

Abstract
Post-transcriptional regulation of messenger RNA contributes to numerous aspects of gene expression. The key component to this level of regulation is the interaction of RNA-binding proteins (RBPs) and their associated target mRNA. Splicing, stability, localization, translational efficiency, and alternate codon use are just some of the post-transcriptional processes regulated by RBPs. Central to our understanding of these processes is the need to characterize the network of RBP-mRNA associations and create a map of this functional post-transcriptional regulatory system. Here we provide a detailed methodology for mRNA isolation using RBP immunoprecipitation (RIP) as a primary partitioning approach followed by microarray (Chip) or next generation sequencing (NGS) analysis. We do this by using specific antibodies to target RBPs for the capture of associated RNA cargo. RIP-Chip/Seq has proven to be is a versatile, genomic technique that has been widely used to study endogenous RBP-RNA associations.
Keywords: RIP-Chip, Rip-Seq, Ribonomics, mRNA localization, RNA-binding proteins (RBPs), Post-Transcription, microarray expression profiling
1. Introduction
The past decade has seen a rapid growth of research in the field of post-transcriptional gene regulation. Recently, with the emergence of siRNA and miRNA [1–5], more attention has been focused on this field as it has begun to compete with transcriptional regulation, as a major focus of gene expression regulation. The significance of post-transcriptional regulation gained prominence as it was demonstrated that the transcriptome was frequently not directly correlated with the proteome [6–8]. The discrepancies of these profiles indicate the contribution and the significance both post-transcriptional and translational processes play in the control of protein expression. Technological advances in both genome-wide and targeted gene expression analysis have reinforced our interpretation and enhanced our understanding of the significance that post-transcriptional mechanisms play in several significant cellular systems [9–13].
Immunoprecipitation of RBPs followed by genomic analysis using microarrays, known as RIP-Chip or more recently using next generation sequencing methods which is termed RIP-Seq can be a powerful in vivo, high-throughput technique for identifying specific associated RBP targets from cell extracts [8, 14–15]. RIP-Chip/Seq compliments many other RNA localization and characterization methods and has been widely used to isolate and identify mRNA and miRNA targets associated with RBPs [10, 14–21]. This article will provide procedural detail on how endogenous ribonucleoprotein (RNP) complexes can be isolated using RIP followed by subsequent extraction and identification of co-purified RNA from these complexes. This method can also target recombinant RBPs in a robust in vitro version of this method [10]. We provide an approach for identifying associated mRNA using high throughput Affymetrix cDNA microarrays, to represent the Chip portion of RIP-Chip and Illumina based high-throughput sequencing for the Seq data, however there are many other commercially available microarray and NGS technologies that are equally well suited for these methods. The nature of these genomic-based readouts necessitates a considerable component of bioinformatic analysis, which is needed for processing raw array data to generate a list of target gene mRNAs (or transcriptional fragments, in the case of RNA sequencing) as well as for statistical interpretation and analysis of the data. Initial data analysis often necessitates further computational and experimental validation of putative identified associated mRNA and potential RBP binding sites.
2. Materials
2.1 Preparation of Cell Lysate
-
Polysome Lysis Buffer (1X PLB): 100 mM KCl, 5 mM MgCl2, 10 mM HEPES, pH 7.0, 0.5% Nonidet P-40 (also known as Igepal CA-630; Sigma Cat. No. I8896). A 10X stock buffer is prepared prior to time of use (see step 2). The 10X PLB stock buffer may be stored at room temperature (RT).
To be added at time of use:
1 mM dithiothreitol (DTT), 200 units/ml RNase OUT (Invitrogen Cat. No. 10777-019), one Complete Mini, EDTA-free Protease Inhibitor Tablet (Roche Cat. No. 11836170001). The protease inhibitor cocktail tablet allows inhibition of serine and cysteine proteases during extractions from human cells.
2.2 Buffers Required for Immunoprecipitation
NT-2 buffer: 50 mM Tris pH 7.4, 150 mM NaCl, 1 mM MgCl2, 0.05% Nonidet P-40. Typically, a 5X concentration of NT-2 stock buffer is prepared. A 1X concentration of NT-2 buffer is utilized for the duration of the RIP protocol. NT-2 buffer is stored at 4°C.
NET-2 buffer (binding buffer for RNP to antibody): 1X NT-2 buffer supplemented with 20 mM EDTA pH 8.0 (Ambion Cat. No. AM9260G), 1 mM DTT, 200 units/ml RNase OUT.
Proteinase K digestion buffer: 1X NT-2 buffer supplemented with 1% sodium dodecyl sulfate (SDS), 1.2 mg/ml Proteinase K (Ambion Cat. No. AM2546).
2.3 Immunoprecipitation Reaction
Dynabeads Protein A (Invitrogen Cat. No. 100-02D), and Dynabeads Protein G (Invitrogen Cat. No. 100-04D).
5 µg of the antibody to the RNA binding protein of interest.
For RNA precipitation: acid phenol-chloroform pH 4.5, with isoamyl alcohol (IAA), (Ambion Cat. No. 9722), chloroform (Fisher Scientific Cat. No. BP1145-1), 5 M ammonium acetate (Ambion Cat. No. 9070G), 7.5 M LiCl (Ambion Cat. No. 9480), 5 mg/ml glycogen (Ambion Cat. No. 9570), 100% ethanol, 80% ethanol.
A magnetic rack (Invitrogen Cat. No. 123-21D or Millipore Cat. No. 20-400) is used to separate Dynabeads during IP. A Labquake™ tube shaker/rotator (Krackeler Cat. No. 16-4002110) is used for mixing during antibody-bead incubation, and following the addition of cell lysate (4°C overnight incubation).
Buffer NT-2 is used to wash the beads during immunoprecipitation, and a chemical duty pump (Millipore Cat. No. WP6111560) is used to aspirate supernatant/waste.
2.4 Western Blotting for Proteins of Interest
Loading buffer: 2X Laemmli buffer (Bio-Rad Cat. No. 161-0737), 2-mercaptoethanol electrophoresis grade (Fisher Scientific Cat. No. BP176-100).
SDS-PAGE buffer: 10X stock, 10X Tris/Glycine/SDS (TGS) (Bio-Rad Cat. No. 161-0772).
Gels: 4–20% Tris-HCl (Bio-Rad Cat. No. 161-1159), 7.5% Tris-HCl (Bio-Rad Cat. No. 161-1172).
SDS-PAGE ladder: Precision Plus Protein WesternC Standards (Bio-Rad Cat. No. 161-0376).
Transfer buffer: 100 ml 10X Tris/Glycine (TG) (Bio-Rad Cat. No. 161-0771), 200 ml methanol, 700 ml deionized water. Keep transfer buffer at 4°C prior to use.
Membrane: Immobilon-PSQ transfer membrane, PVDF 0.2 µm (Millipore Cat. No. ISEQ07850).
Blocking buffer: 5% non-fat milk in TBST (1 L of 10X TBST: 1.37 M NaCl, 250 mM Tris pH 8.0, 0.5% Tween-20, and titrate HCl to pH of 7.5).
Ponceau red: Stock consists of 2% Ponceau-S in 30% trichloroacetic acid and 30% sulfosalicylic acid. Dilute the Ponceau-S stock 1:10.
Primary antibody incubation buffer: Starting Block T20 (Pierce Cat. No 37543), and dilute the antibody of interest to a concentration of 1:1000–1:2000.
Secondary antibody incubation buffer: Starting Block T20 (Pierce Cat. No 37543), and dilute the appropriate secondary antibody-horseradish peroxidase (HRP) conjugate 1:10000. The same dilution applies to Precision Protein StrepTactin-HRP Conjugate (Bio-Rad Cat. No. 161-0380), and is added along with the secondary antibody of interest. The Precision Protein StrepTactin-HRP Conjugate allows for the resolution of all reference bands with sizes ranging from 10 kDa-250 kDa.
3. Methods
The RIP method (Fig.1) described here has been successfully tested on mammalian cells including stem cells (unpublished results), for the isolation of RNA integrally associated with mRNA-protein complexes (mRNPs). Iron oxide magnetic beads linked to specific antibodies through protein A or G were used to target the specific RBPs.
Figure 1.

Process flow for RIP-Chip
3.1 Preparation of Cell Lysate
Cell pellets utilized for lysate preparation are free of media and have been washed with phosphate buffered saline (PBS) prior to freezing at −80 °C. Typically 1–5 × 106 cells are used to make lysate enough for a single RIP. This may vary based on factors specific to any given experiment and also the cytoplasmic volume of the cell type being used [14]. The goal is to get highly concentrated mRNP lysate with approximately 20 – 50 mg/ml total protein in a suitable volume. We typically use 100 µl for an immunoprecipitation reaction [14].
During cell lysate preparations, the cell pellets are allowed to thaw minimally on ice, followed by the addition of an equal volume of 1X PLB. Adding the PLB buffer prior to thawing inhibits any protease activity and improves the quality of the lysate. When preparing the 1X PLB buffer, it is important to prepare a fresh buffer to ensure proper cell lysis. The main determinant of the duration of stability of the 1X PLB buffer is the activity of the protease inhibitor tablet. A solution containing a protease inhibitor tablet(s) is stable for 1–2 weeks when stored at 4°C, or a maximum of 12 weeks at −20°C.
The cell pellet-PLB mixture is occasionally vortexed to promote thawing and subsequently commence lysis. Once the cells are fully thawed, vortex vigorously to ensure proper cell lysis, aliquot and store at −80°C till further use. Poor vortexing may result in low total RNA yields when conducting IPs.
As general precautions on working with RNA, all instruments, items, tips, and cell lysates should be DNase and RNase-free. Gloves, benches, and pipettes may be cleansed using RNase Zap (Ambion Cat. No. 9780) or RNase Away (Molecular BioProducts Cat. No. 7001), followed by airdrying.
3.2 Immobilization of Antibodies on Magnetic beads
Prepare a 1:1 stock mixture of protein A and protein G coated magnetic beads and store at 4°C. The type of beads utilized depends on immunoglobulin isotype and species. It is important to consult the bead manufacturers binding chart to determine the optimal choice of beads for the particular isotype and species of antibody being used. In most instances, mouse monoclonal antibodies have strong affinity for Dynabeads Protein G and rabbit polyclonal antibodies have strong affinity for both Dynabeads Protein G and Dynabeads Protein A. Using a mixture of the beads avoids potential mistakes in selection. Dynabeads Protein A, and Dynabeads Protein G can be mixed (Protein A + Protein G stock), washed with NT-2, and stored at 4°C with 0.02% sodium azide until ready to use.
As the beads tend to aggregate and settle, vortex vigorously and add 75 µl of the protein A/G slurry into a 1.5 ml eppendorf tube. Wash twice with 0.5 ml of NT-2 buffer. Remove the buffer by placing the tube against a magnet and using a vacuum aspiration system to remove the clear liquid. There will be at least two tubes for each experiment - one negative control (i.e. T7 antibody, pre-immune serum, etc) and one experimental condition (antibody to RBP of interest). A positive control can also be included (U1-70k, PABP, etc.). While aspirating supernatants, be sure to change tips between samples and “negative” controls to avoid carryover contamination. Also, when aspirating, place the tip opposite of the bead aggregate to prevent disruption and potential contamination among IP samples.
Resuspend the beads in 100 µl of NT-2 (Optional: NT-2 with 5% bovine serum albumin) and add 5 µg antibody. Use of excess antibodies tends to minimize non specific binding of mRNA to the protein A/G beads, likely by hindering the accessible protein A/G sites.
Vortex and then tumble the tubes in rotator for a minimum of 1 hour at room temperature. Preferably, the incubation should be done at 4°C overnight for maximal binding. The antibody coated beads can be store at 4°C when supplemented with 0.02% sodium azide [14].
3.3 mRNA Localization using RIP
Centrifuge the bead/antibody slurry for 15 seconds at 5000 × g. Aspirate supernatant keeping the tube against the magnet. Add 1 ml of NT-2, mix, spin at 5000 × g for 15 s. Aspirate supernatant. Repeat this step five more times for a total of six washes with NT-2.
During the previous step, thaw the cell lysate on ice, vortex vigorously and centrifuge at 20,000 × g for 10 min at 4°C and then keep in ice.
Resuspend the beads in 900 µl of NET-2 buffer. The EDTA present in the NET-2 buffer disrupts the formation of ribonucleoprotein complexes between ribosomal RNA (rRNA) and ribosomal proteins [22]. This prevents the immunoprecipitation of rRNA.
Add 100 µl of lysate (clear upper layer) to each IP mixture. Invert to mix and give it a quick spin. Keeping the sample on magnet remove 100 µl and place it into a new tube. This is called the ‘total’. Tumble the IP tubes at 4°C overnight. When adding cell lysate to immunoprecipitation samples, it is important to leave the resulting cellular pellet undisturbed. Only use the lysate (“upper”) layer. If there is any lysate remaining, it may be saved for later use.
After overnight incubation, wash six times with 1 ml of ice cold NT-2. If the first RIP attempt shows high background use four washes with NT-2 followed by two washes with NT-2 having 0.5-3 M urea [15, 23]. Before spinning down the beads for the sixth wash, take out 100 µl of the bead slurry (out of 1 ml final volume) for IP Western (Fig.2). Also take out 10 µl from ‘total’, add 10 µl 2X SDS-PAGE loading dye and store at −20°C for Western blotting. Also when washing beads in NT-2 after cell lysate incubation (rotating at 4°C), be sure to vortex samples vigorously to eliminate potential background. Background may be visualized by obtaining a Western blot positive result for a “negative” control precipitation (e.g. IgG Control), due to poor vortexing.
It is important to obtain an aliquot during the sixth wash (prior to Proteinase K digestion), to test the efficiency of immunoprecipitation by Western Blotting. Proteins are eluted off Dynabeads using 1X SDS-PAGE loading dye followed by heating at 95°C. The beads can be centrifuged down at 8,000 × g for 2 min (once allowed to briefly cool), and supernatant directly applied to an SDS-PAGE. When testing an antibody for the first time, it is important to verify that the protein of interest is being precipitated during immunoprecipitation. In some instances, it may be necessary to try several antibodies before selecting an antibody capable of precipitating the target RBP of interest.
During IP, it is essential not to “pool” IP samples before testing to prevent contaminating viable samples. Each individual precipitation must remain separate.
Millipore commercially supplies EZ-Magna RIP™ (Millipore Cat. No. 17-701), a universal RIP kit, which enables immunoprecipitation using antibodies directed against RBP’s to be conducted. The kit supplies control antibodies and primers allowing for RT-PCR validation of precipitated mRNA.
Figure 2.

Examples of IP-Western blots displaying identification of target proteins. Each blot was resolved using Genscript One-hour IP-Western kits. Shown on the top, is an IP in human cervical cancer cell line Hela, using CUG triplet repeat RNA binding protein 1 (CUG-BP1) antibody (Millipore Cat. No. 05-621). Lanes within this image (on top) are: (A) SDS-PAGE Protein Standard; (B) Mouse IgG (Millipore Cat. No. 12-371); (C) CUG-BP1 protein IP; (D) Total obtained during IP. Shown on the bottom, is again, an IP in human cervical cancer cell line Hela, using Drosha antibody (Millipore Cat. No. 07-717). Drosha is a protein responsible for catalyzing the cleavage of dsRNA to siRNA. Lanes are (image on the bottom): (A) Precision Plus WesternC Protein Standard (Bio-Rad Cat. No. 161-0376); (B) Total RNA obtained during IP; (C) Drosha protein IP; (D) IP using Anti-T7-Tag® Monoclonal antibody (Novagen Cat. No. 69522).
3.4 RNA Purification
Resuspend the beads in 150 µl Proteinase K buffer. To each ‘total’ tube add 36 µl NT-2 + 15 µl 10% SDS + 9 µl Proteinase K. Incubate all the IP tubes and total tubes at 55°C for 30 min (use Eppendorf Thermomixer© R dry block heating and cooling shaker).
While incubating, spin down the aliquot that you took for IP Western, remove supernatant and resuspend the beads in 30 µl of 1X SDS-PAGE loading dye, store at −20°C.
After 30 min, give the tubes a quick spin to bring everything down, add an equal volume (150 µl) buffer-saturated-acid phenol-chloroform (pH 4.5) with isoamyl alcohol, vortex and centrifuge at 20,000 × g for 10 min. Remove aqueous (upper) layer carefully without disturbing the beads and proteins settled at the interface, and place in a new tube. Add 150 µl chloroform, vortex and spin. Remove the supernatant (upper layer).
To the supernatant add 50 µl 5 M ammonium acetate, 15 µl of 7.5 M LiCl, 5 µl of 5 mg/ml glycogen and 1 ml of cold 100% ethanol. Keep in −80°C for at least half hour.
Spin at 20,000 × g for 30 min at 4°C. Decant ethanol, leaving the RNA pellet. Wash once with 80% ethanol, spin again for 30 min. Dry in Vacufuge™ concentrator (5 min at RT) and resuspend the pellet in 10 ml of RNase-free water or any other buffer of choice and store it at −80°C. For long-term storage, stop at the previous step.
Typically, optical absorbance of the RNA can be measured using a Nanodrop® spectrophotometer. Ideally we expect both the A260/A280 and A260/A230 ratios to be close to 2.0, implying the purity of RNA and the absence of any contaminating proteins or chemicals. If this ratio is less than 1.8, there may be problems with further downstream applications.
The molecular weight profile of the subset of RNAs immunoprecipitated can be analyzed on a nanochip using Agilent’s BioAnalyzer. Nanochip is a convenient alternative to using formaldehyde-agarose gels. For the total RNA (or ‘input’) it is helpful to evaluate the 18s/28s rRNA ratio. A ratio between 1.6 and 2.0 indicates RNA of good integrity. However for the immunoprecipitated material such ratio cannot be obtained unless rRNAs are known to be targets of the RNA Binding Protein in question. And so, a bioanalyzer profile of immunoprecipitated RNA only serves to provide information about any extensive degradation of RNA [24].
The quality of RNA can also be validated using RT-PCR. An example of HuR IP analysis is shown in Figure 3. The PCR was performed using gene specific primers for GAPDH and β-actin. GAPDH mRNA does not bind to HuR, however it does have a poly(A) tail and should bind to PABP. Therefore we expect to see enrichment for GAPDH in the PABP IP but not in the HuR IP. Beta-actin is a known HuR target and is also polyadenylated; therefore enrichment is expected in both IPs..
Figure 3.

The figure depicts a RIP experiment followed by RT-PCR readout on a 1.5% agarose gel of immunoprecipitations targeting HuR and PABP in K562 cell line. The PCR was performed using gene specific primers for GAPDH (top) and β-actin (bottom). mRNA from prostate carcinoma (PC-3) cells were used as the positive control. The last lane shows RT-PCR done with no template.
3.5 IP Western
Western Blotting is a standard technique used in several labs. To help the readers below is a compilation of useful step by step tips to perform a successful experiment. Figure 2 shows a sample IP Western blot. When using Immobilon-PSQ transfer membrane, PVDF 0.2µm (Millipore Cat. No ISEQ07850), always remember to soak the membrane for 10 seconds in methanol prior to transfer, followed by rinsing in transfer buffer to ensure proper protein transfer. When using nitrocellulose membranes, do not soak in methanol prior to use. It is beneficial to soak nitrocellulose membranes in transfer buffer briefly before preparing the gel sandwich.
In some instances, especially when transferring multiple blots using the same power supply, it is appropriate to extend the time of transfer by 30 minutes.
When attempting to resolve high molecular weight proteins (>100kDa), reducing the methanol percentage to 10% will allow large proteins to travel easier.
When resolving low molecular weight proteins (<100kDa), keep the methanol concentration at 20%.
A 4 – 20% Tris-HCl gel resolves proteins of 10 – 250 kDa, and is usually utilized when conducting an SDS-PAGE electrophoresis. In some instances, it is beneficial to use a 7.5% Tris-HCl gel to resolve proteins of high molecular weight (>100kDa). A 7.5% Tris11 HCl gel is typically used to resolve a protein(s) of molecular weight(s) between 25 – 200kDa.
Always remember to use a positive control lysate (usually the lysate used for immunoprecipitation) to demonstrate protocol efficiency and verify the antibody is capable of identifying target protein which may not be present in experimental (immunprecipitation) samples. If desired, you may load a control with a known molecular weight (i.e. β-actin, GAPDH, Tubulin) to verify that lanes in the gel have been evenly loaded with sample and it is also beneficial when comparing protein expression levels between samples.
After creating the gel sandwich prior to transfer, it is important to remove possible air bubbles from within. This can be done by rolling a 10ml pipet or polystyrene tube over the sandwich. Air bubbles tend to disrupt protein transfer and will minimize transfer efficiency.
When conducting protein transfer, it is important to create a cold environment (it increases the efficiency of protein transfer). Thus, we use ice blocks both within and outside of the gel transfer unit and conduct at 4°C.
To verify protein transfer (optional), Ponceau Red is added to the membrane and incubated while rocking for 5 minutes and wash with deionized water until the protein bands are well defined. A digital image may be obtained and used for comparison after antibody detection.
In some instances, primary antibodies will produce stronger signal when diluted in blocking buffer (5% non-fat milk in TBST), rather than TBST alone. This is not recommended in cases when high background may be an issue.
For some antibodies, signal is stronger when primary antibody incubation is conducted at room temperature rather than 4°C. However, it is recommended to incubate the primary antibody at 4°C to prevent contamination and consequent destruction of protein. Again, this preference varies among antibodies.
Membranes may be saved in a 1X TBST buffer, supplemented with 0.2% sodium azide (NaN3) or may be dried, covered in filter paper, and saved for later use.
3.6 Bioinformatics analysis
The bioinformatic portion of RIP-Chip/Seq begins with the processing of microarrays to produce a list of genes. Alternatively, a list of genes and/or transcriptional fragments may be developed if deep sequencing is used. While particular transcript variants can be derived from RNA sequencing data, localized enrichment of RBP binding site regions (due to fragmentation) in RIP’d RNAs and associated depletion of other segments may artificially affect the identified proportions.
Gene level arrays contain sets of probes designed against sequence from portions of exons in well-annotated genes and are therefore limited to reporting on these genes. RNA Sequencing yields short segment reads for sequences present in a sample. These reads are then mapped against all genomic sequence, regardless of annotation or expected transcriptional activity. This enables a potentially unbiased, full genome interrogation.
3.7 Gene Array Processing
There are a number of options in manufacturer and model of gene level microarrays as well as tools for their analysis. The 'Human Gene 1.0 ST Array' from Affymetrix and Agilent's GeneSpring GX10 software have been widely used. A standard approach for analyzing these arrays is to gather a minimum of triplicate sets for each of total input RNA, treatment (i.e., using antibody for RBP of interest) RIP RNA, and negative control RIP RNA (i.e., antibody for the G10 protein from T7 bacteriophage). Set members are subjected to basic quality control, which involves comparing correlation values and principal component analysis (Fig. 4) among replicate set members.
The iterative PLIER16 algorithm is then used to gather expression results in each group, which are then filtered for the 20th to 100th percentile. PLIER16 is preferred as it has been shown to be the most consistent with RT-PCR results when preprocessing Affymetrix microarray data [25]. When compared with multiple popular algorithms, PLIER16 has also shown superior performance in reducing false positives when analyzing poorly performing probe sets [26].
A list can then be generated using genes that show a minimum two-fold increase of measured expression in treatment versus input sets. A separate list is created for genes that show a similar level of increase in the negative control versus input sets. This second list is then subtracted from the first to remove RNAs that appear to have a general propensity for inclusion by the RIP process (likely due to an affinity for the beads).
A one-way analysis of variance (ANOVA) can then be performed on this RNA subset. Since microarray expression comparison can be viewed as essentially a set of many experiments, each with an associated null hypothesis a multiple testing correction is applied to the ANOVA p-values (for instance, the Benjamini-Hochberg method [27]). The final set of genes is then filtered based on a desired confidence level, such as 95% (p = 0.05).
Figure 4.
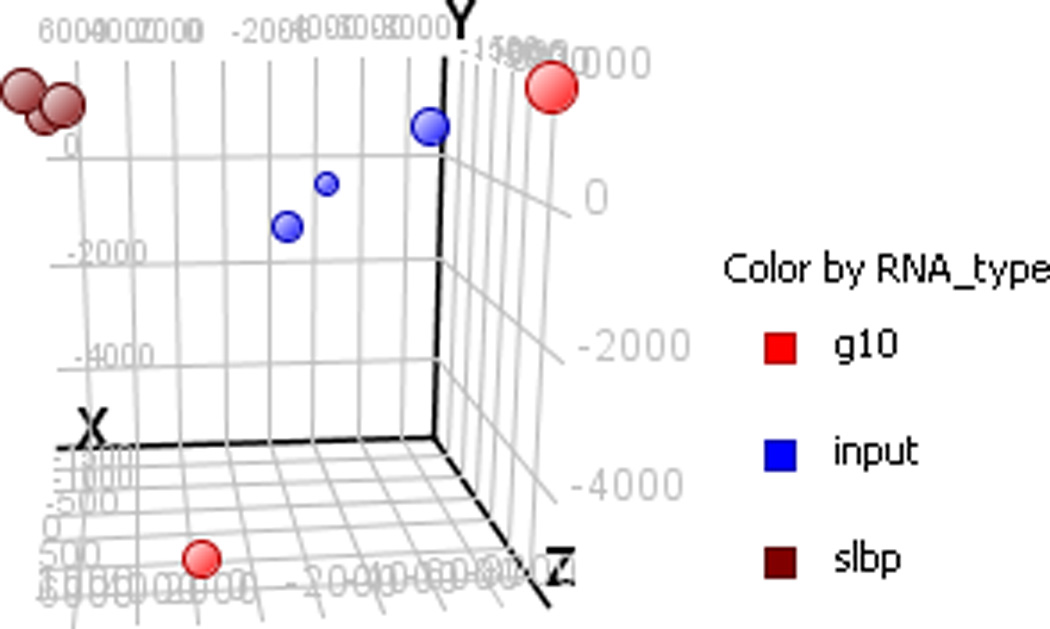
Example of principal component analysis of samples from a stem loop binding protein (SLBP) RIP. PCA provides an indication of similarity amongst samples. This particular image shows some variation in the total transcribed RNA with a tighter correlation amongst the SLBP samples (expected if they represent a specific subset of the overall transcriptome). The G10 samples are poorly correlated, which is to be expected if they reflect mostly random background binding (note there are only two G10 replicates in this example).
3.8 RNA Sequencing Library Processing
Whereas microarrays consist of probe sets engineered to test various segments of exons in well-annotated transcripts, next generation RNA sequencing offers a potentially unbiased interrogation of the genome by providing short oligonucleotide reads that may then be mapped to any matching genomic segment, regardless of annotation. RIP-Seq can allow the examination of RBPs associating with RNA outside the boundaries of traditionally annotated mRNA transcripts including introns and non-coding RNA.
We stress the phrase “potentially unbiased”, as although the mapping is not limited by annotation, the choice of sequencing library preparation protocol can substantially bias what populations (types) of RNA actually get sequenced. For example, a poly(A) separation will reduce much if not most of the signal from non-coding RNAs (ncRNA) or a size exclusion step will remove small ncRNAs. Depth of sequencing (that is, the number of reads sequenced per sample) must also be taken into account to avoid biasing against low abundance RNAs. A static value is not appropriate as the binding profile of the RBP in question (e.g. “what subpopulation of RNA is bound by it?”) will affect the range of signal scale. For example, in our RIP-Seq of the RBP ELAVL1, we observed low double digit values as raw enriched signal for some known targets when library was sequenced at ~30 million reads. We suggest this depth be considered as a baseline for RBPs of moderate or unknown binding activity.
Once sequencing libraries have been produced, Tophat and Bowtie programs can be used to map reads and produce signal by genomic position. This signal is normalized across libraries by number of mapped reads to allow direct quantitative comparison. Various options for peak calling exist. We have previously used an “above threshold, maxgap/min-run” approach where the gap and run numbers are related to the length of our sequencing reads. For example, we typically use a 36 base, single end read library analyzed with maximum gap equal to 2/3 the read length (24) and minimum run equal to 4/3 the read length (48) and a threshold cutoff of genomic wide signal level (e.g., 60th percentile).
Intersect regions of peaks across RIP libraries are identified as “regions of high agreement” and the average signal from these regions in the target RIPs can be compared to those in the negative control RIPs via a t-test and an associated p-value is produced. Following filtering (e.g., p-val < .05), a set of statistically significant transcriptional fragments (transfrags) are produced (Fig. 5).
Alternative mapping tools are available as well as other options for differential analysis. The aforementioned approach is one choice we have used very succesfully.
Figure 5.
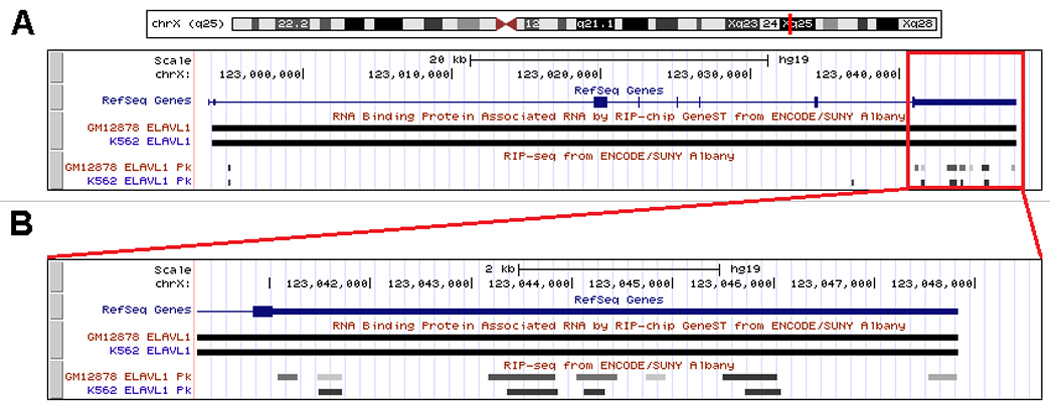
Excerpt from a UCSC Genome Browser window showing ELAVL1(HuR) RIP based tracks for GeneST and Sequencing data in K562 and GM12878 cell lines. Note agreement across both cell lines for full message (GeneST) and transfrag level(RIP-seq) signals in XIAP, an experimentally confirmed [36] target of ELAVL1 binding. (A) Browser view of full XIAP transcription region on Chromosome X. (B) Browser view zoomed into the 3’ UTR region of XIAP.
3.9 Post Analysis
Microarrays and sequencing can essentially be considered "systems biology" tools as they allow high throughput performance of thousands, and potentially millions, of individual experiments simultaneously. The quantity of data produced can be quite substantial and frequently is discernible only by computational means including advanced statistical analysis. This is particularly true in the case of RIP, where protein-protein bridging may precipitate indirectly bound mRNA [10]. For example, histone stem-loop binding protein’s (SLBP) interaction with the translation initiation machinery [29] may make it particularly prone to such relationships with non HSL-motif bearing mRNA. The presence of such indirectly related RNA in RIP samples adds considerable noise when attempting to identify an RNA-binding motif as signal. The complexities of de-novo binding motif characterization, particularly amongst noise, have been described previously [30]. It should be noted, that aside from motif identification, this indirect relationship (at least in such cases as the SLBP example) is a biological reality and therefore legitimate information to use in characterizing the RBP’s system behavior.
Options for secondary analysis of the processed array data are numerous. These include not only looking for potential binding sites, but also methods for examining more general relationships of the RBP with its cellular system. For instance, miRanda [31] and other similar tools can be used to analyze a set of RBP related genes for an overrepresentation of target sites to a particular miRNA.
Associated mRNA may also be programmatically examined for common sequence/secondary structures by leveraging custom programs with output from tools such as those found in the Vienna RNA Secondary Structure Package[32]. Alternatively, transcriptional fragment sets obtained for a given RBP might be used to discover a number of intron hits across diverse genes that share associations with a common GeneOntology (GO) [33] term or Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway [34], perhaps elucidating a role in alternative splicing for the examined RBP in that pathway. These are just a few illustrative examples of further bioinformatic analysis that may be performed on RIP-Chip array results.
4. Conclusion
RIP-Chip, particularly through ribonomic profiling by its in vivo application, offers enormous insight into the post-transcriptional regulatory machinery. Recent research has shown that deep sequencing technology used for RNA-Seq [35] can be used for high throughput transcriptome profiling, and offers some benefits not available through the use of cDNA microarrays. Sequencing technology has already been used for chromatin immunoprecipitation analysis as ChIP-Seq [36]. RIP-Seq protocols are under development and a performance comparison of the RNA-Seq and microarray platforms is forthcoming.
Regardless of the technology used to identify the RNA present in a RIP sample, the results represent a “snap shot” view of RNA sets associated directly or indirectly with a particular RBP. These views provide a means to identify not only RBP-RNA interactions, but also potentially RBP-RBP interactions by the comparison of sets across RIP experiments for different RBPs. Data can also be compared to discern trans-acting modulation of RBP-RNA binding. Ultimately, this approach combined with new advances in RNA based bioinformatic approaches will lead to a more complete understanding of post transcriptional regulatory networks.
Acknowledgments
We thank P. D. Kutscha for assistance with experiments and helpful input. NIH U01 FHG004571, NIH R41 GM097811 and SUNY-RF TAF funding supported FD SJ and SAT.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References (*recommended reading)
- 1.Fire A, et al. Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature. 1998;391:806–811. doi: 10.1038/35888. [DOI] [PubMed] [Google Scholar]
- 2.Moazed D. Small RNAs in transcriptional gene silencing and genome defence. Nature. 2009;457:413–420. doi: 10.1038/nature07756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lewis BP, Burge CB, Bartel DP. Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell. 2005;120:15–20. doi: 10.1016/j.cell.2004.12.035. [DOI] [PubMed] [Google Scholar]
- 4.Carthew RW, Sontheimer EJ. Origins and Mechanisms of miRNAs and siRNAs. Cell. 2009;136:642–655. doi: 10.1016/j.cell.2009.01.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jinek M, Doudna JA. A three-dimensional view of the molecular machinery of RNA interference. Nature. 2009;457:405–412. doi: 10.1038/nature07755. [DOI] [PubMed] [Google Scholar]
- 6.Futcher B, et al. A sampling of the yeast proteome. Mol Cell Biol. 1999;19:7357–7368. doi: 10.1128/mcb.19.11.7357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gygi SP, et al. Correlation between protein and mRNA abundance in yeast. Mol Cell Biol. 1999;19:1720–1730. doi: 10.1128/mcb.19.3.1720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8*.Tenenbaum SA, et al. Identifying mRNA subsets in messenger ribonucleoprotein complexes by using cDNA arrays. Proc Natl Acad Sci U S A. 2000;97:14085–14090. doi: 10.1073/pnas.97.26.14085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Perou CM, et al. Distinctive gene expression patterns in human mammary epithelial cells and breast cancers. Proc Natl Acad Sci U S A. 1999;96:9212–9217. doi: 10.1073/pnas.96.16.9212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.*.Townley-Tilson WH, et al. Genome-wide analysis of mRNAs bound to the histone stem-loop binding protein. RNA. 2006;12:1853–1867. doi: 10.1261/rna.76006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.*.Sanchez-Diaz P, Penalva LO. Post-transcription meets post-genomic: the saga of RNA binding proteins in a new era. RNA Biol. 2006;3:101–109. doi: 10.4161/rna.3.3.3373. [DOI] [PubMed] [Google Scholar]
- 12.Lockhart DJ, Winzeler EA. Genomics, gene expression and DNA arrays. Nature. 2000;405:827–836. doi: 10.1038/35015701. [DOI] [PubMed] [Google Scholar]
- 13.*.Mata J, Marguerat S, Bahler J. Post-transcriptional control of gene expression: a genomewide perspective. Trends Biochem Sci. 2005;30:506–514. doi: 10.1016/j.tibs.2005.07.005. [DOI] [PubMed] [Google Scholar]
- 14.Tenenbaum SA, et al. Ribonomics: identifying mRNA subsets in mRNP complexes using antibodies to RNA-binding proteins and genomic arrays. Methods. 2002;26:191–198. doi: 10.1016/S1046-2023(02)00022-1. [DOI] [PubMed] [Google Scholar]
- 15.*.Keene JD, Komisarow JM, Friedersdorf MB. RIP-Chip: the isolation and identification of mRNAs, microRNAs and protein components of ribonucleoprotein complexes from cell extracts. Nat Protoc. 2006;1:302–307. doi: 10.1038/nprot.2006.47. [DOI] [PubMed] [Google Scholar]
- 16.*.Brown V, et al. Microarray identification of FMRP-associated brain mRNAs and altered mRNA translational profiles in fragile X syndrome. Cell. 2001;107:477–487. doi: 10.1016/s0092-8674(01)00568-2. [DOI] [PubMed] [Google Scholar]
- 17.Darnell JC, et al. Fragile X mental retardation protein targets G quartet mRNAs important for neuronal function. Cell. 2001;107:489–499. doi: 10.1016/s0092-8674(01)00566-9. [DOI] [PubMed] [Google Scholar]
- 18.Wang WX, et al. Anti-Argonaute RIP-Chip shows that miRNA transfections alter global patterns of mRNA recruitment to microribonucleoprotein complexes. RNA. 2001;16:394–404. doi: 10.1261/rna.1905910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.*.Gerber AP, Herschlag D, Brown PO. Extensive Association of Functionally and Cytotopically Related mRNAs with Puf Family RNA-Binding Proteins in Yeast. PLoS Biol. 2004;2:E79. doi: 10.1371/journal.pbio.0020079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.*.Lopez de Silanes I, et al. Identification of a target RNA motif for RNA-binding protein HuR. Proc Natl Acad Sci U S A. 2004;101:2987–2992. doi: 10.1073/pnas.0306453101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mazan-Mamczarz K, et al. Post-transcriptional gene regulation by HuR promotes a more tumorigenic phenotype. Oncogene. 2008;27:6151–6163. doi: 10.1038/onc.2008.215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Okada S, et al. HSP70 and ribosomal protein L2: novel 5S rRNA binding proteins in Escherichia coli. FEBS Lett. 2000;485:153–156. doi: 10.1016/s0014-5793(00)02184-0. [DOI] [PubMed] [Google Scholar]
- 23.Baroni TE, et al. Advances in RIP-chip analysis : RNA-binding protein immunoprecipitationmicroarray profiling. Methods Mol Bio. 2008;419:93–108. doi: 10.1007/978-1-59745-033-1_6. [DOI] [PubMed] [Google Scholar]
- 24.Jain R, et al. RIP-CHIP in drug development. Methods Mol Biol. 2010;632:159–171. doi: 10.1007/978-1-60761-663-4_10. [DOI] [PubMed] [Google Scholar]
- 25.Gyorffy B, et al. Evaluation of microarray preprocessing algorithms based on concordance with RT-PCR in clinical samples. PLoS On. 2009;4:e5645. doi: 10.1371/journal.pone.0005645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Seo J, Hoffman EP. Probe set algorithms: is there a rational best bet? BMC Bioinformatics. 2006;7:395. doi: 10.1186/1471-2105-7-395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Benjamini Y, Hochberg Y. Controlling the false discovery rate: A practical and powerful approach to multiple testing. J. Roy. Statist. Soc. Ser. 1995;57:289–300. [Google Scholar]
- 28.Birney E, et al. Identification and analysis of functional elements in 1% of the human genome by the ENCODE pilot project. Nature. 2007;447:799–816. doi: 10.1038/nature05874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gorgoni B, et al. The stem-loop binding protein stimulates histone translation at an early step in the initiation pathway. RNA. 2005;11:1030–1042. doi: 10.1261/rna.7281305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Doyle F, et al. Bioinformatic tools for studying post-transcriptional gene regulation : The UAlbany TUTR collection and other informatic resources. Methods Mol Biol. 2008;419:39–52. doi: 10.1007/978-1-59745-033-1_3. [DOI] [PubMed] [Google Scholar]
- 31.Enright AJ, et al. MicroRNA targets in Drosophila. Genome Bio. 2003;5:R1. doi: 10.1186/gb-2003-5-1-r1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hofacker IL, Stadler PF. Memory efficient folding algorithms for circular RNA secondary structures. Bioinformatics. 2006;22:1172–1176. doi: 10.1093/bioinformatics/btl023. [DOI] [PubMed] [Google Scholar]
- 33.Ashburner M, et al. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet. 2000;25:25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kanehisa M, Goto S. KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000;28:27–30. doi: 10.1093/nar/28.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mortazavi A, et al. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat Methods. 2008;5:621–628. doi: 10.1038/nmeth.1226. [DOI] [PubMed] [Google Scholar]
- 36.Casolaro V, et al. Posttranscriptional regulation of IL-13 in T cells: role of the RNA-binding protein HuR. J Allergy Clin Immunol. 2008;121:853–894. doi: 10.1016/j.jaci.2007.12.1166. [DOI] [PMC free article] [PubMed] [Google Scholar]