

Abstract
Receptor interacting protein (RIP)-1 kinase activity mediates a novel pathway that signals for regulated necrosis, a form of cell death prominent in traumatic and ischemic brain injury. Recently, we showed that an allosteric inhibitor of RIP-1 kinase activity, necrostatin-1 (Nec-1), provides neuroprotection in the forebrain following neonatal hypoxia–ischemia (HI). Because Nec-1 also prevents early oxidative injury, we hypothesized that mechanisms involved in this neuroprotection may involve preservation of mitochondrial function and prevention of secondary energy failure. Therefore, our objective was to determine if Nec-1 treatment following neonatal HI attenuates oxidative stress and mitochondrial injury. Postnatal day (p) 7 mice exposed to HI were injected intracerebroventricularly with 0.1 μL (80 μmol) of Nec-1 or vehicle. Nec-1 treatment prevented nitric oxide (NO•), inducible nitric oxide synthase (iNOS) and 3-nitrotyrosine increase, and attenuated glutathione oxidation that was found in vehicle-treated mice at 3 h following HI. Similarly, Nec-1 following HI prevented: (i) up-regulation of hypoxia inducible factor-1 alpha (HIF-1α) and BCL2/adenovirus E1B 19 kDa protein-interacting protein 3 (BNIP3) expression, (ii) decline in mitochondrial complex-I activity, (iii) decrease in ATP levels, and (iv) mitochondrial structural pathology in astrocytes and in neurons. Up-regulation of glial fibrillary acidic protein (GFAP) following HI was also prevented by Nec-1 treatment. No differences by gender were observed. We conclude that Nec-1 immediately after HI, is strongly mitoprotective and prevents secondary energy failure by blocking early NO• accumulation, glutathione oxidation and attenuating mitochondrial dysfunction.
Keywords: astrocytes, hypoxia–ischemia, mitochondria, neurons, receptor interacting protein-1 kinase, regulated necrosis
INTRODUCTION
Following neonatal hypoxic–ischemic (HI) brain injury, a broad variety of neuronal death phenotypes are observed in the rodent forebrain (Northington et al., 2007). Although hybrid forms along a continuum between apoptosis and necrosis were originally described in the immature model (Portera-Cailliau et al., 1997), they are also found in adult models and in cell culture (Degterev et al., 2005; Festjens et al., 2006). Regulated necrosis, also called programmed cell necrosis (Galluzzi et al., 2011), has mechanistic similarities to apoptotic–necrotic hybrids and is classically described upon activation of the death receptor (tumor necrosis factor (TNF)-receptor superfamily) in the setting of caspase inhibition and/or mitochondrial failure (Han et al., 2009). Regulated necrosis proceeds with subsequent receptor-interacting protein (RIP)-1 kinase activation and formation of RIP-1–RIP-3 complex (necrosome) (Holler et al., 2000; Cho et al., 2009; Kim et al., 2010).
A selective and potent allosteric inhibitor of RIP-1 kinase, necrostatin-1 (Nec-1), prevents the progression of regulated necrosis in adult murine models of traumatic and ischemic cerebral and ischemic myocardial injury (Degterev et al., 2005; Lim et al., 2007; Smith et al., 2007). Similarly, inhibition of RIP-1 kinase activity provides neuroprotection in the cortex, hippocampus and thalamus at subacute and chronic stages of degeneration following neonatal HI in the mouse model, an effect that appears to be more robust in male mice compared to female mice (Northington et al., 2011a). RIP-1 kinase inhibition immediately after neonatal HI, using Nec-1, also decreases protein oxidative modification and inflammatory markers expression within first 24 h following HI, suggesting that histologic techniques may lack sensitivity to detect smaller acute changes (Northington et al., 2011a). Other intermediate steps between prevention of necrosome formation and neuroprotection are mostly unknown.
Mitochondrial dysfunction and energy failure is a recognized hallmark of necrotic cell death (Eguchi et al., 1997; Nicotera and Lipton, 1999) and well described following neonatal HI (Blomgren and Hagberg, 2006; Northington et al., 2007). Nitric oxide (NO•) inhibits in vitro NADH dehydrogenase (complex I) function, ultimately depleting intracellular adenosine-5′-triphosphate (ATP) and promoting a switch from apoptosis to necrosis (Nicotera et al., 1998; Leist et al., 1999; Riobo et al., 2001). In cell cultures, Nec-1 has no direct antioxidant effects; however, it prevents NO• formation and mitochondrial complex I dysfunction while increasing reduced glutathione (GSH) levels and preventing glutamate-induced programmed necrosis (Xu et al., 2007; Davis et al., 2010). From these data we hypothesized that the neuroprotection afforded by Nec-1 treatment following HI in vivo was mediated by prevention of secondary energy failure via blockade of NO• accumulation with resultant mitochondrial preservation. Our main goal was to determine if Nec-1 would provide biochemical and ultrastructural protection of mitochondrial following neonatal HI. We evaluated levels of NO•, inducible nitric oxide synthase (iNOS) and 3-nytrotyrosine as well as gluthatione oxidation and markers of mitochondrial function (complex I activity and ATP levels) and ultrastructure in mice treated with Nec-1 or vehicle following neonatal HI. We also investigated the expression of BCL2/adenovirus E1B 19 kDa protein-interacting protein 3 (BNIP3) which in conditions triggering ROS accumulation, such as neonatal HI, inserts into the mitochondrial membrane triggering a necrotic-like cell death, similar to regulated necrosis associated with energy failure (Vande Velde et al., 2000; Kubli et al., 2008).
EXPERIMENTAL PROCEDURES
Animals
All experiments were performed with approval by the Institutional Animal Care and Use Committee at Johns Hopkins University – School of Medicine and followed the Guide for the Care and Use of Laboratory Animals provided by the National Institutes of Health, US Department of Health and Human Services (NIH Publications No. 80-23, revised in 1996). All efforts were made to minimize the number of animals used and their suffering.
Neonatal mouse hypoxic–ischemic brain injury model and tissue preparation
We used the Vannucci model adapted for neonatal mice to induce HI in C57B6 mice at postnatal day (p) 7 (ligation of the right common carotid artery under anesthesia with isofluorane and subsequent exposure to FiO2 = 0.08 for 45 min) (Graham et al., 2004). Within 15 min after hypoxia, the mice were exposed to a second brief period of anesthesia with isofluorane followed by intracerebroventricular injection of 0.1 μl of 80μmol of Nec-1 (5-(1H-indol-3-ylmethyl)-3-methyl-2-sulfanylideneimidazolidin-4-1, Calbiochem-EMD Chemicals Group, Gibbstown, NJ), an allosteric inhibitor of RIP-1 kinase, or vehicle (methyl-β-cyclodextrin, Sigma, St. Louis, MO, USA). Pups were returned to the dam until they were killed at 3 h, 24 h (p8) and 96 h (p11) (n = 5–12/treatment/gender/time) for biochemical analysis and perfusion. Controls were age-matched and gender-matched naive littermates not exposed to HI or to treatment with vehicle or Nec-1.
Mice were killed with an exposure to 20% (v/v) mixture of isoflurane in propylene glycol via one-drop exposure method (Markovic and Murasko, 1993). Animals were then decapitated and forebrain tissue was rapidly dissected and frozen in isopentane on dry ice. Pieces of fresh tissue from forebrain were obtained for the experiments described below.
Nitric oxide colorimetric assay
NO• was assessed indirectly by measuring the levels of oxidized forms (nitrites and nitrates) in samples using the Nitric Oxide Colorimetric Assay Kit (ab65328; Abcam, Cambridge, MA, USA). A standard curve was generated to measure levels between 1 and 100 μM of nitrite per well. Within 2 weeks of freezing, tissues were homogenized at 1:10 (w/v) using ice-cold homogenization buffer prepared in 20 mM Tris/HCl, containing 1 mM EDTA, 5 mM EGTA, 0.1 mM PMSF, 10 mM benzamidine and protease inhibitors (Complete® Protease Inhibitor Cocktail tablets, Roche Apply Science, Indianapolis, IN) (n = 6–8 mice/group). A 5-μl aliquot of homogenized tissue was used to determine total protein concentration using Bradford assay. A 200-μL aliquot of homogenized tissue was centrifugated at 10,000g at 4 °C for 2 min and the clarified supernatant recovered. The clarified sample was deproteinated to improve NO• stability by adding ice-cold 5% metaphosphoric acid in a 1:1 ratio (v:v) (Sigma–Aldrich), mixing and spinning at 10,000g for 5 min. Clarified deproteinated samples (supernatants) and standards were exposed to nitrate reductase and cofactors for 1 h at room temperature to transform nitrate to nitrite. Following application of the enhancer supplied by manufacturer, Griess reaction reagents were applied to convert nitrite to a purple azo cromophore compound and developed over 10 min providing a lower limit of detection of 1 μM at 540 nm using a linear model in a microplate reader.
iNOS, 3-nitrotyrosine, GFAP and BNIP3 protein expression
Ipsilateral forebrain samples were obtained and frozen at 3 h, 24 h (p8) and p11 post-HI from naive control, vehicle and Nec-1-treated mice (n = 5–9 mice/treatment/gender/time). Protein homogenates were prepared as previously described (Northington et al., 1996) and concentrations were determined using Bradford assay. Twenty-μg aliquots of homogenized protein were diluted 2:1 (v:v) in loading buffer containing 20% (w/v) glycerol and loaded onto 15% SDS–PAGE. Protein was transferred to nitrocellulose membrane, stained with Ponceau S, blocked with 2.5% nonfat dry milk (or 0.1% bovine serum albumin (BSA) for 3-nitrotyrosine) and with 0.1% Tween-20 in 50 mM Tris-buffered saline (TBST, 50 mM Tris/HCl and 150 mM NaCl, pH 7.4). Nitrocellulose membranes were consecutively incubated overnight at 4 °C with primary antibodies at 1:200 (except for 3-nitrotyrosine at 1:40,000). After exposure to each primary antibody, membranes were washed with 2.5% nonfat dry milk (or TBST for 3-nitrotyrosine), exposed to secondary antibodies for 1 h and then developed with enhanced chemiluminescence using SuperSignal kit (Thermo Scientific, Rockford, IL). To quantify protein immunoreactivity, films were scanned using Adobe Photoshop (Adobe Systems Inc., San Jose, CA), and optical density (OD) was determined with NIH Image J Software (NIH, Bethesda, MD) adjusted for background. The reliability of sample loading and protein transfer was evaluated by staining nitrocellulose membranes with Ponceau S before immunoblotting.
Antibodies
For iNOS immunoblotting, we used a rabbit polyclonal antibody raised against amino acids 2–175 at NOS2 N-terminus (sc-8310, Santa Cruz Biotechnology Inc., Santa Cruz, CA) which detects a single 130 kDa band (1 μg/mL). There is no NOS1/NOS3 cross reactivity with this antibody. For protein nitration, we used a mouse monoclonal antibody raised against 3-nitrotyrosine (Ab52309, Abcam Inc., Cambridge, MA) (0.025 μg/mL). For GFAP immunoblotting, we used a rabbit polyclonal antibody raised against amino acids 1–50 of GFAP (sc-9065) that detects a 50-kDa band and smaller breakdown products (1 μg/mL). For BNIP3, we used a mouse monoclonal antibody raised against amino acids 1–163 (sc-56167) that detects a ~30 kD monomer and ~60 kDa active homodimer (1 μg/mL).
Reduced (GSH) and oxidized (GSSG) glutathione assay
Tissues were washed three times by inversion in 500 μL of ice-cold phosphate-buffered saline (PBS) pH 7.4 with sodium heparin (0.5 units/mL) to remove contaminating blood and stored at −80 °C. Within 2 weeks of freezing, tissues were homogenized at 1:10 (w/v) using ice-cold homogenization buffer as described above. 200-μL aliquots of homogenized tissue were clarified and deproteinated following the procedures described for NO colorimetric assay. Recovered supernatants were used for experiments following the manufacturer’s instructions for the NWLSS Glutathione Assay (NWK-GSH01; Northwest Life Science Specialties, LLC, Vancouver, WA). A standard curve using glutathione disulfide in 1 mM HCl was used to extrapolate optical densitometry to GSH equivalents between 2.5 to 20 μM (GSH) and 1.25 to 10 μM (GSSG). Deproteinated clarified specimens were mixed with 4 N NaOH at 1:20 (v:v) and 4-vinylpyridine (Sigma–Aldrich) in ethanol at 1:10 (v:v) for GSSG procedure, while specimens were diluted with manufacturer’s assay buffer by 20-fold for GSH procedure. Samples and standards were incubated at room temperature for 1 h, placed in a 96-well plate, mixed 1:1 (v:v) with 5–5′-dithiobis (2-nitrobenzoic acid) in phosphate buffer with EDTA (DTNB) and glutathione reductase in assay buffer with protein stabilizer, then incubated again for 5 min at room temperature and mixed at 1:1 (v:v) with reduced β-nicotinamide adenine dinucleotide phosphate. Using a kinetic protocol, the OD at 405 nm at 15 s intervals was measured to determine the linear reduction for GSH (over 3 min) and GSSG (over 15 min). Results were adjusted by milligram of protein (Bradford method) and described as GSH, GSSG and GSH:GSSG ratio.
Mitochondrial complex I activity assay
Experiments were performed according to instructions provided by manufacturer of the Complex I Enzyme Activity Microplate Assay Kit (MS141; Mitoscience, Eugene, OR). Tissue was homogenized with 200 μL of PBS, pH 7.3. Protein concentrations were determined using the standard Bradford method. An aliquot of homogenized tissue was further diluted in PBS to a concentration of 5.5 μg/μl. One hundred-microliter aliquot was solubilized using 10× detergent provided as part of the kit. Specimens were centrifuged for 20 min at 12,000g and supernatants collected. Following a 5-fold dilution using incubation solution (provided by the manufacturer), a 200-μg aliquot of protein/well was incubated for 3 h at room temperature in a 96-well microplate coated with anti-mitochondrial NADH dehydrogenase antibody. The degree of oxidation of NADH to NAD+ by the immunocaptured complex I was determined by the rate of increase in absorbance (mOD/min) at 450 nm. The kinetic analysis at 21°C was linear over 30 min with 60 s intervals to determine maximum rate of increase using microplate reader.
ATP colorimetric assay
Following dissection, tissue was flash frozen by immediate immersion in 100% isopentane (Mallinckrodt Baker Inc., Phillipsburg, NJ) pre-chilled on dry ice. Frozen tissues were immediately retrieved, placed in pre-chilled 1.5 mL microtubes and stored at −80 °C. Tissues were homogenized on dry ice using ice-cold ATP assay buffer provided as part of the ATP colorimetric Assay kit (ab83355; Abcam). A 5-μL aliquot of the homogenized tissue was used to determine protein concentrations using Bradford method. Samples were then deproteinated using the Deproteinizing Sample Preparation Kit (ab93299; Abcam) by mixing with ice-cold perchloric acid at 1:5.5 (v:v), incubating on ice for 5 min, centrifugating at 13,000g for 2 min at 4 °C and recovering supernatant. Specimens were neutralized by adding ice-cold Neutralization Solution (ab93299; Abcam) at 1:24 (v:v), incubating on ice for 5 min, centrifuging at 10,000g for 2 min and mixing with ice-cold ATP Assay Buffer at 1:5 (v:v). Using the ATP standard provided by the manufacturer, a 10-mM ATP stock solution (in ddH2O) was prepared and used to generate a standard curve to estimate the amount of ATP per well between 1 and 20 nmol. Preliminary experiments were performed to assure accuracy and specificity of the ATP standard provided as part of the kit. A second standard curve was generated using a 1 mM ATP solution prepared by reconstituting 1 mg of ATP-lyophilized powder (FLAAS, MW 551.14, Sigma–Aldrich) in 1.8 mL of dH2O. The correlation between both standards was determined at 1–50 nmol/well. PBS pH 7.4 and ATP (5 nmol) treated with Na+ K+ ATPase (A7510, 1 unit, Sigma–Aldrich) reconstituted in 50 mM Tris–HCl, 100 mM NaCl, 20 mM KCl, 3 mM MgCl2 and 0.2 mM EGTA were used as negative controls to confirm that our assay: (i) did not detect inorganic phosphate, and (ii) was specific for ATP. Next, as part of the assay protocol, a reaction mix containing 4% ATP probe, 4% ATP converter and 4% developer in ATP Assay Buffer was mixed in a 1:1 (v:v) ratio with standards and deproteinated neutralized samples and incubated in a 96-well plate for 30 min at room temperature protected from the light. ATP concentrations in the samples were calculated by plotting the measured optical densitometry at 570 nm in a microplate reader versus the linear distribution generated by the standard curve with a final adjustment for protein concentration.
Transmission electron microscopy (EM) and mitochondria ultrastructure
Brains were perfusion fixed with 2% glutaraldehyde/2% paraformaldehyde in 0.1 M PBS for 30 min at 4 ml/min. Ipsilateral sensory motor cortical tissue sections for EM studies were post-fixed in the perfusion solution but not cryoprotected or frozen. EM samples were post-fixed (1 h) in 2% osmium tetroxide, dehydrated, and flat embedded in resin. Plastic-embedded samples were mounted in an Epon block (Electron Microscopy Sciences, Fort Washington, PA) and cut into semithin (1 μm) and ultrathin (70 nm) sections for light microscopy and EM, respectively. All semithin sections were counterstained with Toluidine Blue and screened by light microscopy to select sections for viewing by EM. Ultrathin sections were stained with uranyl acetate and lead citrate and viewed with a Hitachi 7600 transmission electron microscope. Mitochondrial ultrastructure was examined at 5000× and 20,000×.
Determination of gene expression by real time quantitative reverse transcription polymerase chain reaction (qRT-PCR)
Total RNA was extracted from forebrain harvested from mice exposed to neonatal HI followed by Nec-1 or vehicle treatments and naive control at 3 and 24 h (p8) (n = 6 mice/treatment/time/gender). PureLink Micro-to-midi total RNA purification system (Invitrogen, Carlsbad, CA) was used according to specifications. Approximately 1 μg of total RNA was used for generation of complementary DNA (cDNA) using iScript cDNA synthesis kit (BioRad, Hercules, CA). Reverse transcription protocol included 5 min at 25 °C; 30 min at 42 °C and 5 min at 85 °C. cDNA is then used to amplify the hypoxia-inducible factor-1α (HIF-1α) and BNIP3 genes by real time qRT-PCR using 300 nm concentration of specific primers (Table 1). The amplification protocol included 40 cycles of 30 s at 95.0 °C, 1 min at 60.0 °C and 30 s at 72.0 °C. GADPH and β-actin were evaluated as housekeeping genes using the BestKeeper approach to determine stability of gene expression under experimental conditions (Pfaffl et al., 2004). Based on their gene stability, β-actin was selected as the housekeeping gene for calculations (stability coefficients were 1.2 for β-actin vs. 2.0 for GAPDH). Fold difference in gene expression was then corrected to β-actin using the Pfaffl method (Pfaffl, 2001). Melting curves confirm amplification of single PCR products.
Table 1.
Primers used for real time qRT-PCR
| Gene | Direction | Sequence | Base pair |
|---|---|---|---|
| HIF-1α | Sense Antisense |
5′-ATTTTTGGACACTGGTGGCT-3′ 5′-ATGCAATGGTGAAATGCTGA-3′ |
200-bp |
| BNIP3 | Sense Antisense |
5′-GCTCCCAGACACCACAAGAT-3′ 5′-TGAGAGTAGCTGTGCGCTTC-3′ |
222-bp |
| β-actin | Sense Antisense |
5′-CCCAACTTGATGTATGAAGG-3′ 5′-TTGTGTAAGGTAAGGTGTGC-3′ |
119-bp |
| GAPDH | Sense Antisense |
5′-TTGTCAAGCTCATTTCCTGGTATG-3′ 5′-GCCATGTAGGCCATGAGGTC-3′ |
76-bp |
BNIP3, BCL2/adenovirus E1B 19 kDa protein-interacting protein 3; bp, base pair; GAPDH, glyceraldehyde 3-phosphate dehydrogenase; HIF-1α, hypoxia inducible factor-1 alpha.
Statistics
For analysis of multiple groups, analysis of variance (ANOVA) was applied with post hoc pair analysis using Tukey’s test. Stratification by gender was performed and the results were reported as mean±s.e.m. and represented as bar graphs. Due to the non-normal distribution of the gene expression data, non-parametric analysis was applied (Wilcoxon test) and results were represented as box and whisker plots, where the box was limited by the 25th and 75th percentiles and the solid line represented the median. Significance was assigned in all cases by p ≤ 0.05. Analysis was performed using IBM SPSS Statistics 18.0 software (IBM Corporation, Armonk, NY).
RESULTS
Nec-1 treatment immediately after HI attenuated early NO• production, iNOS expression and glutathione oxidation in male and female mice
Following neonatal HI, NO• in forebrain increased by 72% (p = 0.003 vs. naive) at 3 h in vehicle-treated mice and this increase was prevented by post-treatment with Nec-1 (p = 0.04 vs. vehicle). NO• levels in forebrain of vehicle-treated mice significantly declined by 24 h post-HI (p = 0.003 vs. 3 h vehicle) to levels that were similar to those of naive and Nec-1-treated mice (p = 0.001, ANOVA; n = 10–12 mice/group). The increase in NO• in vehicle-treated mice was similar in both genders, males (by 63%, p = 0.008) and females (by 87%, p = 0.006), and was prevented by Nec-1 in both genders (p = 0.002, ANOVA [males]; p = 0.005, ANOVA [females], 5–6 mice/treatment/time/gender; Fig. 1A). Similarly, 3 h following neonatal HI, iNOS expression was up-regulated by 121% in vehicle-treated mice (p<0.001, ANOVA; p<0.001 vs. naive, n = 13 mice/group) and by 24 h, expression returned to levels similar to those in naive mice (p = 0.006 vs. 3 h vehicle). HI-induced iNOS protein up-regulation was blocked by Nec-1 treatment (p = 0.02 vs. 3 h vehicle). Like with NO•, stratification showed no difference by gender, with iNOS expression increased by 96% in males (p = 0.001 vs. naive; p = 0.002, ANOVA; n = 3–7 per treatment/time) and by 149% in females (p = 0.02 vs. naive; p = 0.02, ANOVA; n = 3–6 per treatment/time) and returning to normal in both genders by 24 h (Fig. 1B). Nec-1 similarly prevented iNOS increase at 3 h post-HI in both males (p = 0.05 vs. 3 h vehicle) and females (p = 0.04 vs. 3 h vehicle). Up-regulation of NO• and iNOS was followed by a 40% increase in 3-nitrotyrosine levels by 24 h (33% in males [p = 0.001 vs. naive; ANOVA, p<0.001] and 46% in females [p = 0.004 vs. naive; ANOVA, p<0.001]). This increase was prevented by Nec-1 treatment in both genders (Fig. 1C and 1D).
Fig. 1.

Nec-1 treatment following neonatal HI prevents early nitric oxide (NO•) accumulation, inducible nitric oxide synthase (iNOS) up-regulation and 3-nitrotyrosine production in forebrain of male and female mice. Bar graphs showing (A) NO• levels, (B) iNOS expression, (C) 3-nitrotyrosine levels in ipsilateral forebrain at 3 and 24 h following neonatal HI. Bars represent the mean±SEM measured in naive control (white), vehicle (light gray) and Nec-1 (dark gray)-treated male (solid color, M) and female (hashed color, F) mice. p ≤ 0.05 in all cases (one-way ANOVA).*, vs. naive control; ‡, vs. 3 h vehicle; #, vs. 24 h vehicle; p<0.05 (Tukey’s post hoc). n = 5–6 mice/treatment/time/gender. Representative immunoblots for (i) iNOS (NOS2) showing a single 130 kDa band with corresponding loading controls are shown in B for males and females and (ii) 3-nitrotyrosine levels for male and female mice at 24 h post-HI (D).
Following in vivo neonatal HI, GSH decreased by 38% (p = 0.05 vs. naive) at 3 h and by 64% (p = 0.04 vs. naive) at 24 h post-HI in forebrain of vehicle-treated mice (p<0.001, ANOVA; n = 8–10 animals/group) and inhibition of RIP-1 kinase activity (Nec-1) prevented this decline at 24 h (p = 0.05 vs. 24 h vehicle). GSH effects were similar in both genders (p = 0.05, ANOVA [male], p = 0.006 ANOVA [female]; Fig. 2A). In concert, GSSG increased in forebrain of vehicle-treated mice by~2.5-fold at 3 h post-HI (p = 0.008 vs. naive) and this effect persisted at 24 h (p<0.001 vs. naive) post-HI (p = 0.001, ANOVA; n = 8–10/group). Nec-1-treated mice had 33% lower GSSG levels compared to vehicle-treated mice at 24 h (p = 0.02), and these levels were similar to those in naive mice. Once again similar effects were observed regardless of gender (p = 0.009, ANOVA [male]; p = 0.04, ANOVA [female]; Fig. 2B). Thus, Nec-1 treated mice had a 3.3-fold greater ratio of reduced vs. oxidized glutathione at 24 h after HI (GSH: GSSG; p = 0.04 vs. 24 h vehicle; ANOVA, p<0.001) in both, males (p = 0.02 vs. 24 h vehicle; ANOVA, p<0.001) and females (p = 0.05 vs. 24 h vehicle; ANOVA, p<0.001) (Fig. 2C).
Fig. 2.
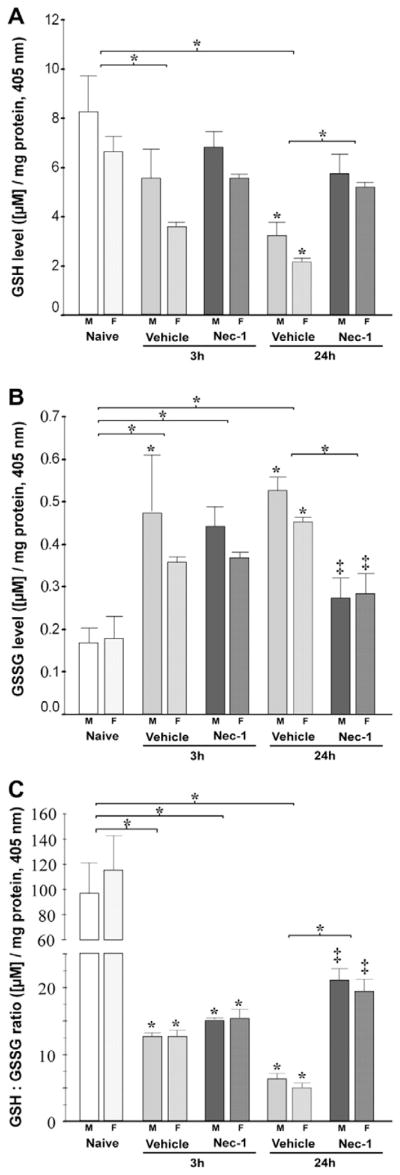
Nec-1 treatment following neonatal HI prevents early glutathione oxidation in the forebrain of male and female mice. Bar graphs showing (A) reduced glutathione (GSH) levels, (B) oxidized glutathione (GSSG) levels, and (C) GSH:GSSG ratio in ipsilateral forebrain at 3 and 24 h following neonatal HI. Bars represent the mean±SEM measured in naive control (white), vehicle (light gray) and Nec-1 (dark gray) treated male (solid color, M) and female (hashed color, F) mice. p ≤ 0.05 in all cases (one-way ANOVA). *, vs. naive control; ‡, vs. 24 h vehicle; p<0.05 (Tukey’s post hoc). n=4–5 mice/treatment/ time/gender. Bracket and *, represent p ≤ 0.05 for the whole pool.
Nec-1 treatment prevented secondary energy failure following neonatal HI regardless of gender
Complex-I activity progressively declined in vehicle-treated mice at 3 and 24 h after HI (p<0.001, ANOVA, n = 9–16/per group). By 24 h post HI, complex-I activity in vehicle-treated mice was 49% lower than naive control (p<0.001) and 44% lower than levels at 3 h (p = 0.01). Nec-1 prevented complex-I activity decline at 24 h (p = 0.04 vs. 24 h vehicle). Complex I activity changes were consistent in both genders (p = 0.01, ANOVA [male]; p = 0.004 ANOVA [female]) demonstrating a significant decline in activity at 24 h, by 48% in males (p = 0.005 vs. naive) and by 49% in females (p = 0.002 vs. naive) with both being prevented by Nec-1 (Fig. 3).
Fig. 3.
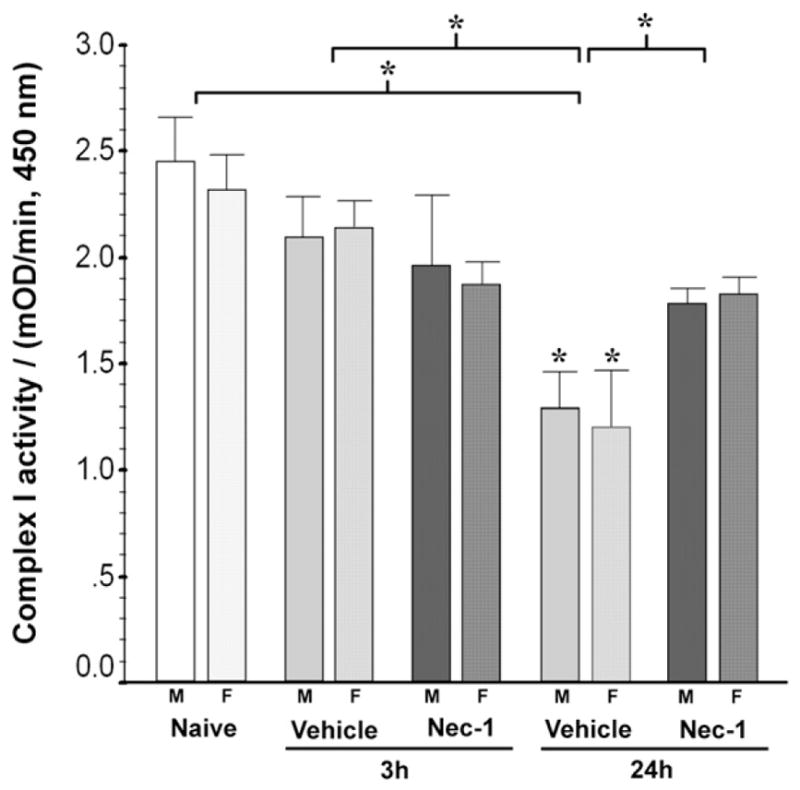
Nec-1 treatment prevents the progressive decline in complex-I activity in the ipsilateral mouse forebrain following neonatal HI in both genders. Bar graph shows the mitochondrial complex-I activity in forebrain at 3 and 24 h following neonatal HI. Bars represent the mean±SEM measured in naive control (white), vehicle (light gray) and Nec-1 (dark gray)-treated male (solid color, M) and female (hashed color, F) mice. p ≤ 0.05 in both genders (one-way ANOVA). *, vs. naive control; p<0.05 (Tukey’s post hoc). n=4–11 mice/ treatment/time/gender. Bracket and *, represent p ≤ 0.05 for the whole pool (ANOVA, p<0.001).
Because Nec-1 immediately after HI prevented the decline in complex-I activity at 24 h, we measured ATP levels in forebrain at 24 h (p8) and again at p11. In a control experiment, a significant correlation was found between known concentrations prepared using lyophilized ATP and the predicted values calculated based on standard curve using the manufacturer’s supplied reagents (R2=0.9678, p<0.001, Fig. 4A). Additionally this method to measure ATP levels did not detect inorganic phosphate and was specific for the detection of ATP as shown by the decline in levels following exposure to Na+ K+ ATPase (Fig. 4A). Twenty-four hours after HI, forebrain of vehicle-treated mice had 51% less ATP (p=0.003 vs. naive), while those from Nec-1-treated mice had 2.3-fold greater ATP than vehicle-treated mice (p<0.001 vs. p8 vehicle; ANOVA, p<0.001, n= 6–12/per group). By p11, a more modest but persistent ATP decline (30%, p<0.001 vs. p11 naive; ANOVA, p<0.001, n=6–12/per group) was observed in vehicle-treated mice, this decline was prevented by Nec-1. The decline in ATP in vehicle-treated mice by 24 h after HI was similar in males (by 46%, p=0.02; ANOVA, p=0.02, n=3–6/treatment) and females (by 56%, p=0.05; ANOVA, p=0.02, n=3–6/treatment) and Nec-1 prevented the decline in both genders (male, p=0.04 vs. p8 vehicle; female p=0.01 vs. p8 vehicle; Fig. 4B). Although no obvious difference in ATP levels was observed between genders at p11, females showed a statistically significant decrease in ATP levels following HI which is prevented by Nec-1 (Fig. 4B). The lack of difference in male mice is likely due to the greater degree of variance observed (Fig. 4B).
Fig. 4.

Following neonatal HI, early ATP depletion and later neuronal mitochondrial disruption are blocked by Nec-1 treatment in both genders. (A) Assay validation experiments demonstrated a significant correlation between two independent ATP standards as well as significantly lower signal with negative controls, PBS ph 7.4 and ATP (5 nmol) treated with Na+ K+ ATPase (1 unit). (B) ATP levels in forebrain at 24 h (p8) and 96 h (p11) following neonatal HI. Bars represent (mean±SEM) naive control (white bar), vehicle (light gray bar) and Nec-1 (dark gray bar)-treated male (solid color, M) and female (hashed color, F) mice. p ≤ 0.05 in both genders (one-way ANOVA). *, vs. naive control; ‡, vs. vehicle; p<0.05 (Tukey’s post hoc). n=3–6 mice/treatment/time/gender. Whole pool ANOVA, p<0.001 for both, p8 and p11. (C) Mitochondria in forebrain neurons were considered disrupted if inner cristae were swollen, inner and outer membrane were separated and/or mitochondria ultrastructure was grossly altered by electron microscopy. Representative examples of the findings for vehicle and Nec-1-treated mice at p8 (C1, C2) and at p11 (C3, C4) are shown in two magnifications. White box=area of magnification; white arrows=intact mitochondria; black arrows=disrupted mitochondria. Scale bar=1 μm.
Mitochondrial ultrastructure is preserved in Nec-1-treated mice following neonatal HI
Because our biochemical experiments suggested preservation of mitochondrial function in Nec-1-treated mice, we evaluated mitochondrial ultrastructure in the sensory-motor cortex by transmission electron microscopy. Male and female mice were evaluated. At p8 (24 h) disrupted neuronal mitochondria could be identified in vehicle-treated mice while mitochondria looked better preserved in Nec-1-treated mice (Fig. 4C.1 and C.2); by p11 clear differences between groups were evident with greater preservation of mitochondrial ultrastructure in mice treated with RIP-1 kinase inhibitor, Nec-1, immediately after HI (Fig. 4C.3 and C.4).
We also investigated whether non-neuronal cells might be responsible for part of the biochemical changes found in our experiments at early stages. Immunoblotting revealed that Nec-1 prevented the 2.4-fold increase in GFAP expression observed in vehicle-treated mice (p=0.01 vs. naive; p=0.01, ANOVA; n=9/per group, Fig. 5A). This biochemical evidence of astrogliosis at p11 in vehicle-treated mice was preceded by ultrastructural changes in astrocyte mitochondria at p8. Astrocytes in forebrain of vehicle-treated mice following HI showed evidence of mitochondrial injury with darkened–condensed mitochondria at p8 compared to astrocytes in Nec-1-treated mice that displayed mostly normal-appearing mitochondria (Fig. 5B).
Fig. 5.

GFAP upregulation and mitochondria disruption in astrocytes were prevented by Nec-1 treatment following neonatal HI. (A) GFAP levels adjusted by loading control in mouse forebrain at p8 and p11 following HI. p=0.02 (one-way ANOVA); *, p<0.05 (Tukey’s post hoc). n=9 mice/ group. Representative 15% SDS–PAGE showing a 50-kDa band and breakdown product at 45 kDa for GFAP is presented. (B) Mitochondria in forebrain astrocytes were classified as disrupted following the same criteria described for Fig. 4. Representative examples of the findings for vehicle (B1) and Nec-1 (B2)-treated mice at p8 are shown in two magnifications. White box=area of magnification; white arrows=intact mitochondria; black arrows=disrupted mitochondria. Scale bar=1 μm.
Nec-1 treatment following neonatal HI prevents HIF-1α and BNIP3 expression
Gene expression of the BNIP3 transcription factor, HIF-1α, was unchanged at 3 h following HI in vehicle and Nec-1-treated mice (vs. naive control). By 24 h following neonatal HI, HIF-1α gene expression was up-regulated by 3-fold in vehicle-treated mice ([1.4–4.3], vs. naive control, p=0.02, n=6), while it was unchanged in Nec-1-treated mice (1.3 [0.9–1.8], p=0.54, Fig. 6A). Coincidently, BNIP3 gene expression was unchanged at 3 h post-HI, while at 24 h expression was up-regulated by 74% ([1.2–3.6], p=0.02) in vehicle-treated mice and Nec-1 prevented the increase (Fig. 6B). Similar findings were also observed for BNIP3 at the protein level by western blot with increased expression of the 60-kDa BNIP3 active homodimer in vehicle-treated mice but not in Nec-1-treated mice (Fig. 6C).
Fig. 6.
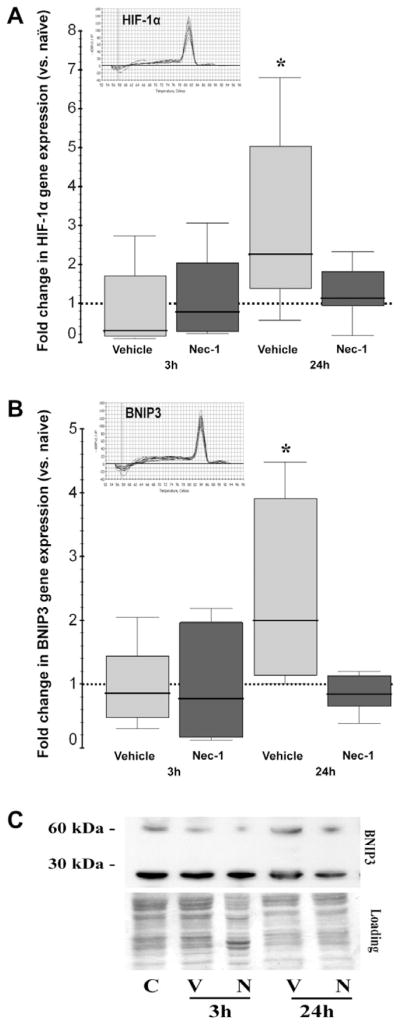
Transcriptional upregulation of HIF-1α and BNIP3 is prevented by Nec-1. Box and whisker plots represent the fold change in (A) HIF-1α (BNIP3 transcription factor) and (B) BNIP3 gene expression using β-actin as housekeeping gene. Boxes are limited by the 25th and 75th percentiles for vehicle (light gray) and Nec-1 (dark gray), solid line inside the box represents the median, discontinuous line sitting at 1 represents the comparison group (naive animals). *, p<0.05 (vs. naive, Wilcoxon test). Melting curves show a single PCR product for HIF-1α and BNIP3. (C) Representative western blot analysis for BNIP3 with loading control shows an increase in active 60 kDa BNIP3 heterodimer by 24 h following HI in vehicle-treated mice.
DISCUSSION
Here, we show that treatment with Nec-1 immediately after in vivo neonatal HI, prevents mitochondrial dysfunction in the forebrain. Nec-1 blocks early NO• production, iNOS expression, protein nitration and modestly attenuates glutathione oxidation. Nec-1 prevents mitochondrial dysfunction and secondary energy failure during early recovery from HI as evidence by the preservation of: (i) complex I activity, (ii) ATP production, and (iii) mitochondrial ultrastructure. Although the determination of the source(s) of these biochemical changes is challenging using an in vivo model of neonatal HI, the preservation of mitochondrial ultrastructure in astrocytes, in addition to neurons, by transmission electron microscopy along with the prevention of iNOS and GFAP expression suggests that astrocytes might also be a target of Nec-1 protection possibly providing additional downstream protection to neurons. Furthermore, we speculate that prevention of BNIP3 up-regulation may also participate in the mitochondrial protection observed following Nec-1 treatment in this model.
Classically programmed or regulated necrosis (Galluzzi et al., 2011) is initiated by the activation of death receptors and executed by the formation of a RIP-1–RIP-3 complex (necrosome) (Holler et al., 2000; Cho et al., 2009). Necrosome formation triggers ROS production via NADPH oxidase activation and mitochondria disruption (Hsu et al., 2009; Kim et al., 2010; Northington et al., 2011b). Nec-1, or inhibition of RIP-3, using silencing RNA (siRNA) or genetic deletion, decreases ROS production and prevents execution of programmed necrosis (Cho et al., 2009). Inhibition of the necrosome formation does not have intrinsic antioxidant effect against hydrogen peroxide-induced cell death; however, it appears to increase GSH production in HT-22 cells independent of glutamate exposure (Xu et al., 2007; Kim et al., 2010). Although our in vivo experiments do not confirm these specific findings, they do show significant prevention of the glutathione oxidation following neonatal HI. This finding may imply an indirect effect of Nec-1 via prevention of ROS production and early protein carbonyl formation after neonatal HI (Northington et al., 2011a) or it may reflect neural cell protection in general and be independent of Nec-1.
Accumulation of NO• and expression of iNOS potentiate glutamate release, N-methyl D-aspartate receptor (NMDAR) activation and necrotic neuronal death, suggesting a critical role of NO• in the progression of excitotoxic injury in vitro (Hewett et al., 1996; Leist et al., 1999; Nicotera and Lipton, 1999). Furthermore, in the setting of increased iNOS expression by astrocytes, NOS inhibition protects against oxygen-glucose deprivation-induced neuronal injury (Hewett et al., 1996). Here, we show that NO• levels, iNOS expression and 3-nitrotyrosine levels are increased following neonatal HI and that these events are prevented by Nec-1 treatment. Nec-1 treatment immediately after neonatal HI attenuates cytokine expression. This is a plausible upstream effect that could block iNOS expression and astrocyte activation and partially explain the neuroprotection afforded by this treatment (Faraco et al., 2007; Northington et al., 2011a).
At low concentrations NO• reversibly inhibits mitochondrial complex I (Beltran et al., 2000; Riobo et al., 2001; Chinta and Andersen, 2006); however, at higher concentrations, this inhibition becomes irreversible resulting in increased free radical production (Beckman and Koppenol, 1996; Davis et al., 2010) and progression of injury following neonatal HI. Here, we report a progressive decline in complex I activity during the first 24 h following neonatal HI. This decline was preceded by an increase in iNOS expression and NO• accumulation and coincided with the increase in 3-nitrotyrosine levels. Davis et al. reported that blockade of necrosome formation in vitro using Nec-1 or RIP-1 siRNA prevents 3-nitrotyrosine accumulation and attenuates complex I activity decline and NO•-dependent necrosis (Davis et al., 2010). Likewise, our data show that complex I activity decline, NO• accumulation and 3-nitrotyrosine production are blocked by Nec-1 treatment immediately after neonatal HI. Furthermore, we show that the significant impairment in ATP production observed at early stages following HI is also prevented by Nec-1 treatment and as expected the most significant ATP changes were observed at 24 h following secondary energy failure in this neonatal HI model (Vannucci et al., 1998).
Neonatal HI is characterized by loss of structurally intact mitochondria (Northington et al., 2007). Biochemical changes indicative of mitochondrial dysfunction occur within the first 24 h after neonatal HI in this model (Yager et al., 1991; Hagberg, 2004). These biochemical changes coincide with presence of damaged mitochondria in neurons at p8 and p11 in vehicle-treated mice as seen directly by EM. Because reactive astrocytes release cytokines and express iNOS (Laird et al., 2008), both of which are blocked by Nec-1 treatment (Northington et al., 2011a), we reasoned that astroglia were also protected by this treatment. Evaluation of astrocytes by electron microscopy suggests that astroglia are also targets of Nec-1 mediated mitochondrial protection following neonatal HI. Furthermore, as a result of the protection of astroglia by Nec-1, GFAP expression is attenuated at later stages following HI suggesting decreased astrogliosis.
Additional mechanisms explaining the mitochondrial-protection afforded by Nec-1 treatment remain unclear. One potential mechanism may include modulation of BNIP3 expression by Nec-1. Cytokine expression (Ghavami et al., 2009), NO• accumulation (Yook et al., 2004), and hypoxia (Bruick, 2000) are the three major stimuli modulating BNIP3 expression. NO• and hypoxia up-regulate HIF-1α, a transcription factor that binds to the hypoxia response element at the BNIP3 promoter (Bruick, 2000; An et al., 2006). Inhibition of RIP-1 kinase activity prevents pro-inflammatory cytokine expression (Northington et al., 2011a) as well as NO• accumulation and downstream HIF-1α up-regulation, thus favoring down-regulation of BNIP3 expression. In the native state, BNIP3 (30 kDa monomer) binds loosely to the mitochondrial membrane (Yook et al., 2004); however, in conditions triggering ROS accumulation (i.e. neonatal HI), BNIP3 dimerizes and firmly inserts into the mitochondrial membrane, opens the mitochondria permeability transition pore, and triggers necrotic-like cell death (Vande Velde et al., 2000; Kubli et al., 2008). Therefore, we speculate that suppression of hypoxia-mediated BNIP3 expression is potentially one of the intermediate steps involved in the mitochondrial protection likely afforded by the inhibition of RIP-1kinase activity by Nec-1 treatment immediately after neonatal HI.
We have previously questioned the influence of gender in the neuroprotection afforded by Nec-1. These differences may lay in intrinsic differences in primary injury pathways explaining the greater degree of variability of injury in female mice vs. the more uniformly severe injury in male mice (Northington et al., 2011a). Although the mechanisms explaining these gender differences are unresolved, they may involve a more significant decline in NAD+ in male mice following PARP-1 activation (Hagberg et al., 2004) and preferential nuclear translocation of AIF (Zhu et al., 2006) found in male rodents following neonatal HI. Stratification of our results by gender does not show differences between male and female mice in response to Nec-1 immediately after HI. We suggest that Nec-1 uniformly provides protection of mitochondria regardless of gender. However, other possible effects of Nec-1 may be gender specific.
These experiments do not resolve several mechanistic facets of the neuroprotection afforded by Nec-1 treatment. First, we have not directly linked the increase in NO• and iNOS expression to inhibition of complex I activity; however, we show a temporal relationship between the iNOS expression and NO• accumulation preceding 3-nitrotyrosine production and complex I activity decline, that is in agreement with previous studies showing that NO•-mediated inhibition of complex I activity is associated with irreversible mitochondrial dysfunction (Davis et al., 2010). Second, treatment with Nec-1 immediately after HI appears to effectively preserve mitochondrial ultrastructure but it is still unclear how this effect is related, directly or indirectly, to RIP-1 kinase inhibition. Although we provide some evidence that part of the mechanism may involve prevention of BNIP3 expression, many inter-mediate steps are still unknown. Third, because these experiments were done in vivo, it is not possible to determine if the cells with disrupted mitochondrial ultrastructure were directly responsible for the observed biochemical changes; nevertheless mitochondrial preservation in astrocytes and neurons, coincides temporarily with the overall early biochemical changes that follows neonatal HI in this model and suggests effects of Nec-1 on both cell types. Lastly, while biochemical studies were performed in homogenized forebrain samples which included cortex, hippocampus and striatum, mitochondria ultrastructure was reviewed in cortex. Therefore, the relative lag of structural findings by electron microscopy in comparison to biochemical changes is perhaps explained by the brain regional differences in susceptibility to HI in the neonatal mouse model.
CONCLUSIONS
Nec-1 treatment affords neuroprotection following neonatal HI in part by preventing early oxidative protein damage and inflammation (Northington et al., 2011a). However, these effects do not fully explain the neuroprotection seen at p11 and later. Nec-1 treatment immediately after neonatal HI in a mouse model prevents iNOS expression, NO• accumulation and protein nitration attenuating progressive mitochondrial complex I dysfunction and ATP decline. These effects were associated with preservation of astrocyte and neuronal mitochondrial ultrastructure following neonatal HI. We conclude that Nec-1 preserves energy supply by preventing mitochondrial dysfunction and secondary energy failure in vivo in both astrocytes and neurons equally in males and females. The translational implications of these findings are significant. Regulated necrosis and necrosome formation may be directly implicated in mitochondrial failure following neonatal HI. Blockade of this pathway provides an important new avenue for neonatal neuroprotection research.
Acknowledgments
SOURCES OF SUPPORT
These experiments were supported in part by the March of Dimes Foundation 6-08-275 (F.J.N.) and NS 059529 (F.J.N.) and AG 016282 (L.J.M.). Supporting agencies did not have any role in study design, collection, analysis and interpretation of data.
The authors thank Dr. Estelle Gauda for her critical evaluation of the paper, Devin Mack for his expert assistance with immunoblotting and Julia Hinojos and Darla Mata for their administrative assistance.
Abbreviations
- ANOVA
analysis of variance
- ATP
adenosine-5′-triphosphate
- BNIP3
BCL2/adenovirus E1B 19 kDa protein-interacting protein 3
- EDTA
ethylenediaminetetraacetic acid
- EGTA
ethylene glycol tetraacetic acid
- EM
electron microscopy
- GAPDH
Glyceraldehyde 3-phosphate dehydrogenase
- GFAP
glial fibrillary acidic protein
- GSH
reduced glutathione
- GSSG
oxidized glutathione
- HI
hypoxia ischemia
- HIF-1α
hypoxia inducible factor-1 alpha
- iNOS
inducible nitric oxide synthase
- NADH
nicotinamide adenine dinucleotide
- Nec-1
necrostatin-1
- NO•
nitric oxide
- OD
optical density
- p
postnatal day
- PBS
phosphate buffered saline
- PCR
Polymerase Chain Reaction
- PMSF
phenylmethanesulfonyl fluoride
- qRT PCR
quantitative reverse transcription PCR
- RIP
receptor interacting protein
- siRNA
silencing RNA
- TNF
tumor necrosis factor
Footnotes
AUTHOR CONTRIBUTIONS
R.C.V. participated in experimental design and execution, carried out biochemical studies, analyzed the data and drafted manuscript. L.J.M. participated in experimental design, carried out treatment injections, critically reviewed results and drafted manuscript. D.L.F. carried out dissections and immunoassays and participated in experimental design. F.J.N. participated in experimental design and execution, performed electron microscopy, reviewed results and drafted manuscript. F.J.N. is responsible for the overall research direction of this and related projects. All authors read and approved the final manuscript.
ETHICS STATEMENT
I have read and have abided by the statement of ethical standards for manuscripts submitted to Neuroscience.
FINAL APPROVAL
All authors have approved the final article.
References
- An HJ, Maeng O, Kang KH, Lee JO, Kim YS, Paik SG, Lee H. Activation of Ras up-regulates pro-apoptotic BNIP3 in nitric oxide-induced cell death. J Biol Chem. 2006;281:33939–33948. doi: 10.1074/jbc.M605819200. [DOI] [PubMed] [Google Scholar]
- Beckman JS, Koppenol WH. Nitric oxide, superoxide, and peroxynitrite: the good, the bad, and ugly. Am J Physiol. 1996;271:C1424–C1437. doi: 10.1152/ajpcell.1996.271.5.C1424. [DOI] [PubMed] [Google Scholar]
- Beltran B, Mathur A, Duchen MR, Erusalimsky JD, Moncada S. The effect of nitric oxide on cell respiration: a key to understanding its role in cell survival or death. Proc Natl Acad Sci U S A. 2000;97:14602–14607. doi: 10.1073/pnas.97.26.14602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blomgren K, Hagberg H. Free radicals, mitochondria, and hypoxia–ischemia in the developing brain. Free Radic Biol Med. 2006;40:388–397. doi: 10.1016/j.freeradbiomed.2005.08.040. [DOI] [PubMed] [Google Scholar]
- Bruick RK. Expression of the gene encoding the proapoptotic Nip3 protein is induced by hypoxia. Proc Natl Acad Sci U S A. 2000;97:9082–9087. doi: 10.1073/pnas.97.16.9082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chinta SJ, Andersen JK. Reversible inhibition of mitochondrial complex I activity following chronic dopaminergic glutathione depletion in vitro: implications for Parkinson’s disease. Free Radic Biol Med. 2006;41:1442–1448. doi: 10.1016/j.freeradbiomed.2006.08.002. [DOI] [PubMed] [Google Scholar]
- Cho YS, Challa S, Moquin D, Genga R, Ray TD, Guildford M, Chan FK. Phosphorylation-driven assembly of the RIP1–RIP3 complex regulates programmed necrosis and virus-induced inflammation. Cell. 2009;137:1112–1123. doi: 10.1016/j.cell.2009.05.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis CW, Hawkins BJ, Ramasamy S, Irrinki KM, Cameron BA, Islam K, Daswani VP, Doonan PJ, Manevich Y, Madesh M. Nitration of the mitochondrial complex I subunit NDUFB8 elicits RIP1- and RIP3-mediated necrosis. Free Radic Biol Med. 2010;48:306–317. doi: 10.1016/j.freeradbiomed.2009.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Degterev A, Huang Z, Boyce M, Li Y, Jagtap P, Mizushima N, Cuny GD, Mitchison TJ, Moskowitz MA, Yuan J. Chemical inhibitor of nonapoptotic cell death with therapeutic potential for ischemic brain injury. Nat Chem Biol. 2005;1:112–119. doi: 10.1038/nchembio711. [DOI] [PubMed] [Google Scholar]
- Eguchi Y, Shimizu S, Tsujimoto Y. Intracellular ATP levels determine cell death fate by apoptosis or necrosis. Cancer Res. 1997;57:1835–1840. [PubMed] [Google Scholar]
- Faraco G, Fossati S, Bianchi ME, Patrone M, Pedrazzi M, Sparatore B, Moroni F, Chiarugi A. High mobility group box 1 protein is released by neural cells upon different stresses and worsens ischemic neurodegeneration in vitro and in vivo. J Neurochem. 2007;103:590–603. doi: 10.1111/j.1471-4159.2007.04788.x. [DOI] [PubMed] [Google Scholar]
- Festjens N, Vanden Berghe T, Vandenabeele P. Necrosis, a well-orchestrated form of cell demise: signalling cascades, important mediators and concomitant immune response. Biochim Biophys Acta. 2006;1757:1371–1387. doi: 10.1016/j.bbabio.2006.06.014. [DOI] [PubMed] [Google Scholar]
- Galluzzi L, Vanden Berghe T, Vanlangenakker N, Buettner S, Eisenberg T, Vandenabeele P, Madeo F, Kroemer G. Programmed necrosis from molecules to health and disease. Int Rev Cell Mol Biol. 2011;289:1–35. doi: 10.1016/B978-0-12-386039-2.00001-8. [DOI] [PubMed] [Google Scholar]
- Ghavami S, Eshraghi M, Kadkhoda K, Mutawe MM, Maddika S, Bay GH, Wesselborg S, Halayko AJ, Klonisch T, Los M. Role of BNIP3 in TNF-induced cell death – TNF upregulates BNIP3 expression. Biochim Biophys Acta. 2009;1793:546–560. doi: 10.1016/j.bbamcr.2009.01.002. [DOI] [PubMed] [Google Scholar]
- Graham EM, Sheldon RA, Flock DL, Ferriero DM, Martin LJ, O’Riordan DP, Northington FJ. Neonatal mice lacking functional Fas death receptors are resistant to hypoxic–ischemic brain injury. Neurobiol Dis. 2004;17:89–98. doi: 10.1016/j.nbd.2004.05.007. [DOI] [PubMed] [Google Scholar]
- Hagberg H. Mitochondrial impairment in the developing brain after hypoxia–ischemia. J Bioenerg Biomembr. 2004;36:369–373. doi: 10.1023/B:JOBB.0000041770.00567.4f. [DOI] [PubMed] [Google Scholar]
- Hagberg H, Wilson MA, Matsushita H, Zhu C, Lange M, Gustavsson M, Poitras MF, Dawson TM, Dawson VL, Northington F, Johnston MV. PARP-1 gene disruption in mice preferentially protects males from perinatal brain injury. J Neurochem. 2004;90:1068–1075. doi: 10.1111/j.1471-4159.2004.02547.x. [DOI] [PubMed] [Google Scholar]
- Han W, Xie J, Li L, Liu Z, Hu X. Necrostatin-1 reverts shikonin-induced necroptosis to apoptosis. Apoptosis. 2009;14:674–686. doi: 10.1007/s10495-009-0334-x. [DOI] [PubMed] [Google Scholar]
- Hewett SJ, Muir JK, Lobner D, Symons A, Choi DW. Potentiation of oxygen-glucose deprivation-induced neuronal death after induction of iNOS. Stroke. 1996;27:1586–1591. doi: 10.1161/01.str.27.9.1586. [DOI] [PubMed] [Google Scholar]
- Holler N, Zaru R, Micheau O, Thome M, Attinger A, Valitutti S, Bodmer JL, Schneider P, Seed B, Tschopp J. Fas triggers an alternative, caspase-8-independent cell death pathway using the kinase RIP as effector molecule. Nat Immunol. 2000;1:489–495. doi: 10.1038/82732. [DOI] [PubMed] [Google Scholar]
- Hsu TS, Yang PM, Tsai JS, Lin LY. Attenuation of cadmium-induced necrotic cell death by necrostatin-1: potential necrostatin-1 acting sites. Toxicol Appl Pharmacol. 2009;235:153–162. doi: 10.1016/j.taap.2008.12.012. [DOI] [PubMed] [Google Scholar]
- Kim S, Dayani L, Rosenberg PA, Li J. RIP1 kinase mediates arachidonic acid-induced oxidative death of oligodendrocyte precursors. Int J Physiol Pathophysiol Pharmacol. 2010;2:137–147. [PMC free article] [PubMed] [Google Scholar]
- Kubli DA, Quinsay MN, Huang C, Lee Y, Gustafsson AB. Bnip3 functions as a mitochondrial sensor of oxidative stress during myocardial ischemia and reperfusion. Am J Physiol Heart Circ Physiol. 2008;295:H2025–H2031. doi: 10.1152/ajpheart.00552.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laird MD, Wakade C, Alleyne CH, Jr, Dhandapani KM. Hemin-induced necroptosis involves glutathione depletion in mouse astrocytes. Free Radic Biol Med. 2008;45:1103–1114. doi: 10.1016/j.freeradbiomed.2008.07.003. [DOI] [PubMed] [Google Scholar]
- Leist M, Single B, Naumann H, Fava E, Simon B, Kuhnle S, Nicotera P. Inhibition of mitochondrial ATP generation by nitric oxide switches apoptosis to necrosis. Exp Cell Res. 1999;249:396–403. doi: 10.1006/excr.1999.4514. [DOI] [PubMed] [Google Scholar]
- Lim SY, Davidson SM, Mocanu MM, Yellon DM, Smith CC. The cardioprotective effect of necrostatin requires the cyclophilin-D component of the mitochondrial permeability transition pore. Cardiovasc Drugs Ther. 2007;21:467–469. doi: 10.1007/s10557-007-6067-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markovic SN, Murasko DM. Anesthesia inhibits interferon-induced natural killer cell cytotoxicity via induction of CD8+ suppressor cells. Cell Immunol. 1993;151:474–480. doi: 10.1006/cimm.1993.1256. [DOI] [PubMed] [Google Scholar]
- Nicotera P, Leist M, Ferrando-May E. Intracellular ATP, a switch in the decision between apoptosis and necrosis. Toxicol Lett. 1998;102–103:139–142. doi: 10.1016/s0378-4274(98)00298-7. [DOI] [PubMed] [Google Scholar]
- Nicotera P, Lipton SA. Excitotoxins in neuronal apoptosis and necrosis. J Cereb Blood Flow Metab. 1999;19:583–591. doi: 10.1097/00004647-199906000-00001. [DOI] [PubMed] [Google Scholar]
- Northington FJ, Chavez-Valdez R, Graham EM, Razdan S, Gauda EB, Martin LJ. Necrostatin decreases oxidative damage, inflammation, and injury after neonatal HI. J Cereb Blood Flow Metab. 2011a;31:178–189. doi: 10.1038/jcbfm.2010.72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Northington FJ, Chavez-Valdez R, Martin LJ. Neuronal cell death in neonatal hypoxia-ischemia. Ann Neurol. 2011b;69:743–758. doi: 10.1002/ana.22419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Northington FJ, Koehler RC, Traystman RJ, Martin LJ. Nitric oxide synthase 1 and nitric oxide synthase 3 protein expression is regionally and temporally regulated in fetal brain. Brain Res Dev Brain Res. 1996;95:1–14. doi: 10.1016/0165-3806(96)00051-x. [DOI] [PubMed] [Google Scholar]
- Northington FJ, Zelaya ME, O’Riordan DP, Blomgren K, Flock DL, Hagberg H, Ferriero DM, Martin LJ. Failure to complete apoptosis following neonatal hypoxia-ischemia manifests as “continuum” phenotype of cell death and occurs with multiple manifestations of mitochondrial dysfunction in rodent forebrain. Neuroscience. 2007;149:822–833. doi: 10.1016/j.neuroscience.2007.06.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfaffl MW. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 2001;29:e45. doi: 10.1093/nar/29.9.e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfaffl MW, Tichopad A, Prgomet C, Neuvians TP. Determination of stable housekeeping genes, differentially regulated target genes and sample integrity: BestKeeper – Excel-based tool using pair-wise correlations. Biotechnol Lett. 2004;26:509–515. doi: 10.1023/b:bile.0000019559.84305.47. [DOI] [PubMed] [Google Scholar]
- Portera-Cailliau C, Price DL, Martin LJ. Excitotoxic neuronal death in the immature brain is an apoptosis–necrosis morphological continuum. J Comp Neurol. 1997;378:70–87. [PubMed] [Google Scholar]
- Riobo NA, Clementi E, Melani M, Boveris A, Cadenas E, Moncada S, Poderoso JJ. Nitric oxide inhibits mitochondrial NADH: ubiquinone reductase activity through peroxynitrite formation. Biochem J. 2001;359:139–145. doi: 10.1042/0264-6021:3590139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith CC, Davidson SM, Lim SY, Simpkin JC, Hothersall JS, Yellon DM. Necrostatin: a potentially novel cardioprotective agent? Cardiovasc Drugs Ther. 2007;21:227–233. doi: 10.1007/s10557-007-6035-1. [DOI] [PubMed] [Google Scholar]
- Vande Velde C, Cizeau J, Dubik D, Alimonti J, Brown T, Israels S, Hakem R, Greenberg AH. BNIP3 and genetic control of necrosis-like cell death through the mitochondrial permeability transition pore. Mol Cell Biol. 2000;20:5454–5468. doi: 10.1128/mcb.20.15.5454-5468.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vannucci RC, Towfighi J, Vannucci SJ. Hypoxic preconditioning and hypoxic-ischemic brain damage in the immature rat: pathologic and metabolic correlates. J Neurochem. 1998;71:1215–1220. doi: 10.1046/j.1471-4159.1998.71031215.x. [DOI] [PubMed] [Google Scholar]
- Xu X, Chua CC, Kong J, Kostrzewa RM, Kumaraguru U, Hamdy RC, Chua BH. Necrostatin-1 protects against glutamate-induced glutathione depletion and caspase-independent cell death in HT-22 cells. J Neurochem. 2007;103:2004–2014. doi: 10.1111/j.1471-4159.2007.04884.x. [DOI] [PubMed] [Google Scholar]
- Yager JY, Brucklacher RM, Vannucci RC. Cerebral oxidative metabolism and redox state during hypoxia–ischemia and early recovery in immature rats. Am J Physiol. 1991;261:H1102–H1108. doi: 10.1152/ajpheart.1991.261.4.H1102. [DOI] [PubMed] [Google Scholar]
- Yook YH, Kang KH, Maeng O, Kim TR, Lee JO, Kang KI, Kim YS, Paik SG, Lee H. Nitric oxide induces BNIP3 expression that causes cell death in macrophages. Biochem Biophys Res Commun. 2004;321:298–305. doi: 10.1016/j.bbrc.2004.06.144. [DOI] [PubMed] [Google Scholar]
- Zhu C, Xu F, Wang X, Shibata M, Uchiyama Y, Blomgren K, Hagberg H. Different apoptotic mechanisms are activated in male and female brains after neonatal hypoxia–ischaemia. J Neurochem. 2006;96:1016–1027. doi: 10.1111/j.1471-4159.2005.03639.x. [DOI] [PubMed] [Google Scholar]