

Abstract
Coherent anti-Stokes Raman scattering (CARS) is an emerging tool for label-free characterization of living cells. Here, unsupervised multivariate analysis of CARS datasets was used to visualize the subcellular compartments. In addition, a supervised learning algorithm based on the “random forest” ensemble learning method as a classifier, was trained with CARS spectra using immunofluorescence images as a reference. The supervised classifier was then used, to our knowledge for the first time, to automatically identify lipid droplets, nucleus, nucleoli, and endoplasmic reticulum in datasets that are not used for training. These four subcellular components were simultaneously and label-free monitored instead of using several fluorescent labels. These results open new avenues for label-free time-resolved investigation of subcellular components in different cells, especially cancer cells.
Introduction
Microscopic imaging of cellular compartments based on various spectroscopic signals is a topic of wide interest. The most common methods for imaging the subcellular organelles are fluorescence microscopy, electron microscopy, and cryoelectron microscopy. Several difficulties are encountered with fluorescence microscopy such as extensive sample preparations and photobleaching. Furthermore, the fluorescent label may be toxic or perturbative and change the biochemical properties of the specimen. The electron microscopic methods are invasive and also require extensive sample preparation and they are not suitable for live cell imaging under physiological conditions.
There is growing interest in applying label-free imaging methods such as autofluorescence (1) and second harmonic generation (2) without introducing any external labels or dyes. However, these techniques are limited to relatively few specific molecular signatures. On the other hand, vibrational microscopy including infrared absorption and Raman scattering have been used as label-free imaging methods based on the identification of molecular vibrations that are characteristic of distinct functional groups (3–12). In cells, vibrational fingerprints arise from the functional groups of proteins, nucleic acids, lipids, phospholipids, and carbohydrates, which are the basic building blocks of cells. Thus, vibrational microscopy probes the molecular composition. Infrared microscopy has a limited spatial resolution (∼10 μm at 1000 cm−1) due to its long wavelengths, whereas Raman microscopy has higher spatial resolution due to shorter excitation wavelengths. Therefore, Raman microscopy is superior for spatial resolving of subcellular components.
Several methods based on Raman scattering have been applied for label-free cell imaging (5,13,14). For instance, it has been shown that spontaneous Raman microscopy may resolve subcellular organelles such as the nucleus or chromatin (4,9) and lipid droplets (7,8,15). Raman imaging of mitochondrial distribution in cells was also reported by Matthäus et al. (5), who presented a combination of Raman, fluorescence, and multivariate analysis. The authors used visible excitation, and surprisingly, the Raman data were free of autofluorescence. In addition, Klein et al. (16) demonstrated recently the Raman imaging of nucleus, mitochondria, endoplasmic reticulum, and Golgi apparatus in an LN-18 glioma cell-line using immunofluorescence as a reference. However, the authors only used unsupervised methods for the data evaluation, and the simultaneous identification of several subcellular organelles by Raman microscopy, to our knowledge, has not yet been reported.
Coherent anti-Stokes Raman scattering (CARS) is a variant of Raman microscopy. It is a nonlinear optical four-wave mixing process, which is resonantly enhanced if the frequency difference between pump and Stokes waves matches a vibrational resonance of the specimen. Because of its coherency, CARS produces a strong and directed signal compared with spontaneous Raman. When coupled to laser scanning microscopes, CARS imaging can be performed at a speed up to a video rate (13,14,17–19). Most of the CARS applications in cell biology were investigations of lipids that play a pivotal role in biological membranes (20–23). CARS detects lipids in cells due to their high local concentration. The abundant C-H2 contents in lipids cause a strong CARS signal near 2850 cm−1. CARS microscopy has also been used to probe live cells undergoing mitosis and live cells in interphase (24). In addition, a combination of spontaneous Raman microspectroscopy, CARS, and fluorescence imaging was used to investigate the macromolecular organization of the nucleus through the cell-cycle (24).
Several studies have combined coherent Raman imaging of cells and tissues with unsupervised multivariate analyses such as principle component analysis (PCA), nonnegative matrix factorization (NMF), and multivariate curve resolution (25–30). For instance, PCA of CARS datasets was used to classify individual mesenchymal stem cells into adipocytes, osteoblasts, and undifferentiated stem cells groups (30). In addition, Pohling et al. (26) have shown that PCA of CARS spectra can be used to differentiate between plant cellular components. However, the ability of CARS microscopy to set a supervised classifier that can be used for automated and simultaneous identification of several subcellular organelles in a bioinformatics approach has not yet been established.
Here, we report CARS hyperspectral images of MIA PaCa-2 pancreatic cancer cells generated via unsupervised multivariate methods such as hierarchical cluster analysis (HCA). We also demonstrate the feasibility of using CARS coupled with HCA to colocalize the subcellular organelles by comparing the HCA images with those obtained using immunofluorescence staining. With these data, a supervised learning algorithm based on the ensemble method “random forest” as a classifier was trained and used, for the first time to our knowledge, to identify simultaneously and automatically the nucleus, nucleolus, lipid droplets, and endoplasmic reticulum.
Materials and Methods
Cell culture
Human pancreatic cancer MIA PaCa-2 (CRL-1420) and colorectal adenocarcinoma HT29 (HTB-38) cells were obtained from the American Type Culture Collection (Manassas, VA). The cells were grown in Dulbecco’s modified Eagle’s medium (Invitrogen, Carlsbad, CA) supplemented with 10% fetal bovine serum (Invitrogen) and 0.1% penicillin/streptomycin, incubated at 37°C in 10% CO2 atmosphere. The cells were plated out on glass coverslips (Menzel Glas, Braunschweig, Germany) for 1 day, fixed with 4% paraformaldehyde (VWR International, Darmstadt, Germany), and subsequently submerged into phosphate-buffered saline (PBS; Invitrogen).
Confocal Raman microscopy
Raman micro-spectroscopy was acquired using a confocal Raman microscope (Alpha300AR; WITec, Ulm, Germany) as described in details given previously in Mavarani et al. (11) and El-Mashtoly et al. (31).
CARS data acquisition
The CARS-images were obtained using a commercial setup consisting of a picosecond-pulsed laser system that generates two synchronized beams collinearly aligned in an inverted confocal microscope (TCS SP5 II CARS; Leica Microsystems, Heidelberg, Germany). A fraction of the fundamental light of an Nd:YVO4 (HighQ Laser, Rankweil, Austria) at 1064 nm is directly coupled into the microscope and used as a Stokes beam in the CARS imaging process. The frequency-doubled output (532 nm) is used to synchronously pump an optical parametric oscillator (picoEmerald; APE, Berlin, Germany), which is tunable in the range of 780–960 nm. The laser beams are focused into the sample using an HCX IRAPO L water immersion objective (40×/1.1 W CORR; Leica Microsystems). The epi- and forward-detected CARS signals are measured simultaneously, via non-descanned detectors. In this study, we will present only the forward-CARS signals.
The mean laser power was measured at the sample position and found to be 28 and 21 mW at 816 and 1064 nm, respectively. The power was kept stable during the wavelength tuning of the laser. A typical pixel dwell time of 200 μs per scan was selected (51 s per image, 512 × 512 pixels covering up to 150 × 150 μm sample area, pixel resolution 200–300 nm). The total time needed to collect a CARS hyperspectral image is ∼50 min, which is much faster than the corresponding spontaneous Raman measurement (∼5 h) with the same pixel resolution (16). Even faster CARS measurements can be achieved by measuring selected wavelengths only. The CARS hyperspectral images of cells were measured between 826–806 nm, corresponding to a vibrational range of 2700–3000 cm−1 with ∼5 cm−1 spectral resolution. In case of live cell imaging, the CARS hyperspectral dataset is measured (2800–3000 cm−1) within 17 min (17 s per image, 512 × 512 pixels covering at ∼150 × 150 μm sample area, pixel resolution 200–300 nm).
Immunofluorescence staining and imaging
The formalin-fixed cells were permeabilized with 0.2% Triton X-100 for 5 min at room temperature. After washing with PBS, cells were blocked with 1% bovine serum albumin for 30 min. The primary antibodies were incubated overnight at 4°C. Rabbit monoclonal anti-Calnexin (Jackson ImmunoResearch, West Grove, PA), mouse monoclonal anti-Syntaxin 6 (BD Biosciences, Franklin Lakes, NJ), and anti-COX IV (Abcam, Cambridge, UK) antibodies were used for endoplasmic reticulum, Golgi apparatus, and mitochondria, respectively. Excess antibodies were removed by washing three times with PBS, followed by the incubation of the secondary antibodies for 1 h at room temperature. The secondary antibodies conjugated to TRITC (tetramethyl rhodamine) and FITC (fluorescein) from Jackson ImmunoResearch were used. Excess antibodies were removed by again washing three times with PBS. Cells (living or fixed) were incubated with DRAQ-5 (1,5-bis{[2-(di-methylamino)ethyl]amino}-4,8-dihydroxyanthracene-9,10-dione; Cell Signaling Technology, Danvers, MA) or LD540 (4,4-difluoro-2,3,5,6-bis-tetramethylene-4-bora-3a,4a-diaza-s-indacene) for 10 and 30 min, respectively, followed by washing with PBS.
Fluorescence measurements were performed all the time sequentially on double- or triple-stained specimen with a confocal laser-scanning microscope (TCS SP5 II; Leica Microsystems) using an HCX IRAPO L (25×/0.95 W) water immersion objective (Leica Microsystems). To enable an optimal match with CARS images, stacks of fluorescence images were recorded and the distance between each layer was 0.5 μm.
Data analysis
The raw data and image processing was implemented in the software MATLAB Ver. 8.1 along with the Image Processing and Statistics Toolboxes (The MathWorks, Inc., Natick, MA). To obtain representative spectra for different cellular compartments, a colocalization approach motivated by fluorescence colocalization statistics was employed (32). Hereby a best-matching cluster is identified in a hierarchical clustering of the normalized spectral image data (S.D. Krauss, D. Petersen, D. Niedieker, E. Freier, S.F. El-Mashtoly, K. Gerwert, and A. Mosig, unpublished). Therefore, HCA images of CARS spectra were manually overlaid with the corresponding fluorescence images. For every possible combination of a cluster and a fluorescence color channel, the degree of colocalization is calculated according to Pearson’s correlation coefficient (PCC) (32–34) and the cluster with the highest PCC value was kept. After that, 100 thresholds were tested on the fluorescence from 1 to 100% intensity, and the image was binarized with the threshold showing the highest correlation with the best matching cluster from the HCA. Spectra were used for training of a random forest method from image positions whenever the fluorescence intensity was above the threshold value and was covered by the best matching cluster.
Supervised classification by random forest method
The comparison of different datasets is disturbed by intensity fluctuation of the laser system caused by tuning the pump laser and requires a correction. The CARS signal intensity is proportional to the third order of the susceptibility χ, the Stokes laser intensity, and the square of the pump laser intensity. Disregarding the nonresonant background, intensity fluctuations can be handled as an additive factor of the logarithmic signal intensity. The intensity of the laser can be measured directly or can be reconstructed by an internal standard, which, in this case, is the PBS buffer around the cell. The logarithmic mean spectrum of the buffer was subtracted from the logarithmic hyperspectral dataset. Afterwards, the data was reexponentiated. The SNR was enhanced by a Gaussian filter in image space with a window size of 3 × 3 pixels.
The spectral data was automatically classified by the random forest method (35). Training spectra were acquired from the colocalization step by selecting the spectra from the overlap of the best matching cluster and the fluorescence. The training spectra for lipid droplets were selected based on the spectral image at 2850 cm−1 if the fluorescence staining for lipid droplets was not performed. The classification was performed on vector-normalized spectral datasets after the correction of the laser fluctuation and was validated by estimating the colocalization with the corresponding fluorescence image.
Results and Discussion
Label-free Raman imaging of pancreatic cancer cells
Fig. 1 depicts the Raman results of MIA PaCa-2 cells. Fig. 1 A displays a univariate image based on the integrated Raman intensities of the C-H stretching vibration (2800–3100 cm−1). This image shows the nucleus in the middle of the cell, three nucleoli within the nucleus, and lipid droplets in the cytoplasm. Fig. 1 B displays the automated computed HCA, which is an unsupervised clustering method and used to generate an index-colored image from the Raman hyperspectral dataset. An algorithm is searching for similar spectra and merges them into a new object designated as a cluster. The merging process is repeated until all Raman spectra are combined into a small number of clusters and each cluster is assigned a color, constructing an index-colored image based on spectral similarity (36). HCA in Fig. 1 B was performed on the spectral range of 1200–1800 and 2800–3100 cm−1. These regions exhibit the most predominant bands of amino acids, protein backbones, nucleotides, nucleic acid backbones, carbohydrates, and lipids. It contains sufficient spectral information to provide excellent clustering results (36,37). Sixteen clusters were chosen to reproduce the position of nucleus with nucleoli, as well as several regions within the cytoplasm, reflecting different compositions of the cytoplasm with various subcellular components. Alternatively, HCA was also performed on either the 1200–1800 cm−1 (Fig. 1 C) or 2800–3100 cm−1 regions (C-H stretching, Fig. 1 D). The results are similar to those of Fig. 1 B, implying that the different spectral ranges (Fig. 1, B–D) at which HCA is performed are sufficient to produce excellent and comparable clustering results. However, the minor differences in Fig. 1, B–D, might originate from the contribution of different spectral regions.
Figure 1.
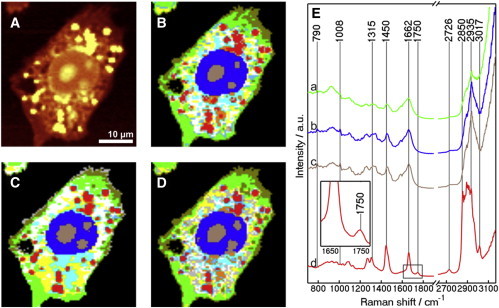
Hierarchical cluster analysis of Raman dataset. (A) Raman image reconstructed from the C-H stretching intensity. HCA is performed in the following ranges: (B) 1200–1800 and 2800–3100 cm−1; (C) 1200–1800 cm−1; and (D) 2800–3100 cm−1. The morphology and topology of some clusters suggest that they are associated with the cytoplasm (green), nucleus (blue), nucleoli (brown), and lipid droplets (red). Other clusters in panels B–D cannot be assigned in this manner. (E) The mean cluster Raman spectra are also shown.
Fig. 1 E shows the mean cluster spectra of only a few subcellular organelles, where morphology and topology of their representative clusters clearly suggest that they are associated with cytoplasm (a), nucleus (b), nucleoli (c), and lipid droplets (d). The most pronounced Raman bands result from the C-H stretching vibrations among 2850 and 3020 cm−1, the carbonyl stretching (amide I) located at ∼1662 cm−1, C-H and CH2 bending deformations at 1450 cm−1, amide III of the peptide linkages between 1200 and 1400 cm−1, and the ring-breathing mode of phenylalanine at 1008 cm−1 (5,38,39).
Spectrum d is significantly different from the others with strong intensity. Note that, due to normalization, the strong intensity is not seen in Fig. 1 E. By comparing the mean cluster spectrum d with the lipid and phospholipid spectra (7,40–42), it is clear that the spectrum d arises from high lipid or phospholipid content. The high C-H stretching intensities in the spectral range of 2850–2935 cm−1 are due to long alkane chains. Only the spectrum of lipid droplets (d) exhibits a Raman band at 1750 cm−1, which is assigned to the C=O stretching vibration of the ester form of fatty acids, probably triglyceride (which is the dominant component of lipid droplets).
The above results indicate that Raman microspectroscopy coupled with HCA has a potential for the label-free identification of subcellular compartments of MIA PaCa-2 cells. This is in agreement with previous studies that had shown the distribution of lipid droplets and mitochondria in Caco-2 and HeLa cells, respectively (5,15). In addition, the 1750 cm−1 band can be used as a marker-free band for imaging of lipid droplets.
Hierarchical cluster analysis of CARS datasets as an unsupervised method
Lipids are rich in C-H2 groups and have high local concentrations. In addition, the CARS signal is proportional to the square of the concentration of the specimen (43). Thus, lipids cause a very strong CARS signal at 2850 cm−1, and imaging of lipid droplets in cells is the most common application of CARS microscopy (13,17,19,44). Fig. 2 A depicts the CARS image taken at 2850 cm−1 of the MIA PaCa-2 cells. It is obvious that lipid droplets significantly emit a stronger CARS signal compared to the other cellular compartments. Fig. 1 E indicates that lipid droplets (spectrum d) exhibit a Raman band at 1750 cm−1, which is assigned to the C=O stretching vibration of the ester form of fatty acids. Thus, we have performed CARS imaging taken at both 2850 and 1750 cm−1 and the results are depicted in Fig. S1 (see the Supporting Material). Identification of these bright spots as lipid droplets was confirmed by confocal fluorescence microscopy, which was performed subsequent to CARS data acquisition. Results of CARS imaging of lipid droplets taken at 2850 cm−1 are consistent with those of previous studies (13,45); however, to the best of our knowledge, we have shown for the first time that the C=O stretching vibration of the ester form of fatty acids can also be used as a label-free marker for imaging the lipid droplets by CARS microscopy.
Figure 2.
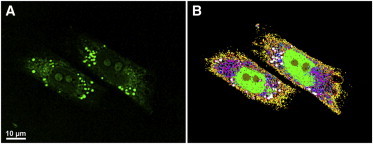
Hierarchical cluster analysis of CARS dataset. (A) CARS imaging taken at 2850 cm−1 of the MIA PaCa-2 cells. (B) HCA of CARS dataset of MIA PaCa-2 cells. Spectra were normalized in the 2700–3000 cm−1 region and HCA was performed on the same spectral range. The morphology and topology of some clusters suggest that they are associated with the cytoplasm (yellow), nucleus (green), nucleoli (brown), and lipid droplets (pink). Other clusters in panel B cannot be assigned in this manner.
The HCA of the spontaneous Raman spectral dataset (Fig. 1) has shown that it is a powerful method to visualize different cellular components and that the spectral region of 2700–3000 cm−1 is sufficient to produce good clustering results. These results were encouraging to perform HCA on CARS datasets in the C-H stretching region only. Restriction to this region has an additional advantage that the broad Raman band of the glass coverslips, on which cells are grown, does not interfere (16).
In CARS measurements, the pump wave was tuned several times between 826 and 806 nm, corresponding to a vibrational range of 2700–3000 cm−1 with ∼5 cm−1 spectral resolution. CARS datasets were obtained with a pixel resolution of 200–300 nm. The region of interest was selected by masking the area outside cells to reduce the number of spectra. The data were vector-normalized in the 2700–3000 cm−1 range and HCA was performed in the same spectral range. Fig. 2 B displays the HCA of a CARS dataset of MIA PaCa-2 cells. Twenty-four clusters were chosen to reproduce the position of the nucleus with nucleoli and lipid droplets, as well as reflecting the compositions of the cytoplasm with several organelles.
Recently, PCA and NMF analyses of CARS datasets were used to visualize cellular components (26,29,30). Further, this study shows the potential of HCA of CARS datasets in visualizing several subcellular organelles and opens an avenue toward their label-free identification. An advantage of HCA over other clustering methods is that it clearly visualizes several cellular components, as shown very recently for the HCA of spontaneous Raman dataset (31).
Automatic identification of subcellular organelles
In this study, we believe that we have established, for the first time, a supervised algorithm for automated identification of pancreatic cancer subcellular organelles by using label-free CARS imaging based on the random forest method as a classifier. Basically, the data analysis involves two stages as depicted in Fig. 3, i.e., the training and validation stages.
Figure 3.
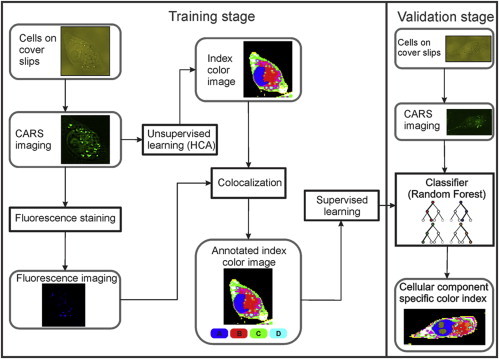
Workflow for spectral image classifier. The procedure consists of two stages—training (left side) and validation (right side) stages.
Training stage
The aim of the training phase is to obtain representative spectra for different cellular compartments to train a supervised classifier. Therefore, the dissimilarity of the normalized space-resolved CARS spectra is determined by HCA.
The PCC value of each combination of the spectra is calculated and a hierarchy of clusters is built up by the use of Ward’s method. In this hierarchy of clusters, an algorithm searches for the cluster best matching with the fluorescence image of the corresponding organelle. This is because the standard method in life sciences for imaging the subcellular organelles and their dynamics is fluorescence microscopy (46). Figs. 4 and 5 show the colocalization of several organelles in the HCA results.
Figure 4.
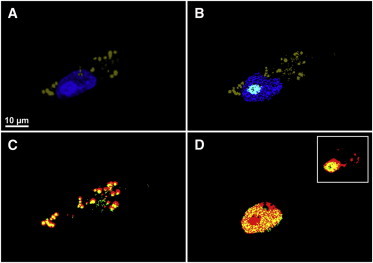
Colocalization of the nucleus and lipid droplets. (A) Fluorescence imaging of the nucleus and lipid droplets. (B) HCA of CARS dataset. Overlaid images of lipid droplets and the nucleus fluorescence (red) from panel A with their corresponding HCA clusters (green) from panel B are displayed in panels C and D, respectively. (yellow) Overlapping regions. (D, inset) Overlay region of the nucleolus between the fluorescence (red) and HCA (green).)
Figure 5.
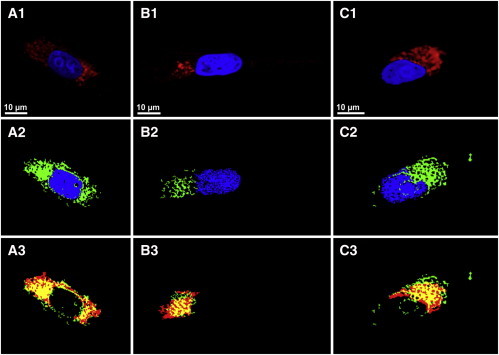
Colocalization of the endoplasmic reticulum, Golgi apparatus, and mitochondria. (A1–C1) Fluorescence images of the nucleus (blue), endoplasmic reticulum (A1), Golgi apparatus (B1), and mitochondria (C1). (A2–C2) HCA of CARS datasets. (A3–C3) Overlaid images of endoplasmic reticulum, Golgi apparatus, and mitochondria staining (red) from panels A1–C1 with their corresponding HCA (green) from A2–C2 and the overlapping regions (yellow).
Fluorescence staining was carried out on the lipid droplets and the nucleus after CARS measurements and the results are depicted in Fig. 4 A. In this study, we used DRAQ-5 to stain the nucleus as well as nucleolus (47), and LD540 fluorescent dye for lipid droplets (48). Lipid droplets are displayed in olive, whereas the nucleus is shown in blue. It is noted that the nucleolus gives stronger fluorescence intensity than the nucleus; the regions above the threshold in the nucleus fluorescence images are classified as nucleolus. Best matching clusters from HCA results according to the overlap with the fluorescence are shown in Fig. 4 B. Fig. 4, C and D, illustrates the overlay of the HCA clusters (green) with their corresponding fluorescence channels (red) for the lipid droplets and the nucleus, respectively, and the regions of agreements are shown in yellow.
The overlapping regions fit well and the PCC values were calculated to be ∼0.6 for lipid droplets and ∼0.78 for the nucleus. Fig. 4 D (inset) also displays similar overlay for the nucleolus and good agreement is observed with a PCC value at ∼0.69. The blue, cyan, and olive clusters in the HCA (Fig. 4 B) are assigned to the nucleus, nucleolus, and lipid droplets, respectively. In addition, the mean CARS spectrum of lipid droplets has a maximum peak at 2850 cm−1 (see Fig. S2), which is in agreement with previous results (13,45).
In a previous spontaneous Raman study (16), the constructed fluorescence images based on selected Raman spectral features in the 80–3040 cm−1 regime contain contributions from a broad glass band near 1100 cm−1. This band obscures the DNA band at ∼1094 cm−1 (39), and it creates artifacts in the spectral images, especially for the nucleus and nucleolus. The constructed images depict regions within the nucleus with strong signal, which were absent in the fluorescence. These regions were assumed to be the nucleoli (16). The CARS measurements, however, were performed on the C-H region, which is free from the glass contribution. The colocalization of the nucleolus here is based on fluorescence.
Furthermore, immunofluorescence staining was performed after CARS measurements for several cells using either anti-Calnexin for endoplasmic reticulum, anti-Syntaxin-6 for Golgi apparatus, or anti-COX IV for mitochondria to illustrate other subcellular organelles in addition to the nucleus. Panels A1–C1 in Fig. 5 show two-channel immunofluorescence images of MIA PaCa-2 cells. In addition to the nucleus (blue), they describe the distribution of endoplasmic reticulum (Fig. 5 A1), Golgi apparatus (Fig. 5 B1), or mitochondria (Fig. 5 C1), as illustrated in red. In general, the endoplasmic reticulum is surrounding the nucleus in high concentration accompanied by Golgi apparatus, whereas the mitochondria are spread in the cytoplasm. A visual assessment indicates that the best matching clusters from the HCA (Fig. 5, A2–C2) share information with their corresponding fluorescence images (Fig. 5, A1–C1). For instance, there is a great match of the blue channels in both HCA and fluorescence staining.
For the colocalization of the cellular components in the HCA results, the fluorescence channels were overlaid with their corresponding HCA clusters in detail. For instance, the matching regions (yellow) of the fluorescence imaging (red) with the HCA (green) for the endoplasmic reticulum, Golgi apparatus, and mitochondria are displayed in Fig. 5, A3–C3. Good overlay is observed with PCC values at ∼0.55 for these three organelles. Furthermore, mitochondrial distribution in glioma and HeLa cells has been detected previously by spontaneous Raman microscopy (5,16). Spectral differences between the mitochondria and the nucleus were observed in the C-H region. Specifically, the shoulder at the region at ∼2850 cm−1 is more distinct for mitochondria than for the nucleus (5). Similar results were observed in the mean CARS spectra for MIA PaCa-2 cells (see Fig. S2). Thus, the above results shown in Figs. 4 and 5 produced a set of spectral training datasets for six classes of subcellular organelles including the nucleus, nucleolus, lipid droplets, endoplasmic reticulum, Golgi apparatus, and mitochondria.
Several reports have demonstrated the feasibility of using CARS microscopy coupled with unsupervised multivariate analyses such as PCA and NMF (25,26,29,30), but none of them have reported the supervised classifications of cellular organelles. Further, in this study, we first applied unsupervised HCA on CARS datasets to visualize cellular components, which are identified using fluorescence images as a reference. More importantly, we have obtained representative CARS spectra for different cellular components to train a supervised classifier, which is an unprecedented step in comparison with previous publications of multivariate analyses of CARS datasets (25,26,29,30). The supervised classifier is then used for an automatic identification of the subcellular organelles, as explained below in the validation stage.
It is noted that the PCC values for the colocalization are higher for the nucleus than the other subcellular organelles, which are lower than those of the nucleus. This might be due to the size of the cellular components, where the nucleus is the largest component in the cell. This implies that the probability of probing a pure component in the confocal volume becomes higher as the size of the subcellular organelles increases. In the case of small organelles, more than one component might be present in the confocal volume. In the case of fluorescence microscopy, even minor components can be still detected due to the selective labeling. On the other hand, in CARS microscopy the spectrum is composed of a combination of the spectra of all components within the confocal volume. If the amount of the components gets lower, the HCA might assign the pixel to another class and thus, the fluorescence only colocalizes with a fraction. The sensitivity of these two methods is limited by different factors. Whereas fluorescence is limited by specificity and efficiency of the dye, CARS is limited by the characteristic spectrum of the target component.
Furthermore, the lower PCC values can be explained in terms of a different confocal volume, especially in the z direction of both CARS and fluorescence measurements that arises from the following:
-
1.
After CARS measurements, the coverslips were removed from the microscope to perform fluorescence staining protocol, and afterwards they were repositioned on the same microscope to acquire fluorescence measurements, which may lead to a slight shift in the z direction between the CARS and fluorescence measurements.
-
2.
Single photon fluorescence imaging was used here—in contrast to CARS, which is a multiphoton process.
-
3.
The wavelengths used for fluorescence imaging are shorter than those used for CARS measurements.
Validation stage
On the basis of the classifications achieved in the training stage, the random forest classifier was trained to perform supervised classification. Random forest is an attractive classifier because it is simple; it does not require a feature selection before training; it is efficient in both training and validation; and it is robust against overfitting (35). We have recently used random forest as a classifier to automatically identify colon tissue types and area of colon adenocarcinoma in IR and Raman spectral datasets (11,12). Using the training dataset described above, a random forest classifier was trained on the preprocessed spectral images using 300 trees. The accuracy of the random forest is ∼90%, independent of the number of trees. Based on this, the spectral images depicted in Fig. 6 A are seen as automatic identifications of subcellular organelles performed by the random forest.
Figure 6.
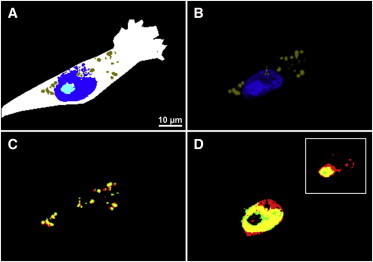
Automatic identification of the nucleus, nucleolus, and lipid droplets. (A) Random forest of CARS dataset. (B) Fluorescence imaging of the nucleus and lipid droplets. Overlaid images of lipid droplets and the nucleus fluorescence (red) from panel B with their corresponding random forest classes (green) from panel A are displayed in panels C (lipid droplets) and D (nucleus), respectively. (yellow) Overlapping region. (D, inset) Overlay region of the nucleolus between the fluorescence (red) and random forest (green).
In Fig. 6 A, the CARS pseudo-color image is displayed based on a random forest of nucleus (blue) and lipid droplets (olive). It also clearly displays the nucleolus (cyan) within the nucleus. To evaluate the quality of the CARS pseudo-color images, the fluorescence channels (red) from Fig. 6 B were overlaid with their counterpart random forest classes (green) and the results are displayed in Fig. 6, C (lipid droplets) and D (nucleus).
The nucleolus is also shown as an inset of Fig. 6 D. Good overlay is observed and the PCC values between the random forest classes and their corresponding fluorescence channels were estimated to be ∼0.65 for lipid droplets and ∼0.76 for the nucleus and nucleolus. These results indicate that the supervised random forest learning algorithm is successfully reproducing the fluorescence results. Thus, three subcellular components are label-free monitored instead of using at least two different fluorescent labels. Although the identification of lipid droplets by CARS imaging at ∼2850 cm−1 has been reported previously using fluorescence staining as a reference (13,44), these results show, for the first time to our knowledge, their automated identification in addition to the nucleus and nucleolus by means of random forest.
Label-free identification of subcellular organelles such as endoplasmic reticulum, Golgi apparatus, and mitochondria is important because these components play major roles in cellular functions (46). We have constructed three selective training datasets for three random forests. Each of them contains the nucleus, nucleolus, and lipid droplets classes in addition to only one class of endoplasmic reticulum, Golgi apparatus, or mitochondria. The random forest results are shown in Fig. 7. In this case, endoplasmic reticulum (Fig. 7 A1), Golgi apparatus (Fig. 7 B1), or mitochondria (Fig. 7 C1) are monitored independent of each other.
Figure 7.
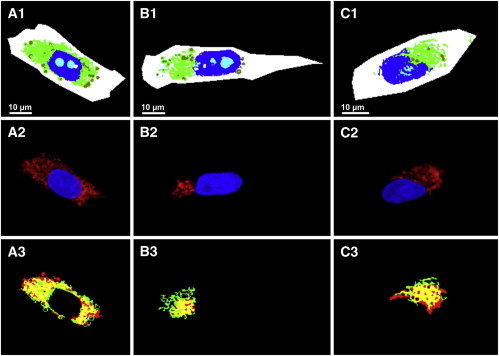
Automatic identification of the endoplasmic reticulum, Golgi apparatus, and mitochondria. (A1–C1) Random forests of CARS datasets. (A2–C2) Fluorescence images of the nucleus (blue), endoplasmic reticulum (A2), Golgi apparatus (B2), and mitochondria (C2). (A3–C3) Overlaid images of endoplasmic reticulum, Golgi apparatus, and mitochondria fluorescence (red) from panels A2–C2 with their corresponding random forest (green) and from panels A1–C1 with overlapping regions (yellow).
The fluorescence-labeled images of the cellular organelles (red) taken from Fig. 7, A2–C2, are overlaid with their corresponding classes of random forest (green) taken from Fig. 7, A1 to C1, and are displayed in Fig. 7, A3–C3. The regions of overlaps are shown in yellow. These data show that random forest classes correlate well with their corresponding fluorescence images of cellular components. This is reflected by the PCC values, which are estimated to be ∼0.61 for endoplasmic reticulum, ∼0.48 for Golgi apparatus, ∼0.61 for mitochondria, and ∼0.86 for the nucleus. Similar results have been observed for the calculated artificial fluorescence images based on Raman data for the subcellular components of glioma cells using a different approach (16).
However, when the classes of different random forests for endoplasmic reticulum, Golgi apparatus, or mitochondria (Fig. 7, A1–C1) are overlaid, the PCC values for the correlation of any two of these classes are found to be quite high (∼0.84). This indicates that these classes are quite similar, and the selective random forests display very little differentiation between any of these three organelles (see also Fig. S3).
To improve the differentiation between these three organelles and identify the subcellular organelles simultaneously, we have created one training dataset that includes all classes for a single random forest. Fig. 8 shows the simultaneous identification of endoplasmic reticulum, Golgi apparatus, and mitochondria in addition to the nucleus, nucleoli, and lipid droplets (see also Fig. S4). The simultaneous identification of several subcellular organelles is one of the believed novel findings in this study and was not reported previously.
Figure 8.
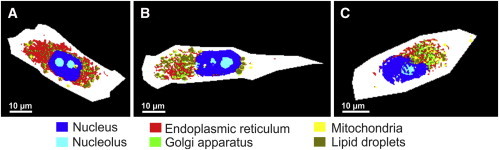
Simultaneous automatic identification of subcellular components in MIA PaCa-2 cells.
Correlation between the random forest classes and their counterpart fluorescence channels is reflected by PCC, which is ∼0.57 for endoplasmic reticulum (Fig. 7 A), ∼0.29 for Golgi apparatus (Fig. 7 B), and ∼0.23 for mitochondria (Fig. 7 C). The average PCC value of the nucleus in Fig. 7, A–C, was estimated to be ∼0.87. Because the staining of lipid droplets was not performed for these cells (Fig. 7), the lipid droplets class was overlaid with the CARS image at 2850 cm−1, which is a lipid droplets marker band (13,17,19).
Perfect overlays were observed (data not shown). Thus, the simultaneous identification of subcellular organelles gives satisfactory results for nucleus, nucleoli, lipid droplets, and endoplasmic reticulum. The random forest is also able to distinguish among endoplasmic reticulum, Golgi apparatus, and mitochondria. However, the PCC values for the correlation among the random forest classes of Golgi apparatus and mitochondria and their corresponding fluorescence channels are lower than expected. Both endoplasmic reticulum and Golgi apparatus belong to the endomembrane system, and are colocalized (49). Endoplasmic reticulum is also colocalized with mitochondria at certain positions in cells (49,50). Indeed, the fluorescence results of MIA PaCa-2 cells and the calculated PCC values have indicated the colocalization of these components (see Fig. S5 and Fig. S6). Thus, the colocalization of these three subcellular organelles makes the simultaneous identification of Golgi apparatus and mitochondria difficult, leading to lower PCC values. This is because the classifier has to decide on one assignment for each pixel even though, in the corresponding confocal volume, more than one component is present.
In this study, we have used CARS microscopy rather than spontaneous Raman measurements because CARS is a much faster method. Even faster time resolution is possible by using instruments capable of line scanning (51) or fast wavelength tunability (52). This point is critical, especially for live cell imaging, due to the viability of cells and the dynamical behavior of most subcellular organelles. CARS imaging of living MIA PaCa-2 cells has been performed in addition to fluorescence imaging of nucleus and lipid droplets (see Fig. S7). The PCC values for the correlation between random forest classes of lipid droplets and nucleus with their corresponding fluorescence channels are calculated to be 0.58 and 0.62, respectively. These results imply that the current trained classifier can also be applied on live cells.
Furthermore, to examine whether this trained classifier can be applied on different cell types, we have performed CARS and fluorescence measurements of human colorectal adenocarcinoma cells, HT29, and the results are depicted in Fig. S8. The PCC values for the correlation between random forest classes of nucleus and lipid droplets with their corresponding fluorescence channels are satisfactory. These results suggest that the trained classifier is probably applicable to a variety of human cancer cells as well.
In summary, the nucleus, nucleoli, lipid droplets, and endoplasmic reticulum can be automatically and marker-free identified. This is based not only on visual inspection, but also on quantitative evaluation, which demonstrates that random forest is a powerful tool to identify the subcellular organelles. The random forest classifier can also differentiate among endoplasmic reticulum, Golgi apparatus, and mitochondria. Methods that can assign a combination of components to each pixel (e.g., NMF) might further improve the correlation (36). In addition, the trained classifier is probably applicable on different human cancer cells.
Prospective of an automated label-free identification of subcellular organelles
An automatic identification of pancreatic cancer subcellular organelles by CARS microscopy opens new avenues for several applications. For instance, lipid droplets are highly dynamical organelles and perform several diverse functions such as regulating storage and release of cholesterol and fatty acids (20–23,53). It was observed that the number of lipid droplets is increased in neoplastic cells and tissues, implying that lipid droplets have a high potential as disease markers (54). It has also been suggested that lipid droplets are potential targets to produce new drugs for cancer treatment (55). Thus, the automatic identification of lipid droplets by CARS microscopy can be used for lipid droplets quantification and to evaluate the response of these new drugs.
Finally, CARS microscopy promises an attractive approach for the label-free characterization of the morphology of nuclei, which are of high relevance in high-throughput microscopy. Contemporary protocols for studying the cell-cycle using quantitative analysis of time-lapse microscopy (56) or tumor microenvironments (57) are based on either fluorescence or hematoxylin-eosin-staining protocols, to extract the morphology of nuclei. CARS microscopy as a label-free approach may greatly simplify these protocols by eliminating the need for staining in high-throughput microscopic studies.
Conclusions
CARS imaging coupled with HCA have been used for what we believe is the first time to visualize the cellular components without the need for external markers. Furthermore, we have established an algorithm for automatic label-free identification of the cellular components such as nucleus, nucleolus, lipid droplets, and endoplasmic reticulum using the random forest method. This study opens an avenue for the label-free simultaneous imaging of the dynamics of several subcellular organelles. Furthermore, in the future, we aim to identify several subcellular components in living cells by CARS imaging by using only a few wavenumbers, to make the measurements much faster than this study. This can be achieved, for instance, using wavenumber feature selection by means of the random forest method.
Acknowledgments
We thank Professor Ralf Erdmann for providing us with LD540 staining material and Hesham K. Yosef for the preparation of HT29 cells.
This research was supported by the Protein Research Unit Ruhr within Europe, Ministry of Innovation, Science and Research of North-Rhine Westphalia, Germany (to K.G.) and the Center for Vibrational Microscopy, European Regional Development Fund, European Union and North-Rhine Westphalia, Germany.
Footnotes
Samir F. El-Mashtoly and Daniel Niedieker contributed equally to this work.
Supporting Material
References
- 1.König K., Ehlers A., Kaatz M. Clinical two-photon microendoscopy. Microsc. Res. Tech. 2007;70:398–402. doi: 10.1002/jemt.20445. [DOI] [PubMed] [Google Scholar]
- 2.Llewellyn M.E., Barretto R.P.J., Schnitzer M.J. Minimally invasive high-speed imaging of sarcomere contractile dynamics in mice and humans. Nature. 2008;454:784–788. doi: 10.1038/nature07104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Marcsisin E.J., Uttero C.M., Diem M. Infrared microspectroscopy of live cells in aqueous media. Analyst (Lond.) 2010;135:3227–3232. doi: 10.1039/c0an00548g. [DOI] [PubMed] [Google Scholar]
- 4.Matthäus C., Boydston-White S., Diem M. Raman and infrared microspectral imaging of mitotic cells. Appl. Spectrosc. 2006;60:1–8. doi: 10.1366/000370206775382758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Matthäus C., Chernenko T., Diem M. Label-free detection of mitochondrial distribution in cells by nonresonant Raman microspectroscopy. Biophys. J. 2007;93:668–673. doi: 10.1529/biophysj.106.102061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Popp J. Wiley-VCH/John Wiley; Weinheim, Germany: 2011. Handbook of Biophotonics: Vol. 2, Photonics for Health Care. [Google Scholar]
- 7.Krafft C., Knetschke T., Salzer R. Studies on stress-induced changes at the subcellular level by Raman microspectroscopic mapping. Anal. Chem. 2006;78:4424–4429. doi: 10.1021/ac060205b. [DOI] [PubMed] [Google Scholar]
- 8.van Manen H.-J., Kraan Y.M., Otto C. Single-cell Raman and fluorescence microscopy reveal the association of lipid bodies with phagosomes in leukocytes. Proc. Natl. Acad. Sci. USA. 2005;102:10159–10164. doi: 10.1073/pnas.0502746102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Uzunbajakava N., Lenferink A., Otto C. Nonresonant confocal Raman imaging of DNA and protein distribution in apoptotic cells. Biophys. J. 2003;84:3968–3981. doi: 10.1016/S0006-3495(03)75124-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Boydston-White S., Romeo M., Diem M. Cell-cycle-dependent variations in FTIR micro-spectra of single proliferating HeLa cells: principal component and artificial neural network analysis. Biochim. Biophys. Acta. 2006;1758:908–914. doi: 10.1016/j.bbamem.2006.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mavarani L., Petersen D., Gerwert K. Spectral histopathology of colon cancer tissue sections by Raman imaging with 532 nm excitation provides label free annotation of lymphocytes, erythrocytes and proliferating nuclei of cancer cells. Analyst (Lond.) 2013;138:4035–4039. doi: 10.1039/c3an00370a. [DOI] [PubMed] [Google Scholar]
- 12.Kallenbach-Thieltges A., Grosserüschkamp F., Gerwert K. Immunohistochemistry, histopathology and infrared spectral histopathology of colon cancer tissue sections. J. Biophoton. 2013;6:88–100. doi: 10.1002/jbio.201200132. [DOI] [PubMed] [Google Scholar]
- 13.Nan X., Cheng J.-X., Xie X.S. Vibrational imaging of lipid droplets in live fibroblast cells with coherent anti-Stokes Raman scattering microscopy. J. Lipid Res. 2003;44:2202–2208. doi: 10.1194/jlr.D300022-JLR200. [DOI] [PubMed] [Google Scholar]
- 14.Saar B.G., Freudiger C.W., Xie X.S. Video-rate molecular imaging in vivo with stimulated Raman scattering. Science. 2010;330:1368–1370. doi: 10.1126/science.1197236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Scalfi-Happ C., Udart M., Rück A. Investigation of lipid bodies in a colon carcinoma cell line by confocal Raman microscopy. Med. Laser Appl. 2011;26:152–157. [Google Scholar]
- 16.Klein K., Gigler A.M., Schlegel J. Label-free live-cell imaging with confocal Raman microscopy. Biophys. J. 2012;102:360–368. doi: 10.1016/j.bpj.2011.12.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cheng J.-X., Jia Y.K., Xie X.S. Laser-scanning coherent anti-Stokes Raman scattering microscopy and applications to cell biology. Biophys. J. 2002;83:502–509. doi: 10.1016/S0006-3495(02)75186-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jüngst C., Winterhalder M.J., Zumbusch A. Fast and long term lipid droplet tracking with CARS microscopy. J. Biophoton. 2011;4:435–441. doi: 10.1002/jbio.201000120. [DOI] [PubMed] [Google Scholar]
- 19.Krafft C., Dietzek B., Popp J. Raman and CARS microspectroscopy of cells and tissues. Analyst (Lond.) 2009;134:1046–1057. doi: 10.1039/b822354h. [DOI] [PubMed] [Google Scholar]
- 20.Welte M.A., Cermelli S., Gross S.P. Regulation of lipid-droplet transport by the perilipin homolog LSD2. Curr. Biol. 2005;15:1266–1275. doi: 10.1016/j.cub.2005.06.062. [DOI] [PubMed] [Google Scholar]
- 21.Fernández M.A., Albor C., Pol A. Caveolin-1 is essential for liver regeneration. Science. 2006;313:1628–1632. doi: 10.1126/science.1130773. [DOI] [PubMed] [Google Scholar]
- 22.Welte M.A. Proteins under new management: lipid droplets deliver. Trends Cell Biol. 2007;17:363–369. doi: 10.1016/j.tcb.2007.06.004. [DOI] [PubMed] [Google Scholar]
- 23.Boström P., Andersson L., Olofsson S.O. SNARE proteins mediate fusion between cytosolic lipid droplets and are implicated in insulin sensitivity. Nat. Cell Biol. 2007;9:1286–1293. doi: 10.1038/ncb1648. [DOI] [PubMed] [Google Scholar]
- 24.Pliss A., Kuzmin A.N., Prasad P.N. Nonlinear optical imaging and Raman microspectrometry of the cell nucleus throughout the cell cycle. Biophys. J. 2010;99:3483–3491. doi: 10.1016/j.bpj.2010.06.069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lin C.-Y., Suhalim J.L., Potma E.O. Picosecond spectral coherent anti-Stokes Raman scattering imaging with principal component analysis of meibomian glands. J. Biomed. Opt. 2011;16:021104. doi: 10.1117/1.3533716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pohling C., Buckup T., Motzkus M. Hyperspectral data processing for chemoselective multiplex coherent anti-Stokes Raman scattering microscopy of unknown samples. J. Biomed. Opt. 2011;16:021105. doi: 10.1117/1.3533309. [DOI] [PubMed] [Google Scholar]
- 27.Chung C.-Y., Potma E.O. Biomolecular imaging with coherent nonlinear vibrational microscopy. Annu. Rev. Phys. Chem. 2013;64:77–99. doi: 10.1146/annurev-physchem-040412-110103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhang D., Wang P., Cheng J.X. Quantitative vibrational imaging by hyperspectral stimulated Raman scattering microscopy and multivariate curve resolution analysis. Anal. Chem. 2013;85:98–106. doi: 10.1021/ac3019119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Masia F., Glen A., Langbein W. Quantitative chemical imaging and unsupervised analysis using hyperspectral coherent anti-Stokes Raman scattering microscopy. Anal. Chem. 2013;85:10820–10828. doi: 10.1021/ac402303g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lee Y.J., Vega S.L., Cicerone M.T. Quantitative, label-free characterization of stem cell differentiation at the single-cell level by broadband coherent anti-Stokes Raman scattering microscopy. Tissue Eng. Part C Methods. 2013 doi: 10.1089/ten.tec.2013.0472. 2013 Dec 31 [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.El-Mashtoly S.F., Petersen D., Gerwert K. Label-free imaging of drug distribution and metabolism in colon cancer cells by Raman microscopy. Analyst (Lond.) 2014;139:1155–1161. doi: 10.1039/c3an01993d. [DOI] [PubMed] [Google Scholar]
- 32.Manders E.M., Stap J., Aten J.A. Dynamics of three-dimensional replication patterns during the S-phase, analyzed by double labeling of DNA and confocal microscopy. J. Cell Sci. 1992;103:857–862. doi: 10.1242/jcs.103.3.857. [DOI] [PubMed] [Google Scholar]
- 33.Manders E.M.M., Verbeek F.J., Aten J.A. Measurement of co-localization of objects in dual-color confocal images. J. Microsc. 1993;169:375–382. doi: 10.1111/j.1365-2818.1993.tb03313.x. [DOI] [PubMed] [Google Scholar]
- 34.Costes S.V., Daelemans D., Lockett S. Automatic and quantitative measurement of protein-protein colocalization in live cells. Biophys. J. 2004;86:3993–4003. doi: 10.1529/biophysj.103.038422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Breiman L. Random forests. Mach. Learn. 2001;45:5–32. [Google Scholar]
- 36.Miljković M., Chernenko T., Diem M. Label-free imaging of human cells: algorithms for image reconstruction of Raman hyperspectral datasets. Analyst (Lond.) 2010;135:2002–2013. doi: 10.1039/c0an00042f. [DOI] [PubMed] [Google Scholar]
- 37.Hedegaard M., Matthäus C., Popp J. Spectral unmixing and clustering algorithms for assessment of single cells by Raman microscopic imaging. Theor. Chem. Acc. 2011;130:1249–1260. [Google Scholar]
- 38.Salzer R. Wiley-VCH; Weinheim, Germany: 2009. Infrared and Raman Spectroscopic Imaging. [Google Scholar]
- 39.Diem M., Chalmers J.M., Griffiths P.R. John Wiley; Hoboken, NJ: 2008. Vibrational Spectroscopy for Medical Diagnosis. [Google Scholar]
- 40.Krafft C., Neudert L., Salzer R. Near infrared Raman spectra of human brain lipids. Spectrochim. Acta A Mol. Biomol. Spectrosc. 2005;61:1529–1535. doi: 10.1016/j.saa.2004.11.017. [DOI] [PubMed] [Google Scholar]
- 41.Huang Y.-S., Karashima T., Hamaguchi H.O. Molecular-level investigation of the structure, transformation, and bioactivity of single living fission yeast cells by time- and space-resolved Raman spectroscopy. Biochemistry. 2005;44:10009–10019. doi: 10.1021/bi050179w. [DOI] [PubMed] [Google Scholar]
- 42.Matthäus C., Krafft C., Popp J. Noninvasive imaging of intracellular lipid metabolism in macrophages by Raman microscopy in combination with stable isotopic labeling. Anal. Chem. 2012;84:8549–8556. doi: 10.1021/ac3012347. [DOI] [PubMed] [Google Scholar]
- 43.Dogariu A., Goltsov A., Scully M.O. Concentration dependence in coherent Raman scattering. J. Mod. Opt. 2008;55:3255–3261. [Google Scholar]
- 44.Paar M., Jüngst C., Wolinski H. Remodeling of lipid droplets during lipolysis and growth in adipocytes. J. Biol. Chem. 2012;287:11164–11173. doi: 10.1074/jbc.M111.316794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rinia H.A., Burger K.N.J., Müller M. Quantitative label-free imaging of lipid composition and packing of individual cellular lipid droplets using multiplex CARS microscopy. Biophys. J. 2008;95:4908–4914. doi: 10.1529/biophysj.108.137737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lakowicz J.R. Springer; New York: 2006. Principles of Fluorescence Spectroscopy. [Google Scholar]
- 47.Smith P.J., Blunt N., Patterson L.H. Characteristics of a novel deep red/infrared fluorescent cell-permeant DNA probe, DRAQ5, in intact human cells analyzed by flow cytometry, confocal and multiphoton microscopy. Cytometry. 2000;40:280–291. doi: 10.1002/1097-0320(20000801)40:4<280::aid-cyto4>3.0.co;2-7. [DOI] [PubMed] [Google Scholar]
- 48.Spandl J., White D.J., Thiele C. Live cell multicolor imaging of lipid droplets with a new dye, LD540. Traffic. 2009;10:1579–1584. doi: 10.1111/j.1600-0854.2009.00980.x. [DOI] [PubMed] [Google Scholar]
- 49.Alberts B. Garland Science; New York: 2008. Molecular Biology of the Cell. [Google Scholar]
- 50.Bravo R., Vicencio J.M., Lavandero S. Increased ER-mitochondrial coupling promotes mitochondrial respiration and bioenergetics during early phases of ER stress. J. Cell Sci. 2011;124:2143–2152. doi: 10.1242/jcs.080762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hamada K., Fujita K., Kawata S. Raman microscopy for dynamic molecular imaging of living cells. J. Biomed. Opt. 2008;13:044027. doi: 10.1117/1.2952192. [DOI] [PubMed] [Google Scholar]
- 52.Ozeki Y., Umemura W., Itoh K. High-speed molecular spectral imaging of tissue with stimulated Raman scattering. Nat. Photon. 2012;6:845–851. [Google Scholar]
- 53.Martin S., Parton R.G. Lipid droplets: a unified view of a dynamic organelle. Nat. Rev. Mol. Cell Biol. 2006;7:373–378. doi: 10.1038/nrm1912. [DOI] [PubMed] [Google Scholar]
- 54.Bozza P.T., Viola J.P.B. Lipid droplets in inflammation and cancer. Prostaglandins Leukot. Essent. Fatty Acids. 2010;82:243–250. doi: 10.1016/j.plefa.2010.02.005. [DOI] [PubMed] [Google Scholar]
- 55.Accioly M.T., Pacheco P., Viola J.P. Lipid bodies are reservoirs of cyclooxygenase-2 and sites of prostaglandin-E2 synthesis in colon cancer cells. Cancer Res. 2008;68:1732–1740. doi: 10.1158/0008-5472.CAN-07-1999. [DOI] [PubMed] [Google Scholar]
- 56.Neumann B., Walter T., Ellenberg J. Phenotypic profiling of the human genome by time-lapse microscopy reveals cell division genes. Nature. 2010;464:721–727. doi: 10.1038/nature08869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Yuan Y., Failmezger H., Markowetz F. Quantitative image analysis of cellular heterogeneity in breast tumors complements genomic profiling. Sci. Transl. Med. 2012;4:157ra143. doi: 10.1126/scitranslmed.3004330. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.