

Abstract
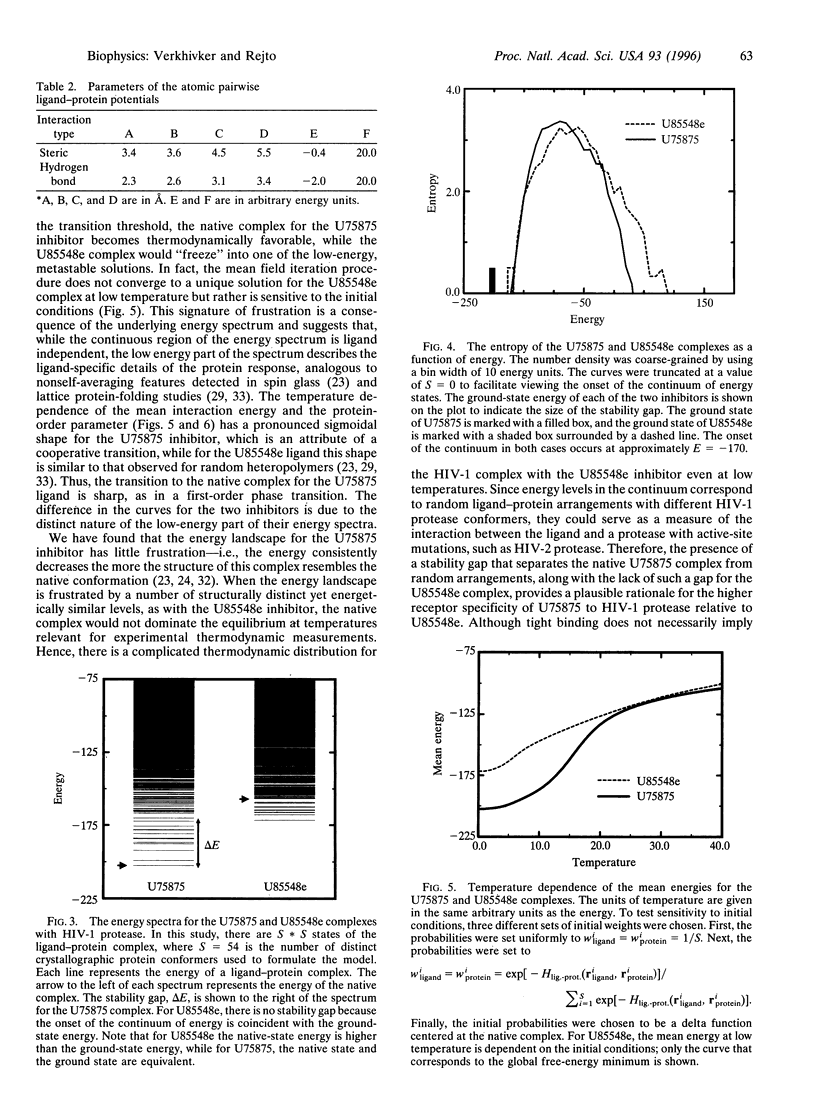
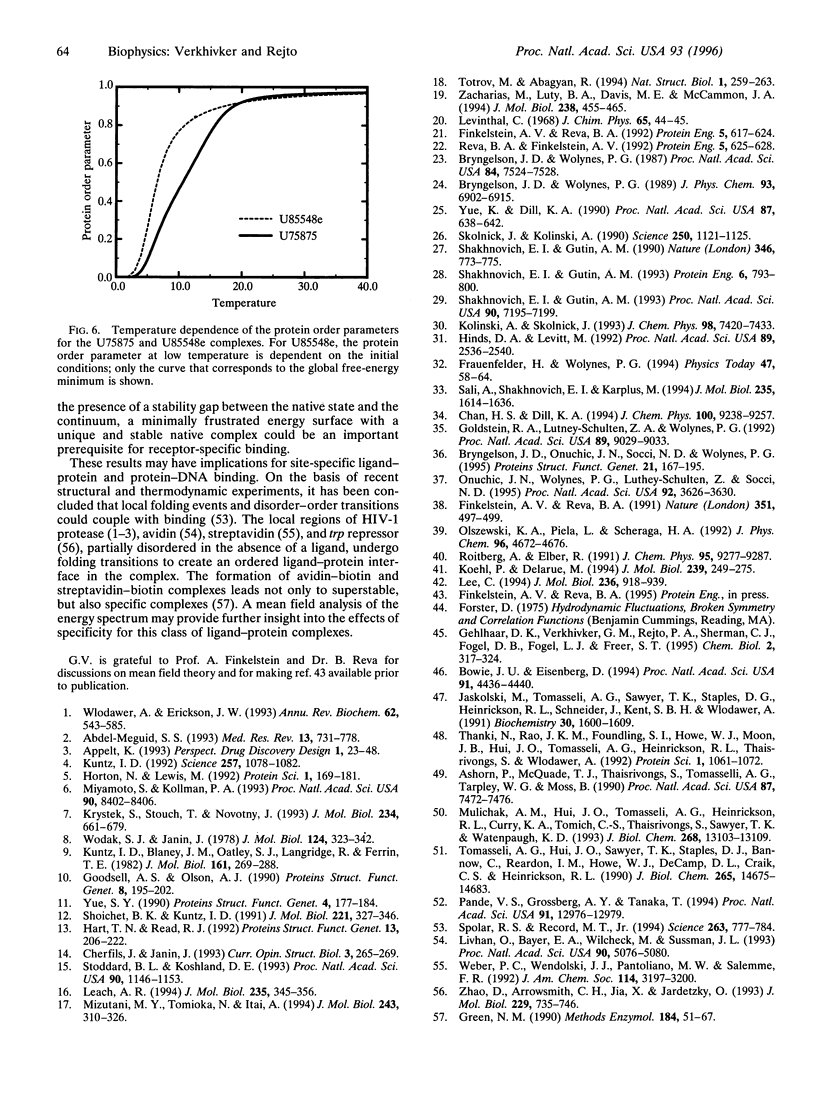
We propose a general mean field model of ligand-protein interactions to determine the thermodynamic equilibrium of a system at finite temperature. The method is employed in structural assessments of two human immuno-deficiency virus type 1 protease complexes where the gross effects of protein flexibility are incorporated by utilizing a data base of crystal structures. Analysis of the energy spectra for these complexes has revealed that structural and thermo-dynamic aspects of molecular recognition can be rationalized on the basis of the extent of frustration in the binding energy landscape. In particular, the relationship between receptor-specific binding of these ligands to human immunodeficiency virus type 1 protease and a minimal frustration principle is analyzed.
Full text
PDF


Images in this article
Selected References
These references are in PubMed. This may not be the complete list of references from this article.
- Abdel-Meguid S. S. Inhibitors of aspartyl proteinases. Med Res Rev. 1993 Nov;13(6):731–778. doi: 10.1002/med.2610130605. [DOI] [PubMed] [Google Scholar]
- Ashorn P., McQuade T. J., Thaisrivongs S., Tomasselli A. G., Tarpley W. G., Moss B. An inhibitor of the protease blocks maturation of human and simian immunodeficiency viruses and spread of infection. Proc Natl Acad Sci U S A. 1990 Oct;87(19):7472–7476. doi: 10.1073/pnas.87.19.7472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowie J. U., Eisenberg D. An evolutionary approach to folding small alpha-helical proteins that uses sequence information and an empirical guiding fitness function. Proc Natl Acad Sci U S A. 1994 May 10;91(10):4436–4440. doi: 10.1073/pnas.91.10.4436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryngelson J. D., Onuchic J. N., Socci N. D., Wolynes P. G. Funnels, pathways, and the energy landscape of protein folding: a synthesis. Proteins. 1995 Mar;21(3):167–195. doi: 10.1002/prot.340210302. [DOI] [PubMed] [Google Scholar]
- Bryngelson J. D., Wolynes P. G. Spin glasses and the statistical mechanics of protein folding. Proc Natl Acad Sci U S A. 1987 Nov;84(21):7524–7528. doi: 10.1073/pnas.84.21.7524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finkelstein A. V., Reva B. A. A search for the most stable folds of protein chains. Nature. 1991 Jun 6;351(6326):497–499. doi: 10.1038/351497a0. [DOI] [PubMed] [Google Scholar]
- Finkelstein A. V., Reva B. A. Search for the stable state of a short chain in a molecular field. Protein Eng. 1992 Oct;5(7):617–624. doi: 10.1093/protein/5.7.617. [DOI] [PubMed] [Google Scholar]
- Gehlhaar D. K., Verkhivker G. M., Rejto P. A., Sherman C. J., Fogel D. B., Fogel L. J., Freer S. T. Molecular recognition of the inhibitor AG-1343 by HIV-1 protease: conformationally flexible docking by evolutionary programming. Chem Biol. 1995 May;2(5):317–324. doi: 10.1016/1074-5521(95)90050-0. [DOI] [PubMed] [Google Scholar]
- Goldstein R. A., Luthey-Schulten Z. A., Wolynes P. G. Protein tertiary structure recognition using optimized Hamiltonians with local interactions. Proc Natl Acad Sci U S A. 1992 Oct 1;89(19):9029–9033. doi: 10.1073/pnas.89.19.9029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodsell D. S., Olson A. J. Automated docking of substrates to proteins by simulated annealing. Proteins. 1990;8(3):195–202. doi: 10.1002/prot.340080302. [DOI] [PubMed] [Google Scholar]
- Green N. M. Avidin and streptavidin. Methods Enzymol. 1990;184:51–67. doi: 10.1016/0076-6879(90)84259-j. [DOI] [PubMed] [Google Scholar]
- Hart T. N., Read R. J. A multiple-start Monte Carlo docking method. Proteins. 1992 Jul;13(3):206–222. doi: 10.1002/prot.340130304. [DOI] [PubMed] [Google Scholar]
- Hinds D. A., Levitt M. A lattice model for protein structure prediction at low resolution. Proc Natl Acad Sci U S A. 1992 Apr 1;89(7):2536–2540. doi: 10.1073/pnas.89.7.2536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horton N., Lewis M. Calculation of the free energy of association for protein complexes. Protein Sci. 1992 Jan;1(1):169–181. doi: 10.1002/pro.5560010117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaskólski M., Tomasselli A. G., Sawyer T. K., Staples D. G., Heinrikson R. L., Schneider J., Kent S. B., Wlodawer A. Structure at 2.5-A resolution of chemically synthesized human immunodeficiency virus type 1 protease complexed with a hydroxyethylene-based inhibitor. Biochemistry. 1991 Feb 12;30(6):1600–1609. doi: 10.1021/bi00220a023. [DOI] [PubMed] [Google Scholar]
- Koehl P., Delarue M. Application of a self-consistent mean field theory to predict protein side-chains conformation and estimate their conformational entropy. J Mol Biol. 1994 Jun 3;239(2):249–275. doi: 10.1006/jmbi.1994.1366. [DOI] [PubMed] [Google Scholar]
- Krystek S., Stouch T., Novotny J. Affinity and specificity of serine endopeptidase-protein inhibitor interactions. Empirical free energy calculations based on X-ray crystallographic structures. J Mol Biol. 1993 Dec 5;234(3):661–679. doi: 10.1006/jmbi.1993.1619. [DOI] [PubMed] [Google Scholar]
- Kuntz I. D., Blaney J. M., Oatley S. J., Langridge R., Ferrin T. E. A geometric approach to macromolecule-ligand interactions. J Mol Biol. 1982 Oct 25;161(2):269–288. doi: 10.1016/0022-2836(82)90153-x. [DOI] [PubMed] [Google Scholar]
- Kuntz I. D. Structure-based strategies for drug design and discovery. Science. 1992 Aug 21;257(5073):1078–1082. doi: 10.1126/science.257.5073.1078. [DOI] [PubMed] [Google Scholar]
- Lau K. F., Dill K. A. Theory for protein mutability and biogenesis. Proc Natl Acad Sci U S A. 1990 Jan;87(2):638–642. doi: 10.1073/pnas.87.2.638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leach A. R. Ligand docking to proteins with discrete side-chain flexibility. J Mol Biol. 1994 Jan 7;235(1):345–356. doi: 10.1016/s0022-2836(05)80038-5. [DOI] [PubMed] [Google Scholar]
- Lee C. Predicting protein mutant energetics by self-consistent ensemble optimization. J Mol Biol. 1994 Feb 25;236(3):918–939. doi: 10.1006/jmbi.1994.1198. [DOI] [PubMed] [Google Scholar]
- Livnah O., Bayer E. A., Wilchek M., Sussman J. L. Three-dimensional structures of avidin and the avidin-biotin complex. Proc Natl Acad Sci U S A. 1993 Jun 1;90(11):5076–5080. doi: 10.1073/pnas.90.11.5076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyamoto S., Kollman P. A. What determines the strength of noncovalent association of ligands to proteins in aqueous solution? Proc Natl Acad Sci U S A. 1993 Sep 15;90(18):8402–8406. doi: 10.1073/pnas.90.18.8402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizutani M. Y., Tomioka N., Itai A. Rational automatic search method for stable docking models of protein and ligand. J Mol Biol. 1994 Oct 21;243(2):310–326. doi: 10.1006/jmbi.1994.1656. [DOI] [PubMed] [Google Scholar]
- Mulichak A. M., Hui J. O., Tomasselli A. G., Heinrikson R. L., Curry K. A., Tomich C. S., Thaisrivongs S., Sawyer T. K., Watenpaugh K. D. The crystallographic structure of the protease from human immunodeficiency virus type 2 with two synthetic peptidic transition state analog inhibitors. J Biol Chem. 1993 Jun 25;268(18):13103–13109. [PubMed] [Google Scholar]
- Onuchic J. N., Wolynes P. G., Luthey-Schulten Z., Socci N. D. Toward an outline of the topography of a realistic protein-folding funnel. Proc Natl Acad Sci U S A. 1995 Apr 11;92(8):3626–3630. doi: 10.1073/pnas.92.8.3626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pande V. S., Grosberg A. Y., Tanaka T. Thermodynamic procedure to synthesize heteropolymers that can renature to recognize a given target molecule. Proc Natl Acad Sci U S A. 1994 Dec 20;91(26):12976–12979. doi: 10.1073/pnas.91.26.12976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reva B. A., Finkelstein A. V. A new approach to the design of a sequence with the highest affinity for a molecular surface. Protein Eng. 1992 Oct;5(7):625–628. doi: 10.1093/protein/5.7.625. [DOI] [PubMed] [Google Scholar]
- Sali A., Shakhnovich E., Karplus M. Kinetics of protein folding. A lattice model study of the requirements for folding to the native state. J Mol Biol. 1994 Feb 4;235(5):1614–1636. doi: 10.1006/jmbi.1994.1110. [DOI] [PubMed] [Google Scholar]
- Shakhnovich E. I., Gutin A. M. A new approach to the design of stable proteins. Protein Eng. 1993 Nov;6(8):793–800. doi: 10.1093/protein/6.8.793. [DOI] [PubMed] [Google Scholar]
- Shakhnovich E. I., Gutin A. M. Engineering of stable and fast-folding sequences of model proteins. Proc Natl Acad Sci U S A. 1993 Aug 1;90(15):7195–7199. doi: 10.1073/pnas.90.15.7195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shakhnovich E. I., Gutin A. M. Implications of thermodynamics of protein folding for evolution of primary sequences. Nature. 1990 Aug 23;346(6286):773–775. doi: 10.1038/346773a0. [DOI] [PubMed] [Google Scholar]
- Shoichet B. K., Kuntz I. D. Protein docking and complementarity. J Mol Biol. 1991 Sep 5;221(1):327–346. doi: 10.1016/0022-2836(91)80222-g. [DOI] [PubMed] [Google Scholar]
- Skolnick J., Kolinski A. Simulations of the folding of a globular protein. Science. 1990 Nov 23;250(4984):1121–1125. doi: 10.1126/science.250.4984.1121. [DOI] [PubMed] [Google Scholar]
- Spolar R. S., Record M. T., Jr Coupling of local folding to site-specific binding of proteins to DNA. Science. 1994 Feb 11;263(5148):777–784. doi: 10.1126/science.8303294. [DOI] [PubMed] [Google Scholar]
- Stoddard B. L., Koshland D. E., Jr Molecular recognition analyzed by docking simulations: the aspartate receptor and isocitrate dehydrogenase from Escherichia coli. Proc Natl Acad Sci U S A. 1993 Feb 15;90(4):1146–1153. doi: 10.1073/pnas.90.4.1146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thanki N., Rao J. K., Foundling S. I., Howe W. J., Moon J. B., Hui J. O., Tomasselli A. G., Heinrikson R. L., Thaisrivongs S., Wlodawer A. Crystal structure of a complex of HIV-1 protease with a dihydroxyethylene-containing inhibitor: comparisons with molecular modeling. Protein Sci. 1992 Aug;1(8):1061–1072. doi: 10.1002/pro.5560010811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomasselli A. G., Hui J. O., Sawyer T. K., Staples D. J., Bannow C., Reardon I. M., Howe W. J., DeCamp D. L., Craik C. S., Heinrikson R. L. Specificity and inhibition of proteases from human immunodeficiency viruses 1 and 2. J Biol Chem. 1990 Aug 25;265(24):14675–14683. [PubMed] [Google Scholar]
- Totrov M., Abagyan R. Detailed ab initio prediction of lysozyme-antibody complex with 1.6 A accuracy. Nat Struct Biol. 1994 Apr;1(4):259–263. doi: 10.1038/nsb0494-259. [DOI] [PubMed] [Google Scholar]
- Wlodawer A., Erickson J. W. Structure-based inhibitors of HIV-1 protease. Annu Rev Biochem. 1993;62:543–585. doi: 10.1146/annurev.bi.62.070193.002551. [DOI] [PubMed] [Google Scholar]
- Wodak S. J., Janin J. Computer analysis of protein-protein interaction. J Mol Biol. 1978 Sep 15;124(2):323–342. doi: 10.1016/0022-2836(78)90302-9. [DOI] [PubMed] [Google Scholar]
- Yue S. Y. Distance-constrained molecular docking by simulated annealing. Protein Eng. 1990 Dec;4(2):177–184. doi: 10.1093/protein/4.2.177. [DOI] [PubMed] [Google Scholar]
- Zacharias M., Luty B. A., Davis M. E., McCammon J. A. Combined conformational search and finite-difference Poisson-Boltzmann approach for flexible docking. Application to an operator mutation in the lambda repressor-operator complex. J Mol Biol. 1994 May 6;238(3):455–465. doi: 10.1006/jmbi.1994.1304. [DOI] [PubMed] [Google Scholar]
- Zhao D., Arrowsmith C. H., Jia X., Jardetzky O. Refined solution structures of the Escherichia coli trp holo- and aporepressor. J Mol Biol. 1993 Feb 5;229(3):735–746. doi: 10.1006/jmbi.1993.1076. [DOI] [PubMed] [Google Scholar]