spectrum of oxidized Rieske proteins is pH-dependent in near-UV and visible regions due to the presence of some deprotonation events.804 Rieske proteins show temperature-dependent MCD spectra with multiple positive and negative bands in the reduced state, but the intense negative band at 300–350 nm and positive band at 275 nm, which is observed in rubredoxins and [2Fe–2S] ferredoxins, is not visible in them due to a blue shift of the bands to higher energies because of the nitrogen ligation from the His ligand.773
Figure 33.

Representative spectra of Rieske centers. (a) UV–vis of the reduced (lower spectrum) and oxidized (upper spectrum) forms. Reprinted with permission from ref (866). Copyright 2004 National Academy of Sciences. (b) EPR of the reduced form. Reprinted with permission from ref (867). Copyright 2007 National Academy of Sciences. (c) Mössbauer of the [2Fe−2S]+ cluster of the Rieske protein from Ps. mendocina at T = 200 K. Reprinted with permission from ref (529). Copyright 1997 American Association for the Advancement of Science.
Mössbauer studies of Rieske proteins show a temperature-independent four-line spectrum resulting from two quadruple doublets of the same intensity (Figure 33). The spectrum of the reduced form is very similar to that of ferredoxins with a more positively shifted δ (0.68 mm/s at 200 K), which is due to the less electron-donating nature of the His ligands.529,778,847 While the Fe3+ state shows quite isotropic features, the Fe2+ state has an anisotropic A tensor. The electric field gradient tensor is symmetric around x axis of the A tensor for Fe2+, with the largest component being positive.773
Resonance Raman studies of Rieske proteins using laser excitation at different wavelengths showed features very similar to those of ferredoxins in both the reduced and oxidized states, with some shifts in the bands and additional vibrations due to the presence of the His ligands.848 The higher number of bands in the 250–450 cm–1 region is an indicator of a lower symmetry of the Rieske proteins than those of all cysteinate [2Fe–2S] ferredoxins (C2v vs D2h or C2h symmetry). Rieske proteins feature a weak peak at 266–270 cm–1 that is assigned to the Fe(III)–N(His) stretching mode, which is thought to have some Fe–Fe mixing character. The peak is shifted 8 cm–1 up in more basic pH, consistent with deprotonation of the His ligand. The peaks at 260–261 cm–1 are assigned to Fe–His bending modes and are also very sensitive to 15N substitution. A peak at 357–360 cm–1 corresponds mainly to Fe(III)–Sterminal stretching (B2t) mode.848 This peak is very similar to that of ferredoxins, only upshifted due to either a different H-bond pattern or Fe–Sγ–Cα–Cβ dihedral angles, which is a sign of similar Fe3+ environments in the two classes of proteins. This peak can be observed at 319–328 cm–1 after reduction.725 pH-dependent studies in the 250–450 cm–1 region show that there are no resonance Raman-detectable changes at the pKa of the first His ligand and changes are only observed above the pKa of the second His ligand. These changes arise, however, from additional factors such as protonation of some amide backbones and not solely in regions related to the Fe–Nimid vibrational frequency. A lack of changes at physiological pH can ensure rapid proton-coupled ET.849 No significant change was observed for Rieske-type proteins. Most resonance Raman features are due to the Fe–S stretch. The kinematic coupling observed by resonance Raman and rigidity of the H-bond network around the cluster help minimize the reorganization energy and hence facilitate ET.850 resonance Raman studies were also performed to analyze the role of the H-bonding network in Rieske proteins. It has been shown that the presence or removal of the S···Tyr H-bond shows significant changes in resonance Raman bands at 320–400 cm–1, whereas removal of the S···Ser H-bond does not show a detectable resonance Raman change.849
XAS analysis showed very similar geometries of clusters in Rieske proteins and ferredoxins and also indicated the contraction of the site upon oxidation. Early XAS analyses were hampered by the fact that the presence of His ligands was not known. XAS studies of Rieske oxygenases showed a small but significant change in the Fe–S bond length upon reduction. A larger increase in the Fe–Nimid bond distance (0.1 A) was observed through reduction, which can facilitate ET between the Rieske center and its redox partner. The edge feature has a shift toward lower energies upon reduction.847,851
EPR spectroscopy is one of the first techniques used to identify Fe–S proteins. The g values of Rieske proteins are significantly lower than those of ferredoxins (1.9–1.91 vs 1.945–1.975) due to the presence of nitrogen ligands (Figure 33). This EPR signal is mainly due to Fe3+ and its His ligands and environment.773 EPR signals vary significantly among different groups of Rieske proteins, with gz = 2.008–2.042, gy = 1.888–1.92, and gx = 1.72–1.834. The rhombicity changes between 51% in the z axis and 100–59% in the x axis.773 In Rieske proteins all g values correlate with rhombicity, indicating that EPR properties are influenced mainly by the protein environment. Changes in the EPR signal upon binding to quinone or inhibitors will change the shape of the EPR signal and g values. These effects can also be correlated to rhombicity parameters.773 An EPR study of a Rieske protein at pH 14 showed increased g values with broadened features. The appearance of these new features can be assigned to a decrease in the energy difference between reductions of the Fe with two His ligands and the one with two Cys ligands due to deprotonation of both His ligands.852
ENDOR and ESEEM studies support the presence of two nitrogen ligands in both Rieske and Rieske-type proteins.853 Studies with 15N-labeled protein further support the presence of nitrogen ligands.845,854−858 X-band 14N hyperfine sub-level correlation (HYSCORE) spectroscopy of reduced Rieske and Rieske-type proteins is dominated by two histidine Nd ligands with hyperfine couplings of ∼4–5 MHz. A combination of site-specific 14/15N labeling together with orientation-selective HYSCORE studies was used to gain more insight into the nature of the H-bonding network around the cluster and through-bond electrostatic effects.814 ESEEM studies coupled with isotope exchange with H2O were used to understand the proton environment around Rieske proteins from Rb. sphaeroides.859 The magnetic and structural features of the Cys and His ligand protons and the protons involved in the H-bonding network were analyzed.8591H ENDOR analysis of the Rieske proteins from the bovine mitochondrial bc1 complex showed three peaks from orientation behavior: two from β protons of Cys ligands and one from the β proton of the His141 ligand. The direction of gmax lies in the FeS plane with the largest proton coupling along gint.2006
NMR studies have been applied to different Rieske and Rieske-type proteins.861,862 Cysteines coordinated to Fe3+ show four strongly downshifted signals between 50 and 110 ppm. Temperature-dependent studies of Hβ protons of the cysteines show that they follow Curie law. Hε1 of one of the histidine ligands shows a sharp resonance at 25 ppm, showing a weak Curie-temperature-dependent behavior. There are still complications in assigning all the resonances in NMR spectra due to the unique features of Rieske NMR. NMR studies were used to monitor the H-bonding patterns863 and solvent accessibility.864 NMR studies on Rieske proteins from T. thermophilus revealed slight conformational changes that are dependent on both the oxidation state and ligand binding. 1H, 15N, and 13C NMR analyses showed that two of the observable prolyl backbones change from the trans to the cis mode upon reduction.865
3.4.5. HiPIPs
3.4.5.1. Introduction/History
HiPIPs are a well-defined superfamily of Fe–S proteins found mainly in photosynthetic anaerobic bacteria, although proteins from aerobic bacteria have also been reported. HiPIPs were expressed in both aerobic and anaerobic conditions.868 HiPIPs contain a [4Fe–4S] cluster as with ferredoxins. However, the higher reduction potential of HiPIPs results in one less electron in both the reduced and oxidized states of these proteins compared to ferredoxins, meaning a [4Fe–4S]2+/3+ state.872,873
3.4.5.2. Structural Aspects
HiPIPs are usually small proteins (6–11 kDa). The [4Fe–4S] cluster is embedded within a characteristic fold of HiPIPs. HiPIPs are highly charged, either acidic or basic depending on the organism from which they have been purified. Despite low sequence homology, the structures of all HiPIPs share similar features, especially in loop regions. HiPIPs were the first iron–sulfur proteins for which a crystal structure in both the oxidized and reduced forms was obtained. The small size of the protein requires that the [4Fe–4S] cluster occupies a large portion of the total volume of the protein. Their structures mainly consist of loops with two small α-helices and five β-strands. The cluster is positioned in the C-terminal domain of the protein (Figure 34). A conserved Tyr in most HiPIPs is located in a small helix in N-terminal packs against the cluster and interacts with one of the inorganic sulfurs, S3. Two of the Cys ligands are in two β-strands in a twisted β-sheet, and two hairpins provide the other two. Three of the four cysteines form H-bonds with the backbone amides of residues i + 2. Aromatic side chains from a C-terminal loop together with the conserved Tyr from the N-terminal form a hydrophobic pocket that further shields the cluster from solvent. HiPIPs share the consensus motif of Cys-(Xxx)2-Cys-(Xxx)8–16-Cys-(Xxx)10–13-Gly-Trp/Tyr-Cys to coordinate the [4Fe–4S] cluster. Several loops around the protein make a hydrophobic pocket for the protein to accommodate the cluster. In some cases conserved water ligands have been shown to be important for stabilizing the structure.870
Figure 34.
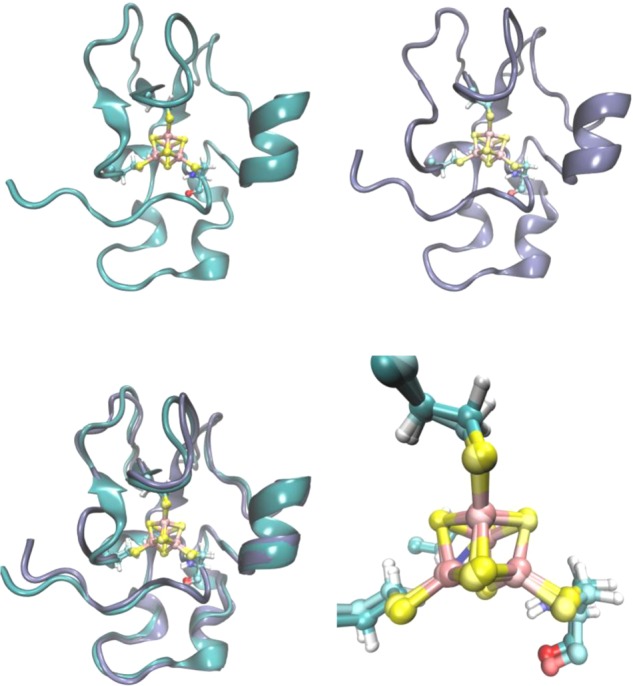
Structure of reduced (PDB ID 1HRR) and oxidized (PDB ID 1NER) HiPIP from Ch. vinosum (top left and top right, respectively). The overlay of the structures and zoom-in of the Fe–S cluster are shown at the bottom. As shown, only slight structural changes occurred upon reduction.
The [4Fe–4S] cluster, as with ferredoxins, has a cubane structure in which each iron is coordinated with three inorganic sulfurs and one thiolate from cysteine. All the irons have tetrahedral geometry. Fe–Fe distances are significantly shorter than S–S distances (2.72 vs 3.58 Å), resulting in lower accessibility to the iron atoms. The spin coupling between pairs of irons leads to Jahn–Teller distortion and a D2d state rather than a Td point group symmetry. There is also a conserved Gly close to the conserved Tyr in most HiPIPs, which is believed to mainly play a role in steric control.9008
Mutational analysis of conserved aromatic residues in HiPIPs confirmed a protective role for these residues against hydrolysis of the cluster by decreasing solvent accessibility.715 Removal of this protection resulted in degradation of the cluster through a [3Fe–4S] intermediate as evidenced by heteronuclear multiple quantum coherence (HMQC) NMR.875 Some HiPIPs form higher quaternary structures; HiPIP from Tb. ferrooxidans, for example, was isolated in a tetrameric state.874 There are several aromatic residues in close proximity to the Fe–S cluster in HiPIPs. These residues have been hypothesized to play a role in ET, reduction potential determination, and cluster stability. Several mutational studies suggest that these residues play a major supportive role against degradation.873,875,877
3.4.5.3. Function
The HiPIPs appear to be unique to the bacterial kingdom, and higher organisms replaced them by other more sophisticated ET proteins. Despite thorough characterization of these proteins, their function is not yet fully understood. HiPIPs act as soluble periplasmic electron carriers in photosynthetic bacteria between the photosynthetic reaction center and the cytochrome bc1 complex. Other functions have been reported, such as an iron oxidizing enzyme in Acidithiobacillus ferrooxidans,878 an electron donor to cytochrome cd-type nitrate reductase in Paracoccus(868) species or to terminal oxidases in Rhodothermus marinus,879 or a role in thiosulfate oxidation.880 The relative distribution of HiPIPs and their redox behavior suggest an overlapping role of these proteins with cytochrome c2 as a final electron acceptor in the photocycle.872 However, other studies have shown a role for HiPIPs distinct from that of cytochrome c.881 HiPIPs are also found in the membrane of some thermophilic organisms.879 HiPIPs are mainly found in organisms with a photosynthetic reaction center having a tetraheme cytochrome (THC) subunit. Multiple studies have shown that HiPIPs could be the preferred electron carrier in purple sulfur bacteria. Crystal structure analysis, molecular docking studies, and computational modeling have suggested that the hydrophobic patch of HiPIPs can interact with a hydrophobic patch in THC so that it plays a role as a redox partner to this protein.873,882,883
3.4.5.4. Important Structural Elements
HiPIPs have three ferric ions and one ferrous ion that occur as a pair of two Fe3+ ions and a pair of two Fe2.5+ ions. In the reduced state, the cluster has two ferric and two ferrous ions, mainly existing as a set of mixed-valence Fe2.5+.543,884 The reduction potentials of HiPIPs are very high, occupying a range of 100–500 mV. Several methods have been applied to measure the reduction potential of HiPIPs, including redox titration monitored by EPR,879,881 chemical redox titration,876 and direct electrochemistry.715,799 Some studies have suggested further delineation of HiPIPs into two categories: the first with a narrow reduction potential range of around 330 mV and the second with a broader range that depends on protein charges. However, only a few studies currently support this classification.872,885
Two classes of factors should be considered while studying the reduction potential of HiPIPs. The first class includes factors that differentiate the reduction potentials of HiPIPs from those of ferredoxins. The main explanation for the difference in reduction potential between the HiPIPs and ferredoxins has been well established now as the different redox states employed by the two proteins. While the ferredoxins go through a [4Fe–4S]1+/2+ transition, the HiPIPs have a [4Fe–4S]2+/3+ state. This oxidation state has an intrinsically higher reduction potential.719 It has been reported, however, that HiPIPs can form a super-reduced state of [4Fe–4S]1+ if unfolded in 80% Me2SO or by pulse radiolysis. The reduction potential of this [4Fe–4S]2+/1+ state was calculated to be 400–600 mV lower than that of the same pair in ferredoxins.886 There are studies in support of the importance of the overall structural and backbone conformation in determining the overall potential range of the protein.887 Also, these studies demonstrated the role of the protein environment in ET not only by manipulating the driving force and reduction potential but also through changing the activation energy via environmental reorganization.887 Resonance Raman, X-ray crystal structure analysis, computational analysis, and spin echo studies have all revealed an important role of solvent accessibility in the higher reduction potential of HiPIPs vs ferredoxins.872,873 Moreover, crystal structure analyses of HiPIPs have revealed conserved NHamide···S H-bonds to the coordinating sulfurs.719,872,873 These H-bonds stabilize the reduced form of the protein by decreasing the electron density on sulfurs, thereby increasing the reduction potential. This effect was demonstrated by using chemically synthesized peptides in which the peptide amide bond was replaced with an ester linkage, thus removing the H-bond between the amide and Cys sulfu.888 Ferredoxins have more of these amide H-bonds, resulting in the alternate oxidation state of the [4Fe–4S] cluster (Table 6).93,617,618,719,887 When elongated or compressed, the [4Fe–4S] cubanes have different spin topologies; however, sulfur K-edge XAS, 2D NMR, and DFT calculations have shown that the orientation of [Fe2S2]+ subclusters is very similar in both ferredoxins and HiPIPs, suggesting a localized oxidation or reduction in only one of the two subclusters889 and making cluster spin topology an unlikely source of redox-state differentiation.
Table 6. Reduction Potential of Different Rieske and Rieske-Type Proteinsa.
| protein | organism | Em (mV) | ref |
|---|---|---|---|
| Rieske Proteins | |||
| bc1 complex | pigeon heart | 285 | (825) |
| bc1 complex | beef heart | 290 | (806) |
| bc1 complex | beef heart | 304 | (826) |
| bc1 complex | beef heart | 312 | (798) |
| bc1 complex | beef heart | 306 | (827) |
| bc1 complex | beef heart | 315 | (828) |
| bc1 complex | yeast | 262 | (779) |
| bc1 complex | yeast | 286 | (829) |
| bc1 complex | yeast | 285 | (781) |
| bc1 complex | Paracoccus denitrificans | 298 | (815) |
| bc1 complex | Paracoccus denitrificans | 280 | (830) |
| bc1 complex | Rhodobacter capsulatus | 310 | (831) |
| bc1 complex | Rhodobacter capsulatus | 321 | (832) |
| bc1 complex | Rhodobacter capsulatus | 294 | (832) |
| bc1 complex | Rhodobacter sphaeroides | 285 | (831) |
| bc1 complex | Rhodobacter sphaeroides | 300 | (796) |
| bc1 complex | Rhodobacter sphaeroides | 300 | (796) |
| bc1 complex | Chromatium vinosum | 285 | (833) |
| b6f complex | spinach | 320 | (834) |
| b6f complex | spinach | 375 | (835) |
| b6f complex | spinach | 320 | (835) |
| bc1 complex | Nostoc | 321 | (836) |
| bc complex | Chlorobium limicola | 160 | (837) |
| bc complex | Bacillus alcalophilus | 150 | (838) |
| bc complex | Heliobacterium chlorum | 120 | (839) |
| bc complex | Bacillus PS3 | 165 | (837) |
| bc complex | Bacillus firmus | 105 | (840) |
| Rieske protein | Thermus thermophilus | 140 | (841) |
| SoxFII | Sulfolobus acidocaldarius | 375 | (842) |
| Rieske-Type Proteins | |||
| FdBED | Pseudomonas putida | –155 | (843) |
| FdBED | Pseudomonas putida | –156 | (844) |
| FdBED | Pseudomonas putida | –155 | (809) |
| benzene dioxygenase | Pseudomonas putida | –112 | (843) |
| 2-halobenzoate 1,2-dioxygenase | Burkholderia cepacia | –125 | (845) |
| 2-oxo-1,2-dihydoquinoline 8-monooxygenase | Burkholderia cepacia | –100 | (846) |
Reprinted with permission from ref (773). Copyright 1999 Elsevier.
Specific interactions between hydrophobic residues are also considered a source of variation in reduction potential between HiPIPs and ferredoxins. While in HiPIPs aromatic···S interactions are through face of the aromatic ring, leading to interactions between the highest occupied orbital of the cluster and the lowest unoccupied Tyr orbital, ferredoxins have an interaction via the edge of Tyr with the highest occupied Tyr orbital interacting with the lowest unoccupied cluster orbital.872 Some studies have suggested that the main role of the conserved Tyr is to stabilize the cluster through these aromatic and H-bond interactions and not to have any profound effect on the reduction potential;877 however, because the Tyr in different proteins tends to take a different alignment, this hypothesis cannot be generalized to all HiPIPs.540
The second class of factors of important influence on the reduction potential of HiPIPs includes interactions that fine-tune the reduction potential. This class has not yet been fully elucidated; however, solvation and net charges on the protein are postulated to play a role in this class of proteins.220,885,890,891 No correlation was found between the orientation of aromatic residues in the protein and its reduction potential.892 Different factors including the net surface charge of the protein, partial charges of certain residues, atomic polarizability of protein atoms, and solvent dipoles have been thoroughly studied in a number of HiPIPs, and the only factor determined to correlate with the reduction potential was the net charge on the protein surface (Table 7).873,890
Table 7. Effect of the Net Charge on the Reduction Potential of Some HiPIPsa.
| protein source | Em (mV) | net charge | ref |
|---|---|---|---|
| Chromatium purpuratum | 390 | (902) | |
| Chromatium tepidum | 323 | –4 | (903) |
| Thiocapsa roseopersicina | 346 or 325 | –6 | (904) |
| (905) | |||
| (906) | |||
| (907) | |||
| Chromatium warmingii Bart | 355 | –4 | (908) |
| Chromatium uinosum | 356 | –5 | (909) |
| Chromatium gracile | 350 | –7 | (906) |
| (910) | |||
| Thiocapsa pfennigii | 350 | –9 | (911) |
| Ectothiorhodospira halophile | 120 (iso I) | –12 | (896) |
| (912) | |||
| (913) | |||
| Ectothiorhodospira uacuolata | 260 (iso I), 150 (iso II) | –5 (iso I), −8 (iso II) | (914) |
| (896) | |||
| Ectothiorhodospira shaposhnikouii | 270 (iso I), 155 (iso II) | –6 (iso I), −8 (iso II) | (914) |
| Rhodoferar fermentans | 351 | (915) | |
| (882) | |||
| (916) | |||
| Rhodopila globiformis | 450 | –3 | (917) |
| (896) | |||
| Rhodospirillum salinarum | 265 (iso I) | –5 (iso I), −1 (iso II) | (914) |
| (918) | |||
| Rhodopseudomonas marina | 345 | 5 | (918) |
| Rhodocyclus tenuis | 300 | 2 | (914) |
| (917) | |||
| (919) | |||
| Rhodocyclus gelatinosus | 332 | 3 | (896) |
| (920) | |||
| (884) | |||
| Paracoccus halodenitricans | 282 | –13 | (921) |
| Thiobacillus ferrooxidans | 380 | 1 | (878) |
| (878) | |||
| (917) | |||
| (874) |
Reprinted with permission from ref (873). Copyright 1998 Elsevier.
The roles of different parameters involved in determining the reduction potential of HiPIPs have been explored through mutational studies. In one such study, mutation of the Cys77 ligand of Ch. vinosum to Ser was analyzed by NMR, which found negligible conformational changes in this mutant.894 The role of the conserved Phe66 in the same protein was likewise investigated, finding that mutation to polar residues had minimal effects (<25 mV) on the reduction potential.799,876 Mutations in buried polar groups have indicated a role for these groups in the reduction potential as well. Mutation of Ser79Pro in Ch. vinosum HiPIP resulted in a 104 mV decrease in reduction potential. It has been suggested that the different electrostatic properties of the amide group between Ser and Phe and hence the ability to H-bond are the main reasons for the observed effect.9009 Mutations of conserved hydrophobic residues around the Fe–S cluster (making the site more solvent-accessible) resulted in minimal changes in the midpoint potential as well as entropy and enthalpy of reduction.875 Mutation of a conserved Phe to Lys showed similar marginal changes in the reduction potential. However, a 15-fold decrease in the self-exchange rate was observed upon addition of positive charge to the protein surface. The same protective roles have also been reported by mutation of conserved Tyr19 from Ch. vinosum.873
A CD analysis of different HiPIPs has shown that the pH dependence of the reduction potential in HiPIPs is very dependent on the proximity of a His residue to the cluster. In HiPIPs from Thiocapsa roseopersicina, which has His49, strong pH dependence was observed, while in HiPIPs from Ch. vinosum and Rhodopseudomonas gelatinosa, which have His42, show smaller pH dependence. In cases with no His, the reduction potential was independent of the pH.896 Recently, computational studies have been used to locate residues that cause the pH dependence of a Ch. vinosum HiPIP and identified His42 as a candidate, which is consistent with previous observations.897
Studies have shown a more prominent role of enthalpy in determining the reduction potential of HiPIPs, noting a favorable change in bonding upon reduction. These proteins also show a negative entropy change. Increased loss of both entropy and enthalpy results from increasing temperature, mainly due to elongation and breakage of H-bonds in the oxidized state.873 The covalency of the Fe–S bond and geometry of the ligands in the structure have been shown to play a role in different redox states and the reduction potential between HiPIPs and ferredoxins (Table 8).898 DFT and potential energy surface (PES) studies have further shown that this difference in covalency is mainly due to different arrangements of the ligands of the cluster.899 Ligand K-edge XAS studies have also shown large differences in Fe–S covalency between HiPIPs and ferredoxins. The primary transition of the K-edge is 1s → 4p; however, the covalent mixing from ligand 3p orbitals into unoccupied metal 3d orbitals results in an additional observable 1s → 3p transition. XAS studies demonstrated that the redox-active molecular orbital (RAMO) in HiPIPs is the HOMO of the [4Fe–4S]2+ resting state and has 50% sulfur ligand character. This results in a better superexchange rate from cluster to surface, which is necessary for the buried cluster in HiPIPs to transfer electrons.900 Another XAS study found that the difference in charge donation is due to different H-bonds to sulfur ligands between HiPIPs and ferredoxins. A more recent XAS study suggested hydration of the clusters as the main reason for the difference. This study showed that removal of water from ferredoxins results in higher covalency. In a similar way, exposure of the HiPIP cluster by unfolding decreases the covalency.901
Table 8. Redox Potential of Some HiPIPs and Some Ferredoxins with the Number of Their NH···S H-Bond Contactsa.
| protein | Em (mV) | no. of H-bond contacts | ref |
|---|---|---|---|
| Ectothiorhodospira halophila I HiPIP | 120 | 5 | (922) |
| Ectothiorhodspira vacuolata I1 HiPIP | 150 | 5 | (892) |
| Chromatium vinosum HiPIP | 360 | 5 | (923) |
| Rhodocyclus tenuis HiPIP | 303 | 5 | (919) |
| Bacillus thermoproteolyticus Fd′ | –280 | 8 | (924) |
| Peptococcus aerogenes Fdf | –430 | 8 | (923) |
| Azotobacter vinelandii Fd Ib | –650 | 8 | (925) |
Reprinted with permission from ref (873). Copyright 1998 Elsevier.
3.4.5.5. Spectroscopic Features
The HiPIPs have a brown-green color with a prominent band at 388 nm, with an R/Z ratio of ∼0.5, which is bleached after oxidation.872,926 The oxidized form has a very broad band with shoulders at 450, 735, and 350 nm. Both forms have 280 nm absorptions that are much higher than what is expected from aromatic contents, indicating that the cluster has some absorption in that region as well.926 CD spectroscopy in both visible and far/near-UV region has been used to probe the effect of the protein environment on the properties of HiPIPs. It has been shown that visible CD spectra of reduced HiPIPs are very similar, implying strong homology in their cluster environment. Most of the spectra show a positive feature at 450 nm and two distinct negative features in the 350 and 390 nm regions, with some of them showing a positive ellipticity at 330 nm. A group of HiPIPs show completely different features, having two positive bands between 350 and 440 nm and a negative feature at around 460 nm. CD studies indicate that the maximum band observable in absorption spectroscopy consists of several transitions, mainly a S → Fe charge transition. Visible CD of oxidized HiPIPs is usually featureless with broad maxima at 350, 400, and 450 nm. Near-UV CD spectra are very dependent on the position of aromatic residues in the protein. Far-UV CD spectra showed ∼12–20% α-helical content in the protein structure and slight changes upon oxidation and reduction.926
HiPIPs were the first class of paramagnetic proteins for which a thorough solution NMR study was able to determine the structure in both the reduced and oxidized forms.9271H NMR studies confirmed the mixed-valence state in HiPIPs884 and provided additional structural insights for these proteins.928,929 NMR was also used to find Fe–S–Cα–Cβ dihedral angles on the basis of hyperfine shifts of β protons and α carbons.930 Differences in the electronic features of iron pairs in the oxidized and reduced forms cause a significant hyperfine shift of 1H and 13C of the cysteine ligands of the cluster. Similar shifts of β carbons in the reduced state confirm the notion that they all have similar electronic features. Most HiPIPs show at least two isomeric electronic states apparent by room temperature NMR studies. The best explanation for this phenomenon is that the mixed-valence pair can switch from an iron(II/III) pair to an iron(III/IV) pair. The reduction potential of irons in the cluster usually follows this trend: Fe(III) > Fe(IV) ≈ Fe(II) > Fe(I), so only two states are observable in the oxidized state of HiPIPs, which explains the presence of two electronic isomers observed in NMR and EPR.884 NMR of the oxidized pair shows two downfield signals arising from the mixed-valence pair and two upfield signals (or extrapolated upfield, which is two downfield signals with anti-Curie temperature dependence) assigned to the ferric pair with inverted electron polarization.895,9311H 2D exchange spectroscopy (EXSY) NMR studies have analyzed self-exchange rates for HiPIP from Ch. vinosum and its aromatic mutants. An exchange rate of 2.3 × 104 M–1 S–1 was observed for the native protein at 298 K, with rates within 2-fold for the mutants. This study ruled out the role of aromatic residues in ET.876 β protons from cysteine ligands of the cluster experience large contact shifts. Eight signals from +110 to −40 ppm can be assigned to eight protons from four β-CH2 Cys ligands. The assignment of protons that are involved in amide–S H-bonds is more difficult due to their broad features that overlap with other protons.929,932 NMR experiments have also been used to assess water accessibility of the cluster and its mutants through analyzing the H2O/D2O exchange rates. 1H–13C heteronuclear correlation (HETCOR) NMR was used to show that the oxidized cluster has an overall shorter relaxation time than the reduced state.933
EPR of HiPIPs shows a nearly axial signal with g values at 2.13 and 2.03 that result from an S = 1/2 ground state in the oxidized form.934 In contrast to ferredoxins, HiPIPs are EPR-silent in their reduced state. Some HiPIPs show heterogeneous signals, probably due to sample preparation or dimerization of the cluster.799 ENDOR studies confirmed the presence of two pairs of irons in the oxidized form of the protein.935,936 EPR of most HiPIPs has shown at least two populations. Four species can be observed by EPR of HiPIPs with g⊥ = 2.15–2.13, 2.13–2.11, 2.06–2.08, and maybe 2.09–2.11, with the first two often being the most dominant species.872 Assignment of these two species can be performed by correlating the EPR data with room temperature 1H NMR.
Zero-field Mössbauer studies of HiPIPs at temperatures above 100 K show a broad quadruple splitting, indicative of fast electronic relaxation, with δ = 0.29–0.33 mm/s and quadruple splitting values of 0.74–0.80 mm/s. At lower temperature (4.2 K) the spectra show two nonequivalent iron pairs, one of which increases quadruple splitting with increased applied field, whereas the other decreases quadruple splitting. The subsets are assigned to a ferric pair (δ = 0.27 mm/s, with a −0.87 mm/s splitting) and a ferric–ferrous pair (δ = 0.37 mm/s with a splitting value of −0.94 mm/s).895 Mössbauer shows nondistinguishable iron atoms in reduced HiPIPs. Mössbauer studies of mutated Cys → Ser HiPIP have shown loss of covalent iron features due to replacement of S with O and a different spectrum of the Ser-bound iron in the reduced form, suggesting the importance of Cys residues in maintaining the mixed-valence state of the cluster.937 Mössbauer analyses of partially unfolded HiPIPs have found a slight increase in Fe–S bond distances without significant changes in the core cluster, indicating that the cluster is not denatured in early steps of unfolding.529,938
EXAFS analysis of the structure of the core cluster of HiPIPs and Fe–S distances has found a small temperature dependence. Analyses of Cys → Ser mutants reveal slight changes to the core structure and the Fe–S distances of intact cysteines, while the Fe–O bond is shortened, suggesting that the entire cluster is shifted toward the Ser ligand.937 Ligand K-edge XAS studies have also elucidated some of the differences between HiPIPs and ferredoxins.900
3.4.6. Complex Fe–S Centers
3.4.6.1. Hydrogenases
3.4.6.1.1. [NiFe] Hydrogenase Cluster
[NiFe] hydrogenases catalyze interconversion of H2 and H+ in microorganisms and ultimately provide electrons for ATP synthesis. [NiFe] hydrogenases from different sources have a conserved large domain of ∼60 kDa, containing the binuclear Ni–Fe active site and a small Fe–S cluster domain for ET. [NiFe] hydrogenase from Dv. gigas contains two [4Fe–4S] clusters and one [3Fe–4S] cluster, supported by EPR, Mössbauer,939 and crystallographic studies.940,941 The reduction potentials are −70 mV for the [3Fe–4S]+,0 cluster and −290 and −340 mV for the two flanking [4Fe–4S]2+,1+ clusters. The fully oxidized state of the two clusters ([4Fe–4S]2+) gives an isomer shift of 0.35 mm/s and quadruple splitting of 1.10 mm/s. Upon reduction, the two clusters are separated. Cluster I gives an isomer shift of 0.525 mm/s and quadruple splitting of 1.15 mm/s, and cluster II gives 0.47 and 1.35 mm/s, respectively. The parameters of [3Fe–4S]1+ are δ = 0.47 mm/s and ΔEQ = 1.67 mm/s, and those of [3Fe–4S]0 are δ = 0.39 mm/s and ΔEQ = 0.38 mm/s. The three Fe–S clusters are arranged linearly in the 3-D structure, with one [4Fe–4S] cluster proximal to the Ni–Fe–S catalytic center, the other [4Fe–4S] cluster at the surface, and the [3Fe–4S] cluster in the middle of them (Figure 35),940,941 suggesting the existence of an ET pathway.
Figure 35.
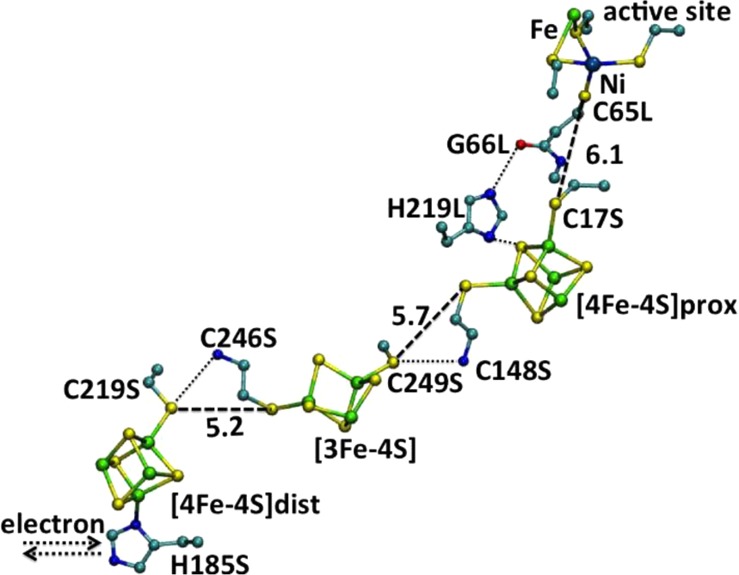
Proposed ET pathway in Dv. gigas [NiFe] hydrogenase. Selected distances are given in angstroms. PDB ID 1FRV. Color code: Fe, green; Ni, gray blue; C, cyan; S, yellow, O, red; N, blue. Reprinted with permission from ref (940). Copyright 1995 Macmillan Publishers Ltd.
[NiSeFe] hydrogenase, a subclass of [NiFe] hydrogenases, contains three [4Fe–4S] clusters.942,943 The crystal structure reveals that a cysteine residue near the middle cluster, as opposed to proline usually observed in [NiFe] hydrogenases, serves as an extra ligand and results in a [4Fe–4S] cluster instead of a [3Fe–4S] cluster .
[NiFe] hydrogenase from Dv. fructosovorans is structurally similar to that from Dv. gigas.944 On the basis of observations made with respect to [NiSeFe] hydrogenases, a Pro238Cys mutation has been made. The [3Fe–4S]1+,0 cluster was successfully converted to a [4Fe–4S]2+,1+ cluster and resulted in a 300 mV decrease of the reduction potential with little influence on activity, indicating that the [3Fe–4S]1+,0 cluster is not essential in the ET pathway of [NiFe] hydrogenase.
Recently, a new type of [NiFe] hydrogenase was discovered. Unlike the usually air-sensitive members of the family, [NiFe] hydrogenases from the bacteria Ralstonia eutropha, Ralstonia metallidurans, Hydrogenovibrio marinus, and Aquifex aeolicus could tolerate O2 to a limited extent.947 The oxygen tolerance arises from neither modification of the [Ni–Fe] active site nor limited access to O2. Crystal structures of the proteins have revealed a novel Fe–S cluster proximal to the Ni–Fe center (Figure 36a).948,949 Instead of the normal proximal [4Fe–4S] cluster coordinated by four cysteines from the protein, this cluster is a plastic [4Fe–3S] cluster bound by six cysteines with a flexible glutamic acid residue nearby. Upon oxidation, the backbone amide of the coordinating Cys26 is deprotonated by the nearby glutamic carboxylate and replaces the bridging Cys25 (Figure 36b,c), analogous to the P cluster in nitrogenases. The negative charge of amide will help to stabilize the oxidized state. As a result, the [4Fe–3S] cluster could transfer two electrons in a window of 200 mV and remain stable in three oxidation states.950 DFT calculations have revealed that the supernumerary coordination frame provided by the six cysteines and the flexible coordination sphere of the Cys26-bound Fe lead to plasticity of the unique proximal [4Fe–3S] cluster and, consequently, low reorganization energy in the reduced state.945 Hence, the proximal cluster could not only transfer electrons efficiently from the active site during H2 oxidation, but also rapidly supply two electrons to the active sites upon O2 binding, which in combination with one electron from the middle [3Fe–4S] cluster would efficiently reduce O2 to H2O and prevent formation of an inactive [Ni3+– –OOH–Fe2+] cluster, the so-called Ni-A state, and overoxidation by O2.951−953
Figure 36.
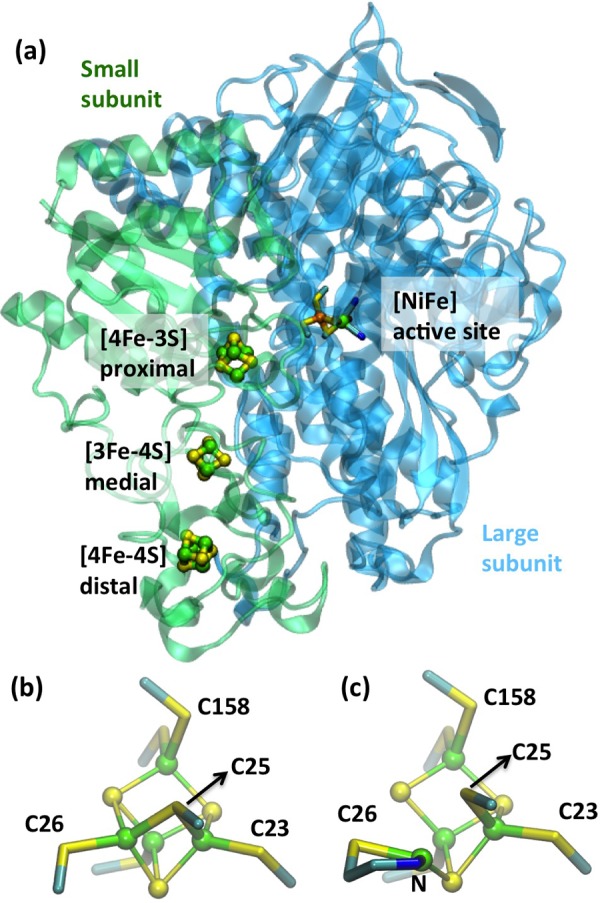
(a) Crystal structure of O2-tolerant membrane-bound hydrogenase from Ralstonia eutropha (PDB ID 3RGW). Reprinted from ref (945). Copyright 2013 American Chemical Society. (b) Reduced [4Fe–3S] cluster from MBH (PDB ID 3AYX) (Reprinted with permission from ref (946). Copyright 2012 Wiley-VCH) and (c) oxidized [4Fe–3S] cluster from MBH (PDB ID 3AYZ). Reprinted with permission from ref (946). Copyright 2012 Wiley-VCH. Color code: Fe, green; C, cyan; S, yellow; N, blue; Ni, orange.
3.4.6.1.2. [FeFe] Hydrogenases
[FeFe] hydrogenases share a conserved catalytic subunit binding metal cluster, called the H-cluster, as the catalytic site and have various Fe–S subunits harboring different Fe–S clusters for ET to and from the H-cluster. The Fe–S domains are usually located at the N-terminus of the catalytic domain and contain [4Fe–4S] or [2Fe–2S] binding motifs similar to those of ferredoxins.954−956 For example, [FeFe] hydrogenase from Dv. desulfuricans ATCC 7757 possesses two [4Fe–4S] clusters for ET,957 and the protein from Cl. pasteurianum contains one [2Fe–2S] cluster and three [4Fe–4S] clusters.958 The Fe–S clusters in Cl. pasteurianum [FeFe] hydrogenase are separated by 8–11 Å, indicating potential ET pathways through covalent bonds or a H-bonding network (Figure 37). The FS4C and FS2 near the protein surface possibly function as the initial electron acceptors of external electron donors and transfer electrons to the FS4B at the junction position. The FS4A is 10 Å from cluster FS4B and 9 Å from the H-cluster and could mediate sequential ET to and from the catalytic site.
Figure 37.
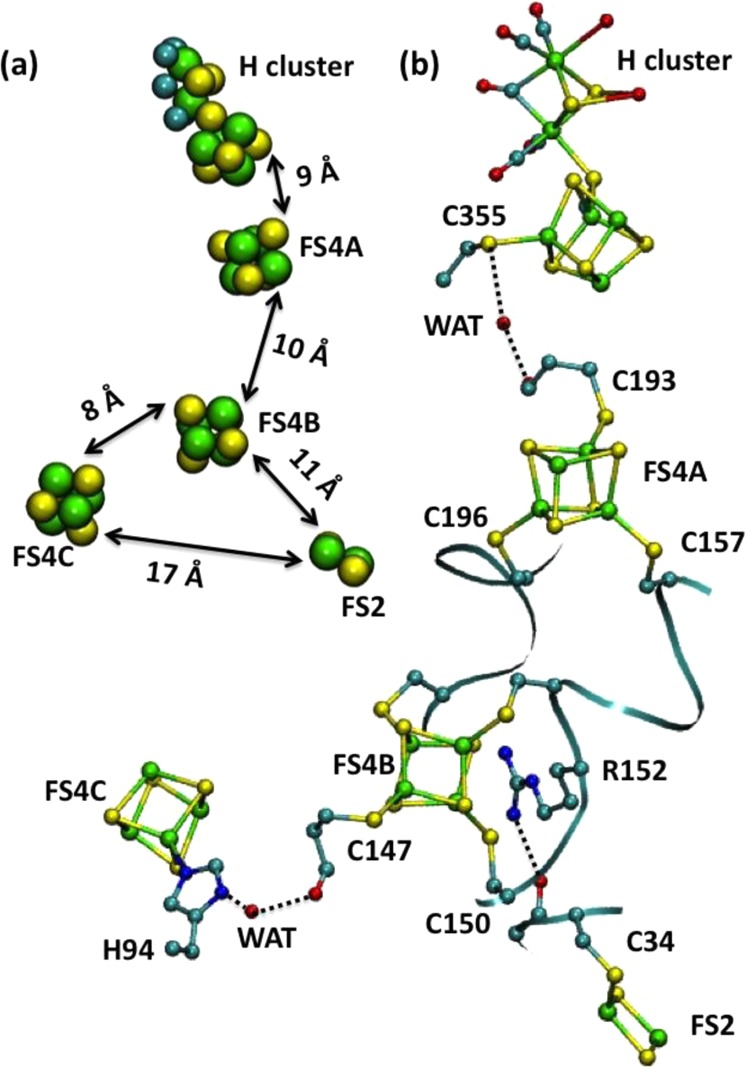
(a) Location of Fe–S clusters in [FeFe] hydrogenase (PDB ID 1FEH). (b) Proposed ET pathways for [FeFe] hydrogenase. Reprinted with permission from ref (958). Copyright 1998 American Association for the Advancement of Science.
3.4.6.2. Molybdonum-Containing Enzymes273
3.4.6.2.1. [4Fe–4S] Cluster and P-Cluster in Nitrogenase
Four types of nitrogenases have been discovered: two containing Mo and Fe, one containing V and Fe, and one containing only Fe in the catalytic site in a large domain with a molar mass of 220–250 kDa. Among them, [FeMo] nitrogenase has been the most extensively studied (Figure 38a). Besides the active site, all nitrogenases contain an iron protein as α2 dimers with a molar mass of 60–70 kDa. It contains a single [4Fe–4S] cluster between the two monomers, which is coordinated by one conserved cysteine from each monomer and is exposed to water.959 The cluster transfers electrons efficiently via a MgATP hydrolysis reaction at the larger domain containing a catalytic site, along with other functions, including involvement in biosynthesis and insertion of FeMoco into [FeMo] nitrogenase and regulation of biosynthesis in other nitrogenases.960
Figure 38.
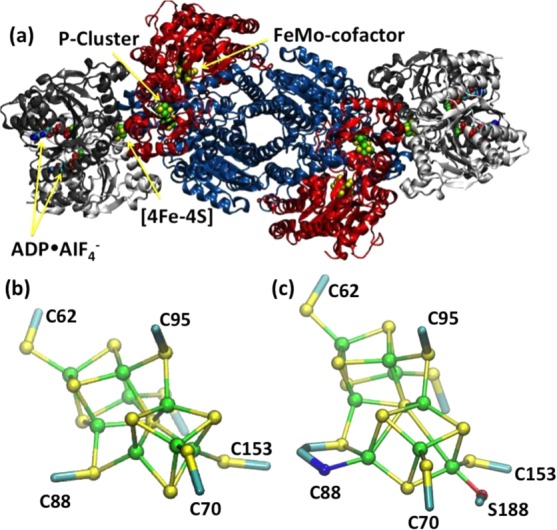
(a) Overall structure of nitrogenase (PDB ID 1N2C). Cofactors are shown as spheres and denoted. Reprinted with permission from ref (965). Copyright 1997 Macmillan Publishers Ltd. (b) Reduced P cluster from nitrogenase (PDB ID 3U7Q) (Reprinted with permission from ref (946). Copyright 2012 Wiley-VCH.) and (c) oxidized P cluster from nitrogenase (PDB ID 2MIN). Reprinted with permission from ref (946). Copyright 2012 Wiley-VCH.
Three oxidation states, +2, +1, and 0, have been observed for the [4Fe–4S] cluster, indicating that the cluster could transfer one or two electrons to the catalytic domain. The reduction potential to achieve an all-ferrous [4Fe–4S]0 cluster is −460 mV, and this is the first example of this oxidation state for [4Fe–4S] clusters, both in proteins and in model complexes.961−963 EXAFS studies show that changes of the Fe–S and Fe–Fe distances are less than 0.02 Å from the [4Fe–4S]2+ cluster to the [4Fe–4S]1+ cluster.964
The Fe protein can bind 2 equiv of MgATP or MgADP, each in a Walker A motif on one monomer. The Walker A binding site is 15–20 Å away from the [4Fe–4S] cluster with a series of salt bridges and H-bonds in between. However, the reduction potential of the [4Fe–4S] cluster decreases ∼100 mV upon binding of either nucleotide, possibly arising from protein conformational changes induced by binding and hydrolysis reactions.965−970 The reduction potential change is proposed to be the driving force for ET.968 UV–vis, resonance Raman, and EPR spectroscopic studies indicate that the [4Fe–4S] cluster could reversibly cycle between a regular [4Fe–4S] cluster in the reduced state and two [2Fe–2S] clusters in the oxidized state.971
The [FeMo] domain contains the FeMoco cluster and a P-cluster. The FeMoco cluster is the catalytic center and will not be discussed here. The P-cluster is situated at the interface of the α and β subunits of the [FeMo] domain. It is an [8Fe–7S] cluster, with a 6-coordinate sulfur at the center. The structure of the P-cluster changes with the oxidation state. The dithionite reduced P cluster (PN) is bound by six cysteines from the protein, four of which coordinate a single iron, and the remaining two function as bridging ligands (Figure 38b).972 After two-electron oxidation of PN, a form called Pox is obtained. In the Pox cluster, the coordination between the center 6-coordinate sulfur and two irons associated with the β subunit is replaced by the amide N of Cys88 of the α subunit and side chain hydroxyl of Ser188 of the β subunit (Figure 38c), similar to the changes of oxygen-tolerant [NiFe] hydrogenases mentioned above (see Figure 36). The changes are proposed to be related to the proton-coupled electron transfer process in nitrogenases.972−974
3.4.6.2.2. Aldehyde Oxidoreductases
Aldehyde oxidoreductase belongs to the molybdoflavoenzymes. It is a homodimer and usually requires Fe–S clusters, a molybdopterin or tungstopterin site, and sometimes an FAD cofactor for substrate oxidation. Aldehyde oxidoreductase from Dv. gigas is composed of four domains, including two small N-terminal domains binding two types of [2Fe–2S] clusters and two large domains containing the molybdopterin cofactors.975,976 The first Fe–S domain (residue 1–76) is similar to that of spinach ferredoxins, and the [2Fe–2S] cluster is coordinated by Cys40, Cys45, Cys47, and Cys60. The second Fe–S domain (residues 84–156) is a four-helix bundle, and the [2Fe–2S] cluster is coordinated by Cys100, Cys103, Cys137, and Cys139. The molybdopterin is 15 Å from the surface and 14.9 Å from the Fe–S cluster of the second domain. Recently, the crystal structure of aldehyde oxidase of mouse liver has been reported. The overall fold is very similar to that from Dv. gigas, but that of the mammalian protein has an additional FAD domain.977
EPR studies revealed two types of [2Fe–2S] clusters, named Fe–SI and Fe–SII.978−981 Fe–SI is observable at 77 K with g values of 2.021, 1.938, and 1.919, while Fe–SII is only observable below 40 K with g values of 2.057, 1.970, and 1.900. The reduction potentials of Fe–SI and Fe–SII are −260 and −280 mV, respectively.
In the presence of the substrate benzaldehyde, partial reduction of the Fe–S clusters has been detected in Mössbauer studies, indicating participation of the Fe–S clusters in the catalytic reaction and fast ET from the molybdopterin center.982
3.4.6.3. Ni-Containing CO Dehydrogenase and Hybrid Cluster Protein
3.4.6.3.1. Ni-Containing CO Dehydrogenase
CO dehydrogenases (CODHs) catalyze oxidation of CO to CO2 along with dehydrogenation of water and release of protons and electrons. It is important in the oxygen-based respiratory process in hydrogenogenic bacteria. There are two types of CODHs. One is Mo-based CODHs with a mono-Mo cofactor coordinated by dithiolene sulfurs of a pterin ligand found in aerobic organisms, which is beyond the scope of this review but has been reviewed extensively in other papers.983,984 The other is Ni-containing CODHs with a Ni–Fe–S cluster as well as multiple Fe–S clusters found in anaerobic organisms985−987 and will be discussed briefly below.
Ni CODHs are β2 homodimers.988,989 Each monomer contains a Ni–Fe–S cluster (cluster C) as the catalytic site and a [4Fe–4S] cluster (cluster B). In addition, another [4Fe–4S] cluster (cluster D) is situated at the interface of the two monomers and coordinated by residues from both monomers (Figure 39a). Clusters B and D transfer electrons between cluster C and external redox regents. They also bind acetyl-CoA synthases to form α2β2 bifunctional enzymes acetyl-CoA synthases/carbon monoxide dehydrogenases (ACSs/CODHs).990 Two additional [4Fe–4S] clusters, E and F, have been found in an extra subunit of the ACS/CODH complex.991 The crystal structure of Ni CODH from Carboxydothermus hydrogenoformans reveals that cluster C is a [Ni–4Fe–5S] cluster (Figure 39b). The geometries of the irons are approximately tetrahedral, and that of Ni is close to square planar. It is associated with the protein through four cysteines and one histidine.988 On the other hand, the structures of Rhodospirillum rubrum Ni CODHs989 and the M. thermoacetica ACS/CODH complex991 show cluster C as [Ni–4Fe–4S], coordinated similarly by five cysteines and one histidine from the protein (Figure 39c). The Ni is also coordinated by an external nonprotein ligand.
Figure 39.
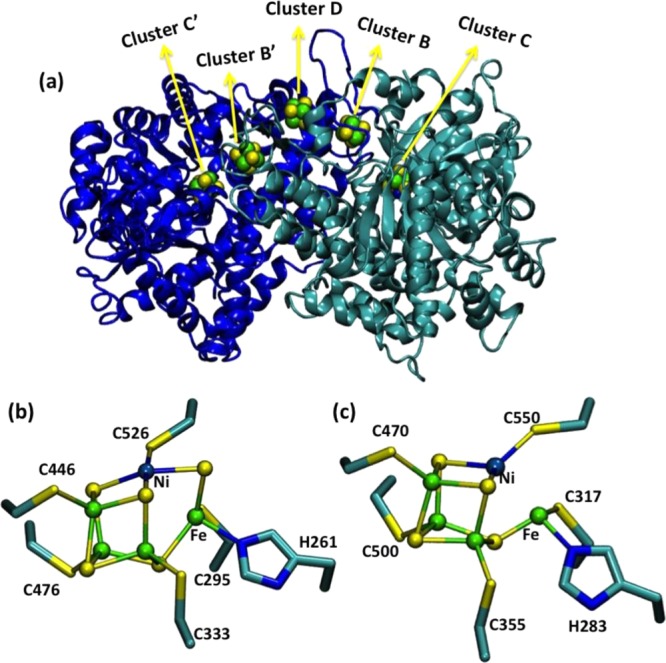
(a) Crystal structure of Rs. rubrum Ni CODH. Clusters are shown as spheres. PDB ID 1JQK. (b) [4Fe–5S–Ni] cluster C of Ca. hydrogenoformans Ni CODH. PDB ID 1SU8. (c) [4Fe–4S–Ni] cluster C of M. thermoacetica Ni CODH. PDB ID 1MJG. Reprinted with permission from ref (990). Copyright 2011 Elsevier.
3.4.6.3.2. Hybrid Cluster Proteins
Hybrid cluster proteins (HCPs) are a type of Fe–S proteins with unknown functions. However, they have been detected in more than 15 bacteria and archaea. There are three categories of HCPs. The first is found in anaerobic bacteria such as Dv. vulgaris and Dv. desulfuricans or methanogen archeon Methanococcus jannaschii, with coordinating cysteines arranged in the sequence Cys-(Xxx)2-Cys-(Xxx)7–8-Cys-(Xxx)5-Cys. The second is found in facultative anaerobic Gram-negative bacteria such as E. coli, Morganella morganii, or Tb. ferrooxidans, with the sequence Cys-(Xxx)2-Cys-(Xxx)11-Cys-(Xxx)6-Cys. The third is found in (hyper)thermophilic bacteria or archaea, including Methanobacterium thermoautotrophicum, Pyrococcus abyssi, or Tt. maritima, with the same sequence arrangement as the first category but with smaller size due to residue deletion downstream of the N-terminal cysteine region.
HCP from Dv. vulgaris contains three domains (Figure 40a).992,993 A [4Fe–4S] cluster is bound to domain 1 by Cys3, Cys6, Cys15, and Cys21 from the N-terminal region, similar to the cubane cluster in ferredoxins except that no cysteine is from the C-terminal region. This Cys-(Xxx)2-Cys-(Xxx)8-Cys-(Xxx)5-Cys motif is conserved in all HCPs, and HCPs from both categories 1 and 3 contain a [4Fe–4S] cluster linked by this motif. HCPs from category 2, on the other hand, might instead have two [2Fe–2S] clusters at this position.994
Figure 40.

Hybrid clusters in HCP. (a) Overall structure of as-isolated Dv. vulgaris HCP. Metal clusters are shown as spheres. PDB ID 1W9M. (b) Superposition of Dv. vulgaris HCP (cyan) and NiCODH (red, PDB code 1SU7). (c) Hybrid cluster in the as-isolated oxidized form of Dv. vulgaris HCP prepared anaerobically. PDB ID 1W9M. (d) Hybrid cluster in the reduced form of Dv. vulgaris HCP. PDB ID 1OA1. Residue backbones are omitted for clarity. Bonds inside the cluster are shown as dotted lines, and bonds between residues and the cluster are shown as solid lines. Color code: Fe, green; C, cyan; S, yellow; O, red; N, blue. Reprinted with permission from ref (995). Copyright 2008 International Union of Crystallography.
HCPs also contain a unique hybrid cluster, [4Fe–2S–3O], which was isolated in the oxidized form from Dv. vulgaris HCP (Figure 40c),995 and [4Fe–3S] with a water molecule between Glu494 and His244 in the reduced form (Figure 40d).996 In the former state, the cluster is linked to the protein by Cys12, Cys434, Cys459, thio-Cys406 (Cys with an additional S on the S(Cys), called Css406), His244, Glu268, and Glu494, and in the latter case Css406 is reduced to cysteine. The EPR signal of HCP is similar to that of the prismane model complex (Et4N)3[Fe6S6(SC6H4-p-Me)6]3+.997 Therefore, the four oxidation states of the hybrid cluster are named analogously to those of the prismane complex as “3+”, “4+”, “5+”, and “6+”. The midpoint reduction potentials of the Dv. vulgaris HCP hybrid cluster range from −200 to +300 mV at pH 7.5.998
It is noteworthy that HCPs demonstrate a high degree of similarity to Ni CODHs.992,993,999 They not only share similar overall folding, but also exhibit similar cluster positions and structures inside the monomer (Figure 40b). The closest distance between the [4Fe–4S] cluster and hybrid cluster is 10.9 Å, with Tyr493, Thr71, Asn72, and Glu494 in between. In addition, two tryptophan residues, Trp292 and Trp293, are located between the hybrid cluster and the protein surface. The arrangements indicate possible ET pathways, yet no involvement in such processes has been detected so far. The protein can be reduced by NAD(P)H oxidoreductase,994 but there is no genomic evidence for the existence of a similar redox partner in the sources from which HCP has been detected or isolated.
3.4.6.4. Siroheme Fe–S Proteins
Siroheme is an iron-containing reduced tetrahydroporphyrin of the isobacteriochlorin class (Figure 41a). Siroheme proteins are a type of iron–sulfur protein containing a siroheme conjugated to a [4Fe–4S] cluster through a thiolate bridge.1000 Siroheme is the catalytic center, and the [4Fe–4S] cluster serves as an electron trapping and storage site. Siroheme proteins includes sulfite reductases and nitrite reductases, and they are important in assimilation and dissimilation of sulfite and nitrite.1001,1002
Figure 41.
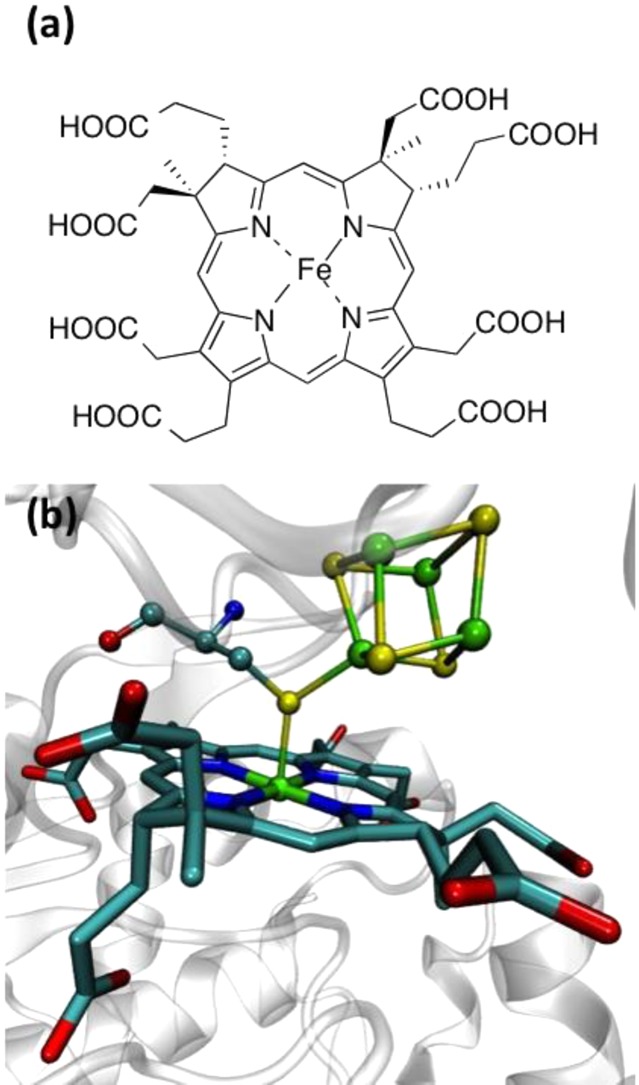
(a) Structure of siroheme. (b) Siroheme and the [4Fe–4S] cluster of spinach nitrite reductase. PDB ID 2AKJ. Color code: Fe, green; C, cyan; S, yellow; O, red; N, blue.
3.4.6.4.1. Nitrite Reductase
NiR catalyzes the six-electron reduction of nitrite to ammonia. It exists in both eukaryotes and prokaryotes. There are two types of NiR categorized by the physiological electron donor: ferredoxin-dependent NiR in photosynthetic organisms and NAD(P)H-dependent NiR in most heterotrophic organisms.276,1003−1005 Ferredoxin-dependent NiR contains a siroheme and a [4Fe–4S] cluster, while NAD(P)H-dependent NiR contains an additional FAD cofactor bound at an extended N-terminal region.276
Spinach nitrite reductase is a type of ferredoxin-dependent NiR isolated from higher plants. It is composed of 594 amino acids divided into three α/β domains. The siroheme cofactor is situated at the interface of the three domains and bridged to the [4Fe–4S] cluster via Cys486 (Figure 41b). The [4Fe–4S] cluster is also coordinated by Cys441, Cys447, and Cys482. The midpoint reduction potentials are −290 mV for the siroheme and −365 mV for the [4Fe–4S] cluster. Although the two cofactors are magnetically coupled with a distance of 4.2 Å, they are independent in redox titration processes.1006,1007 Spinach NiR can form a 1:1 complex with ferredoxin with electrostatic interactions between acidic residues from NiR and basic residues from ferredoxin. The interprotein ET chain has been established as from photoexcited photosystem I via the [2Fe–2S] cluster of ferredoxin to the [4Fe–4S] cluster of NiR followed by intraprotein transfer to the siroheme.1006−1008
3.4.6.4.2. Sulfite Reductase
Sulfite reductase catalyzes the six-electron reduction of sulfite to sulfide in biological systems and can be categorized as assimilatory sulfite reductase (aSiR) or dissimilatory sulfite reductase (dSiR). aSiR reduces sulfite directly to sulfide, while dSiR provides a mixture of sulfide, trithionate, and thiosulfate in in vitro experiments.1009
The aSiRs are found in archaebacteria, bacteria, fungi, and plants.1010,1011 Assimilatory ferredoxin-dependent sulfite reductases from plant chloroplasts and cyanobacteria are soluble monomeric proteins with molar masses of ∼65 kDa. They contain a siroheme linked to a [4Fe–4S] cluster structurally similar to those in nitrite reductase, and they undergo reduction by ferredoxin from photoreduced photosystem I as well.1002 They can also catalyze the reduction of nitrite to ammonia, the reaction catalyzed by NiR, but with a higher KM for nitrite than sulfite, further demonstrating the significant similarity of the two types of enzymes.1002,1012,1013 For maize sulfite reductase, the midpoint potentials of siroheme and the [4Fe–4S] cluster have been determined to be −285 ± 5 and −400 ± 5 mV, respectively, at pH 7.5 in Tris buffer by redox titrations. Although the E° of the [4Fe–4S] cluster is more negative than that of spinach nitrite reductase (E° = −375 ± 10 mV at pH 7.5 in Tris buffer), reduction by ferredoxin (E° = −430 mV) is still a thermodynamically favorable process. In the presence of cyanide, the E° of siroheme shifts positively to −155 ± 5 mV, while that of the [4Fe–4S] cluster shifts negatively to −455 ± 10 mV, possibly due to decreased affinity of the enzyme for cyanide upon reduction of the [4Fe–4S] cluster. Similar trends are observed in spinach nitrite reductase as well.1014 The aSiR from E. coli is a 780 kDa hemeoflavoprotein with an α8β4 arrangement. The α subunit, known as sulfite reductase flavoprotein, contains FAD and FMN, while the β unit, named sulfite reductase hemoprotein, harbors the associated [4Fe–4S] cluster and siroheme. The ET pathway is in the FAD–FMN–[4Fe–4S]–siroheme sequence, with NADPH as the initial donor and sulfite as the terminal acceptor.1015
dSiRs exist in sulfate reducing microorganisms.1010,1011 dSiR is composed of two types of subunits, DsrA and DsrB, generally in a heterotetrametric α2β2 arrangement with similar overall folds for all dSiRs from different sources.1016,1017 Some dSiRs form a complex with two additional subunits of DsrC and result in an α2β2γ2 arrangement. The dSiR contains eight [4Fe–4S] clusters together with four sirohemes or two sirohemes and two sirohydrochlorins (the metal-free form of siroheme) (Figure 42a,b), and only two of the four sites are catalytically active. In Dv. gigas, desulfoviridin, a subcategory of dSiR, a [3Fe–4S] cluster is associated with the siroheme instead of a [4Fe–4S] cluster in one active form, DsrII (Figure 42c). The relative position of siroheme and the [4Fe–4S] cluster is similar to that in aSiRs, and both the [4Fe–4S] clusters proximal to and remote from the siroheme are coordinated by four cysteines from the protein.1018−1020
Figure 42.
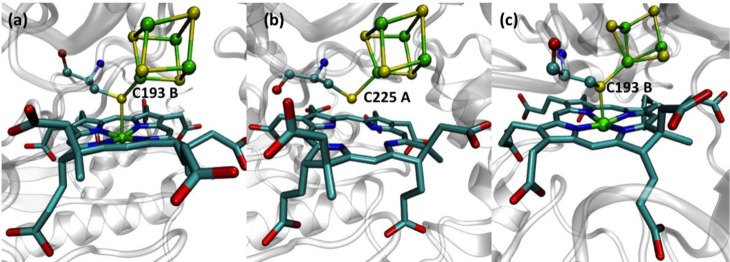
(a) Siroheme group and [4Fe–4S] cluster of DsrI. PDB ID 3OR1. (b) Sirohydrochlorin group and [4Fe–4S] cluster of DsrII. PDB ID 3OR2. (c) Siroheme group and [3Fe–4S] cluster of DsrII. PDB ID 3OR2. Color code: Fe, green; C, cyan; S, yellow; O, red; N, blue.
3.4.6.5. Respiratory Complex Chain
The mitochondrial respiratory system is the main energy producer in eukaryotic cells.1021,1022 It consists of five membrane complexes, complex I,1023 complex II (succinate dehydrogenase),1024,1025 complex III (cytochrome bc1 complex),1026−1029 complex IV (cytochrome c oxidase complex),1030,1031 and complex V (ATPase).1032 The first four complexes are located on the inner membrane and function by transferring electrons from electron donors, NADH and succinate, to the final electron acceptor, oxygen, and meanwhile pump protons across the membrane. This proton gradient is utilized by ATPase to generate ATP.
3.4.6.5.1. Respiratory Complex I
Respiratory complex I (CI), also known as NADH:ubiquinone oxidoreductase or NADH dehydrogenase, is involved in one of the ET pathways of the respiratory chain. It is composed of the following steps: (1) NADH donates electrons through CI to reduce ubiquinone to ubiquinol. (2) Ubiquinol transfers electrons through complex III to cytochrome c. (3) Cytochrome c is oxidized by complex IV and transfers electrons to O2 to produce water. In this process, each electron transferred is associated with five protons pumped from the matrix to the inner membrane space.
Although CI is the most complicated complex in the mitochondrial respiratory chain, important breakthroughs have been achieved, and multiple structures have been reported recently.1023,1033−1036 Mammalian CI (∼980 kDa) is composed of up to 45 different subunits, including 7 subunits in hydrophilic parts harboring one FMN and eight Fe–S clusters, 7 subunits in transmembrane parts, and ∼30 accessory subunits.1022,1037 Bacterial NADH dehydrogenase (∼550 kDa) only contains 13–16 subunits, which is sufficient for complete CI function as well.1023,1038−1040 The crystal structure of the hydrophilic part of complex I from T. thermophilus(1023) reveals for the first time the main ET pathway of the protein as shown in Figure 43: electrons from NADH are transferred through FMN to N3, followed by N1b, N4, N5, N6a, and N6b sequentially, and finally through N2 to ubiquinone coupled with proton translocation.1022
Figure 43.
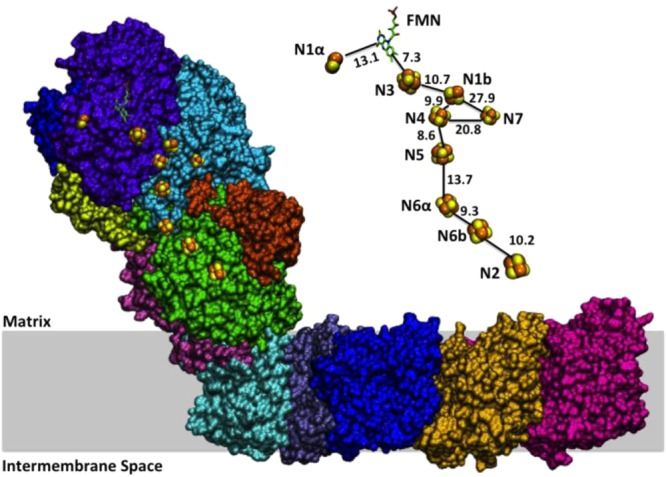
Crystal structure of mitochondrial respiratory complex I from T. thermophilus. PDB ID 4HEA. Cofactors involved in the ET pathway are shown on the right side with distances and directions denoted. Reprinted with permission from ref (1022). Copyright 2013 Elsevier.
3.4.6.5.2. Respiratory Complex II (Succinate Dehydrogenase) and Fumarate Reducatse
Complex II in the respiratory chain (CII), also known as succinate dehydrogenase (SDH) or succinate:quinone reductase, is a membrane-bound protein involved in the citric acid cycle and the second ET pathway in the mitochondrial respiratory chain. In the mitochondrial respiratory chain, electrons are transferred from succinate to ubiquinone through complex II, then to cytochrome c through complex III, and finally to O2 through complex IV. This process is less efficient than the process associated with complex I, and each electron transferred will pump only three protons across the membrane.
CII catalyzes oxidation of succinate to fumarate by a hydrophilic catalytic domain composed of a large flavoprotein (Fp; 65–79 kDa) with a covalently bound FAD cofactor and an iron–sulfur protein (Ip; 25–37 kDa) containing [2Fe–2S] (center S1), [4Fe–4S] (center S2), and [3Fe–4S] (center S3) clusters.1024,1025,1041 The catalytic domain is anchored to the membrane by one or two hydrophobic domains (CybL, CybS) harboring usually b-type cytochromes (Figure 44). The [2Fe–2S] center is coordinated by four cysteines close to the N-terminus, and the [4Fe–4S] and [3Fe–4S] clusters are coordinated near the C-terminus by two cysteine-containing sequences: Cys-(Xxx)2-Cys-(Xxx)2-Cys-(Xxx)3-Pro and Cys-(Xxx)2-Xxx-(Xxx)2-Cys-(Xxx)3–Cys-Pro (Xxx = Ile, Val, Leu, or Ala), similar to 7Fe ferredoxins. The [4Fe–4S] cluster usually has a low reduction potential and functions as the energy barrier of the ET process to direct the electron flow and, consequently, the reaction pathway.1042 The [3Fe–4S] cluster is involved in a direct ET process from the initial electron donor quinones.1043−1045 The midpoint reduction potential of the [3Fe–4S]1+,0 cluster is in the range of +60 to +90 mV, and the potential of the initial electron donor ubiquinone is +65 mV.1046 SDH from Sl. acidocaldarius contains a [4Fe–4S] center instead of a [3Fe–4S] center for cluster S2 and displays poor reactivity toward caldariella quinone.1047
Figure 44.

Crystal structure of mitochondrial respiratory complex II. FAD binding protein (Fp) is shown in blue, iron–sulfur protein (Ip) is shown in cream, hydrophobic domains are shown in pink and orange, and the putative membrane is shown in gray shading. PDB ID 1ZOY. Cofactors involved in the ET pathway are shown on the right side, with distances, reduction potential, and directions denoted. Reprinted with permission from ref (1024). Copyright 2005 Elsevier.
It is noteworthy that heme b (E° = +35 mV) in the hydrophobic domain of SDH is not involved in the ET pathway mentioned above. It is proposed that heme b in SDH of E. coli functions as an electron sink and reduces ROS to protect FAD and Fe–S clusters.1025 However, the reduction potential of heme b in SDH of porcine is −185 mV,1048 much lower than that of E. coli. Therefore, the electron sink mechanism is less effective in this case and needs further investigation.
Fumarate reductase is a member of the succinate–ubiquinone oxidoreductase superfamily as well. It catalyzes the reduction of fumarate to succinate, the reverse reaction of SDH. It is very similar to SDH in subunit composition and cofactors.1049,1050 Its three iron–sulfur clusters are linked to the protein by cysteine residues in E. coli, which are conserved in other fumarate reductases too. The midpoint reduction potential is between −70 and −20 mV, and that of the initial electron donor menaquinol is −74 mV.1046
3.5. Engineered Fe–S Proteins
3.5.1. Artificial Rubredoxins
A rubredoxin-like [FeCys4] center has been constructed into thioredoxin by computational design. The first coordination sphere is composed of two cysteines, Cys32 and Cys35, which form a disulfide bond in wild-type thioredoxin, as well as two cysteines introduced by mutation, Trp28Cys and Ile75Cys. The resulting monoiron center resembles Rd in UV–vis and EPR spectra, and the mimic protein is able to undergo three cycles of air oxidation and β-mercaptoethanol reduction.1051
The redox process of rubredoxin is not fully reversible due to the instability of the reduced form. Nanda et al. have constructed a minimal rubredoxin mimic, RM1, on the basis of computational design for a more restrained tertiary structure derived from PfRd. RM1 is a domain-swapped dimer fused with a highly stable hairpin motif tryptophan zipper and displays spectroscopic properties very similar to those of native Rd’s. Moreover, it shows a reduction potential of 55 mV vs SHE and maintains redox activity for up to 16 cycles under aerobic conditions.1051
3.5.2. Artificial [4Fe–4S] Clusters
There have been numerous studies focusing on making model compounds of ferredoxins1052−1054 and using those models to elucidate features of natural Fe–S clusters using several methods.803,1055,1056,1057 In addition to synthetic models of ferredoxins that are discussed in a review in this journal,2007 protein and peptide models of ferredoxins have also been made. These models have been discussed in detail in another review in this thematic issue,3000 and we will discuss them here only briefly.
Almost all of these mimics are modeled after [4Fe–4S] clusters, usually made by placing the conserved motif within a scaffold. These model systems have been used for unraveling the minimal structures required for binding of Fe–S clusters.732,1058,1059,1061
A 16 amino acid peptide has been modeled to incorporate a low-potential [4Fe–4S] cluster. More detailed sequence alignments resulted in design of peptides with better cluster binding features that mimic FA and FB of photosystem I.705 Other peptide models have also been made to analyze reduction potential properties of different Fe–S clusters, including [4Fe–4S] clusters, [2Fe–2S] clusters, and rubredoxins.717
Four-helix bundle models of [4Fe–4S] clusters are among the most common systems to build and study these clusters. Both a single [4Fe–4S] cluster and a [4Fe–4S] cluster together with a heme cofactor have been designed in such four-helix bundles.1061,1062 Recently, a “metal first” approach has been taken to introduce a [4Fe–4S] cluster into a non-natural α-helical coiled coil structure. The design then went through further optimization and addition of secondary sphere interactions to stabilize the reduced form and prevent aggregation. Such designs that are independent of structural motifs can be used as a platform for the future design of multiclusters to be used as biological “wires” that transfer electrons through a chain of proteins.1063
3.6. Cluster Interconversion
Although the Fe–S clusters are mostly classified on the basis of the number of iron atoms in the center, there are several cases in which changing one cluster to another type has been observed. These cluster interconversions can happen through three types of processes: natural changes in the environment of the cluster, chemical treatments of the cluster, or amino acid replacements.
One of the most common types of cluster interconversion is the change from a [4Fe–4S] cluster to a [2Fe–2S] cluster. This kind of conversion has been observed in hydrogenases and nitrogenases. While CD and MCD analyses show that MgATP/ADP binding to the [4Fe–4S] cluster of Fe hydrogenase does not result in conversion to a [2Fe–2S] cluster,1064 addition of α,α′-dipyridyl to the [4Fe–4S] cluster of nitrogenase resulted in formation of a [2Fe–2S] cluster in the presence of MgATP.1065,1066 The [4Fe–4S] to [2Fe–2S] cluster conversion has been observed in enzymes such as ribonucleotide reductase1067 and pyruvate formate activating enzyme1068 as well, usually upon oxidation in air or chemical treatment.
A very well studied case of the role of [4Fe–4S] to [2Fe–2S] cluster conversion in regulating cellular responses is that of fumarate nitrate reduction transcription factor. It has been shown that this protein undergoes the conversion upon O2 stress. The excess oxygen will oxidize S ligands and generate disulfide cysteines. The formation of a disulfide Cys-ligated [2Fe–2S] cluster will result in a monomerization of the fumarate nitrite reduction transcription factor dimer, hence unbinding from DNA.1069,1070 The conversion is composed of two steps: first, the [4Fe–4S] cluster undergoes a one-electron oxidation to form a [3Fe–4S]1+ intermediate after releasing an Fe2+. Second, the [3Fe–4S]1+ cluster converts to a [2Fe–2S] cluster and releases an Fe3+ and two sulfide ions.1071,1072 Mutating Ser24 into Phe and shielding Cys23 could inhibit step 1.1073 Chelators of both Fe2+ and Fe3+ could accelerate step 2 significantly.1074
Another very common interconversion is [4Fe–4S] to [3Fe–4S] interconversion. The [4Fe–4S] clusters are very sensitive to air, and oxidation in air can remove one of the irons, resulting in a 3Fe cluster.1075 The most well studied case of this interconversion is the enzyme aconitase. Aconitase has a [4Fe–4S] cluster in its active form, which is very sensitive to air. Aerobic purification of the protein causes formation of an inactive enzyme with a 3Fe cluster. Addition of extra Fe, however, can reverse the conversion and reactivate the enzyme.1076 Exposure of the [3Fe–4S] aconitase to high pH (>9.0) will result in the formation of a purple species that has been attributed to a linear [3Fe–4S] cluster. This purple protein can be activated again through reduction in the presence of Fe.1077
While more often clusters of higher iron number convert into clusters with fewer iron atoms, the reverse case has also been observed. In biotin synthase, there are two [2Fe–2S] clusters that can convert to a [4Fe–4S] cluster after reduction. UV–vis and EPR studies reveal that the conversion process occurs through dissociation of Fe from the protein followed by slow reassociation.1078 Ferredoxin II of Dv. gigas has a [3Fe–3S] cluster that can convert into a [4Fe–4S] cluster through incubation with excess Fe, presumably through a non-Cys ligand.1079 The [3Fe–4S]1+ and [2Fe–2S]2+ clusters in isolated pyruvate formate–lyase can both be converted to [4Fe–4S] clusters with mixed valences of +1 and +2 upon dithionite reduction.1080
Interconversion between [4Fe–4S] and [3Fe–4S] clusters has been investigated through mutational studies. Removal of Cys ligands in [4Fe–4S] clusters results in the formation of [3Fe–4S] clusters. Replacement of the conserved Asp in [3Fe–4S] clusters with a ligating residue such as His or Cys causes formation of [4Fe–4S] clusters.735,944,1081,1082 In [NiFe] hydrogenase, mutating a conserved Pro residue into Cys near the [3Fe–4S] cluster has successfully converted it to a [4Fe–4S] cluster accompanied by a 300 mV decrease in the reduction potential,944 while in F420 reducing hydrogenase of Methanococcus voltae the [4Fe–4S] to [3Fe–4S] conversion has been achieved by replacing a Cys residue, producing a ∼400 mV increase in the reduction potential.1081
Addition of other metal ions in place of the fourth iron into a [3Fe–4S] cluster is sometimes also called interconversion. There are multiple reports of the formation of such hybrid clusters with Zn, Tl, and other metal ions.1083,1084
3.7. Structural Features Controlling the Redox Chemistry of Fe–S Proteins
The Fe–S proteins cover a wide range of reduction potentials, mostly in the lower or negative end of the range. Several parameters are known to be important in the ability of Fe–S proteins to accommodate such a wide range of reduction potentials. Unique electronic structures of iron in different clusters and different protein environments are among the most important factors. The ability of each iron to go through 2+ to 3+ oxidation states will allow multiple states for the core cluster, each of which having a different reduction potential range. This factor is more evident in the case of HiPIPs vs ferredoxins. Solvent accessibility, H-bonding patterns around the cluster, the net charge of the protein, partial charges around the cluster, and the identity of the ligands are among the other features that contribute to fine-tuning the reduction potential. Detailed examples of the role of each feature are discussed in section 3.4.3.3.3, “Important Structural Elements”. Below is a summary of these features and their effects in different Fe–S proteins.
3.7.1. Roles of the Geometry and Redox State of the Cluster
As with other redox-active metal centers, the primary coordination sphere of a metal ion plays an important role in its redox properties. The iron center(s) has the same distorted tetrahedral structure in almost all Fe–S proteins; however, it has been shown that slight changes in this structure will result in changes in the reduction potentials. Differences in the Fe–S–Cα–Cβ torsion angle618,731,1085 and distortion of the cuboidal structure in some [3Fe–4S] clusters1086 are examples of this distortion. Different geometries can lead to slight differences in electronic structures that will affect the redox properties of the protein.
Another important feature that influences the reduction potential is the number of redox centers in the cluster and the redox state of the cluster. While rubredoxin has only one iron that simply switches between Fe2+ and Fe3+ states, the same transition differs significantly in a [4Fe–4S] cluster in an environment with three more irons and a mixed-valence state (e.g., 2Fe3+–2Fe2.5+ and Fe2.5+). Even the same cluster can undergo different redox transitions, as has been observed in the case of HiPIPs and ferredoxins.719
3.7.2. Role of Ligands
While sulfurs are the most dominant ligands in Fe–S proteins, it has been shown that other ligands can replace sulfurs in some cases and that these ligands play a prominent role in fine-tuning the reduction potential of the proteins.541 Generally speaking, ligands that are less electron-donating than sulfur will increase the reduction potentials by selectively destabilizing the oxidized state. A well-established example of this principle is the increased reduction potential of [2Fe–2S] clusters in Rieske proteins compared to ferredoxins due to replacement of two of the Cys ligands with His residues. Mutational studies on Cys ligands, mostly replacement with Ser, have shown an increased reduction potential compared to that of the wild-type (WT) proteins.721,750,773,1087
3.7.3. Role of the Cellular Environment
As mentioned earlier in this review, some Fe–S proteins such as vertebrate ferredoxins and certain [3Fe–4S] clusters and Rieske proteins show pH-dependent redox behavior. This behavior can be due to the presence of a protonable residue such as Asp or His residue as a ligand or near the active site.712,746,801 Therefore, proteins in the presence of different pH values in different cellular compartments should demonstrate different reduction potentials. Another effect of the environment is indirect through evolution: as shown in the case of ferredoxins, organisms subjected to extreme environments will undergo changes in the overall charges of proteins, which will affect the reduction potentials.823 Peptide models of different Fe–S clusters have demonstrated the impact of solvent composition in ET features of the cluster.717
3.7.4. Role of the Protein Environment
Several studies have shown the importance of the protein environment in fine-tuning the reduction potentials of metal centers. The protein environment is one of the, if not the, most important factors determining the reduction potential in Fe–S proteins because the general geometry and primary coordination of iron are very similar in this family of proteins. The protein environment conveys its effect via several routes.
3.7.4.1. Solvent Accessibility/Cluster Burial
Solvent accessibility has been shown to be a very important factor in the reduction potential for different metal centers, including Cu centers, hemes, and Fe–S clusters. As a general rule of thumb, the more buried a cluster, the higher or more positive the reduction potential will be. This is mainly due to the electrostatic destabilization of more positive charges in the clusters. Being more buried is proposed to be one of the most important reasons behind the difference between the reduction potentials of the [4Fe–4S] clusters in HiPIPs vs ferredoxins.618,749,752 Hydration of the cluster can influence the covalency of Fe–S bonds, hence affecting the reduction potential.901
Cluster burial can be accomplished through physical positioning of the cluster by covering it with more secondary structure elements or partially via more hydrophobic residues around the cluster. As discussed earlier, there are exceptions to this trend, and there are clusters that are significantly more solvent-exposed, but little reduction potential change is observed for them.875 It should be noted that cluster burial is dependent on the size of the protein, the location of the cluster, and the extent of solvent interaction, so it is difficult to make a fair comparison of the effect of cluster burial among different proteins.92
3.7.4.2. Secondary Coordination Sphere
While ligands in the primary coordination sphere are very important in tuning the reduction potentials of the Fe–S centers, the role of secondary coordination sphere interactions cannot be ignored. A mounting number of studies support the essential roles of these interactions in fine-tuning the reduction potentials.1088 In the case of Fe–S proteins, secondary coordination interactions are the major cause of differences in the reduction potentials within a class of proteins.887 The number of backbone to amide H-bonds has been shown to be important in redox potential differences between HiPIPs and ferredoxins.617,618 As described in each section, a conserved H-bonding pattern is observed in each subclass of ferredoxins, and this pattern differs from one subclass to another.718,719 Removal of some conserved H-bonds from this pattern is shown to be one of the main causes of different reduction potentials between different types of ferredoxins.718,719 Removal of conserved H-bonds in several cases resulted in a decrease in the reduction potential.773,780 It is important to mention that although H-bonds are important, they are not the sole cause of differences in the reduction potentials. Moreover, their analyses are complicated in some cases due to ambiguity in their assignment and variation in their number based on the environmental condition.92
3.7.4.3. Electrostatics and Local Charges
Local charges can selectively stabilize either the reduced or oxidized form of the cluster and influence the reduction potential. Many studies of the Fe–S proteins showed that although these proteins usually have conserved charged residues (such as positive charges in ferredoxins), these charges are mainly important for interaction with the redox partner, and usually their mutations do not cause significant changes in the reduction potential.749 In cases where these residues are very close to the cluster, unpredictable effects have been observed.611 However, the total charge of the cluster has been suggested to be an important factor influencing the higher reduction potential of Rieske proteins compared to ferredoxins.773 Mutational analysis on rubredoxins and thioredoxin-like ferredoxins confirmed an important role for the charges around the cluster in the reduction potential of the protein. There is convincing evidence for the role of backbone amides and partial positive charges in the reduction potential of Fe–S centers.887 It has been proposed that the diploes induced by the these backbone amides can influence the reduction potential of different clusters, such as HiPIPs and ferredoxins. The net protein charge and the dipole induced from backbone amides have been shown to be important in determining the reduction potential of HiPIPs.752,873,890
While all these features are important, it should be noted that none of them are the sole determinants of the reduction potential in Fe–S proteins, and it has been found that different features act as the major contributors to differences in the reduction potential between different classes of the Fe–S proteins. Even among members of a class, the same factor might not play the same role.
3.7.5. Computational Analysis of the Reduction Potentials of Fe–S Proteins
To further understand factors influencing the reduction potentials, computational methods have been developed for calculating the reduction potential of Fe–S proteins on the basis of their structures.591,887 One of these methods uses Gunner’s multiconformational continuum electrostatics method and has been calibrated using proteins with known structure and reduction potential.780 In another method a combined quantum-chemical and electrostatic calculation was used to generate predictions for reduction potentials. Poisson–Boltzmann electrostatic methods in combination with QM/MM studies have also been used to analyze the reduction potentials of Fe–S proteins.93 The PDLP method was applied to HiPIPs to analyze the effects of solvent accessibility on the reduction potentials of these proteins.92,719 B3LYP density functional methods have been used in combination with broken symmetry to analyze factors that are important in tuning the reduction potential of Rieske proteins.800 Broken symmetry in combination with hybrid density functional theory has also been used to characterize Rieske proteins.1089
4. Copper Redox Centers in Electron Transfer Processes
4.1. Introduction to Copper Redox Centers
Copper is the second most abundant transition metal in biological systems, next to iron.1090 In addition to their critical role in electron transfer process, copper-containing proteins catalyze a variety of reactions. In this section, we focus on copper proteins that merely function as ET mediators, which include blue or type 1 (T1) copper and CuA centers. A number of reviews on these two centers have appeared in the literature.94−104
Despite the lack of modern structural and computational methods, initial attempts to understand the structure and function of copper redox centers were very successful. This success was in part due to the strong colors and interesting magnetic properties displayed by these redox centers that allowed various spectroscopic studies. The blue copper proteins were so-named because they display an intense blue color, due to a strong absorption around 600 nm, first observed in the 1960s.1091,1092 It was found that this T1 copper protein also displayed an unusual EPR spectrum with narrow hyperfine splittings, suggesting the presence of Cu in a different ground state compared to the normal copper complexes.1093 The electronic structure of the blue copper center was further elucidated with low-temperature absorption, CD, MCD, single-crystal EPR, XAS, and computational studies.96,99,1094,1095 The results of all these studies demonstrated that the 600 nm band is associated with a S → Cu charge transfer transition and that the highly covalent nature of the Cu–S bond is responsible for the narrow hyperfine splitting in the EPR spectra. The crystal structure of poplar plastocyanin later confirmed that T1 copper proteins contain a copper site with an unusual geometry.1096
Although the existence of copper in cytochrome c oxidases (CcOs) has been known since the 1930s, the nature of the CuA centers was not established until much later due to the presence of heme cofactors that complicated interpretation of the spectroscopic results.1097 EPR and elemental analyses have revealed that two copper-binding sites exist in CcOs.1098−1100 MCD studies by Thomson and co-workers showed features at 475, 525, and 830 nm corresponding to a CuA center.1101,1102 Kinetic measurement of reoxidation of reduced CcO, performed by a flow-flash technique, indicated that the CuA is the ET center in CcO.1103,1104 From 1987 to 1993, Buse and co-workers performed chemical analysis of CcO with inductively coupled plasma atomic emission spectroscopy, leading to the conclusion that three copper atoms exist in one protein along with two hemes.1105,1106 Later, resonance Raman,1107 EXAFS,1108 and finally crystal structures1030,1109 revealed an unusual dinuclear copper structure for the CuA center, which will be discussed in detail in section 4.5.
4.2. Classification of Copper Proteins
As a diverse family of proteins, copper proteins could be divided into several types according to ligand sets, spectroscopic features, and functions (Table 9).1110,1111 Mononuclear T1 copper centers and dinuclear CuA centers are the two types which act only as ET mediators. T1 copper centers and CuA centers share several common features. First, both centers contain Cu–thiolate bond(s), which are highly covalent and display rich spectroscopic signatures.99,1095,1112−1115 Second, both centers are located in a cupredoxin fold.94,100,103 Finally, they are highly optimized for ET, showing low reorganization energies and high ET rate constants. These two types of copper proteins are collectively called cupredoxins, analogous to ferredoxin for Fe–S-based ET centers.1116 Other types of copper proteins may also involve ET as part of their enzymatic reactions, including peptidylglycine α-hydroxylating monooxygenase and dopamine β-monooxygenase,1117 but will not be discussed here.
Table 9. Different Types of Copper Proteinsa.
| mononuclear |
dinuclear |
tetranuclear | |||
|---|---|---|---|---|---|
| type 1 | type 2 | type 3 | CuA | CuZ | |
| UV–vis spectrum | strong absorption, ∼600 nm and (in some proteins) 450 nm | weak absorption, ∼700 nm | 300–400 nm | strong absorption, ∼480 and 530 nm | strong absorption, ∼640 nm |
| EPR spectrum | four-line (A|| < 80 × 10–4 cm–1) | four-line (A|| ≈ (130–180) × 10–4 cm–1) | nondetectable | seven-line (A|| ≈ 30–40 × 10–4 cm–1) | 2 × four-line (A|| ≈ 61 × 10–4 cm–1 and A|| ≈ 24 × 10–4 cm–1) |
| common ligands | His, Cys (Met) | His, Asp (Tyr) | His (Tyr) | His, Cys (Met) | His, S2– |
| active site geometry | trigonal pyramidal or distorted tetrahedral | distorted tetragonal | tetragonal | trigonal planar | m4-S2– tetracopper cluster |
| examples | azurin | superoxide | hemocyanin | cyt c oxidase | N2O reductase |
| plastocyanin | dismutase | tyrosinase | N2O reductase | ||
| stellacyanin | galactose oxidase | catechol | menaquinol NO reductase | ||
| nitrite reductase | amine oxidase | oxidase | |||
| nitrite reductase | laccase | ||||
| laccase | laccase | ||||
Reprinted with permission from ref (98). Copyright 2004 Elsevier.
4.3. Native Type 1 Copper Proteins
Exclusively serving as ET centers, T1 copper proteins are distinct from other copper proteins because of their unique geometry and ligand sets. The copper ion is normally coordinated to two histidines and one cysteine in a trigonal plane with the axial position often occupied by a methionine at a relatively longer distance. They contain a highly covalent copper–thiolate bond that imparts an intense blue color to the T1 centers, due to absorption at ∼600 nm, and narrow four-line hyperfine splitting in the EPR spectra.99,1118
The T1 copper centers reside in either single- or multiple-domain proteins.1119 The former includes the most common T1 copper proteins, such as plastocyanin, azurin, and amicyanin, while the latter includes stellacyanin, uclacyanin, and dicyanin. The T1 copper centers are also found in multicopper centers involving other types of copper centers, such as in nitrite reductases, laccases, and ascorbate oxidases. We will discuss the T1 copper centers in single- and multiple-domain proteins in this section, while the T1 copper centers in multicopper proteins will be discussed in section 4.3.4.
The T1 copper proteins are found in archaea, bacteria, and plants. In addition to the cupredoxin fold, genes containing the T1 copper proteins may contain other components (Figure 45). All T1 copper proteins have an N-terminal signal peptide or transit peptide. With the signal peptide, the T1 copper proteins from bacteria or archaea are directed into the periplasmic space. Their counterparts in plants, on the other hand, are transported to the extracellular milieu and anchored to the cell surface through an additional C-terminal hydrophobic sequence.1119 Plastocyanin is guided to the chloroplast in plant cells by a transit peptide sequence that is cleaved in the mature protein.1120
Figure 45.

Domain arrangement of type 1 copper protein. Reprinted with permission from ref (1119). Copyright 2006 Wiley-VCH.
4.3.1. Structures of the Type 1 Copper Proteins
The first crystal structure of the T1 copper protein, plastocyanin from poplar leaves (Populus nigra var. italica), was reported in 1978.1096 Since then, crystal structures of many other T1 copper proteins have been reported, as listed in Table 10. Despite the fact that sequence identity between the T1 copper proteins is less than 20%,1154 the overall structural folds of different T1 copper proteins are highly conserved. This common fold is called cupredoxin fold, which consists of eight β-strands arranged into a Greek key β-barrel as shown in Figures 46 and 47.94 There are also one to two α-helices in different locations outside the core fold of the protein. This fold is present not only in T1 copper proteins and the CuA domain,1155 but also in other copper proteins, such as Cu–Zn SOD,94,1156 and in proteins without metal cofactors, such as immunoglobins.94,1157
Table 10. Properties of T1 Copper Proteins.
| name | organism isolated from | first reported | PDB code for first structure | ligand set | Em (mV) | redox partner |
|---|---|---|---|---|---|---|
| Single Domain | ||||||
| azurin | bacteria | 19621121 | 1AZU | 1Cys, 2His, 1Met, 1 carbonyl oxygen | 3101122 | |
| amicyanin | methylotrophic bacteria | 19811123 | 1MDA | 1Cys, 2His, 1Met | 2601124 | methylamine dehydrogenase, cytochrome c551 |
| plastocyanin | plant/algae/cyanobacteria | 19601125 | 1PLC | 1Cys, 2His, 1Met | 3701126 | cytochrome f, P700+ |
| pseudoazurin | denitrifying bacteria and methylotrophs | 19731127 | 1PAZ | 1Cys, 2His, 1Met | 2801128 | nitrite reductase |
| rusticyanin | acidophilic bacteria | 19751129 | 1RCY | 1Cys, 2His, 1Met | 6701130 | cytochrome c, cytochrome c4 |
| auracyanin | photosynthetic bacteria | 19921131 | 1QHQ | 1Cys, 2His, 1Met | 2401131 | |
| plantacyanin | plants | 19741132 | 2CBP | 1Cys, 2His, 1Met | 3101133 | |
| halocyanin | haloalkaliphilic archaea Natronobacterium pharaonis | 19931134 | 1Cys, 2His, 1Met | 1831134 | ||
| sulfocyanin | acidophilic archaea Sulfolobus acidocaldarius | 20011135 | 1Cys, 2His, 1Met | 3001135 | ||
| nitrosocyanin | autotrophic bacteria | 20011136 | 1IBY | 1Cys, 2His, 1Glu, 1H2O | 851137 | |
| Multidomain Protein with T1 Center | ||||||
| stellacyanin | plants | 19671138 | 1JER | 1Cys, 2His, 1Gln | 1901133 | |
| uclacyanin | plants | 19981139 | 1Cys, 2His, 1Met | 3201139 | ||
| dicyanin | plants | 20001140 | 1Cys, 2His, 1Gln | |||
| Multidomain Protein with T1 Center and Other Copper Center | ||||||
| laccase | fungi | 1A65 | 1Cys, 2His (1Leu/Phe) | 465–7781141−1143 | ||
| Pplants | 1Cys, 2His, 1Met | 4341144,1145 | ||||
| ascorbate oxidase | plants | 1AOZ | 1Cys, 2His, 1Met | 3501146 | ||
| ceruloplasmin | animals | 19481147 | 1KCW | 1Cys, 2His (1Leu) | >10001148(redox-inactive) | |
| ceruloplasmin | 1Cys, 2His, 1Met | 4481149(redox-active) | ||||
| hephaestin | mammals | 19991150 | ||||
| Fet3p | yeast | 19941151 | 1ZPU | 1Cys, 2His | 4271152 | |
| nitrite reductase | plants, bacteria | 1NIA | 1Cys, 2His, 1Met, 1 carbonyl oxygen | 2601153 | ||
Figure 46.

Crystal structures of the T1 copper proteins. The secondary structure (α-helix and β-sheet) is shown in cartoon format, copper is shown as a purple ball, and ligands are shown in stick format. The name of the protein and its PDB ID are given below each structure.
Figure 47.
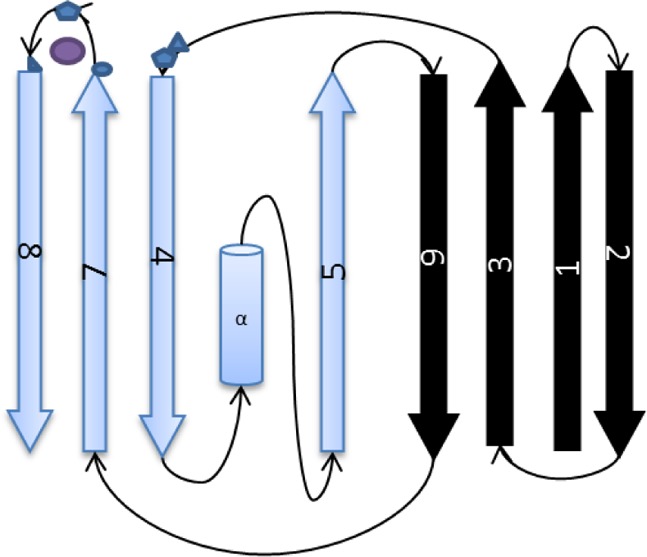
Topology diagram showing the scheme of the secondary structure of azurin. β-Strands are shown as arrows, and the α-helix is shown as a cylinder. Copper ligands between β-strands 3 and 4 and between β-strands 7 and 8 are shown as blue polygons, while copper is shown as a purple circle.
Most of the ligands to the T1 copper center resides at the C-terminal end of the cupredoxin fold. As shown in Figure 47, one of the His ligands is the first residue of the fourth β-strand and is referred to as N-terminal His. Carbonyl oxygen, the fifth ligand of azurin, is located in the loop between the third and fourth β-strands. Other ligands, including Cys, the second His on the trigonal plane, and the axial ligand, are located in or adjacent to the loop between the seventh and eighth β-strands, close to the C-terminus of the protein. Cys is the last residue of the seventh β-strand, while the second His is in the middle of the loop and is referred as the C-terminal His. Met is the first residue of the eighth β-strand. The three ligands are arranged in Cys-(Xxx)n-His-(Xxx)m-Met fashion, where n and m could vary between 2 and 4 in different T1 copper proteins. This variation in length and amino acid composition is important for the function of T1 copper proteins. In section 4.4.5 we discuss the implications of the variations based on loop-directed mutagenesis results.
While X-ray crystallography could give a fairly good description of the overall structure, EXAFS is more accurate in determining the metal–ligand distance because it is sensitive to oxidation state of the metal ion.1158 The short Cu–S distance was first revealed by EXAFS.99,1159 By comparing data from oxidized and reduced plastocyanin and azurin, it was found that an average increase of ∼0.06 and ∼0.08 Å for Cu–N(His) and Cu–S(Cys), respectively, happens upon reduction.99 These small changes upon reduction are consistent with data from crystallography and suggest a small reorganization energy for the redox process.
4.3.1.1. Copper Ligands
Even though the amino acid sequences and overall structures vary among different T1 copper proteins, the ligand composition, ligand–metal distance, and geometry of the T1 copper centers are almost identical (Figure 48).94,95,99 As the most conserved structural feature, T1 copper centers invariably contain two His residues and one Cys residue as equatorial copper ligands. In T1 copper proteins, the His coordinates with copper through Nδ, in contrast to Nε used by T2 and most other copper proteins. The Cu–His bond length is about 2.0 Å in T1 copper proteins, which is normal for such types of bonds. On the other hand, the Cu–Cys bond lengths range from 2.07 to 2.26 Å, which is short compared to those of normal copper complexes and other copper proteins (Table 11). The short Cu–S distance is key to the unique spectroscopic properties of T1 copper and is maintained through extensive H-bonding within the protein scaffold, as will be discussed later in this section. The 2N and 1S from His and Cys, respectively, form a pseudotrigonal plane, with average bond angles in the Cu(II) state being 101°, 117°, and 134° with RMS deviations of 2.5°, 4.1°, and 2.8°, calculated from crystal structures with resolution of 2.0 Å or higher.1119 The Cu–Sγ–Cβ–Cα and Sγ–Cβ–Cα–N dihedral angles are also consistently close to 180°, making the Cu–Sγ bond coplanar with the Cys side chain and backbone.
Figure 48.
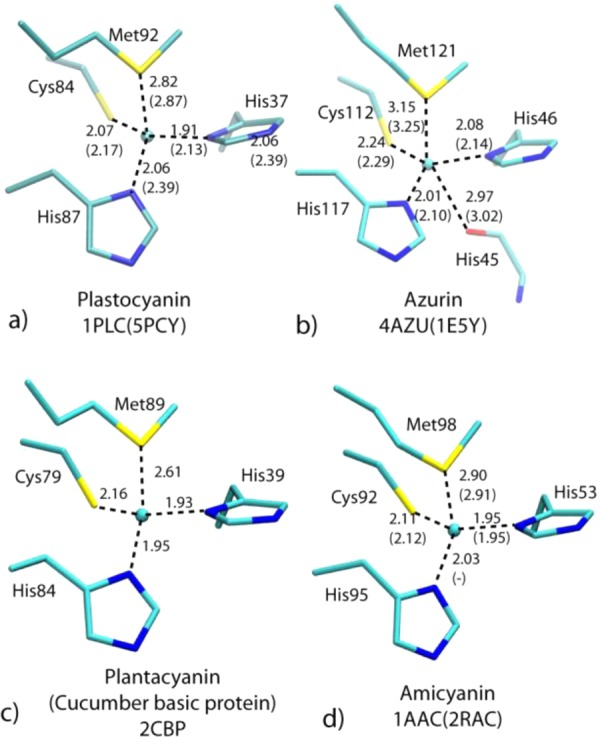
T1 copper centers in plastocyanin, azurin, plantacyanin, and amicyanin. Reprinted with permission from ref (1119). Copyright 2006 Wiley-VCH.
Table 11. Distances (Å) between Cu or Other Substituted Metals and Ligands in T1 Copper Proteinsa.
| P. aeruginosa azurin | pH | Cu–Nδ (His46)b | Cu–S (Cys112)b | Cu–Nδ (His117)b | Cu–S (Met121)b | Cu–O (Gly45)b | resolution (Å) | PDB ID | ref |
|---|---|---|---|---|---|---|---|---|---|
| Cu(II) | 5.5 | 2.08(6) | 2.24(5) | 2.01(7) | 3.15(7) | 2.97(10) | 1.9 | 4AZU | (1160) |
| Cu(I) | 5.5 | 2.14(9) | 2.29(2) | 2.10(9) | 3.25(7) | 3.02(8) | 2.0 | 1E5Y | |
| Cu(II) | 9.0 | 2.06(6) | 2.26(4) | 2.03(4) | 3.12(7) | 2.94(11) | 1.9 | 5AZU | (1160) |
| Cu(I) | 9.0 | 2.20(11) | 2.30(23) | 2.21(12) | 3.16(9) | 3.11(11) | 2.0 | 1E5Z | |
| T. ferrooxidans rusticyanin | pH | Cu–Nδ (His85) | Cu–S (Cys138) | Cu–Nδ (His143) | Cu–S (Met148) | – | resolution (Å) | PDB ID | ref |
| Cu(II) | 4.6 | 2.04 | 2.26 | 1.89 | 2.88 | – | 1.9 | 1RCY | (1161) |
| Cu(I) | 4.6 | 2.22 | 2.25 | 1.96 | 2.75 | – | 2.0 | 1A3Z | |
| P. nigra plastocyanin | pH | Cu–Nδ (His37) | Cu–S (Cys84) | Cu–Nδ (His87) | Cu–S (Met92) | – | resolution (Å) | PDB ID | ref |
| Cu(II) | 6.0 | 1.91 | 2.07 | 2.06 | 2.82 | – | 1.33 | 1PLC | (1162) |
| Cu(I) | 7.0 | 2.13 | 2.17 | 2.39 | 2.87 | – | 1.80 | 5PCY | (1163) |
| P. denitrificans amicyanin | pH | Cu–Nδ (His53) | Cu–S (Cys92) | Cu–Nδ (His95) | Cu–S (Met98) | – | resolution (Å) | PDB ID | ref |
| Cu(II) | 6.0 | 1.95 | 2.11 | 2.03 | 2.90 | – | 1.31 | 1AAC | (1164) |
| Cu(I) | 7.7 | 1.95 | 2.12 | unbound | 2.91 | – | 1.30 | 2RAC | (1165) |
| C. sativus cucumber basic protein | pH | Cu–Nδ (His39) | Cu–S (Cys79) | Cu–Nδ (His84) | Cu–S (Met89) | – | resolution (Å) | PDB ID | ref |
| Cu(II) | 6.0 | 1.93 | 2.16 | 1.95 | 2.61 | – | 1.80 | 2CBP | (1166) |
| C. sativus stellacyanin | pH | Cu–Nδ (His46) | Cu–S (Cys89) | Cu–Nδ (His94) | – | Cu–O (Gln89) | resolution (Å) | PDB ID | ref |
| Cu(II) | 7.0 | 1.96 | 2.18 | 2.04 | – | 2.21 | 1.60 | 1JER | (1167) |
Adapted with permission from ref (104). Copyright 2012 Elsevier.
Average of distances for four molecules in the asymmetric unit. Errors are 1 standard deviation.
The axial ligand in the T1 copper center is less conserved. A Met is present at 2.6–3.2 Å in this axial position in most proteins, while a Gln is found in stellacyanin and dicyanin. In the T1 center of fungal laccase and ceruloplasmin, a noncoordinating ligand such as Phe or Leu takes this axial position. In azurin, there is an additional backbone carbonyl oxygen at the opposite end of the axial position to Met, giving the T1 copper site a trigonal bipyrimidal geometry.
4.3.1.2. Secondary Coordination Sphere
While the above mentioned ligands exert significant influence on the properties of T1 copper centers, the protein scaffold should not be viewed as a passive entity to hold the copper site. On the contrary, it can play important roles. First, it can shield the copper site from water, raising the reduction potential and lowering the reorganization energy for ET. More importantly, the extensive H-bond network surrounding it can fine-tune the properties of the T1 copper site.94,98
As shown in Figure 49, the Cys112 in azurin forms two hydrogen bonds with adjacent backbone amide groups of Asn47 and Phe114 at ∼3.5 Å. Together with S–Cu and S–Cβ covalent bonds, these H-bonds form a tetrahedral geometry around Sγ of Cys (Figure 49A). Plastocyanin, pseudoazurin, and amicyanin have only one H-bond around the Cys as a Pro in the site eliminates the other amide bond. Additionally, cucumber basic protein has a very weak H-bond at 3.7–3.8 Å. These H-bonds modulate the electron density of S on Cys, which is crucial for the highly covalent nature of the Cu–S bond.
Figure 49.
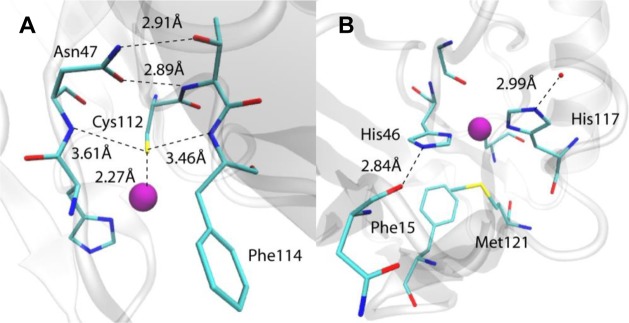
H-bonding around Cys112 (A) and other ligands (B) of azurin. PDB ID 4AZU.
In azurin, the N-terminal His coordinates with Cu through Nδ, whereas Nε is hydrogen-bonded to the carbonyl oxygen of Phe15. The same His is hydrogen-bonded to the Gln49 side chain in amicyanin, the side chain of Asn80 in rusticyanin, and a water molecule in phytocyanins. The C-terminal His is in a hydrophobic patch of the protein packed against other residues. The Nε of C-terminal His is hydrogen-bonded to a water molecule. The axial Met/Gln usually packs against aromatic side chains such as Phe15 in azurin (Figure 49). In azurin, the carbonyl oxygen is held in place by the secondary structure of the loop and packs with Phe114.
There are more H-bonding interactions beyond the copper center. For example, an Asn close to the N-terminal His in the first ligand loop is hydrogen-bonded to residues from the other ligand loop. This interaction, acting like a zipper, further holds the copper site together.
Extensive H-bonding around the copper site in T1 copper proteins has important functional implications, as we will address in section 4.4.2.
4.3.1.3. Comparison of Structures in Different States
As suggested by the “rack mechanism” 1168,1169 or entatic state,1170 the active site structure is predetermined by the protein scaffold. Thus, there is little change in the structures of T1 copper proteins at different oxidation states, with different metals, or even in the absence of metal ions.
As shown in Table 11, compared to the same protein with Cu(II), the metal to ligand bonds elongated by 0.1 Å or less in protein containing Cu(I). Similar results were obtained by EXAFS, which provides a more accurate determination of the bond length.99 The small change in bond length is crucial for the low reorganization energy of the T1 copper site and, thus, fast ET for its function. However, bond lengths in X-ray crystal structures should be interpreted with caution, as it has been shown that Cu(II) ions in protein undergo photoreduction during X-ray exposure.1171,1172 It will be useful to conduct single-crystal microspectrophotometry concurrent with X-ray diffraction to make sure that the oxidized protein is not reduced during diffraction.1173 On the other hand, the oxidation state of the Cu ion can be easily monitored at the edge and XANES regions of its X-ray absorption spectrum. Bond lengths derived from carefully designed and conducted EXAFS should reflect the actual bond lengths at the corresponding oxidation states.
Besides structures with copper in oxidized or reduced states, crystal structures of apo and metal-substituted T1 copper proteins also shed light on how proteins interact with copper. Structures of apo forms of azurin,1174,1175 plastocyanin,1176 pseudoazurin,1177 and amicyanin1178 show little difference (0.1–0.3 Å) from that of the copper-bound form, confirming the entatic state hypothesis.
Metal substitution is useful in spectroscopic studies, such as electronic absorption1118,1179 and NMR.1180 Due to the different sizes and ligand affinities of different metals, the bond length and overall geometry are changed upon substitution, but only to a small extent due to confinement of the protein scaffold.1181−1183
4.3.2. Spectroscopy and Electronic Structure
Intense (∼5000 M–1 cm–1) electronic absorption at ∼600 nm is the hallmark of T1 copper proteins (Figure 50). Solomon and co-workers attributed the origin of the ∼600 nm absorption to the S(Cys)pπ → Cux2–y2 LMCT transition.1094,1184,1185 Another feature at ∼450 nm is not prominent in plastocyanin or azurin, but is more pronounced in a perturbed T1 copper sites such as that of cucumber basic protein. This absorption is attributed to S(Cys)pπ → Cux2–y2 LMCT. The geometry of the copper site is believed to be important for the ratio between the two peaks at ∼600 and ∼450 nm.1095,1186 A series of weak absorption peaks from 650 to 1050 nm are attributed to a d → d transition or ligand field transition.1184
Figure 50.
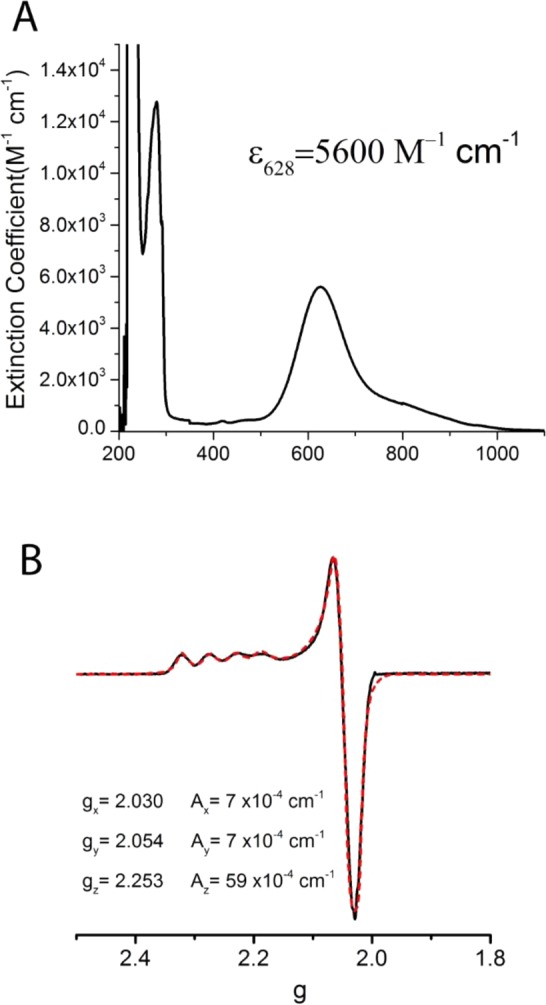
Electronic absorption (A) and EPR (B) spectra of azurin.
EPR provides a sensitive way to determine the copper site geometry. T1 copper proteins exhibit a distinctive small hyperfine splitting (<100 × 10–4 cm–1) in the EPR spectrum, as opposed to that of T2 copper and other complexes (>150 × 10–4 cm–1).1119 Through S K-edge XAS, Solomon and co-workers showed that the small hyperfine splitting is due to high covalency between Cu and S, which delocalizes unpaired electrons onto S, thus decreasing the electron density on Cu.1187
Other spectroscopic techniques, such as resonance Raman spectroscopy and Cu L-edge and S K-edge XAS, have also been important in deciphering the electronic structures of T1 copper proteins. They are beyond the scope of this review, but there are excellent reviews elsewhere1095,1119 and in this issue that cover more details about these techniques.2000
4.3.3. Redox Chemistry of Type 1 Copper Protein
As a class of proteins dedicated to ET, T1 copper proteins display various features for facile redox chemistry.
4.3.3.1. Redox Partner
T1 copper proteins shuttle electrons between donor and acceptor proteins as redox partners. So far five T1 copper proteins with known physiological redox partners have been identified: plastocyanin, amicyanin, rusticyanin, pseudoazurin, and azurin. As an electron carrier in chloroplasts in plants, plastocyanin accepts electrons from cytochrome f of membrane-bound cytochrome b6f complex and transfers them to P700+ in photosystem I.256,1188−1192 Amicyanin accepts electrons from methylamine dehydrogenase and transfers them to cytochrome c oxidase via a c-type cytochrome.279,1193−1200 Rusticyanin is suggested to shuttle electrons between cytochrome c and cytochrome c4.1201,1202 Pseudoazurin reduces nitrite reductase, but its electron donor is not yet known.1203−1207 Azurin is likely to interact with aromatic amine dehydrogenase in vivo, as suggested by coexpression, the kinetics of reduction, and the crystal structure.1208−1210
Interaction between a T1 copper protein and its redox partner is generally weak and transient. NMR and crystallographic studies have revealed a structural basis for this interaction. Interactions between plastocyanin from various organisms and cyt f have been extensively studied by NMR spectroscopy (Figure 51). Chemical shift analysis and rigid-body structure calculations have demonstrated that the hydrophobic patch around His87, the C-terminal His ligand to copper, mediates the interaction between plastocyanin and cyt f.1211,1212 Besides that, two acidic patches around Tyr83 have been shown to interact with positively charged residues of cyt f.1213 Mutation of Tyr83 to Phe or Leu drastically decreases the ET rate between the two proteins, indicating that Tyr83 is involved in binding to cyt f and ET.1214 The absence of acidic patches also demolishes ET activity at low ionic strength, showing they are involved in the interaction with cyt f.1215,1216 However, interaction between acidic patches and cyt f is not very specific as small changes in acidic patches have a minimal effect on the interaction between two proteins.1216,1217
Figure 51.
Structures of plastocyanin (left) and the complex of plastocyanin and cyt f (right). Left: copper ion is represented as a purple ball, His87 and Tyr 83 are represented in licorice format, and residues in two acidic patches are represented as ball and stick models. Right: plastocyanin is colored cyan, and cyt f is orange. The copper ion and His87 from plastocyanin and heme from cyt f are also shown.
Another demonstration of the interaction between the T1 copper proteins and their redox partners comes from X-ray crystallography. The structures of the amicyanin–methylamine dehydrogenase complex and methylamine dehydrogenase–amicyanin–cytochrome c551 ternary complex have been determined.279,1196 These structures further confirmed that the hydrophobic patch surrounding His95 (the C-terminal His ligand equivalent to His87 in plastocyanin and His117 in azurin) interacts with a hydrophobic patch on methylamine dehydrogenase. An ET pathway from Trp57 and Trp108 in methylamine dehydrogenase to His95 in amicyanin and eventually to copper has been proposed from these structures.
Recently, the crystal structure of the azurin and aromatic amine dehydrogenase complex from Alcaligenes faecalis has been solved.1208 In this structure, only one azurin molecule is present in complex with four molecules of aromatic amine dehydrogenase. The B factor of the azurin structure is high except for those residues in the interface. This result is consistent with the transient nature of the interaction between the T1 copper proteins and their redox partners. The interaction is very similar to the one between amicyanin and methylamine dehydrogenase.
The T1 copper proteins show promiscuity in reacting with proteins other than their physiological redox partners,64,1218 including small inorganic complexes such as [Fe(CN)6]3– and [Co(phen)3]3+,31,44,1219 small molecules such as flavins and ascorbate, and the proteins themselves through electron self-exchange reactions.100 Gray and co-workers have used Ru derivatives of T1 copper proteins as a model to study long-range ET in biological systems.24,31,44,2005
4.3.3.2. Electron Transfer Rate
T1 copper proteins are involved in long-range ET in vivo and in vitro. For a more detailed review of long-range ET, please refer to the review in this issue by Gray et al.2005 The process can be described by the semiclassical Marcus equation:
| 1 |
In this equation, ΔE° is the difference in reduction potential between the donor and acceptor sites (also known as the driving force), HAB is the donor–acceptor electron coupling or electron matrix coupling element, and λ is the reorganization energy required for ET. Under the same driving force, the rate is maximized when HAB is large and λ is small. In long-range ET, there is little direct coupling between the donor and the acceptor. The coupling is mediated by intervening atoms via the superexchange mechanism. HAB is determined by the distance between the donor and acceptor and the covalency of the metal–ligand bond.1220−1222
Electron transfer rates between T1 copper proteins and their redox partners have been measured by kinetic UV–vis spectroscopy or cyclic voltammetry.1223−1226 The kET between plastocyanin and cyt f has been determined to be 2.8–62 s–1,1227−1229while the constant between plastocyanin and P700+ has been determined to be 38–58 s–1.1191,1192,1230,1231 Davidson and co-workers have used kinetic UV–vis spectroscopy to measure the kET between amicyanin and methylamine dehydrogenase, which was determined to be ∼10 s–1.1232,1233 Suzuki and co-workers have determined the kET between pseudoazurin and nitrite reductase to be (0.8–7) × 105 M–1 s–1 by kinetic UV–vis spectroscopy or cyclic voltammetry.1204,1224,1234−1236
As several studies have pointed out, the rate constant measurement for interprotein ET processes is complicated by other processes, such as multiple binding sites of the two proteins, transient formation of conformational intermediates, and protonation/deprotonation processes.1225,1237 There are two methods to measure the ET rate in T1 copper proteins without involvement of a redox partner: pulse radiolysis and NMR. Pulse radiolysis1238 uses a short pulse (typically 0.1–1 μs) of high-energy (2–10 MeV) electrons to excite and decompose solvent molecules. A typical reaction generates the CO2•– radical:
Radicals generated in solvent molecules trigger downstream reactions. In azurin, CO2– can reduce either Cu(II) or the disulfide bond between Cys3 and Cys26 at a nearly diffusion-controlled rate. Molecules with a reduced disulfide bond (RSSR–) can further reduce Cu(II) in the same protein via intramolecular ET:101
By monitoring absorbance changes at 410 nm (RSS•R–) and 625 nm (Cu(II)), a fast reduction process corresponding to reduction of Cu(II) or RSS•R– by CO2•– and a slower process of intramolecular ET between RSSR and Cu(II) can be resolved. The ET rate and driving force (ΔG°) can be calculated from the kinetics of intramolecular ET. By running experiments at different temperatures, the activation enthalpy and activation entropy of the ET process can be calculated.
Using this method, Farver and Pecht determined the rate constant of intramolecular ET of WT azurin to be 44 ± 7 s–1 at pH 7.0 and 25 °C with a driving force ΔG° = −68.9 kJ mol–1. The activation enthalpy and activation entropy were calculated to be 47.5 ± 4.0 kJ mol–1 and −56.5 ± 7.0 J K–1 mol–1.1239 ET rates for azurin of different origins and mutations have been measured and reviewed by Farver and Pecht.101
Electron self-exchange is an intrinsic property of all redox systems.1240 Exchange of electrons happens to two molecules of the same complex at different oxidation states. Only one redox couple is involved, and there is no driving force for this reaction. Measuring electron self-exchange rate constants by NMR provides a more universal way to measure ET transfer activity as it is carried out in T1 copper centers1241−1249 (reviewed in ref (100)) as well as in other redox centers.1250−1252 Electron self-exchange rate constants (kSES) of T1 copper proteins range from 103 to 106 M–1 s–1 at moderate to low ionic strength. The electron self-exchange is thought to happen through a hydrophobic patch as the rate constant is affected by the presence of an acidic patch1248 or basic residues1253 close to the hydrophobic patch.
4.3.3.3. Reduction Potential
T1 copper proteins have reduction potentials ranging from 183 to 800 mV (see Table 10). Compared to the aqueous Cu(I)/Cu(II) couple (which has a reduction potential of ∼150 mV), copper complexes, and other copper proteins, T1 copper proteins have unusually high reduction potentials. Their potentials also span a wide range (>600 mV), nearly half the range of biologically relevant potentials (Figure 1). Within the T1 copper proteins, groups of proteins are apparent when sorted on the basis of the midpoint reduction potential (Em). Nitrite reductases,1153 stellacyanins,1133 amicyanins,1124 and pseudoazurins1128 natively have substantially lower (∼100 mV) Em values as compared to azurin.98 Azurin and umecyanins have moderate Em values natively around 200–300 mV vs SHE. On the other end of the scale, rusticyanins have Em values ∼400 mV higher than that of azurin. Understanding the origin of this variance and the structural features involved in tuning the reduction potential are of great importance. By comparing the native proteins with different axial ligands (Table 12), it is revealed that proteins with Gln as an axial ligand generally have lower reduction potentials (190–320 mV), proteins with Met axial ligands have higher potentials (183–670 mV), and proteins with a noncoordinating ligand in multicopper proteins have the highest potentials (354–800 mV). This trend is further confirmed by mutagenesis studies that are discussed in section 4.4.1.
Table 12. Dependence of E° on the Axial Ligand in Blue Cu Proteinsa.
|
E° (mV) |
||||
|---|---|---|---|---|
| Phe/Leu/Thr | Met | Gln | ref | |
| fungal laccase | 770 | 680 | (1254−1256) | |
| azurin | 412 | 310 | 285 | (1122, 1257) |
| cuc. stellacyanin | 500 | 420 | 260 | (1139) |
| nitrite reductase | 354 | 247 | (1258) | |
| rusticyanin | 800 | 667 | 563 | (1259) |
| mavicyanin | 400 | 213 | (1260) | |
| amicyanin | 250 | 165 | (1261) | |
Reprinted from ref (99). Copyright 2004 American Chemical Society.
Variation within proteins containing the same axial ligand indicates that there are more factors affecting the reduction potentials of the T1 copper center. These factors have been uncovered by mutagenesis studies and engineering of copper proteins and are discussed in section 4.4.
4.3.4. T1 Copper Center in Multicopper Proteins
The T1 copper center exists not only in single-domain proteins, but also in multidomain proteins with multiple copper cofactors. These proteins include multicopper oxidases and nitrite reductases (Table 9). The former contain a T1 copper (blue copper), a type II copper (abbreviated as T2), and a pair of type III copper centers (Figure 53).1262−1266 The latter contain T1 and T2 copper centers and are evolutionarily related to the multicopper oxidases.1265−1267 As shown in Figure 52, multicopper oxidases and nitrite reductases are closely related and are composed of two, three, or six domains.1265 In multicopper oxidases, the T1 copper center resides in the cupredoxin-like domain while the T2 and T3 copper centers are located between domains.
Figure 53.
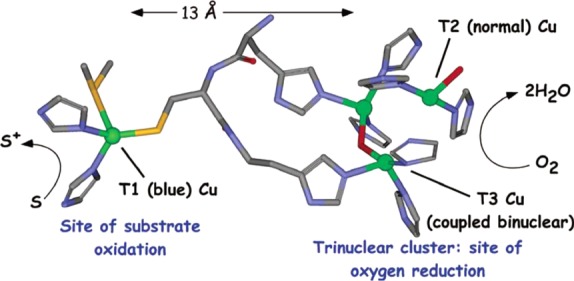
Active site of the multicopper oxidases. Cu sites are shown as green spheres. Figure generated from the crystal structure of ascorbate oxidase (PDB ID 1AOZ). Reprinted from ref (1264). Copyright 2007 American Chemical Society.
Figure 52.

Domain organization and copper center distribution in multicopper oxidases. Reprinted with permission from ref (1265). Copyright 2011 Wiley-VCH.
T1 copper centers in multicopper oxidases (MCOs) are very similar to those in single-domain T1 copper proteins. The copper ion is coordinated by one Cys residue and two His residues at its equatorial positions. In plant laccases, ascorbate oxidases, and nitrite reductases, axial Met coordinates with copper and forms a trigonal pyramidal geometry. In fungal laccase, ceruloplasmin, and Fet3p, the axial ligand is a noncoordinating Leu or Phe, leaving equatorial ligands and copper in a more trigonal geometry.1262,1265,1266 One noticeable feature for T1 copper centers in MCOs is their high reduction potential compared with that of single-domain T1 copper proteins. Ceruloplasmin has the highest reduction potential1148 (>1000 mV) reported in T1 centers, while TvLac has the second highest reduction potential1141−1143 (778 mV). The high reduction potential is partially attributed to the more hydrophobic axial ligand, while other factors such as hydrogen bonding around the T1 Cu centers may contribute too.1268
4.3.5. A Novel Red Copper Protein—Nitrosocyanin
Recently, a mononuclear red copper protein, nitrosocyanin from N. europaea, an ammonia oxidizing bacterium, was isolated and structurally characterized (Figure 54).1137,1269−1271 The crystal structure shows that the copper ion is coordinated by two His residues, one S(Cys), and a side chain O(Glu) and has an additional fifth water ligand in the oxidized form, but not in the reduced form. Nitrosocyanin shows a strong absorption band at 390 nm (ε = 7000 M–1 cm–1), a large hyperfine splitting value (147 × 10–4 cm–1) in the EPR spectrum, and a very low reduction potential of 85 mV (compared with those of the T1 copper proteins, which are in the range of 150–800 mV).1137,1271 With an exogenous water ligand, the reorganization energy of this protein is calculated to be 2.2 eV, significantly higher than those of T1 copper proteins.1271 Similar to T1 copper proteins, nitrosocyanin has copper–thiolate coordination and strong UV–vis absorbance. However, the water ligand in nitrosocyanin has not been observed in T1 copper proteins before. Its copper site geometry and absorption at ∼400 nm are also different from those of T1 copper proteins. Its EPR spectrum, reorganization energy, and reduction potential more closely resemble those of T2 copper proteins. Solomon and co-workers attribute these properties to the relative orientation of the Cu-N-N-S and Cu-S-Cβ planes, which in turn is due to “coupled distortion” between the axial ligand and the whole copper center.1095,1186,1271
Figure 54.
Crystal structures of (A) the oxidized red copper site in nitrosocyanin, (B) the oxidized T1 copper site in plastocyanin, and (C) the reduced red copper site in nitrosocyanin. Reprinted from ref (1271). Copyright 2005 American Chemical Society.
The biological role of this protein, however, has not yet been identified. It has been proposed that it might be involved in ET or serve some as-yet-unknown catalytic function due to the presence of the open coordination site.1269,1270
4.4. Structural Features Controlling the Redox Chemistry of Type 1 Copper Proteins
Although the study of native proteins provides valuable information about the structure, spectroscopy, and function of T1 copper centers, it is difficult to draw any conclusion only by comparing copper centers from different scaffolds with low sequence homology. With the advancement of modern molecular biology, powerful tools such as mutagenesis are available to research groups, allowing the amino acid sequence to be modified at will. Methods of unnatural amino acid mutagenesis have further expanded the toolbox for bioinorganic chemists.1272−1274 With these methods, not only amino acid residues directly coordinating to copper, but also residues beyond the first coordination sphere have been changed. Mutagenesis reveals how different components of the protein contribute to the structure, spectroscopy, and function, especially in reduction potential tuning.
4.4.1. Role of Axial Met
The T1 copper center has highly conserved equatorial ligands, two His residues and one Cys residue. The axial position of the T1 copper center shows more variation, as Met, Gln, and noncoordinating residues can all be found in the native proteins. Mutagenesis of the axial ligand has been carried out in azurin,1122,1275−1278 nitrite reductase,1235,1258,1279 amicyanin,1261 rusticyanin,1259 pseudoazurin,1234 laccase,1256 and stellacyanin.1139,1280,1281 Mutation of the axial ligand in different T1 copper proteins generally results in a protein that retains copper-binding ability but with a different reduction potential or altered spectroscopic properties. An early work replaced Met121 in azurin with all other 19 amino acids with minimal alteration of the T1 character of the copper center.1276 While changing the axial ligand to hydrophobic ligands such as Ala, Val, Leu, or Ile increases the reduction potential by 40–160 mV,1122 substitution with Glu or Gln decreases the reduction potential by 100–260 mV.1122,1257 As the axial ligand is changed from Gln to Met to more hydrophobic residues, the reduction potential of the protein increases. Theoretical studies have suggested that the axial ligand is involved in tuning the potential.1282,1283 To test the role of the axial ligand in tuning the reduction potential of the T1 copper protein, Lu and co-workers incorporated unnatural amino acid analogs of Met with different hydrophobicities at the axial position in azurin.1284,1285 The reduction potential varied from 222 to 449 mV at pH 4.0. Such a replacement of Met with its iso-structural analogs allowed conclusive identification of hydrophobicity of the axial ligand as the major factor in tuning reduction potentials, because a linear correlation was found between the reduction potential and hydrophobicity of the axial ligand. Likewise, Dennison and co-worker mutated the axial Met of cucumber basic protein to Gln and Val. As the axial ligand was changed from Gln to Met to Val, the electron self-exchange rate increased by 1 order of magnitude, and the reduction potential increased by ∼350 mV.1286 These studies have firmly established a correlation between hydrophobicity of the axial ligand and reduction potential, providing a better understanding of the role of the axial ligand in reduction potential tuning.
Within T1 copper proteins, there are two classes of proteins with slightly different spectroscopic features. Typical T1 copper proteins, such as plastocyanin and azurin, have absorption at ∼600 nm and an axial EPR signal, whereas “perturbed” T1 copper proteins or green copper proteins have an additional ∼400 nm absorption peak in their UV–vis spectra, as well as rhombic EPR signals. At the same time, the perturbed T1 copper proteins have longer Cu–S(Cys) distances and shorter Cu–axial ligand distances.1283 A more extreme case comes from the newly discovered protein nitrosocyanin, which has a cysteine ligand and dominant ∼400 nm absorption in its UV–vis spectrum, resulting in a red color.1137,1271 Although the strong absorption and 1Cys/2His/1Glu ligand set resembles those of T1 copper proteins, nitrosocyanin has large hyperfine splittings (A|| ≈ 150 × 10–4cm–1) in its EPR spectrum and a low reduction potential (85 mV), which falls into the range of T2 copper proteins.1136,1137,1271 Solomon and co-workers proposed coupled distortion theory on the basis of a suite of spectroscopic studies in combination with theoretical calculations to explain the variance in electronic absorption and concomitant color change from blue to green to red in native proteins. This theory states that shorter Cu–axial ligand distances result in distortion of the T1 copper geometry toward tetragonal, which elongates the Cu–S(Cys) distance.1283 This distortion renders the pσ(Cys)–Cu CT more favorable than pπ(Cys)–Cu CT, which causes an increase in the ∼400 nm absorption over the ∼600 nm absorption in the UV–vis spectrum. Mutational studies on the axial ligand of various T1 copper proteins have validated the coupled distortion theory. By changing a weak Met to a stronger His1277,1287,1288 or Glu ligand,1289−1291 the blue copper protein azurin can be converted to a green copper protein. By mutating Met to a weaker ligand such as Thr, the natively green copper protein, nitrite reductase, has been converted to a blue copper protein.1292 Recently, the axial Met was mutated to Cys, a strong ligand, and then to the unnatural amino acid homocysteine (Hcy), a strong ligand with a longer side chain. The resulting Met121Cys azurin has an additional ∼450 nm absorption, while in Met121Hcy the ∼410 nm peak dominates over the ∼625 nm peak. Together with EPR evidence, it was shown that, within the same scaffold, blue copper protein azurin was converted to a green copper protein and then to a red copper protein.1293 Interestingly, the engineered red copper protein, Met121Hcy azurin, has a low reduction potential (113 mV) similar to that of nitrosocyanin (85 mV).
4.4.2. Role of His Ligands
Although equatorial His residues are highly conserved in T1 copper proteins, their mutation does not impair the copper binding ability of the protein. Canters and co-workers mutated two His residues into Gly separately, and the resulting protein still had T1 characteristics.1294,1295 As His to Gly mutation creates extra space around copper, exogenous ligands such as halides, azides, and imidazoles could diffuse into His46Gly and His117Gly azurins and coordinate with copper. Depending on the type of external ligand, the mutants will be either T1 or T2 copper proteins.1294−1296 His117Gly and His46Gly mutations also changed solvent exposure of the copper site. Without external ligands, His117Gly azurin has a reduction potential of 670 mV, much higher than that of WT azurin (310 mV). The high reduction potential is due to loss of a water ligand during reduction. Addition of external ligands will lower the reduction potential.1297 The open coordination site of His117Gly makes it possible to study ET using imidazole-modified complexes.1298,1299 The mutants generally exhibit a lower ET rate. As the properties of exogenous imidazoles affect the ET rate, it has been suggested that His is also important in the WT protein.1300
4.4.3. Role of Cys Ligands
As the Cu–S(Cys) bond defines the properties of type I copper sites,99 mutation of Cys to other natural amino acids will dramatically alter the copper site in T1 copper proteins (Figure 55). Substitution of any other amino acid for Cys will result in loss of the intense LMCT bands, which is due to the interaction of the Cys S with copper. As an isostructural analogue of Cys, selenocysteine (SeC) can replace Cys without major structural perturbation. This strategy has been employed as a spectroscopic probe for T1 copper centers.1301−1303 The protein Cys112SeC azurin showed a reduction potential similar to that of WT azurin (328 mV vs 316 mV at pH 4) and a red-shifted LMCT band at 677 nm.1301 So far, only Cys112Asp mutation in azurin has been characterized. Mutation of Cys to Asp makes azurin a T2 copper protein, as evidenced by large hyperfine splitting (A|| ≈ 152 × 10–4 cm–1) in the EPR spectrum and slow ET.1304−1307 Addition of another mutation at the axial position, Met121Leu (Phe/Ile), results in a novel copper center called type 0 copper, which has the small parallel hyperfine splittings and rapid ET characteristic of T1 copper centers but no longer fits the classification of T1 copper due to the loss of the copper–thiolate interaction.1308−1311 Moreover, there is only a slight increase of the reorganization energy to 0.9–1.1 eV compared with that of WT azurin, much less than that of T2 copper proteins. The ET rate of type 0 copper protein is 100-fold faster than that of the Cys112Asp mutant, a typical T2 protein.1308,1309,1311
Figure 55.
Active sites of type 2, type 1, and the newly constructed type 0 copper. In the center, a plot shows (in the shaded ovals) the typical values of two electron paramagnetic resonance spectroscopy parameters, A∥ and g∥, for type 1 (lower) and type 2 (upper) copper sites and the values of type 0 copper (green, red, and black points, right center), showing that type 0 copper does not fall into the typical ranges for these other kinds of sites. Reprinted with permission from ref (1308). Copyright 2009 Macmillan Publishers Ltd.
4.4.4. Role of Structural Features in the Secondary Coordination Sphere
Copper ligands exert a significant influence on the spectroscopic features and reduction potentials of T1 copper proteins. However, copper ligands cannot fully account for variation in the reduction potentials of T1 copper proteins. Mutation of copper ligands usually results in loss of T1 characteristics or reduction of ET activity. For the limited mutations that maintain T1 characteristics and ET activity, the reduction potential is tuned over a 227 mV range by introducing Met analogues at the axial position, which is far less than the 600 mV range reported in native proteins.1285 As discussed in section 4.3.3, the H-bonding network beyond the T1 copper center plays an important role in maintaining the structure and function of T1 copper centers. Mutagenesis studies focusing on changes of hydrogen bonds have revealed important information about how the reduction potential and other properties are tuned in T1 copper proteins.
Rusticyanin has a higher potential relative to other T1 copper proteins. By sequence comparison, it is identified that there is a Ser in rusticyanin at the position corresponding to Asn that “zips” two ligand loops together. Asn has been proposed to decrease the Em by strengthening the H-bonding interactions between two ligand-containing loops. Mutating Ser86 in rusticyanin to Asn established such a hydrogen bond and lowered the Em by 77 mV.1312 On the other hand, changing Asn in azurin to Ser eliminates one hydrogen bond between two loops (Figure 56) and results in a protein with a 131 mV higher reduction potential.1293
Figure 56.
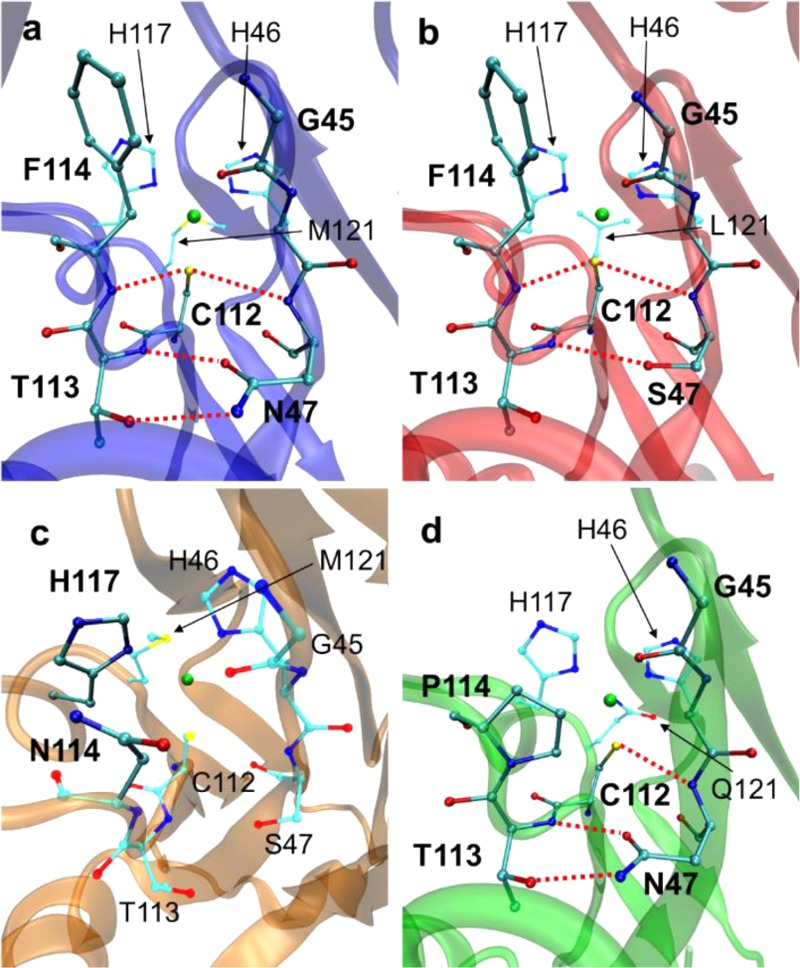
X-ray structures of Az and selected variants. (a) Native azurin (PDB ID 4AZU). (b) N47S/M121L azurin (PDB ID 3JT2). (c) N47S/F114N azurin (PDB ID 3JTB). (d) F114P/M121Q azurin (PDB ID 3IN0). Copper is shown in green, carbon in cyan, nitrogen in blue, oxygen in red, and sulfur in yellow. Hydrogen-bonding interactions are shown by dashed red lines. Reprinted with permission from ref (1088). Copyright 2009 Macmillan Publishers Ltd.
By comparing certain cupredoxins that natively have lower Em than the rest of the family, it is observed that they share a conserved Pro residue that is two residues after the copper-ligating Cys.114,1313 The backbone amide in the equivalent residue in azurin hydrogen bonds to the thiolate of Cys112.1160 Placing a Pro in this position converts this secondary amide to a tertiary amide, which is incapable of donating a hydrogen bond. The Phe114Pro mutant has a lower reduction potential.114 It is proposed that deleting the hydrogen bond to the thiolate gives Cys112 more conformational freedom, and it allows for the electron density that was previously tied up in a hydrogen bond to contribute to the Cu–SCys interaction.114
Another examination of cupredoxin crystal structures reveals the presence of backbone carbonyl oxygen from Gly45 near the copper ion in azurin, which is missing in other cupredoxins such as rusticyanin.97,98,1314 This ionic interaction in azurin is proposed to result in higher electron density near the copper, preferentially stabilizing the Cu(II) form of the protein and, therefore, lowering the Em.98,481,1315 Phe114Asn mutation was made to hydrogen bond with Gly45 backbone carbonyl and decrease the effect of carbonyl oxygen in Az. The mutant showed a 129 mV higher reduction potential compared to that of the wild type.1088
With all of these individual factors in mind, Lu and co-workers combined mutations on both the copper ligands and residues in the secondary coordination sphere. These mutations showed an additive effect on the reduction potential in azurin. With different combinations, the reduction potential was tuned from 90 to 640 mV, which is beyond the reported range of native T1 copper proteins and their mutants (Figure 57).1088
Figure 57.
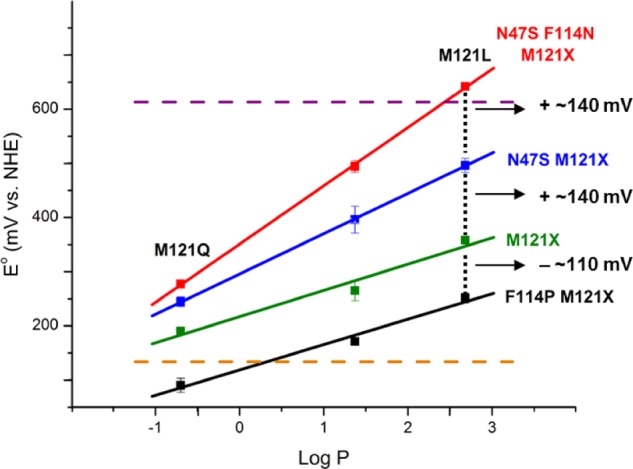
Reduction potentials for a number of Az mutants versus a measure of the hydrophobicity (log P), revealing the linear trend with respect to the axial position (residue 121). Reprinted with permission from ref (1088). Copyright 2009 Macmillan Publishers Ltd.
Unlike mutations on the copper ligands, mutations of residues in the secondary coordination sphere are less likely to change the T1 characteristics according to UV–vis, EPR,1293 and resonance Raman1316 spectroscopy. DFT studies were able to separate the effects of covalent interaction and nonlocal electrostatic components; while the covalent and nonlocal electrostatic contributions can be significant and additive for active H-bonds, they can be additive or oppose one another for dipoles (Figure 58).1317
Figure 58.
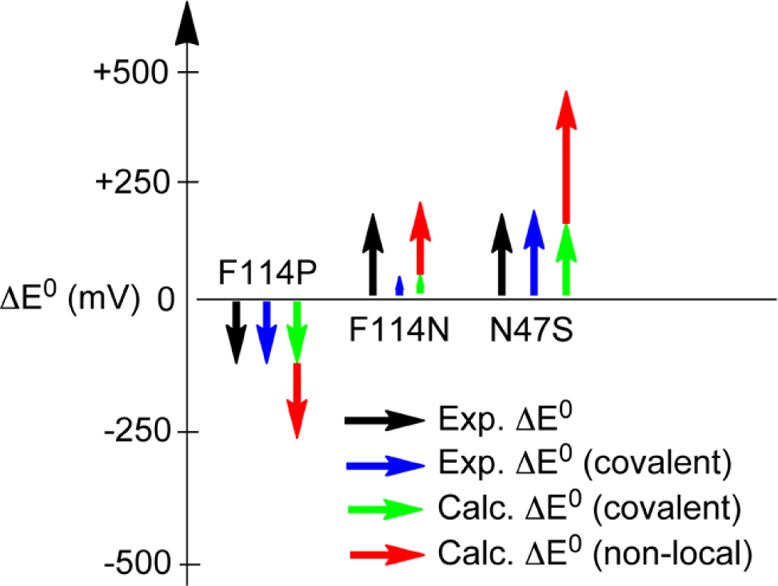
Illustration of the experimentally derived covalent and nonlocal electrostatic contributions to E° for the variants of Az relative to WT Az and their comparison to calculations. Reprinted from ref (1316). Copyright 2012 American Chemical Society.
Lower reorganization energies in the ET process generally increase the ET rate constants and efficiency. However, rational design of ET centers to lower the reorganization energy has so far not been demonstrated. Such a task is particularly challenging for ET proteins such as the blue copper protein azurin that have already been shown to possess very low reorganization energies in comparison to the majority of the other proteins. A study of intramolecular ET by pulse radiolytically produced disulfide radicals to Cu(II) in the above rationally designed azurin mutants showed that the reorganization energies of the designed mutants are lower than that of WT azurin, increasing the intramolecular ET rate constants almost 10-fold.1317 More interestingly, analysis of structural parameters of these mutants suggested that this lowering in reorganization energy is correlated with increased flexibility of the copper center.
4.4.5. Role of Ligand Loop
Besides directly mutating individual ligands, loop-directed mutagenesis containing the ligands to the copper center enables manipulation of copper center by changing the protein structure on a broader scale. T1 copper proteins and CuA domains in heme–copper oxidases share the same cupredoxin fold, with three ligands of T1 copper and four ligands of CuA residing in the so-called “ligand loop” (Figure 59). By careful design, it is possible to transplant the ligand loop of one protein into another, enabling interconversion between T1 copper and CuA and between different T1 copper proteins (section 4.5.3).
Figure 59.

Ligand and loop structure in different T1 copper proteins, CuA from T. thermophilus heme–copper oxidase, and red copper protein nitrosocyanin: (A) amicyanin (PDB ID 1AAC); (B) pseudoazurin (PDB ID 1PAZ); (C) plastocyanin (PDB ID 1PLC); (D) azurin (PDB ID 2AZA); (E) rusticyanin (PDB ID 1RCY); (F) CuA from T. thermophilus heme–copper oxidase (PDB ID 1CUA); (G) nitrosocyanin (PDB ID 1IBY).
An early example of loop-directed mutagenesis comes from interconversion between different copper centers, as two research groups independently installed a ligand loop from the CuA domain of cytochrome c oxidases on amicyanin and azurin, converting the T1 copper proteins to a CuA protein,1318,1319 discussed in detail in section 4.5. Recently, Berry and co-workers transplanted the ligand loop of nitrosocyanin, a newly discovered red copper protein, to azurin.1320 The resulting protein, NCAz, has UV–vis and EPR features similar to those of nitrosocyanin despite having His instead of Glu as the fourth ligand.
Although the T1 copper proteins have a conserved ligand set (section 4.3.1.1), the ligand loops from different proteins show variation in length and sequence (Figure 59). Loop-directed mutagenesis has been carried out between different T1 copper proteins. Ligand loops from azurin, pseudoazurin, plastocyanin, rusticyanin, and nitrite reductase were introduced into the amicyanin scaffold to create loop elongation mutants.1321−1324 Later, the ligand loop from amicyanin, which is the shortest among T1 copper proteins, was introduced into azurin, pseudoazurin, and plastocyanin scaffolds to create loop contraction mutants.1325,1326 The ligand loop from plastocyanin was introduced into the azurin scaffold as well.1327 All of the loop-directed mutants maintain T1 copper spectroscopic characteristics, indicating a similar structure in the Cu(II) state. On the other hand, the loop length has been shown to affect the pKa of C-terminal His and the Cu(I)–N(His) distance.1326,1327 It has been observed that introducing the short loop of amicyanin into pseudoazurin and plastocyanin increases the pKa of C-terminal His, probably due to an entropically favored Cu(I)–N(His) interaction with a longer, more flexible loop.1324−1326
As expected, the reduction potentials of loop-directed mutants are between the reduction potentials of donors of the loops and scaffolds. Amicyanin has the second lowest reduction potential in T1 copper proteins (see Table 10). Introducing the amicyanin loop into other copper protein scaffolds decreases their reduction potentials by 30–60 mV.1326 On the other hand, introducing loops of other T1 copper proteins into amicyanin increases its reduction potential.1322−1324
The ET activity of loop-directed mutants has been measured by the electron self-exchange rate constant (kSES). The loop elongation mutants generally have 10-fold lower kSES values, while loop contraction has less influence on kSES.1322,1323,1326 All the studies indicate that, T1 copper proteins can accommodate changes in loops and assume the same active site structure, consistent with the “rack” or entatic state of the T1 copper center.95,1168,1170
4.5. CuA Centers
4.5.1. Overview of the CuA Centers
The CuA is a binuclear copper center bridged by two cysteine ligands to form a Cu2S2 “diamond-core” structure, which has been found naturally in CcOs,1030,1109,1328 nitrous oxide reductases (N2ORs),1329,1330 the oxidase from Sl. acidocaldarius (SoxH),1331 and a nitric oxide reductase (qCuANOR)1332,1333 to date (Figure 60). Interestingly, all of these proteins are terminal electron acceptors of ET processes; e.g., CcO is the terminal electron acceptor in aerobic respiration, and N2OR is the terminal electron acceptor in anaerobic respiration. One of the most important features of the CuA center is that the two copper ions form a direct metal–metal bond. Therefore, the unpaired electron is delocalized between two copper ions, and the resting state of the CuA center is a Cu(+1.5)–Cu(+1.5) state rather than a Cu(+2)–Cu(+1) state. This is the first example of a metal–metal bond found in biology, which makes it unique compared to centers of other metalloproteins. In addition to the bridging Cys ligands, the copper ions are coordinated by a His from the equatorial position to form a trigonal NS2 coordination. There is a weak distal axial ligand on each copper ion. The axial ligands are a methionine at one copper and a backbone carbonyl at the other. Considering only each copper ion, the CuA center is very similar to the T1 blue copper center with an overall distorted tetrahedral geometry. Hence, the CuA center can be treated as two T1 copper centers joined together with a Cu–Cu bond in between, suggesting an evolutionary relationship between these two centers. Indeed, such a relationship has been proposed on the basis of three-dimensional structure comparison and construction of phylogenetic trees, indicating that T1 copper and CuA proteins share a common ancestor and are developed in part by divergent evolution.1334,1335
Figure 60.

Crystal structures of cytochrome c oxidase (PDB ID 3HB3) and nitrous oxide reductase (PDB ID 1FWX). The CuA sites are highlighted (copper is in green, sulfur is in yellow, nitrogen is in blue, and carbon is in cyan).
The UV–vis absorption spectrum of CuA shows two intense absorbance bands at ∼480 and 530 nm that arise from S(Cys) → Cu charge transfer in the visible region and also a broad band at ∼760–800 nm that arises from Cu(+1.5)–Cu(+1.5) intervalence charge transfer.860,1113−1115 The reduced Cu(I)–Cu(I) form is colorless because of the d10 electronic configuration at each copper center. The more oxidized Cu(II)–Cu(II) state has not been observed in proteins to date.1336,1337 Attempts to oxidize the CuA site normally give an irreversible anodic current at around 1 V, probably due to oxidation of the bridging dithiolate to disulfide.1337,1338 Therefore, the CuA site acts as a one-ET center under physiological condition.72
The Cu–Cu bond in CuA sites has been the subject of extensive debate.1353 Later, the structure of the CuA site was confirmed by different spectroscopic methods. Blackburn et al. reported the extended EXAFS studies of the CuA binding domain of B. subtilis CcO, which showed a strong Cu–Cu interaction of ∼2.5 Å together with a short 2.2 Å Cu–S interaction.1108 The Cu–Cu bond distance is nearly identical to the results from EXAFS studies of native CcO from bovine heart mitochondria, which is 2.46 Å.1354 The dinuclear nature and the unusually short Cu–Cu distance of ∼2.55 Å were further established by X-ray crystal structures of CcO from Pa. denitrificans and bovine heart mitochondria,1030,1109 as well as an engineered CuA center in CyoA.1349 Similar structural features were also observed in the crystal structure of N2OR from Ps. nautica.1329,1330 The most intense bands at 339, 260, and 138 cm–1 observed in resonance Raman spectroscopy of the Pa. denitrificans CcO CuA domain were assigned to symmetric stretches involving primarily the Cu–S(Cys), Cu–N(His), and Cu–Cu bonds, respectively.1107
The Cu–Cu bond in the CuA site causes a valence delocalization between the two copper ions and produces a seven-line hyperfine splitting pattern in the EPR spectra. This unique EPR pattern can be explained by the delocalized unpaired electron coupled with two equivalent copper ions with a nuclear spin I = 3/2.1112,1355,1356 Compared to centers in T1 blue copper proteins, CuA centers show even smaller A∥ on the basis of EPR simulations,1114,1339,1342,1345,1346,1357 reflecting greater covalent interaction and unpaired electron delocalization between the copper ions and the bridging Cys residues.
4.5.2. CuA Centers in Water-Soluble Domains Truncated from Native Proteins
Historically, studying the biochemical role and probing the unique structure of CuA centers have not been easy due to complications arising by overlapping spectroscopic features of other metal centers present in the native proteins containing the CuA center. For instance, the CcO is a membrane protein containing two heme groups (heme a and heme a3), two copper centers (CuA and CuB), a zinc ion, and a magnesium ion. To overcome these inherent difficulties in studying native CuA centers, two strategies are developed: producing truncates of native proteins containing CuA sites742,1331,1339,1341,1342,1345,1352,1358−1361 and designing CuA centers into small, soluble proteins.1318,1362,1363
In the first strategy, the sequence of the CuA subunit from CcO or SoxH was isolated and recombinantly expressed without the helices that normally anchor this domain to the membrane. This way, a water-soluble protein containing only the CuA site was obtained. Such truncates have been constructed for CcO from B. subtilis,1345Pa. dentrificans,742,1339,1358,1361Procambarus versutus,1360Synechocystis PCC 6803,1352 and T. thermophilus(1341,1342,1359,1361) and for SoxH from Sl. acidocaldarius.1331 The UV–vis, EPR, and EXAFS spectroscopic characterizations as well as the reduction potentials measurments for these soluble truncates are consistent with each other (Table 13).742,1339,1358,1361 To date, only the truncate from T. thermophilus has been successfully crystallized.1359
Table 13. Summary of the Spectroscopic Parameters of CuA Sites in Different Proteins.
| CuA site containing protein | organism | λmax (nm) (extinction coefficient, M–1cm–1) | reduction potential vs SHE (mV) | ERP params (gx, gy, gz) | Cu–Cu distance (Å) | ref |
|---|---|---|---|---|---|---|
| subunit II of cytochrome c oxidase | Paracoccus denitrificans | 363 (1200), 480 (3000), 530, 808 (1600) (pH 7) | 240 | gx = gy = 2.03, gz = 2.18, Az = 3.5 mT | 2.6 | (742, 1109, 1339) |
| subunit II of cytochrome ba3 | Thermus thermophilus | 363 (1300), 480 (3100), 530 (3200), 790 (1900) | 250 (pH 8.1), 240 (pH 8), 297 (pH 4.6) | gx = 1.99, gy = 2.00, gz = 2.17, Az = 3.1 mT | 2.43 | (1337, 1341−1344) |
| subunit II of caa3-type cytochrome c oxidase | Bacillus subtilis | 365, 480, 530, 775–800 | gx = gy = 1.99–2.03, gz = 2.178, Az = 3.82 mT | 2.44 | (1344, 1345) | |
| nitrous oxide reductase | Paracoccus dentrificans | 480, 540 (1700), 800 | (1330) | |||
| nitrous oxide reductase | Pseudomonas stutzeri | 480, 540 | gx = gy = 2.03, gz = 2.18, Az = 3.83 mT | 2.44 | (1346) | |
| nitrous oxide reductase | Achromobacter cycloclastes | 350, 481 (5200), 534 (5300), 780 (2900) | gx = gy = 2.045 | (1347) | ||
| biosynthetic model in CyoA protein | Escherichia coli | 360, 538 (2000) | gx = 2.03, gy = 2.03, gz = 2.18, Az = 6.8, 5.3 mT | 2.48 | (1114, 1348, 1349) | |
| biosynthetic model in amicyanin | 360, 483, 532, 790 | gx = gy = 1.99–2.02, gz = 2.18, Az = 3.24 mT | (1318) | |||
| biosynthetic model in azurin | 360 (550), 485 (3730), 530 (3370), 770 (1640) | gx = gy = 2.06, gz = 2.17, Az = 5.5 mT | 2.39 | (1319, 1350) | ||
| nitrous oxide reductase | Pseudomonas nautica 617 | 480, 540, 800 | 260 | gx = gy = 2.021, gz = 2.178, Az = 7 mT | (1351) | |
| subunit II of SoxM | Sulfolobus acidocaldrius | 361 (2300), 478 (3200), 538 (3700), 789 (2400) | 237 | gx = gy = 2.01, gz = 2.20 | (1331) | |
| subunit II of cytochrome c oxidase | Synechocystis PCC 6803 | 359 (1580), 482 (2820), 535 (3080), 785 (1840) | 216 (pH 7) | (1352) |
4.5.3. Engineered CuA Centers
The second strategy to study CuA sites is designing this site into other proteins, first accomplished in a quinol oxidase.1362 The authors aligned subunit II of cytochrome c and quinol oxidases and found that the C-terminal of both proteins contained a subdomain in a Greek key β-barrel scaffold. This alignment suggested that both proteins contain a basic structural motif characteristic of cupredoxins. The CyoA lacked the putative ligands for the formation of the CuA in CcO. The CuA ligand set was thus introduced by extensive mutagenesis of the isolated cupredoxin domain. This engineered CyoA bound copper and showed two strong peaks at 358 and 536 nm, a shoulder at 475 nm, and a broad peak between 750 and 780 nm, as well as an EPR pattern similar to that observed in native CuA from CcO. Later, the crystal structure of CyoA was reported with 2.3 Å resolution.1349 The distance between the two coppers is 2.5 Å. Shortly after the release of the purple CyoA study, two other research groups independently developed designed CuA centers in T1 copper proteins.1318,1319 Dennison et al. replaced the C-terminal loop of the blue copper protein amicyanin, which contained three of the four T1 Cu-binding ligands, with a CuA binding loop. After copper binding, a purple protein was produced with UV–vis absorbance at 360, 483, and 532 nm and a broad absorption at approximately 790 nm, almost identical to that of the native CuA domain of CcO from B. subtilis.1318 The EPR spectrum of the CuA amicyanin contained signals from two Cu(II) species; a distinctive T2 copper site, and a CuA center.1364
Hay et al. constructed a CuA protein from a recombinant T1 copper protein, Ps. aeruginosa azurin, by replacing the loop containing the three ligands to the blue copper center with the corresponding loop of the CuA site in CcO from Pa. dentrificans.1319 The UV–vis and EPR spectra of this protein (CuAAz) were remarkably similar to those of native CuA sites in CcO from Pa. dentrificans (Figure 61). The UV–vis absorption spectrum of CuAAz features two S(Cys) → Cu CT bands at 485 (ε ≈ 3700 M–1 cm–1) and 530 nm (ε ≈ 3400 M–1 cm–1),1113,1350 compared to 480–485 and 530–540 nm for native CuA centers.98 CuAAz also showed a broad band centered at 760–800 nm (ε ≈ 2000 M–1 cm–1), typical of the Cu–Cu ψ → ψ* transition, suggesting that CuAAz had reproduced the Cu–Cu bond. Additionally, the EPR spectrum of CuAAz displayed a seven-line hyperfine splitting pattern, demonstrating that this biosynthetic model duplicated the mixed-valence ground state of native CuA centers.1319,1350 EXAFS, CD, MCD, and resonance Raman analyses of the CuAAz also suggested a high level of electronic and structural identity with CuA centers from CcO.1113,1319,1350,1364,1366 The X-ray crystal structure of CuAAz showed a very similar arrangement of ligands around the copper ions and a Cu–Cu distance that was even slightly shorter than the native CuA center in CcO, confirming the presence of a Cu–Cu bond.1367
Figure 61.

(A) Crystal structure of the biosynthetic model of the CuA site in azurin (PDB ID 1CC3). (B) Comparison of UV–vis spectra between the soluble CuA domain in cytochrome c oxidase (green line), wild-type azurin (blue line), and the biosynthetic CuA model in azurin (purple line). (C) Comparison of X-band CW EPR between wild-type azurin (blue line) and the biosynthetic CuA model in azurin (purple line), four-line splitting vs seven-line splitting. Reprinted with permission from ref (1365). Copyright 2010 Springer-Verlag.
4.5.4. Mutations of the Axial Met
The weaker axial methionine ligand has been investigated through mutagenesis in CcO from Pa. denitrificans and Rb. sphaeroides. The Met227Ile mutation in CcO from Pa. denitrificans resulted in a protein with unchanged stoichiometry of the metals. However, the two copper ions in the CuA site were no longer equivalent and converted from a delocalized Cu(+1.5)–Cu(+1.5) system to a localized Cu(+1)–Cu(+2) system on the basis of EPR and near-IR studies.1368 The ET from cytochrome c to CuA was not affected, but the rate of ET to heme a was significantly diminished in the mutant protein compared with the wild-type protein due to an altered reduction potential of the CuA site. It was concluded that the weak axial Met was not essential for copper binding, but it was important for maintaining the mixed-valence electronic structure of the CuA site. The Met263Leu mutation in CcO from Rb. sphaeroides also showed the binding of two copper ions and proton pumping activity. Multifrequency EPR studies showed that the two copper ions in the CuA site were still electronically coupled. While all the other metals remained unchanged on the basis of UV–vis, EPR, and FTIR spectroscopy, the mutant only maintained 10% of the activity1369 of the native enzyme. The kinetic analysis of ET showed a decrease of ET rate from heme c to CuA to 16 000 s–1 in the mutant, compared to 40 000 s–1 in the wild type. The rate constant for the reverse reaction was increased to 66 000 s–1, compared to 17 000 s–1 in the wild type. This result was attributed to an increased reduction potential of 120 mV relative to that of the native enzyme.1370
The perturbation of the weak axial methionine ligand was also tested in the soluble CuA-containing subunit of cytochrome ba3 from T. thermophilus.1357 The mutants, Met160Gln and Met160Glu, affected the gz region of the EPR spectra and the Cu hyperfine became more resolved and larger in both mutants. Notably, the Az values of both mutants were increased from 3.1 to 4.2 mT, larger than most of characterized native CuA sites. The UV–vis spectra showed enhanced intensity and a blue shift relative to that of the wild type. The EPR and UV–vis data suggested that the axial ligand to copper interaction became stronger, moving from WT to Met160Gln and then to Met160Glu. The effects of both mutations were further studied by pulsed EPR/ENDOR spectroscopy.1371 The results from this study showed an increase of A∥, larger hyperfine coupling, and reduction in the isotropic hyperfine interaction and the axial g tensor. All these effects were associated with an increase in the Cu–Cu distance and changes in the geometry of the Cu2S2 core structure. The mutant Met160Gln was also studied by paramagnetic 1H NMR spectroscopy.1372 The fast nuclear relaxation in this mutant suggested that a low-lying excited state had shifted to higher energies compared to that of the wild-type protein.
Blackburn et al. reported a selenomethionine-substituted T. thermophilus cytochrome ba3 and characterized it with Cu K-edge EXAFS.1373 Interestingly, the optical and EPR spectra of the selenomethionine-substituted CuA site were essentially identical to those of the native CuA site as was the reduction potential. These data suggested that whatever role the S(Met) atom played in the electronic structure of the CuA site was also carried out by the Se(Met) atom.
The axial Met in CuAAz was mutated to Asp, Glu, and Leu, covering the entire range of the hydrophobicity among the natural amino acids. The measured reduction potentials for these axial Met variants showed very little change, only about 20 mV, from that of the original CuAAz, despite some visible perturbation to the UV–vis and EPR spectra of these mutants. The significantly smaller effect of axial ligand in tuning reduction potential of CuAAz compared with WT-Az may reflect the resilience of the diamond core of CuA. In other words, the stability of the interactions making up the diamond core—the bridging Cys thiolates and copper–copper bond—may lead to greater resistance to perturbations arising from the axial position.1374 Recently, a different set of axial Met mutants was generated in the truncated water-soluble CuA domain from T. thermophilus.1375 By introducing Gln, His, Ser, Tyr, and Leu at the axial Met position, a change of about 200 mV in reduction potential was observed. The difference between the results from the truncated CuA domain and CuAAz was attributed to the difference in Cu–S(Met) bond lengths in these two systems: 2.47 Å in the truncated CuA domain vs 3.07 Å in CuAAz. Another explanation is that CuAAz contains the shortest Cu–Cu bond length (∼2.4 Å), hence enhances resistance of the diamond-core structure toward ligand changes.
It is interesting to note that the reduction potentials of the native CuA site from the soluble fragment of subunit II of T. thermophilusba3 at different pH values showed no significant changes.1376 However, the engineered CuA site in azurin exhibited strong pH dependence of the redox properties. This difference might be caused by protonation and dissociation of one of the histidine ligands in the engineered CuA center.
4.5.5. Mutations of the Equatorial His Ligands
The equatorial His ligands bind to the copper ions with a bond length of ∼2.0 Å. In principle, mutations at this His position would result in a significant perturbation of the CuA site. This assumption has been proven to be true in the native system. The His260Asn mutant in cytochrome c oxidase from Rb. sphaeroides only exhibited 1% of the wild-type activity.1369 The 850 nm band was shifted, and the extinction coefficient was diminished to around 1230 M–1 cm–1, compared with 1900 M–1 cm–1 in the wild type. No apparent hyperfine splitting pattern was observed in the EPR spectrum. The kinetic analysis of ET rates showed that the rate constant for ET from CuA to heme c was decreased to 11 000 s–1, compared to 40 000 s–1 in the wild type. The ET rate from CuA to heme c was decreased to 45 s–1, compared with 90 000 s–1 in the wild type. An increase of 90 mV in the reduction potential was also observed.1370
However, dramatic differences were observed in the biosynthetic model of CuA in azurin. The mutation of His120 to Ala yielded a UV–vis spectrum similar to that of the original CuAAz, including the Cu–Cu ψ → ψ* band at ∼760 nm.1377,1378 The EPR spectrum of His120Ala only showed a four-line hyperfine splitting pattern, suggesting that the active site had undergone a transformation to trapped valence, although a Q-band ENDOR study of His120Ala CuAAz showed evidence for the CuA site still being delocalized.1379 Xie et al. applied a series of spectroscopic techniques, including EPR, UV–vis, MCD, resonance Raman, and XAS to both CuAAz and His120Ala CuAAz and correlated the results with DFT calculations.1380 The surprising conclusion of this work was that a minute, 1% mixing of the 4s orbital of one copper ion into the ground-state spin wave function caused the collapse to a four-line hyperfine splitting pattern in the EPR spectrum of His120Ala, not a change from valence-delocalized to trapped valence. The resonance Raman and MCD spectra both demonstrated that the valence delocalization of the CuA center was still intact, although slightly perturbed, despite the loss of His120 as a ligand. The authors attributed the ability of CuA in azurin to remain valence-delocalized, even with the loss of such a strong ligand, to the large electronic coupling matrix element, which arises from the strong and direct Cu–Cu bond. Thus, the diamond core of CuA plays an immense role in the robust nature of this center.
4.5.6. Mutations of the Bridging Cys Ligands
Mutagenesis studies of the CuA binding ligands in native CcO from Pa. denitrificans and N2OR from Ps. stutzeri have demonstrated that the cysteine ligands play an important role in the functions of the enzymes and the spectroscopic features of CuA. Mutating one of the two bridging cysteines to serine, Cys216Ser, in CcO from Pa. denitrificans resulted in a type 1 blue copper site with four-line EPR hyperfine splitting rather than the seven-line EPR signal observed in the CuA site, and only retained below 1% of the wild-type activity. The Cys216Ser mutant no longer exhibited the near-IR absorption in the optical spectrum, indicating the loss of the Cu–Cu bond. Mutation of the second cysteine, Cys220Ser, resulted in 5–10% of the wild-type activity. The higher activity in Cys220Ser is suggested to be due to the intact binuclear copper site on the basis of the metal/protein ratio and copper/iron ratio.1381 The Cys618Asp mutant in N2OR resulted in almost complete loss of activity. The copper was bound only weakly and was hardly detectable on the gel filtration column. In contrast to the Cys618Asp mutant, the Cys622Asp mutant retained some copper binding ability and activity, although the characteristic multiline feature of the mixed-valence CuA was no longer resolved in EPR.1382
Similar to the studies in the native system, the bridging Cys ligands were also individually mutated to Ser in the biosynthetic model of CuA in azurin.1383 Although the resulting mutants still bound to the copper ions, the features of the Cu–Cu bond were completely lost in that the Cys112Ser mutant resulting in two T2 copper sites. The Cys116Ser mutation resulted in a T1 copper site. To account for the loss of symmetry in a single Cys to Ser mutant, a double Cys to Ser construct was made.1384 At high pH, the double mutant indeed bound two coppers, but the EPR spectrum showed that the two copper ions were in two distinct T2 copper sites rather than a mixed-valence site with seven-line hyperfine splitting.
4.5.7. Tuning the CuA Center through Noncovalent Interactions
The H-bonding and hydrophobic interactions around the active site of copper proteins can significantly tune the ET process.1088 Two mutations, Asn47Ser and Glu114Pro, were made in CuAAz.1385 Both the Asn47Ser and Phe114Pro mutations alter H-bonding interactions near the Cys112 ligated to a copper ion, but the Phe114Pro mutation decreases the reduction potential by deleting the hydrogen bond between Cys112 and the backbone NH group,114 while the Asn47Ser mutation increases the reduction potential by affecting the rigidity of the copper binding site and most likely forming a direct hydrogen bonds between the protein backbone and Cys112 (Figure 62).1088 Interestingly, by placing both CuA and T1 blue copper centers in the same scaffold of azurin, Lu and coworkers were able to demonstrate that the same mutations in the secondary coordination sphere resulted in similar decease or increase of the reduction potentials of the copper centers, but the magnitude of the effect is much smaller in CuA center, probably because its “diamond core” structure is more resistant to the perturbation (Figure 62).1088
Figure 62.
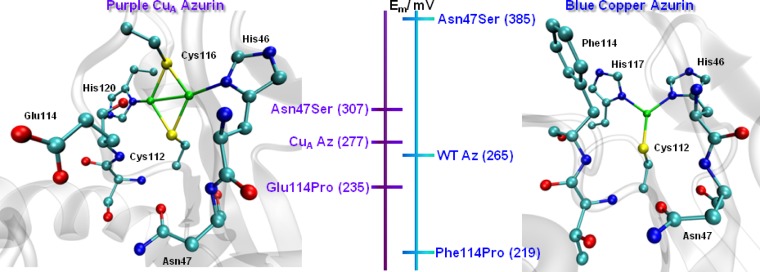
Tuning the reduction potential at blue copper azurin and CuA azurin by redesigning the second coordination sphere. The effects of these mutants are in the same direction, but the magnitude is smaller in the CuA site due to the electron delocalization between the two copper ions. Adapted with permission from ref (1385). Copyright 2012 The Royal Society of Chemistry.
4.5.8. Electron Transfer Properties of the CuA Center
The CuA site is the point of entry of the electrons from cytochrome c. In CcO, the CuA receives electrons from cytochrome c and transfers them to cytochrome a. However, in N2OR, the CuA is believed to transfer electrons between cytochrome c and the catalytic site where nitrous oxide is reduced. The characterization of the ET between cytochrome c and cytochrome c oxidase has been a difficult problem. The stopped-flow method has been used to study the kinetics of electron transfer but does not have sufficient time resolution to monitor such a rapid ET process.
The electron transfers between bovine cytochrome c oxidase and horse cytochrome c labeled with (dicarboxybipyridine)bis(bipyridine)ruthenium(II) were studied by laser flash photolysis.1386 The electron was transferred from Lys25 ruthenium-labeled cytochrome c to the CuA site with a rate constant of 11 000 s–1. The CuA site then transferred an electron to cytochrome a with a rate constant of 23 000 s–1. Lys7, Lys39, Lys55, and Lys60 ruthenium-labeled derivatives showed nearly the same kinetics.
The ET between the CuA site and heme a in bovine cytochrome c oxidase was measured by pulse radiolysis.1387 The rate constant of ET was 13 000 s–1 from the CuA site to heme a, and 3700 s–1 for the reverse process. From this study a low activation barrier was observed, suggesting a small reorganization energy during the ET process. The method was also applied to study the electron transfer between the CuA site and heme a in cytochrome c oxidase from Pa. denitrificans.1340 The ET rates were found to be 20 400 and 10 030 s–1 for the forward and reverse reactions, respectively.
The T1 blue copper sites and CuA sites are commonly used as ET centers found in many biological systems. However, direct comparison between the ET rates of these two centers is difficult to achieve due to different protein scaffolds and redox partners. The engineered CuA site in azurin provides a great opportunity to eliminate the protein structure contribution to the ET process since the ET rates are measured in the same azurin scaffold.1388 The authors first radiolytically reduced the disulfide bond within the azurin scaffold and then measured the long-range ET rate from the reduced disulfide bond to the oxidized CuA center. The rate constant of this intramolecular ET process in CuAAz is ∼650 s–1. Although CuAAz has a smaller driving force (0.69 eV for CuAAz vs 0.76 eV for blue copper azurin), the ET rate of CuAAz is almost 3-fold faster than for the same process in the WT-Az (∼250 s–1). The calculated reorganization energy of the CuA center is only ∼0.4 eV, which is 50% of that found for the blue copper azurin. The low reorganization energy of CuA was also observed in the truncated soluble CuA domain of CcO from T. thermophilus.1337 Farver et al. studied the ET rates and reorganization energies of the mixed-valence CuAAz site and trapped-valence His120Ala CuAAz.1389 They found that changing from the mixed-valence to the trapped-valence state increased the reorganization energy by 0.18 eV, but lowering the pH from 8.0 to 4.0 resulted in a ∼0.4 eV decrease in the reorganization energy, suggesting that the mixed-valence state only played a secondary role in controlling the ET property.
4.5.9. pH-Dependent Effects
As an electron entry site for cytochrome c oxidase, the CuA center receives electrons from cytochrome c and transfers the electrons to the heme a site. The electrons are finally transferred to the heme a3–CuB site where dioxygen reduction takes place. The reduction results in a proton gradient, which in turns drives the synthesis of ATP. For the CcO to function well, a regulator is needed for initiating and shutting down the whole ET process and dioxygen reduction reaction. A pH-dependent study on engineered CuAAz suggested that the CuA site may play such a role.1390 CuAAz displayed a seven-line EPR hyperfine with a mixed-valence state. When the pH was decreased from 7.0 to 4.0, the absorption at 760 nm shifted to 810 nm; at the same time, a four-line EPR hyperfine was observed. The pH dependence was reversible, and the mixed-valence state was restored when the pH was increased back to 7.0. A dramatic increase in reduction potential was also observed from 160 to 340 mV when the pH was decreased from 7.0 to 4.0. It was identified that the protonation of C-terminal His120 caused such a pH-dependent transition, as the His120Ala mutation completely abolished this observation. A feedback mechanism was proposed to explain how the CuA site regulated the function of cytochrome c oxidase. The pumped proton may result in protonation of the C-terminal His and cause a different valence state of the CuA site. The increased reduction potential in the new state will stop the whole ET process and proton pumping (Figure 63). This hypothesis is further supported by ET studies in the His260Asn mutant in cytochrome c oxidase from Rb. sphaeroides which showed that protonation of the C-terminal histidine resulted in a change in the valence state and an increase of the reduction potential by 90 mV.1370 The ET rate from the CuA site to heme a decreased by over 4 orders of magnitude. The His260 in cytochrome c oxidase corresponds to His120 in CuAAz.
Figure 63.
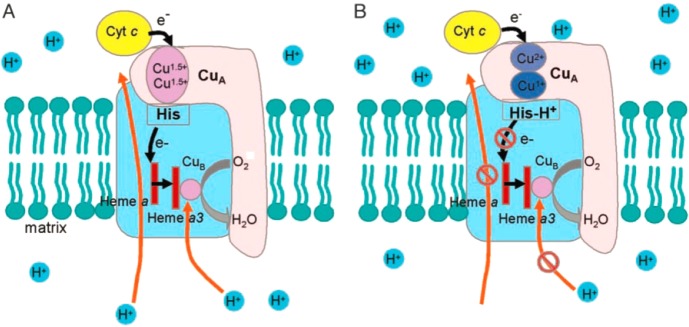
Schematic model of different states of the CuA center in cytochrome c oxidase: (A) mixed-valence form at neutral pH and (B) trapped-valence form at low pH. Subunit I is in light blue, and subunit II is in pink. Black arrows represent the flow of electrons, and orange arrows represent the flow of protons. Reprinted with permission from ref (1390). Copyright 2004 National Academy of Sciences.
4.5.10. Copper Incorporation into the CuA Center
While numerous studies have established the structural features of CuA, the question of how copper ions are delivered into the CuA sites in vivo is still poorly understood. In the cytoplasm, copper levels are rigorously regulated, and free copper levels are extremely low and estimated to be at the attomolar level.1391−1397 Although it has been proposed that a metallochaperone called Sco is responsible for metalation of the CuA site, delivering the copper ions to the CuA site in CcO by Sco proteins has not been demonstrated.1398
Besides the delivery of copper ions by Sco proteins, another possibility is unmediated metalation. The CcOs from eukaryotes are located in mitochondrial membranes.1399 In Gram-negative bacteria, CuA in CcO is exposed to the periplasmic space. However, in Gram-positive bacteria, CuA in CcO is exposed to the extracellular space.1109,1393,1400,1401 In addition, the N2OR is a soluble protein also located in the periplasmic space.1402 In periplasmic and extracellular spaces, copper levels are not regulated as rigorously as inside the cell, and the free copper ion concentration could be much higher. In fact, unmediated CuA metalation has been considered as a possibility for CuA metalation in N2OR.1403−1405 From this view, the studies of free copper ion incorporation into CuA sites in vitro may provide important insights into this process, although they do not perfectly reflect the process in vivo.
In an early study of CuAAz, the metalation of apo-CuAAz by adding a 10-fold excess of CuSO4 was studied by stopped-flow UV–vis spectroscopy.1406 A single intermediate with intense absorbance at 385 nm was observed, which is characteristic of the Cys S → Cu CT bands of tetragonal T2 copper centers.98,1095 This T2 copper intermediate formed with kobsd = 1.2 × 103 s–1 and subsequently decayed with kobs = 3.1 s–1; meanwhile the absorptions corresponding to the CuA site increased. An isosbestic point between the ∼385 nm band and the ∼485 nm band of the CuA site was observed, indicating the T2 copper intermediate was converted to CuA. Because only Cu(II) ion was added during metalation, a reducing agent must be supplied by the system itself to form a Cu(+1.5)–Cu(+1.5) site, indicating that the free thiols in apo-CuAAz were providing electrons by forming disulfide bonds.1407−1409 Adding ascorbate or Cu(I) salt increased the yield of CuA center formation.
A similar study was investigated in N2OR from Pa. denitrificans.1410 Different from the previous study, two intermediates were observed upon adding Cu(II) salt. These two intermediates formed within a similar time scale and also decayed at the same time with simultaneous formation of CuA sites. Two isosbestic points were present between the absorption bands of both intermediates and the CuA absorption bands, strongly suggesting conversion of these intermediates to CuA. One of these two intermediates has spectral features typical of T2 copper centers with thiolate ligation, and another shows the characteristics of a T1 copper center. These observations suggested that the purple CuA site contained the essential elements of T1 and T2 copper centers and provided experimental evidence for a previously proposed evolutionary link between the cupredoxin proteins.1334,1335
Guided by the observation of both T1 copper and T2 copper intermediates in the metalation of the CuA site in N2OR, the metalation of CuAAz was revisited by varying both the copper concentration and pH.1411 When the CuAAz concentration was greater than the CuSO4 concentration, both T2 copper and T1 copper intermediates were observed, similar to the results obtained for N2OR. Global fitting of the UV–vis absorption kinetic data and time-dependent EPR together with previously studied mutants of CuAAz provided valuable information about the mechanism of copper incorporation where a new intermediate, Ix, was observed. When Cys112 was mutated to Ser, a T2 copper site formed, with UV–vis and EPR spectra similar to those of the T2 copper intermediate. From this study it was inferred that the T2 copper intermediate is a capture complex with Cys116, which is also supported by the greater solution accessibility of this residue, compared to Cys112. Conversely, when Cys116 was changed to Ser, a T1 copper center formed, with UV–vis and EPR spectra nearly identical to those of the T1 copper intermediate (Figure 64).1383
Figure 64.

Proposed mechanism of copper incorporation into the biosynthetic CuA model in azurin. Reprinted with permission from ref (103). Copyright 2012 Elsevier.
4.5.11. Synthetic Models of the CuA Center
Another approach to study the CuA center is to synthesize small-molecule mimics of CuA.1412 This has been proven to be a difficult task because of the formation of disulfide bonds between free thiols mediated by copper ions.1338 Also, the most important feature in the CuA site, the diamond-core structure with Cu–Cu bond bridging by thiolates, is difficult to achieve. Besides the first coordination sphere, the second coordination sphere has also proven to be important in tuning the properties of the CuA site, which is even harder to mimic in small-molecule compounds.1385 However, model compounds have met with varying degrees of success.369,1413−1428
Houser et al. reported a fully delocalized mixed-valence dicopper complex with bis(thiolate) bridging which was the first closet small-molecule CuA mimic. The crystal structure of this model complex showed that the Cu2S2 core is planar with an average Cu–Cu distance of 2.92 Å. However, it is still longer than the Cu–Cu distance (2.46 Å by EXAFS1354 and 2.55 Å by X-ray crystal structures1030,1109) in native CuA centers.1416 The EPR spectrum recorded at 4.2 K clearly showed the seven-line hyperfine splitting indicating the fully delocalized electronic structure.
More recently, Gennari et al. reported a new bis(μ-thiolato)dicopper complex that mimicked most of the important spectroscopic features of the CuA site.1429 Notably, this dicopper complex is the first CuA model with a Cu2S2 core that can be reversibly oxidized or reduced between the Cu(+1.5)–Cu(+1.5) state and the Cu(+1)–Cu(+1) state. However, the short Cu(+1)–Cu(+1) distance (2.64 Å) and long Cu(+1.5)–Cu(+1.5) distance (2.93 Å) significantly increased the reorganization energy of ET, which was much higher compared to the reorganization energy observed in the water-soluble CuA domain of T. thermophilus cytochrome ba3.1337
4.6. Structural Features Controlling the Redox Chemistry of the Cupredoxins
4.6.1. Role of the Ligands
As the immediate residues that coordinate to the copper centers, the ligands exert a huge influence on the redox properties of cupredoxins. The strong Cu–thiolate bond(s) playd the dominant role in defining T1 Cu and CuA centers in both their electronic structures and ET functions. Except for a few unnatural amino acids, mutation of Cys will change the T1 copper character. The same happens in the CuA center in that mutation of Cys to Ser will result in either T1 or T2 center.
The His residues are important for shielding the copper center from the solvent and for directing ET. C-terminal His is on a hydrophobic patch of T1 copper proteins. The hydrophobic patch directly interacts with redox partners of T1 copper proteins. Mutation of either His to Gly creates an open binding site, where external ligands could coordinate with copper and influence the properties of T1 copper proteins. Due to the open binding site, the His to Gly mutant exhibited a high reorganization energy and low ET rate.
The axial Met is less conserved in T1 copper proteins. Besides Met, native T1 copper proteins could have the more hydrophilic Gln or the more hydrophobic, noncoordinating Leu/Phe at the axial position. There is a general trend that proteins with Gln as their axial ligand have the lowest reduction potentials, proteins with Met have intermediate reduction potentials, and proteins with Leu/Phe have the highest potentials. The reduction potential tuning role of the axial ligand has been further confirmed by mutagenesis studies. The correlation between the hydrophobicity of the axial ligand and the reduction potential has been established by incorporation of a series of Met analogues. The role of the highly conserved axial methionine ligand was performed by glutamate, aspartate, and leucine in the engineered CuAAz.1374 In contrast to the same substitutions in the structurally related blue copper azurin, much smaller changes (∼20 mV) in reduction potential were observed, indicating that the diamond-core structure of the CuA site is much more resistant to variation in axial ligand interactions than the distorted tetrahedral structure of the blue copper protein.
4.6.2. Role of the Protein Environment
The first coordination sphere directly affects the spectroscopic properties and ET of the T1 copper proteins. Beyond the first coordination sphere, the protein scaffold holds copper ligands together and forces trigonal geometry regardless of the oxidation state of copper, as suggested by the rack mechanism1168 or the entatic state.1170 Furthermore, the environment around the primary coordination sphere can fine-tune the electronic structure and redox properties of the copper centers by noncovalent interactions such as a H-bonding network to the copper ligands.94,1119,1430 Through manipulation of H-bonding networks in the secondary coordination sphere, Marshall et al. managed to tune the reduction potential of azurin over the natural range while maintaining T1 character in the copper center.1088 The same mutations that affected the noncovalent interactions in azurin were introduced to tune the reduction potentials of engineered CuAAz.1385 The effects of these mutations were in the same direction, but with smaller magnitude in the CuA site due to dissipation of the effects by two copper ions rather than the single copper ion in blue copper proteins.
All these findings are important in understanding the different roles of the two cupredoxins. Since the T1 blue copper proteins are used in a wide range of ET processes, the reduction potentials of the blue copper proteins need to be tuned to fit a wide range. Such a tuning is mainly achieved by changing the axial ligands and H-bonding network in the secondary coordination sphere.95,1088 However, the CuA sites are only found in terminal electron acceptors with very small potential differences between redox partners where a wide range of reduction potentials is not preferred. The diamond-core structure of CuA sites decreases the reorganization energies and enables fast ET processes.
4.6.3. Blue Type I Copper Centers vs Purple CuA Centers
The type I blue copper centers are widely found as ET centers common in many biological systems. However, the CuA centers are only found in CcOs, N2ORs, and the oxidase from Sl. acidocaldarius (SoxH). Several key questions that have been raised regarding these sites are concerned with how such a mixed-valence binuclear copper site was selected, what the advantage of such a site compared to T1 blue copper sites is, and why the CuA sites are only found in terminal electron acceptors. To answer these questions, a direct comparison of the ET rates of these two centers is required. The engineered CuA site in azurin provides a great opportunity to eliminate the protein structure contribution to the ET process since the ET rates are measured in the same azurin scaffold.1388 The CuAAz demonstrated that CuA is a more efficient ET site even with a smaller driving force between the reduced disulfide and CuA site than between the reduced disulfide and blue copper site. The calculated reorganization energy of the CuA site is only half that of the blue copper site, which is due to the rigid structure of the diamond core in the CuA site. Both CcOs and N2ORs are large enzymes that contain multiple ET sites. As the electrons are transferred along the chain, the difference in reduction potentials as the driving force must fall within a narrow range of values. In this case, the ET sites with lower reorganization energy would be preferred because the driving force might be small.
5. Enzymes Employing a Combination of Different Types of Electron Transfer Centers
5.1. Enzymes Using Both Heme and Cu as Electron Transfer Centers
5.1.1. Cytochrome c and CuA as Redox Partners to Cytochrome c Oxidases
The CcO is a terminal protein complex in the respiratory electron transport chain located in the bacterial or mitochondrial membranes. This large protein complex receives four electrons from cyt c that are used to efficiently reduce molecular oxygen to water with the help of four protons from the aqueous phase without producing any reactive oxygen species such as superoxide and peroxide. In addition, it translocates four protons across the membrane, which establishes an electrochemical potential gradient used for ATP synthesis.
Out of many different types of CcOs from various different organisms, the families involved in aerobic respiration that generally use cyt c as their biological electron donors are caa3, aa3, cbb3, ba3, co, bb3, cao, and bd oxidases.1431 Cyts caa3 and cbb3 oxidases contain a distinct cyt c domain integrated into the cyt c oxidase enzyme complex. Cyt aa3 oxidase is the mitochondrial counterpart of cyt caa3 except that it does not contain the cyt c domain at the C-terminal end of subunit II (Cox2) of the enzyme complex. Subunit II also contains the binuclear CuA center. Cyt cbb3 oxidases do not contain the CuA center, but they contain both a monocytochrome c subunit (FixO or CcoO) and a dicytochrome c subunit (FixP or CcoP).79,1432 Many facultative anaerobes use bo and bo3 oxidases which use quinol as the substrate instead of cyts c. Depending on the organism, the cyts c are associated with the enzyme complex by either covalent or noncovalent interactions.1433 For example, in the bacterium PS3, cyt c binds covalently to the protein complex at the C-terminal end of subunit II.1434−1438 In Pa. denitrificans, the cyt c subunit is tightly bound to the oxidase subunit by covalent interactions and can be removed by treatment of a high concentration of detergent. In eukaryotes, cyts c bind to the cyt c oxidase loosely, which can be removed at high salt concentrations. Mammalian cyt c oxidases have been shown to bind one molecule of cyt c at a high-affinity site, which serves as the electron entry point.1439−1441 There is evidence of the presence of a second low-affinity site, but the role of such secondary interactions between cyt c and the oxidase is not well-known. It has been shown that cyts c use a series of several (six or seven) positively charged lysines near the heme edge which form complementary electrostatic interactions with negatively charged carboxylates on the high-affinity site of subunit II of the oxidase. Such electrostatic interactions are important for placing the substrate in the correct orientation to bind to the oxidase complex.1442,1443
Available data suggest that electrons are transferred from reduced cyt c, one at a time, to the oxidized CuA.1444,1445 Then internal ET takes place from the reduced CuA to the LS heme a and to the binuclear active site consisting of HS heme a3 and CuB where the dioxygen reduction takes place (Figure 65). It has been measured that the ET rate constant from CuA to heme a is 20 400 s–1 and the rate of the reverse process, from heme a to CuA, is 10 030 s–1 in Pa. denitrificans cytochrome c oxidase by pulse radiolysis.1340 A similar study was also applied to cytochrome ba3 from T. thermophilus, and the first-order rate constants are 11 200 and 770 s–1, respectively.1340 Electron transfer from cyt c to CuA and CuA to heme a is fast,1445,1446 while the ET from heme a to the heme a3/CuB site is slow and has been proven to be the rate-limiting step of the reaction.1447,1448 It has also been shown that the presence of CuA is not required for the oxidase activity as the deletion of the CuA gene from beef heart cyt c oxidase slows down the ET rate, but still maintains some oxidase activity.1449,1450
Figure 65.
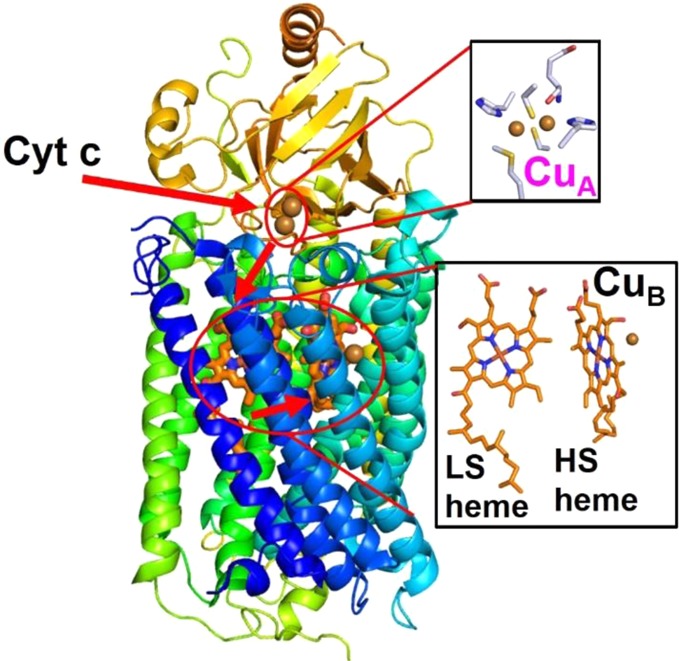
Cyt c oxidase from Pa. denitrificans (PDB ID 3HB3). The ET pathway is shown with arrows.
Binding of cyt c to the oxidase causes conformational changes in both the protein partners.1451,1452 The major changes are observed upon reduction of the CuA and heme a centers. It has been proposed that the reduction of these two redox centers causes a conformational change of the binuclear active site from a closed to an open state that facilitates the intramolecular ET that couples the subsequent redox reaction and proton translocation.1453−1456 NRVS on cyt c552 from Hydrogenobacter thermophilus has indicated the presence of strong vibrational dynamic coupling between the heme and the conserved -Cys-Xxx-Xxx-Cys-His- motif of the polypeptide chain.1457 Such vibrational coupling has been proposed to lower the energy barrier for ET by either transferring the vibration energy released upon protein–protein complex formation or by modulating the heme vibrations.
A recent NMR study has shown that the hydrophobic residues near the heme of cyt c form hydrophobic interactions with cyt c oxidase and are major contributors to the complex formation, while the charged residues near the hydrophobic core dictate the alignment and orientation of cyt c with the enzyme to ensure efficient ET.1458 The affinity of oxidized cyt c for complex formation with CcO is significantly lower, suggesting that ET is gated by the dissociation of oxidized cyt c from CcO. The rate of dissociation of oxidized cyt c is dictated by the affinity of oxidized cyt c for CcO that provides facile ET.
5.1.2. CuA and Heme b as Redox Partners to Nitric Oxide Reductases
Although the NORs from Gram-negative bacteria use cyt c as the biological electron donor to the heme c, one NOR (qCuANOR) purified from the Gram-positive bacterium B. azotoformans shows the presence of a quinol binding site and uses the binuclear CuA site as an electron acceptor instead of heme c.1332,1333 This family of NORs use melaquinol as the physiological electron donor to the CuA site instead of cyt c. Electrons are passed from melaquinol to the CuA site and are then transferred to the LS heme b and onto the binuclear active site consisting of a HS heme b3 and a nonheme FeB site.
5.1.3. Cytochrome c and CuA as Redox Partners to Nitrous Oxide Reductases
The N2OR is the last enzyme in the denitrification pathway which reduces nitric oxide to dinitrogen.1329,1330,1459 N2ORs are homodimeric periplasmic enzymes containing the binuclear ET site CuA which receives electrons from cyt c and a tetranuclear catalytic site, CuZ. A unique N2OR has been reported from Wolinella succinogenes which has a C-terminal cytochrome c domain that is suggested to be the biological electron donor to the CuA center.1460
5.2. Enzymes Using Both Heme and Iron–Sulfur Clusters as Electron Transfer Centers
5.2.1. As Redox Partners to the Cytochrome bc1 Complex
The coenzyme Q–cytochrome c oxidoreductase, also called the cytochrome bc1 complex or complex III, is the third complex in the electron transport chain playing a crucial role in oxidative phosphorylation or ATP generation. The bc1 complex is a multisubunit transmembrane protein complex located at the mitochondrial and bacterial inner membrane that catalyzes the oxidation of ubihydroquinone and the reduction of cyt c(1461) coupled to the proton translocation from the matrix to the cytosol. The catalytic core of the bc1 complex consists of three respiratory subunits: (1) subunit cyt b that contains two b-type hemes, bL and bH, (2) subunit cyt c, containing a heme c1, and (3) iron–sulfur protein subunit containing a Rieske-type [2Fe–2S] cluster (Figure 66). While in some α proteobacteria such as Paracoccus, Rs. rubrum, and Rb. capsulatus, this enzymatic core containing the three subunits is catalytically active, several additional (seven or eight) subunits are present in the mitochondrial cytochrome bc1 complexes.86,1462
Figure 66.
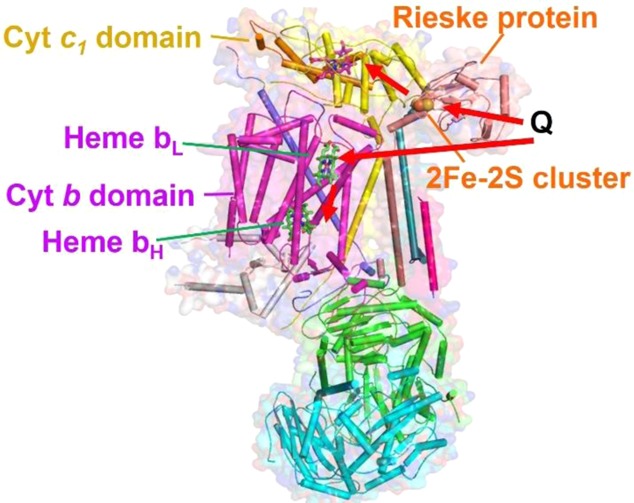
Bovine cytochrome bc1 complex (PDB ID 1BE3). Different ET domains and their cofactors are shown. bL = low-potential heme, bH = high-potential heme, and Q = ubiquinol. Electron transfer pathways are shown with arrows.
Structures of the bc1 complex from various resources such as yeast, chicken,1029 rabbit,1029 and cow1026,1029,1463 show that the cyt b subunit consists of eight transmembrane helices designated as A–H. Hemes bL and bH are contained in a four-helix bundle formed by helices A–D and are separated by a distance of 8.2 Å. The axial ligands for both hemes are all His and are located in helices B and D. His83 and His182 are bound to heme bL, while His97 and His196 are axial ligands for heme bH. The cyt c subunit containing cyt c1 is anchored to the membrane by a cytoplasmic domain and belongs to the Ambler type 1 cyt c based on the protein fold and the presence of the signature sequence -Cys-Xxx-Xxx-Cys-His-. Electron transfer has been proposed to occur through the exposed “front” face of the corner of the pyrrole II ring.1029 One of the His residues that acts as a ligand to the [2Fe–2S] cluster is 4.0 Å from an oxygen atom of heme propionate-6 and 8.2 Å from the C3D atom of the heme edge of cyt c1. Such proximity of the heme group and the Rieske-type cluster has been proposed to facilitate ET. Using this distance of 8.2 Å, a rough estimation of the ET rate from the iron–sulfur protein to cyt c1 has been calculated to be 4.8–80 × 106 s–1.
On the basis of the relative orientations of the prosthetic groups as discussed above, an ET pathway has been proposed where in round I an electron is transferred from a bound ubiquinol to the Rieske-type cluster into the cyt c1 via its heme propionate-6 and out of cyt c1 via its pyrrole II heme edge to the cyt c (not the same as cyt c1).78,1029 At the same time the low-potential heme (bL) pulls an electron from the ubiquinol and transfers it to the high-potential heme (bH), which is ultimately picked up by an oxidized ubiquinone. The same cycle is repeated in round II.
Mitochondrial cyt c or bacterial cyt c2 connects the bc1 complex with the photosynthetic reaction center or cyt c oxidase.80,1464 The mode of interaction between cyt c (or c2) with its redox partners has been proposed to involve docking of cyt c with its solvent-exposed heme edge (called the “front” side). There are multiple dynamic H-bonding and salt bridge interactions between the cyt c and cyt c1 of the bc1 complex.1465 The front side is composed of a ring of positively charged Lys residues near the exposed heme edge. The opposite side, called the “back” side, is composed of several negatively charged residues. This charge separation creates a dipole moment in both bacterial cyts c2 and mitochondrial cyt c.1466,1467 The positively charged front side forms complementary interactions with the negatively charged surface of its partner, which orients the electron donor in proper alignment for facile ET. EPR experiments with cyt c2 from Rb. capsulatus have demonstrated that the dipolar nature of cyt c2 influences its orientations, which facilitate ET to its partner under physiological conditions.1468−1470
Rieske protein can accommodate three conformations in the complex: The first is the c1 position in which the His ligand is H-bonded to propionate of heme in cyt c, and fast ET (60 000 s–1)1471 between the two will occur.1026 At this state the cluster is far from the quinone binding site. The b position allows interaction between the cluster and quinone. This position was stabilized by interaction of H161 with the inhibitor stigmatellin that mimics the H-bond pattern of semiquinone.223,1029 The final conformation is an intermediate state in which the Rieske protein cannot interact with either cytochrome or quinone.865
The cycle starts from an intermediate state (Figure 67). Upon binding of reduced hydroquinone, the Rieske protein will move to state b and an electron will be transferred to hydroquinone, generating a semiquinone, which binds tightly to the Rieske protein. This tight interaction will become loose by transfer of a second electron from semiquinone to heme bL and generation of quinone. The thermodynamically disfavored reduction of heme bL by semiquinone is coupled to favorable oxidation of hydroquinone by the Rieske center. As a result the reduction potential of the Rieske center is of significant importance to the rate of reduction of heme bL. Reduction of the Rieske center and heme bL happens within a half-life of 250 μs as evident by freeze quench EPR. The semiquinone intermediate has a very high affinity for the Rieske protein. This tight binding will increase the reduction potential of the Rieske center by 250 mV. This binding mode and increased reduction potential will ensure that the Rieske center will not reduce cyt c before heme bL is reduced and quinone is formed. The reduced Rieske center will then move to its c1 state and transfer an electron to cyt c. After complete transfer of both electrons, the Rieske protein will go back to its intermediate state for the second cycle.773,787,865 The binding of quinone and Rieske protein is redox-dependent. While the kinetics of ET to cyt c is pH-dependent due to the pH dependence of the reduction potential, it has been proposed that the rate-limiting step in this reaction is mostly the transition from one state (e.g., state b) to another state (e.g., state c1) of the Rieske center and not the ET, considering the same rate observed in mutants with different reduction potentials.1078
Figure 67.
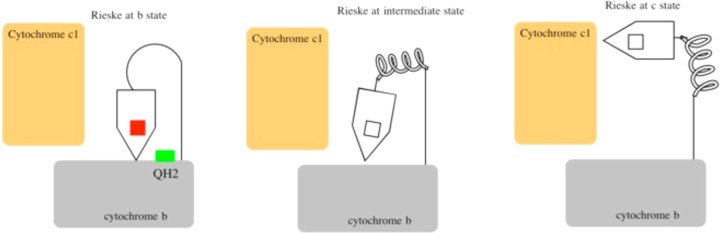
Schematic cycle of Rieske positions in the bc1 complex. Reprinted from ref (865). Copyright 2013 American Chemical Society.
Although the mechanism of proton transfer is not very well understood in this system, evidence suggested that the two protons are bound to the Rieske center, one to each His in the reduced state. The oxidized state can have no protons, one proton, or two protons depending on the pH. It has been shown that removal or mutation of the Rieske cluster will result in a proton-permeable bc1 complex, suggesting a role as a proton gate for the Rieske protein.1472 NMR was used to calculate the pKa of His ligands in the T. thermophilus Rieske protein. In this study, residue-selective labeling was used to unambiguously assign the NMR shifts. The results were consistent with other pH-dependent studies of Rieske proteins, showing that one of the water-exposed His ligands that is close to quinone undergoes large redox-dependent ionization changes. Their system also supports proton-coupled ET in the Rieske–quinone system.864 Analysis of driving forces using a Marcus–Bronsted method in mutants that had distorted H-bonding due to mutation of either conserved Ser or Tyr resulted in the proposal of a proton-first-then-electron mechanism in which the ET follows the transfer of a proton between hydroquinone and the imidazole ligand of the Rieske cluster.814
5.2.2. As Redox Partners to the Cytochrome b6f Complex
Cyt b6f (plastoquinol–plastocyanin or cyt c6 oxidoreductase) is a protein complex belonging to a “Rieske–cytochrome b” family of energy-transducing protein complexes found in the thylakoid membrane in the chloroplasts of green algae, cyanobacteria, and plants and catalyzes ET from plastoquinol to plastocyanin or cyt c6 (PSII to PSI) coupled with the proton translocation across the membrane for ATP generation.282,1473−1476 It is located in between the PSII and PSI reaction centers in oxygenic photosynthesis (Figure 68). The b6f complex is analogous to the bc1 complex of the mitochondrial electron transport chain. The b6f complex comprises seven subunits: a cyt b6 with a low-potential (bp) and a high-potential (bn) heme, a cyt f, a Rieske iron–sulfur protein, subunit IV, and three low molar mass (∼4 kDa) transmembrane subunits.1473 There are a total of seven prosthetic groups that are found in the b6f complex: cyt f, hemes bn and bp, a Rieske [Fe2–S2] cluster, chlorophyll a, β-carotene, and a c-type heme designated as cn, cx, or ci. This heme, located close to the quinone reductase site near the electronegative side of the membrane, is linked to the protein via a single thioether linkage, lacks any axial ligands, and has been shown to be critical for function of the b6f complex.225,1478−1481 The cyt b6 subunit contains two bis-His-ligated hemes, a high-potential heme (−45 mV) on the luminal side and a low-potential heme (−150 mV) on the stromal side of the thylakoid membrane. EPR and Mössbauer data reveal that both hemes are 6cLS and have His planes that are perpendicular. Cyt b6 and subunit IV of the b6f complex are structurally similar to cyt c of the bc1 complex,184 while there is no structural similarity between cyt f and cyt c1 even though they are functionally similar.123,1029 The cyt b6f complex takes part in linear electron flow between PSII and PSI where it links the plastoquinone pool of PSII to plastocyanin or cyt c6 to PSI as well as in cyclic electron flow within PSI (Figure 68). The linear electron flow path involves oxidation of quinol to quinone from PSII to PSI coupled to the generation of ATP and reduced ferredoxin, which reduces NADP+ to NADPH via an oxidoreductase FNR. Cyclic electron flow in PSI involves electron flow via the b6f complex back to the P700 reaction center of PSI. In both the cases two electrons are passed from plastoquinol at the quinol oxidation site (QP) near the lumenal, electropositive site of the membrane to the one-electron acceptor plastocyanin, which is coupled to the “Q-cycle” 1482,1483 involving proton translocation across the membrane. One of the electrons from plastoquinol is transferred to PSI via the high-potential chain, while the second electron is passed onto the low-potential, transmembrane chain on the electronegative side of the membrane where plastoquinone reduction takes place.
Figure 68.
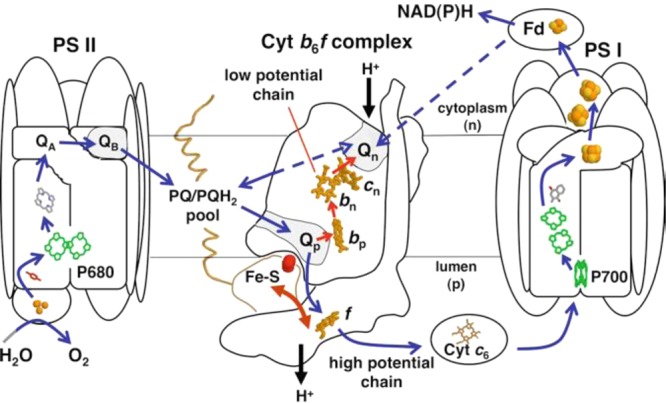
Cyt b6f complex in the photosynthetic electron transport chain. P680 = reaction center chlorophylls of PSII, QA, QB = quinones of PSII, PQ/PQH2 pool = plastoquinone/plastoquinol pool, Fe–S = Rieske cluster, f = cyt f of the high-potential chains (blue arrows), Qp, Qn = plastoquinol oxidation and plastoquinone reduction sites, bp, bn, cn = hemes of the low-potential chain (red arrows), Fd = ferredoxin, and P700 = reaction center chlorophylls of PSI. The domain movement of the Rieske protein is shown by a two-sided arrow. The direction of proton translocation across the membrane is shown by proton arrows. The electronegative (cytoplasmic) (n) and electropositive (luminal) (p) sides of the membrane are labeled, and ET pathways are shown by arrows. A possible direct ET path from PSI to the cyt b6f complex is shown as the dashed line from Fd to the Qn site. Reprinted with permission from ref (1477). Copyright 2012 Springer Science+Business Media.
On the His ligation side of the heme, a chain of five conserved water molecules oriented in an L-shaped manner have been identified from the X-ray structure, which form hydrogen bonds with ten amino acid residues from the protein, seven of which are conserved.1473,1484,1485 These water molecules have been proposed to act as “proton wires” in coupling of the ET with proton transfer across the membrane.1485,1486 The heme of cyt f is located in a hydrophobic environment and is protected from the solvent by Tyr1, Pro2, Ile3, and Phe4 (or Trp4 in cyanobacteria).161 The side chain of residue 4 is located close to the heme edge and oriented almost perpendicular to the heme plane (Figure 69).1485 This edge-to-face interaction of the Trp4 and the heme has been proposed to be responsible for tuning the reduction potential of the heme by interaction with the porphyrin π molecular orbitals. Such edge-to-face interactions have been observed in cyt b5 (Phe58, Phe35),141,366 cyt b562 (Phe61),382 and peptide-sandwich mesoheme model systems reported by Benson and co-workers (Trp or Phe).423,1487 In these peptide mesoheme sandwich complexes the heme–Trp interaction has been shown to be important to stabilize the α-helical scaffold as well as the ferric state of the heme iron.1488 Such interactions also stabilize the ferric state of the heme iron in the cyanobacterium cyt f.
Figure 69.
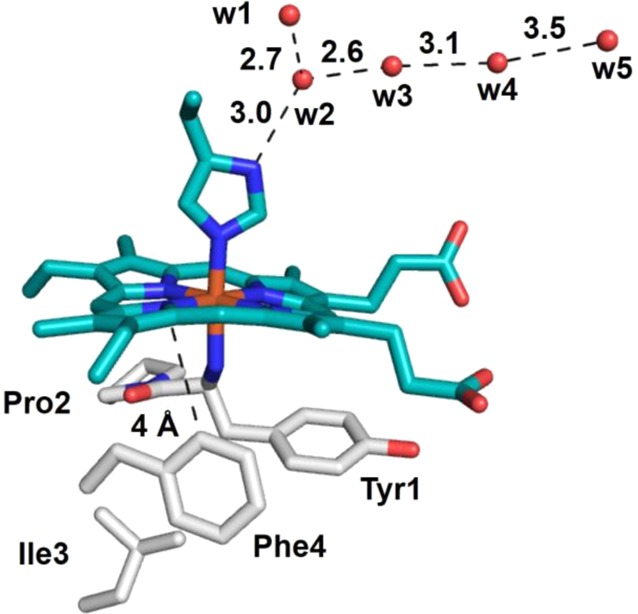
Environment around the heme of cyt f (PDB ID 1HCZ). Hydrophobic residues are shown as gray sticks. The “edge-to-face” interaction at 4 Å between Phe4 and the heme that is proposed to be important to tune the reduction potential of the heme iron is shown. The five conserved molecules that have been proposed to act as “proton wires” that couple ET with proton transfer are shown as red spheres. Residue numbering of waters is arbitrary.
The chloroplast Rieske proteins work in the same way. It has been shown that the movement of these Rieske proteins will also function as a redox-state sensor that can balance the light capacity of the two photosystems. This state transition can also act as a switch between cyclic and linear electron flow.1489
5.2.3. As Redox Centers in Formate Dehydrogenases
Formate dehydrogenases (Fdh’s) catalyze decomposition of formate to CO2. They exist in both prokaryotes and eukaryotes. Fdh’s are mainly NAD+-dependent in aerobic organisms and NAD+-independent in anaerobic prokaryotes, donating electrons from formate to a terminal electron acceptor other than O2.1490 Structural studies reveal that Fdh’s contain one to three subunits with either W or Mo in the active site.1491−1493
Fdh-N from E. coli is among the most well studied Fdh’s. It is important in the nitrate respiratory pathway under anaerobic conditions. It is a membrane-bound trimer (α3β3γ3) with a molar mass of 510 kDa. It harbors a Mo-bis-MGD cofactor and a [4Fe–4S] cluster in the catalytic α subunit, four [4Fe–4S] clusters in the β subunit, and two heme b groups in the γ subunit (Figure 70).1492 The β subunit transfers electrons between the α and γ subunits, similar to other membrane-bound oxidoreductases that bind four [4Fe–4S] clusters, such as nitrate reductases, [NiFe] hydrogenases, DMSO reductase, and thiosulfate reductase.1494
Figure 70.
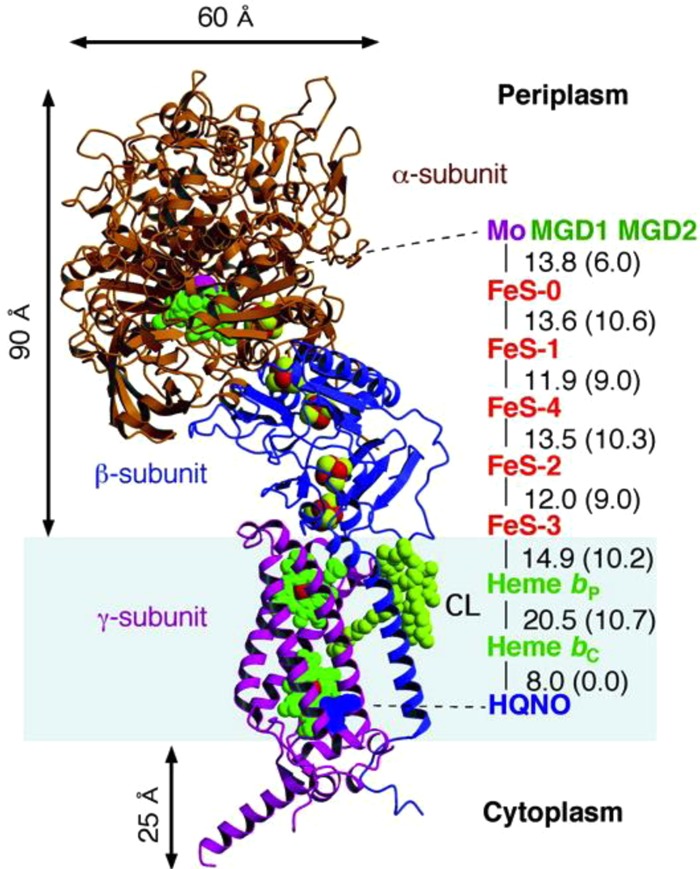
Overall structure of Fdh-N from E. coli. Cofactors are displayed as spheres and denoted accordingly on the right. The putative membrane is shown in gray shading. PDB ID 1KQF. Reprinted with permission from ref (1492). Copyright 2002 American Association for the Advancement of Science.
Fdh from Dv. desulfuricans is an αβγ protein with a molar mass of ∼150 kDa. It contains four different types of redox centers, including four heme c centers, two [4Fe–4S] clusters, and a molybdopterin.1495 EPR studies showed the existence of two types of Fe–S clusters after reduction, i.e., center I with g values of 2.050, 1.947, and 1.896 and center II with g values of 2.071, 1.926, and 1.865. Midpoint reduction potentials of the two Fe–S clusters are −350 ± 5 mV for center I and −335 ± 5 mV for center II.
Fdh from Dv. gigas is an αβ protein1493 containing tungsten instead of molybdenum. It also possesses two [4Fe–4S] clusters similar to Fdh from Dv. desulfuricans.981,1496
5.2.4. As Redox Centers in Nitrate Reductase
NARs reduce nitrate to nitrite, a vital component in the nitrogen respiratory cycle. Most NARs isolated so far contain three subunits, NarG (112–140 kDa), NarH (52–64 kDa), and NarI (19–25 kDa). NarG harbors a Mo-bis-MGD cofactor and a [4Fe–4S] cluster, NarH contains one [3Fe–4S] cluster and three [4Fe–4S] clusters, and NarI immersed in the membrane binds two b-type hemes (Figure 71).1497−1502 The overall folding and cofactor positions are strongly homologous to those of Fdh from E. coli.1503 The eight redox centers are separated by 12–15 Å from each other and form an ET pathway about 90 Å long. NAR from Cupriavidus necator does not contain the NarH domain and harbors two c-type hemes in the small subunit.1504
Figure 71.
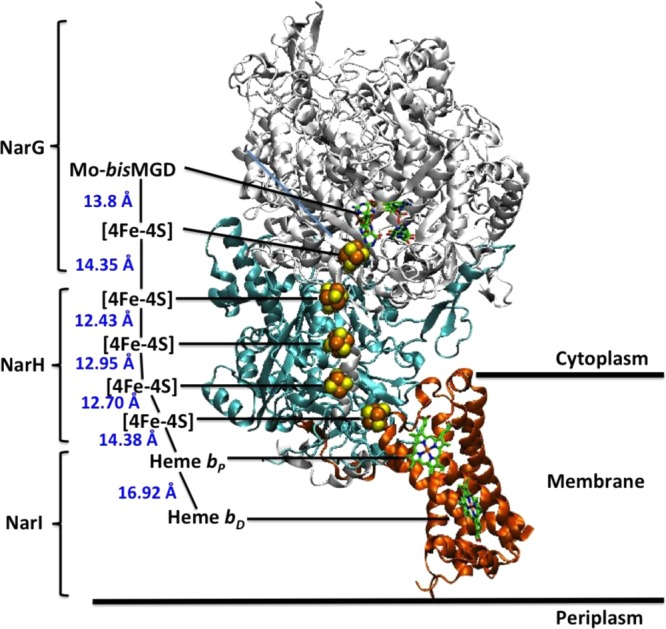
Overall three-dimensional structure of NarGHI from E. coli K12. PDB ID 1Q16. Subunit and cofactor names are denoted. Reprinted with permission from ref (1505). Copyright 2006 Elsevier.
6. Summary and Outlook
This review summarizes three important classes of redox centers involved in ET processes. Although each class spans a wide range of reduction potentials, none of them can cover the whole range needed for biological processes. Together, however, they can cover the whole range, with cytochromes in the middle, Fe–S centers toward the lower end, and the cupredoxins toward the higher end (Figure 1). All three redox centers have structural features that make them unique, and yet they also show many similarities that make them excellent choices for ET processes.
Because the redox-active iron is fixed into a rigid porphyrin that accounts for four of the iron’s six coordination sites, most of the electronic structure and redox properties remain similar between different cytochromes. In completing the primary coordination sphere of the iron, cytochromes typically use a combination of nitrogen and sulfur ligations from histidine or methionine side chains, respectively; terminal amine ligation has also been observed. In general, mutagenesis studies reveal that methionine ligation raises the reduction potential by 100–200 mV, relative to histidine ligation, primarily due to the lower affinity of thioether to the higher oxidation state of the heme, and that the effect is generally additive.192,386,461−463,465 Heme puckering or flexing has been demonstrated to tune the reduction potentials by up to 200 mV.513 Changes in the heme type between b and c would be expected to change the electronic properties of the heme; however, the effect on the reduction potential is small and varies depending on the systems studied.446,448 It is clear, on the other hand, that the electron-withdrawing formyl group on heme a appears to be responsible for the increase in the reduction potential by ∼160 mV.459,460
For iron–sulfur proteins, the reduction potential ranges are influenced to some extent by the number of irons because it affects the redox states and transitions. In the case of clusters with the same number of irons, the higher the redox pair, the higher the reduction potentials (e.g., HiPIPs have a [4Fe–4S]2+/3+ pair, while ferredoxins have a [4Fe–4S]1+/2+ pair).719 In addition, the cluster geometry such as Fe–Sγ–Cα–Cβ torsional angles, the Fe–Fe distance, and covalency of Fe–S bonds also play important roles in some proteins.618,901,1085,1506 Electron delocalization of the cluster and the net charge of the cluster are also important. For example, it has been shown that the net charge of the protein is the main factor determining the reduction potential within HiPIPs. Electrostatic effects of the charged residues in the secondary coordination sphere can influence the solvent accessibility and consequently the dielectric constant around the metal center. However, the effects are usually complicated and difficult to rationalize by just Coulomb’s law. For example, in rubredoxin from Cl. pasteurianum, replacement of a neutral surface residue by a positively charged Arg or a negatively charged Asp has led to an increase of reduction potentials in both cases.611,612 Finally, the direct ligands to iron and H-bonding interactions with the direct ligands make significant contributions to the reduction potential.541 When the common Cys thiolate ligand was replaced with a His imidazole ligand, naturally in the Rieske proteins, or with Ser by site-directed mutagenesis, the reduction potentials changed accordingly.721,773,1087 The multiple NH···S H-bonding interactions in rubredoxin render the reduction potential of the [FeCys4] center to fall in the range of −100 to +50 mV, while reduction potential of the corresponding model complexes without the H-bonding networks is around 1 V.92,588−590 The NH···S H-bonds have also been shown to be important in determining reduction potentials between different ferredoxins as well as ferredoxins vs HiPIPs.617,618,718,719
For cupredoxins, the metal centers cannot be easily fixed like in either porphyrin or thermodynamically stable iron–sulfur clusters and proteins play a more prominent role in enforcing the unique trigonal geometry and strong copper–thiolate bond to maintain a low reorganization energy for the ET function. In this class of proteins, both the geometry and the ligands, particularly the strictly conserved Cys, play a dominant role in controlling the redox properties. In T1 copper protein azurin, changing axial Met to a stronger cysteine or homocysteine induced a geometry change and weakened the Cu–S bond. These changes in turn resulted in a >100 mV decrease in the reduction potential.1293 Deleting the H-bonding to Cys, realized through the Phe114Pro mutation in azurin, affected the covalency of the Cu–S bond and lowered the reduction potential of azurin.114,1088,1316
Despite the differences in the primary coordination spheres, all three redox centers employ noncovalent secondary coordination interactions in fine-tuning the redox properties.
The first common feature is the control of the degree of solvent exposure; the deeper the redox centers are buried into the hydrophobic center of the protein, the higher the reduction potential and the smaller the changes in the reorganization energy due to influences by the solvent. For example, redox center burial is considered to be one of the main factors for differences in reduction potentials between different HiPIPs and ferredoxins.618,719,749,752 Furthermore, a computational study of heme proteins over an 800 mV range has attributed the greatest correlation with the reduction potential to solvent exposure.457
The second common feature is the electrostatic interactions. For example, the net charge of protein is shown to be the only factor that correlates with the reduction potentials of different HiPIPs.715,752,890 The number of amide dipoles and not necessarily H-bonding is shown to be important in reduction potential determination in ferredoxins.718,719 In myoglobin, Val68, which was in the van der Waals interaction distance with the heme group, was replaced by Glu, Asp, and Asn. A 200 mV decrease in reduction potential was observed for the Glu and Asp mutants compared to the wild type.481 This study demonstrated that replacement of hydrophobic Val68 by charged and polar residues led to substantial changes in the reduction potential of the heme iron. In a number of different cytochromes, electrostatic polar and charged groups near the heme were shown to vary the potential by 100–200 mV.169,479,481,482 For instance, in cyts c6 and c6A, the glutamine at positions 52 and 51, respectively, were shown to raise the potential ∼100 mV,479 and in cyt c, the Tyr48Lys mutation raised the potential 117 mV;480 all these effects can be attributed to charge compensation in the heme pocket. Similarly, replacing Met121 with Glu or Asp in T1 copper azurin resulted in 100 and 20 mV decreases in the reduction potentials, respectively.1278,1289 Beyond copper ligands, mutating Met44 in azurin to Lys destabilizes Cu(II), causing a 40 mV increase of the reduction potential.1507
The final common feature is the presence of a hydrogen-bonding network around the ligands to the metal center, especially those to the ligand that dominates the metal–ligand interactions. For example, the NHamide···Scys H-bonds are known to be important in different reduction potentials between rubredoxins, HiPIPs, and ferredoxins.617,618,718,719 They are also shown to play a role in different reduction potentials of different ferredoxins. Other than backbone amide H-bonds, H-bonds from side chains are also important. A good example of such is H-bonds from conserved Ser and Tyr in Rieske proteins and a lack of thereof in Rieske-type proteins, hence differences in the reduction potential.781 In cytochromes, H-bonding interactions with the axial ligands can tune the potential by up to 100 mV.474,476,477,1508 For instance, increasing the imidazolate character of the axial His ligand in cyt c by strengthening H-bonding from the H to the Nε increased the potential by nearly 100 mV,474 and disrupting the hydrogen bond donation from Tyr67 to the axial Met resulted in a 56 mV decrease in potential.476,1508 Similarly, the H-bonding interactions to the Cys in cupredoxins are known be responsible for their reduction potential differences.114
A test of how much we understand these structural features responsible for the redox properties is to start with a native redox center and use the above knowledge to fine-tune the redox properties. A pioneering work in this area is the demonstration of a ∼200 mV decrease in the reduction potential of myoglobin when a buried ionizable amino acid (Glu) was introduced into the distal pocket of the protein, and such a change has been attributed to electrostatic interactions.481 Since then, not many examples have shown similar magnitude changes of reduction potentials by electrostatic interactions, perhaps due to the compensation effect by ions in the buffer or other ionizable residues nearby. Instead, hydrophobicity and H-bonding network have been shown to play increasing roles, and a combination of these effects has been shown to fine-tune the reduction potentials of T1 copper azurins by more than 700 mV, beyond its natural range.1088 These features were further shown to be additive, making reduction potential tuning predictable. Such rational design also allowed the lowering of the reorganization energy of azurin,1317 which is already known to be very low in comparison to those of other redox centers. With more such successful examples in other systems, we will be able to achieve a deeper understanding of ET reactivity in proteins and facilitate de novo design of ET centers for applications such as advanced energy conversions.
Acknowledgments
The research in the Lu group was supported by grants from the National Science Foundation (CHE1058959) and the National Institutes of Health (GM062211).
Glossary
Abbreviations
- ACS
acetyl-CoA synthase
- Adx
adrenodoxin
- Az
Azurin
- CcO
cytochrome c oxidase
- CcP
cytochrome c peroxidase
- CD
circular dichroism
- CI
complex I in respiratory chain
- CII
complex II in respiratory chain
- CODH
carbon monoxide dehydrogenase
- CV
cyclic voltammetry
- cyt
cytochrome
- DEAE
(diethylamino) ethanol
- DFT
density functional theory
- Dfx
desulfoferredoxin
- Dx
desulforedoxin
- ENDOR
electron−nuclear double resonance
- EPR
electron paramagnetic resonance
- ESEEM
electron spin echo envelope modulation
- ET
electron transfer
- EXAFS
X-ray absorption fine structure
- FAD
flavin adenine dinucleotide
- Fd
ferredoxin
- Fdh
formate dehydrogenase
- FMN
flavin mononucleotide
- FNR
ferredoxin:NADP reductase
- FTR
ferredoxin:thioredoxin reductase
- HAO
hydroxylamine oxidoreductase
- H-bond
hydrogen bond
- HCO
heme copper oxidase
- HCP
hybrid cluster protein
- HiPIP
high-potential iron–sulfur protein
- hp
high-potential
- HS
high-spin
- ITC
isothermal titration calorimetry
- LMCT
ligand to metal charge transfer
- lp
low-potential
- LS
low-spin
- MCD
magnetic circular dichroism
- MCO
multicopper oxidase
- MO
molecular orbital
- NDO
naphthalene dioxygenase
- NHE
normal hydrogen electrode
- NiR
nitrite reductase
- NMR
nuclear magnetic resonance
- NOR
nitric oxide reductase
- N2OR
nitrous oxide reductase
- NR
nitrate reductase
- NRVS
nuclear resonance vibrational spectroscopy
- OM
outer membrane
- ORD
optical rotatory dispersion
- PCMH
p-cresol methylhydroxylase
- PDLP
protein dipole Langevine dipole
- PES
potential energy surface
- PLFP
plant ferredoxin-like protein
- PQQ
pyrroloquinoline quinone
- PSI
photosystem I
- PSII
photosystem II
- PSM
peptide-sandwiched mesoheme
- RC
reaction center
- Rd
rubredoxin
- ROS
reactive oxygen species
- Rr
rubrerythrin
- SDH
succinate dehydrogenase
- SHE
standard hydrogen electrode
- SHP
sphaeroides heme protein
- SiR
sulfite reductase
- SOR
superoxide reductase
- STC
small tetraheme cyt c
- TASP
template-assisted synthetic protein
- THC
tetraheme cytochrome
- T1
type 1
- T2
type 2
- WT
wild type
- XANES
X-ray absorption near edge structure
- XAS
X-ray absorption spectroscopy
- 6cLS
6-coordinate low-spin
Biographies

Jing Liu was born in Inner Mongolia, China. She obtained her B.A. degree in Chemistry from Nankai University (China) in 2003. Then she moved to Peking University (China) and worked under the guidance of Prof. Zhen Yang and Prof. Jiahua Chen, focusing on mechanistic and kinetic investigation of sulfur-containing ligands in palladium catalysis. During her graduate studies, she also worked with Prof. Todd B. Marder at Durham University (United Kingdom) and Prof. Aiwen Lei at Wuhan University (China). She completed her Ph.D in 2008. Afterward, she joined Prof. Yi Lu’s group as a postdoctoral researcher at the University of Illinois at Urbana-Champaign. Her current research focuses on engineering artificial nonheme iron enzymes and developing biocatalysts.

Saumen Chakraborty earned his B.Sc. in Chemistry (Hons.) from the University of Calcutta in 2004 and M.Sc. in Chemistry from the Indian Institute of Technology, Madras, in 2006. He went to the University of Michigan in 2006 for graduate research and joined Prof. Vincent Pecoraro’s group, working in the area of de novo metalloprotein design using three-stranded coiled coils and three-helix bundles with emphasis on creation of novel metal coordination sites, understanding how to control and fine-tune metal ion properties within a protein matrix, and determining ligand and metal exchange dynamics. After completing his Ph.D. in 2011, he joined Prof. Yi Lu’s group at the University of Illinois at Urbana-Champaign as a postdoctoral researcher. His current research interests include biosynthetic metalloprotein design, preparation of protein-based structural and functional analogues of various enzymes, biochemical mechanisms, nonheme iron centers, and cobalt metalloproteins. He enjoys traveling and photography.

Parisa Hosseinzadeh was born in Neyshabour, Iran. She received her B.Sc degree in Biotechnology from the University of Tehran in 2010. She is a Ph.D. candidate in Biochemistry at the University of Illinois at Urbana-Champaign. Her research in Dr. Yi Lu’s laboratory focuses on rationally tuning protein features using secondary coordination sphere interactions and characterizing engineered proteins with several biochemical and bioinorganic techniques.

Yang Yu was born in Xinzhou, China. He received his B.S. in Biology from Peking University in 2008. He is pursuing a Ph.D. degree in Biophysics and Computational Biology at the University of Illinois at Urbana-Champaign under the supervision of Dr. Yi Lu. His current research interests include using unnatural amino acids for metalloprotein engineering and X-ray absorption spectroscopy for metalloproteins.

Shiliang Tian received his B.S. in Chemistry in 2005 from the School of Chemistry and Chemical Engineering, Nanjing University, China. He then joined Prof. Zhang-Jie Shi’s laboratory at Peking University, working on C–H activation using transition metals. After receiving his Masters degree in Organic Chemistry, he joined Prof. Yi Lu’s laboratory at the University of Illinois at Urbana-Champaign in 2010. He is currently a Ph.D. candidate, working on designing the metalloenzyme for biocatalysis, primarily focusing on copper proteins and nonheme iron proteins.

Igor Petrik was born in L’viv, Ukraine, and grew up in Philadelphia, PA. From 2006 to 2009, he attended the University of the Sciences in Philadelphia, where he investigated the physicochemical properties of ionic liquids by NMR and MD techniques under the mentorship of Prof. Guillermo Moyna. After graduating with a B.S. in Chemistry and a minor in Forensics, he began his Ph.D. studies in Chemical Biology in the laboratory of Prof. Yi Lu at the University of Illinois at Urbana-Champaign. He is interested in rational design of metalloenzymes and is focusing on understanding and improving the activity of biosynthetic models of terminal oxidases.

Ambika Bhagi was born in Moradabad, India. She received her B.Sc. from St. Stephen’s College, Delhi, in 2009, followed by a Masters degree in Chemistry from the Indian Institute of Technology, Delhi, in 2011. She is currently a Ph.D. candidate in Chemical Biology at the University of Illinois at Urbana-Champaign. Her research focuses on elucidating the role of the heme iron redox potential toward tuning oxygen reduction rates in terminal oxidases.

Yi Lu received his B.S. degree from Peking University in 1986 and Ph.D. degree from the University of California at Los Angeles in 1992 under Professor Joan S. Valentine. After 2 years of postdoctoral research in Professor Harry B. Gray’s group at the California Institute of Technology, Lu started his own independent career in the Department of Chemistry at the University of Illinois at Urbana-Champaign in 1994. He is now a Jay and Ann Schenck Professor of Chemistry in the Departments of Chemistry, Biochemistry, Bioengineering, and Materials Science and Engineering. He is also a member of the Center for Biophysics and Computational Biology and Beckman Institute for Advanced Science and Technology. His research interests lie at the interface between chemistry and biology. His group is developing new chemical approaches to provide deeper insight into biological systems. At the same time, they take advantage of recently developed biological tools to advance many areas in chemistry. Lu has received numerous research and teaching awards, including the Early Career Award, Society of Biological Inorganic Chemistry (2007), Howard Hughes Medical Institute Professor Award (2002), Camile Dreyfus Teacher-Scholar Award (1999), Alfred P. Sloan Research Fellowship (1998), Research Corporation Cottrell Scholars Award (1997), and Beckman Young Investigators Award (1996), and was named a Fellow of the American Association for the Advancement of Science (2007).
Author Contributions
∥ J.L., S.C., P.H., and Y.Y. contributed equally to this work.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
References
- Malkin R.; Rabinowitz J. C. Annu. Rev. Biochem. 1967, 36, 113. [DOI] [PubMed] [Google Scholar]
- Adman E. T. Biochim. Biophys. Acta, Rev. Bioenerg. 1979, 549, 107. [DOI] [PubMed] [Google Scholar]
- Brunori M.; Colosimo A.; Silvestrini M. C. Pure Appl. Chem. 1983, 55, 1049. [Google Scholar]
- Thomson A. J. Nachr. Chem., Tech. Lab. 1984, 32, 326. [Google Scholar]
- Chiara S.; Maria; Brunori M. Life Chem. Rep. 1987, 5, 155. [Google Scholar]
- Gil’manshin R. I.; Lazarev P. I. Mater. Sci. 1987, 13, 71. [Google Scholar]
- Armstrong F. A.; Hill H. A. O.; Walton N. J. Acc. Chem. Res. 1988, 21, 407. [Google Scholar]
- Cusanovich M. A.; Meyer T. E.. Heme Proteins; Elsevier: Amsterdam, 1988; Vol. 7. [Google Scholar]
- McLendon G. Acc. Chem. Res. 1988, 21, 160. [Google Scholar]
- Huber R. Prix Nobel 1989, 189. [Google Scholar]
- Meyer T. E.; Cusanovich M. A. Biochim. Biophys. Acta, Bioenerg. 1989, 975, 1. [DOI] [PubMed] [Google Scholar]
- Gray H. B.; Malmstroem B. G. Biochemistry 1989, 28, 7499. [DOI] [PubMed] [Google Scholar]
- Bowler B. E.; Raphael A. L.; Gray H. B. Prog. Inorg. Chem. 1990, 38, 259. [Google Scholar]
- Malmstroem B. G. Biol. Met. 1990, 3, 64. [Google Scholar]
- Blondin G.; Girerd J. J. Chem. Rev. 1990, 90, 1359. [Google Scholar]
- Gray H. B. Aldrichimica Acta 1990, 23, 87. [Google Scholar]
- Lee H.; DeFelippis M. H.; Faraggi M.; Klapper M. H.. Oxidative Damage and Repair; Pergamon: Oxford, U.K., 1991; p 246. [Google Scholar]
- Glusker J. P. Adv. Protein Chem. 1991, 42, 1. [DOI] [PubMed] [Google Scholar]
- Mauk A. G. Struct. Bonding (Berlin, Ger.) 1991, 75, 131. [Google Scholar]
- Isied S. S. Met. Ions Biol. Syst. 1991, 27, 1. [Google Scholar]
- Kostic N. M. Met. Ions Biol. Syst. 1991, 27, 129. [PubMed] [Google Scholar]
- Beratan D. N.; Onuchic J. N.; Winkler J. R.; Gray H. B. Science 1992, 258, 1740. [DOI] [PubMed] [Google Scholar]
- Canters G. W.; Van d. K. M. Curr. Opin. Struct. Biol. 1992, 2, 859. [Google Scholar]
- Winkler J. R.; Gray H. B. Chem. Rev. 1992, 92, 369. [Google Scholar]
- Mathews F. S.; White S. Curr. Opin. Struct. Biol. 1993, 3, 902. [Google Scholar]
- Farid R. S.; Moser C. C.; Dutton P. L. Curr. Opin. Struct. Biol. 1993, 3, 225. [Google Scholar]
- Friesner R. A. Structure (London, U. K.) 1994, 2, 339. [DOI] [PubMed] [Google Scholar]
- Dong S.; Niu J.; Cotton T. M. Methods Enzymol. 1995, 246, 701. [DOI] [PubMed] [Google Scholar]
- Chen L.; Liu M.-Y.; LeGall J. Biotechnol. Handb. 1995, 8, 113. [Google Scholar]
- Chapman S. K.; Mount A. R. Nat. Prod. Rep. 1995, 12, 93. [Google Scholar]
- Gray H. B.; Winkler J. R. Annu. Rev. Biochem. 1996, 65, 537. [DOI] [PubMed] [Google Scholar]
- Mathews F. S.; Durley R. C. E. In Protein Electron Transfer; Bendall D. S., Ed.; BIOS Scientific Publishers Ltd.: Oxford, U.K., 1996; p 99. [Google Scholar]
- Bendall D. S.Protein Electron Transfer; BIOS Scientific Publishers Ltd.: Oxford, U.K., 1996. [Google Scholar]
- Hill H. A. O.; Moore C. B.; NabiRahni D. M. A. Bioelectrochem.: Princ. Pract. 1997, 5, 183. [Google Scholar]
- Hayashi T.; Ogoshi H. Chem. Soc. Rev. 1997, 26, 355. [Google Scholar]
- Beratan D. N.; Skourtis S. S. Curr. Opin. Chem. Biol. 1998, 2, 235. [DOI] [PubMed] [Google Scholar]
- Malmstrom B. G.; Wittung-Stafshede P. Coord. Chem. Rev. 1999, 185–186, 127. [Google Scholar]
- Kennedy M. L.; Gibney B. R. Curr. Opin. Struct. Biol. 2001, 11, 485. [DOI] [PubMed] [Google Scholar]
- Ichiye T.Computational Biochemistry and Biophysics; Marcel Dekker, Inc.: New York, 2001; p 393. [Google Scholar]
- Bellelli A.; Brunori M.; Brzezinski P.; Wilson M. T. Methods 2001, 24, 139. [DOI] [PubMed] [Google Scholar]
- Davidson V. L. Biochemistry 2002, 41, 14633. [DOI] [PubMed] [Google Scholar]
- Gray H. B.; Winkler J. R. Q. Rev. Biophys. 2003, 36, 341. [DOI] [PubMed] [Google Scholar]
- Winkler J. R.; Gray H. B.; Prytkova T. R.; Kurnikov I. V.; Beratan D. N. In Bioelectronics; Willner I., Katz E., Eds.; Wiley-VCH: Weinheim, Germany, 2005; p 15. [Google Scholar]
- Gray H. B.; Winkler J. R. Proc. Natl. Acad. Sci. U.S.A. 2005, 102, 3534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunner M. R.; Mao J.; Song Y.; Kim J. Biochim. Biophys. Acta, Bioenerg. 2006, 1757, 942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banci L.; Bertini I.; Luchinat C.; Turano P. In Bioinorganic Chemistry; Bertini I., Gray H. B., Stiefel E. I., Valentine J. S., Eds.; University Science Books: Sausalito, CA, 2007. [Google Scholar]
- Farver O.; Pecht I. Prog. Inorg. Chem. 2007, 55, 1. [Google Scholar]
- Bollinger J. M. Jr Science 2008, 320, 1730. [DOI] [PubMed] [Google Scholar]
- Hammes-Schiffer S.; Soudackov A. V. J. Phys. Chem. B 2008, 112, 14108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brittain T. Protein Pept. Lett. 2008, 15, 556. [DOI] [PubMed] [Google Scholar]
- Cordes M.; Giese B. Chem. Soc. Rev. 2009, 38, 892. [DOI] [PubMed] [Google Scholar]
- Gray H. B.; Winkler J. R. Chem. Phys. Lett. 2009, 483, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bashir Q.; Scanu S.; Ubbink M. FEBS J. 2011, 278, 1391. [DOI] [PubMed] [Google Scholar]
- Choi M.; Davidson V. L. Metallomics 2011, 3, 140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de la Lande A.; Babcock N. S.; Rezac J.; Levy B.; Sanders B. C.; Salahub D. R. Phys. Chem. Chem. Phys. 2012, 14, 5902. [DOI] [PubMed] [Google Scholar]
- Giese B.; Eckhardt S.; Lauz M.. Encyclopedia of Radicals in Chemistry, Biology and Materials; John Wiley & Sons, Ltd.: Hoboken, NJ, 2012. [Google Scholar]
- Marshall N. M.; Miner K. D.; Wilson T. D.; Lu Y.. Coordination Chemistry in Protein Cages; John Wiley & Sons, Inc.: Hoboken, NJ, 2013; p 111. [Google Scholar]
- Wherland S.; Gray H. B. In Biological Aspects of Inorganic Chemistry; Dolphin D., Ed.; John Wiley: New York, 1977; p 289. [Google Scholar]
- Sutin N.; Brunschwig B. S. Adv. Chem. Ser. 1990, 226, 65. [Google Scholar]
- McLendon G. Met. Ions Biol. Syst. 1991, 27, 183. [Google Scholar]
- McLendon G.; Hake R.; Zhang Q.; Corin A. Mol. Cryst. Liq. Cryst. 1991, 194, 225. [Google Scholar]
- Tollin G.; Hazzard J. T. Arch. Biochem. Biophys. 1991, 287, 1. [DOI] [PubMed] [Google Scholar]
- Vervoort J. Curr. Opin. Struct. Biol. 1991, 1, 889. [Google Scholar]
- McLendon G.; Hake R. Chem. Rev. 1992, 92, 481. [Google Scholar]
- Onuchic J. N.; Beratan D. N.; Winkler J. R.; Gray H. B. Annu. Rev. Biophys. Biomol. Struct. 1992, 21, 349. [DOI] [PubMed] [Google Scholar]
- Wampler J. E. Methods Enzymol. 1994, 243, 559. [DOI] [PubMed] [Google Scholar]
- Moser C. C.; Dutton P. L. In Protein Electron Transfer; Bendall D. S., Ed.; BIOS Scientific Publishers Ltd.: Oxford, U.K., 1996; p 1. [Google Scholar]
- Telford J. R.; Wittung-Stafshede P.; Gray H. B.; Winkler J. R. Acc. Chem. Res. 1998, 31, 755. [Google Scholar]
- Sharp R. E.; Chapman S. K. Biochim. Biophys. Acta, Protein Struct. Mol. Enzymol. 1999, 1432, 143. [DOI] [PubMed] [Google Scholar]
- Sticht H. Recent Res. Dev. Biochem. 1999, 1, 1. [Google Scholar]
- Supkowski R. M.; Bolender J. P.; Smith W. D.; Reynolds L. E. L.; Horrocks W. D. Jr Coord. Chem. Rev. 1999, 185–186, 307. [Google Scholar]
- Randall D. W.; Gamelin D. R.; LaCroix L. B.; Solomon E. I. JBIC, J. Biol. Inorg. Chem. 2000, 5, 16. [DOI] [PubMed] [Google Scholar]
- Page C. C.; Moser C. C.; Dutton P. L. Curr. Opin. Chem. Biol. 2003, 7, 551. [DOI] [PubMed] [Google Scholar]
- Wheeler R. A. ACS Symp. Ser. 2004, 883, 1. [Google Scholar]
- Noodleman L.; Han W.-G. JBIC, J. Biol. Inorg. Chem. 2006, 11, 674. [DOI] [PubMed] [Google Scholar]
- Solomon E. I.; Xie X.; Dey A. Chem. Soc. Rev. 2008, 37, 623. [DOI] [PubMed] [Google Scholar]
- Prabhulkar S.; Tian H.; Wang X.; Zhu J.-J.; Li C.-Z. Antioxid. Redox Signaling 2012, 17, 1796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodgers K. R.; Lukat-Rodgers G. S. In Comprehensive Coordination Chemistry II; McCleverty J., Meyer T. J., Eds.; Elsevier Pergamon: Amsterdam, 2004; Vol. 8, p 17. [Google Scholar]
- Bertini I.; Cavallaro G.; Rosato A. Chem. Rev. 2006, 106, 90. [DOI] [PubMed] [Google Scholar]
- Moore G. R.; Pettigrew G. W.. Cytochromes c: Evolutionary, Structural and Physicochemical Aspects; Springer-Verlag: Berlin, 1990. [Google Scholar]
- Reedy C. J.; Gibney B. R. Chem. Rev. 2004, 104, 617. [DOI] [PubMed] [Google Scholar]
- Simonneaux G.; Bondon A. Chem. Rev. 2005, 105, 2627. [DOI] [PubMed] [Google Scholar]
- Walker F. A. Chem. Rev. 2004, 104, 589. [DOI] [PubMed] [Google Scholar]
- Weiss R.; Gold A.; Terner J. Chem. Rev. 2006, 106, 2550. [DOI] [PubMed] [Google Scholar]
- Schultz B. E.; Chan S. I. Annu. Rev. Biophys. Biomol. Struct. 2001, 30, 23. [DOI] [PubMed] [Google Scholar]
- Berry E. A.; Guergova-Kuras M.; Huang L. S.; Crofts A. R. Annu. Rev. Biochem. 2000, 69, 1005. [DOI] [PubMed] [Google Scholar]
- Brayer G. D.; Murphy M. E. P. In Cytochrome c: A Multidisciplinary Approach; Scott R. A., Mauk A. G., Eds.; University Science Books: Sausalito, CA, 1996; p 103. [Google Scholar]
- Meyer T. E. In Cytochrome c: A Multidisciplinary Approach; Scott R. A., Mauk A. G., Eds.; University Science Books: Sausalito, CA, 1996; p 33. [Google Scholar]
- Ambler R. P.From Cyclotrons to Cytochromes; Academic Press: Weinheim, Germany, 1982. [Google Scholar]
- Meyer T. E.; Kamen M. D. Adv. Protein Chem. 1982, 35, 105. [DOI] [PubMed] [Google Scholar]
- Meyer J. JBIC, J. Biol. Inorg. Chem. 2008, 13, 157. [DOI] [PubMed] [Google Scholar]
- Stephens P. J.; Jollie D. R.; Warshel A. Chem. Rev. 1996, 96, 2491. [DOI] [PubMed] [Google Scholar]
- Noodleman L.; Pique M. E.; Roberts V. A.. Wiley Encyclopedia of Chemical Biology; John Wiley & Sons, Inc.: Hoboken, NJ, 2009; Vol. 2, p 458. [Google Scholar]
- Adman E. T. Adv. Protein Chem. 1991, 42, 145. [DOI] [PubMed] [Google Scholar]
- Gray H. B.; Malmstrom B. G.; Williams R. J. JBIC, J. Biol. Inorg. Chem. 2000, 5, 551. [DOI] [PubMed] [Google Scholar]
- Solomon E. I.; Randall D. W.; Glaser T. Coord. Chem. Rev. 2000, 200–202, 595. [Google Scholar]
- Vila A. J.; Fernandez C. O. In Handbook on Metalloproteins; Bertini I., Sigel A., Sigel H., Eds.; Marcel Dekker: New York, 2001; p 813. [Google Scholar]
- Lu Y.Electron Transfer: Cupredoxins , In Comprehensive Coordination Chemistry II; McCleverty J. A., Meyer T. J., Eds.; Elsevier: Oxford, U.K., 2003; Vol. 8 (Biocoordination Chemistry; Que, J. L., Tolman, W. B. Eds.), p 91122. [Google Scholar]
- Solomon E. I.; Szilagyi R. K.; DeBeer George S.; Basumallick L. Chem. Rev. 2004, 104, 419. [DOI] [PubMed] [Google Scholar]
- Dennison C. Coord. Chem. Rev. 2005, 249, 3025. [Google Scholar]
- Farver O.; Pecht I.. Progress in Inorganic Chemistry; John Wiley & Sons, Inc.: Hoboken, NJ, 2008; p 1. [Google Scholar]
- Kolczak U.; Dennison C.; Messerschmidt A.; Canters G. W.. Encyclopedia of Inorganic and Bioinorganic Chemistry; John Wiley & Sons, Ltd.: Hoboken, NJ, 2011. [Google Scholar]
- Wilson T. D.; Yu Y.; Lu Y. Coord. Chem. Rev. 2012, 257, 260. [Google Scholar]
- Warren J. J.; Lancaster K. M.; Richards J. H.; Gray H. B. J. Inorg. Biochem. 2012, 115, 119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noodleman L.; Han W.-G. JBIC, J. Biol. Inorg. Chem. 2006, 11, 674. [DOI] [PubMed] [Google Scholar]
- Szilagyi R. K.; Solomon E. I. Curr. Opin. Chem. Biol. 2002, 6, 250. [DOI] [PubMed] [Google Scholar]
- Solomon E. I.; Hadt R. G. Coord. Chem. Rev. 2011, 255, 774. [Google Scholar]
- Neese F. Coord. Chem. Rev. 2009, 253, 526. [Google Scholar]
- Solomon E. I.; Xie X.; Dey A. Chem. Soc. Rev. 2008, 37, 623. [DOI] [PubMed] [Google Scholar]
- Yoon J.; Solomon E. I. Biol. Magn. Reson. 2009, 28, 471. [Google Scholar]
- Solomon E. I.; Heppner D. E.; Johnston E. M.; Ginsbach J. W.; Cirera J.; Qayyum M.; Kieber-Emmons M. T.; Kjaergaard C. H.; Hadt R. G.; Tian L.. Chem. Rev. 2014, 114 (7), DOI: 10.1021/cr400327t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blomberg M. R. A.; Borowski T.; Himo F.; Liao R.-Z.; Siegbahn P. E. M.. Chem. Rev. 2014, 114 (7), DOI: 10.1021/cr400388t. [DOI] [PubMed] [Google Scholar]
- Layfield J. P.; Hammes-Schiffer S.. Chem. Rev. 2014, 114 (7), DOI: 10.1021/cr400400p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margoliash E.; Schejter A. Trends Biochem. Sci. 1984, 9, 364. [Google Scholar]
- Keilin D. Proc. R. Soc. London, Ser. B 1925, 98, 312. [Google Scholar]
- Yaoi H.; Tamiya H. Proc. Imp. Acad. (Tokyo) 1928, 4, 436. [Google Scholar]
- Yanagisawa S.; Banfield M. J.; Dennison C. Biochemistry 2006, 45, 8812. [DOI] [PubMed] [Google Scholar]
- Jiang X. J.; Wang X. D. Annu. Rev. Biochem. 2004, 73, 87. [DOI] [PubMed] [Google Scholar]
- Yamanaka T.The Biochemistry of Bacterial Cytochromes; Japan Scientific Societies Press: Tokyo; Springer-Verlag: New York, 1992. [Google Scholar]
- Vonjagow G.; Sebald W. Annu. Rev. Biochem. 1980, 49, 281. [DOI] [PubMed] [Google Scholar]
- Kamen M. D.; Horio T. Annu. Rev. Biochem. 1970, 39, 673. [DOI] [PubMed] [Google Scholar]
- Puustinen A.; Wikstrom M. Proc. Natl. Acad. Sci. U.S.A. 1991, 88, 6122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frolow F.; Kalb A. J.; Yariv J. Nat. Struct. Biol. 1994, 1, 453. [DOI] [PubMed] [Google Scholar]
- LeBrun N. E.; Thomson A. J.; Moore G. R. In Metal Sites in Proteins and Models; Hill H. A. O., Sadler P. J., Thomson A. J., Eds.; Springer: Berlin, Heidelberg, 1997; Vol. 88, p 103. [Google Scholar]
- Iverson T. M.; Arciero D. M.; Hsu B. T.; Logan M. S. P.; Hooper A. B.; Rees D. C. Nat. Struct. Biol. 1998, 5, 1005. [DOI] [PubMed] [Google Scholar]
- Martinez S. E.; Huang D.; Szczepaniak A.; Cramer W. A.; Smith J. L. Structure (London, U. K.) 1994, 2, 95. [DOI] [PubMed] [Google Scholar]
- Liu X. S.; Kim C. N.; Yang J.; Jemmerson R.; Wang X. D. Cell 1996, 86, 147. [DOI] [PubMed] [Google Scholar]
- Yang J.; Liu X. S.; Bhalla K.; Kim C. N.; Ibrado A. M.; Cai J. Y.; Peng T. I.; Jones D. P.; Wang X. D. Science 1997, 275, 1129. [DOI] [PubMed] [Google Scholar]
- Kluck R. M.; BossyWetzel E.; Green D. R.; Newmeyer D. D. Science 1997, 275, 1132. [DOI] [PubMed] [Google Scholar]
- Mauk A. G. Struct. Bonding (Berlin, Ger.) 1991, 75, 132. [Google Scholar]
- Wilson M. T.; Greenwood C.. Cytochrome c: A Multidisciplinary Approach; University Science Books: Sausalito, CA, 1996. [Google Scholar]
- Mauk A. G.; Moore G. R. JBIC, J. Biol. Inorg. Chem. 1997, 2, 119. [Google Scholar]
- McGee W. A.; Rosell F. I.; Liggins J. R.; RodriguezGhidarpour S.; Luo Y. G.; Chen J.; Brayer G. D.; Mauk A. G.; Nall B. T. Biochemistry 1996, 35, 1995. [DOI] [PubMed] [Google Scholar]
- Rafferty S. P.; Guillemette J. G.; Berghuis A. M.; Smith M.; Brayer G. D.; Mauk A. G. Biochemistry 1996, 35, 10784. [DOI] [PubMed] [Google Scholar]
- Bjerrum M. J.; Casimiro D. R.; Chang I. J.; Di Bilio A. J.; Gray H. B.; Hill M. G.; Langen R.; Mines G. A.; Skov L. K.; Winkler J. R.; et al. J. Bioenerg. Biomembr. 1995, 27, 295. [DOI] [PubMed] [Google Scholar]
- Therien M. J.; Chang J.; Raphael A. L.; Bowler B. E.; Gray H. B. Struct. Bonding (Berlin, Ger.) 1991, 75, 109. [Google Scholar]
- Brzezinski P.; Wilson M. T. Proc. Natl. Acad. Sci. U.S.A. 1997, 94, 6176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dopner S.; Hildebrandt P.; Rosell F. I.; Mauk A. G.; von Walter M.; Buse G.; Soulimane T. Eur. J. Biochem. 1999, 261, 379. [DOI] [PubMed] [Google Scholar]
- Hildebrandt P.; Heimburg T.; Marsh D.; Powell G. L. Biochemistry 1990, 29, 1661. [DOI] [PubMed] [Google Scholar]
- Hildebrandt P.; Vanhecke F.; Heibel G.; Mauk A. G. Biochemistry 1993, 32, 14158. [DOI] [PubMed] [Google Scholar]
- Weber C.; Michel B.; Bosshard H. R. Proc. Natl. Acad. Sci. U.S.A. 1987, 84, 6687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durham B.; Fairris J. L.; McLean M.; Millett F.; Scott J. R.; Sligar S. G.; Willie A. J. Bioenerg. Biomembr. 1995, 27, 331. [DOI] [PubMed] [Google Scholar]
- Mauk A. G.; Mauk M. R.; Moore G. R.; Northrup S. H. J. Bioenerg. Biomembr. 1995, 27, 311. [DOI] [PubMed] [Google Scholar]
- Rodriguez-Maranon M. J.; Qiu F.; Stark R. E.; White S. P.; Zhang X.; Foundling S. I.; Rodriguez V.; Schilling C. L. 3rd; Bunce R. A.; Rivera M. Biochemistry 1996, 35, 16378. [DOI] [PubMed] [Google Scholar]
- Ullmann G. M.; Kostic N. M. J. Am. Chem. Soc. 1995, 117, 4766. [Google Scholar]
- Crnogorac M. M.; Ullmann G. M.; Kostic N. M. J. Am. Chem. Soc. 2001, 123, 10789. [DOI] [PubMed] [Google Scholar]
- Pletneva E. V.; Fulton D. B.; Kohzuma T.; Kostic N. M. J. Am. Chem. Soc. 2000, 122, 1034. [Google Scholar]
- Zhou J. S.; Kostic N. M. J. Am. Chem. Soc. 1992, 114, 3562. [Google Scholar]
- Hoffman B. M.; Natan M. J.; Nocek J. M.; Wallin S. A. Struct. Bonding (Berlin, Ger.) 1991, 75, 85. [Google Scholar]
- Liang N.; Mauk A. G.; Pielak G. J.; Johnson J. A.; Smith M.; Hoffman B. M. Science 1988, 240, 311. [DOI] [PubMed] [Google Scholar]
- McLendon G. Struct. Bonding (Berlin, Ger.) 1991, 75, 159. [Google Scholar]
- Nocek J. M.; Zhou J. S.; DeForest S.; Priyadarshy S.; Beratan D. N.; Onuchic J. N.; Hoffman B. M. Chem. Rev. 1996, 96, 2459. [DOI] [PubMed] [Google Scholar]
- Ferguson S. J.; Stevens J. M.; Allen J. W. A.; Robertson I. B. Biochim. Biophys. Acta, Bioenerg. 2008, 1777, 980. [DOI] [PubMed] [Google Scholar]
- Stevens T. H.; Martin C. T.; Wang H.; Brudvig G. W.; Scholes C. P.; Chan S. I. J. Biol. Chem. 1982, 257, 12106. [PubMed] [Google Scholar]
- Ambler R. P. Biochim. Biophys. Acta, Bioenerg. 1991, 1058, 42. [DOI] [PubMed] [Google Scholar]
- Arciero D. M.; Hooper A. B. J. Biol. Chem. 1994, 269, 11878. [PubMed] [Google Scholar]
- Shimizu H.; Schuller D. J.; Lanzilotta W. N.; Sundaramoorthy M.; Arciero D. M.; Hooper A. B.; Poulos T. L. Biochemistry 2001, 40, 13483. [DOI] [PubMed] [Google Scholar]
- Menin L.; Schoepp B.; Garcia D.; Parot P.; Vermeglio A. Biochemistry 1997, 36, 12175. [DOI] [PubMed] [Google Scholar]
- Bertrand P.; Mbarki O.; Asso M.; Blanchard L.; Guerlesquin F.; Tegoni M. Biochemistry 1995, 34, 11071. [DOI] [PubMed] [Google Scholar]
- Gunner M. R.; Honig B. Proc. Natl. Acad. Sci. U.S.A. 1991, 88, 9151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caffrey M. S.; Cusanovich M. A. Biochim. Biophys. Acta, Bioenerg. 1994, 1187, 277. [DOI] [PubMed] [Google Scholar]
- Caffrey M. S.; Daldal F.; Holden H. M.; Cusanovich M. A. Biochemistry 1991, 30, 4119. [DOI] [PubMed] [Google Scholar]
- Zhao D. Z.; Hutton H. M.; Meyer T. E.; Walker F. A.; MacKenzie N. E.; Cusanovich M. A. Biochemistry 2000, 39, 4053. [DOI] [PubMed] [Google Scholar]
- Ponamarev M. V.; Schlarb B. G.; Howe C. J.; Carrell C. J.; Smith J. L.; Bendall D. S.; Cramer W. A. Biochemistry 2000, 39, 5971. [DOI] [PubMed] [Google Scholar]
- Grönberg K. L.; Roldán M. D.; Prior L.; Butland G.; Cheesman M. R.; Richardson D. J.; Spiro S.; Thomson A. J.; Watmough N. J. Biochemistry 1999, 38, 13780. [DOI] [PubMed] [Google Scholar]
- Ellfolk N.; Ronnberg M.; Aasa R.; Andreasson L. E.; Vanngard T. Biochim. Biophys. Acta, Protein Struct. Mol. Enzymol. 1983, 743, 23. [DOI] [PubMed] [Google Scholar]
- Sawaya M. R.; Krogmann D. W.; Serag A.; Ho K. K.; Yeates T. O.; Kerfeld C. A. Biochemistry 2001, 40, 9215. [DOI] [PubMed] [Google Scholar]
- Hickey D. R.; Berghuis A. M.; Lafond G.; Jaeger J. A.; Cardillo T. S.; McLendon D.; Das G.; Sherman F.; Brayer G. D.; McLendon G. J. Biol. Chem. 1991, 266, 11686. [PubMed] [Google Scholar]
- Döpner S.; Hildebrandt P.; Rosell F. I.; Mauk A. G. J. Am. Chem. Soc. 1998, 120, 11246. [Google Scholar]
- Rosell F. I.; Ferrer J. C.; Mauk A. G. J. Am. Chem. Soc. 1998, 120, 11234. [Google Scholar]
- Cutler R. L.; Davies A. M.; Creighton S.; Warshel A.; Moore G. R.; Smith M.; Mauk A. G. Biochemistry 1989, 28, 3188. [DOI] [PubMed] [Google Scholar]
- Lett C. M.; Berghuis A. M.; Frey H. E.; Lepock J. R.; Guillemette J. G. J. Biol. Chem. 1996, 271, 29088. [DOI] [PubMed] [Google Scholar]
- Pearce L. L.; Gartner A. L.; Smith M.; Mauk A. G. Biochemistry 1989, 28, 3152. [DOI] [PubMed] [Google Scholar]
- Rafferty S. P.; Pearce L. L.; Barker P. D.; Guillemette J. G.; Kay C. M.; Smith M.; Mauk A. G. Biochemistry 1990, 29, 9365. [DOI] [PubMed] [Google Scholar]
- Rosell F. I.; Harris T. R.; Hildebrand D. P.; Dopner S.; Hildebrandt P.; Mauk A. G. Biochemistry 2000, 39, 9047. [DOI] [PubMed] [Google Scholar]
- Komarpanicucci S.; Bixler J.; Bakker G.; Sherman F.; McLendon G. J. Am. Chem. Soc. 1992, 114, 5443. [Google Scholar]
- Raphael A. L.; Gray H. B. J. Am. Chem. Soc. 1991, 113, 1038. [Google Scholar]
- Springs S. L.; Bass S. E.; McLendon G. L. Biochemistry 2000, 39, 6075. [DOI] [PubMed] [Google Scholar]
- Barker P. D.; Butler J. L.; de Oliveira P.; Hill H. A. O.; Hunt N. I. Inorg. Chim. Acta 1996, 252, 71. [Google Scholar]
- Walker F. A.; Emrick D.; Rivera J. E.; Hanquet B. J.; Buttlaire D. H. J. Am. Chem. Soc. 1988, 110, 6234. [DOI] [PubMed] [Google Scholar]
- Fantuzzi A.; Sadeghi S.; Valetti F.; Rossi G. L.; Gilardi G. Biochemistry 2002, 41, 8718. [DOI] [PubMed] [Google Scholar]
- Funk W. D.; Lo T. P.; Mauk M. R.; Brayer G. D.; Macgillivray R. T. A.; Mauk A. G. Biochemistry 1990, 29, 5500. [DOI] [PubMed] [Google Scholar]
- Rodgers K. K.; Sligar S. G. J. Am. Chem. Soc. 1991, 113, 9419. [Google Scholar]
- Xue L. L.; Wang Y. H.; Xie Y.; Yao P.; Wang W. H.; Qian W.; Huang Z. X.; Wu J.; Xia Z. X. Biochemistry 1999, 38, 11961. [DOI] [PubMed] [Google Scholar]
- Grein F.; Venceslau S. S.; Schneider L.; Hildebrandt P.; Todorovic S.; Pereira I. A.; Dahl C. Biochemistry 2010, 49, 8290. [DOI] [PubMed] [Google Scholar]
- Klarskov K.; Van Driessche G.; Backers K.; Dumortier C.; Meyer T. E.; Tollin G.; Cusanovich M. A.; Van Beeumen J. J. Biochemistry 1998, 37, 5995. [DOI] [PubMed] [Google Scholar]
- Widger W. R.; Cramer W. A.; Herrmann R. G.; Trebst A. Proc. Natl. Acad. Sci. U.S.A. 1984, 81, 674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guerrero F.; Sedoud A.; Kirilovsky D.; Rutherford A. W.; Ortega J. M.; Roncel M. J. Biol. Chem. 2011, 286, 5985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siponen M.; Adryanczyk G.; Ginet N.; Arnoux P.; Pignol D. Biochem. Soc. Trans. 2012, 40, 1319. [DOI] [PubMed] [Google Scholar]
- Rivera M.; Seetharaman R.; Girdhar D.; Wirtz M.; Zhang X.; Wang X.; White S. Biochemistry 1998, 37, 1485. [DOI] [PubMed] [Google Scholar]
- Rodriguez J. C.; Rivera M. Biochemistry 1998, 37, 13082. [DOI] [PubMed] [Google Scholar]
- Kay C. J.; Barber M. J.; Solomonson L. P. Biochemistry 1988, 27, 6142. [DOI] [PubMed] [Google Scholar]
- Spence J. T.; Barber M. J.; Solomonson L. P. Biochem. J. 1988, 250, 921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kostanjevecki V.; Leys D.; Van Driessche G.; Meyer T. E.; Cusanovich M. A.; Fischer U.; Guisez Y.; Van Beeumen J. J. Biol. Chem. 1999, 274, 35614. [DOI] [PubMed] [Google Scholar]
- Dolla A.; Florens L.; Bianco P.; Haladjian J.; Voordouw G.; Forest E.; Wall J.; Guerlesquin F.; Bruschi M. J. Biol. Chem. 1994, 269, 6340. [PubMed] [Google Scholar]
- Musveteau I.; Dolla A.; Guerlesquin F.; Payan F.; Czjzek M.; Haser R.; Bianco P.; Haladjian J.; Rappgiles B. J.; Wall J. D.; Voordouw G.; Bruschi M. J. Biol. Chem. 1992, 267, 16851. [PubMed] [Google Scholar]
- Shibata N.; Iba S.; Misaki S.; Meyer T. E.; Bartsch R. G.; Cusanovich M. A.; Morimoto Y.; Higuchi Y.; Yasuoka N. J. Mol. Biol. 1998, 284, 751. [DOI] [PubMed] [Google Scholar]
- Tahirov T. H.; Misaki S.; Meyer T. E.; Cusanovich M. A.; Higuchi Y.; Yasuoka N. J. Mol. Biol. 1996, 259, 467. [DOI] [PubMed] [Google Scholar]
- Fujii S.; Yoshimura T.; Kamada H.; Yamaguchi K.; Suzuki S.; Shidara S.; Takakuwa S. Biochim. Biophys. Acta, Protein Struct. Mol. Enzymol. 1995, 1251, 161. [DOI] [PubMed] [Google Scholar]
- Lamar G. N.; Jackson J. T.; Dugad L. B.; Cusanovich M. A.; Bartsch R. G. J. Biol. Chem. 1990, 265, 16173. [PubMed] [Google Scholar]
- Tsan P.; Caffrey M.; Daku M. L.; Cusanovich M.; Marion D.; Gans P. J. Am. Chem. Soc. 2001, 123, 2231. [DOI] [PubMed] [Google Scholar]
- Zahn J. A.; Arciero D. M.; Hooper A. B.; DiSpirito A. A. Eur. J. Biochem. 1996, 240, 684. [DOI] [PubMed] [Google Scholar]
- Korszun Z. R.; Bunker G.; Khalid S.; Scheidt W. R.; Cusanovich M. A.; Meyer T. E. Biochemistry 1989, 28, 1513. [DOI] [PubMed] [Google Scholar]
- Monkara F.; Bingham S. J.; Kadir F. H. A.; McEwan A. G.; Thomson A. J.; Thurgood A. G. P.; Moore G. R. Biochim. Biophys. Acta, Bioenerg. 1992, 1100, 184. [DOI] [PubMed] [Google Scholar]
- Yoshimura T.; Suzuki S.; Nakahara A.; Iwasaki H.; Masuko M.; Matsubara T. Biochim. Biophys. Acta, Protein Struct. Mol. Enzymol. 1985, 831, 267. [DOI] [PubMed] [Google Scholar]
- Maltempo M. M. J. Chem. Phys. 1974, 61, 2540. [Google Scholar]
- Ambler R. P.; Bartsch R. G.; Daniel M.; Kamen M. D.; McLellan L.; Meyer T. E.; Vanbeeumen J. Proc. Natl. Acad. Sci. U.S.A. 1981, 78, 6854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ambler R. P.; Meyer T. E.; Bartsch R. G.; Cusanovich M. A. Arch. Biochem. Biophys. 2001, 388, 25. [DOI] [PubMed] [Google Scholar]
- Bartsch R. G.; Ambler R. P.; Meyer T. E.; Cusanovich M. A. Arch. Biochem. Biophys. 1989, 271, 433. [DOI] [PubMed] [Google Scholar]
- Assfalg M.; Banci L.; Bertini I.; Bruschi M.; Giudici-Orticoni M. T.; Turano P. Eur. J. Biochem. 1999, 266, 634. [DOI] [PubMed] [Google Scholar]
- Banci L.; Bertini I.; Bruschi M.; Sompornpisut P.; Turano P. Proc. Natl. Acad. Sci. U.S.A. 1996, 93, 14396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matias P. M.; Coelho R.; Pereira I. A. C.; Coelho A. V.; Thompson A. W.; Sieker L. C.; Le Gall J.; Carrondo M. A. Structure (London, U. K.) 1999, 7, 119. [DOI] [PubMed] [Google Scholar]
- Pereira I. A. C.; Pacheco I.; Liu M. Y.; Legall J.; Xavier A. V.; Teixeira M. Eur. J. Biochem. 1997, 248, 323. [DOI] [PubMed] [Google Scholar]
- Pettigrew G. W.; Moore G. R.. Cytochromes c: Biological Aspects; Springer: Berlin, 1987. [Google Scholar]
- Ferguson S. J. Biochim. Biophys. Acta, Bioenerg. 2012, 1817, 1754. [DOI] [PubMed] [Google Scholar]
- Louro R. O.; Catarino T.; LeGall J.; Xavier A. V. J. Biol. Inorg. Chem. 1997, 2, 488. [Google Scholar]
- Paixão V. B.; Vis H.; Turner D. L. Biochemistry 2010, 49, 9620. [DOI] [PubMed] [Google Scholar]
- Haser R.; Pierrot M.; Frey M.; Payan F.; Astier J. P.; Bruschi M.; Legall J. Nature 1979, 282, 806. [DOI] [PubMed] [Google Scholar]
- Higuchi Y.; Kusunoki M.; Matsuura Y.; Yasuoka N.; Kakudo M. J. Mol. Biol. 1984, 172, 109. [DOI] [PubMed] [Google Scholar]
- Morais J.; Palma P. N.; Frazao C.; Caldeira J.; LeGall J.; Moura I.; Moura J. J.; Carrondo M. A. Biochemistry 1995, 34, 12830. [DOI] [PubMed] [Google Scholar]
- Pierrot M.; Haser R.; Frey M.; Payan F.; Astier J. P. J. Biol. Chem. 1982, 257, 4341. [PubMed] [Google Scholar]
- Sieker L. C.; Jensen L. H.; Legall J. FEBS Lett. 1986, 209, 261. [DOI] [PubMed] [Google Scholar]
- Banci L.; Bertini I.; Quacquarini G.; Walter O.; Diaz A.; Hervas M.; delaRosa M. A. JBIC, J. Biol. Inorg. Chem. 1996, 1, 330. [Google Scholar]
- Roden E. E.; Lovley D. R. Appl. Environ. Microbiol. 1993, 59, 734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiechtner M. D.; Kassner R. J. Biochim. Biophys. Acta, Protein Struct. 1979, 579, 269. [DOI] [PubMed] [Google Scholar]
- Lancaster C. R. D.; Michel H. Structure (London, U. K.) 1997, 5, 1339. [DOI] [PubMed] [Google Scholar]
- Iverson T. M.; Arciero D. M.; Hooper A. B.; Rees D. C. JBIC, J. Biol. Inorg. Chem. 2001, 6, 390. [DOI] [PubMed] [Google Scholar]
- Stroebel D.; Choquet Y.; Popot J. L.; Picot D. Nature 2003, 426, 413. [DOI] [PubMed] [Google Scholar]
- Allen J. W. A.; Ginger M. L.; Ferguson S. J. Biochem. J. 2004, 383, 537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettigrew G. W.; Leaver J. L.; Meyer T. E.; Ryle A. P. Biochem. J. 1975, 147, 291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ginger M. L.; Sam K. A.; Allen J. W. A. Biochem. J. 2012, 448, 253. [DOI] [PubMed] [Google Scholar]
- Murphy M. E.; Nall B. T.; Brayer G. D. J. Mol. Biol. 1992, 227, 160. [DOI] [PubMed] [Google Scholar]
- Berghuis A. M.; Brayer G. D. J. Mol. Biol. 1992, 223, 959. [DOI] [PubMed] [Google Scholar]
- Bushnell G. W.; Louie G. V.; Brayer G. D. J. Mol. Biol. 1990, 214, 585. [DOI] [PubMed] [Google Scholar]
- Louie G. V.; Brayer G. D. J. Mol. Biol. 1990, 214, 527. [DOI] [PubMed] [Google Scholar]
- Ochi H.; Hata Y.; Tanaka N.; Kakudo M.; Sakurai T.; Aihara S.; Morita Y. J. Mol. Biol. 1983, 166, 407. [DOI] [PubMed] [Google Scholar]
- Takano T.; Dickerson R. E. Proc. Natl. Acad. Sci. U.S.A. 1980, 77, 6371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takano T.; Dickerson R. E. J. Mol. Biol. 1981, 153, 79. [DOI] [PubMed] [Google Scholar]
- Banci L.; Bertini I.; Rosato A.; Varani G. JBIC, J. Biol. Inorg. Chem. 1999, 4, 824. [DOI] [PubMed] [Google Scholar]
- Qi P. X.; Urbauer J. L.; Fuentes E. J.; Leopold M. F.; Wand A. J. Nat. Struct. Biol. 1994, 1, 378. [DOI] [PubMed] [Google Scholar]
- Qi P. X. R.; Beckman R. A.; Wand A. J. Biochemistry 1996, 35, 12275. [DOI] [PubMed] [Google Scholar]
- Benning M. M.; Wesenberg G.; Caffrey M. S.; Bartsch R. G.; Meyer T. E.; Cusanovich M. A.; Rayment I.; Holden H. M. J. Mol. Biol. 1991, 220, 673. [DOI] [PubMed] [Google Scholar]
- Cordier F.; Caffrey M.; Brutscher B.; Cusanovich M. A.; Marion D.; Blackledge M. J. Mol. Biol. 1998, 281, 341. [DOI] [PubMed] [Google Scholar]
- Gooley P. R.; Zhao D.; Mackenzie N. E. J. Biomol. NMR 1991, 1, 145. [DOI] [PubMed] [Google Scholar]
- Zhao D. Z.; Hutton H. M.; Cusanovich M. A.; Mackenzie N. E. Protein Sci. 1996, 5, 1816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banci L.; Bertini I.; De la Rosa M. A.; Koulougliotis D.; Navarro J. A.; Walter O. Biochemistry 1998, 37, 4831. [DOI] [PubMed] [Google Scholar]
- Frazao C.; Soares C. M.; Carrondo M. A.; Pohl E.; Dauter Z.; Wilson K. S.; Hervas M.; Navarro J. A.; Delarosa M. A.; Sheldrick G. M. Structure (London, U. K.) 1995, 3, 1159. [DOI] [PubMed] [Google Scholar]
- Harrenga A.; Reincke B.; Ruterjans H.; Ludwig B.; Michel H. J. Mol. Biol. 2000, 295, 667. [DOI] [PubMed] [Google Scholar]
- Rajendran C.; Ermler U.; Ludwig B.; Michel H. Acta Crystallogr,. D: Biol. Crystallogr. 2010, 66, 850. [DOI] [PubMed] [Google Scholar]
- Reincke B.; Thony-Meyer L.; Dannehl C.; Odenwald A.; Aidim M.; Witt H.; Ruterjans H.; Ludwig B. Biochim. Biophys. Acta, Bioenerg. 1999, 1411, 114. [DOI] [PubMed] [Google Scholar]
- Anthony C. Biochim. Biophys. Acta, Bioenerg. 1992, 1099, 1. [PubMed] [Google Scholar]
- Read J.; Gill R.; Dales S. L.; Cooper J. B.; Wood S. P.; Anthony C. Protein Sci. 1999, 8, 1232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Than M. E.; Hof P.; Huber R.; Bourenkov G. P.; Bartunik H. D.; Buse G.; Soulimane T. J. Mol. Biol. 1997, 271, 629. [DOI] [PubMed] [Google Scholar]
- Atack J. M.; Kelly D. J. Adv. Microb. Physiol. 2007, 52, 73. [DOI] [PubMed] [Google Scholar]
- Erman J. E.; Vitello L. B. Biochim. Biophys. Acta, Protein Struct. Mol. Enzymol. 2002, 1597, 193. [DOI] [PubMed] [Google Scholar]
- Ronnberg M.; Araiso T.; Ellfolk N.; Dunford H. B. J. Biol. Chem. 1981, 256, 2471. [PubMed] [Google Scholar]
- De Smet L.; Pettigrew G. W.; Van Beeumen J. J. Eur. J. Biochem. 2001, 268, 6559. [DOI] [PubMed] [Google Scholar]
- Fulop V.; Ridout C. J.; Greenwood C.; Hajdu J. Structure (London, U. K.) 1995, 3, 1225. [DOI] [PubMed] [Google Scholar]
- Crowley P. B.; Ubbink M. Acc. Chem. Res. 2003, 36, 723. [DOI] [PubMed] [Google Scholar]
- Hart S. E.; Schlarb-Ridley B. G.; Delon C.; Bendall D. S.; Howe C. J. Biochemistry 2003, 42, 4829. [DOI] [PubMed] [Google Scholar]
- Koh M.; Meyer T. E.; De Smet L.; Van Beeumen J. J.; Cusanovich M. A. Arch. Biochem. Biophys. 2003, 410, 230. [DOI] [PubMed] [Google Scholar]
- Alves T.; Besson S.; Duarte L. C.; Pettigrew G. W.; Girio F. M. F.; Devreese B.; Vandenberghe I.; Van Beeumen J.; Fauque G.; Moura I. Biochim. Biophys. Acta, Protein Struct. Mol. Enzymol. 1999, 1434, 248. [DOI] [PubMed] [Google Scholar]
- Pettigrew G. W.; Goodhew C. F.; Cooper A.; Nutley M.; Jumel K.; Harding S. E. Biochemistry 2003, 42, 2046. [DOI] [PubMed] [Google Scholar]
- Pettigrew G. W.; Prazeres S.; Costa C.; Palma N.; Krippahl L.; Moura I.; Moura J. J. J. Biol. Chem. 1999, 274, 11383. [DOI] [PubMed] [Google Scholar]
- Berks B. C.; Ferguson S. J.; Moir J. W.; Richardson D. J. Biochim. Biophys. Acta, Bioenerg. 1995, 1232, 97. [DOI] [PubMed] [Google Scholar]
- Ferguson S. J. Curr. Opin. Chem. Biol. 1998, 2, 182. [DOI] [PubMed] [Google Scholar]
- Philippot L. Biochim. Biophys. Acta, Gene Struct. Expression 2002, 1577, 355. [DOI] [PubMed] [Google Scholar]
- Cheesman M. R.; Ferguson S. J.; Moir J. W.; Richardson D. J.; Zumft W. G.; Thomson A. J. Biochemistry 1997, 36, 16267. [DOI] [PubMed] [Google Scholar]
- Nurizzo D.; Silvestrini M.-C.; Mathieu M.; Cutruzzolà F.; Bourgeois D.; Fülöp V.; Hajdu J.; Brunori M.; Tegoni M.; Cambillau C. Structure (London, U. K.) 1997, 5, 1157. [DOI] [PubMed] [Google Scholar]
- Dodd F. E.; Van Beeumen J.; Eady R. R.; Hasnain S. S. J. Mol. Biol. 1998, 282, 369. [DOI] [PubMed] [Google Scholar]
- Murphy M. E. P.; Turley S.; Adman E. T. J. Biol. Chem. 1997, 272, 28455. [DOI] [PubMed] [Google Scholar]
- Lopes H.; Besson S. p.; Moura I.; Moura J. J. JBIC, J. Biol. Inorg. Chem. 2001, 6, 55. [DOI] [PubMed] [Google Scholar]
- Moenne-Loccoz P. Nat. Prod. Rep. 2007, 24, 610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiro Y.; Sugimoto H.; Tosha T.; Nagano S.; Hino T. Philos. Trans. R. Soc., B 2012, 367, 1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hino T.; Matsumoto Y.; Nagano S.; Sugimoto H.; Fukumori Y.; Murata T.; Iwata S.; Shiro Y. Science 2010, 330, 1666. [DOI] [PubMed] [Google Scholar]
- Hille R. Chem. Rev. 1996, 96, 2757. [DOI] [PubMed] [Google Scholar]
- Hille R. Trends Biochem. Sci. 2002, 27, 360. [DOI] [PubMed] [Google Scholar]
- Kay C. J.; Solomonson L. P.; Barber M. J. Biochemistry 1990, 29, 10823. [DOI] [PubMed] [Google Scholar]
- Campbell W. H.; Kinghorn J. R. Trends Biochem. Sci. 1990, 15, 315. [DOI] [PubMed] [Google Scholar]
- Chen K.; Bonagura C. A.; Tilley G. J.; McEvoy J. P.; Jung Y.-S.; Armstrong F. A.; Stout C. D.; Burgess B. K. Nat. Struct. Biol. 2002, 9, 188. [DOI] [PubMed] [Google Scholar]
- Cunane L. M.; Chen Z.-W.; Shamala N.; Mathews F. S.; Cronin C. n. N.; McIntire W. S. J. Mol. Biol. 2000, 295, 357. [DOI] [PubMed] [Google Scholar]
- Chen L.; Durley R.; Mathews F.; Davidson V. Science 1994, 264, 86. [DOI] [PubMed] [Google Scholar]
- Nitschke W.; Rutherford A. W. Trends Biochem. Sci. 1991, 16, 241. [DOI] [PubMed] [Google Scholar]
- Shen J. R.; Ikeuchi M.; Inoue Y. J. Biol. Chem. 1997, 272, 17821. [DOI] [PubMed] [Google Scholar]
- Kerfeld C. A.; Krogmann D. W. Annu. Rev. Plant Biol. 1998, 49, 397. [DOI] [PubMed] [Google Scholar]
- Ferreira K. N.; Iverson T. M.; Maghlaoui K.; Barber J.; Iwata S. Science 2004, 303, 1831. [DOI] [PubMed] [Google Scholar]
- Goussias C.; Boussac A.; Rutherford A. W. Philos. Trans. R. Soc., B 2002, 357, 1369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alam J.; Sprinkle J.; Hermodson M.; Krogmann D. Biochim. Biophys. Acta, Bioenerg. 1984, 766, 317. [Google Scholar]
- Holton R. W.; Myers J. Science 1963, 142, 234. [DOI] [PubMed] [Google Scholar]
- Navarro J. A.; Hervás M.; De la Cerda B.; De la Rosa M. A. Arch. Biochem. Biophys. 1995, 318, 46. [DOI] [PubMed] [Google Scholar]
- Roncel M.; Boussac A.; Zurita J. L.; Bottin H.; Sugiura M.; Kirilovsky D.; Ortega J. M. JBIC, J. Biol. Inorg. Chem. 2003, 8, 206. [DOI] [PubMed] [Google Scholar]
- Vrettos J. S.; Reifler M. J.; Kievit O.; Lakshmi K.; de Paula J. C.; Brudvig G. W. JBIC, J. Biol. Inorg. Chem. 2001, 6, 708. [DOI] [PubMed] [Google Scholar]
- Roncel M.; Kirilovsky D.; Guerrero F.; Serrano A.; Ortega J. M. Biochim. Biophys. Acta, Bioenerg. 2012, 1817, 1152. [DOI] [PubMed] [Google Scholar]
- Chitnis P. R.; Xu Q.; Chitnis V. P.; Nechushtai R. Photosynth. Res. 1995, 44, 23. [DOI] [PubMed] [Google Scholar]
- Ho K.; Krogmann D. Biochim. Biophys. Acta, Bioenerg. 1984, 766, 310. [Google Scholar]
- Navarro J.; Hervás M.; De la Rosa M. JBIC, J. Biol. Inorg. Chem. 1997, 2, 11. [Google Scholar]
- De la Rosa M. A.; Navarro J.; Díaz-Quintana A.; De la Cerda B.; Molina-Heredia F. P.; Balme A.; Murdoch P. d. S.; Díaz-Moreno I.; Durán R.; Hervás M. Bioelectrochemistry 2002, 55, 41. [DOI] [PubMed] [Google Scholar]
- Malakhov M. P.; Malakhova O. A.; Murata N. FEBS Lett. 1999, 444, 281. [DOI] [PubMed] [Google Scholar]
- Molina-Heredia F. P.; Balme A.; Hervás M.; Navarro J.; De la Rosa M. A. FEBS Lett. 2002, 517, 50. [DOI] [PubMed] [Google Scholar]
- Leys D.; Backers K.; Meyer T. E.; Hagen W. R.; Cusanovich M. A.; Van Beeumen J. J. J. Biol. Chem. 2000, 275, 16050. [DOI] [PubMed] [Google Scholar]
- Blakemore R.; Maratea D.; Wolfe R. J. Bacteriol. 1979, 140, 720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richardson D. J. Microbiology 2000, 146, 551. [DOI] [PubMed] [Google Scholar]
- Czjzek M.; Arnoux P.; Haser R.; Shepard W. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2001, 57, 670. [DOI] [PubMed] [Google Scholar]
- Dantas J. M.; Saraiva I. H.; Morgado L.; Silva M. A.; Schiffer M.; Salgueiro C. A.; Louro R. O. Dalton Trans. 2011, 40, 12713. [DOI] [PubMed] [Google Scholar]
- Morgado L.; Bruix M.; Pessanha M.; Londer Y. Y.; Salgueiro C. A. Biophys. J. 2010, 99, 293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pokkuluri P.; Londer Y.; Yang X.; Duke N.; Erickson J.; Orshonsky V.; Johnson G.; Schiffer M. Biochim. Biophys. Acta, Bioenerg. 2010, 1797, 222. [DOI] [PubMed] [Google Scholar]
- Dahl C.; Engels S.; Pott-Sperling A. S.; Schulte A.; Sander J.; Lubbe Y.; Deuster O.; Brune D. C. J. Bacteriol. 2005, 187, 1392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roldάn M. D.; Sears H. J.; Cheesman M. R.; Ferguson S. J.; Thomson A. J.; Berks B. C.; Richardson D. J. J. Biol. Chem. 1998, 273, 28785. [DOI] [PubMed] [Google Scholar]
- Dobbin P.; Butt J.; Powell A.; Reid G.; Richardson D. Biochem. J. 1999, 342, 439. [PMC free article] [PubMed] [Google Scholar]
- Igarashi N.; Moriyama H.; Fujiwara T.; Fukumori Y.; Tanaka N. Nat. Struct. Mol. Biol. 1997, 4, 276. [DOI] [PubMed] [Google Scholar]
- Einsle O.; Messerschmidt A.; Stach P.; Bourenkov G. P.; Bartunik H. D.; Huber R.; Kroneck P. M. Nature 1999, 400, 476. [DOI] [PubMed] [Google Scholar]
- Kemp G.; Clarke T.; Marritt S.; Lockwood C.; Poock S.; Hemmings A.; Richardson D.; Cheesman M.; Butt J. Biochem. J. 2010, 431, 73. [DOI] [PubMed] [Google Scholar]
- Bamford V.; Dobbin P. S.; Richardson D. J.; Hemmings A. M. Nat. Struct. Mol. Biol. 1999, 6, 1104. [DOI] [PubMed] [Google Scholar]
- Leys D.; Tsapin A. S.; Nealson K. H.; Meyer T. E.; Cusanovich M. A.; Van Beeumen J. J. Nat. Struct. Mol. Biol. 1999, 6, 1113. [DOI] [PubMed] [Google Scholar]
- Taylor P.; Pealing S. L.; Reid G. A.; Chapman S. K.; Walkinshaw M. D. Nat. Struct. Mol. Biol. 1999, 6, 1108. [DOI] [PubMed] [Google Scholar]
- Field S. J.; Dobbin P. S.; Cheesman M. R.; Watmough N. J.; Thomson A. J.; Richardson D. J. J. Biol. Chem. 2000, 275, 8515. [DOI] [PubMed] [Google Scholar]
- Simon J. r.; Pisa R.; Stein T.; Eichler R.; Klimmek O.; Gross R. Eur. J. Biochem. 2001, 268, 5776. [DOI] [PubMed] [Google Scholar]
- Bewley K.; FirerSherwood M.; Mock J.; Ando N.; Drennan C.; Elliott S. Biochem. Soc. Trans. 2012, 40, 1268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke T. A.; Edwards M. J.; Gates A. J.; Hall A.; White G. F.; Bradley J.; Reardon C. L.; Shi L.; Beliaev A. S.; Marshall M. J.; Wang Z.; Watmough N. J.; Fredrickson J. K.; Zachara J. M.; Butt J. N.; Richardson D. J. Proc. Natl. Acad. Sci. U.S.A. 2011, 108, 9384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gralnick J. A.; Vali H.; Lies D. P.; Newman D. K. Proc. Natl. Acad. Sci. U.S.A. 2006, 103, 4669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lovley D. R. Annu. Rev. Microbiol. 1993, 47, 263. [DOI] [PubMed] [Google Scholar]
- Myers C. R.; Myers J. M. J. Bacteriol. 1997, 179, 1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myers C. R.; Myers J. M. Biochim. Biophys. Acta, Biomembr. 1997, 1326, 307. [DOI] [PubMed] [Google Scholar]
- Myers C. R.; Nealson K. H. Science 1988, 240, 1319. [DOI] [PubMed] [Google Scholar]
- Nealson K. H.Prokaryotes; Springer: Berlin, 2006. [Google Scholar]
- Schwalb C.; Chapman S. K.; Reid G. A. Biochemistry 2003, 42, 9491. [DOI] [PubMed] [Google Scholar]
- Shi L.; Richardson D. J.; Wang Z.; Kerisit S. N.; Rosso K. M.; Zachara J. M.; Fredrickson J. K. Environ. Microbiol. Rep. 2009, 1, 220. [DOI] [PubMed] [Google Scholar]
- Myers J. M.; Myers C. R. J. Bacteriol. 2000, 182, 67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dohnalkova A. C.; Marshall M. J.; Arey B. W.; Williams K. H.; Buck E. C.; Fredrickson J. K. Appl. Environ. Microbiol. 2011, 77, 1254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Firer-Sherwood M.; Pulcu G. S.; Elliott S. J. JBIC, J. Biol. Inorg. Chem. 2008, 13, 849. [DOI] [PubMed] [Google Scholar]
- Hartshorne R. S.; Reardon C. L.; Ross D.; Nuester J.; Clarke T. A.; Gates A. J.; Mills P. C.; Fredrickson J. K.; Zachara J. M.; Shi L.; Beliaev A. S.; Marshall M. J.; Tien M.; Brantley S.; Butt J. N.; Richardson D. J. Proc. Natl. Acad. Sci. U.S.A. 2009, 106, 22169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pitts K. E.; Dobbin P. S.; Reyes-Ramirez F.; Thomson A. J.; Richardson D. J.; Seward H. E. J. Biol. Chem. 2003, 278, 27758. [DOI] [PubMed] [Google Scholar]
- Firer-Sherwood M. A.; Ando N.; Drennan C. L.; Elliott S. J. J. Phys. Chem. B 2011, 115, 11208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fonseca B. M.; Paquete C. M.; Neto S. E.; Pacheco I.; Soares C. M.; Louro R. O. Biochem. J. 2013, 449, 101. [DOI] [PubMed] [Google Scholar]
- Coursolle D.; Gralnick J. A. Mol. Microbiol. 2010, 77, 995. [DOI] [PubMed] [Google Scholar]
- Shi L.; Squier T. C.; Zachara J. M.; Fredrickson J. K. Mol. Microbiol. 2007, 65, 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breuer M.; Zarzycki P.; Blumberger J.; Rosso K. M. J. Am. Chem. Soc. 2012, 134, 9868. [DOI] [PubMed] [Google Scholar]
- Breuer M.; Zarzycki P.; Shi L.; Clarke T. A.; Edwards M. J.; Butt J. N.; Richardson D. J.; Fredrickson J. K.; Zachara J. M.; Blumberger J.; Rosso K. M. Biochem. Soc. Trans. 2012, 40, 1198. [DOI] [PubMed] [Google Scholar]
- Beliaev A. S.; Saffarini D. A. J. Bacteriol. 1998, 180, 6292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma J. G.; Zhang J.; Franco R.; Jia S. L.; Moura I.; Moura J. J.; Kroneck P. M.; Shelnutt J. A. Biochemistry 1998, 37, 12431. [DOI] [PubMed] [Google Scholar]
- Pieulle L.; Haladjian J.; Bonicel J.; Hatchikian E. C. Biochim. Biophys. Acta, Bioenerg. 1996, 1273, 51. [DOI] [PubMed] [Google Scholar]
- Coutinho I. B.; Xavier A. V. Methods Enzymol. 1994, 243, 119. [DOI] [PubMed] [Google Scholar]
- Louro R. O.; Catarino T.; LeGall J.; Turner D. L.; Xavier A. V. ChemBioChem 2001, 2, 831. [DOI] [PubMed] [Google Scholar]
- Martel P. J.; Soares C. M.; Baptista A. M.; Fuxreiter M.; Naray-Szabo G.; Louro R. O.; Carrondo M. A. JBIC, J. Biol. Inorg. Chem. 1999, 4, 73. [DOI] [PubMed] [Google Scholar]
- Park J. S.; Ohmura T.; Kano K.; Sagara T.; Niki K.; Kyogoku Y.; Akutsu H. Biochim. Biophys. Acta, Protein Struct. Mol. Enzymol. 1996, 1293, 45. [DOI] [PubMed] [Google Scholar]
- Brennan L.; Turner D. L.; Messias A. C.; Teodoro M. L.; LeGall J.; Santos H.; Xavier A. V. J. Mol. Biol. 2000, 298, 61. [DOI] [PubMed] [Google Scholar]
- Harada E.; Fukuoka Y.; Ohmura T.; Fukunishi A.; Kawai G.; Fujiwara T.; Akutsu H. J. Mol. Biol. 2002, 319, 767. [DOI] [PubMed] [Google Scholar]
- Salgueiro C. A.; da Costa P. N.; Turner D. L.; Messias A. C.; van Dongen W. M.; Saraiva L. M.; Xavier A. V. Biochemistry 2001, 40, 9709. [DOI] [PubMed] [Google Scholar]
- Matias P. M.; Pereira I. A.; Soares C. M.; Carrondo M. A. Prog. Biophys. Mol. Biol. 2005, 89, 292. [DOI] [PubMed] [Google Scholar]
- Norager S.; Legrand P.; Pieulle L.; Hatchikian C.; Roth M. J. Mol. Biol. 1999, 290, 881. [DOI] [PubMed] [Google Scholar]
- Pieulle L.; Morelli X.; Gallice P.; Lojou E.; Barbier P.; Czjzek M.; Bianco P.; Guerlesquin F.; Hatchikian E. C. J. Mol. Biol. 2005, 354, 73. [DOI] [PubMed] [Google Scholar]
- Quintas P. O.; Oliveira M. S.; Catarino T.; Turner D. L. Biochim. Biophys. Acta, Bioenerg. 2013, 1827, 502. [DOI] [PubMed] [Google Scholar]
- Lederer F. Biochimie 1994, 76, 674. [DOI] [PubMed] [Google Scholar]
- Qian W.; Wang Y.-H.; Wang W.-H.; Yao P.; Zhuang J.-H.; Xie Y.; Huang Z.-X. J. Electroanal. Chem. 2002, 535, 85. [Google Scholar]
- Schenkman J. B.; Jansson I. Pharmacol. Ther. 2003, 97, 139. [DOI] [PubMed] [Google Scholar]
- Vergeres G.; Waskell L. Biochimie 1995, 77, 604. [DOI] [PubMed] [Google Scholar]
- Hultquist D. E.; Passon P. G. Nat. New Biol. 1971, 229, 252. [DOI] [PubMed] [Google Scholar]
- Sannes L. J.; Hultquist D. E. Biochim. Biophys. Acta, Gen. Subj. 1978, 544, 547. [DOI] [PubMed] [Google Scholar]
- Strittmatter P.; Spatz L.; Corcoran D.; Rogers M. J.; Setlow B.; Redline R. Proc. Natl. Acad. Sci. U.S.A. 1974, 71, 4565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parthasarathy S.; Altuve A.; Terzyan S.; Zhang X.; Kuczera K.; Rivera M.; Benson D. R. Biochemistry 2011, 50, 5544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altuve A.; Silchenko S.; Lee K. H.; Kuczera K.; Terzyan S.; Zhang X.; Benson D. R.; Rivera M. Biochemistry 2001, 40, 9469. [DOI] [PubMed] [Google Scholar]
- Borgese N.; D’Arrigo A.; De Silvestris M.; Pietrini G. FEBS Lett. 1993, 325, 70. [DOI] [PubMed] [Google Scholar]
- Fukushima K.; Sato R. J. Biochem. 1973, 74, 161. [DOI] [PubMed] [Google Scholar]
- Kuroda R.; Ikenoue T.; Honsho M.; Tsujimoto S.; Mitoma J. Y.; Ito A. J. Biol. Chem. 1998, 273, 31097. [DOI] [PubMed] [Google Scholar]
- Mitoma J.; Ito A. EMBO J. 1992, 11, 4197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivera M.; Wells M. A.; Walker F. A. Biochemistry 1994, 33, 2161. [DOI] [PubMed] [Google Scholar]
- Ito A.; Hayashi S.; Yoshida T. Biochem. Biophys. Res. Commun. 1981, 101, 591. [DOI] [PubMed] [Google Scholar]
- Nishino H.; Ito A. J. Biochem. 1986, 100, 1523. [DOI] [PubMed] [Google Scholar]
- Durley R. C.; Mathews F. S. Acta Crystallogr., D: Biol. Crystallogr. 1996, 52, 65. [DOI] [PubMed] [Google Scholar]
- Alam S.; Yee J.; Couture M.; Takayama S. J.; Tseng W. H.; Mauk A. G.; Rafferty S. Metallomics 2012, 4, 1255. [DOI] [PubMed] [Google Scholar]
- La Mar G. N.; Burns P. D.; Jackson J. T.; Smith K. M.; Langry K. C.; Strittmatter P. J. Biol. Chem. 1981, 256, 6075. [PubMed] [Google Scholar]
- Kababya S.; Nelson J.; Calle C.; Neese F.; Goldfarb D. J. Am. Chem. Soc. 2006, 128, 2017. [DOI] [PubMed] [Google Scholar]
- Keller R.; Groudinsky O.; Wuthrich K. Biochim. Biophys. Acta, Protein Struct. 1976, 427, 497. [DOI] [PubMed] [Google Scholar]
- Keller R. M.; Wuthrich K. Biochim. Biophys. Acta, Protein Struct. 1972, 285, 326. [DOI] [PubMed] [Google Scholar]
- Keller R. M.; Wuthrich K. Biochim. Biophys. Acta, Protein Struct. 1980, 621, 204. [DOI] [PubMed] [Google Scholar]
- McLachlan S. J.; La Mar G. N.; Burns P. D.; Smith K. M.; Langry K. C. Biochim. Biophys. Acta, Protein Struct. Mol. Enzymol. 1986, 874, 274. [DOI] [PubMed] [Google Scholar]
- Lee K. B.; La Mar G. N.; Kehres L. A.; Fujinari E. M.; Smith K. M.; Pochapsky T. C.; Sligar S. G. Biochemistry 1990, 29, 9623. [DOI] [PubMed] [Google Scholar]
- Rivera M.; Barillas-Mury C.; Christensen K. A.; Little J. W.; Wells M. A.; Walker F. A. Biochemistry 1992, 31, 12233. [DOI] [PubMed] [Google Scholar]
- Lee K.; La Mar G. N.; Pandey R. K.; Rezzano I. N.; Mansfield K. E.; Smith K. M.; Pochapsky T. C.; Sligar S. G. Biochemistry 1991, 30, 1878. [DOI] [PubMed] [Google Scholar]
- Guiles R. D.; Basus V. J.; Kuntz I. D.; Waskell L. Biochemistry 1992, 31, 11365. [DOI] [PubMed] [Google Scholar]
- Lee K. B.; La Mar G. N.; Mansfield K. E.; Smith K. M.; Pochapsky T. C.; Sligar S. G. Biochim. Biophys. Acta, Protein Struct. Mol. Enzymol. 1993, 1202, 189. [DOI] [PubMed] [Google Scholar]
- McLachlan S. J.; La Mar G. N.; Lee K. B. Biochim. Biophys. Acta, Protein Struct. 1988, 957, 430. [DOI] [PubMed] [Google Scholar]
- Veitch N. C.; Whitford D.; Williams R. J. FEBS Lett. 1990, 269, 297. [DOI] [PubMed] [Google Scholar]
- Arnesano F.; Banci L.; Bertini I.; Faraone-Mennella J.; Rosato A.; Barker P. D.; Fersht A. R. Biochemistry 1999, 38, 8657. [DOI] [PubMed] [Google Scholar]
- Hamada K.; Bethge P. H.; Mathews F. S. J. Mol. Biol. 1995, 247, 947. [DOI] [PubMed] [Google Scholar]
- Wittung-Stafshede P.; Gray H. B.; Winkler J. R. J. Am. Chem. Soc. 1997, 119, 9562. [Google Scholar]
- Bixler J.; Bakker G.; McLendon G. J. Am. Chem. Soc. 1992, 114, 6938. [Google Scholar]
- Pascher T.; Chesick J. P.; Winkler J. R.; Gray H. B. Science 1996, 271, 1558. [DOI] [PubMed] [Google Scholar]
- Tezcan F. A.; Winkler J. R.; Gray H. B. J. Am. Chem. Soc. 1998, 120, 13383. [Google Scholar]
- Winkler J. R.; Wittung-Stafshede P.; Leckner J.; Malmstrom B. G.; Gray H. B. Proc. Natl. Acad. Sci. U.S.A. 1997, 94, 4246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wittung-Stafshede P.; Lee J. C.; Winkler J. R.; Gray H. B. Proc. Natl. Acad. Sci. U.S.A. 1999, 96, 6587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu F.; Cangelosi V. M.; Zastrow M. L.; Tegoni M.; Plegaria J. S.; Tebo A. G.; Mocny C. S.; Ruckthong L.; Qayyum H.; Pecoraro V. L. Chem. Rev. 2014, 10.1021/cr400458x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choma C. T.; Lear J. D.; Nelson M. J.; Dutton P. L.; Robertson D. E.; DeGrado W. F. J. Am. Chem. Soc. 1994, 116, 856. [Google Scholar]
- DeGrado W. F.; Regan L.; Ho S. P. Cold Spring Harbor Symp. Quant. Biol. 1987, 52, 521. [DOI] [PubMed] [Google Scholar]
- Regan L.; DeGrado W. F. Science 1988, 241, 976. [DOI] [PubMed] [Google Scholar]
- DeGrado W. F.; Wasserman Z. R.; Lear J. D. Science 1989, 243, 622. [DOI] [PubMed] [Google Scholar]
- Robertson D. E.; Farid R. S.; Moser C. C.; Urbauer J. L.; Mulholland S. E.; Pidikiti R.; Lear J. D.; Wand A. J.; DeGrado W. F.; Dutton P. L. Nature 1994, 368, 425. [DOI] [PubMed] [Google Scholar]
- Kalsbeck W. A.; Robertson D. E.; Pandey R. K.; Smith K. M.; Dutton P. L.; Bocian D. F. Biochemistry 1996, 35, 3429. [DOI] [PubMed] [Google Scholar]
- Shifman J. M.; Gibney B. R.; Sharp R. E.; Dutton P. L. Biochemistry 2000, 39, 14813. [DOI] [PubMed] [Google Scholar]
- Ishida M.; Dohmae N.; Shiro Y.; Oku T.; Iizuka T.; Isogai Y. Biochemistry 2004, 43, 9823. [DOI] [PubMed] [Google Scholar]
- Gibney B. R.; Rabanal F.; Skalicky J. J.; Wand A. J.; Dutton P. L. J. Am. Chem. Soc. 1997, 119, 2323. [Google Scholar]
- Gibney B. R.; Rabanal F.; Reddy K. S.; Dutton P. L. Biochemistry 1998, 37, 4635. [DOI] [PubMed] [Google Scholar]
- Chen X.; Discher B. M.; Pilloud D. L.; Gibney B. R.; Moser C. C.; Dutton P. L. J. Phys. Chem. B 2001, 106, 617. [Google Scholar]
- Privett H. K.; Reedy C. J.; Kennedy M. L.; Gibney B. R. J. Am. Chem. Soc. 2002, 124, 6828. [DOI] [PubMed] [Google Scholar]
- Gibney B. R.; Isogai Y.; Rabanal F.; Reddy K. S.; Grosset A. M.; Moser C. C.; Dutton P. L. Biochemistry 2000, 39, 11041. [DOI] [PubMed] [Google Scholar]
- Reddi A. R.; Reedy C. J.; Mui S.; Gibney B. R. Biochemistry 2007, 46, 291. [DOI] [PubMed] [Google Scholar]
- Huang S. S.; Koder R. L.; Lewis M.; Wand A. J.; Dutton P. L. Proc. Natl. Acad. Sci. U.S.A. 2004, 101, 5536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Discher B. M.; Noy D.; Strzalka J.; Ye S. X.; Moser C. C.; Lear J. D.; Blasie J. K.; Dutton P. L. Biochemistry 2005, 44, 12329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghirlanda G.; Osyczka A.; Liu W.; Antolovich M.; Smith K. M.; Dutton P. L.; Wand A. J.; DeGrado W. F. J. Am. Chem. Soc. 2004, 126, 8141. [DOI] [PubMed] [Google Scholar]
- Farid T. A.; Kodali G.; Solomon L. A.; Lichtenstein B. R.; Sheehan M. M.; Fry B. A.; Bialas C.; Ennist N. M.; Siedlecki J. A.; Zhao Z.; Stetz M. A.; Valentine K. G.; Anderson J. L. R.; Wand A. J.; Discher B. M.; Moser C. C.; Dutton P. L. Nat. Chem. Biol. 2013, 9, 826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costanzo L.; Geremia S.; Randaccio L.; Nastri F.; Maglio O.; Lombardi A.; Pavone V. JBIC, J. Biol. Inorg. Chem. 2004, 9, 1017. [DOI] [PubMed] [Google Scholar]
- Bender G. M.; Lehmann A.; Zou H.; Cheng H.; Fry H. C.; Engel D.; Therien M. J.; Blasie J. K.; Roder H.; Saven J. G.; DeGrado W. F. J. Am. Chem. Soc. 2007, 129, 10732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korendovych I. V.; Senes A.; Kim Y. H.; Lear J. D.; Fry H. C.; Therien M. J.; Blasie J. K.; Walker F. A.; DeGrado W. F. J. Am. Chem. Soc. 2010, 132, 15516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson J. L. R.; Armstrong C. T.; Kodali G.; Lichtenstein B. R.; Watkins D. W.; Mancini J. A.; Boyle A. L.; Farid T. A.; Crump M. P.; Moser C. C.; Dutton P. L. Chem. Sci. 2014, 5, 507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang X.; Pistor E.; Farid R. S.; Farid H. Protein Sci. 2000, 9, 403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Z.; Farid R. S. Protein Sci. 2001, 10, 236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hecht M. H.; Vogel K. M.; Spiro T. G.; Rojas N. R. L.; Kamtekar S.; Simons C. T.; McLean J. E.; Farid R. S. Protein Sci. 1997, 6, 2512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei Y. N.; Liu T.; Sazinsky S. L.; Moffet D. A.; Pelczer I.; Hecht M. H. Protein Sci. 2003, 12, 92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moffet D. A.; Foley J.; Hecht M. H. Biophys. Chem. 2003, 105, 231. [DOI] [PubMed] [Google Scholar]
- Das A.; Trammell S. A.; Hecht M. H. Biophys. Chem. 2006, 123, 102. [DOI] [PubMed] [Google Scholar]
- Rau H. K.; Haehnel W. J. Am. Chem. Soc. 1998, 120, 468. [Google Scholar]
- Rau H. K.; DeJonge N.; Haehnel W. Angew. Chem., Int. Ed. 2000, 39, 250. [PubMed] [Google Scholar]
- Albrecht T.; Li W. W.; Ulstrup J.; Haehnel W.; Hildebrandt P. ChemPhysChem 2005, 6, 961. [DOI] [PubMed] [Google Scholar]
- Albrecht T.; Li W.-W.; Haehnel W.; Hildebrandt P.; Ulstrup J. Bioelectrochemistry 2006, 69, 193. [DOI] [PubMed] [Google Scholar]
- Benson D. R.; Hart B. R.; Zhu X.; Doughty M. B. J. Am. Chem. Soc. 1995, 117, 8502. [Google Scholar]
- Arnold P. A.; Benson D. R.; Brink D. J.; Hendrich M. P.; Jas G. S.; Kennedy M. L.; Petasis D. T.; Wang M. Inorg. Chem. 1997, 36, 5306. [Google Scholar]
- Liu D.; Williamson D. A.; Kennedy M. L.; Williams T. D.; Morton M. M.; Benson D. R. J. Am. Chem. Soc. 1999, 121, 11798. [Google Scholar]
- Liu D.; Lee K.-H.; R. Benson D. Chem. Commun. 1999, 1205. [Google Scholar]
- Cowley A. B.; Kennedy M. L.; Silchenko S.; Lukat-Rodgers G. S.; Rodgers K. R.; Benson D. R. Inorg. Chem. 2006, 45, 9985. [DOI] [PubMed] [Google Scholar]
- Nastri F.; Lombardi A.; Morelli G.; Maglio O.; Dauria G.; Pedone C.; Pavone V. Chem.—Eur. J. 1997, 3, 340. [Google Scholar]
- D’Auria M.; Racioppi R. Tetrahedron 1997, 53, 17307. [Google Scholar]
- Lombardi A.; Nastri F.; Sanseverino M.; Maglio O.; Pedone C.; Pavone V. Inorg. Chim. Acta 1998, 276, 301. [Google Scholar]
- Lombardi A.; Nastri F.; Marasco D.; Maglio O.; De Sanctis G.; Sinibaldi F.; Santucci R.; Coletta M.; Pavone V. Chem.—Eur. J. 2003, 9, 5643. [DOI] [PubMed] [Google Scholar]
- Arnold P. A.; Shelton W. R.; Benson D. R. J. Am. Chem. Soc. 1997, 119, 3181. [Google Scholar]
- Huffman D. L.; Rosenblatt M. M.; Suslick K. S. J. Am. Chem. Soc. 1998, 120, 6183. [Google Scholar]
- Huffman D. L.; Suslick K. S. Inorg. Chem. 2000, 39, 5418. [DOI] [PubMed] [Google Scholar]
- Rosenblatt M. M.; Huffman D. L.; Wang X.; Remmer H. A.; Suslick K. S. J. Am. Chem. Soc. 2002, 124, 12394. [DOI] [PubMed] [Google Scholar]
- Rosenblatt M. M.; Wang J.; Suslick K. S. Proc. Natl. Acad. Sci. U.S.A. 2003, 100, 13140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isogai Y.; Ota M.; Fujisawa T.; Izuno H.; Mukai M.; Nakamura H.; Iizuka T.; Nishikawa K. Biochemistry 1999, 38, 7431. [DOI] [PubMed] [Google Scholar]
- Isogai Y.; Ishii A.; Fujisawa T.; Ota M.; Nishikawa K. Biochemistry 2000, 39, 5683. [DOI] [PubMed] [Google Scholar]
- Isogai Y.; Ishida M. Biochemistry 2009, 48, 8136. [DOI] [PubMed] [Google Scholar]
- Qin J.; La Mar G. N.; Dou Y.; Admiraal S. J.; Ikeda-Saito M. J. Biol. Chem. 1994, 269, 1083. [PubMed] [Google Scholar]
- Lloyd E.; Hildebrand D. P.; Tu K. M.; Mauk A. G. J. Am. Chem. Soc. 1995, 117, 6434. [Google Scholar]
- Dou Y.; Admiraal S. J.; Ikeda-Saito M.; Krzywda S.; Wilkinson A. J.; Li T.; Olson J. S.; Prince R. C.; Pickering I. J.; George G. N. J. Biol. Chem. 1995, 270, 15993. [DOI] [PubMed] [Google Scholar]
- Shinde S.; Cordova J. M.; Woodrum B. W.; Ghirlanda G. JBIC, J. Biol. Inorg. Chem. 2012, 17, 557. [DOI] [PubMed] [Google Scholar]
- Wilson J. R.; Caruana D. J.; Gilardi G. Chem. Commun. 2003, 0, 356. [DOI] [PubMed] [Google Scholar]
- Di Nardo G.; Di Venere A.; Mei G.; Sadeghi S. J.; Wilson J. R.; Gilardi G. JBIC, J. Biol. Inorg. Chem. 2009, 14, 497. [DOI] [PubMed] [Google Scholar]
- Cordova J. M.; Noack P. L.; Hilcove S. A.; Lear J. D.; Ghirlanda G. J. Am. Chem. Soc. 2007, 129, 512. [DOI] [PubMed] [Google Scholar]
- Ibrahim S. M.; Nakajima H.; Ohta T.; Ramanathan K.; Takatani N.; Naruta Y.; Watanabe Y. Biochemistry 2011, 50, 9826. [DOI] [PubMed] [Google Scholar]
- Tomlinson E. J.; Ferguson S. J. J. Biol. Chem. 2000, 275, 32530. [DOI] [PubMed] [Google Scholar]
- Tomlinson E. J.; Ferguson S. J. Proc. Natl. Acad. Sci. U.S.A. 2000, 97, 5156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barker P. D.; Ferrer J. C.; Mylrajan M.; Loehr T. M.; Feng R.; Konishi Y.; Funk W. D.; MacGillivray R. T.; Mauk A. G. Proc. Natl. Acad. Sci. U.S.A. 1993, 90, 6542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barker P. D.; Nerou E. P.; Freund S. M.; Fearnley I. M. Biochemistry 1995, 34, 15191. [DOI] [PubMed] [Google Scholar]
- Wain R.; Redfield C.; Ferguson S. J.; Smith L. J. J. Biol. Chem. 2004, 279, 15177. [DOI] [PubMed] [Google Scholar]
- Arnesano F.; Banci L.; Bertini I.; Ciofi-Baffoni S.; Woodyear T. L.; Johnson C. M.; Barker P. D. Biochemistry 2000, 39, 1499. [DOI] [PubMed] [Google Scholar]
- Watt G. D.; Frankel R. B.; Papaefthymiou G. C.; Spartalian K.; Stiefel E. I. Biochemistry 1986, 25, 4330. [Google Scholar]
- Lember R.; Barrett J.. Cytochromes; Academic Press: London, 1973. [Google Scholar]
- Barker P. D.; Ferguson S. J. Structure (London, U. K.) 1999, 7, R281. [DOI] [PubMed] [Google Scholar]
- Allen J. W. A.; Barker P. D.; Daltrop O.; Stevens J. M.; Tomlinson E. J.; Sinha N.; Sambongi Y.; Ferguson S. J. Dalton Trans. 2005, 0, 3410. [DOI] [PubMed] [Google Scholar]
- Bowman S. E. J.; Bren K. L. Nat. Prod. Rep. 2008, 25, 1118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng Z.; Gunner M. R. Proteins: Struct., Funct., Bioinf. 2009, 75, 719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Contreras-Zentella M.; Mendoza G.; Membrillo-Hernández J.; Escamilla J. E. J. Biol. Chem. 2003, 278, 31473. [DOI] [PubMed] [Google Scholar]
- Zhuang J.; Amoroso J. H.; Kinloch R.; Dawson J. H.; Baldwin M. J.; Gibney B. R. Inorg. Chem. 2006, 45, 4685. [DOI] [PubMed] [Google Scholar]
- Zhuang J.; Reddi A. R.; Wang Z.; Khodaverdian B.; Hegg E. L.; Gibney B. R. Biochemistry 2006, 45, 12530. [DOI] [PubMed] [Google Scholar]
- Raphael A. L.; Gray H. B. Proteins: Struct., Funct., Bioinf. 1989, 6, 338. [DOI] [PubMed] [Google Scholar]
- Hay S.; Wydrzynski T. Biochemistry 2004, 44, 431. [DOI] [PubMed] [Google Scholar]
- Miller G. T.; Zhang B.; Hardman J. K.; Timkovich R. Biochemistry 2000, 39, 9010. [DOI] [PubMed] [Google Scholar]
- Sligar S. G.; Egeberg K. D.; Sage J. T.; Morikis D.; Champion P. M. J. Am. Chem. Soc. 1987, 109, 7896. [Google Scholar]
- Barker P. D.; Nerou E. P.; Cheesman M. R.; Thomson A. J.; de Oliveira P.; Hill H. A. O. Biochemistry 1996, 35, 13618. [DOI] [PubMed] [Google Scholar]
- Barker P. D.; Freund S. M. Biochemistry 1996, 35, 13627. [DOI] [PubMed] [Google Scholar]
- Cheesman M. R.; Kadir F. H.; al-Basseet J.; al-Massad F.; Farrar J.; Greenwood C.; Thomson A. J.; Moore G. R. Biochem. J. 1992, 286Part 2361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kassner R. J. Proc. Natl. Acad. Sci. U.S.A. 1972, 69, 2263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Churg A. K.; Warshel A. Biochemistry 1986, 25, 1675. [DOI] [PubMed] [Google Scholar]
- Battistuzzi G.; Borsari M.; Cowan J. A.; Ranieri A.; Sola M. J. Am. Chem. Soc. 2002, 124, 5315. [DOI] [PubMed] [Google Scholar]
- Stellwagen E. Nature 1978, 275, 73. [DOI] [PubMed] [Google Scholar]
- Paoli M.; Marles-Wright J.; Smith A. DNA Cell Biol. 2002, 21, 271. [DOI] [PubMed] [Google Scholar]
- Warshel A.; Papazyan A.; Muegge I. JBIC, J. Biol. Inorg. Chem. 1997, 2, 143. [Google Scholar]
- Bowman S. E. J.; Bren K. L. Inorg. Chem. 2010, 49, 7890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banci L.; Bertini I.; Turano P.; Tien M.; Kirk T. K. Proc. Natl. Acad. Sci. U.S.A. 1991, 88, 6956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berghuis A. M.; Guillemette J. G.; Smith M.; Brayer G. D. J. Mol. Biol. 1994, 235, 1326. [DOI] [PubMed] [Google Scholar]
- Ye T.; Kaur R.; Wen X.; Bren K. L.; Elliott S. J. Inorg. Chem. 2005, 44, 8999. [DOI] [PubMed] [Google Scholar]
- Ozawa K.; Takayama Y.; Yasukawa F.; Ohmura T.; Cusanovich M. A.; Tomimoto Y.; Ogata H.; Higuchi Y.; Akutsu H. Biophys. J. 2003, 85, 3367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Worrall J. A. R.; Schlarb-Ridley B. G.; Reda T.; Marcaida M. J.; Moorlen R. J.; Wastl J.; Hirst J.; Bendall D. S.; Luisi B. F.; Howe C. J. J. Am. Chem. Soc. 2007, 129, 9468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lett C. M.; Guillemette J. G. Biochem. J. 2002, 362, 281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varadarajan R.; Zewert T.; Gray H.; Boxer S. Science 1989, 243, 69. [DOI] [PubMed] [Google Scholar]
- Springs S. L.; Bass S. E.; Bowman G.; Nodelman I.; Schutt C. E.; McLendon G. L. Biochemistry 2002, 41, 4321. [DOI] [PubMed] [Google Scholar]
- Tai H.; Mikami S.-i.; Irie K.; Watanabe N.; Shinohara N.; Yamamoto Y. Biochemistry 2009, 49, 42. [DOI] [PubMed] [Google Scholar]
- Takayama Y.; Taketa-Sato M.; Komori H.; Morita K.; Kang S.-J.; Higuchi Y.; Akutsu H. Bull. Chem. Soc. Jpn. 2011, 84, 1096. [Google Scholar]
- Moore G. R. FEBS Lett. 1983, 161, 171. [DOI] [PubMed] [Google Scholar]
- Mikami S.-i.; Tai H.; Yamamoto Y. Biochemistry 2009, 48, 8062. [DOI] [PubMed] [Google Scholar]
- Tai H.; Udagawa T.; Mikami S.-i.; Sugimoto A.; Yamamoto Y. J. Inorg. Biochem. 2012, 108, 182. [DOI] [PubMed] [Google Scholar]
- Louro R. O.; Catarino T.; Salgueiro C. A.; LeGall J.; Xavier A. V. JBIC, J. Biol. Inorg. Chem. 1996, 1, 34. [Google Scholar]
- Matsuno T.; Morishita N.; Yamazaki K.; Inoue N.; Sato Y.; Ichise N.; Hara I.; Hoshino T.; Matsuyama H.; Yoshimune K.; Yumoto I. J. Biosci. Bioeng. 2007, 103, 247. [DOI] [PubMed] [Google Scholar]
- Shibamoto T.; Kato Y.; Watanabe T. FEBS Lett. 2008, 582, 1490. [DOI] [PubMed] [Google Scholar]
- Moore G. R.; Pettigrew G. W.; Pitt R. C.; Williams R. J. P. Biochim. Biophys. Acta, Bioenerg. 1980, 590, 261. [DOI] [PubMed] [Google Scholar]
- Takayama S. J.; Mikami S.; Terui N.; Mita H.; Hasegawa J.; Sambongi Y.; Yamamoto Y. Biochemistry 2005, 44, 5488. [DOI] [PubMed] [Google Scholar]
- Pettigrew G. W.; Bartsch R. G.; Meyer T. E.; Kamen M. D. Biochim. Biophys. Acta, Bioenerg. 1978, 503, 509. [DOI] [PubMed] [Google Scholar]
- Shifman J. M.; Moser C. C.; Kalsbeck W. A.; Bocian D. F.; Dutton P. L. Biochemistry 1998, 37, 16815. [DOI] [PubMed] [Google Scholar]
- Reid L. S.; Mauk M. R.; Mauk A. G. J. Am. Chem. Soc. 1984, 106, 2182. [Google Scholar]
- Hunter C. L.; Lloyd E.; Eltis L. D.; Rafferty S. P.; Lee H.; Smith M.; Mauk A. G. Biochemistry 1997, 36, 1010. [DOI] [PubMed] [Google Scholar]
- Chapman S. K.; Daff S.; Munro A. W. In Metal Sites in Proteins and Models; Hill H. A. O., Sadler P. J., Thomson A. J., Eds.; Springer: Berlin, Heidelberg, 1997; Vol. 88, p 39. [Google Scholar]
- Fan K.; Akutsu H.; Kyogoku Y.; Niki K. Biochemistry 1990, 29, 2257. [DOI] [PubMed] [Google Scholar]
- Santos H.; Moura J. J. G.; Moura I.; Legall J.; Xavier A. V. Eur. J. Biochem. 1984, 141, 283. [DOI] [PubMed] [Google Scholar]
- Coletta M.; Catarino T.; Legall J.; Xavier A. V. Eur. J. Biochem. 1991, 202, 1101. [DOI] [PubMed] [Google Scholar]
- Correia I. J.; Paquete C. M.; Coelho A.; Almeida C. C.; Catarino T.; Louro R. O.; Frazão C.; Saraiva L. M.; Carrondo M. A.; Turner D. L.; Xavier A. V. J. Biol. Chem. 2004, 279, 52227. [DOI] [PubMed] [Google Scholar]
- Louro R. O. JBIC, J. Biol. Inorg. Chem. 2007, 12, 1. [DOI] [PubMed] [Google Scholar]
- Teixeira V. H.; Soares C. M.; Baptista A. M. JBIC, J. Biol. Inorg. Chem. 2002, 7, 200. [DOI] [PubMed] [Google Scholar]
- Louro R. O.; Catarino T.; Turner D. L.; Piçarra-Pereira M. A.; Pacheco I.; LeGall J.; Xavier A. V. Biochemistry 1998, 37, 15808. [DOI] [PubMed] [Google Scholar]
- Yoshikawa S.; Muramoto K.; Shinzawa-Itoh K. Annu. Rev. Biophys. 2011, 40, 205. [DOI] [PubMed] [Google Scholar]
- Michel H. Biochemistry 1999, 38, 15129. [DOI] [PubMed] [Google Scholar]
- Kaila V. R. I.; Verkhovsky M. I.; Wikström M. Chem. Rev. 2010, 110, 7062. [DOI] [PubMed] [Google Scholar]
- Senge M. O. Chem. Commun. 2006, 0, 243. [DOI] [PubMed] [Google Scholar]
- Roder B.; Buchner M.; Ruckmann I.; Senge M. O. Photochem. Photobiol. Sci. 2010, 9, 1152. [DOI] [PubMed] [Google Scholar]
- Jentzen W.; Ma J.-G.; Shelnutt J. A. Biophys. J. 1998, 74, 753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Senge M. O. J. Photochem. Photobiol., B 1992, 16, 3. [Google Scholar]
- Hobbs J.; Shelnutt J. J. Protein Chem. 1995, 14, 19. [DOI] [PubMed] [Google Scholar]
- Olea C.; Kuriyan J.; Marletta M. A. J. Am. Chem. Soc. 2010, 132, 12794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Can M.; Zoppellaro G.; Andersson K. K.; Bren K. L. Inorg. Chem. 2011, 50, 12018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- A. Shelnutt J.; Song X.-Z.; Ma J.-G.; Jia S.-L.; Jentzen W.; J. Medforth C. Chem. Soc. Rev. 1998, 27, 31. [Google Scholar]
- Ma J.-G.; Laberge M.; Song X.-Z.; Jentzen W.; Jia S.-L.; Zhang J.; Vanderkooi J. M.; Shelnutt J. A. Biochemistry 1998, 37, 5118. [DOI] [PubMed] [Google Scholar]
- Patra R.; Chaudhary A.; Ghosh S. K.; Rath S. P. Inorg. Chem. 2008, 47, 8324. [DOI] [PubMed] [Google Scholar]
- Ravikanth M.; Chandrashekar T. K.. Coordination Chemistry; Springer: Berlin, Heidelberg, 1995; Vol. 82, p 105. [Google Scholar]
- MacGowan S. A.; Senge M. O. Inorg. Chem. 2013, 52, 1228. [DOI] [PubMed] [Google Scholar]
- Beinert H.; Sands R. H. Biochem. Biophys. Res. Commun. 1960, 3, 41. [Google Scholar]
- Mortenson L. E.; Valentine R. C.; Carnahan J. E. Biochem. Biophys. Res. Commun. 1962, 7, 448. [DOI] [PubMed] [Google Scholar]
- Tagawa K.; Arnon D. I. Nature 1962, 195, 537. [DOI] [PubMed] [Google Scholar]
- Imlay J. A. Mol. Microbiol. 2006, 59, 1073. [DOI] [PubMed] [Google Scholar]
- Johnson D. C.; Dean D. R.; Smith A. D.; Johnson M. K. Annu. Rev. Biochem. 2005, 74, 247. [DOI] [PubMed] [Google Scholar]
- Dos S.; Patricia C.; Dean D. R. Proc. Natl. Acad. Sci. U.S.A. 2008, 105, 11589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beinert H.; Meyer J.; Lill R.. Encyclopedia of Biological Chemistry; Elsevier Ltd.: Amsterdam, 2004; Vol. 2, p 482. [Google Scholar]
- Lill R. Nature 2009, 460, 831. [DOI] [PubMed] [Google Scholar]
- Flint D. H.; Allen R. M. Chem. Rev. 1996, 96, 2315. [DOI] [PubMed] [Google Scholar]
- Bian S.; Cowan J. A. Coord. Chem. Rev. 1999, 190–192, 1049. [Google Scholar]
- Broderick J. B.Comprehensive Coordination Chemistry II; Elsevier Ltd.: Amsterdam, 2004; Vol. 8, p 739. [Google Scholar]
- Beinert H.; Holm R. H.; Munck E. Science 1997, 277, 653. [DOI] [PubMed] [Google Scholar]
- Py B.; Moreau P. L.; Barras F. Curr. Opin. Microbiol. 2011, 14, 218. [DOI] [PubMed] [Google Scholar]
- Beinert H.; Kiley P. J. Curr. Opin. Chem. Biol. 1999, 3, 152. [DOI] [PubMed] [Google Scholar]
- Brzoska K.; Meczynska S.; Kruszewski M. Acta Biochim. Pol. 2006, 53, 685. [PubMed] [Google Scholar]
- Rouault T. A.; Klausner R. D. Trends Biochem. Sci. 1996, 21, 174. [PubMed] [Google Scholar]
- Lukianova O. A.; David S. S. Curr. Opin. Chem. Biol. 2005, 9, 145. [DOI] [PubMed] [Google Scholar]
- IUPAC-IUB Commission on Biochemical Nomenclature (CBN). Eur. J. Biochem. 1973, 35, 1. [PubMed] [Google Scholar]
- Beinert H.; Kennedy M. C. Eur. J. Biochem. 1989, 186, 5. [DOI] [PubMed] [Google Scholar]
- Bill E. Hyperfine Interact. 2012, 205, 139. [Google Scholar]
- Orme-Johnson W. H. Annu. Rev. Biochem. 1973, 42, 159. [DOI] [PubMed] [Google Scholar]
- Tsibris J. C. M.; Woody R. W. Coord. Chem. Rev. 1970, 5, 417. [Google Scholar]
- Sticht H.; Rosch P. Prog. Biophys. Mol. Biol. 1998, 70, 95. [DOI] [PubMed] [Google Scholar]
- Balk J.; Lill R. ChemBioChem 2004, 5, 1044. [DOI] [PubMed] [Google Scholar]
- Noodleman L.; Lovell T.; Liu T.; Himo F.; Torres R. A. Curr. Opin. Chem. Biol. 2002, 6, 259. [DOI] [PubMed] [Google Scholar]
- Beinert H. JBIC, J. Biol. Inorg. Chem. 2000, 5, 409. [DOI] [PubMed] [Google Scholar]
- Hong J.-S.; Rabinowitz J. C. Biochem. Biophys. Res. Commun. 1967, 29, 246. [DOI] [PubMed] [Google Scholar]
- Malkin R.; Rabinowitz J. C. Biochem. Biophys. Res. Commun. 1966, 23, 822. [DOI] [PubMed] [Google Scholar]
- Rouault T. A. Dis. Models Mech. 2012, 5, 155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frazzon J.; Fick J. R.; Dean D. R. Biochem. Soc. Trans. 2002, 30, 680. [DOI] [PubMed] [Google Scholar]
- Py B.; Barras F. Nat. Rev. Microbiol. 2010, 8, 436. [DOI] [PubMed] [Google Scholar]
- Peters J. W.; Broderick J. B. Annu. Rev. Biochem. 2012, 81, 429. [DOI] [PubMed] [Google Scholar]
- Bandyopadhyay S.; Chandramouli K.; Johnson M. K. Biochem. Soc. Trans. 2008, 36, 1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayala-Castro C.; Saini A.; Outten F. W. Microbiol. Mol. Biol. Rev. 2008, 72, 110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roche B.; Aussel L.; Ezraty B.; Mandin P.; Py B.; Barras F. Biochim. Biophys. Acta, Bioenerg. 2013, 1827, 455. [DOI] [PubMed] [Google Scholar]
- Lill R.; Mühlenhoff U. Trends Biochem. Sci. 2005, 30, 133. [DOI] [PubMed] [Google Scholar]
- Day M. W.; Hsu B. T.; Joshua-Tor L.; Park J. B.; Zhou Z. H.; Adams M. W. W.; Rees D. C. Protein Sci. 1992, 1, 1494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dauter Z.; Wilson K. S.; Sieker L. C.; Moulis J.-M.; Meyer J. Proc. Natl. Acad. Sci. U.S.A. 1996, 93, 8836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LeGall J.; Liu M. Y.; Gomes C. M.; Braga V.; Pacheco I.; Regalla M.; Xavier A. V.; Teixeira M. FEBS Lett. 1998, 429, 295. [DOI] [PubMed] [Google Scholar]
- Hiller R.; Zhou Z. H.; Adams M. W. W.; Englander S. W. Proc. Natl. Acad. Sci. U.S.A. 1997, 94, 11329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vondrasek J.; Bendova L.; Klusak V.; Hobza P. J. Am. Chem. Soc. 2005, 127, 2615. [DOI] [PubMed] [Google Scholar]
- Riley K. E.; Merz K. M. Jr J. Phys. Chem. B 2006, 110, 15650. [DOI] [PubMed] [Google Scholar]
- Sieker L. C.; Stenkamp R. E.; LeGall J. Methods Enzymol. 1994, 243, 203. [DOI] [PubMed] [Google Scholar]
- Xiao Z.; Lavery M. J.; Ayhan M.; Scrofani S. D. B.; Wilce M. C. J.; Guss J. M.; Tregloan P. A.; George G. N.; Wedd A. G. J. Am. Chem. Soc. 1998, 120, 4135. [Google Scholar]
- Westler W. M.; Lin I. J.; Perczel A.; Weinhold F.; Markley J. L. J. Am. Chem. Soc. 2011, 133, 1310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertini I.; Luchinat C. Nature 2011, 470, 469. [DOI] [PubMed] [Google Scholar]
- Zartler E. R.; Jenney F. E. Jr.; Terrell M.; Eidsness M. K.; Adams M. W. W.; Prestegard J. H. Biochemistry 2001, 40, 7279. [DOI] [PubMed] [Google Scholar]
- Min T.; Ergenekan C. E.; Eidsness M. K.; Ichiye T.; Kang C. Protein Sci. 2001, 10, 613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park I. Y.; Youn B.; Harley J. L.; Eidsness M. K.; Smith E.; Ichiye T.; Kang C. JBIC, J. Biol. Inorg. Chem. 2004, 9, 423. [DOI] [PubMed] [Google Scholar]
- Kennepohl P.; Solomon E. I. Inorg. Chem. 2003, 42, 696. [DOI] [PubMed] [Google Scholar]
- Lee H. J.; Basran J.; Scrutton N. S. Biochemistry 1998, 37, 15513. [DOI] [PubMed] [Google Scholar]
- Perry A.; Tambyrajah W.; Grossmann J. G.; Lian L.-Y.; Scrutton N. S. Biochemistry 2004, 43, 3167. [DOI] [PubMed] [Google Scholar]
- Lode E. T.; Coon M. J. J. Biol. Chem. 1971, 246, 791. [PubMed] [Google Scholar]
- Adman E. T.; Sieker L. C.; Jensen L. H. J. Mol. Biol. 1991, 217, 337. [DOI] [PubMed] [Google Scholar]
- Van Beilen J. B.; Neuenschwander M.; Smits T. H. M.; Roth C.; Balada S. B.; Witholt B. J. Bacteriol. 2002, 184, 1722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bau R.; Rees D. C.; Kurtz D. M. Jr.; Scott R. A.; Huang H.; Adams M. W. W.; Eidsness M. K. JBIC, J. Biol. Inorg. Chem. 1998, 3, 484. [Google Scholar]
- Hageluekent G.; Wiehlmannt L.; Adams T. M.; Kolmar H.; Heinz D. W.; Tuemmler B.; Schubert W.-D. Proc. Natl. Acad. Sci. U.S.A. 2007, 104, 12276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Page C. C.; Moser C. C.; Chen X.; Dutton P. L. Nature 1999, 402, 47. [DOI] [PubMed] [Google Scholar]
- Santos H.; Fareleira P.; Xavier A. V.; Chen L.; Liu M. Y.; LeGall J. Biochem. Biophys. Res. Commun. 1993, 195, 551. [DOI] [PubMed] [Google Scholar]
- Victor B. L.; Vicente J. B.; Rodrigues R.; Oliveira S.; Rodrigues-Pousada C.; Frazao C.; Gomes C. M.; Teixeira M.; Soares C. M. JBIC, J. Biol. Inorg. Chem. 2003, 8, 475. [DOI] [PubMed] [Google Scholar]
- Gomes C. M.; Silva G.; Oliveira S.; LeGall J.; Liu M.-Y.; Xavier A. V.; Rodrigues-Pousada C.; Teixeira M. J. Biol. Chem. 1997, 272, 22502. [DOI] [PubMed] [Google Scholar]
- Frazao C.; Silva G.; Gomes C. M.; Matias P.; Coelho R.; Sieker L.; Macedo S.; Liu M. Y.; Oliveira S.; Teixeira M.; Xavier A. V.; Rodrigues-Pousada C.; Carrondo M. A.; Le Gall J. Nat. Struct. Biol. 2000, 7, 1041. [DOI] [PubMed] [Google Scholar]
- Fareleira P.; Santos B. S.; Antonio C.; Moradas-Ferreira P.; LeGall J.; Xavier A. V.; Santos H. Microbiology 2003, 149, 1513. [DOI] [PubMed] [Google Scholar]
- Jenney F. E. Jr.; Verhagen M. F. J. M.; Cui X.; Adams M. W. W. Science 1999, 286, 306. [DOI] [PubMed] [Google Scholar]
- Coulter E. D.; Kurtz D. M. Jr Arch. Biochem. Biophys. 2001, 394, 76. [DOI] [PubMed] [Google Scholar]
- Yoon K.-S.; Hille R.; Hemann C.; Tabita F. R. J. Biol. Chem. 1999, 274, 29772. [DOI] [PubMed] [Google Scholar]
- Shimizu F.; Ogata M.; Yagi T.; Wakabayashi S.; Matsubara H. Biochimie 1989, 71, 1171. [DOI] [PubMed] [Google Scholar]
- Wastl J.; Duin E. C.; Iuzzolino L.; Dorner W.; Link T.; Hoffmann S.; Sticht H.; Dau H.; Lingelbach K.; Maier U.-G. J. Biol. Chem. 2000, 275, 30058. [DOI] [PubMed] [Google Scholar]
- Gaillard J.; Zhuang-Jackson H.; Moulis J.-M. Eur. J. Biochem. 1996, 238, 346. [DOI] [PubMed] [Google Scholar]
- Holm R. H.; Kennepohl P.; Solomon E. I. Chem. Rev. 1996, 96, 2239. [DOI] [PubMed] [Google Scholar]
- Meyer J.; Moulis J.-M. Handb. Metalloproteins 2001, 1, 505. [Google Scholar]
- Xiao Z.; Wedd A. G. Mod. Coord. Chem. 2002, 288. [Google Scholar]
- Swartz P. D.; Beck B. W.; Ichiye T. Biophys. J. 1996, 71, 2958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swartz P. D.; Ichiye T. Biophys. J. 1997, 73, 2733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan M.-L.; Kang C.; Ichiye T. Proteins: Struct., Funct., Bioinf. 2006, 62, 708. [DOI] [PubMed] [Google Scholar]
- Sulpizi M.; Raugei S.; VandeVondele J.; Carloni P.; Sprik M. J. Phys. Chem. B 2007, 111, 3969. [DOI] [PubMed] [Google Scholar]
- Gamiz-Hernandez A. P.; Kieseritzky G.; Ishikita H.; Knapp E. W. J. Chem. Theory Comput. 2011, 7, 742. [DOI] [PubMed] [Google Scholar]
- Rose K.; Shadle S. E.; Eidsness M. K.; Kurtz D. M. Jr.; Scott R. A.; Hedman B.; Hodgson K. O.; Solomon E. I. J. Am. Chem. Soc. 1998, 120, 10743. [Google Scholar]
- Swartz P. D.; Ichiye T. Biochemistry 1996, 35, 13772. [DOI] [PubMed] [Google Scholar]
- Gilles de Pelichy L. D.; Smith E. T. Biochemistry 1999, 38, 7874. [DOI] [PubMed] [Google Scholar]
- Sundararajan M.; Hillier I. H.; Burton N. A. J. Phys. Chem. A 2006, 110, 785. [DOI] [PubMed] [Google Scholar]
- Sun N.; Dey A.; Xiao Z.; Wedd A. G.; Hodgson K. O.; Hedman B.; Solomon E. I. J. Am. Chem. Soc. 2010, 132, 12639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuemmerle R.; Zhuang-Jackson H.; Gaillard J.; Moulis J.-M. Biochemistry 1997, 36, 15983. [DOI] [PubMed] [Google Scholar]
- Eidsness M. K.; Burden A. E.; Richie K. A.; Kurtz D. M. Jr.; Scott R. A.; Smith E. T.; Ichiye T.; Beard B.; Min T.; Kang C. Biochemistry 1999, 38, 14803. [DOI] [PubMed] [Google Scholar]
- Yoo S. J.; Meyer J.; Achim C.; Peterson J.; Hendrich M. P.; Munck E. JBIC, J. Biol. Inorg. Chem. 2000, 5, 475. [DOI] [PubMed] [Google Scholar]
- Ayhan M.; Xiao Z.; Lavery M. J.; Hamer A. M.; Nugent K. W.; Scrofani S. D. B.; Guss M.; Wedd A. G. Inorg. Chem. 1996, 35, 5902. [Google Scholar]
- Xiao Z.; Maher M. J.; Cross M.; Bond C. S.; Guss J. M.; Wedd A. G. JBIC, J. Biol. Inorg. Chem. 2000, 5, 75. [DOI] [PubMed] [Google Scholar]
- Ayhan M.; Xiao Z.; Lavery M. J.; Hammer A. M.; Scrofani S. D. B.; Wedd A. G. ACS Symp. Ser. 1996, 653, 40. [Google Scholar]
- Mukherjee M. Acta Crystallogr., Sect. D: Biol. Crystallogr. 1999, D55, 820. [DOI] [PubMed] [Google Scholar]
- Lin I. J.; Gebel E. B.; Machonkin T. E.; Westler W. M.; Markley J. L. J. Am. Chem. Soc. 2003, 125, 1464. [DOI] [PubMed] [Google Scholar]
- Park I. Y.; Eidsness M. K.; Lin I. J.; Gebel E. B.; Youn B.; Harley J. L.; Machonkin T. E.; Frederick R. O.; Markley J. L.; Smith E. T.; Ichiye T.; Kang C. Proteins: Struct., Funct., Bioinf. 2004, 57, 618. [DOI] [PubMed] [Google Scholar]
- Lin I. J.; Gebel E. B.; Machonkin T. E.; Westler W. M.; Markley J. L. Proc. Natl. Acad. Sci. U.S.A. 2005, 102, 14581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng Q.; Smith E. T.; Kurtz D. M. Jr.; Scott R. A. Inorg. Chim. Acta 1996, 242, 245. [Google Scholar]
- Zheng H.; Kellog S. J.; Erickson A. E.; Dubauskie N. A.; Smith E. T. JBIC, J. Biol. Inorg. Chem. 2003, 8, 12. [DOI] [PubMed] [Google Scholar]
- Low D. W.; Hill M. G. J. Am. Chem. Soc. 1998, 120, 11536. [Google Scholar]
- Xiao Y.; Wang H.; George S. J.; Smith M. C.; Adams M. W. W.; Jenney F. E. Jr.; Sturhahn W.; Alp E. E.; Zhao J.; Yoda Y.; Dey A.; Solomon E. I.; Cramer S. P. J. Am. Chem. Soc. 2005, 127, 14596. [DOI] [PubMed] [Google Scholar]
- Cheney B. V.; Schulz M. W.; Cheney J. Biochim. Biophys. Acta, Protein Struct. Mol. Enzymol. 1989, 996, 116. [Google Scholar]
- Reid K. S. C.; Lindley P. F.; Thornton J. M. FEBS Lett. 1985, 190, 209. [Google Scholar]
- Adman E.; Watenpaugh K. D.; Jensen L. H. Proc. Natl. Acad. Sci. U.S.A. 1975, 72, 4854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Backes G.; Mino Y.; Loehr T. M.; Meyer T. E.; Cusanovich M. A.; Sweeney W. V.; Adman E. T.; Sanders-Loehr J. J. Am. Chem. Soc. 1991, 113, 2055. [Google Scholar]
- Okamura T.-A.; Takamizawa S.; Ueyama N.; Nakamura A. Inorg. Chem. 1998, 37, 18. [DOI] [PubMed] [Google Scholar]
- Atherton N. M.; Garbett K.; Gillard R. D.; Mason R.; Mayhew S. J.; Peel J. L.; Stangroom J. E. Nature 1966, 212, 590. [DOI] [PubMed] [Google Scholar]
- Zawadzke L. E.; Berg J. M. J. Am. Chem. Soc. 1992, 114, 4002. [Google Scholar]
- Eidsness M. K.; Richie K. A.; Burden A. E.; Kurtz D. M. Jr.; Scott R. A. Biochemistry 1997, 36, 10406. [DOI] [PubMed] [Google Scholar]
- Vrajmasu V. V.; Bominaar E. L.; Meyer J.; Muenck E. Inorg. Chem. 2002, 41, 6358. [DOI] [PubMed] [Google Scholar]
- Barra A. L.; Hassan A. K.; Janoschka A.; Schmidt C. L.; Schunemann V. Appl. Magn. Reson. 2006, 30, 385. [Google Scholar]
- Rao K. K.; Evans M. C. W.; Cammack R.; Hall D. O.; Thompson C. L.; Jackson P. J.; Johnson C. E. Biochem. J. 1972, 129, 1063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng P.; Li H. J. Am. Chem. Soc. 2011, 133, 6791. [DOI] [PubMed] [Google Scholar]
- Zheng P.; Takayama S.-i. J.; Mauk A. G.; Li H. J. Am. Chem. Soc. 2012, 134, 4124. [DOI] [PubMed] [Google Scholar]
- Bergmann U.; Sturhahn W.; Linn D. E. Jr.; Jenney F. E. Jr.; Adams M. W. W.; Rupnik K.; Hales B. J.; Alp E. E.; Mayse A.; Cramer S. P. J. Am. Chem. Soc. 2003, 125, 4016. [DOI] [PubMed] [Google Scholar]
- Wilkens S. J.; Xia B.; Weinhold F.; Markley J. L.; Westler W. M. J. Am. Chem. Soc. 1998, 120, 4806. [Google Scholar]
- Xia B.; Wilkens S. J.; Westler W. M.; Markley J. L. J. Am. Chem. Soc. 1998, 120, 4893. [Google Scholar]
- Volkman B. F.; Wilkens S. J.; Lee A. L.; Xia B.; Westler W. M.; Beger R.; Markley J. L. J. Am. Chem. Soc. 1999, 121, 4677. [Google Scholar]
- Wasserfallen A.; Ragettli S.; Jouanneau Y.; Leisinger T. Eur. J. Biochem. 1998, 254, 325. [DOI] [PubMed] [Google Scholar]
- Gardner A. M.; Helmick R. A.; Gardner P. R. J. Biol. Chem. 2002, 277, 8172. [DOI] [PubMed] [Google Scholar]
- Drake Harold L.; Daniel Steven L. Res. Microbiol. 2004, 155, 869. [DOI] [PubMed] [Google Scholar]
- Gomes C. M.; Vicente J. B.; Wasserfallen A.; Teixeira M. Biochemistry 2000, 39, 16230. [DOI] [PubMed] [Google Scholar]
- Vicente J. B.; Teixeira M. J. Biol. Chem. 2005, 280, 34599. [DOI] [PubMed] [Google Scholar]
- Vicente J. B.; Scandurra F. M.; Rodrigues J. V.; Brunori M.; Sarti P.; Teixeira M.; Giuffre A. FEBS J. 2007, 274, 677. [DOI] [PubMed] [Google Scholar]
- Silaghi-Dumitrescu R.; Coulter E. D.; Das A.; Ljungdahl L. G.; Jameson G. N. L.; Huynh B. H.; Kurtz D. M. Jr Biochemistry 2003, 42, 2806. [DOI] [PubMed] [Google Scholar]
- Zanello P. Coord. Chem. Rev. 2013, 257, 1777. [Google Scholar]
- van Beilen J. B.; Wubbolts M. G.; Witholt B. Biodegradation 1994, 5, 161. [DOI] [PubMed] [Google Scholar]
- Lee H. J.; Lian L.-Y.; Scrutton N. S. Biochem. J. 1997, 328, 131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moura J. J. G.; Goodfellow B. J.; Romao M. J.; Rusnak F.; Moura I. Comments Inorg. Chem. 1996, 19, 47. [Google Scholar]
- Archer M.; Huber R.; Tavares P.; Moura I.; Moura J. J. G.; Carrondo M. A.; Sieker L. C.; LeGall J.; Romao M. J. J. Mol. Biol. 1995, 251, 690. [DOI] [PubMed] [Google Scholar]
- Hoff A. J., Ed. Advanced EPR: Applications in Biology and Biochemistry; Elsevier: New York, 1989; p 813. [Google Scholar]
- Moura I.; Huynh B. H.; Hausinger R. P.; Le Gall J.; Xavier A. V.; Munck E. J. Biol. Chem. 1980, 255, 2493. [PubMed] [Google Scholar]
- Yu L.; Kennedy M.; Czaja C.; Tavares P.; Moura J. J. G.; Moura I.; Rusnak F. Biochem. Biophys. Res. Commun. 1997, 231, 679. [DOI] [PubMed] [Google Scholar]
- Tavares P.; Ravi N.; Moura J. J. G.; LeGall J.; Huang Y.-H.; Crouse B. R.; Johnson M. K.; Huynh B. H.; Moura I. J. Biol. Chem. 1994, 269, 10504. [PubMed] [Google Scholar]
- Auchere F.; Pauleta S. R.; Tavares P.; Moura I.; Moura J. J. G. JBIC, J. Biol. Inorg. Chem. 2006, 11, 433. [DOI] [PubMed] [Google Scholar]
- Rodrigues J. V.; Saraiva L. M.; Abreu I. A.; Teixeira M.; Cabelli D. E. JBIC, J. Biol. Inorg. Chem. 2007, 12, 248. [DOI] [PubMed] [Google Scholar]
- Riebe O.; Fischer R.-J.; Bahl H. FEBS Lett. 2007, 581, 5605. [DOI] [PubMed] [Google Scholar]
- Devreese B.; Tavares P.; Lampreia J.; Van Damme N.; Le Gall J.; Moura J. J. G.; Van Beeumen J.; Moura I. FEBS Lett. 1996, 385, 138. [DOI] [PubMed] [Google Scholar]
- Coelho A. V.; Matias P.; Fulop V.; Thompson A.; Gonzalez A.; Carrondo M. A. JBIC, J. Biol. Inorg. Chem. 1997, 2, 680. [Google Scholar]
- Verhagen M. F. J. M.; Voorhorst W. G. B.; Kolkman J. A.; Wolbert R. B. G.; Hagen W. R. FEBS Lett. 1993, 336, 13. [DOI] [PubMed] [Google Scholar]
- Cordas C. M.; Raleiras P.; Auchere F.; Moura I.; Moura J. J. G. Eur. Biophys. J. 2012, 41, 209. [DOI] [PubMed] [Google Scholar]
- Ascenso C.; Rusnak F.; Cabrito I.; Lima M. J.; Naylor S.; Moura I.; Moura J. J. G. JBIC, J. Biol. Inorg. Chem. 2000, 5, 720. [DOI] [PubMed] [Google Scholar]
- Berthomieu C.; Dupeyrat F.; Fontecave M.; Vermeglio A.; Niviere V. Biochemistry 2002, 41, 10360. [DOI] [PubMed] [Google Scholar]
- Emerson J. P.; Cabelli D. E.; Kurtz D. M. Jr Proc. Natl. Acad. Sci. U.S.A. 2003, 100, 3802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abreu I. A.; Saraiva L. M.; Carita J.; Huber H.; Stetter K. O.; Cabelli D.; Teixeira M. Mol. Microbiol. 2000, 38, 322. [DOI] [PubMed] [Google Scholar]
- Weinberg M. V.; Jenney F. E. Jr.; Cui X.; Adams M. W. W. J. Bacteriol. 2004, 186, 7888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- deMare F.; Kurtz D. M. Jr.; Nordlund P. Nat. Struct. Biol. 1996, 3, 539. [DOI] [PubMed] [Google Scholar]
- Jin S.; Kurtz D. M. Jr.; Liu Z.-J.; Rose J.; Wang B.-C. J. Am. Chem. Soc. 2002, 124, 9845. [DOI] [PubMed] [Google Scholar]
- LeGall J.; Prickril B. C.; Moura I.; Xavier A. V.; Moura J. J. G.; Huynh Boi H. Biochemistry 1988, 27, 1636. [DOI] [PubMed] [Google Scholar]
- Pierik A. J.; Wolbert R. B. G.; Portier G. L.; Verhagen M. F. J. M.; Hagen W. R. Eur. J. Biochem. 1993, 212, 237. [DOI] [PubMed] [Google Scholar]
- Luo Y.; Ergenekan C. E.; Fischer J. T.; Tan M.-L.; Ichiye T. Biophys. J. 2010, 98, 560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin S.; Kurtz D. M. Jr.; Liu Z.-J.; Rose J.; Wang B.-C. Biochemistry 2004, 43, 3204. [DOI] [PubMed] [Google Scholar]
- Pinto A. F.; Todorovic S.; Hildebrandt P.; Yamazaki M.; Amano F.; Igimi S.; Romao C. V.; Teixeira M. JBIC, J. Biol. Inorg. Chem. 2011, 16, 501. [DOI] [PubMed] [Google Scholar]
- Lumppio H. L.; Shenvi N. V.; Garg R. P.; Summers A. O.; Kurtz D. M. Jr J. Bacteriol. 1997, 179, 4607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coulter E. D.; Shenvi N. V.; Kurtz D. M. Jr Biochem. Biophys. Res. Commun. 1999, 255, 317. [DOI] [PubMed] [Google Scholar]
- Matsubara H.; Saeki K. Adv. Inorg. Chem. 1992, 38, 223. [Google Scholar]
- Shethna Y. I.; Wilson P. W.; Hansen R. E.; Beinert H. Proc. Natl. Acad. Sci. U.S.A. 1964, 52, 1263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchanan B. B.; Lovenberg W.; Rabiowitz J. C. Proc. Natl. Acad. Sci. U.S. 1963, 49, 345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoch D. C.; Valentine R. C. Annu. Rev. Microbiol. 1972, 26, 139. [DOI] [PubMed] [Google Scholar]
- Bruschi M.; Guerlesquin F. FEMS Microbiol. Rev. 1988, 54, 155. [DOI] [PubMed] [Google Scholar]
- Sieker L. C.; Adman E. T. In Encyclopedia of Inorganic and Bioinorganic Chemistry; John Wiley & Sons, Ltd.: Hoboken, NJ, 2011. [Google Scholar]
- Valentine R. C. Bacteriol. Rev. 1964, 28, 497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lovenberg W.; Buchanan B. B.; Rabinowitz J. C. J. Biol. Chem. 1963, 238, 3899. [PubMed] [Google Scholar]
- Zanetti G.; Binda C.; Aliverti A. In Encyclopedia of Inorganic and Bioinorganic Chemistry; John Wiley & Sons, Ltd.: Hoboken, NJ, 2011. [Google Scholar]
- Hurley J. K.; Weber-Main A. M.; Stankovich M. T.; Benning M. M.; Thoden J. B.; Vanhooke J. L.; Holden H. M.; Chae Y. K.; Xia B.; Cheng H.; Markley J. L.; Martinez-Julvez M.; Gomez-Moreno C.; Schmeit J. L.; Tollin G. Biochemistry 1997, 363711100. [DOI] [PubMed] [Google Scholar]
- Hurley J. K.; Weber-Main A. M.; Hodges A. E.; Stankovich M. T.; Benning M. M.; Holden H. M.; Cheng H.; Xia B.; Markley J. L.; Genzor C.; Gomez-Moreno C.; Hafezi R.; Tollin G. Biochemistry 1997, 36, 15109. [DOI] [PubMed] [Google Scholar]
- De Pascalisa A. R.; Antonio R.; Jelesarov I.; Ackermann F.; Koppenol W. H.; Hirasawa M.; Knaff D. B.; Bosshard H. R. Protein Sci. 1993, 2, 1126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Pascalisa A. R.; Schuermann P.; Bosshard H. R. FEBS Lett. 1994, 337, 217. [DOI] [PubMed] [Google Scholar]
- Muller J. J.; Muller A.; Rottmann M.; Bernhardt R.; Heinemann U. J. Mol. Biol. 1999, 294, 501. [DOI] [PubMed] [Google Scholar]
- Zanetti G.; Pandini V.. Encyclopedia of Biological Chemistry; Elsevier Ltd.: Amsterdam, 2004; Vol. 2, p 104. [Google Scholar]
- Fukuyama K. Photosynth. Res. 2004, 81, 289. [DOI] [PubMed] [Google Scholar]
- Holden H. M.; Jacobson B. L.; Hurley J. K.; Tollin G.; Oh B. H.; Skjeldal L.; Chae Y. K.; Cheng H.; Xia B.; Markley J. L. J. Bioenerg. Biomembr. 1994, 26, 67. [DOI] [PubMed] [Google Scholar]
- Morales R.; Chron M.-H.; Hudry-Clergeon G.; Petillot Y.; Norager S.; Medina M.; Frey M. Biochemistry 1999, 38, 15764. [DOI] [PubMed] [Google Scholar]
- Bes M. T.; Parisini E.; Inda L. A.; Saraiva L. M.; Peleato M. L.; Sheldrick G. M. Structure (London, U. K.) 1999, 7, 1201. [DOI] [PubMed] [Google Scholar]
- ; Hardy R., Knight E. Jr., McDonald C., D’Eustachio A., Eds.; Non-Heme Iron Proteins: Role in Energy Conversion. Antioch Press: Yellow Springs, OH, 1965. [Google Scholar]
- Meyer J.; Moulis J. M.; Lutz M. Biochem. Biophys. Res. Commun. 1984, 119, 828. [DOI] [PubMed] [Google Scholar]
- Meyer J.; Andrade S. L. A.; Einsle O. In Encyclopedia of Inorganic and Bioinorganic Chemistry; John Wiley & Sons: Hoboken, NJ, 2011. [Google Scholar]
- Shergill J. K.; Golinelli M.-P.; Cammack R.; Meyer J. Biochemistry 1996, 35, 12842. [DOI] [PubMed] [Google Scholar]
- Golinelli M.-P.; Akin L. A.; Crouse B. R.; Johnson M. K.; Meyer J. Biochemistry 1996, 35, 8995. [DOI] [PubMed] [Google Scholar]
- Golinelli M.-P.; Chatelet C.; Duin E. C.; Johnson M. K.; Meyer J. Biochemistry 1998, 37, 10429. [DOI] [PubMed] [Google Scholar]
- Meyer J. FEBS Lett. 2001, 509, 1. [DOI] [PubMed] [Google Scholar]
- Hurley J. K.; Tollin G.; Medina M.; Gomez-Moreno C. Adv. Photosynth. Respir. 2006, 24Photosystem I455. [Google Scholar]
- Yeh A. P.; Chatelet C.; Soltis S. M.; Kuhn P.; Meyer J.; Rees D. C. J. Mol. Biol. 2000, 300, 587. [DOI] [PubMed] [Google Scholar]
- Hase T.; Schuermann P.; Knaff D. B. In Photosystem I; Goldbeck J. H., Ed.; Springer: Dordrecht, The Netherlands, 2006. [Google Scholar]
- Hurley J. K.; Medina M.; Gomez-Moreno C.; tollin G. Arch. Biochem. Biophys. 1994, 312, 480. [DOI] [PubMed] [Google Scholar]
- Hurley J. K.; Schmeits J. L.; Genzor C.; Gomez-Moreno C.; Tollin G. Arch. Biochem. Biophys. 1996, 333, 243. [DOI] [PubMed] [Google Scholar]
- Hurley J. K.; Salamon Z.; Meyer T. E.; Fitch J. C.; Cusanovich M. A.; Markley J. L.; Cheng H.; Xia B.; Chae Y. K. Biochemistry 1993, 32, 9346. [DOI] [PubMed] [Google Scholar]
- Aliverti A.; Livragh A.; Piubelli L.; Zanetti G. Biochim. Biophys. Acta 1997, 1342, 45. [DOI] [PubMed] [Google Scholar]
- Hurley J. K.; Tollin G.; Medina M.; Gomez-Moreno C. Adv. Photosynth. Respir. 2006, 24, 455. [Google Scholar]
- Kimata-Ariga Y.; Kubota-Kawai H.; Lee Y.-H.; Muraki N.; Ikegami T.; Kurisu G.; Hase T. Biochem. Biophys. Res. Commun. 2013, 434, 867. [DOI] [PubMed] [Google Scholar]
- Dai S.; Schwendtmayer C.; Schurmann P.; Ramaswamy S.; Eklund H. Science 2000, 287, 655. [DOI] [PubMed] [Google Scholar]
- Schmitz S.; Navarro F.; Kutzki C. K.; Florencio F. J.; Boehme H. Biochim. Biophys. Acta, Bioenerg. 1996, 1277, 135. [DOI] [PubMed] [Google Scholar]
- Garcia-Sanchez M. I.; Gotor C.; Jacquot E.-P.; Stein M.; Suzuki A.; Vega J. M. Eur. J. Biochem. 1997, 250, 364. [DOI] [PubMed] [Google Scholar]
- Akashi T.; Matsumura T.; Ideguchi T.; Iwakiri K.-I.; Kawakatsu T.; Taniguchi I.; Hase T. J. Biol. Chem. 1999, 274, 29399. [DOI] [PubMed] [Google Scholar]
- Antonkine M. L.; Koay M. S.; Epel B.; Breitenstein C.; Gopta O.; Gaertner W.; Bill E.; Lubitz W. Biochim. Biophys. Acta, Bioenerg. 2009, 1787, 995. [DOI] [PubMed] [Google Scholar]
- Ewen K. M.; Kleser M.; Bernhardt R. Biochim. Biophys. Acta, Proteins Proteomics 2011, 1814, 111. [DOI] [PubMed] [Google Scholar]
- Vickery L. E. Steroids 1997, 62, 124. [DOI] [PubMed] [Google Scholar]
- Ziegler G. A.; Vonrhein C.; Hanukoglu I.; Schulz G. E. J. Mol. Biol. 1999, 289, 981. [DOI] [PubMed] [Google Scholar]
- Muller J. J.; Lapko A.; Bourenkov G.; Ruckpaul K.; Heinemann U. J. Biol. Chem. 2001, 276, 2786. [DOI] [PubMed] [Google Scholar]
- Chatelet C.; Meyer J. Biochim. Biophys. Acta, Protein Struct. Mol. Enzymol. 2001, 1549, 32. [DOI] [PubMed] [Google Scholar]
- Skjeldal L.; Markley J. L.; Coghlan V. M.; Vickery L. E. Biochemistry 1991, 30, 9078. [DOI] [PubMed] [Google Scholar]
- Lambeth J. D.; Kamin H. J. Biol. Chem. 1979, 254, 2766. [PubMed] [Google Scholar]
- Vidakovic M.; Fraczkiewicz G.; Germanas J. P. J. Biol. Chem. 1996, 271. [DOI] [PubMed] [Google Scholar]
- Agarwall A.; Li D.; Cowan J. A. Proc. Natl. Acad. Sci. U.S.A. 1995, 92, 9440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duff J. L. C.; Breton J. L. J.; Butt J. N.; Armstrong F. A.; Thomson A. J. J. Am. Chem. Soc. 1996, 118, 8593. [Google Scholar]
- Nakamura A.; Ueyama N. ACS Symp. Ser. 1988, 372, 292. [Google Scholar]
- Chen K.; Tilley G. J.; Sridhar V.; Prasad G. S.; Stout C. D.; Armstrong F. A.; Burgess B. K. J. Biol. Chem. 1999, 274, 36479. [DOI] [PubMed] [Google Scholar]
- Langen R.; Jensen G. M.; Jacob U.; Stephens P. J.; Warshel A. J. Biol. Chem. 1992, 267, 25625. [PubMed] [Google Scholar]
- Werth M. T.; Cecchini G.; Manodori A.; Ackrell B. A. C.; Schroder I.; Gunsalus R. P.; Johnson M. K. Proc. Natl. Acad. Sci. U.S.A. 1990, 87, 8965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollocher T. C.; Solomon F.; Ragland T. E. J. Biol. Chem. 1966, 241, 3452. [PubMed] [Google Scholar]
- Dervartanian D. V.; Orme-Johnson W. H.; Hansen R. E.; Beinert H.; Tsai R. L.; Tsibris J. C. M.; Bartholomaus R. C.; Gunsalus I. C. Biochem. Biophys. Res. Commun. 1967, 26, 569. [DOI] [PubMed] [Google Scholar]
- Crouse B. R.; Meyer J.; Johnson M. K. J. Am. Chem. Soc. 1995, 117, 9612. [Google Scholar]
- Fu W.; Drozdzewski P. M.; Davies M. D.; Sligar S. G.; Johnson M. K. J. Biol. Chem. 1992, 267, 15502. [PubMed] [Google Scholar]
- Dunham W. R.; Palmer G.; Sands R. H.; Bearden A. J. Biochim. Biophys. Acta, Bioenerg. 1971, 253, 373. [DOI] [PubMed] [Google Scholar]
- Cheng H.; Markley J. L. Annu. Rev. Biophys. Biomol. Struct. 1995, 24, 209. [DOI] [PubMed] [Google Scholar]
- Meyer J.; Clay M. D.; Johnson M. K.; Stubna A.; Muenck E.; Higgins C.; Wittung-Stafshede P. Biochemistry 2002, 41, 3096. [DOI] [PubMed] [Google Scholar]
- Otaka E.; Ooi T. J. Mol. Evol. 1987, 26, 257. [DOI] [PubMed] [Google Scholar]
- Gaillard J.; Quinkal I.; Moulis J. M. Biochemistry 1993, 32, 9881. [DOI] [PubMed] [Google Scholar]
- Dauter Z.; Wilson K. S.; Sieker L. C.; Meyer J.; Moulis J.-M. Biochemistry 1997, 36, 16065. [DOI] [PubMed] [Google Scholar]
- Mulholland S. E.; Gibney B. R.; Rabanal F.; Dutton P. L. Biochemistry 1999, 38, 10442. [DOI] [PubMed] [Google Scholar]
- Bott A. W. Curr. Sep. 1999, 18, 47. [Google Scholar]
- Hannan J. P.; Busch J. L. H.; Breton J.; James R.; Thomson A. J.; Moore G. R.; Davy S. L. JBIC, J. Biol. Inorg. Chem. 2000, 5, 432. [DOI] [PubMed] [Google Scholar]
- Busch J. L.; Breton J. L.; Bartlett B. M.; Armstrong F. A.; James R.; Thomson A. J. Biochem. J. 1997, 323, 95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macedo A. L.; Rodrigues P.; Goodfellow B. J. Coord. Chem. Rev. 1999, 190–192, 871. [Google Scholar]
- Stout C. D. In Encyclopedia of Inorganic and Bioinorganic Chemistry; John Wiley & Sons, Ltd.: Hoboken, NJ, 2011. [Google Scholar]
- Quinkal I.; Kyritsis P.; Kohzuma T.; Im S.-C.; Sykes A. G.; Moulis J.-M. Biochim. Biophys. Acta, Protein Struct. Mol. Enzymol. 1996, 1295, 201. [DOI] [PubMed] [Google Scholar]
- Fish A.; Danieli T.; Ohad I.; Nechushtai R.; Livnah O. J. Mol. Biol. 2005, 350, 599. [DOI] [PubMed] [Google Scholar]
- Bertini I.Biological Inorganic Chemistry: Structure and Reactivity; University Science Books: Sausalito, CA, 2007; p 244. [Google Scholar]
- Fukuyama K. In Encyclopedia of Inorganic and Bioinorganic Chemistry; John Wiley & Sons, Ltd.: Hoboken, NJ, 2011. [Google Scholar]
- O’Keefe D. P.; Gibson K. J.; Emptage M. H.; Lenstra R.; Romesser J. A.; Litle P. J.; Omer C. A. Biochemistry 1991, 30, 447. [DOI] [PubMed] [Google Scholar]
- Marritt S. J.; Farrar J. A.; Breton J. L.; Hagen W. R.; Thomson A. J. Eur. J. Biochem. 1995, 2322501. [DOI] [PubMed] [Google Scholar]
- Kyritsis P.; Hatzfeld O. M.; Link T. A.; Moulis J.-M. J. Biol. Chem. 1998, 273, 15404. [DOI] [PubMed] [Google Scholar]
- Kyritsis P.; Huber J. G.; Quinkal I.; Gaillard J.; Moulis J.-M. Biochemistry 1997, 36, 7839. [DOI] [PubMed] [Google Scholar]
- Breton J. L.; Duff J. L. C.; Butt J. N.; Armstrong F. A.; George S. J.; Petillot Y.; Forest E.; Schaefer G.; Thomson A. J. Eur. J. Biochem. 1995, 233, 937. [DOI] [PubMed] [Google Scholar]
- Shen B.; Martin L. L.; Butt J. N.; Armstrong F. A.; Stout C. D.; Jensen G. M.; Stephens P. J.; La M.; Gerd N.; Gorst C. M.; Burgess B. K. J. Biol. Chem. 1993, 268, 25928. [PubMed] [Google Scholar]
- Beck B. W.; Xie Q.; Ichiye T. Biophys. J. 2001, 812601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camba R.; Armstrong F. A. Biochemistry 2000, 39, 10587. [DOI] [PubMed] [Google Scholar]
- Brereton P. S.; Maher M. J.; Tregloan P. A.; Wedd A. G. Biochim. Biophys. Acta, Protein Struct. Mol. Enzymol. 1999, 1429, 307. [DOI] [PubMed] [Google Scholar]
- Shen B.; Jollie D. R.; Stout C. D.; Diller T. C.; Artstring F. A.; Gorst C. M.; La Mar G. N.; Stephens P. J.; Burgess B. K. J. Biol. Chem. 1994, 269118564. [PubMed] [Google Scholar]
- Iismaa S. E.; Vazquez A. E.; Jensen G. M.; Stephens P. J.; Butt J. N.; Armstrong F. A.; Burgess B. K. J. Biol. Chem. 1991, 266, 21563. [PubMed] [Google Scholar]
- Moulis J. M.; Meyer J.; Lutz M. Biochemistry 1984, 23, 6605. [Google Scholar]
- Jensen G. M.; Warshel A.; Stephens P. J. Biochemistry 1994, 33, 10911. [DOI] [PubMed] [Google Scholar]
- Kyritsis P.; Kuemmerle R.; Huber J. G.; Gaillard J.; Guigliarelli B.; Popescu C.; Muenck E.; Moulis J.-M. Biochemistry 1999, 38, 6335. [DOI] [PubMed] [Google Scholar]
- Sweeney W. V.; Rabinowitz J. C. Annu. Rev. Biochem. 1980, 49, 139. [DOI] [PubMed] [Google Scholar]
- Busse S. C.; La Mar G. N.; Yu L. P.; Howard J. B.; Smith E. T.; Zhou Z. H.; Adams M. W. W. Biochemistry 1992, 31, 11952. [DOI] [PubMed] [Google Scholar]
- Kummerle R.; Kyritsis P.; Gaillard J.; Moulis J.-M. J. Inorg. Biochem. 2000, 79, 83. [DOI] [PubMed] [Google Scholar]
- Wildegger G.; Bentrop D.; Ejchart A.; Alber M.; Hage A.; Sterner R.; Roesch P. Eur. J. Biochem. 1995, 229, 658. [DOI] [PubMed] [Google Scholar]
- Loehr T. M. J. Raman Spectrosc. 1992, 23, 531. [Google Scholar]
- Mitra D.; Pelmenschikov V.; Guo Y.; Case D. A.; Wang H.; Dong W.; Tan M.-L.; Ichiye T.; Jenney F. E.; Adams M. W. W.; Yoda Y.; Zhao J.; Cramer S. P. Biochemistry 2011, 50, 5220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagedoorn P. L.; Chen T.; Schroeder I.; Piersma S. R.; de V.; Simon; Hagen W. R. JBIC, J. Biol. Inorg. Chem. 2005, 10, 259. [DOI] [PubMed] [Google Scholar]
- Duderstadt R. E.; Brereton P. S.; Adams M. W. W.; Johnson M. K. FEBS Lett. 1999, 454, 21. [DOI] [PubMed] [Google Scholar]
- Pereira M. M.; Jones K. L.; Campos M. G.; Melo A. M. P.; Saraiva L. M.; Louro R. O.; Wittung-Stafshede P.; Teixeira M. Biochim. Biophys. Acta, Proteins Proteomics 2002, 1601, 1. [DOI] [PubMed] [Google Scholar]
- Lin Y.-H.; Huang H.-E.; Chen Y.-R.; Liao P.-L.; Chen C.-L.; Feng T.-Y. Phytopathology 2011, 101, 741. [DOI] [PubMed] [Google Scholar]
- Klipp W.; Reilander H.; Schluter A.; Krey R.; Puhler A. Mol. Gen. Genet. 1989, 216, 293. [DOI] [PubMed] [Google Scholar]
- Masepohl B.; Kutsche M.; Riedel K. U.; Schmehl M.; Klipp W.; Puhler A. Mol. Gen. Genet. 1992, 233, 33. [DOI] [PubMed] [Google Scholar]
- Sjoblom S.; Harjunpaa H.; Brader G.; Palva E. T. Mol. Plant-Microbe Interact. 2008, 21, 967. [DOI] [PubMed] [Google Scholar]
- Huang H.-E.; Ger M.-J.; Chen C.-Y.; Yip M.-K.; Chung M.-C.; Feng T.-Y. Plant Sci. (Amsterdam, Neth.) 2006, 171, 17. [Google Scholar]
- Kwon D.-H.; El-Zaatari F. A. K.; Kato M.; Osato M. S.; Reddy R.; Yamaoka Y.; Graham D. Y. Antimicrob. Agents Chemother. 2000, 44, 2133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yip M.-K.; Huang H.-E.; Ger M.-J.; Chiu S.-H.; Tsai Y.-C.; Lin C.-I.; Feng T.-Y. Plant Cell Rep. 2007, 26, 449. [DOI] [PubMed] [Google Scholar]
- Rieske J. S.; MacLennan D. H.; Coleman R. Biochem. Biophys. Commun. 1964, 15, 388. [Google Scholar]
- Schneider D.; Schmidt C. L. Biochim. Biophys. Acta, Bioenerg. 2005, 1710, 1. [DOI] [PubMed] [Google Scholar]
- Harnisch U.; Weiss H.; Sebald W. Eur. J. Biochem. 1985, 149, 95. [DOI] [PubMed] [Google Scholar]
- Link T. A. Adv. Inorg. Chem. 1999, 47, 83. [Google Scholar]
- Iwata S.; Saynovits M.; Link T. A.; Michel H. Structure (London, U. K.) 1996, 4, 567. [DOI] [PubMed] [Google Scholar]
- Graham L. A.; Trumpower B. L. J. Biol. Chem. 1991, 266, 22485. [PubMed] [Google Scholar]
- Davidson E.; Ohnishi T.; Atta-Asafo-Adjei E.; Daldal F. Biochemistry 1992, 31, 3342. [DOI] [PubMed] [Google Scholar]
- Schmidt C. L.; Anemuller S.; Schafer G. FEBS Lett. 1996, 388, 43. [DOI] [PubMed] [Google Scholar]
- Fee J. A.; Findling K. L.; Yoshida T.; Hille R.; Tarr G. E.; Hearshen D. O.; Dunham W. R.; Day E. P.; Kent T. A.; Munck E. J. Biol. Chem. 1984, 259, 124. [PubMed] [Google Scholar]
- Gatti D. L.; Meinhardt S. W.; Onishi T.; Tzagoloff A. J. Mol. Biol. 1989, 205, 421. [DOI] [PubMed] [Google Scholar]
- Brown E. N.; Friemann R.; Karlsson A.; Parales J. V.; Couture M. M.-J.; Eltis L. D.; Ramaswamy S. JBIC, J. Biol. Inorg. Chem. 2008, 13, 1301. [DOI] [PubMed] [Google Scholar]
- Denke E.; Merbitz-Zahradnik T.; Hatzfeld O. M.; Snyder C. H.; Link T. A.; Trumpower B. L. J. Biol. Chem. 1998, 273, 9085. [DOI] [PubMed] [Google Scholar]
- Lebrun E.; Santini J. M.; Brugna M.; Ducluzeau A.-L.; Ouchane S.; Schoepp-Cothenet B.; Baymann F.; Nitschke W. Mol. Biol. Evol. 2006, 23, 1180. [DOI] [PubMed] [Google Scholar]
- Botelho H. M.; Leal S. S.; Veith A.; Prosinecki V.; Bauer C.; Frohlich R.; Kletzin A.; Gomes C. M. JBIC, J. Biol. Inorg. Chem. 2010, 15, 271. [DOI] [PubMed] [Google Scholar]
- Bonisch H.; Schmidt C. L.; Schafer G.; Ladenstein R. J. Mol. Biol. 2002, 319, 791. [DOI] [PubMed] [Google Scholar]
- Kauppi B.; Lee K.; Carredano E.; Parales R. E.; Gibson D. T.; Eklund H.; Ramaswamy S. Structure (London, U. K.) 1998, 6, 571. [DOI] [PubMed] [Google Scholar]
- Schmidt C. L. J. Bioenerg. Biomembr. 2004, 36, 107. [DOI] [PubMed] [Google Scholar]
- Link T. A. FEBS Lett. 1997, 412, 257. [DOI] [PubMed] [Google Scholar]
- Mason J. R.; Cammack R. Annu. Rev. Microbiol. 1992, 46, 277. [DOI] [PubMed] [Google Scholar]
- Parales R. E.; Parales J. V.; Gibson D. T. J. Bacteriol. 1999, 181, 1831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarasev M.; Pinto A.; Kim D.; Elliott S. J.; Ballou D. P. Biochemistry 2006, 45, 10208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinto A.; Tarasev M.; Ballou D. P. Biochemistry 2006, 45, 9032. [DOI] [PubMed] [Google Scholar]
- Martins B. M.; Svetlitchnaia T.; Dobbek H. Structure (London, U. K.) 2005, 13, 817. [DOI] [PubMed] [Google Scholar]
- Ferraro D. J.; Gakhar L.; Ramaswamy S. Biochem. Biophys. Res. Commun. 2005, 338, 175. [DOI] [PubMed] [Google Scholar]
- Snyder C. H.; Merbitz-Zahradnik T.; Link T. A.; Trumpower B. L. J. Bioenerg. Biomembr. 1999, 31, 235. [DOI] [PubMed] [Google Scholar]
- Kletzin A.; Ferreira A. S.; Hechler T.; Bandeiras T. M.; Teixeira M.; Gomes C. M. FEBS Lett. 2005, 579, 1020. [DOI] [PubMed] [Google Scholar]
- Ugulava N. B.; Crofts A. R. FEBS Lett. 1998, 440, 409. [DOI] [PubMed] [Google Scholar]
- Verhagen M. F. J. M.; Link T. A.; Hagen W. R. FEBS Lett. 1995, 361, 75. [DOI] [PubMed] [Google Scholar]
- Link T. A.; Hagen W. R.; Pierik A. J.; Assmann C.; von Jagow G. Eur. J. Biochem. 1992, 208, 685. [DOI] [PubMed] [Google Scholar]
- Couture M. M.-J.; Auger M.; Rosell F.; Mauk A. G.; Boubour E.; Lennox R. B.; Eltis L. D. Biochim. Biophys. Acta, Protein Struct. Mol. Enzymol. 1999, 1433, 159. [DOI] [PubMed] [Google Scholar]
- Kuznetsov A. M.; Zueva E. M.; Masliy A. N.; Krishtalik L. I. Biochim. Biophys. Acta, Bioenerg. 2010, 1797, 347. [DOI] [PubMed] [Google Scholar]
- Brugna M.; Albouy D.; Nitschke W. J. Bacteriol. 1998, 180, 3719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anemueller S.; Schmidt C. L.; Schaefer G.; Bill E.; Trautwein A. X.; Teixeira M. Biochem. Biophys. Res. Commun. 1994, 202, 252. [DOI] [PubMed] [Google Scholar]
- Albers A.; Bayer T.; Demeshko S.; Dechert S.; Meyer F. Chem.—Eur. J. 2013, 19, 10101. [DOI] [PubMed] [Google Scholar]
- Konkle M. E.; Muellner S. K.; Schwander A. L.; Dicus M. M.; Pokhrel R.; Britt R. D.; Taylor A. B.; Hunsicker-Wang L. M. Biochemistry 2009, 48, 9848. [DOI] [PubMed] [Google Scholar]
- Matsuura K.; Bowyer J. R.; Ohnishi T.; Dutton P. L. J. Biol. Chem. 1983, 258, 1571. [PubMed] [Google Scholar]
- von Jagow G.; Ohnishi T. FEBS Lett. 1985, 185, 311. [DOI] [PubMed] [Google Scholar]
- Konkle M. E.; Elsenheimer K. N.; Hakala K.; Robicheaux J. C.; Weintraub S. T.; Hunsicker-Wang L. M. Biochemistry 2010, 49, 7272. [DOI] [PubMed] [Google Scholar]
- Zu Y.; Couture M. M.-J.; Kolling D. R. J.; Crofts A. R.; Eltis L. D.; Fee J. A.; Hirst J. Biochemistry 2003, 42, 12400. [DOI] [PubMed] [Google Scholar]
- Link T. A.; Hatzfeld O. M.; Unalkat P.; Shergill J. K.; Cammack R.; Mason J. R. Biochemistry 1996, 35, 7546. [DOI] [PubMed] [Google Scholar]
- Colbert C. L.; Couture M. M.; Eltis L. D.; Bolin J. T. Structure (London, U. K.) 2000, 8, 1267. [DOI] [PubMed] [Google Scholar]
- Lhee S. Ph.D. Thesis, University of Illinois at Urbana-Champaign, 2007. [Google Scholar]
- Klingen A. R.; Ullmann G. M. Biochemistry 2004, 43, 12383. [DOI] [PubMed] [Google Scholar]
- Dikanov S. A.; Kolling D. J.; Endeward B.; Samoilova R. I.; Prisner T. F.; Nair S. K.; Crofts A. R. J. Biol. Chem. 2006, 281, 27416. [DOI] [PubMed] [Google Scholar]
- Lhee S.; Kolling D. R. J.; Nair S. K.; Dikanov S. A.; Crofts A. R. J. Biol. Chem. 2010, 285, 9233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroter T.; Hatzfeld O. M.; Gemeinhardt S.; Korn M.; Friedrich T.; Ludwig B.; Link T. A. Eur. J. Biochem. 1998, 255, 100. [DOI] [PubMed] [Google Scholar]
- Kolling D. J.; Brunzelle J. S.; Lhee S.; Crofts A. R.; Nair S. K. Structure 2007, 15129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brasseur G.; Sled V.; Liebl U.; Ohnishi T.; Daldal F. Biochemistry 1997, 36, 11685. [DOI] [PubMed] [Google Scholar]
- Leggate E. J.; Hirst J. Biochemistry 2005, 44, 7048. [DOI] [PubMed] [Google Scholar]
- Cooley J. W.; Roberts A. G.; Bowman M. K.; Kramer D. M.; Daldal F. Biochemistry 2004, 43, 2217. [DOI] [PubMed] [Google Scholar]
- Merbitz-Zahradnik T.; Zwicker K.; Nett J. H.; Link T. A.; Trumpower B. L. Biochemistry 2003, 42, 13637. [DOI] [PubMed] [Google Scholar]
- Zu Y.; Fee J. A.; Hirst J. Biochemistry 2002, 41, 14054. [DOI] [PubMed] [Google Scholar]
- Beharry Z. M.; Eby D. M.; Coulter E. D.; Viswanathan R.; Neidle E. L.; Phillips R. S.; Kurtz D. M. Jr Biochemistry 2003, 42, 13625. [DOI] [PubMed] [Google Scholar]
- Brugna M.; Nitschke W.; Asso M.; Guigliarelli B.; Lemesle-Meunier D.; Schmidt C. J. Biol. Chem. 1999, 274, 16766. [DOI] [PubMed] [Google Scholar]
- Uchiyama T.; Kounosu A.; Sato T.; Tanaka N.; Iwasaki T.; Kumasaka T. Acta Cryst. Section D 2004, D60, 1487. [DOI] [PubMed] [Google Scholar]
- Wilson D. F.; Erecinska M.; Brocklehurst E. S. Arch. Biochem. Biophys. 1972, 151, 180. [DOI] [PubMed] [Google Scholar]
- Link T. A.; Hatzfeld O. M.; Saynovits M. In Bioinorganic Chemistry; Trautwein A. X., Ed.; Wiley-VCH: Weinheim, Germany, 1997; p 312. [Google Scholar]
- Link T. A.; Saynovits M.; Assmann C.; Iwata S.; Ohnishi T.; von Jagow G. Eur. J. Biochem. 1996, 237, 71. [DOI] [PubMed] [Google Scholar]
- Bertrand P.; Guigliarelli B.; Gayda J. P.; Beardwood P.; Gibson J. F. Biochim. Biophys. Acta, Protein Struct. Mol. Enzymol. 1985, 831, 261. [Google Scholar]
- T’sai A. L.; Palmer G. Biochim. Biophys. Acta, Bioenerg. 1983, 722, 349. [DOI] [PubMed] [Google Scholar]
- Hatzfeld O. M. Ph.D. Thesis, Universitätsklinikum Frankfurt, Frankfurt, 1998. [Google Scholar]
- Prince R. C.; Lindsay J. G.; Dutton P. L. FEBS Lett. 1975, 51, 108. [DOI] [PubMed] [Google Scholar]
- Sharp R. E.; Palmitessa A.; Gibney B. R.; Moser C. C.; Daldal F.; Dutton P. L. FEBS Lett. 1998, 431, 423. [DOI] [PubMed] [Google Scholar]
- Evans M. C. W.; Lord A. V.; Reeves S. G. Biochem. J. 1974, 138, 177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nitschke W.; Joliot P.; Liebl U.; Rutherford A. W.; Hauska G.; Mueller A.; Riedel A. Biochim. Biophys. Acta, Bioenerg. 1992, 1102, 266. [Google Scholar]
- Zhang H.; Carrell C. J.; Huang D.; Sled V.; Ohnishi T.; Smith J. L.; Cramer W. A. J. Biol. Chem. 1996, 271, 31360. [DOI] [PubMed] [Google Scholar]
- Holton B.; Wu X.; Tsapin A. I.; Kramer D. M.; Malkin R.; Kallas T. Biochemistry 1996, 35, 15485. [DOI] [PubMed] [Google Scholar]
- Liebl U.; Pezennec S.; Riedel A.; Kellner E.; Nitschke W. J. Biol. Chem. 1992, 267, 14068. [PubMed] [Google Scholar]
- Lewis R. J.; Prince R. C.; Dutton P. L.; Knaff D. B.; Krulwich T. A. J. Biol. Chem. 1981, 256, 10543. [PubMed] [Google Scholar]
- Liebl U.; Rutherford A. W.; Nitschke W. FEBS Lett. 1990, 261, 427. [Google Scholar]
- Riedel A.; Kellner E.; Grodzitzki D.; Liebl U.; Hauska G.; Mueller A.; Rutherford A. W.; Nitschke W. Biochim. Biophys. Acta, Bioenerg. 1993, 1183, 263. [Google Scholar]
- Kuila D.; Fee J. A. J. Biol. Chem. 1986, 261, 2768. [PubMed] [Google Scholar]
- Schmidt C. L.; Hatzfeld O. M.; Petersen A.; Link T. A.; Schaefer G. Biochem. Biophys. Res. Commun. 1997, 234, 283. [DOI] [PubMed] [Google Scholar]
- Geary P. J.; Saboowalla F.; Patil D.; Cammack R. Biochem. J. 1984, 217, 667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertrand P.; Gayda J. P. Biochim. Biophys. Acta, Protein Struct. 1979, 579, 107. [DOI] [PubMed] [Google Scholar]
- Shergill J. K.; Joannou C. L.; Mason J. R.; Cammack R. Biochemistry 1995, 34, 16533. [DOI] [PubMed] [Google Scholar]
- Rosche B.; Fetzner S.; Lingens F.; Nitschke W.; Riedel A. Biochim. Biophys. Acta, Protein Struct. Mol. Enzymol. 1995, 1252, 177. [DOI] [PubMed] [Google Scholar]
- Tsang H. T.; Batie C. J.; Ballou D. P.; Penner-Hahn J. E. Biochemistry 1989, 28, 7233. [DOI] [PubMed] [Google Scholar]
- Kuila D.; Schoonover J. R.; Dyer R. B.; Batie C. J.; Ballou D. P.; Fee J. A.; Woodruff W. H. Biochim. Biophys. Acta, Bioenerg. 1992, 1140, 175. [DOI] [PubMed] [Google Scholar]
- Iwasaki T.; Kounosu A.; Kolling D. R. J.; Crofts A. R.; Dikanov S. A.; Jin A.; Imai T.; Urushiyama A. J. Am. Chem. Soc. 2004, 126, 4788. [DOI] [PubMed] [Google Scholar]
- Rotsaert F. A. J.; Pikus J. D.; Fox B. G.; Markley J. L.; Sanders-Loehr J. JBIC, J. Biol. Inorg. Chem. 2003, 8, 318. [DOI] [PubMed] [Google Scholar]
- Cosper N. J.; Eby D. M.; Kounosu A.; Kurosawa N.; Neidle E. L.; Kurtz D. M. Jr.; Iwasaki T.; Scott R. A. Protein Sci. 2002, 11, 2969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tiago d. O. F.; Bominaar E. L.; Hirst J.; Fee J. A.; Munck E. J. Am. Chem. Soc. 2004, 126, 5338. [DOI] [PubMed] [Google Scholar]
- Cline J. F.; Hoffman B. M.; Mims W. B.; LaHaie E.; Ballou D. P.; Fee J. A. J. Biol. Chem. 1985, 260, 3251. [PubMed] [Google Scholar]
- Britt R. D.; Sauer K.; Klein M. P.; Knaff D. B.; Kriauciunas A.; Yu C. A.; Yu L.; Malkin R. Biochemistry 1991, 30, 1892. [DOI] [PubMed] [Google Scholar]
- Gurbiel R. J.; Ohnishi T.; Robertson D. E.; Daldal F.; Hoffman B. M. Biochemistry 1991, 30, 11579. [DOI] [PubMed] [Google Scholar]
- Gurbiel R. J.; Doan P. E.; Gassner G. T.; Macke T. J.; Case D. A.; Ohnishi T.; Fee J. A.; Ballou D. P.; Hoffman B. M. Biochemistry 1996, 35, 7834. [DOI] [PubMed] [Google Scholar]
- Shergill J. K.; Cammack R. Biochim. Biophys. Acta, Bioenerg. 1994, 1185, 35. [DOI] [PubMed] [Google Scholar]
- Dikanov S. A.; Xun L.; Karpiel A. B.; Tyryshkin A. M.; Bowman M. K. J. Am. Chem. Soc. 1996, 118, 8408. [Google Scholar]
- Kolling D. R. J.; Samoilova R. I.; Shubin A. A.; Crofts A. R.; Dikanov S. A. J. Phys. Chem. A 2009, 113, 653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kappl R.; Ebelshaueser M.; Hannemann F.; Bernhardt R.; Huettermann J. Appl. Mag. Res. 2006, 30, 427. [Google Scholar]
- Neese F.; Kappl R.; Huttermann J.; Zumft W. G.; Kroneck P. M. H. JBIC, J. Biol. Inorg. Chem. 1998, 3, 53. [Google Scholar]
- Holz R. C.; Small F. J.; Ensign S. A. Biochemistry 1997, 36, 14690. [DOI] [PubMed] [Google Scholar]
- de Vries S.; Cherepanov A.; Berg A.; Canters G. W. Inorg. Chim. Acta 1998, 275–276, 493. [Google Scholar]
- Xia B.; Pikus J. D.; Xia W.; McClay K.; Steffan R. J.; Chae Y. K.; Westler W. M.; Markley J. L.; Fox B. G. Biochemistry 1999, 38, 727. [DOI] [PubMed] [Google Scholar]
- Hsueh K.-L.; Westler W. M.; Markley J. L. J. Am. Chem. Soc. 2010, 132, 7908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsueh K.-L.; Tonelli M.; Cai K.; Westler W. M.; Markley J. L. Biochemistry 2013, 52, 2862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leggate E. J.; Bill E.; Essigke T.; Ullmann G. M.; Hirst J. Proc. Natl. Acad. Sci. U.S.A. 2004, 101, 10913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu J.; Egawa T.; Yeh S.-R.; Yu L.; Yu C.-A. Proc. Natl. Acad. Sci. U.S.A. 2007, 104, 4864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hori K. J. Biochem. 1961, 50, 481. [DOI] [PubMed] [Google Scholar]
- Dus K.; De Klerk H.; Sletten K.; Bartsch R. G. Biochim. Biophys. Acta, Protein Struct. 1967, 140, 291. [DOI] [PubMed] [Google Scholar]
- Parisini E.; Capozzi F.; Lubini P.; Lamzin V.; Luchinat C.; Sheldrick G. M. Acta Crystallogr., Sect. D: Biol. Crystallogr. 1999, D55, 1773. [DOI] [PubMed] [Google Scholar]
- Carter C. W. Jr. Ed.; Lovenberg W. Iron-Sulfur Proteins 1977, 3, 157. [Google Scholar]
- Kim J. D.; Rodriguez-Granillo A.; Case D. A.; Nanda V.; Falkowski P. G. PLoS Comput. Biol. 2012, 8, e1002463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter C. W., Jr.Handbook of Metalloproteins; John Wiley & Sons, Ltd.: Hoboken, NJ, 2006. [Google Scholar]
- Cowan J. A.; Lui S. M. Adv. Inorg. Chem. 1998, 45, 313. [Google Scholar]
- Cavazza C.; Guigliarelli B.; Bertrand P.; Bruschi M. FEMS Microbiol. Lett. 1995, 130, 193. [Google Scholar]
- Bian S.; Hemann C. F.; Hille R.; Cowan J. A. Biochemistry 1996, 35, 14544. [DOI] [PubMed] [Google Scholar]
- Soriano A.; Li D.; Bian S.; Agarwal A.; Cowan J. A. Biochemistry 1996, 35, 12479. [DOI] [PubMed] [Google Scholar]
- Iwagami S. G.; Creagh A. L.; Haynes C. A.; Borsari M.; Felli I. C.; Piccioli M.; Eltis L. D. Protein Sci. 1995, 4, 2562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kusano T.; Takeshima T.; Sugawara K.; Inoue C.; Shiratori T.; Yano T.; Fukumori Y.; Yamanaka T. J. Biol. Chem. 1992, 267, 11242. [PubMed] [Google Scholar]
- Pereira M. M.; Antunes A. M.; Nunes O. C.; da C.; Milton S.; Teixeira M. FEBS Lett. 1994, 352, 327. [DOI] [PubMed] [Google Scholar]
- Fukumori Y.; Yamanaka T. Curr. Microbiol. 1979, 3, 117. [Google Scholar]
- Lieutaud C.; Alric J.; Bauzan M.; Nitschke W.; Schoepp-Cothenet B. Proc. Natl. Acad. Sci. U.S.A. 2005, 102, 3260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hochkoeppler A.; Kofod P.; Ferro G.; Ciurli S. Arch. Biochem. Biophys. 1995, 322, 313. [DOI] [PubMed] [Google Scholar]
- Ciurli S.; Musiani F. Photosynth. Res. 2005, 85, 115. [DOI] [PubMed] [Google Scholar]
- Banci L.; Bertini I.; Briganti F.; Luchinat C.; Scozzafava A.; Oliver M. V. Inorg. Chem. 1991, 30, 4517. [Google Scholar]
- Heering H. A.; Bulsink Y. B. M.; Hagen W. R.; Meyer T. E. Biochemistry 1995, 34, 14675. [DOI] [PubMed] [Google Scholar]
- Heering H. A.; Bulsink Y. B. M.; Hagen W. R.; Meyer T. E. Eur. J. Biochem. 1995, 232, 811. [PubMed] [Google Scholar]
- Ichiye T. AIP Conf. Proc. 1999, 492. [Google Scholar]
- Low D. W.; Hill M. G. J. Am. Chem. Soc. 2000, 122, 11039. [Google Scholar]
- Dey A.; Roche C. L.; Walters M. A.; Hodgson K. O.; Hedman B.; Solomon E. I. Inorg. Chem. 2005, 44, 8349. [DOI] [PubMed] [Google Scholar]
- Banci L.; Bertini I.; Ciurli S.; Luchinat C.; Pierattelli R. Inorg. Chim. Acta 1995, 240, 251. [Google Scholar]
- Bertini I.; Borsari M.; Bosi M.; Eltis L. D.; Felli I. C.; Luchinat C.; Piccioli M. JBIC, J. Biol. Inorg. Chem. 1996, 1, 257. [Google Scholar]
- Benning M. M.; Meyer T. E.; Rayment I.; Holden H. M. Biochemistry 1994, 33, 2476. [DOI] [PubMed] [Google Scholar]
- Sticht H.; Wildegger G.; Bentrop D.; Darimont B.; Sterner R.; Rosch P. Eur. J. Biochem. 1996, 237, 726. [DOI] [PubMed] [Google Scholar]
- Agarwal A.; Li D.; Cowan J. A. J. Am. Chem. Soc. 1996, 118, 927. [Google Scholar]
- Babini E.; Borsari M.; Capozzi F.; Eltis L. D.; Luchinat C. JBIC, J. Biol. Inorg. Chem. 1999, 46692. [DOI] [PubMed] [Google Scholar]
- Dilg A. W. E.; Mincione G.; Achterhold K.; Iakovleva O.; Mentler M.; Luchinat C.; Bertini I.; Parak F. G. JBIC, J. Biol. Inorg. Chem. 1999, 4, 727. [DOI] [PubMed] [Google Scholar]
- Luchinat C.; Capozzi F.; Borsari M.; Battistuzzi G.; Sola M. Biochem. Biophys. Res. Commun. 1994, 203, 436. [DOI] [PubMed] [Google Scholar]
- Perrin B. S.; Ichiye T. Biochemistry 2013, 52, 3022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glaser T.; Bertini I.; Moura J. J. G.; Hedman B.; Hodgson K. O.; Solomon E. I. J. Am. Chem. Soc. 2001, 123, 4859. [DOI] [PubMed] [Google Scholar]
- Niu S.; Ichiye T. J. Am. Chem. Soc. 2009, 131, 5724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dey A.; Glaser T.; Couture M. M.-J.; Eltis L. D.; Holm R. H.; Hedman B.; Hodgson K. O.; Solomon E. I. J. Am. Chem. Soc. 2004, 126, 8320. [DOI] [PubMed] [Google Scholar]
- Dey A.; Jenney F. E. Jr.; Adams M. W. W.; Babini E.; Takahashi Y.; Fukuyama K.; Hodgson K. O.; Hedman B.; Solomon E. I. Science 2007, 318, 1464. [DOI] [PubMed] [Google Scholar]
- Kerfeld C. A.; Chan C.; Hirasawa M.; Kleis-SanFrancisco S.; Yeates T. O.; Knaff D. B. Biochemistry 1996, 35, 7812. [DOI] [PubMed] [Google Scholar]
- Moulis J. M.; Scherrer N.; Gagnon J.; Forest E.; Petillot Y.; Garcia D. Arch. Biochem. Biophys. 1993, 305, 186. [DOI] [PubMed] [Google Scholar]
- Ciszewska H.; Bagyinka C.; Tigyi G.; Kovacs K. L. Acta Biochim. Biophys. Hung. 1989, 24, 361. [PubMed] [Google Scholar]
- Zorin N. A.; Gogotov I. N. Biokhimiya (Moscow) 1983, 48, 1181. [PubMed] [Google Scholar]
- Tedro S. M.; Meyer T. E.; Bartsch R. G.; Kamen M. D. J. Biol. Chem. 1981, 256, 731. [PubMed] [Google Scholar]
- Ciszewska H.; Bagyinka C.; Kovacs K. L. Biokonvers. Soln. Energ., Mater. Mezhdunar. Simp. 1984, 330. [Google Scholar]
- Wermter U.; Fischer U. Z. Naturforsch., C: J. Biosci. 1983, 38C, 968. [DOI] [PubMed] [Google Scholar]
- Bartsch R. G. Methods Enzymol. 1978, 53, 329. [DOI] [PubMed] [Google Scholar]
- Sola M.; Cowan J. A.; Gray H. B. Biochemistry 1989, 28, 5261. [DOI] [PubMed] [Google Scholar]
- Tedro S. M.; Meyer T. E.; Kamen M. D. J. Biol. Chem. 1974, 249, 1182. [PubMed] [Google Scholar]
- Tedro S. M.; Meyer T. E.; Kamen M. D. Arch. Biochem. Biophys. 1985, 241, 656. [DOI] [PubMed] [Google Scholar]
- Bhattacharyya A. K.; Meyer T. E.; Cusanovich M. A.; Tollin G. Biochemistry 1987, 26, 758. [DOI] [PubMed] [Google Scholar]
- Meyer T. E. Methods Enzymol. 1994, 243, 435. [DOI] [PubMed] [Google Scholar]
- Hochkoeppler A.; Zannoni D.; Ciurli S.; Meyer T. E.; Cusanovich M. A.; Tollin G. Proc. Natl. Acad. Sci. U.S.A. 1996, 93, 6998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hochkoeppler A.; Kofod P.; Zannoni D. FEBS Lett. 1995, 375, 197. [DOI] [PubMed] [Google Scholar]
- Ambler R. P.; Meyer T. E.; Kamen M. D. Arch. Biochem. Biophys. 1993, 306, 215. [DOI] [PubMed] [Google Scholar]
- Meyer T. E.; Cannac V.; Fitch J.; Bartsch R. G.; Tollin D.; Tollin G.; Cusanovich M. A. Biochim. Biophys. Acta, Bioenerg. 1990, 1017, 125. [DOI] [PubMed] [Google Scholar]
- Rayment I.; Wesenberg G.; Meyer T. E.; Cusanovich M. A.; Holden H. M. J. Mol. Biol. 1992, 228, 672. [DOI] [PubMed] [Google Scholar]
- Schoepp B.; Parot P.; Menin L.; Gaillard J.; Richaud P.; Vermeglio A. Biochemistry 1995, 34, 11736. [DOI] [PubMed] [Google Scholar]
- Tedro S. M.; Meyer T. E.; Kamen M. D. J. Biol. Chem. 1977, 252, 7826. [PubMed] [Google Scholar]
- Breiter D. R.; Meyer T. E.; Rayment I.; Holden H. M. J. Biol. Chem. 1991, 266, 18660. [DOI] [PubMed] [Google Scholar]
- Stombaugh N. A.; Sundquist J. E.; Burris R. H.; Orme-Johnson W. H. Biochemistry 1976, 15, 2633. [DOI] [PubMed] [Google Scholar]
- Fukuyama K.; Matsubara H.; Tsukihara T.; Katsube Y. J. Mol. Biol. 1989, 210, 383. [DOI] [PubMed] [Google Scholar]
- Sweeney W. V.; Rabinowitz J. C.; Yoch D. C. J. Biol. Chem. 1975, 250, 7842. [PubMed] [Google Scholar]
- Przysiecki C. T.; Meyer T. E.; Cusanovich M. A. Biochemistry 1985, 24, 2542. [DOI] [PubMed] [Google Scholar]
- Bertini I.; Capozzi F.; Luchinat C.; Piccioli M. Eur. J. Biochem. 1993, 212, 69. [DOI] [PubMed] [Google Scholar]
- Banci L.; Bertini I.; Ferretti S.; Luchinat C.; Piccioli M. J. Mol. Struct. 1993, 292, 207. [DOI] [PubMed] [Google Scholar]
- Bertini I.; Capozzi F.; Luchinat C.; Piccioli M.; Vicens Oliver M. Inorg. Chim. Acta 1992, 198–200, 483. [Google Scholar]
- Ciurli S.; Cremonini A.; Kofod P.; Luchinat C. Eur. J. Biochem. 1996, 236, 405. [DOI] [PubMed] [Google Scholar]
- Bertini I.; Capozzi F.; Luchinat C. ACS Symp. Ser. 2003, 858, 272. [Google Scholar]
- Gaillard J.; Albrand J. P.; Moulis J. M.; Wemmer D. E. Biochemistry 1992, 31, 5632. [DOI] [PubMed] [Google Scholar]
- Bertini I.; Donaire A.; Felli I. C.; Luchinat C.; Rosato A. Inorg. Chem. 1997, 36, 4798. [DOI] [PubMed] [Google Scholar]
- Priem A. H.; Klaassen A. A. K.; Reijerse E. J.; Meyer T. E.; Luchinat C.; Capozzi F.; Dunham W. R.; Hagen W. R. JBIC, J. Biol. Inorg. Chem. 2005, 10, 417. [DOI] [PubMed] [Google Scholar]
- Anderson R. E.; Anger G.; Petersson L.; Ehrenberg A.; Cammack R.; Hall D. O.; Mullinger R.; Rao K. K. Biochim. Biophys. Acta, Bioenerg. 1975, 376, 63. [DOI] [PubMed] [Google Scholar]
- Antanaitis B. C.; Moss T. H. Biochim. Biophys. Acta, Protein Struct. 1975, 405, 262. [DOI] [PubMed] [Google Scholar]
- Dilg A. W. E.; Grantner K.; Iakovleva O.; Parak F. G.; Babini E.; Bertini I.; Capozzi F.; Luchinat C.; Meyer-Klaucke W. JBIC, J. Biol. Inorg. Chem. 2002, 7, 691. [DOI] [PubMed] [Google Scholar]
- Bentrop D.; Bertini I.; Iacoviello R.; Luchinat C.; Niikura Y.; Piccioli M.; Presenti C.; Rosato A. Biochemistry 1999, 38, 4669. [DOI] [PubMed] [Google Scholar]
- Teixeira M.; Moura I.; Xavier A. V.; Moura J. J. G.; LeGall J.; DerVartanian D. V.; Peck H. D. Jr.; Huynh Boi H. J. Biol. Chem. 1989, 264, 16435. [PubMed] [Google Scholar]
- Volbeda A.; Charon M.-H.; Piras C.; Hatchikian E. C.; Frey M.; Fontecilla-Camps J. C. Nature 1995, 373, 580. [DOI] [PubMed] [Google Scholar]
- Montet Y.; Amara P.; Volbeda A.; Vernede X.; Hatchikian E. C.; Field M. J.; Frey M.; Fontecilla-Camps J. C. Nat. Struct. Biol. 1997, 4, 523. [DOI] [PubMed] [Google Scholar]
- Garcin E.; Vernede X.; Hatchikian E. C.; Volbeda A.; Frey M.; Fontecilla-Camps J. C. Structure (London, U. K.) 1999, 7, 557. [DOI] [PubMed] [Google Scholar]
- Marques M. C.; Coelho R.; De Lacey A. L.; Pereira I. A. C.; Matias P. M. J. Mol. Biol. 2010, 396, 893. [DOI] [PubMed] [Google Scholar]
- Rousset M.; Montet Y.; Guigliarelli B.; Forget N.; Asso M.; Bertrand P.; Fontecilla-Camps J. C.; Hatchikian E. C. Proc. Natl. Acad. Sci. U.S.A. 1998, 95, 11625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pelmenschikov V.; Kaupp M. J. Am. Chem. Soc. 2013, 135, 11809. [DOI] [PubMed] [Google Scholar]
- Grubel K.; Holland P. L. Angew. Chem., Int. Ed. 2012, 51, 3308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ludwig M.; Cracknell J. A.; Vincent K. A.; Armstrong F. A.; Lenz O. J. Biol. Chem. 2009, 284, 465. [DOI] [PubMed] [Google Scholar]
- Fritsch J.; Scheerer P.; Frielingsdorf S.; Kroschinsky S.; Friedrich B.; Lenz O.; Spahn C. M. T. Nature 2011, 479, 249. [DOI] [PubMed] [Google Scholar]
- Shomura Y.; Yoon K.-S.; Nishihara H.; Higuchi Y. Nature 2011, 479, 253. [DOI] [PubMed] [Google Scholar]
- Pandelia M.-E.; Nitschke W.; Infossi P.; Giudici-Orticoni M.-T.; Bill E.; Lubitz W. Proc. Natl. Acad. Sci. U.S.A. 2011, 108, 6097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goris T.; Wait A. F.; Saggu M.; Fritsch J.; Heidary N.; Stein M.; Zebger I.; Lendzian F.; Armstrong F. A.; Friedrich B.; Lenz O. Nat. Chem. Biol. 2011, 7, 310. [DOI] [PubMed] [Google Scholar]
- Pandelia M.-E.; Fourmond V.; Tron-Infossi P.; Lojou E.; Bertrand P.; Leger C.; Giudici-Orticoni M.-T.; Lubitz W. J. Am. Chem. Soc. 2010, 132, 6991. [DOI] [PubMed] [Google Scholar]
- Lukey M. J.; Roessler M. M.; Parkin A.; Evans R. M.; Davies R. A.; Lenz O.; Friedrich B.; Sargent F.; Armstrong F. A. J. Am. Chem. Soc. 2011, 133, 16881. [DOI] [PubMed] [Google Scholar]
- Vignais P. M.; Billoud B.; Meyer J. FEMS Microbiol. Rev. 2001, 25, 455. [DOI] [PubMed] [Google Scholar]
- Dubini A.; Mus F.; Seibert M.; Grossman A. R.; Posewitz M. C. J. Biol. Chem. 2009, 284, 7201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulder D. W.; Shepard E. M.; Meuser J. E.; Joshi N.; King P. W.; Posewitz M. C.; Broderick J. B.; Peters J. W. Structure (London, U. K.) 2011, 19, 1038. [DOI] [PubMed] [Google Scholar]
- Nicolet Y.; Piras C.; Legrand P.; Hatchikian C. E.; Fontecilla-Camps J. C. Structure (London, U. K.) 1999, 7, 13. [DOI] [PubMed] [Google Scholar]
- Peters J. W.; Lanzilotta W. N.; Lemon B. J.; Seefeldt L. C. Science 1998, 282, 1853. [DOI] [PubMed] [Google Scholar]
- Georgiadis M. M.; Komiya H.; Chakrabarti P.; Woo D.; Kornuc J. J.; Rees D. C. Science 1992, 257, 1653. [DOI] [PubMed] [Google Scholar]
- Smith B. E. Adv. Inorg. Chem. 1999, 47, 159. [Google Scholar]
- Watt G. D.; Reddy K. R. N. J. Inorg. Biochem. 1994, 53, 281. [Google Scholar]
- Angove H. C.; Yoo S. J.; Munck E.; Burgess B. K. J. Biol. Chem. 1998, 273, 26330. [DOI] [PubMed] [Google Scholar]
- Strop P.; Takahara P. M.; Chiu H.-J.; Angove H. C.; Burgess B. K.; Rees D. C. Biochemistry 2001, 40, 651. [DOI] [PubMed] [Google Scholar]
- Lindahl P. A.; Boon Keng T.; Orme-Johnson W. H. Inorg. Chem. 1987, 26, 3912. [Google Scholar]
- Schindelin H.; Kisker C.; Schlessman J. L.; Howard J. B.; Rees D. C. Nature 1997, 387, 370. [DOI] [PubMed] [Google Scholar]
- Jang S. B.; Seefeldt L. C.; Peters J. W. Biochemistry 2000, 39, 14745. [DOI] [PubMed] [Google Scholar]
- Jeong M. S.; Jang S. B. Mol. Cells 2004, 18, 374. [PubMed] [Google Scholar]
- Jang S. B.; Seefeldt L. C.; Peters J. W. Biochemistry 2000, 39, 641. [DOI] [PubMed] [Google Scholar]
- Sen S.; Krishnakumar A.; McClead J.; Johnson M. K.; Seefeldt L. C.; Szilagyi R. K.; Peters J. W. J. Inorg. Biochem. 2006, 100, 1041. [DOI] [PubMed] [Google Scholar]
- Tezcan F. A.; Kaiser J. T.; Mustafi D.; Walton M. Y.; Howard J. B.; Rees D. C. Science 2005, 309, 1377. [DOI] [PubMed] [Google Scholar]
- Sen S.; Igarashi R.; Smith A.; Johnson M. K.; Seefeldt L. C.; Peters J. W. Biochemistry 2004, 43, 1787. [DOI] [PubMed] [Google Scholar]
- Peters J. W.; Stowell M. H. B.; Soltis S. M.; Finnegan M. G.; Johnson M. K.; Rees D. C. Biochemistry 1997, 36, 1181. [DOI] [PubMed] [Google Scholar]
- Surerus K. K.; Hendrich M. P.; Christie P. D.; Rottgardt D.; Orme-Johnson W. H.; Munck E. J. Am. Chem. Soc. 1992, 114, 8579. [Google Scholar]
- Pierik A. J.; Wassink H.; Haaker H.; Hagen W. R. Eur. J. Biochem. 1993, 212, 51. [DOI] [PubMed] [Google Scholar]
- Huber R.; Hof P.; Duarte R. O.; Moura J. J. G.; Moura I.; Liu M.-Y.; LeGall J.; Hille R.; Archer M.; Romao M. J. Proc. Natl. Acad. Sci. U.S.A. 1996, 93, 8846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rebelo J. M.; Dias J. M.; Huber R.; Moura J. J. G.; Romao M. J. JBIC, J. Biol. Inorg. Chem. 2001, 6, 791. [DOI] [PubMed] [Google Scholar]
- Coelho C.; Mahro M.; Trincao J.; Carvalho A. T. P.; Ramos M. J.; Terao M.; Garattini E.; Leimkuehler S.; Romao M. J. J. Biol. Chem. 2012, 287, 40690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner N.; Barata B.; Bray R. C.; Deistung J.; Le Gall J.; Moura J. J. G. Biochem. J. 1987, 243, 755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bray R. C.; Turner N. A.; Le Gall J.; Barata B. A. S.; Moura J. J. G. Biochem. J. 1991, 280, 817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moura J. J. G.; Xavier A. V.; Hatchikian E. C.; Le Gall J. FEBS Lett. 1978, 89, 177. [DOI] [PubMed] [Google Scholar]
- Moura I.; Pereira A. S.; Tavares P.; Moura J. J. G. Adv. Inorg. Chem. 1999, 47, 361. [Google Scholar]
- Barata B. A. S.; Liang J.; Moura I.; Legall J.; Moura J. J. G.; Huynh Boi H. Eur. J. Biochem. 1992, 204, 773. [DOI] [PubMed] [Google Scholar]
- Dobbek H.; Gremer L.; Meyer O.; Huber R. Proc. Natl. Acad. Sci. U.S.A. 1999, 96, 8884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer O.; Gremer L.; Ferner R.; Ferner M.; Dobbek H.; Gnida M.; Meyer-Klaucke W.; Huber R. Biol. Chem. 2000, 381, 865. [DOI] [PubMed] [Google Scholar]
- Ferry J. G. Annu. Rev. Microbiol. 1995, 49, 305. [DOI] [PubMed] [Google Scholar]
- Ragsdale S. W.; Kumar M. Chem. Rev. 1996, 96, 2515. [DOI] [PubMed] [Google Scholar]
- Ermler U.; Grabarse W.; Shima S.; Goubeaud M.; Thauer R. K. Curr. Opin. Struct. Biol. 1998, 8, 749. [DOI] [PubMed] [Google Scholar]
- Dobbek H.; Svetlitchnyi V.; Gremer L.; Huber R.; Meyer O. Science 2001, 293, 1281. [DOI] [PubMed] [Google Scholar]
- Drennan C. L.; Heo J.; Sintchak M. D.; Schreiter E.; Ludden P. W. Proc. Natl. Acad. Sci. U.S.A. 2001, 98, 11973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kung Y.; Drennan C. L. Curr. Opin. Chem. Biol. 2011, 15, 276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong W.; Hao B.; Wei Z.; Ferguson D. J. Jr.; Tallant T.; Krzycki J. A.; Chan M. K. Proc. Natl. Acad. Sci. U.S.A. 2008, 105, 9558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arendsen A. F.; Hadden J.; Card G.; McAlpine A. S.; Bailey S.; Zaitsev V.; Duke E. H. M.; Lindley P. F.; Krockel M.; Trautwein A. X.; Feiters M. C.; Charnock J. M.; Garner C. D.; Marritt S. J.; Thomson A. J.; Kooter I. M.; Johnson M. K.; Van Den Berg W. A. M.; Van Dongen W. M. A. M.; Hagen W. R. JBIC, J. Biol. Inorg. Chem. 1998, 3, 81. [Google Scholar]
- Cooper S. J.; Garner C. D.; Hagen W. R.; Lindley P. F.; Bailey S. Biochemistry 2000, 39, 15044. [DOI] [PubMed] [Google Scholar]
- Van den Berg W. A. M.; Hagen W. R.; Van Dongen W. M. A. M. Eur. J. Biochem. 2000, 267, 666. [DOI] [PubMed] [Google Scholar]
- Aragao D.; Mitchell E. P.; Frazao C. F.; Carrondo M. A.; Lindley P. F. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2008, 64, 665. [DOI] [PubMed] [Google Scholar]
- Aragao D.; Macedo S.; Mitchell E. P.; Romao C. V.; Liu M. Y.; Frazao C.; Saraiva L. M.; Xavier A. V.; LeGall J.; van Dongen W. M. A. M.; Hagen W. R.; Teixeira M.; Carrondo M. A.; Lindley P. JBIC, J. Biol. Inorg. Chem. 2003, 8, 540. [DOI] [PubMed] [Google Scholar]
- Kanatzidis M. G.; Hagen W. R.; Dunham W. R.; Lester R. K.; Coucouvanis D. K. J. Am. Chem. Soc. 1985, 107, 953. [Google Scholar]
- Stokkermans J. P. W. G.; Houba P. H. J.; Pierik A. J.; Hagen W. R.; Van Dongen W. M. A. M.; Veeger C. Eur. J. Biochem. 1992, 210, 983. [DOI] [PubMed] [Google Scholar]
- Arendsen A. F.; Lindley P. F. Adv. Inorg. Chem. 1999, 47, 219. [Google Scholar]
- Crane B. R.; Siegel L. M.; Getzoff E. D. Science 1995, 270, 59. [DOI] [PubMed] [Google Scholar]
- Crane B. R.; Getzoff E. D. Curr. Opin. Struct. Biol. 1996, 6, 744. [DOI] [PubMed] [Google Scholar]
- Nakayama M.; Akashi T.; Hase T. J. Inorg. Biochem. 2000, 82, 27. [DOI] [PubMed] [Google Scholar]
- Wessels J. G. H. Adv. Microb. Physiol. 1997, 38, 1. [DOI] [PubMed] [Google Scholar]
- Moreno-Vivian C.; Cabello P.; Martinez-Luque M.; Blasco R.; Castillo F. J. Bacteriol. 1999, 181, 6573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richardson D. J.; Berks B. C.; Russell D. A.; Spiro S.; Taylor C. J. Cell. Mol. Life Sci. 2001, 58, 165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirasawa M.; Tollin G.; Salamon Z.; Knaff D. B. Biochim. Biophys. Acta, Bioenerg. 1994, 1185, 336. [DOI] [PubMed] [Google Scholar]
- Swamy U.; Wang M.; Tripathy J. N.; Kim S.-K.; Hirasawa M.; Knaff D. B.; Allen J. P. Biochemistry 2005, 44, 16054. [DOI] [PubMed] [Google Scholar]
- Setif P.; Hirasawa M.; Cassan N.; Lagoutte B.; Tripathy J. N.; Knaff D. B. Biochemistry 2009, 48, 2828. [DOI] [PubMed] [Google Scholar]
- Peck H. D. Jr.; LeGall J. Philos. Trans. R. Soc., B 1982, 298, 443. [DOI] [PubMed] [Google Scholar]
- Stroupe M. E.; Getzoff E. D. Handb. Metalloproteins 2001, 1, 471. [Google Scholar]
- Kopriva S.; Koprivova A. J. Exp. Bot. 2004, 55, 1775. [DOI] [PubMed] [Google Scholar]
- Krueger R. J.; Siegel L. M. Biochemistry 1982, 21, 2892. [DOI] [PubMed] [Google Scholar]
- Janick P. A.; Rueger D. C.; Krueger R. J.; Barber M. J.; Siegel L. M. Biochemistry 1983, 22, 396. [DOI] [PubMed] [Google Scholar]
- Hirasawa M.; Nakayama M.; Hase T.; Knaff D. B. Biochim. Biophys. Acta, Bioenerg. 2004, 1608, 140. [DOI] [PubMed] [Google Scholar]
- Zeghouf M.; Fontecave M.; Coves J. J. Biol. Chem. 2000, 275, 37651. [DOI] [PubMed] [Google Scholar]
- Molitor M.; Dahl C.; Molitor I.; Schafer U.; Speich N.; Huber R.; Deutzmann R.; Truper H. G. Microbiology (Reading, U. K.) 1998, 144, 529. [DOI] [PubMed] [Google Scholar]
- Schiffer A.; Parey K.; Warkentin E.; Diederichs K.; Huber H.; Stetter K. O.; Kroneck P. M. H.; Ermler U. J. Mol. Biol. 2008, 379, 1063. [DOI] [PubMed] [Google Scholar]
- Oliveira T. F.; Vonrhein C.; Matias P. M.; Venceslau S. S.; Pereira I. A. C.; Archer M. J. Biol. Chem. 2008, 283, 34141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsieh Y.-C.; Liu M.-Y.; Wang V. C. C.; Chiang Y.-L.; Liu E.-H.; Wu W.-g.; Chan S. I.; Chen C.-J. Mol. Microbiol. 2010, 78, 1101. [DOI] [PubMed] [Google Scholar]
- Oliveira Tania F.; Franklin E.; Afonso Jose P.; Khan Amir R.; Oldham Neil J.; Pereira Ines A. C.; Archer M. Front. Microbiol. 2011, 2, 71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saraste M. Science 1999, 283, 1488. [DOI] [PubMed] [Google Scholar]
- Sun F.; Zhou Q.; Pang X.; Xu Y.; Rao Z. Curr. Opin. Struct. Biol. 2013, 23, 526. [DOI] [PubMed] [Google Scholar]
- Baradaran R.; Berrisford J. M.; Minhas G. S.; Sazanov L. A. Nature 2013, 494, 443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun F.; Huo X.; Zhai Y.; Wang A.; Xu J.; Su D.; Bartlam M.; Rao Z. Cell 2005, 121, 1043. [DOI] [PubMed] [Google Scholar]
- Yankovskaya V.; Horsefield R.; Toernroth S.; Luna-Chavez C.; Miyoshi H.; Leger C.; Byrne B.; Cecchini G.; Iwata S. Science 2003, 299, 700. [DOI] [PubMed] [Google Scholar]
- Iwata S.; Lee J. W.; Okada K.; Lee J. K.; Iwata M.; Rasmussen B.; Link T. A.; Ramaswamy S.; Jap B. K. Science 1998, 281, 64. [DOI] [PubMed] [Google Scholar]
- Lange C.; Hunte C. Proc. Natl. Acad. Sci. U.S.A. 2002, 99, 2800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia D.; Yu C.-A.; Kim H.; Xia J.-Z.; Kachurin A. M.; Zhang L.; Yu L.; Deisenhofer J. Science 1997, 277, 60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z.; Huang L.; Shulmeister V. M.; Chi Y.-I.; Kim K. K.; Hung L.-W.; Crofts A. R.; Berry E. A.; Kim S.-H. Nature 1998, 392, 677. [DOI] [PubMed] [Google Scholar]
- Tsukihara T.; Aoyama H.; Yamashita E.; Tomizaki T.; Yamaguchi H.; Shinzawa-Itoh K.; Nakashima R.; Yaono R.; Yoshikawa S. Science 1995, 269, 1069. [DOI] [PubMed] [Google Scholar]
- Tsukihara T.; Aoyama H.; Yamashita E.; Tomizaki T.; Yamaguchi H.; Shinzawa-Itoh K.; Nakashima R.; Yaono R.; Yoshikawa S. Science 1996, 272, 1136. [DOI] [PubMed] [Google Scholar]
- Abrahams J. P.; Leslie A. G. W.; Lutter R.; Walker J. E. Nature 1994, 370, 621. [DOI] [PubMed] [Google Scholar]
- Sazanov L. A.; Hinchliffe P. Science 2006, 311, 1430. [DOI] [PubMed] [Google Scholar]
- Efremov R. G.; Baradaran R.; Sazanov L. A. Nature 2010, 465, 441. [DOI] [PubMed] [Google Scholar]
- Efremov R. G.; Sazanov L. A. Nature 2011, 476, 414. [DOI] [PubMed] [Google Scholar]
- Hunte C.; Zickermann V.; Brandt U. Science 2010, 329, 448. [DOI] [PubMed] [Google Scholar]
- Carroll J.; Fearnley I. M.; Skehel J. M.; Shannon R. J.; Hirst J.; Walker J. E. J. Biol. Chem. 2006, 281, 32724. [DOI] [PubMed] [Google Scholar]
- Sazanov L. A.; Carroll J.; Holt P.; Toime L.; Fearnley I. M. J. Biol. Chem. 2003, 278, 19483. [DOI] [PubMed] [Google Scholar]
- Yagi T.; Matsuno-Yagi A. Biochemistry 2003, 42, 2266. [DOI] [PubMed] [Google Scholar]
- Sazanov L. A. Biochemistry 2007, 46, 2275. [DOI] [PubMed] [Google Scholar]
- Huang L.-s.; Sun G.; Cobessi D.; Wang A. C.; Shen J. T.; Tung E. Y.; Anderson V. E.; Berry E. A. J. Biol. Chem. 2006, 281, 5965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng V. W. T.; Ma E.; Zhao Z.; Rothery R. A.; Weiner J. H. J. Biol. Chem. 2006, 281, 27662. [DOI] [PubMed] [Google Scholar]
- Cecchini G.; Sices H.; Schroeder I.; Gunsalus R. P. J. Bacteriol. 1995, 177, 4587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haegerhaell C.; Sled V.; Hederstedt L.; Ohnishi T. Biochim. Biophys. Acta, Bioenerg. 1995, 1229, 356. [DOI] [PubMed] [Google Scholar]
- Manodori A.; Cecchini G.; Schroder I.; Gunsalus R. P.; Werth M. T.; Johnson M. K. Biochemistry 1992, 31, 2703. [DOI] [PubMed] [Google Scholar]
- Haegerhaell C. Biochim. Biophys. Acta, Bioenerg. 1997, 1320, 107. [DOI] [PubMed] [Google Scholar]
- Janssen S.; Schafer G.; Anemuller S.; Moll R. J. Bacteriol. 1997, 179, 5560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu L.; Xu J. X.; Haley P. E.; Yu C. A. J. Biol. Chem. 1987, 262, 1137. [PubMed] [Google Scholar]
- Iverson T. M.; Luna-Chavez C.; Cecchini G.; Rees D. C. Science 1999, 284, 1961. [DOI] [PubMed] [Google Scholar]
- Lancaster C. R. D.; Kroger A.; Auer M.; Michel H. Nature 1999, 402, 377. [DOI] [PubMed] [Google Scholar]
- Nanda V.; Rosenblatt M. M.; Osyczka A.; Kono H.; Getahun Z.; Dutton P. L.; Saven J. G.; DeGrado W. F. J. Am. Chem. Soc. 2005, 127, 5804. [DOI] [PubMed] [Google Scholar]
- Ogino H.; Inomata S.; Tobita H. Chem. Rev. 1998, 98, 2093. [DOI] [PubMed] [Google Scholar]
- Rao P. V.; Holm R. H. Chem. Rev. 2004, 104, 527. [DOI] [PubMed] [Google Scholar]
- Saouma C. T.; Kaminsky W.; Mayer J. M. J. Am. Chem. Soc. 2012, 134, 7293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mouesca J.-M.; Lamotte B. Coord. Chem. Rev. 1998, 178–180, 1573. [Google Scholar]
- Anand B. N. Resonance 1998, 3, 52. [Google Scholar]
- Lee S. C.; Wayne L.; Holm R. H.. Chem. Rev. 2014, 114 (7), DOI: 10.1021/cr4004067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holm R. H. Compr. Coord. Chem. II 2004, 8, 61. [Google Scholar]
- Gibney B. R. Chemtracts 2003, 16, 263. [Google Scholar]
- Kennedy M. L.; Gibney B. R. J. Am. Chem. Soc. 2002, 124, 6826. [DOI] [PubMed] [Google Scholar]
- Gibney B. R.; Mulholland S. E.; Rabanal F.; Dutton P. L. Proc. Natl. Acad. Sci. U.S.A. 1996, 93, 15041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grzyb J.; Xu F.; Nanda V.; Luczkowska R.; Reijerse E.; Lubitz W.; Noy D. Biochim. Biophys. Acta, Bioenerg. 2012, 1817, 1256. [DOI] [PubMed] [Google Scholar]
- Grzyb J.; Xu F.; Weiner L.; Reijerse E. J.; Lubitz W.; Nanda V.; Noy D. Biochim. Biophys. Acta, Bioenerg. 2010, 1797, 406. [DOI] [PubMed] [Google Scholar]
- Ryle M. J.; Lanzilotta W. N.; Seefeldt L. C.; Scarrow R. C.; Jensen G. M. J. Biol. Chem. 1996, 271, 1551. [DOI] [PubMed] [Google Scholar]
- Anderson G. L.; Howard J. B. Biochemistry 1984, 23, 2118. [DOI] [PubMed] [Google Scholar]
- Fu W. G.; Morgan T. V.; Mortenson L. E.; Johnson M. K. FEBS Lett. 1991, 284, 165. [DOI] [PubMed] [Google Scholar]
- Mulliez E.; Ollagnier-de Choudens S.; Meier C.; Cremonini M.; Luchinat C.; Trautwein A. X.; Fontecave M. JBIC, J. Biol. Inorg. Chem. 1999, 4, 614. [DOI] [PubMed] [Google Scholar]
- Yang J.; Naik S. G.; Ortillo D. O.; Garcia-Serres R.; Li M.; Broderick W. E.; Huynh B. H.; Broderick J. B. Biochemistry 2009, 48, 9234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang B.; Crack J. C.; Subramanian S.; Green J.; Thomson A. J.; Le B.; Nick E.; Johnson M. K. Proc. Natl. Acad. Sci. U.S.A. 2012, 109, 15734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicolet Y.; Rohac R.; Martin L.; Fontecilla-Camps J. C. Proc. Natl. Acad. Sci. U.S.A. 2013, 110, 7188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crack J. C.; Green J.; Le Brun N. E.; Thomson A. J. J. Biol. Chem. 2006, 281, 18909. [DOI] [PubMed] [Google Scholar]
- Khoroshilova N.; Popescu C.; Munck E.; Beinert H.; Kiley P. J. Proc. Natl. Acad. Sci. U.S.A. 1997, 94, 6087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jervis A. J.; Crack J. C.; White G.; Artymiuk P. J.; Cheesman M. R.; Thomson A. J.; Le Brun N. E.; Green J. Proc. Natl. Acad. Sci. U.S.A. 2009, 106, 4659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crack J. C.; Gaskell A. A.; Green J.; Cheesman M. R.; Le Brun N. E.; Thomson A. J. J. Am. Chem. Soc. 2008, 130, 1749. [DOI] [PubMed] [Google Scholar]
- Kent T. A.; Emptage M. H.; Merkle H.; Kennedy M. C.; Beinert H.; Muenck E. J. Biol. Chem. 1985, 260, 6871. [PubMed] [Google Scholar]
- Kent T. A.; Dreyer J. L.; Kennedy M. C.; Boi H. H.; Emptage M. H.; Beinert H.; Muenck E. Proc. Natl. Acad. Sci. U.S.A. 1982, 79, 1096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kennedy M. C.; Kent T. A.; Emptage M.; Merkle H.; Beinert H.; Munck E. J. Biol. Chem. 1984, 259, 14463. [PubMed] [Google Scholar]
- Guergova-Kuras M.; Kuras R.; Ugulava N.; Hadad I.; Crofts A. R. Biochemistry 2000, 39, 7436. [DOI] [PubMed] [Google Scholar]
- Moura J. J. G.; Moura I.; Kent T. A.; Lipscomb J. D.; Huynh B. H.; LeGall J.; Xavier A. V.; Munck E. J. Biol. Chem. 1982, 257, 6259. [PubMed] [Google Scholar]
- Krebs C.; Henshaw T. F.; Cheek J.; Huynh B. H.; Broderick J. B. J. Am. Chem. Soc. 2000, 122, 12497. [Google Scholar]
- Bingemann R.; Klein A. Eur. J. Biochem. 2000, 267, 6612. [DOI] [PubMed] [Google Scholar]
- Hannan J. P.; Busch J. L. H.; James R.; Thomson A. J.; Moore G. R.; Davy S. L. FEBS Lett. 2000, 468, 161. [DOI] [PubMed] [Google Scholar]
- Finnegan M. G.; Conover R. C.; Park J.-B.; Zhou Z. H.; Adams M. W. W.; Johnson M. K. Inorg. Chem. 1995, 34, 5358. [Google Scholar]
- Butt J. N.; Sucheta A.; Armstrong F. A.; Breton J.; Thomson A. J.; Hatchikian E. C. J. Am. Chem. Soc. 1991, 113, 8948. [Google Scholar]
- Duee E. D.; Fanchon E.; Vicat J.; Sieker L. C.; Meyer J.; Moulis J.-M. J. Mol. Biol. 1994, 243, 683. [DOI] [PubMed] [Google Scholar]
- Stout C. D.; Stura E. A.; Mcree D. E. J. Mol. Biol. 1998, 278, 629. [DOI] [PubMed] [Google Scholar]
- Cheng H.; Xia B.; Reed G. H.; Markley J. L. Biochemistry 1994, 33, 3155. [DOI] [PubMed] [Google Scholar]
- Marshall N. M.; Garner D. K.; Wilson T. D.; Gao Y.-G.; Robinson H.; Nilges M. J.; Lu Y. Nature 2009, 462, 113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shoji M.; Isobe H.; Takano Y.; Kitagawa Y.; Yamanaka S.; Okumura M.; Yamaguchi K. J. Quantum Chem. 2007, 107, 3250. [Google Scholar]
- Underwood E. J.Trace Elements in Human and Animal Nutrition, 4th ed.; Academic Press: New York, 1977. [Google Scholar]
- Williams R. J. P. In Proceedings of the 5th International Congress on Biochemistry; Desnuelle P. A. E., Ed.; Pergamon Press: Oxford, U.K., 1963; Vol. IV, p 133. [Google Scholar]
- Williams R. J. P. Inorg. Chim. Acta, Rev. 1971, 5, 137. [Google Scholar]
- Malmstrom B. G.; Vanngard T. J. Mol. Biol. 1960, 2, 118. [DOI] [PubMed] [Google Scholar]
- Solomon E. I.; Hare J. W.; Gray H. B. Proc. Natl. Acad. Sci. U.S.A. 1976, 73, 1389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solomon E. I. Inorg. Chem. 2006, 45, 8012. [DOI] [PubMed] [Google Scholar]
- Colman P. M.; Freeman H. C.; Guss J. M.; Murata M.; Norris V. A.; Ramshaw J. A. M.; Venkatappa M. P. Nature 1978, 272, 319. [Google Scholar]
- Beinert H. Eur. J. Biochem. 1997, 245, 521. [DOI] [PubMed] [Google Scholar]
- Beinert H.; Griffiths D. E.; Wharton D. C.; Sands R. H. J. Biol. Chem. 1962, 237, 2337. [PubMed] [Google Scholar]
- Beinert H.; Palmer G. J. Biol. Chem. 1964, 239, 1221. [PubMed] [Google Scholar]
- Beinert H.; Palmer G. Adv. Enzymol. Relat. Areas Mol. Biol. 1965, 27, 105. [DOI] [PubMed] [Google Scholar]
- Greenwood C.; Hill B. C.; Barber D.; Eglinton D. G.; Thomson A. J. Biochem. J. 1983, 215, 303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomson A. J.; Greenwood C.; Peterson J.; Barrett C. P. J. Inorg. Biochem. 1986, 28, 195. [DOI] [PubMed] [Google Scholar]
- Gibson Q. H.; Greenwood C. Biochem. J. 1963, 86, 541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibson Q. H.; Greenwood C. J. Biol. Chem. 1965, 240, 2694. [PubMed] [Google Scholar]
- Buse G.; Steffens G. C. J. Bioenerg. Biomembr. 1991, 23, 269. [DOI] [PubMed] [Google Scholar]
- Steffens G. C.; Soulimane T.; Wolff G.; Buse G. Eur. J. Biochem. 1993, 213, 1149. [DOI] [PubMed] [Google Scholar]
- Andrew C. R.; Fraczkiewicz R.; Czernuszewicz R. S.; Lappalainen P.; Saraste M.; Sanders-Loehr J. J. Am. Chem. Soc. 1996, 118, 10436. [Google Scholar]
- Blackburn N. J.; Barr M. E.; Woodruff W. H.; van der Ooost J.; de Vries S. Biochemistry 1994, 33, 10401. [DOI] [PubMed] [Google Scholar]
- Iwata S.; Ostermeier C.; Ludwig B.; Michel H. Nature 1995, 376, 660. [DOI] [PubMed] [Google Scholar]
- Malkin R.; Malmstrom B. G. Adv. Enzymol. Relat. Subj. Biochem. 1970, 33, 177. [DOI] [PubMed] [Google Scholar]
- Fee J. A. Struct. Bonding (Berlin, Ger.) 1975, 33, 1. [Google Scholar]
- Neese F.; Zumft W. G.; Antholine W. E.; Kroneck P. M. H. J. Am. Chem. Soc. 1996, 118, 8692. [Google Scholar]
- Gamelin D. R.; Randall D. W.; Hay M. T.; Houser R. P.; Mulder T. C.; Canters G. W.; de V. S.; Tolman W. B.; Lu Y.; Solomon E. I. J. Am. Chem. Soc. 1998, 120, 5246. [Google Scholar]
- Farrar J. A.; Neese F.; Lappalainen P.; Kroneck P. M. H.; Saraste M.; Zumft W. G.; Thomson A. J. J. Am. Chem. Soc. 1996, 118, 11501. [Google Scholar]
- Olsson M. H.; Ryde U. J. Am. Chem. Soc. 2001, 123, 7866. [DOI] [PubMed] [Google Scholar]
- Adman E. In Metalloproteins; Macmillan: London, 1985; Vol. 1, p 1. [Google Scholar]
- Prigge S. T.; Kolhekar A. S.; Eipper B. A.; Mains R. E.; Amzel L. M. Science 1997, 278, 1300. [DOI] [PubMed] [Google Scholar]
- Gray H. B.; Solomon E. I. In Copper Proteins; Spiro T. G., Ed.; Wiley: New York, 1981; Vol. 3, p 1. [Google Scholar]
- Hart P. J.; Nersissian A. M.; George S. D. In Encyclopedia of Inorganic Chemistry; John Wiley & Sons, Ltd.: Hoboken, NJ, 2006. [Google Scholar]
- Smeekens S.; De G. M.; Van B. J.; Weisbeek P. Nature 1985, 317, 456. [Google Scholar]
- Sutherland I. W.; Wilkinson J. F. J. Gen. Microbiol. 1963, 30, 105. [DOI] [PubMed] [Google Scholar]
- Pascher T.; Karlsson B. G.; Nordling M.; Malmstroem B. G.; Vaenngaard T. Eur. J. Biochem. 1993, 212, 289. [DOI] [PubMed] [Google Scholar]
- Tobari J.; Harada Y. Biochem. Biophys. Res. Commun. 1981, 101, 502. [DOI] [PubMed] [Google Scholar]
- van Houwelingen T.; Canters G. W.; Stobbelaar G.; Duine J. A.; Frank J. Jr.; Tsugita A. Eur. J. Biochem. 1985, 153, 75. [DOI] [PubMed] [Google Scholar]
- Katoh S. Nature 1960, 186, 533. [PubMed] [Google Scholar]
- Katoh S.; Shiratori I.; Takamiya A. J. Biochem. 1962, 51, 32. [DOI] [PubMed] [Google Scholar]
- Iwasaki H.; Matsubara T. J. Biochem. 1973, 73, 659. [DOI] [PubMed] [Google Scholar]
- Sakurai T.; Ikeda O.; Suzuki S. Inorg. Chem. 1990, 29, 4715. [Google Scholar]
- Cobley J. G.; Haddock B. A. FEBS Lett. 1975, 60, 29. [DOI] [PubMed] [Google Scholar]
- Lappin A. G.; Lewis C. A.; Ingledew W. J. Inorg. Chem. 1985, 24, 1446. [Google Scholar]
- McManus J. D.; Brune D. C.; Han J.; Sanders-Loehr J.; Meyer T. E.; Cusanovich M. A.; Tollin G.; Blankenship R. E. J. Biol. Chem. 1992, 267, 6531. [PubMed] [Google Scholar]
- Markossian K. A.; Aikazyan V. T.; Nalbandyan R. M. Biochim. Biophys. Acta, Protein Struct. 1974, 359, 47. [DOI] [PubMed] [Google Scholar]
- Battistuzzi G.; Borsari M.; Loschi L.; Righi F.; Sola M. J. Am. Chem. Soc. 1999, 121, 501. [Google Scholar]
- Scharf B.; Engelhard M. Biochemistry 1993, 32, 12894. [DOI] [PubMed] [Google Scholar]
- Komorowski L.; Schafer G. FEBS Lett. 2001, 487, 351. [DOI] [PubMed] [Google Scholar]
- Arciero D. M.; Hooper A. B.; Rosenzweig A. C. Biochemistry 2001, 40, 5674. [DOI] [PubMed] [Google Scholar]
- Arciero D. M.; Pierce B. S.; Hendrich M. P.; Hooper A. B. Biochemistry 2002, 41, 1703. [DOI] [PubMed] [Google Scholar]
- Peisach J.; Levine W. G.; Blumberg W. E. J. Biol. Chem. 1967, 242, 2847. [PubMed] [Google Scholar]
- Nersissian A. M.; Immoos C.; Hill M. G.; Hart P. J.; Williams G.; Herrmann R. G.; Valentine J. S. Protein Sci. 1998, 7, 1915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nersissian A. M.; Valentine J. S. Unpublished results.
- Malmstrom B. G.; Reinhammar B.; Vanngard T. Biochim. Biophys. Acta, Gen. Subj. 1968, 156, 67. [DOI] [PubMed] [Google Scholar]
- Xu F.; Shin W.; Brown S. H.; Wahleithner J. A.; Sundaram U. M.; Solomon E. I. Biochim. Biophys. Acta, Protein Struct. Mol. Enzymol. 1996, 1292, 303. [DOI] [PubMed] [Google Scholar]
- Ducros V.; Brzozowski A. M.; Wilson K. S.; Brown S. H.; Ostergaard P.; Schneider P.; Yaver D. S.; Pedersen A. H.; Davies G. J. Nat. Struct. Biol. 1998, 5, 310. [DOI] [PubMed] [Google Scholar]
- Reinhammar B. R. M. Biochim. Biophys. Acta, Bioenerg. 1972, 275, 245. [DOI] [PubMed] [Google Scholar]
- Reinhammar B. R.; Vanngard T. I. Eur. J. Biochem. 1971, 18, 463. [DOI] [PubMed] [Google Scholar]
- Kroneck Peter M H.; Armstrong Fraser A.; Merkle H.; Marchesini A.. Ascorbic Acid: Chemistry, Metabolism, and Uses; American Chemical Society: Washington, DC, 1982; Vol. 200, p 223. [Google Scholar]
- Holmberg C. G.; Laurell C. B. Acta Chem. Scand. 1948, 2, 550. [DOI] [PubMed] [Google Scholar]
- Machonkin T. E.; Zhang H. H.; Hedman B.; Hodgson K. O.; Solomon E. I. Biochemistry 1998, 37, 9570. [DOI] [PubMed] [Google Scholar]
- Machonkin T. E.; Solomon E. I. J. Am. Chem. Soc. 2000, 122, 12547. [Google Scholar]
- Vulpe C. D.; Kuo Y.-M.; Murphy T. L.; Cowley L.; Askwith C.; Libina N.; Gitschier J.; Anderson G. J. Nat. Genet. 1999, 21, 195. [DOI] [PubMed] [Google Scholar]
- Askwith C.; Eide D.; Van Ho A.; Bernard P. S.; Li L.; Davis-Kaplan S.; Sipe D. M.; Kaplan J. Cell 1994, 76, 403. [DOI] [PubMed] [Google Scholar]
- Machonkin T. E.; Quintanar L.; Palmer A. E.; Hassett R.; Severance S.; Kosman D. J.; Solomon E. I. J. Am. Chem. Soc. 2001, 123, 5507. [DOI] [PubMed] [Google Scholar]
- Masuko M.; Iwasaki H.; Sakurai T.; Suzuki S.; Nakahara A. J. Biochem. 1984, 96, 447. [DOI] [PubMed] [Google Scholar]
- De Rienzo F.; Gabdoulline R. R.; Menziani M. C.; Wade R. C. Protein Sci. 2000, 9, 1439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wittung P.; Källebring B.; Malmström B. G. FEBS Lett. 1994, 349, 286. [DOI] [PubMed] [Google Scholar]
- Valentine J. S.; Doucette P. A.; Zittin Potter S. Annu. Rev. Biochem. 2005, 74, 563. [DOI] [PubMed] [Google Scholar]
- Stevens F. J. J. Mol. Recognit. 2008, 21, 20. [DOI] [PubMed] [Google Scholar]
- Hasnain S. S.; Hodgson K. O. J. Synchrotron Radiat. 1999, 6, 852. [Google Scholar]
- Tullius T. D.; Frank P.; Hodgson K. O. Proc. Natl. Acad. Sci. U.S.A. 1978, 75, 4069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nar H.; Messerschmidt A.; Huber R.; Vandekamp M.; Canters G. W. J. Mol. Biol. 1991, 221, 765. [DOI] [PubMed] [Google Scholar]
- Walter R. L.; Ealick S. E.; Friedman A. M.; Blake R. C.; Proctor P.; Shoham M. J. Mol. Biol. 1996, 263, 730. [DOI] [PubMed] [Google Scholar]
- Guss J. M.; Bartunik H. D.; Freeman H. C. Acta Crystallogr., Sect. B: Struct. Sci. 1992, 48, 790. [DOI] [PubMed] [Google Scholar]
- Guss J. M.; Harrowell P. R.; Murata M.; Norris V. A.; Freeman H. C. J. Mol. Biol. 1986, 192, 361. [DOI] [PubMed] [Google Scholar]
- Cunane L. M.; Chen Z. W.; Durley R. C. E.; Mathews F. S. Acta Crystallogr., Sect. D: Biol. Crystallogr. 1996, 52, 676. [DOI] [PubMed] [Google Scholar]
- Zhu Z. Y.; Cunane L. M.; Chen Z. W.; Durley R. C. E.; Mathews F. S.; Davidson V. L. Biochemistry 1998, 37, 17128. [DOI] [PubMed] [Google Scholar]
- Guss J. M.; Merritt E. A.; Phizackerley R. P.; Freeman H. C. J. Mol. Biol. 1996, 262, 686. [DOI] [PubMed] [Google Scholar]
- Hart P. J.; Nersissian A. M.; Herrmann R. G.; Nalbandyan R. M.; Valentine J. S.; Eisenberg D. Protein Sci. 1996, 5, 2175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malmström B. G. In Oxidases and Related Redox Systems; King T. E., Mason H. S., Morrison M., Eds.; Wiley: New York, 1965; p 207. [Google Scholar]
- Gray H. B.; Malmstroem B. G. Comments Inorg. Chem. 1983, 2, 203. [Google Scholar]
- Vallee B. L.; Williams R. J. Proc. Natl. Acad. Sci. U.S.A. 1968, 59, 498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penner-Hahn J. E.; Murata M.; Hodgson K. O.; Freeman H. C. Inorg. Chem. 1989, 28, 1826. [Google Scholar]
- Carugo O.; Carugo K. D. Trends Biochem. Sci. 2005, 30, 213. [DOI] [PubMed] [Google Scholar]
- Orville A. M.; Lountos G. T.; Finnegan S.; Gadda G.; Prabhakar R. Biochemistry 2009, 48, 720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nar H.; Messerschmidt A.; Huber R.; Van d. K. M.; Canters G. W. FEBS Lett. 1992, 306, 119. [DOI] [PubMed] [Google Scholar]
- Shepard W. E. B.; Kingston R. L.; Anderson B. F.; Baker E. N. Acta Crystallogr., Sect. D: Biol. Crystallogr. 1993, D49, 331. [DOI] [PubMed] [Google Scholar]
- Garrett T. P. J.; Clingeleffer D. J.; Guss J. M.; Rogers S. J.; Freeman H. C. J. Biol. Chem. 1984, 259, 2822. [DOI] [PubMed] [Google Scholar]
- Petratos K.; Papadovasilaki M.; Dauter Z. FEBS Lett. 1995, 368, 432. [DOI] [PubMed] [Google Scholar]
- Durley R.; Chen L.; Scott Mathews F.; Lim L. W.; Davidson V. L. Protein Sci. 1993, 2, 739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMillin D. R.; Holwerda R. A.; Gray H. B. Proc. Nat. Acad. Sci. U.S.A. 1974, 71, 1339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engeseth H. R.; McMillin D. R.; Otvos J. D. J. Biol. Chem. 1984, 259, 4822. [PubMed] [Google Scholar]
- Church W. B.; Guss J. M.; Potter J. J.; Freeman H. C. J. Biol. Chem. 1986, 261, 234. [DOI] [PubMed] [Google Scholar]
- Baker E. N.; Anderson B. F.; Blackwell K. E.; Kingston R. L.; Norris G. E.; Shepard W. E. B. J. Inorg. Biochem. 1991, 43, 162. [Google Scholar]
- Nar H.; Huber R.; Messerschmidt A.; Filippou A. C.; Barth M.; Jaquinod M.; Van d. K. M.; Canters G. W. Eur. J. Biochem. 1992, 205, 1123. [DOI] [PubMed] [Google Scholar]
- Gewirth A. A.; Solomon E. I. J. Am. Chem. Soc. 1988, 110, 3811. [Google Scholar]
- Solomon E. I.; Penfield K. W.; Gewirth A. A.; Lowery M. D.; Shadle S. E.; Guckert J. A.; LaCroix L. B. Inorg. Chim. Acta 1996, 243, 67. [Google Scholar]
- LaCroix L. B.; Randall D. W.; Nersissian A. M.; Hoitink C. W. G.; Canters G. W.; Valentine J. S.; Solomon E. I. J. Am. Chem. Soc. 1998, 120, 9621. [Google Scholar]
- Shadle S. E.; Penner-Hahn J. E.; Schugar H. J.; Hedman B.; Hodgson K. O.; Solomon E. I. J. Am. Chem. Soc. 1993, 115, 767. [Google Scholar]
- Wood P. M.; Bendall D. S. Biochim. Biophys. Acta, Bioenerg. 1975, 387, 115. [DOI] [PubMed] [Google Scholar]
- Haehnel W. Annu. Rev. Plant Physiol. 1984, 35, 659. [DOI] [PubMed] [Google Scholar]
- Hervas M.; Navarro J. A.; Diaz A.; Bottin H.; De la Rosa M. A. Biochemistry 1995, 34, 11321. [DOI] [PubMed] [Google Scholar]
- Sigfridsson K. Photosynth. Res. 1998, 57, 1. [DOI] [PubMed] [Google Scholar]
- Hope A. B. Biochim. Biophys. Acta, Bioenerg. 2000, 1456, 5. [DOI] [PubMed] [Google Scholar]
- Tobari J.; Harada Y. Biochem. Biophys. Res. Commun. 1981, 101, 502. [DOI] [PubMed] [Google Scholar]
- Gray K. A.; Davidson V. L.; Knaff D. B. J. Biol. Chem. 1988, 263, 13987. [PubMed] [Google Scholar]
- van Spanning R. J.; Wansell C. W.; Reijnders W. N.; Oltmann L. F.; Stouthamer A. H. FEBS Lett. 1990, 275, 217. [DOI] [PubMed] [Google Scholar]
- Chen L.; Durley R.; Poliks B. J.; Hamada K.; Chen Z.; Mathews F. S.; Davidson V. L.; Satow Y.; Huizinga E. Biochemistry 1992, 31, 4959. [DOI] [PubMed] [Google Scholar]
- Brooks H. B.; Davidson V. L. J. Am. Chem. Soc. 1994, 116, 11201. [Google Scholar]
- Dennison C.; Canters G. W.; Vries S. D.; Vijgenboom E.; Spanning R. J. V. In Advances in Inorganic Chemistry; Sykes A. G., Ed.; Academic Press: New York, 1998; Vol. Vol. 45, p 351. [Google Scholar]
- Davidson V. L.; Sun D. P. J. Am. Chem. Soc. 2003, 125, 3224. [DOI] [PubMed] [Google Scholar]
- Meschi F.; Wiertz F.; Klauss L.; Cavalieri C.; Blok A.; Ludwig B.; Heering H. A.; Merli A.; Rossi G. L.; Ubbink M. J. Am. Chem. Soc. 2010, 132, 14537. [DOI] [PubMed] [Google Scholar]
- Giudici-Orticoni M.-T.; Guerlesquin F.; Bruschi M.; Nitschke W. J. Biol. Chem. 1999, 274, 30365. [DOI] [PubMed] [Google Scholar]
- Appia-Ayme C.; Guiliani N.; Ratouchniak J.; Bonnefoy V. Appl. Environ. Microbiol. 1999, 65, 4781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kakutani T.; Watanabe H.; Arima K.; Beppu T. J. Biochem. 1981, 89, 463. [DOI] [PubMed] [Google Scholar]
- Kohzuma T.; Takase S.; Shidara S.; Suzuki S. Chem. Lett. 1993, 149. [Google Scholar]
- Moir J. W. B.; Baratta D.; Richardson D. J.; Ferguson S. J. Eur. J. Biochem. 1993, 212, 377. [DOI] [PubMed] [Google Scholar]
- Kukimoto M.; Nishiyama M.; Ohnuki T.; Turley S.; Adman E. T.; Horinouchi S.; Beppu T. Protein Eng. 1995, 8, 153. [DOI] [PubMed] [Google Scholar]
- Kukimoto M.; Nishiyama M.; Tanokura M.; Adman E. T.; Horinouchi S. J. Biol. Chem. 1996, 271, 13680. [DOI] [PubMed] [Google Scholar]
- Sukumar N.; Chen Z.-w.; Ferrari D.; Merli A.; Rossi G. L.; Bellamy H. D.; Chistoserdov A.; Davidson V. L.; Mathews F. S. Biochemistry 2006, 45, 13500. [DOI] [PubMed] [Google Scholar]
- Edwards S. L.; Davidson V. L.; Hyun Y.-L.; Wingfield P. T. J. Biol. Chem. 1995, 270, 4293. [DOI] [PubMed] [Google Scholar]
- Hyun Y.-L.; Davidson V. L. Biochemistry 1995, 34, 12249. [DOI] [PubMed] [Google Scholar]
- Ubbink M.; Ejdebäck M.; Karlsson B. G.; Bendall D. S. Structure (London, U. K.) 1998, 6, 323. [DOI] [PubMed] [Google Scholar]
- Crowley P. B.; Otting G.; Schlarb-Ridley B. G.; Canters G. W.; Ubbink M. J. Am. Chem. Soc. 2001, 123, 10444. [DOI] [PubMed] [Google Scholar]
- Sykes A. G.Long-Range Electron Transfer in Biology; Springer: Berlin, Heidelberg, 1991; Vol. 75, p 175. [Google Scholar]
- He S.; Modi S.; Bendall D. S.; Gray J. C. EMBO J. 1991, 10, 4011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlarb-Ridley B. G.; Bendall D. S.; Howe C. J. Biochemistry 2003, 42, 4057. [DOI] [PubMed] [Google Scholar]
- Sato K.; Kohzuma T.; Dennison C. J. Am. Chem. Soc. 2004, 126, 3028. [DOI] [PubMed] [Google Scholar]
- Crowley P. B.; Hunter D. M.; Sato K.; McFarlane W.; Dennison C. Biochem. J. 2004, 378, 45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takayama S. J.; Irie K.; Tai H. L.; Kawahara T.; Hirota S.; Takabe T.; Alcaraz L. A.; Donaire A.; Yamamoto Y. JBIC, J. Biol. Inorg. Chem. 2009, 14, 821. [DOI] [PubMed] [Google Scholar]
- Sykes A. G. Chem. Soc. Rev. 1985, 14, 283. [Google Scholar]
- Winkler J. R.; Gray H. B. Chem. Rev. 2014, 10.1021/cr4004715. [DOI] [Google Scholar]
- Newton M. D. Chem. Rev. 1991, 91, 767. [Google Scholar]
- Lowery M. D.; Guckert J. A.; Gebhard M. S.; Solomon E. I. J. Am. Chem. Soc. 1993, 115, 3012. [Google Scholar]
- Regan J. J.; Di Bilio A. J.; Langen R.; Skov L. K.; Winkler J. R.; Gray H. B.; Onuchic J. N. Chem. Biol. 1995, 2, 489. [DOI] [PubMed] [Google Scholar]
- Holwerda R. A.; Wherland S.; Gray H. B. Annu. Rev. Biophys. Bioeng. 1976, 5, 363. [DOI] [PubMed] [Google Scholar]
- Suzuki S.; Kataoka K.; Yamaguchi K.; Inoue T.; Kai Y. Coord. Chem. Rev. 1999, 190–192, 245. [Google Scholar]
- Rorabacher D. B. Chem. Rev. 2004, 104, 651. [DOI] [PubMed] [Google Scholar]
- Gray H. B.; Winkler J. R. Biochim. Biophys. Acta, Bioenerg. 2010, 1797, 1563. [DOI] [PubMed] [Google Scholar]
- Modi S.; He S.; Gray J. C.; Bendall D. S. Biochim. Biophys. Acta, Bioenerg. 1992, 1101, 64. [Google Scholar]
- Meyer T. E.; Zhao Z. G.; Cusanovich M. A.; Tollin G. Biochemistry 1993, 32, 4552. [DOI] [PubMed] [Google Scholar]
- Kannt A.; Young S.; Bendall D. S. Biochim. Biophys. Acta, Bioenerg. 1996, 1277, 115. [PubMed] [Google Scholar]
- Nordling M.; Sigfridsson K.; Young S.; Lundberg L. G.; Hansson Ö. FEBS Lett. 1991, 291, 327. [DOI] [PubMed] [Google Scholar]
- Haehnel W.; Jansen T.; Gause K.; Klosgen R. B.; Stahl B.; Michl D.; Huvermann B.; Karas M.; Herrmann R. G. EMBO J. 1994, 13, 1028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brooks H. B.; Davidson V. L. Biochemistry 1994, 33, 5696. [DOI] [PubMed] [Google Scholar]
- Davidson V. L.; Jones L. H. Biochemistry 1996, 35, 8120. [DOI] [PubMed] [Google Scholar]
- Kataoka K.; Kondo A.; Yamaguchi K.; Suzuki S. J. Inorg. Biochem. 2000, 82, 79. [DOI] [PubMed] [Google Scholar]
- Kataoka K.; Yamaguchi K.; Sakai S.; Takagi K.; Suzuki S. Biochem. Biophys. Res. Commun. 2003, 303, 519. [DOI] [PubMed] [Google Scholar]
- Kataoka K.; Yamaguchi K.; Kobayashi M.; Mori T.; Bokui N.; Suzuki S. J. Biol. Chem. 2004, 279, 53374. [DOI] [PubMed] [Google Scholar]
- Davidson V. L. Acc. Chem. Res. 1999, 33, 87. [DOI] [PubMed] [Google Scholar]
- Adams G. E.; Fielden E. M.; Michael B. D.. Fast Processes in Radiation Chemistry and Biology; John Wiley & Sons, Inc.: New York, 1975. [Google Scholar]
- Farver O.; Pecht I. Proc. Natl. Acad. Sci. U.S.A. 1989, 86, 6968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcus R. A.; Sutin N. Biochim. Biophys. Acta, Rev. Bioenerg. 1985, 811, 265. [Google Scholar]
- Dahlin S.; Reinhammar B.; Wilson M. T. Biochem. J. 1984, 218, 609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armstrong F. A.; Driscoll P. C.; Hill H. A. O. FEBS Lett. 1985, 190, 242. [Google Scholar]
- Lommen A.; Canters G. W. J. Biol. Chem. 1990, 265, 2768. [PubMed] [Google Scholar]
- Kyritsis P.; Dennison C.; Ingledew W. J.; McFarlane W.; Sykes A. G. Inorg. Chem. 1995, 34, 5370. [Google Scholar]
- Hunter D. M.; McFarlane W.; Sykes A. G.; Dennison C. Inorg. Chem. 2001, 40, 354. [DOI] [PubMed] [Google Scholar]
- Dennison C.; Lawler A. T.; Kohzuma T. Biochemistry 2002, 41, 552. [DOI] [PubMed] [Google Scholar]
- Sato K.; Dennison C. Biochemistry 2002, 41, 120. [DOI] [PubMed] [Google Scholar]
- Sato K.; Kohzuma T.; Dennison C. J. Am. Chem. Soc. 2003, 125, 2101. [DOI] [PubMed] [Google Scholar]
- Sato K.; Crowley P. B.; Dennison C. J. Biol. Chem. 2005, 280, 19281. [DOI] [PubMed] [Google Scholar]
- Sola M.; Cowan J. A.; Gray H. B. J. Am. Chem. Soc. 1989, 111, 6627. [Google Scholar]
- Concar D. W.; Whitford D.; Pielak G. J.; Williams R. J. P. J. Am. Chem. Soc. 1991, 113, 2401. [Google Scholar]
- Ubbink M.; Canters G. W. Biochemistry 1993, 32, 13893. [DOI] [PubMed] [Google Scholar]
- Dennison C.; Kohzuma T. Inorg. Chem. 1999, 38, 1491. [Google Scholar]
- Xu F.; Berka R. M.; Wahleithner J. A.; Nelson B. A.; Shuster J. R.; Brown S. H.; Palmer A. E.; Solomon E. I. Biochem. J. 1998, 334, 63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yaver D. S.; Xu F.; Golightly E. J.; Brown K. M.; Brown S. H.; Rey M. W.; Schneider P.; Halkier T.; Mondorf K.; Dalboge H. Appl. Environ. Microbiol. 1996, 62, 834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu F.; Palmer A. E.; Yaver D. S.; Berka R. M.; Gambetta G. A.; Brown S. H.; Solomon E. I. J. Biol. Chem. 1999, 274, 12372. [DOI] [PubMed] [Google Scholar]
- Romero A.; Hoitink C. W. G.; Nar H.; Huber R.; Messerschmidt A.; Canters G. W. J. Mol. Biol. 1993, 229, 1007. [DOI] [PubMed] [Google Scholar]
- Olesen K.; Veselov A.; Zhao Y.; Wang Y.; Danner B.; Scholes C. P.; Shapleigh J. P. Biochemistry 1998, 37, 6086. [DOI] [PubMed] [Google Scholar]
- Hall J. F.; Kanbi L. D.; Strange R. W.; Hasnain S. S. Biochemistry 1999, 38, 12675. [DOI] [PubMed] [Google Scholar]
- Kataoka K.; Nakai M.; Yamaguchi K.; Suzuki S. Biochem. Biophys. Res. Commun. 1998, 250, 409. [DOI] [PubMed] [Google Scholar]
- Diederix R. E. M.; Canters G. W.; Dennison C. Biochemistry 2000, 39, 9551. [DOI] [PubMed] [Google Scholar]
- Solomon E. I.; Sundaram U. M.; Machonkin T. E. Chem. Rev. 1996, 96, 2563. [DOI] [PubMed] [Google Scholar]
- Sakurai T.; Kataoka K. Cell. Mol. Life Sci. 2007, 64, 2642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quintanar L.; Stoj C.; Taylor A. B.; Hart P. J.; Kosman D. J.; Solomon E. I. Acc. Chem. Res. 2007, 40, 445. [DOI] [PubMed] [Google Scholar]
- Lawton T. J.; Rosenzweig A. C. In Encyclopedia of Inorganic and Bioinorganic Chemistry; John Wiley & Sons, Ltd.: Hoboken, NJ, 2011. [Google Scholar]
- Sakurai T.; Kataoka K. In Copper-Oxygen Chemistry; John Wiley & Sons, Inc.: Hoboken, NJ, 2011; p 131. [Google Scholar]
- Wasser I. M.; de Vries S.; Moënne-Loccoz P.; Schröder I.; Karlin K. D. Chem. Rev. 2002, 102, 1201. [DOI] [PubMed] [Google Scholar]
- Hong G.; Ivnitski D. M.; Johnson G. R.; Atanassov P.; Pachter R. J. Am. Chem. Soc. 2011, 133, 4802. [DOI] [PubMed] [Google Scholar]
- Whittaker M.; Bergmann D.; Arciero D.; Hooper A. B. Biochim. Biophys. Acta, Bioenerg. 2000, 1459, 346. [DOI] [PubMed] [Google Scholar]
- Lieberman R. L.; Arciero D. M.; Hooper A. B.; Rosenzweig A. C. Biochemistry 2001, 40, 5674. [DOI] [PubMed] [Google Scholar]
- Basumallick L.; Sarangi R.; DeBeer George S.; Elmore B.; Hooper A. B.; Hedman B.; Hodgson K. O.; Solomon E. I. J. Am. Chem. Soc. 2005, 127, 3531. [DOI] [PubMed] [Google Scholar]
- Dawson P. E.; Kent S. B. H. Annu. Rev. Biochem. 2000, 69, 923. [DOI] [PubMed] [Google Scholar]
- Muir T. W. Annu. Rev. Biochem. 2003, 72, 249. [DOI] [PubMed] [Google Scholar]
- Liu C. C.; Schultz P. G. Annu. Rev. Biochem. 2010, 79, 413. [DOI] [PubMed] [Google Scholar]
- Karlsson B. G.; Aasa R.; Malmstroem B. G.; Lundberg L. G. FEBS Lett. 1989, 253, 99. [Google Scholar]
- Karlsson B. G.; Nordling M.; Pascher T.; Tsai L.-C.; Sjolin L.; Lundberg L. G. Protein Eng. 1991, 4, 343. [DOI] [PubMed] [Google Scholar]
- Kroes S. J.; Hoitink C. W. G.; Andrew C. R.; Sanders-Loehr J.; Messerschmid A.; Hagen W. R.; Canters G. W. Eur. J. Biochem. 1996, 240, 342. [DOI] [PubMed] [Google Scholar]
- Murphy L. M.; Strange R. W.; Karlsson B. G.; Lundberg L. G.; Pascher T.; Reinhammar B.; Hasnain S. S. Biochemistry 1993, 32, 1965. [DOI] [PubMed] [Google Scholar]
- Hough M. A.; Ellis M. J.; Antonyuk S.; Strange R. W.; Sawers G.; Eady R. R.; Samar H. S. J. Mol. Biol. 2005, 350, 300. [DOI] [PubMed] [Google Scholar]
- DeBeer George S.; Basumallick L.; Szilagyi R. K.; Randall D. W.; Hill M. G.; Nersissian A. M.; Valentine J. S.; Hedman B.; Hodgson K. O.; Solomon E. I. J. Am. Chem. Soc. 2003, 125, 11314. [DOI] [PubMed] [Google Scholar]
- Harrison M. D.; Dennison C. Proteins: Struct., Funct., Bioinf. 2004, 55, 426. [DOI] [PubMed] [Google Scholar]
- Frank P.; Licht A.; Tullius T. D.; Hodgson K. O.; Pecht I. J. Biol. Chem. 1985, 260, 5518. [PubMed] [Google Scholar]
- LaCroix L. B.; Shadle S. E.; Wang Y.; Averill B. A.; Hedman B.; Hodgson K. O.; Solomon E. I. J. Am. Chem. Soc. 1996, 118, 7755. [Google Scholar]
- Berry S. M.; Ralle M.; Low D. W.; Blackburn N. J.; Lu Y. J. Am. Chem. Soc. 2003, 125, 8760. [DOI] [PubMed] [Google Scholar]
- Garner D. K.; Vaughan M. D.; Hwang H. J.; Savelieff M. G.; Berry S. M.; Honek J. F.; Lu Y. J. Am. Chem. Soc. 2006, 128, 15608. [DOI] [PubMed] [Google Scholar]
- Yanagisawa S.; Dennison C. J. Am. Chem. Soc. 2005, 127, 16453. [DOI] [PubMed] [Google Scholar]
- Messerschmidt A.; Prade L.; Kroes S. J.; Sanders-Loehr J.; Huber R.; Canters G. W. Proc. Natl. Acad. Sci. U.S.A. 1998, 95, 3443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Gestel M.; Canters G. W.; Krupka H.; Messerschmid A.; de Waal E. C.; Warmerdam G. C. M.; Groenen E. J. J. J. Am. Chem. Soc. 2000, 122, 2322. [Google Scholar]
- Karlsson B. G.; Tsai L. C.; Nar H.; SandersLoehr J.; Bonander N.; Langer V.; Sjolin L. Biochemistry 1997, 36, 4089. [DOI] [PubMed] [Google Scholar]
- Strange R. W.; Murphy L. M.; Karlsson B. G.; Reinhammar B.; Hasnain S. S. Biochemistry 1996, 35, 16391. [DOI] [PubMed] [Google Scholar]
- Webb M. A.; Kiser C. N.; Richards J. H.; Di Bilio A. J.; Gray H. B.; Winkler J. R.; Loppnow G. R. J. Phys. Chem. B 2000, 104, 10915. [Google Scholar]
- Basumallick L.; Szilagyi R. K.; Zhao Y.; Shapleigh J. P.; Scholes C. P.; Solomon E. I. J. Am. Chem. Soc. 2003, 125, 14784. [DOI] [PubMed] [Google Scholar]
- Clark K. M.; Yu Y.; Marshall N. M.; Sieracki N. A.; Nilges M. J.; Blackburn N. J.; van der Donk W. A.; Lu Y. J. Am. Chem. Soc. 2010, 132, 10093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Den Blaauwen T.; Van de Kamp M.; Canters G. W. J. Am. Chem. Soc. 1991, 113, 5050. [Google Scholar]
- van Pouderoyen G.; Andrew C. R.; Loehr T. M.; Sanders-Loehr J.; Mazumdar S.; Hill H. A.; Canters G. W. Biochemistry 1996, 35, 1397. [DOI] [PubMed] [Google Scholar]
- Den Blaauwen T.; Canters G. W. J. Am. Chem. Soc. 1993, 115, 1121. [Google Scholar]
- Jeuken L. J. C.; van Vliet P.; Verbeet M. P.; Camba R.; McEvoy J. P.; Armstrong F. A.; Canters G. W. J. Am. Chem. Soc. 2000, 122, 12186. [Google Scholar]
- Gorren A. C. F.; denBlaauwen T.; Canters G. W.; Hopper D. J.; Duine J. A. FEBS Lett. 1996, 381, 140. [DOI] [PubMed] [Google Scholar]
- Alagaratnam S.; Meeuwenoord N. J.; Navarro J. A.; Hervas M.; De la Rosa M. A.; Hoffmann M.; Einsle O.; Ubbink M.; Canters G. W. FEBS J. 2011, 278, 1506. [DOI] [PubMed] [Google Scholar]
- Canters G. W.; Kolczak U.; Armstrong F.; Jeuken L. J.; Camba R.; Sola M. Faraday Discuss. 2000, 205. [DOI] [PubMed] [Google Scholar]
- Berry S. M.; Gieselman M. D.; Nilges M. J.; van der Donk W. A.; Lu Y. J. Am. Chem. Soc. 2002, 124, 2084. [DOI] [PubMed] [Google Scholar]
- Ralle M.; Berry S. M.; Nilges M. J.; Gieselman M. D.; van der Donk W. A.; Lu Y.; Blackburn N. J. J. Am. Chem. Soc. 2004, 126, 7244. [DOI] [PubMed] [Google Scholar]
- Sarangi R.; Gorelsky S. I.; Basumallick L.; Hwang H. J.; Pratt R. C.; Stack T. D. P.; Lu Y.; Hodgson K. O.; Hedman B.; Solomon E. I. J. Am. Chem. Soc. 2008, 130, 3866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizoguchi T. J.; Di Bilio A. J.; Gray H. B.; Richards J. H. J. Am. Chem. Soc. 1992, 114, 10076. [Google Scholar]
- Piccioli M.; Luchinat C.; Mizoguchi T. J.; Ramirez B. E.; Gray H. B.; Richards J. H. Inorg. Chem. 1995, 34, 737. [Google Scholar]
- Faham S.; Mizoguchi T. J.; Adman E. T.; Gray H. B.; Richards J. H.; Rees D. C. JBIC, J. Biol. Inorg. Chem. 1997, 2, 464. [Google Scholar]
- DeBeer S.; Kiser C. N.; Mines G. A.; Richards J. H.; Gray H. B.; Solomon E. I.; Hedman B.; Hodgson K. O. Inorg. Chem. 1999, 38, 433. [DOI] [PubMed] [Google Scholar]
- Lancaster K. M.; George S. D.; Yokoyama K.; Richards J. H.; Gray H. B. Nat. Chem. 2009, 1, 711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lancaster K. M.; Yokoyama K.; Richards J. H.; Winkler J. R.; Gray H. B. Inorg. Chem. 2009, 48, 1278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lancaster K. M.; Sproules S.; Palmer J. H.; Richards J. H.; Gray H. B. J. Am. Chem. Soc. 2010, 132, 14590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lancaster K. M.; Farver O.; Wherland S.; Crane E. J. III; Richards J. H.; Pecht I.; Gray H. B. J. Am. Chem. Soc. 2011, 133, 4865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanbi L. D.; Antonyuk S.; Hough M. A.; Hall J. F.; Dodd F. E.; Hasnain S. S. J. Mol. Biol. 2002, 320, 263. [DOI] [PubMed] [Google Scholar]
- Libeu C. A. P.; Kukimoto M.; Nishiyama M.; Horinouchi S.; Adman E. T. Biochemistry 1997, 36, 13160. [DOI] [PubMed] [Google Scholar]
- Botuyan M. V.; ToyPalmer A.; Chung J.; Blake R. C.; Beroza P.; Case D. A.; Dyson H. J. J. Mol. Biol. 1996, 263, 752. [DOI] [PubMed] [Google Scholar]
- Lowery M. D.; Solomon E. I. Inorg. Chim. Acta 1992, 198, 233. [Google Scholar]
- Hadt R. G.; Sun N.; Marshall N. M.; Hodgson K. O.; Hedman B.; Lu Y.; Solomon E. I. J. Am. Chem. Soc. 2012, 134, 16701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farver O.; Marshall N. M.; Wherland S.; Lu Y.; Pecht I. Proc. Natl. Acad. Sci. U.S.A. 2013, 110, 10536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dennison C.; Vijgenboom E.; de Vries S.; van der Oost J.; Canters G. W. FEBS Lett. 1995, 365, 92. [DOI] [PubMed] [Google Scholar]
- Hay M.; Richards J. H.; Lu Y. Proc. Natl. Acad. Sci. U.S.A. 1996, 93, 461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berry S. M.; Bladholm E. L.; Mostad E. J.; Schenewerk A. R. JBIC, J. Biol. Inorg. Chem. 2011, 16, 473. [DOI] [PubMed] [Google Scholar]
- Dennison C.; Vijgenboom E.; Hagen W. R.; Canters G. W. J. Am. Chem. Soc. 1996, 118, 7406. [Google Scholar]
- Buning C.; Canters G. W.; Comba P.; Dennison C.; Jeuken L.; Melter M.; Sanders-Loehr J. J. Am. Chem. Soc. 1999, 122, 204. [Google Scholar]
- Remenyi R.; Jeuken L. J.; Comba P.; Canters G. W. JBIC, J. Biol. Inorg. Chem. 2001, 6, 23. [DOI] [PubMed] [Google Scholar]
- Battistuzzi G.; Borsari M.; Canters G. W.; di Rocco G.; de Waal E.; Arendsen Y.; Leonardi A.; Ranieri A.; Sola M. Biochemistry 2005, 44, 9944. [DOI] [PubMed] [Google Scholar]
- Yanagisawa S.; Dennison C. J. Am. Chem. Soc. 2003, 125, 4974. [DOI] [PubMed] [Google Scholar]
- Yanagisawa S.; Dennison C. J. Am. Chem. Soc. 2004, 126, 15711. [DOI] [PubMed] [Google Scholar]
- Li C.; Banfield M. J.; Dennison C. J. Am. Chem. Soc. 2007, 129, 709. [DOI] [PubMed] [Google Scholar]
- Wikstrom M. Biochim. Biophys. Acta, Bioenerg. 2004, 1655, 241. [DOI] [PubMed] [Google Scholar]
- Brown K.; Tegoni M.; Prudencio M.; Pereira A. S.; Besson S.; Moura J.; Moura I.; Cambillau C. Nat. Struct. Mol. Biol. 2000, 7, 191. [DOI] [PubMed] [Google Scholar]
- Haltia T.; Brown K.; Tegoni M.; Cambillau C.; Saraste M.; Mattila K.; Djinovic-Carugo K. Biochem. J. 2003, 369, 77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komorowski L.; Anemuller S.; Schafer G. J. Bioenerg. Biomembr. 2001, 33, 27. [DOI] [PubMed] [Google Scholar]
- Suharti; Heering H. A.; de Vries S. Biochemistry 2004, 43, 13487. [DOI] [PubMed] [Google Scholar]
- Suharti; Strampraad M. J.; Schröder I.; de Vries S. Biochemistry 2001, 40, 2632. [DOI] [PubMed] [Google Scholar]
- Ryden L. G.; Hunt L. T. J. Mol. Evol. 1993, 36, 41. [DOI] [PubMed] [Google Scholar]
- Abolmaali B.; Taylor H. V.; Weser U. Struct. Bonding (Berlin, Ger.) 1998, 91, 91. [Google Scholar]
- Blair D. F.; Ellis W. R. Jr.; Wang H.; Gray H. B.; Chan S. I. J. Biol. Chem. 1986, 261, 11524. [PubMed] [Google Scholar]
- Immoos C.; Hill M. G.; Sanders D.; Fee J. A.; Slutter S. E.; Richards J. H.; Gray H. B. JBIC, J. Biol. Inorg. Chem. 1996, 1, 529. [Google Scholar]
- Ghosh S. K.; Saha S. K.; Ghosh M. C.; Bose R. N.; Reed J. W.; Gould E. S. Inorg. Chem. 1992, 31, 3358. [Google Scholar]
- Lappalainen P.; Aasa R.; Malmstrom B. G.; Saraste M. J. Biol. Chem. 1993, 268, 26416. [PubMed] [Google Scholar]
- Farver O.; Chen Y.; Fee J. A.; Pecht I. FEBS Lett. 2006, 580, 3417. [DOI] [PubMed] [Google Scholar]
- Slutter C. E.; Langen R.; Sanders D.; Lawrence S. M.; Wittung P.; Di Bilio A. J.; Hill M. G.; Fee J. A.; Richards J. H.; et al. Inorg. Chim. Acta 1996, 243, 141. [Google Scholar]
- Slutter C. E.; Sanders D.; Wittung P.; Malmstrom B. G.; Aasa R.; Richards J. H.; Gray H. B.; Fee J. A. Biochemistry 1996, 35, 3387. [DOI] [PubMed] [Google Scholar]
- Fujita K.; Nakamura N.; Ohno H.; Leigh B. S.; Niki K.; Gray H. B.; Richards J. H. J. Am. Chem. Soc. 2004, 126, 13954. [DOI] [PubMed] [Google Scholar]
- Blackburn N. J.; de Vries S.; Barr M. E.; Houser R. P.; Tolman W. B.; Sanders D.; Fee J. A. J. Am. Chem. Soc. 1997, 119, 6135. [Google Scholar]
- von Wachenfeldt C.; de Vries S.; van der Oost J. FEBS Lett. 1994, 340, 109. [DOI] [PubMed] [Google Scholar]
- Riester J.; Zumft W. G.; Kroneck P. M. H. Eur. J. Biochem. 1989, 178, 751. [DOI] [PubMed] [Google Scholar]
- Hulse C. L.; Averill B. A. Biochem. Biophys. Res. Commun. 1990, 166, 729. [DOI] [PubMed] [Google Scholar]
- Kelly M.; Lappalainen P.; Talbo G.; Haltia T.; van d. O. J.; Saraste M. J. Biol. Chem. 1993, 268, 16781. [PubMed] [Google Scholar]
- Wilmanns M.; Lappalainen P.; Kelly M.; Sauer-Eriksson E.; Saraste M. Proc. Natl. Acad. Sci. U.S.A. 1995, 92, 11955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hay M. T.; Ang M. C.; Gamelin D. R.; Solomon E. I.; Antholine W. E.; Ralle M.; Blackburn N. J.; Massey P. D.; Wang X.; Kwon A. H.; Lu Y. Inorg. Chem. 1998, 37, 191. [Google Scholar]
- Prudencio M.; Pereira A. S.; Tavares P.; Besson S.; Cabrito I.; Brown K.; Samyn B.; Devreese B.; Van B. J.; Rusnak F.; Fauque G.; Moura J. J.; Tegoni M.; Cambillau C.; Moura I. Biochemistry 2000, 39, 3899. [DOI] [PubMed] [Google Scholar]
- Paumann M.; Lubura B.; Regelsberger G.; Feichtinger M.; Koellensberger G.; Jakopitsch C.; Furtmueller P. G.; Peschek G. A.; Obinger C. J. Biol. Chem. 2004, 279, 10293. [DOI] [PubMed] [Google Scholar]
- Larsson S.; Kaellebring B.; Wittung P.; Malmstroem B. G. Proc. Natl. Acad. Sci. U.S.A. 1995, 92, 7167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henkel G.; Mueller A.; Weissgraeber S.; Buse G.; Soulimane T.; Steffens G. C. M.; Nolting H.-F. Angew. Chem., Int. Ed. Engl. 1995, 34, 1488. [Google Scholar]
- Froncisz W.; Scholes C. P.; Hyde J. S.; Wei Y. H.; King T. E.; Shaw R. W.; Beiner H. J. Biol. Chem. 1979, 254, 7482. [PubMed] [Google Scholar]
- Hoffman B. M.; Roberts J. E.; Swanson M.; Speck S. H.; Margoliash E. Proc. Natl. Acad. Sci. U.S.A. 1980, 77, 1452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slutter C. E.; Gromov I.; Richards J. H.; Pecht I.; Goldfarb D. J. Am. Chem. Soc. 1999, 121, 5077. [DOI] [PubMed] [Google Scholar]
- Lappalainen P.; Watmough N. J.; Greenwood C.; Saraste M. Biochemistry 1995, 34, 5824. [DOI] [PubMed] [Google Scholar]
- Williams P. A.; Blackburn N. J.; Sanders D.; Bellamy H.; Stura E. A.; Fee J. A.; McRee D. E. Nat. Struct. Biol. 1999, 6, 509. [DOI] [PubMed] [Google Scholar]
- Song A.; Li L.; Yu T.; Chen S.; Huang Z. Protein Eng. 2003, 16, 435. [DOI] [PubMed] [Google Scholar]
- Maneg O.; Ludwig B.; Malatesta F. J. Biol. Chem. 2003, 278, 46734. [DOI] [PubMed] [Google Scholar]
- van der Oost J.; Lappalainen P.; Musacchio A.; Warne A.; Lemieux L.; Rumbley J.; Gennis R. B.; Aasa R.; Pascher T.; Malmstrom B. G.; et al. EMBO J. 1992, 11, 3209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones L. H.; Liu A.; Davidson V. L. J. Biol. Chem. 2003, 278, 47269. [DOI] [PubMed] [Google Scholar]
- Andrew C. R.; Lappalainen P.; Saraste M.; Hay M. T.; Lu Y.; Dennison C.; Canters G. W.; Fee J. A.; Nakamura N.; Sanders-Loehr J. J. Am. Chem. Soc. 1995, 117, 10759. [Google Scholar]
- Savelieff M. G.; Lu Y. JBIC, J. Biol. Inorg. Chem. 2010, 15, 461. [DOI] [PubMed] [Google Scholar]
- DeBeer G. S.; Metz M.; Szilagyi R. K.; Wang H.; Cramer S. P.; Lu Y.; Tolman W. B.; Hedman B.; Hodgson K. O.; Solomon E. I. J. Am. Chem. Soc. 2001, 123, 5757. [DOI] [PubMed] [Google Scholar]
- Robinson H.; Ang M. C.; Gao Y. G.; Hay M. T.; Lu Y.; Wang A. H. Biochemistry 1999, 38, 5677. [DOI] [PubMed] [Google Scholar]
- Zickermann V.; Verkhovsky M.; Morgan J.; Wikstroem M.; Anemueller S.; Bill E.; Steffens G. C. M.; Ludwig B. Eur. J. Biochem. 1995, 234, 686. [DOI] [PubMed] [Google Scholar]
- Zhen Y.; Schmidt B.; Kang U. G.; Antholine W.; Ferguson-Miller S. Biochemistry 2002, 41, 2288. [DOI] [PubMed] [Google Scholar]
- Wang K.; Geren L.; Zhen Y.; Ma L.; Ferguson-Miller S.; Durham B.; Millett F. Biochemistry 2002, 41, 2298. [DOI] [PubMed] [Google Scholar]
- Slutter C. E.; Gromov I.; Epel B.; Pecht I.; Richards J. H.; Goldfarb D. J. Am. Chem. Soc. 2001, 123, 5325. [DOI] [PubMed] [Google Scholar]
- Fernandez C. O.; Cricco J. A.; Slutter C. E.; Richards J. H.; Gray H. B.; Vila A. J. J. Am. Chem. Soc. 2001, 123, 11678. [DOI] [PubMed] [Google Scholar]
- Blackburn N. J.; Ralle M.; Gomez E.; Hill M. G.; Pastuszyn A.; Sanders D.; Fee J. A. Biochemistry 1999, 38, 7075. [DOI] [PubMed] [Google Scholar]
- Hwang H. J.; Berry S. M.; Nilges M. J.; Lu Y. J. Am. Chem. Soc. 2005, 127, 7274. [DOI] [PubMed] [Google Scholar]
- Ledesma G. N.; Murgida D. H.; Ly H. K.; Wackerbarth H.; Ulstrup J.; Costa-Filho A. J.; Vila A. J. J. Am. Chem. Soc. 2007, 129, 11884. [DOI] [PubMed] [Google Scholar]
- Alvarez-Paggi D.; Abriata L. A.; Murgida D. H.; Vila A. J. Chem. Commun. 2013, 49, 5381. [DOI] [PubMed] [Google Scholar]
- Wang X.; Berry S. M.; Xia Y.; Lu Y. J. Am. Chem. Soc. 1999, 121, 7449. [Google Scholar]
- Berry S. M.; Wang X.; Lu Y. J. Inorg. Biochem. 2000, 78, 89. [DOI] [PubMed] [Google Scholar]
- Lukoyanov D.; Berry S. M.; Lu Y.; Antholine W. E.; Scholes C. P. Biophys. J. 2002, 82, 2758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie X.; Gorelsky S. I.; Sarangi R.; Garner D. K.; Hwang H. J.; Hodgson K. O.; Hedman B.; Lu Y.; Solomon E. I. J. Am. Chem. Soc. 2008, 130, 5194. [DOI] [PubMed] [Google Scholar]
- Zickermann V.; Wittershagen A.; Kolbesen B. O.; Ludwig B. Biochemistry 1997, 36, 3232. [DOI] [PubMed] [Google Scholar]
- Charnock J. M.; Dreusch A.; Korner H.; Neese F.; Nelson J.; Kannt A.; Michel H.; Garner C. D.; Kroneck P. M.; Zumft W. G. Eur. J. Biochem. 2000, 267, 1368. [DOI] [PubMed] [Google Scholar]
- Hwang H. J.; Nagraj N.; Lu Y. Inorg. Chem. 2006, 45, 102. [DOI] [PubMed] [Google Scholar]
- Savelieff M. G.; Lu Y. Inorg. Chim. Acta 2008, 361, 1087. [Google Scholar]
- New S. Y.; Marshall N. M.; Hor T. S. A.; Xue F.; Lu Y. Chem. Commun. 2012, 48, 4217. [DOI] [PubMed] [Google Scholar]
- Pan L. P.; Hibdon S.; Liu R. Q.; Durham B.; Millett F. Biochemistry 1993, 32, 8492. [DOI] [PubMed] [Google Scholar]
- Farver O.; Einarsdottir O.; Pecht I. Eur. J. Biochem. 2000, 267, 950. [DOI] [PubMed] [Google Scholar]
- Farver O.; Lu Y.; Ang M. C.; Pecht I. Proc. Natl. Acad. Sci. U.S.A. 1999, 96, 899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farver O.; Hwang H. J.; Lu Y.; Pecht I. J. Phys. Chem. B 2007, 111, 6690. [DOI] [PubMed] [Google Scholar]
- Hwang H. J.; Lu Y. Proc. Natl. Acad. Sci. U.S.A. 2004, 101, 12842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim B.-E.; Nevitt T.; Thiele D. J. Nat. Chem. Biol. 2008, 4, 176. [DOI] [PubMed] [Google Scholar]
- Leary S. C.; Winge D. R.; Cobine P. A. Biochim. Biophys. Acta, Mol. Cell Res. 2009, 1793, 146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boal A. K.; Rosenzweig A. C. Chem. Rev. 2009, 109, 4760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banci L.; Bertini I.; Cantini F.; Ciofi-Baffoni S. Cell. Mol. Life Sci. 2010, 67, 2563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banci L.; Bertini I.; McGreevy K. S.; Rosato A. Nat. Prod. Rep. 2010, 27, 695. [DOI] [PubMed] [Google Scholar]
- Robinson N. J.; Winge D. R. Annu. Rev. Biochem. 2010, 79, 537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertini I.; Cavallaro G.; McGreevy K. S. Coord. Chem. Rev. 2010, 254, 506. [Google Scholar]
- Banci L.; Bertini I.; Cavallaro G.; Ciofi-Baffoni S. FEBS J. 2011, 278, 2244. [DOI] [PubMed] [Google Scholar]
- Richter O. M. H.; Ludwig B. Rev. Physiol., Biochem. Pharmacol. 2003, 147, 47. [DOI] [PubMed] [Google Scholar]
- Ostermeier C.; Iwata S.; Ludwig B.; Michel H. Nat. Struct. Biol. 1995, 2, 842. [DOI] [PubMed] [Google Scholar]
- Soulimane T.; Buse G.; Bourenkov G. P.; Bartunik H. D.; Huber R.; Than M. E. EMBO J. 2000, 19, 1766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zumft W. G. Microbiol. Mol. Biol. Rev. 1997, 61, 533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zumft W. G.; Kroneck P. M. H. Adv. Microb. Physiol. 2007, 52, 107. [DOI] [PubMed] [Google Scholar]
- Wunsch P.; Herb M.; Wieland H.; Schiek U. M.; Zumft W. G. J. Bacteriol. 2003, 185, 887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zumft W. G. J. Mol. Microbiol. Biotechnol. 2005, 10, 154. [DOI] [PubMed] [Google Scholar]
- Wang X.; Ang M. C.; Lu Y. J. Am. Chem. Soc. 1999, 121, 2947. [Google Scholar]
- Cavallini D.; De M. C.; Dupre S.; Rotilio G. Arch. Biochem. Biophys. 1969, 130, 354. [DOI] [PubMed] [Google Scholar]
- Cavallini D.; De M. C.; Dupre S. Arch. Biochem. Biophys. 1968, 124, 18. [DOI] [PubMed] [Google Scholar]
- Pecci L.; Montefoschi G.; Musci G.; Cavallini D. Amino Acids 1997, 13, 355. [Google Scholar]
- Savelieff M. G.; Wilson T. D.; Elias Y.; Nilges M. J.; Garner D. K.; Lu Y. Proc. Natl. Acad. Sci. U.S.A. 2008, 105, 7919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson T. D.; Savelieff M. G.; Nilges M. J.; Marshall N. M.; Lu Y. J. Am. Chem. Soc. 2011, 133, 20778. [DOI] [PubMed] [Google Scholar]
- Belle C.; Rammal W.; Pierre J.-L. J. Inorg. Biochem. 2005, 99, 1929. [DOI] [PubMed] [Google Scholar]
- Harding C.; McKee V.; Nelson J. J. Am. Chem. Soc. 1991, 113, 9684. [Google Scholar]
- Barr M. E.; Smith P. H.; Antholine W. E.; Spencer B. J. Chem. Soc., Chem. Commun. 1993, 1649. [Google Scholar]
- Harding C.; Nelson J.; Symons M. C. R.; Wyatt J. J. Chem. Soc., Chem. Commun. 1994, 2499. [Google Scholar]
- Houser R. P.; Young V. G. Jr.; Tolman W. B. J. Am. Chem. Soc. 1996, 118, 2101. [Google Scholar]
- LeCloux D. D.; Davydov R.; Lippard S. J. J. Am. Chem. Soc. 1998, 120, 6810. [DOI] [PubMed] [Google Scholar]
- Houser R. P.; Tolman W. B. Inorg. Chem. 1995, 34, 1632. [Google Scholar]
- Miskowski V. M.; Franzen S.; Shreve A. P.; Ondrias M. R.; Wallace-Williams S. E.; Barr M. E.; Woodruff W. H. Inorg. Chem. 1999, 38, 2546. [DOI] [PubMed] [Google Scholar]
- He C.; Lippard S. J. Inorg. Chem. 2000, 39, 5225. [DOI] [PubMed] [Google Scholar]
- Franzen S.; Miskowski V. M.; Shreve A. P.; Wallace-Williams S. E.; Woodruff W. H.; Ondrias M. R.; Barr M. E.; Moore L.; Boxer S. G. Inorg. Chem. 2001, 40, 6375. [DOI] [PubMed] [Google Scholar]
- Gupta R.; Zhang Z. H.; Powell D.; Hendrich M. P.; Borovik A. S. Inorg. Chem. 2002, 41, 5100. [DOI] [PubMed] [Google Scholar]
- Harkins S. B.; Peters J. C. J. Am. Chem. Soc. 2004, 126, 2885. [DOI] [PubMed] [Google Scholar]
- Zhao Q.; Wang X.; Liu Y.; Fang R. Chem. Lett. 2004, 33, 138. [Google Scholar]
- Wen Y.-H.; Cheng J.-K.; Zhang J.; Li Z.-J.; Kang Y.; Yao Y.-G. Inorg. Chem. Commun. 2004, 7, 1120. [Google Scholar]
- Patel D. V.; Mihalcik D. J.; Kreisel K. A.; Yap G. P. A.; Zakharov L. N.; Kassel W. S.; Rheingold A. L.; Rabinovich D. Dalton Trans. 2005, 2410. [DOI] [PubMed] [Google Scholar]
- Feng Y.; Han Z.; Peng J.; Hao X. J. Mol. Struct. 2005, 734, 171. [Google Scholar]
- Jiang X.; Bollinger J. C.; Baik M.-H.; Lee D. Chem. Commun. 2005, 1043. [DOI] [PubMed] [Google Scholar]
- Gennari M.; Pécaut J.; DeBeer S.; Neese F.; Collomb M.-N.; Duboc C. Angew. Chem., Int. Ed. 2011, 50, 5662. [DOI] [PubMed] [Google Scholar]
- Li H.; Webb S. P.; Ivanic J.; Jensen J. H. J. Am. Chem. Soc. 2004, 126, 8010. [DOI] [PubMed] [Google Scholar]
- Garcia-Horsman J. A.; Barquera B.; Rumbley J.; Ma J.; Gennis R. B. J. Bacteriol. 1994, 176, 5587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pereira M. M.; Santana M.; Teixeira M. Biochim. Biophys. Acta, Bioenerg. 2001, 1505, 185. [DOI] [PubMed] [Google Scholar]
- Capaldi R. A. Annu. Rev. Biochem. 1990, 59, 569. [DOI] [PubMed] [Google Scholar]
- Ferguson-Miller S.; Brautigan D.; Margoliash E. J. Biol. Chem. 1978, 253, 149. [PubMed] [Google Scholar]
- Berry E. A.; Trumpower B. L. J. Biol. Chem. 1985, 260, 2458. [PubMed] [Google Scholar]
- Fergusonmiller S.; Brautigan D. L.; Margoliash E. J. Biol. Chem. 1976, 251, 1104. [PubMed] [Google Scholar]
- Fee J. A.; Kuila D.; Mather M. W.; Yoshida T. Biochim. Biophys. Acta, Rev. Bioenerg. 1986, 853, 153. [DOI] [PubMed] [Google Scholar]
- Sone N.; Yokoi F.; Fu T.; Ohta S.; Metso T.; Raitio M.; Saraste M. J. Biochem. 1988, 103, 606. [DOI] [PubMed] [Google Scholar]
- Capaldi R. A.; Darleyusmar V.; Fuller S.; Millett F. FEBS Lett. 1982, 138, 1. [DOI] [PubMed] [Google Scholar]
- Rieder R.; Bosshard H. R. J. Biol. Chem. 1980, 255, 4732. [PubMed] [Google Scholar]
- Staudenmayer N.; Ng S.; Smith M. B.; Millett F. Biochemistry 1977, 16, 600. [DOI] [PubMed] [Google Scholar]
- Osheroff N.; Borden D.; Koppenol W. H.; Margoliash E. J. Biol. Chem. 1980, 255, 1689. [PubMed] [Google Scholar]
- Speck S. H.; Dye D.; Margoliash E. Proc. Natl. Acad. Sci. U.S.A. 1984, 81, 347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antalis T. M.; Palmer G. J. Biol. Chem. 1982, 257, 6194. [PubMed] [Google Scholar]
- Greenwood C.; Brittain T.; Wilson M.; Brunori M. Biochem. J. 1976, 157, 591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veerman E. C. I.; Wilms J.; Casteleijn G.; Vangelder B. F. Biochim. Biophys. Acta, Bioenerg. 1980, 590, 117. [DOI] [PubMed] [Google Scholar]
- Sarti P.; Antonini G.; Malatesta F.; Vallone B.; Brunori M. Ann. N. Y. Acad. Sci. 1988, 550, 161. [DOI] [PubMed] [Google Scholar]
- Wilson M. T.; Peterson J.; Antonini E.; Brunori M.; Colosimo A.; Wyman J. Proc. Natl. Acad. Sci. U.S.A. 1981, 78, 7115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nilsson T.; Copeland R. A.; Smith P. A.; Chan S. I. Biochemistry 1988, 27, 8254. [DOI] [PubMed] [Google Scholar]
- Nilsson T.; Gelles J.; Li P. M.; Chan S. I. Biochemistry 1988, 27, 296. [DOI] [PubMed] [Google Scholar]
- Kornblatt J. A.; Luu H. A. Eur. J. Biochem. 1986, 159, 407. [DOI] [PubMed] [Google Scholar]
- Michel B.; Bosshard H. R. J. Biol. Chem. 1984, 259, 85. [PubMed] [Google Scholar]
- Copeland R. A.; Smith P. A.; Chan S. I. Biochemistry 1987, 26, 7311. [DOI] [PubMed] [Google Scholar]
- Jensen P.; Wilson M. T.; Aasa R.; Malmstrom B. G. Biochem. J. 1984, 224, 829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones M. G.; Bickar D.; Wilson M. T.; Brunori M.; Colosimo A.; Sarti P. Biochem. J. 1984, 220, 57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scholes C. P.; Malmstrom B. G. FEBS Lett. 1986, 198, 125. [DOI] [PubMed] [Google Scholar]
- Galinato M. G. I.; Kleingardner J. G.; Bowman S. E. J.; Alp E. E.; Zhao J.; Bren K. L.; Lehnert N. Proc. Natl. Acad. Sci. U.S.A. 2012, 109, 8896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakamoto K.; Kamiya M.; Imai M.; Shinzawa-Itoh K.; Uchida T.; Kawano K.; Yoshikawa S.; Ishimori K. Proc. Natl. Acad. Sci. U.S.A. 2011, 108, 12271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasmussen T.; Berks B. C.; Sanders-Loehr J.; Dooley D. M.; Zumft W. G.; Thomson A. J. Biochemistry 2000, 39, 12753. [DOI] [PubMed] [Google Scholar]
- Simon J. r.; Einsle O.; Kroneck P. M.; Zumft W. G. FEBS Lett. 2004, 569, 7. [DOI] [PubMed] [Google Scholar]
- Crofts A. R. Annu. Rev. Physiol. 2004, 66, 689. [DOI] [PubMed] [Google Scholar]
- Hunte C.; Koepke J.; Lange C.; Rossmanith T.; Michel H. Structure (London, U. K.) 2000, 8, 669. [DOI] [PubMed] [Google Scholar]
- Xia D.; Yu C. A.; Kim H.; Xian J. Z.; Kachurin A. M.; Zhang L.; Yu L.; Deisenhofer J. Science 1997, 277, 60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunter C. N.; Daldal F.. The Purple Phototrophic Bacteria; Springer: Dordrecht, The Netherlands, 2009. [Google Scholar]
- Kokhan O.; Wraight C. A.; Tajkhorshid E. Biophys. J. 2010, 99, 2647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koppenol W. H.; Rush J. D.; Mills J. D.; Margoliash E. Mol. Biol. Evol. 1991, 8, 545. [DOI] [PubMed] [Google Scholar]
- Tiede D. M.; Vashishta A. C.; Gunner M. R. Biochemistry 1993, 32, 4515. [DOI] [PubMed] [Google Scholar]
- Sarewicz M.; Borek A.; Daldal F.; Froncisz W.; Osyczka A. J. Biol. Chem. 2008, 283, 24826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarewicz M.; Szytula S.; Dutka M.; Osyczka A.; Froncisz W. Eur. Biophys. J. Biophys. Lett. 2008, 37, 483. [DOI] [PubMed] [Google Scholar]
- Sarewicz M.; Pietras R.; Froncisz W.; Osyczka A. Metallomics 2011, 3, 404. [DOI] [PubMed] [Google Scholar]
- Sadoski R. C.; Engstrom G.; Tian H.; Zhang L.; Yu C.-A.; Yu L.; Durham B.; Millett F. Biochemistry 2000, 39, 4231. [DOI] [PubMed] [Google Scholar]
- Gurung B.; Yu L.; Xia D.; Yu C.-A. J. Biol. Chem. 2005, 280, 24895. [DOI] [PubMed] [Google Scholar]
- Cramer W. A.; Soriano G. M.; Ponomarev M.; Huang D.; Zhang H.; Martinez S. E.; Smith J. L. Annu. Rev. Plant Physiol. Plant Mol. Biol. 1996, 47, 477. [DOI] [PubMed] [Google Scholar]
- Kallas T.Photosynthesis: Plastid Biology, Energy Conversion and Carbon Assimilation; Advances in Photosynthesis and Respiration; Springer: New York, 2012; Vol. 34. [Google Scholar]
- Baniulis D.; Yamashita E.; Zhang H.; Hasan S. S.; Cramer W. A. Photochem. Photobiol. 2008, 84, 1349. [DOI] [PubMed] [Google Scholar]
- Cramer W. A.; Yamashita E.; Baniulis D.; Hassan S. S. In Handbook of Metalloproteins; Messerschmidt A., Ed.; John Wiley: Chichester, U.K., 2011. [Google Scholar]
- Kallas T. In Photosynthesis: Plastid Biology, Energy Conversion and Carbon Assimilation; Eaton-Rye J. J., Tripathy B. C., Sharkey T. D., Eds.; Springer: London, 2012; Vol. 34, p 501. [Google Scholar]
- Baniulis D.; Yamashita E.; Whitelegge J. P.; Zatsman A. I.; Hendrich M. P.; Hasan S. S.; Ryan C. M.; Cramer W. A. J. Biol. Chem. 2009, 284, 9861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurisu G.; Zhang H.; Smith J. L.; Cramer W. A. Science 2003, 302, 1009. [DOI] [PubMed] [Google Scholar]
- Yamashita E.; Zhang H.; Cramer W. J. Mol. Biol. 2007, 370, 39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan J.; Kurisu G.; Cramer W. A. Proc. Natl. Acad. Sci. U.S.A. 2006, 103, 69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crofts A. R.; Meinhardt S.; Jones K. R.; Snozzi M. Biochim. Biophys. Acta, Bioenerg. 1983, 723, 202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell P. J. Theor. Biol. 1976, 62, 327. [DOI] [PubMed] [Google Scholar]
- Carrell C. J.; Schlarb B. G.; Bendall D. S.; Howe C. J.; Cramer W. A.; Smith J. L. Biochemistry 1999, 38, 9590. [DOI] [PubMed] [Google Scholar]
- Martinez S. E.; Huang D.; Ponomarev M.; Cramer W. A.; Smith J. L. Protein Sci. 1996, 5, 1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ponamarev M. V.; Cramer W. A. Biochemistry 1998, 37, 17199. [DOI] [PubMed] [Google Scholar]
- Williamson D. A.; Benson D. R. Chem. Commun. 1998, 0, 961. [Google Scholar]
- Kennedy M. L.; Silchenko S.; Houndonougbo N.; Gibney B. R.; Dutton P. L.; Rodgers K. R.; Benson D. R. J. Am. Chem. Soc. 2001, 123, 4635. [DOI] [PubMed] [Google Scholar]
- de Vitry C.; Ouyang Y.; Finazzi G.; Wollman F.-A.; Kallas T. J. Biol. Chem. 2004, 279, 44621. [DOI] [PubMed] [Google Scholar]
- Jormakka M.; Byrne B.; Iwata S. Curr. Opin. Struct. Biol. 2003, 13, 418. [DOI] [PubMed] [Google Scholar]
- Boyington J. C.; Gladyshev V. N.; Khangulov S. V.; Stadtman T. C.; Sun P. D. Science 1997, 275, 1305. [DOI] [PubMed] [Google Scholar]
- Jormakka M.; Tornroth S.; Byrne B.; Iwata S. Science 2002, 295, 1863. [DOI] [PubMed] [Google Scholar]
- Raaijmakers H.; Macieira S.; Dias J. M.; Teixeira S.; Bursakov S.; Huber R.; Moura J. J. G.; Moura I.; Romao M. J. Structure (London, U. K.) 2002, 10, 1261. [DOI] [PubMed] [Google Scholar]
- Berks B. C.; Page M. D.; Richardson D. J.; Reilly A.; Cavil A.; Outen F.; Ferguson S. J. Mol. Microbiol. 1995, 15, 319. [DOI] [PubMed] [Google Scholar]
- Costa C.; Teixeira M.; LeGall J.; Moura J. J. G.; Moura I. JBIC, J. Biol. Inorg. Chem. 1997, 2, 198. [Google Scholar]
- Almendra M. J.; Gavel O.; Duarte R. O.; Caldeira J.; Bursakov S. A.; Costa C.; Moura I.; Moura J. J. G. J. Inorg. Biochem. 1997, 67, 16. [Google Scholar]
- Jormakka M.; Richardson D.; Byrne B.; Iwata S. Structure (London, U. K.) 2004, 12, 95. [DOI] [PubMed] [Google Scholar]
- Bertero M. G.; Rothery R. A.; Palak M.; Hou C.; Lim D.; Blasco F.; Weiner J. H.; Strynadka N. C. J. Nat. Struct. Biol. 2003, 10, 681. [DOI] [PubMed] [Google Scholar]
- Arnoux P.; Sabaty M.; Alric J.; Frangioni B.; Guigliarelli B.; Adriano J.-M.; Pignol D. Nat. Struct. Biol. 2003, 10, 928. [DOI] [PubMed] [Google Scholar]
- Dias J.; Than M.; Humm A.; Bourenkov G. P.; Bartunik H. D.; Bursakov S.; Calvete J.; Caldeira J.; Carneiro C.; Moura J. J.; Moura I.; Romao M. J. Structure (London, U. K.) 1999, 7, 65. [DOI] [PubMed] [Google Scholar]
- Najmudin S.; Gonzalez P. J.; Trincao J.; Coelho C.; Mukhopadhyay A.; Cerqueira N. M. F. S. A.; Romao C. C.; Moura I.; Moura J. J. G.; Brondino C. D.; Romao M. J. JBIC, J. Biol. Inorg. Chem. 2008, 13, 737. [DOI] [PubMed] [Google Scholar]
- Jepson B. J. N.; Mohan S.; Clarke T. A.; Gates A. J.; Cole J. A.; Butler C. S.; Butt J. N.; Hemmings A. M.; Richardson D. J. J. Biol. Chem. 2007, 282, 6425. [DOI] [PubMed] [Google Scholar]
- Schindelin H.; Kisker C.; Hilton J.; Rajagopalan K. V.; Rees D. C. Science 1996, 272, 1615. [DOI] [PubMed] [Google Scholar]
- Coelho C.; Gonzalez P. J.; Moura J. G.; Moura I.; Trincao J.; Romao M. J. J. Mol. Biol. 2011, 408, 932. [DOI] [PubMed] [Google Scholar]
- Gonzalez P. J.; Correia C.; Moura I.; Brondino C. D.; Moura J. J. G. J. Inorg. Biochem. 2006, 100, 1015. [DOI] [PubMed] [Google Scholar]
- Dauter Z.; Wilson K. S.; Sieker L. C.; Meyer J.; Moulis J. M. Biochemistry 1997, 36, 16065. [DOI] [PubMed] [Google Scholar]
- Van de Kamp M.; Floris R.; Hali F. C.; Canters G. W. J. Am. Chem. Soc. 1990, 112, 907. [Google Scholar]
- Berghuis A. M.; Brayer G. D. J. Mol. Biol. 1992, 223, 959. [DOI] [PubMed] [Google Scholar]