

Abstract

This review will summarize our structural and kinetic studies of RB69 DNA polymerase (RB69pol) as well as selected variants of the wild-type enzyme that were undertaken to obtain a deeper understanding of the exquisitely high fidelity of B family replicative DNA polymerases. We discuss how the structures of the various RB69pol ternary complexes can be used to rationalize the results obtained from pre-steady-state kinetic assays. Our main findings can be summarized as follows. (i) Interbase hydrogen bond interactions can increase catalytic efficiency by 5000-fold; meanwhile, base selectivity is not solely determined by the number of hydrogen bonds between the incoming dNTP and the templating base. (ii) Minor-groove hydrogen bond interactions at positions n – 1 and n – 2 of the primer strand and position n – 1 of the template strand in RB69pol ternary complexes are essential for efficient primer extension and base selectivity. (iii) Partial charge interactions among the incoming dNTP, the penultimate base pair, and the hydration shell surrounding the incoming dNTP modulate nucleotide insertion efficiency and base selectivity. (iv) Steric clashes between mismatched incoming dNTPs and templating bases with amino acid side chains in the nascent base pair binding pocket (NBP) as well as weak interactions and large gaps between the incoming dNTPs and the templating base are some of the reasons that incorrect dNTPs are incorporated so inefficiently by wild-type RB69pol. In addition, we developed a tC°–tCnitro Förster resonance energy transfer assay to monitor partitioning of the primer terminus between the polymerase and exonuclease subdomains.
Replicative DNA polymerases (pols) are vital for transferring genetic information from parent to progeny.1,2 They do so with extremely high fidelity, exhibiting an error frequency in the range of 1 in 106–109 mistakes per incorporation event depending on the presence or absence of an intrinsic exonuclease activity.3,4 As might be expected, deregulation of normal modes of DNA replication has a profound effect on the rates of cell division. This can lead to uncontrolled cell proliferation and is the underlying cause of a number of pathological states, especially those associated with malignancy.5,6 When mutations affecting base discrimination are in the replicative pol itself, misincorporation events are more frequent and, if not corrected, can cause cell death because of error catastrophe.7 Thus, it is important to understand the basis of pol fidelity as it will contribute to our knowledge of how malignancies arise and will provide a theoretical underpinning for the development of drugs that can be used to treat malignancies and a number of viral, microbial, and parasitic infections.8,9
DNA pols have been grouped into seven different families [A–D, X, Y, and reverse transcriptase (RT)] on the basis of amino acid sequence homology.10,11 Although the ability of pols to select a correct dNTP varies dramatically among different families, they share a number of properties. For example, (i) they have an overall architecture that resembles a right hand,12 as discussed below, and (ii) they require two divalent metal ions to catalyze the nucleotidyl transfer reaction during which the hydroxyl group at the 3′ terminus of the primer strand attacks the α-phosphorus atom of an incoming dNTP.
We have chosen to study the DNA pol from bacteriophage RB69 for several reasons. First, it is extensively similar in sequence and structure to human and other B family eukaryotic replicative pols,13 and unlike eukaryotic replicative pols, RB69pol can be expressed in high yield in Escherichia coli with full activity.14 Second, unlike the closely related DNA pol from bacteriophage T4, it can be readily crystallized either in an apo form or in ternary complexes with a primer/template (P/T) and an incoming dNTP.15 There is also a wealth of kinetic and structural data that makes this pol an excellent model for studying structure–function relationships and fidelity mechanisms. In addition, RB69pol has been recently used as a surrogate enzyme for the discovery and development of antiherpetic drugs.16 This review will focus on the studies of RB69pol, particularly those from our own lab, and from others. For a broader and more comprehensive review of structure–function relationships among replicative DNA polymerases, readers should consult refs (3), (14), and (17−25) as well as refs (26−29).
Structure and Function of RB69pol
RB69pol is primarily responsible for the replication of the RB69 bacteriophage genome.30 The first crystal structure of the apo form of this pol was reported in 1997.31 Subsequently, Franklin et al. published the structure of a ternary complex of an exonuclease deficient variant of RB69pol that contained a correct incoming dNTP and a dideoxy-terminated primer/template with a resolution of 2.6 Å.15 In 2011, we determined the crystal structure of a wild-type (wt) RB69pol ternary complex at a higher resolution (1.8 Å).32 This latter structure captured the pol in a conformational state that may exist just prior to the transfer of an incoming dNTP to the 3′ terminus of the primer strand and allowed us to visualize important details that were not visible in the 2.6 Å resolution structure. However, a possible caveat that applies to all pol complexes is that the crystal structure of the complex may not actually be on the reaction pathway between substrates and products. Like many other DNA polymerases, the overall structure of RB69pol resembles a right hand and consists of five conserved regions: the N-terminus, exonuclease, palm, fingers, and thumb.31 As shown in Figure 1A, these subdomains are arranged in a disklike structure with a central cavity. The P/T duplex is bound in a groove formed between the palm and thumb (Figure 1A,B).
Figure 1.

Overall structure of the dCTP/dG-containing ternary complex of wt RB69pol. (A) The pol structure is colored by subdomain: blue for the N-terminal subdomain (residues 1–108 and 340–382), orange for the exonuclease subdomain (residues 109–339), yellow for the palm subdomain (residues 383–468 and 573–729), red for the fingers subdomain (residues 469–572), and green for the thumb subdomain (residues 730–903). The primer/template duplex is shown as sticks, with the template colored black and the primer pink. (B) Orthogonal view of panel A. (C) Space-filling model depicting the interaction of the S565 hydroxyl group with the purine base of the templating dG. (D) Minor groove HB network that includes five ordered water molecules. (E) Ca2+ coordination in the dUpNpp-containing ternary complex. (F) The 3.45 Å distance between the Cα hydrogen and the N3 hydrogen acceptor is consistent with the presence of HBs.
The N-terminal subdomain is composed of two nonadjacent segments in the amino acid sequence (residues 1–108 and 340–382). Although the function of this subdomain has not been clearly defined, the classic βαββαβ topology of the N-terminal subdomain has also often been found in RNA binding proteins.33,34 Interestingly, both RB69pol and T4pol bind to their own mRNAs, specifically repressing translation, thus exerting tight feedback control on the level of DNA polymerase that is produced in the bacterial cell after phage infection.35−37 The N-terminal subdomain has a specific guanine binding pocket. We took advantage of this feature by designing a P/T duplex with a single overhanging dG at the 3′ end of the template strand that binds tightly to a G specific pocket in RB69pol and facilitates crystallization of the ternary complex. O6 and N2 of the guanine form hydrogen bonds (HBs) with the amide nitrogen of I380 and the carbonyl oxygen of K378 from a symmetrically related molecule in the crystal lattice.32 However, the biological relevance of this guanine binding pocket is still not known.
The exonuclease (exo) subdomain lies between the N-terminal and thumb subdomains. When an incorrect dNTP is incorporated onto the 3′ terminus of the primer strand, the pol helps to switch the primer terminus from the pol to the exo subdomain, facilitating cleavage of the 3′-terminal nucleotide residue.38,39 The exo subdomain of RB69pol is strikingly similar to the exo subdomain of the Klenow Fragment (KF), both consisting of five-stranded β-sheets surrounded by a distribution of α-helices.31,40 However, the exo activity of RB69pol is almost 3 orders of magnitude greater than that of KF.22,40−42 The active site of the exo subdomain is centered around a conserved DEDD motif (D114, E116, D222, and D327). The carboxylate groups of these residues are coordinated to a magnesium ion that is essential for catalyzing excision of the misincorporated base. When one or more of these acidic residues are replaced with Ala, the exo activity is either completely lost or decreased by at least 4 orders of magnitude.43
The palm subdomain harbors the catalytic core responsible for pol activity. Two highly conserved acidic residues (D411 and D623) serve as ligands for metal ions A and B, which are crucial for catalyzing the nucleotidyl transfer reaction.44 From the 1.8 Å resolution structure, we were able to observe a rigid HB network located at the minor groove of the P/T junction.32 As shown in Figure 1D, this HB network, consisting of five ordered water molecules, was linked to pol residues that interact with the O2 and N3 HB acceptors in the P/T duplex. It is worth noting that three of the five water molecules (w1, w3, and w4 in Figure 1D) have classic tetrahedral geometry but the other two (w2 and w5) have an unusual trigonal geometry. This HB network provides an excellent example of how ordered water molecules can serve as extensions of protein side chains to mediate non-sequence specific pol–DNA interactions.
Overall, the palm subdomain is structurally and topologically similar to palm subdomains found in all other pols for which structures exist, except for pol β.12 In contrast, the fingers subdomain of RB69pol bears little resemblance to the fingers subdomain of pols in other families. It consists of two long antiparallel α-helices that extend perpendicularly from the pol surface (Figure 1B). A fingertip region unique to RB69pol is composed of a nine-residue helix and an 18-residue loop that joins the two long helices. It has been suggested that this fingertip region might be involved in pol–pol dimerization in the RB69 replisome.45b The fingers subdomain contains a conserved dNTP binding motif (KX3NSXTG) analogous to the KX4NSXTG motif located in helix O of KF.46−49 Each phosphate group of the incoming dNTP is coordinated to a protein side chain in the following wa.: (i) The ε-amino group of K560 is hydrogen bonded to the nonbridging α- and γ-phosphate oxygens. (ii) The amide side chain of N564 forms a HB to the nonbridging oxygen of the β-phosphate via an ordered water molecule. (iii) The δ-guanidino group of R482 and the ε-amino group of K486 are hydrogen bonded to the oxygens of the γ-phosphate. The triphosphate tail of the incoming dNTP is coordinated to metal ion B in the classic α,β,γ tridendate state. As shown in Figure 1E, metal ion B is coordinated to the three oxygen ligands from the triphosphate tail of the nonhydrolyzable dNTP analogue, dUpNpp, the carboxylates of D411 and D623, and the backbone carbonyl oxygen of L412.50 Metal ion A is coordinated to D411, D623, and the 3′-hydroxyl group of dC at the 3′ end of the primer strand, as well as to a nonbridging oxygen of the α-phosphate of dUpNpp. Metal ion A is responsible for bringing the 3′-OH group at the primer terminus close to the α-phosphorus atom of the incoming dNTP, allowing phosphodiester bond formation.39 Metal ion B stabilizes the pentacoordinate transition state and assists in the departure of the pyrophosphate group.39 A third metal ion with partial occupancy was observed coordinated to E686 and E683. Substitution of these residues with Ala caused a dramatic decrease in pol activity. However, full pol activity could be recovered when Mn2+ was substituted for Mg2+ 53. Similar results were recently reported for ϕ29 DNApol45a. In addition, two unusual interactions were observed in the fingers subdomain: one between the Cα hydrogen of G568 and N3 of the templating dG (Figure 1F) and the other between the aromatic face of the templating base and the hydroxyl group of S565 (Figure 1C). These interactions anchor the templating base tightly within the nascent base pair binding pocket. The thumb subdomain of RB69pol also differs topologically from the thumb subdomain of pols in other families.39 It consists of three long α-helices protruding from the palm subdomain. There is a microsubdomain composed of three short β-strands and two α-helices at the tip of the thumb that bridge the thumb and exo subdomains. The 11 carboxyl-terminal residues of RB69pol extend directly out from the structure and are bound within a deep cleft in the ring-shaped sliding clamp (gp45).51 This sliding clamp increases the processivity of T4pol, and very likely RB69pol, by tethering them at the P/T junction.23 Overall, RB69pol has proven to be a useful model for understanding structure–function relationships in B family pols.25
Relationship between the Kinetics and Structure of RB69pol Variants
With the availability of a high-resolution structure of the RB69pol ternary complex, the next challenge was to relate its structure to function in ways that could account for the high efficiency of DNA synthesis and its exquisite base selectivity. To this end, various RB69pol mutants have been constructed on the basis of its ternary structure and previously determined apo and binary structures of RB69pol.15,31,52 Kinetic studies have been conducted with these mutants to determine the functional role of the many highly conserved residues. The majority of the single-amino acid substitutions were restricted to the palm, fingers, and thumb subdomains (Table 1). In the palm subdomain, replacing D411 and D623, which coordinate the catalytic magnesium ions, with Ala abolished the pol activity. Seven residues near the metal ion binding sites (D621, T622, S624, Y619, E686, and K706 in the palm subdomain and Y708 in the fingers subdomain) were also replaced to determine their roles in the wt enzyme as shown in Table 1. Results from single-turnover experiments showed that the values of the maximal turnover rate (kpol) for all the mutants were greatly reduced.53 These changes can be rationalized on the basis of various crystal structures as follows. (i) The side chains of D621, T622, and K706 are part of a rigid HB network in the minor groove of the P/T junction. (ii) The hydroxyl group of S624 and the carboxyl group of E686 are hydrogen bonded to metal ion A via an ordered water molecule. (iii) Residues Y416 and Y708 form HBs with the nonbridging oxygens in the terminal phosphodiester bond of the primer via a single water molecule. Thus, these residues are all directly or indirectly involved in the correct alignment of the 3′-hydroxyl group at the primer’s 3′ terminus with the α-phosphorus atom of the incoming dNTP. Replacement of any of these residues with Ala or other amino acids disturbs the optimal alignment of the reactive groups in the catalytic center and can account for the dramatic reduction in the corresponding kpol values of these variants. In addition, Y416 acts as a “sugar gate” that sterically blocks the 2′-hydroxyl group of an incoming rNTP. The level of discrimination against rNTPs is decreased by ∼3000-fold when Y416 is replaced with Ala.54 It is worth pointing out that Y416 is highly conserved in B family pols,55 while the same function is performed by a conserved Glu residue in A family pols.56,57
Table 1. Summary of Pre-Steady-State Kinetic Parameters and Functional Consequences of Various RB69pol Variants.
| subdomain | residue | functional interactions with RB69pol | amino acid substitution | kpol (s–1) or kexo (s–1)a | Kd (μM) | kpol/Kd (μM–1 s–1) | ref |
|---|---|---|---|---|---|---|---|
| palm | Y391 | HB to Y567, HB network at minor groove | Y → A | 155 | 980 | 1.5 × 10–1 | (60) |
| Y → F | 312 | 90 | 3.5 | ||||
| D411 | ligands to metal ion at pol site | D → A | inactive | (53) | |||
| L415 | HB to β-phosphate of the incoming dNTP | L → A | 11 | 3 | 3.7 | (82) | |
| L → G | 58 | 9 | 6.4 | ||||
| L → M | 375 | 12 | 31.3 | ||||
| Y416 | “sugar gate”, sterically blocks the 2′-OH group of an incoming rNTP | Y → A | 22 | 70 | 3.1 × 10–1 | (54) | |
| Y → F | 36 | 209 | 1.7 × 10–1 | ||||
| T587 | HB to Y567, HB network at minor groove | T → A | 0.8 | 0.7 | 1.1 × 10–1 | (60) | |
| Y619 | HB to a water molecule, HB network at minor groove | Y → F | 14 | NDb | NDb | (53) | |
| D621 | HB to K706, HB network at the minor groove | D → A | 1.3 | 166 | 7.8 × 10–3 | (53) | |
| T622 | HB to D621, anchors the side chain of D623 | T → A | 0.2 | NDb | NDb | (53) | |
| D623 | ligands to metal ion at pol site | D → A | inactive | (53) | |||
| S624 | ligands to metal ion at pol site via a water molecule | S → C | 4.5 | 18 | 2.5 × 10–1 | (53) | |
| E686 | ligands to metal ion at pol site via a water molecule | E → A | 7 | 58 | 1.2 × 10–1 | (53) | |
| K706 | HB to D621 and base at position n – 2 of primer strand | K → R | 0.1 | NDb | NDb | (53) | |
| fingers | R482 | HB to γ-phosphate of the incoming dNTP | R → A | 0.2 | 1100 | 1.8 × 10–4 | (58) |
| K486 | HB to γ-phosphate of dNTP via a water molecule | K → A | 3.3 | 71 | 4.6 × 10–2 | (58) | |
| K560 | HB to α,γ-phosphate of the incoming dNTP | K → A | 0.8 | 790 | 1.0 × 10–3 | (58) | |
| L561 | anchoring the templating base at the major groove | L → A | 101 | 50 | 2.0 | (61) | |
| N564 | HB to β-phosphate of dNTP | N → A | 23 | 250 | 9.2 × 10–2 | (58) | |
| S565 | anchoring the templating base | S → G | 157 | 48 | 3.3 | (76) | |
| Y567 | HB to Y391, HB network at minor groove | Y → A | 213 | 68 | 3.1 | (60) | |
| Y → F | 7.5 | 2400 | 3.1 × 10–3 | ||||
| thumb | Y708 | HB to 3′-terminal phosphodiester bond (primer strand) | Y → A | 6 | 34 | 1.8 × 10–1 | (53) |
| E716 | ligands to a third metal ion at the pol site | E → A | 200 | NDb | NDb | (53) | |
| exonuclease | E116 | ligands to metal ion at the exo site | E → A | 2.8 × 10–4a | (42) | ||
| D222 | ligands to metal ion at the exo site | D → A | inactivea | (42) | |||
| K302 | anchors the side chain of D327 | K → A | 1.0 × 10–2a | (42) | |||
| Y323 | HB to Q171, anchors the backbone of D327 | Y → F | 5.8 × 10–3a | (42) | |||
| D327 | ligands to metal ion at the exo site | D → A | inactivea | (42) |
Indicates the excision rate for RB69pol variants.
Not determined.
Residues in the dNTP binding motif (KX3NSXTG) of the fingers subdomain are highly conserved in pols of all families.12 Replacing the residues that coordinate the bridging and nonbridging oxygens in the triphosphate tail of the incoming dNTP, such as R482, K486, K560, and N564, with Ala causes the catalytic efficiency of the altered RB69pol to decrease by 2–5 orders of magnitude.58 The ε-amino group of K560, in addition to its role in anchoring the dNTP triphosphate tail, also serves as a proton donor during the nucleotidyl transfer reaction.59 As a consequence, the Lys to Ala substitution at this position causes a 200-fold decrease in kpol.21 Residues L561 and Y567 are located at the major and minor grooves adjacent to the templating base, respectively (Figure 2A). Together, these two residues rigidly anchor the templating base in an optimal position for hydrogen bonding to the base of the incoming dNTP. Replacing either residue with Ala reduces the base selectivity by up to 2 orders of magnitude (Table 2).60,61 When the L561A and Y567A substitutions were combined in the same RB69pol variant, the resulting double mutant (dm) was 3–40-fold more likely to incorporate an incorrect dNTP than either of the single RB69pol mutants.62 This result suggested that increasing the amount of space in the NBP caused a decrease in base selectivity. To further investigate the apparent correlation between an increased NBP volume and the loss of base selectivity, the S565G substitution was added to the dm, creating the L561A/S565G/Y567A triple mutant (tm).62 As shown in Table 2, the probability of accommodating a purine/pyrimidine mispair, a pyrimidine/pyrimidine mispair, or a purine/purine mispair by the tm is increased by (i) 3-fold, (ii) 2–5-fold, or (iii) 4–8-fold, respectively, compared to that exhibited by the dm.62 Because of the diminished base selectivity, we were able to obtain crystal structures of RB69pol ternary complexes with 8 of the 12 possible mispairs using this tm. To obtain the structures of the four missing purine/purine mispairs, an additional substitution, L415A, was introduced into the tm (Figure 2B). Residue L415 is located just below the triphosphate tail of the incoming dNTP (Figure 2A). The resulting quadruple mutant (qm) allowed us to obtain high-resolution crystal structures with all of the 12 mismatches.63 Pre-steady-state kinetic results with the qm showed that the catalytic efficiencies for incorporation of purine dNTPs opposite a purine templating base increased by 28–74-fold compared to efficiencies observed for the tm (Table 2).63 This reduction in selectivity against purine/purine mismatches allowed us to capture the missing purine/purine mispairs in crystals of the qm ternary complexes. A surprising feature of the qm is that the maximal turnover rate for a purine dNTP opposite a purine templating base was greater than 300 s–1, which is faster than the kpol for incorporation of a correct dNTP by the wild-type (wt) RB69pol.63 In summary, we found that incremental expansion of the space in the NBP of RB69pol resulted in a progressive increase in the incorporation efficiency of incorrect dNTPs. On the basis of this finding, we propose that RB69pol exerts base discrimination via a “negative selection” against mispairs by using residues in the NBP of the wt to allow rapid and efficient incorporation of only correct dNTPs.
Figure 2.

Critical residues that comprise the NBP in the dCTP/dG-containing RB69pol ternary complex: (A) wt RB69pol and (B) RB69pol qm. dCTP/dG is shown as yellow sticks, and protein side chains are shown in space-filling mode. L415/A415 is colored green, L561/A561 blue, S565/G565 gray, and Y567/A567 red.
Table 2. Summary of the Catalytic Efficiencies for RB69pol wt and Its Single, Double, Triple, and Quadruple Mutants.
|
kpol/Kd (μM–1 s–1) |
|||||||
|---|---|---|---|---|---|---|---|
| base pair | wt | L561A | Y567A | dma | tmb | qmc | |
| Pu-Py | dATP/dC | 4.4 × 10–5 | 2.2 × 10–3 | 7.6 × 10–3 | 9.1 × 10–2 | 2.7 × 10–1 | 8.5 × 10–1 |
| dCTP/dA | 1.7 × 10–4 | 4.8 × 10–4 | 1.4 × 10–3 | 3.5 × 10–3 | 9.0 × 10–3 | 1.7 × 10–1 | |
| dGTP/dT | 5.4 × 10–5 | 7.2 × 10–4 | 1.5 × 10–3 | 4.0 × 10–2 | 1.2 × 10–1 | 1.0 × 10–2 | |
| dTTP/dG | 7.0 × 10–5 | 3.4 × 10–4 | 3.1 × 10–4 | 4.3 × 10–2 | 4.2 × 10–2 | 2.4 × 10–1 | |
| Py-Py | dCTP/dT | 6.0 × 10–6 | 9.3 × 10–5 | 3.2 × 10–4 | 1.7 × 10–3 | 8.8 × 10–3 | 4.5 × 10–1 |
| dTTP/dC | 5.2 × 10–5 | 1.6 × 10–3 | 3.7 × 10–3 | 5.6 × 10–2 | 9.0 × 10–2 | 5.0 × 10–1 | |
| dCTP/dC | 3.0 × 10–7 | 1.5 × 10–5 | 4.2 × 10–5 | 4.6 × 10–4 | 1.1 × 10–3 | 1.4 × 10–1 | |
| dTTP/dT | 1.0 × 10–5 | 6.2 × 10–4 | 5.0 × 10–4 | 7.2 × 10–3 | 2.2 × 10–2 | 3.2 × 10–1 | |
| Pu-Pu | dATP/dG | 4.4 × 10–4 | 9.7 × 10–5 | 1.1 × 10–4 | 3.0 × 10–3 | 2.3 × 10–2 | 1.1 |
| dGTP/dA | 3.8 × 10–6 | 4.3 × 10–5 | 2.0 × 10–4 | 8.7 × 10–4 | 2.7 × 10–3 | 2.0 × 10–1 | |
| dATP/dA | 1.6 × 10–4 | 1.9 × 10–4 | 1.3 × 10–3 | 4.4 × 10–3 | 1.7 × 10–2 | 4.8 × 10–1 | |
| dGTP/dG | 1.0 × 10–5 | 6.9 × 10–4 | 4.0 × 10–5 | 2.2 × 10–3 | 1.4 × 10–2 | 5.5 × 10–1 | |
| W-C | dATP/dT | 4.4 | 3.7 | 7.2 | 3.7 | 5.0 | 1.0 |
| dTTP/dA | 6.4 | 2.4 | 12.8 | 3.3 | 5.4 | 1.7 | |
| dCTP/dG | 2.9 | 2.6 | 3.1 | 4.5 | 6.0 | 0.8 | |
| dGTP/dC | 2.4 | 3.4 | 4.8 | 5.5 | 5.0 | 1.0 | |
Double mutant (L561A/Y567A).
Triple mutant (L561A/S565G/Y567A).
Quadruple mutant (L415A/L561A/S565G/Y567A).
Factors That Contribute to the dNTP Insertion Efficiency and Fidelity of Wild-Type RB69pol
Studies conducted with many pols have led to various hypotheses about how replicative pols achieve such an extraordinary degree of base selectivity. Waston and Crick originally proposed that HBs between complementary bases provided the specificity for accurate DNA replication.64 However, this explanation failed to account for the huge difference in incorporation efficiencies between dNTPs that form Watson–Crick base pairs and those that do not. Showalter and Tsai attributed base discrimination to the differences in the energy barriers of the transition state between correct and mismatched base pairs.65 Johnson proposed that a rate-limiting conformational change takes place before chemistry and acts as a fidelity checkpoint.21 This was followed by a report by Joyce and Benkovic that further expanded on this notion.3 However, it is now generally accepted, because of the work of Arndt et al,66 Rothwell et al,67 and Luo et al,68 that the open to closed conformational change is not rate-limiting and that it is not possible to ascribe a given degree of selectivity to a particular step in the nucleotidyl transfer reaction pathway.69 In fact, it was Tsai and Johnson who proposed that base discrimination is determined by the relative magnitudes of the forward and reverse rates of the prechemistry conformational change (fingers closing) compared to the rate of the chemical step that completely changed the concept prevailing at the time concerning base selectivity.70 Our studies of RB69pol have shown that nucleotide insertion efficiency and base selectivity are determined by a number of factors, including interbase HBs, minor groove HBs, base stacking, the geometry of the base pair as it fits into the NBP, conformational changes, and the partitioning of the primer terminus between the pol and exo subdomains.32,50,62,63,71−82 Subsequently, we showed that the results obtained by Tsai and Johnson also applied to RB69pol.83 In addition to the results obtained from kinetic and structural studies, molecular dynamics simulation approaches have further enhanced our understanding of conformational dynamics of the polymerase-catalyzed nucleotidyl transfer reaction that results in base selectivity.84,85 We discuss each of them in turn in the following sections.
Role of Interbase HB in Insertion Efficiency and Base Selectivity
There is an ongoing debate about which features of a nascent base pair and the NBP are most important for pols to maintain high base selectivity.86 Several groups have made important contributions that have provided insights into this issue.87−94 Kool’s group challenged Watson and Crick’s argument about the basis for nucleotide discrimination by using an array of nonpolar nucleoside analogues, such as 2′-deoxyribo-4-methylbenzimidazole (dZ), 2′-deoxyribo-2,4-difluorotoluene (dF), and 2′-deoxyribo-9-methyl-1H-imidazole-[(4,5)-β]-pyridine (dQ), in primer extension assays to show that base pairing was not as important as shape for base selectivity.86,95−97 The analogue that created the most controversy was dF, which was reported to be a nonpolar, non-hydrogen bonding isostere of dT.86 As shown in Figure 3A, dF is isosteric to dT but differs from it by having four-atom substitutions, F2 and F4 for O2 and O4, respectively, and C1 and C3 for N1 and N3, respectively. The NMR structure of a dF/dA pair in a DNA duplex showed that dF did not perturb the configurations of B-form DNA as the overall shapes of dF/dA and dT/dA pairs are very similar.86 In addition, when dF is in the templating position, KF preferentially incorporates dATP over dCTP, dGTP, and dTTP. Similarly, when dA is the templating base, dFTP is preferred over dATP, dCTP, and dGTP by KF.98,99 Because dF is considered to be nonpolar according to classic polarity measurements, Kool proposed that HBs between base pairs are not absolutely required for efficient nucleotide insertion.97 Furthermore, they advanced a steric hypothesis, namely that a snug fit between nascent base pair and nearest pol residues would lead to the rapid insertion of dNTPs.100−102 The first quantitative study using dF was conducted by Lee et al. in 2008 using human mitochondrial DNA polymerase (pol γ).103 They showed that the shape of the base analogue alone was not sufficient for base selectivity but that H-bonding was essential for proper primer extension and proofreading. To investigate the contribution of interbase HBs during DNA synthesis by RB69pol, we determined the pre-steady-state kinetic parameters for incorporation of all four dNTPs opposite dF by wt RB69pol. Similar to what was observed with KF, our results showed that there is a strong preference for dATP incorporation compared to the three other dNTPs.79 The catalytic efficiency for incorporation of dATP opposite dF is 1450-, 1120-, and 360-fold greater than those for incorporation of dCTP, dGTP, and dTTP respectively. In contrast, when dT is the templating base, the catalytic efficiency for the insertion of dATP opposite dT is 5057-fold greater than the catalytic efficiency for the incorporation of dATP opposite dF. Therefore, the two direct interbase HBs between the dATP/dT base pair make a big difference with respect to efficient nucleotide insertion.79
Figure 3.

(A) Chemical structures of dT and dF. (B) Superposition of four dNTP/dF-containing structures (dATP/dF, pink; dCTP/dF, green; dGTP/dF, blue; dTTP/dF, yellow) shows a conserved hydration network of four ordered water molecules. (C) dATP/dF pair. (D) dATP/dT pair. (E) dGTP/dF pair. (F) dGTP/dT pair. (G) dCTP/dF pair. (H) dCTP/dT pair. (I) dTTP/dF pair. (J) dTTP/dT pair. Hydrogen bonding atoms are linked with a dashed red line; non-hydrogen bonding atoms are linked with a black line to show the distance.
To obtain a more complete understanding of the properties of the dF isostere, we determined the pre-steady-state kinetic parameters for incorporating all four dNTPs opposite dF in the presence of the RB69pol tm because this variant has the same kinetic parameters for inserting correct dNTPs as wt RB69pol.79 In addition, its enlarged NBP allowed us to obtain high-quality crystals of RB69pol ternary complexes with all four dNTPs opposite dF and dT. We found that the catalytic efficiency for incorporation of dATP opposite dF, using the RB69pol tm, was 120-, 180-, and 60-fold higher than that for the insertion of dCTP, dGTP, and dTTP, respectively.79 We then determined and compared the structures of eight RB69pol ternary complexes, four with each of the dNTP/dF pairs and another four with each of the dNTP/dT pairs. As shown in panels C and D of Figure 3, the geometry of the dATP/dF pair is very similar to that of the dATP/dT pair. The distance between F4 of dF and the N6-H group of dATP is 3.96 Å, which is too great for a HB. Surprisingly, a direct HB was observed between F2 of dF and the N2-H group of dGTP, and between F4 of dF and the N3-H group of dTTP (Figure 3E,I). In addition, the F4 atoms of the four dNTP/dF pairs have four ordered water molecules located within 3.2 Å of its radius (Figure 3B). Superimposition of all four dNTP/dF-containing structures reveals a conserved hydration network around the F4 atoms (Figure 3B). Compared to the structures of dNTP/dT-containing ternary complexes, the shapes of the nascent dNTP/dF pairs differ from those of the dNTP/dT pairs. In particular, the dGTP/dF pair has a direct interbase HB, while the dGTP/dT pair adopts wobble geometry. No ordered water molecules were observed between the dCTP/dF pair, but three ordered water molecules were located at the interface of the dCTP/dT pair mediating their interbase HBs. On the basis of these structures, it seems that dF does not behave as would be predicted for a nonpolar, non-hydrogen bonding isostere of dT.79 It should be noted that there is evidence from NMR studies of the existence of a HF–C hydrogen bond,102 although this bond is likely to be rather weak.104 Our observations challenge the view, which has been debated for the past 15 years, namely that dF, because of its nonpolar nature, is incapable of forming H-bonds.86
One puzzling question concerns the reason for the preferential incorporation of dATP opposite dF compared to the insertion of dGTP and dTTP opposite dF because the latter two dNTPs have direct interbase HBs between each of them and a templating dF whereas dATP does not. In the case of the dGTP/dF pair, the distance between C3 of dF and N1 of dGTP is 3.32 Å (Figure 3E). To avoid a repulsive interaction that would occur between the C3 hydrogen of dF and the N1 hydrogen of dGTP, the templating dF twists away from its normal position, causing a distortion in substrate alignment. This is consistent with the poor electron density around dF and the relatively large B factors for the templating dF. As for the dTTP/dF pair, the base of the incoming dTTP did not stack nearly as well with the penultimate base pair compared to the adenine base of the dATP/dF pair.79 The pyrimidine base of dTTP is not in the same plane as the base of the templating dF. Therefore, base selectivity is not solely determined, in these cases, by the number of HBs between the incoming dNTP and the templating base.
Role of Minor Groove HBs in Insertion Efficiency and Base Selectivity
The lone pair of electrons carried by N3 of purines and O2 of pyrimidines are competent HB acceptors and can form HBs with amino acid side chains of the pol.15,95,105,106 To explore the role of minor groove HBs in nucleotide insertion efficiency and base selectivity, we used 3-deaza-2′-deoxyadenosine (3DA), an analogue of adenine in which N3 of dA is replaced with a carbon atom (Figure 4A).78 As shown in Figure 4B, 3DA has the same hydrogen bonding pattern with dT as dA but lacks hydrogen bonding capability at the C3 position, making it an ideal analogue for probing the role of minor groove HBs. Our 1.8 Å resolution structure of the wt RB69pol ternary complex has shown that (i) the hydroxyl group of Y567 forms a HB with N3 of dG at position n – 1 of the template strand via a water molecule, (ii) the side chain of T622 is hydrogen bonded to O2 of dT at position n – 1 of the primer strand via a water molecule, and (iii) the ε-amino nitrogen of K706 forms a HB with N3 of dA at position n – 2 of the primer strand. Substitution of dA with 3DA at position n – 1 of the template strand results in a 90-fold decrease in catalytic efficiency.32 The effect is more dramatic when the 3DA substitution is in the primer strand. Replacing dA with 3DA at positions n – 1 and n – 2 in the primer strand causes the catalytic efficiency to decrease by 144- and 900-fold, respectively.78
Figure 4.
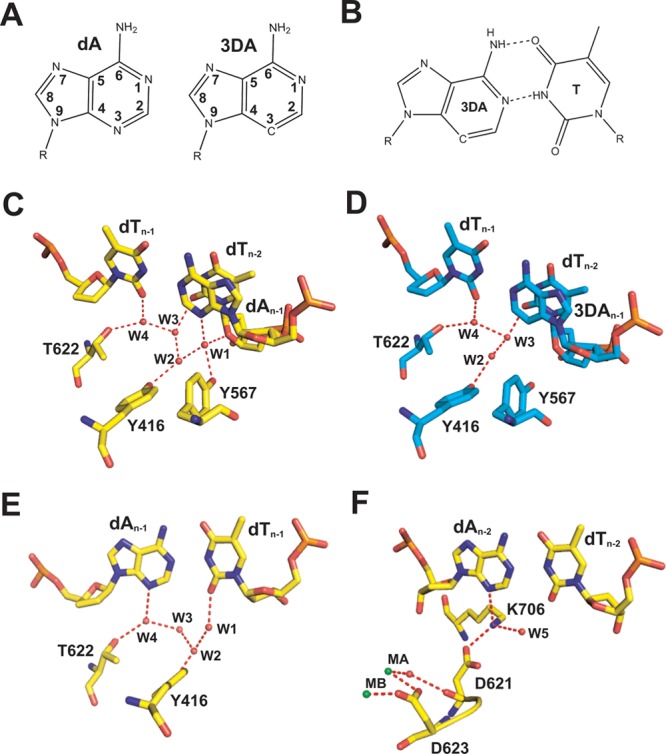
(A) Chemical structures of dA and 3DA. (B) 3DA/dT base pair. (C) Minor groove HB network at the P/T junction of wt RB69pol. (D) Minor groove HB network at the P/T junction of wt RB69pol with 3DA at position n – 1 of the template strand. (E) Minor groove HB network at position n – 1 of the primer strand. (F) Minor groove HB network at position n – 2 of the primer strand.
To provide a structural basis for these observations, we determined the structure of wt RB69pol with 3DA at position n – 1 of the template strand. It is of interest that water molecule w1, which mediates the HB between the hydroxyl group of Y567 and N3 of dA, when dA is at position n – 1 of the template strand, is missing in this structure (Figure 4C,D). In addition, water molecule w2 shifted laterally by 1.2 Å away from the side chain of Y567. The disruption of the rigid HB network at position n – 1 of the template strand could be the reason for the 90-fold decrease in the efficiency of nucleotide incorporation. Although we do not have structures of RB69pol ternary complexes with 3DA at position n – 1 or n – 2 of the primer strand, the structure of the wt RB69pol ternary complex with a P/T control that has dA at positions n – 1 and n – 2 of the primer strand provides insight into the kinetic behavior of these complexes. As shown in Figure 4E, water molecule w4 mediates the HB between the hydroxyl group of T622 and N3 of dA at position n – 1 of the primer strand. Replacing dA with 3DA at this position creates a steric clash between the C3 hydrogen of 3DA and water molecule w4, further disrupting the HB network at position n – 1 of the primer strand. Because 3DA is located at the primer terminus where the 3′-OH attacks the α-phosphorus atom of the incoming dNTP, perturbing the minor groove hydrogen bonding would be expected to have a greater impact on catalysis than the situation in which disruption of the minor groove HB network affects the templating base. This is consistent with our kinetic results. It is worth noting that the catalytic efficiency decreases by almost 3 orders of magnitude when the 3DA substitution is at position n – 2 of the primer strand.78 The sp3-hybridized ε-amino nitrogen is in perfect tetrahedral geometry as it is hydrogen bonded to N3 of dA, the carboxyl group of D621, and an ordered water molecule, w5 (Figure 4F). The carbonyl oxygen of D621 is coordinated to metal ion A via a water molecule. The adjacent D623 residue is essential for catalysis. We speculate that the 3DA substitution at position n – 2 in the primer causes a reorientation of the K706 side chain, which prevents a steric clash between the ε-amino group of K706 and the C3 hydrogen of 3DA. Repositioning K706 would directly affect the rotomer conformation of D621 and would further interfere with the optimal coordination of catalytic metal ion A with its ligands. This could explain why there is a 900-fold decrease in catalytic efficiency when 3DA is at position n – 2 of the primer strand. For these reasons, we have proposed that minor groove HB interactions at position n – 2 of the primer strand in RB69pol complexes are critical for maintaining the correct rotomer conformations of K706 and D621 as well as for positioning of metal ion A so that they are all optimally aligned for nucleotide insertion. In addition, our fluorescence lifetime analysis shows that the minor groove HB interactions also help stabilize the P/T in the pol subdomain. For mismatched base pairs at the P/T junction, the primer terminus of the P/T duplex is more likely to shift to the exo subdomain without the stabilizing influence of minor groove HB interactions of the DNA duplex with amino acid side chains of the pol subdomain.78 On the basis of our kinetic and structural results, we believe that minor groove HB interactions at positions n – 1 and n – 2 of the primer strand and position n – 1 of the template strand in RB69pol ternary complexes are essential for efficient primer extension and base selectivity.
Interactions between the Base at the Primer’s 3′ Terminus and the Base of the Incoming dNTP Affect Insertion Efficiency and Base Selectivity Opposite an Abasic Site
Watson–Crick hydrogen bonding between complementary bases and base stacking are two important factors responsible for the stability of the DNA double helix.106 To evaluate the contribution of base stacking between the incoming dNTP and the base at the 3′ end of the primer to nucleotide insertion efficiency and base selectivity, we determined pre-steady-state kinetic parameters for incorporation of dNTPs opposite an abasic tetrahydrofuran (THF) site by both wt RB69pol and the tm.81 As shown in Figure 5A, THF is an abasic site mimic and has been used in studies of translesion synthesis by repair pols.107−111 Because there is no templating base, the insertion efficiency and fidelity of incorporation of a nucleotide opposite THF are not influenced by the direct interbase HB interactions, but instead by base stacking between the incoming dNTP and the penultimate base pair (pBP). Our pre-steady-state kinetic experiments have shown that RB69pol preferentially inserts dAMP opposite THF when dC/dG is in the pBP position.81 The order of catalytic efficiency for incorporation of different dNTPs opposite THF exhibited by the tm follows the same pattern that is observed with wt RB69pol, but the differences in incorporation efficiencies among the incoming dNTPs are smaller. These results are consistent with the generally accepted “A-rule”, namely that most replicative pols preferentially incorporate dAMP opposite an abasic site.107,112−116 In addition, we also determined the pre-steady-state kinetic parameters for bypassing dN/THF lesions. We found that both wt RB69pol and the tm can bypass a dA/THF lesion more efficiently than other dN/THF lesions. Similar results have been obtained with other B family pols. On the basis of these results, we propose that the A-rule can be extended to state that replicative pols will preferentially incorporate dATP opposite an abasic site but will also bypass a dA/abasic lesion more efficiently than other dN/abasic lesions. To determine the effect of nearest neighbor base stacking on insertion efficiency and fidelity, we varied the pBP adjacent to the templating THF using all 16 possible base pair combinations and then determined the corresponding pre-steady-state kinetic parameters. Unexpectedly, we found that there was a 210-fold difference in catalytic efficiency depending on the identity of the incoming dNTP and the pBP. For a given pBP, dATP was always incorporated more efficiently than the other three dNTPs, again supporting the notion that RB69pol follows the A-rule. For any given incoming dNTP, the incorporation efficiency when the pBP is a G·C pair (dG/dC or dC/dG) is higher than when the pBP is an A·T pair (dA/dT or dT/dA). This is consistent with the fact that more “breathing” is observed for DNA duplexes with A/T rich sequences at the P/T junction. For purine dNTPs, the incorporation efficiency is highest when dC/dG is the pBP and lowest when dA/dT is the pBP.81 In general, although dATP is most often incorporated more efficiently than other dNTPs, adherence to the A-rule is dependent both on the particular DNApol and on the sequence context preceding the abasic site.117
Figure 5.

(A) Chemical structure of THF. (B) Superposition of the four dNTP/THF-containing structures (dATP/THF, red; dCTP/THF, blue; dGTP/THF, orange; dTTP/THF, gray). The PB’s ddC/dG is shown as a space-filling model. (C) Base stacking of an incoming dCTP with ddG/dC. (D) Base stacking of an incoming dCTP with ddA/dT. (E) Base stacking of an incoming dATP with ddC/dG. (F) Base stacking of an incoming dATP with ddA/dT.
To provide a structural basis for these nearest neighbor effects, we determined high-resolution structures of tm RB69pol ternary complexes that had the highest and lowest incorporation efficiencies for each incoming dNTP. Superimposition of these four structures with different incoming dNTPs but with the same pBP (dC/dG) showed that the triphosphate tail and ribosyl moieties of the dNTPs could be overlaid and appeared to be congruent with one another (Figure 5B). As might be expected, incoming dNTPs containing purines stack better against pBPs than dNTPs containing pyrimidines. It is of interest that there was a positive correlation between the incorporation efficiency and favorable partial charge interactions between the incoming dNTP and the pBP. For example, when dCTP was the incoming dNTP, the catalytic efficiency was greatest when the pBP was dG/dC and poorest when the pBP was dA/dT. As shown in Figure 5C, the partial negatively charged O2 of dCTP lies on top of the partial positively charged N2 of dG, and the partial positively charged N4 of dCTP that is situated right over the partial negatively charged O6 of dG. These favorable positive and negative partial charge interactions help to stabilize the ternary complex. In contrast, the partial negatively charged O2 of dCTP is positioned just adjacent to the partial negatively charged O2 of dT, and the partial positively charged N4 of dCTP is positioned right above the partial positively charged N6 of dA (Figure 5D). The resulting repulsive interactions would be expected to destabilize the ternary complex.
When the incoming dNTP contains purine, the hydration network around the purine base could be an important determinant for insertion efficiency. As shown in Figure 5E, when dATP is the incoming dNTP and dC/dG is the pBP, the ordered water molecules are located just above the N3 atoms of the pBP and would be expected to help stabilize the ternary complex. In contrast, when dA/dT is the pBP, the ordered water molecules are located just above the C atoms of the pBP and should destabilize the ternary complex (Figure 5F). This supposition is consistent with kinetic results that showed that when dATP was the incoming dNTP, the catalytic efficiency was greatest when the pBP was dC/dG and greatly diminished when the pBP was dA/dT. Overall, this was the first example in which partial charge interactions among the incoming dNTP, the pBP, and the hydration shell surrounding the incoming dNTP appear to modulate nucleotide insertion efficiency and base selectivity.
How Does the Shape of the Nascent Base Pair and the Shape of the NBP Affect RB69pol Fidelity?
Current hypotheses that attempt to rationalize the fidelity of replicative pols concur that closed ternary complexes with mismatched base pairs are destabilized. Therefore, being able to account for this destabilization in structural and dynamics terms is critical for a mechanistic understanding of base selectivity. Clearly, structures of the 12 mismatches in closed ternary complexes are essential for this purpose.63 For this reason, we used the RB69pol qm variant, mentioned previously, to capture all 16 combinations of base pairs in the NBP of RB69pol.63 As shown in Figure 6A–D, the geometries of the four Watson–Crick base pairs are quite similar, but the C1′–C1′ distances and the glycosidic dihedral bond angles vary slightly among the Watson–Crick base pairs. For purine/pyrimidine mismatches, the dTTP/dG, dGTP/dT, and dCTP/dA pairs all adopt wobble geometry, with the purine bases receding into the minor groove (Figure 6E–G). In contrast, an inverted wobble base pair was observed for the dATP/dC pair (Figure 6H). For pyrimidine/pyrimidine mismatches, two highly ordered water molecules were located at the base pair interface mediating the HB between dCTP and dT and also between dTTP and dC (Figure 6I,J). Only one ordered water molecule was observed at the interface of the dCTP/dC pair (Figure 6K). No water molecules were found between dTTP and dT. Instead, the dTTP/dT pair adopts wobble geometry with the templating dT displaced toward the minor groove (Figure 6L). For purine/purine mispairs, the templating dA bases were flipped 180° from their normal position so that they are now located opposite an incoming dATP or dGTP (Figure 6M,N). Similarly, the templating dG has shifted 30° toward the DNA major groove in the dATP/dG and dGTP/dA pairs (Figure 6O,P).
Figure 6.

Sixteen nascent base pairs in the NBP of the RB69pol qm: (A) dATP/dT pair, (B) dTTP/dA pair, (C) dCTP/dG pair, (D) dGTP/dC pair, (E) dTTP/dG pair, (F) dGTP/dT pair, (G) dCTP/dA pair, (H) dATP/dC pair, (I) dCTP/dT pair, (J) dTTP/dC pair, (K) dCTP/dC pair, (L) dTTP/dT pair, (M) dATP/dA pair, (N) dGTP/dA pair, (O) dATP/dG pair, and (P) dGTP/dG pair.
Next we superimposed all 12 mismatch-containing structures with the structure of a wt RB69pol ternary complex containing dCTP/dG to see if the mismatches would fit into the NBP of wt RB69pol. To our surprise, we found that only 3 of the 12 mismatches (dTTP/dG, dGTP/dT, and dTTP/dT) clashed with side chains in the NBP while the rest of the nascent base pairs could be modeled perfectly well into the NBP of the wt RB69pol. The question then arises as to what prevented the nine remaining mismatched dNTPs from being incorporated if their geometries were compatible with the closed ternary complex of wt RB69pol.
We have proposed four reasons for this. First, there were steric clashes between mismatches and protein side chains, e.g., the three instances mentioned previously. Second, there were only very weak interactions between the incoming dNTP and the templating base. The four purine/purine mismatches fall into this category, as the templating base was either flipped out of the NBP completely or projected 30° toward the major groove. Third, large gaps were observed between some of the incoming dNTPs and the templating bases. For example, the large gap among the dCTP/dT, dTTP/dC, and dCTP/dC pairs would very likely destabilize the closed ternary complex. Fourth, it appears that the purine base of dA in the dCTP/dA pair and the pyrimidine base of dC in the dATP/dC pair are protonated (Figure 7A,B), which would be expected to be disfavored in the highly structured environment of the NBP, and should destabilize the ternary complex.
Figure 7.

Protonation states of (A) dCTP/dA and (B) dATP/dC in the NBP of the RB69pol qm.
When we superimposed all 16 structures of dNTP/dN-containing qm RB69pols, the incoming dNTPs could be overlaid perfectly with one another, particularly with respect to their triphosphate tails and sugar moieties, but the conformation of the templating bases varied dramatically. In the NBP of wt RB69pol, residues L561 and Y567 are located right above and below the templating base. Replacing these two residues with Ala generated large cavities directly above and below the templating base, providing the phosphodiester backbone and the glycosidic bond of the templating base with greater degrees of freedom, allowing the templating base to shift its position so that it can pair with different mismatched incoming dNTPs. Thus, repositioning of the templating base is a critical feature that allows the NBP of the RB69pol qm to accommodate all 12 mismatches. This movement is consistent with structural observations from other pols. For example, in the NBP of T7 DNA pol, a model family A replicative pol, two highly conserved residues, K522 and Y530, are located right above and below the templating base,111 whereas there are no protein side chains located directly above or below the templating base in the NBP of repair pols. Thus, shifting of the templating base to accommodate incorrect dNTPs is a likely reason that the base selectivity of repair pols is so low.118,119 It should be noted, however, that the glycosidic bond can rotate freely and that the sugar pucker can change. If these events occur, then various positions of the base of an incorrect incoming dNTP could be accommodated. In the NBP of wt RB69pol, the side chains of L561 and Y567 restrict the movement of the templating base; consequently, the same mismatch now requires that the incoming incorrect dNTP shift its position, resulting in misalignment of the 3′-hydroxyl group at the primer terminus, thus accounting for the observed low insertion efficiencies. An alternative possibility is that when the bases of either the template or the dNTPs are in the wrong position, as they would be in a mispair, the amino acid side chains around the active site could move to try to maintain interactions with misplaced bases, and it is these altered amino acid positions that could cause polymerization to be inhibited.
Is the Conformational Change upon dNTP Binding a Critical Fidelity Checkpoint for RB69pol?
Structures of the binary and ternary RB69pol complexes show that the fingers subdomain rotates 60° toward the palm subdomain upon the formation of the closed ternary complex (Figure 8A). On the basis of studies with an MDCC-labeled T7 DNA pol, Tsai and Johnson proposed that base selectivity is determined by not only the relative rate of chemistry (k3) and the rate of the forward conformational change (k2) but also by the relative rates of k3 and the rates of reversal of the forward conformational change (k–2) (Scheme 1).70 For example, when T7 DNA pol encounters a correct dNTP, the value of k3 is much greater than the value of k–2, which leads to rapid nucleotide insertion. However, when T7 DNA pol encounters an incorrect dNTP, the value of k–2 is much greater than that of k3, leading to nucleotide release. To see whether this hypothesis for rationalizing base selectivity can be applied to RB69pol, we used 2-aminopurine (2AP), a fluorescent adenine analogue (Figure 8B), as the templating base to estimate the rate of the reverse conformational change.83,120 We have interpreted the fluorescence quenching of 2AP by an incoming dTTP as a consequence of 2AP stacking with the penultimate base pair as the fingers subdomain closes. In addition, we took advantage of two observations reported by Bakhtina et al.122 (i) Rh3+ can be used as an exchange-inert metal ion to form a complex with an incoming dNTP. It fills the B metal ion site without compromising insertion efficiency. (ii) The occupancy of Rh·dNTP at the B metal ion site is sufficient to ensure the formation of a closed ternary complex.122 We found that the rate of product formation was only 4 s–1 when Mg2+ was added after the formation of the RB69pol/P/T/Rh·dTTP ternary complex, when 2AP was the templating base.83 This rate of 4 s–1 was much lower than the rate of 80 s–1 observed when Mg2+ was present in the mixture initially and was interpreted as reopening of the fingers subdomain prior to chemistry.83 In addition, we measured the rate of 2AP quenching in the presence of a catalytically inert Ca2+ with different dTTP concentrations. We obtained an approximate value for k–2 of 4–30 s–1 by extrapolating the hyperbolic curve of the 2AP quenching rate versus the dTTP concentration. Although it was difficult to obtain an accurate estimate of the intercept at the Y axis, the reverse conformational change rate was clearly much slower than the rate of chemistry. A similar experiment was performed with various dCTP concentrations, an incorrect dNTP opposite a templating 2AP. The results were simulated using KinTekSim to obtain an estimate of k–2, which turned out to be greater than 1000 s–1. As an independent check, another fluorescent analogue, tC°, which can form three HBs with guanine (Figure 8C), was used in the same type of experiment.77 The quenching of the tC° fluorescence depends on both base stacking with the penultimate base pair and hydrogen bonding with the templating base. Similar values of k–2 were obtained (S. Xia et al., unpublished results). Thus, our kinetic results with 2AP or tC° quenching were consistent with the proposal advanced by Tsai and Johnson.70 However, it is important to point out that the interpretation of the 2AP and tC° quenching experiments was based on the assumption that the stacking of 2AP or tC° with the penultimate base pair happens concurrently with closing of the fingers subdomain. Our recent kinetic studies with the L415A variant of RB69pol have shown that the rate of 2AP quenching for this mutant was greater than 500 s–1 but that the corresponding value of kpol from chemical quenching was only 11 s–1.82 If our assumption about 2AP quenching is correct, these results strongly suggest that a rate-limiting step exists after fingers subdomain closing but before chemistry, which is likely to be a rearrangement of the active site residues. Ideally, using a fluorescently labeled RB69pol to determine the conformational change rates for correct and incorrect dNTPs would be a more direct measurement of k–2. Unfortunately, RB69pol has eight Cys residues, some of which are critical for pol activity because substitution of all the Cys residues with Ala or Ser inactivated the enzyme (T. Christian et al., unpublished results). However, even if we were able to label RB69pol with a fluorescent probe, it still might not report on the putative rearrangement of active site residues that could be the real rate-limiting step for dNTP insertion. Other approaches will be required to unambiguously address the relationship between conformational changes and base selectivity exhibited by RB69pol.
Figure 8.

(A) Superposition of the structure of the wt RB69pol closed ternary complex [Protein Data Bank (PDB) entry 3NCI] with the structure of the binary complex in an editing mode (PDB entry 1CLQ). (B) Chemical structure of the dAP/dT pair. (C) Chemical structure of the tC°/dG pair.
Scheme 1.

Partitioning of the Primer Terminus between the Pol and Exo Subdomains
When a pol incorporates an incorrect dNTP, it favors switching of the primer terminus from the pol to the exo subdomain, allowing for excision of the misincorporated base. The fidelity of a pol is increased by additional 102–103-fold when it harbors an exo subdomain.4 Thus, the pol to exo partitioning is an important checkpoint that enhances the extraordinarily high fidelity of replicative pols. To further investigate this issue, we developed a novel tC°-tCnitro Förster resonance energy transfer (FRET) assay to monitor pol to exo partitioning.82 As shown in Figure 9A, both tC° (donor) and tCnitro (acceptor) are cytosine analogues and can form HBs with guanine. We designed two sets of duplex P/Ts with tCnitro as the templating base and tC° at position n – 7 of the primer strand (Figure 9B). One set was a fully matched P/T duplex that served as a control; the other set had a mismatched base pair (dA/dA) at the P/T junction (Figure 9B). The relative orientation of the tC°-tCnitro FRET pair, when embedded in the P/T, was revealed by the structure of the tC°-tCnitro FRET-containing RB69pol ternary complex (Figure 9C; S. Xia and M. Wood, unpublished results).
Figure 9.

(A) Chemical structures of tC° and the tCnitro/dG pair. (B) P/T sequences used in the tC°-tCnitro FRET assay. (C) Structure of the tC°-tCnitro FRET pair-containing ternary complex of RB69pol. tC° is colored green and tCnitro red.
Our rationale is that FRET efficiency decreases if the duplex at the P/T junction unwinds because of the partitioning of the primer terminus from the pol to the exo subdomain. The results were exactly what we expected, namely, that FRET efficiencies for the control P/T and mismatched P/T, alone, were 0.43 and 0.32, respectively. When the P/T duplex binds to wt RB69pol, the FRET efficiency for the control P/T decreased slightly to 0.37 but the FRET efficiency for mismatched P/Ts decreased by almost 50% to 0.18. This strongly suggested that RB69pol promoted switching of the mismatched primer terminus from the pol to the exo subdomain; hence, the distance between the FRET pair increased. This separation does not occur unless the mismatched P/T is in a binary complex with the pol. Clearly, this tC°-tCnitro FRET method is sensitive enough to detect unwinding of duplex DNA at the P/T junction and can be used to monitor partitioning of the primer between the pol and exo subdomains.
Using this method, we found that the RB69pol L415A or L415G variant cannot fully distinguish complementary P/Ts from mismatched P/Ts, as the corresponding FRET values for both P/Ts were not statistically different from one another. This is consistent with our kinetic data in that both L415A and L415G mutants are 100-fold more efficient for incorporating a nucleotide residue beyond a mispair. Because residue L415 is located right below the triphosphate tail of the incoming dNTP and does not interact with the P/T, we were puzzled as to why replacing L415 with Ala or Gly would interfere with pol to exo partitioning. The structures of L415A and L415G ternary complexes show that replacing L415 with Ala or Gly greatly enlarges the size of a cavity located at the tip of the L415 side chain. As a consequence, nearby residues, such as L412, D623, and M683, together with a water molecule move in to partially occupy the vacated space. Trapping a water molecule in a hydrophobic pocket is energetically unfavorable and restricts its rotation.123−126 In addition, D623 is located at the tip of a β-hairpin, which has been shown to play a role in the alignment of the primer terminus with an incoming dNTP. We believe that the HB interaction between the trapped water molecule and D623 would likely restrict the movement of this β-hairpin, further affecting the stabilization of the primer terminus when it is located in the pol subdomain. Although our interpretation is speculative, the fact that alteration of the size of the cavity adjacent to the NBP of RB69pol changes its conformational dynamics is clear. Our tC°-tCnitro FRET assay is simple and easy and can be applied to pols from other families (S. Xia and M. Wood, unpublished data), but the studies with the RB69pol L415 variant suggest that ensemble kinetic experiments and static crystal structures alone are not sufficient to address pol–exo dynamics in detail, which are highly relevant for understanding mechanisms that determine base selectivity.
Future Research Will Require a Transition from Studying Structure–Function Relationships to Pol Dynamics
Several experimental results have made us realize that pol dynamics plays a major role in base discrimination.50,63,75−82 For example, (i) conformational changes of the pol upon substrate binding could be a key checkpoint for pol fidelity, (ii) rearrangement of the protein side chains in the NBP could be rate-limiting for nucleotide insertion, (iii) residues that do not interact with the P/T duplex directly can affect pol–exo partitioning, and (iv) mutations quite far from the NBP have dramatic effects on dNTP insertion kinetics. Because of these results, we have shifted our emphasis from studying structure–function relationships in RB69pol to investigating pol dynamics. Accordingly, we attempted to explore the dynamics of conformational changes that contribute to fidelity using time-lapse X-ray crystallography and single-molecule Förster resonance energy transfer (smFRET). Pioneering work using time-lapse X-ray crystallography with human pol η, conducted by Yang’s group, enabled them to observe sequential structural changes of pol η complexes at various stages of primer extension.127 Several interesting results emerged, including the change of the sugar pucker conformation and the presence of a third metal ion. Subsequently, Wilson’s group used the same technique to explore the base selectivity of pol β by capturing structures of the enzyme at various stages along its reaction pathway when it incorporates either a correct or an incorrect incoming dNTP.121 Both pol η and pol β are repair pols, where the insertion rates are quite slow in contrast to that of RB69pol. To see if the same approach could be applied to RB69pol, we produced an RB69pol variant L415G with kpol and Kd,app values greatly reduced compared to those of wt RB69pol, permitting high-quality crystals of a ternary complex to be obtained. Attempts to perform time-lapse crystallography with this variant failed because the crystals were gradually deformed during the primer extension reaction. As an alternative, we have initiated a collaboration with Paul Carey’s group, which has shown that Raman crystallography is also an excellent method for probing pol structure and dynamics during the nucleotidyl transfer reaction.128 Preliminary results from the Carey lab with the L415G variant at low temperatures are encouraging in that they show well-defined changes in the Raman spectra as the nucleotidyl transfer reaction proceeds. Thus, it should be possible to determine the rates of appearance and disappearance of reaction intermediates caused by rearrangements of the side chains in the NBP of the L415G variant. However, neither of these approaches will provide information about the rates of transition between different conformational states in solution, which is crucial for understanding mechanisms of base selectivity, so we are using smFRET with RB69pol to obtain this information. Because we could not remove all eight Cys residues and still retain activity, we had to employ a biorthogonal labeling procedure, introducing a p-acetylphenylalanine (p-Ac-Phe) residue into RB69pol. The p-Ac-Phe residue serves as a unique site for modification by hydroxamate-functionalized fluorescent dyes (Alexa 647, etc.). We have also labeled several different P/T constructs with Cy3B using standard methods. Our preliminary results show that different FRET states are observed depending on (i) the base pairing status at the P/T junction and (ii) the presence of a correct incoming dNTP. The addition of a correct nucleotide to the pol-P/T binary complex also resulted in an altered pattern of FRET efficiencies (M. Wood, unpublished results).
In parallel with our smFRET studies of RB69pol, we are also using Pfu pol, a thermostable B family DNA pol that is fully active even though its resident Cys residues have been replaced with Ser. With the des-Cys Pfu pol variant, we have introduced pairs of Cys residues at different locations. After labeling with donor and acceptor dyes, we expect that the labeled pol will report on conformational changes when dNTPs are added to the pol-P/T binary complex. Our preliminary results suggest the presence of a partially closed (ajar) conformation that could serve as a fidelity checkpoint for nucleotide insertion (M. Wood, unpublished results). An “ajar” conformation has been observed with Bst1pol, an A family DNApol.129 Results with the doubly dye labeled Pfu pol should provide information that will complement results obtained with the RB69pol-P/T ternary complexes in which the acceptor dye is on the pol and the donor dye is on the DNA. We believe that, with the p-Ac-Phe labeling strategy and the dual-Cys Pfu labeling strategy, we have a chance of answering questions about the conformational dynamics of both dNTP discrimination (fidelity) and the pol–exo transition in B family pols.
Summary
RB69pol has proven to be an excellent model for B family replicative DNA polymerases because of (i) sequence similarities in their highly conserved regions, (ii) the ease of crystallization of wt RB69 and many of its mutants, and (iii) extensive kinetic studies that have been correlated with its structure and function. Insights have been obtained about base discrimination that are likely to be applicable to other B family DNA pols. The kinetics for primer extension and editing are consistent with the scheme proposed by Tsai and Johnson that could also hold for other replicative DNApols. While the structures and kinetics of RB69pol have provided valuable information about base selectivity and editing, there are still a number of issues that require further investigation. (i) How does sequence context affect the rate of dNTP incorporation? (ii) How do accessory proteins (sliding clamp, gp45, single-strand binding protein, and gp32) affect the fidelity and rates of nucleotide addition and excision? (iii) Are there intermediate conformational states that can be identified by smFRET and/or Raman spectroscopy on crystals of DNA pol ternary complexes? Clearly, dynamics play a major role in fidelity, and the tools for investigating this are now becoming available. It will be interesting to see how the results obtained with these techniques will influence our current views about the mechanisms that ensure faithful copying of genomic information.
Acknowledgments
This review is dedicated to the memory of Dr. Shuangluo Xia.
Glossary
Abbreviations
- pol
polymerase
- P/T
primer/template
- HB
hydrogen bond
- exo
exonuclease
- RB69pol
RB69 DNA polymerase
- KF
Klenow Fragment
- NBP
nascent base pair binding pocket
- dm
double mutant
- tm
triple mutant
- qm
quadruple mutant
- wt
wild type
- dZ
2′-deoxy-2′-deoxyribo-4-methylbenzimidazole
- dF
2′-deoxyribo-2,4-difluorotoluene
- dQ
2′-deoxyribo-9-methyl-1H-imidazole-[(4,5)-β]-pyridine
- 2AP
2-aminopurine
- 3DA
3-deaza-2′-deoxyadenosine
- THF
tetrahydrofuran
- pBP
penultimate base pair
- FRET
Förster resonance energy transfer
- smFRET
single-molecule Förster resonance energy transfer
- p-Ac-Phe
p-acetylphenylalanine.
Author Present Address
† S.X.: iRND3, Mountain View, CA 94043.
This work is supported by National Institutes of Health Grant GM 063276−09.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
References
- Kunkel T. A.; Bebenek K. (1988) Recent studies of the fidelity of DNA synthesis. Biochim. Biophys. Acta 951, 1–15. [DOI] [PubMed] [Google Scholar]
- Drake J. W. (1991) A constant rate of spontaneous mutation in DNA-based microbes. Proc. Natl. Acad. Sci. U.S.A. 88, 7160–7164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joyce C. M.; Benkovic S. J. (2004) DNA polymerase fidelity: Kinetics, structure, and checkpoints. Biochemistry 43, 14317–14324. [DOI] [PubMed] [Google Scholar]
- Kunkel T. A. (2004) DNA replication fidelity. J. Biol. Chem. 279, 16895–16898. [DOI] [PubMed] [Google Scholar]
- Umar A.; Kunkel T. A. (1996) DNA-replication fidelity, mismatch repair and genome instability in cancer cells. Eur. J. Biochem. 238, 297–307. [DOI] [PubMed] [Google Scholar]
- Sweasy J. B.; Lauper J. M.; Eckert K. A. (2006) DNA polymerases and human diseases. Radiat. Res. 166, 693–714. [DOI] [PubMed] [Google Scholar]
- Bebenek A.; Carver G. T.; Dressman H. K.; Kadyrov F. A.; Haseman J. K.; Petrov V.; Konigsberg W. H.; Karam J. D.; Drake J. W. (2002) Dissecting the fidelity of bacteriophage RB69 DNA polymerase: Site-specific modulation of fidelity by polymerase accessory proteins. Genetics 162, 1003–1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loeb L. A.; Monnat R. J. Jr. (2008) DNA polymerases and human disease. Nat. Rev. Genet. 9, 594–604. [DOI] [PubMed] [Google Scholar]
- Lange S. S.; Takata K.; Wood R. D. (2011) DNA polymerases and cancer. Nat. Rev. Cancer 11, 96–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodman M. F.; Tippin B. (2000) The expanding polymerase universe. Nat. Rev. Mol. Cell Biol. 1, 101–109. [DOI] [PubMed] [Google Scholar]
- Filee J.; Forterre P.; Sen-Lin T.; Laurent J. (2002) Evolution of DNA polymerase families: Evidences for multiple gene exchange between cellular and viral proteins. J. Mol. Evol. 54, 763–773. [DOI] [PubMed] [Google Scholar]
- Joyce C. M.; Steitz T. A. (1995) Polymerase structures and function: Variations on a theme?. J. Bacteriol. 177, 6321–6329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braithwaite D. K.; Ito J. (1993) Compilation, alignment, and phylogenetic relationships of DNA polymerases. Nucleic Acids Res. 21, 787–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karam J. D.; Konigsberg W. H. (2000) DNA polymerase of the T4-related bacteriophages. Prog. Nucleic Acid Res. Mol. Biol. 64, 65–96. [DOI] [PubMed] [Google Scholar]
- Franklin M. C.; Wang J.; Steitz T. A. (2001) Structure of the replicating complex of a pol α family DNA polymerase. Cell 105, 657–667. [DOI] [PubMed] [Google Scholar]
- Bennett N.; Gotte M. (2013) Utility of the bacteriophage RB69 polymerase gp43 as a surrogate enzyme for herpesvirus orthologs. Viruses 5, 54–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loeb L. A.; Kunkel T. A. (1982) Fidelity of DNA synthesis. Annu. Rev. Biochem. 51, 429–457. [DOI] [PubMed] [Google Scholar]
- Echols H.; Goodman M. F. (1991) Fidelity mechanisms in DNA replication. Annu. Rev. Biochem. 60, 477–511. [DOI] [PubMed] [Google Scholar]
- Young M. C.; Reddy M. K.; von Hippel P. H. (1992) Structure and function of the bacteriophage T4 DNA polymerase holoenzyme. Biochemistry 31, 8675–8690. [DOI] [PubMed] [Google Scholar]
- Goodman M. F.; Creighton S.; Bloom L. B.; Petruska J. (1993) Biochemical basis of DNA replication fidelity. Crit. Rev. Biochem. Mol. Biol. 28, 83–126. [DOI] [PubMed] [Google Scholar]
- Johnson K. A. (1993) Conformational coupling in DNA polymerase fidelity. Annu. Rev. Biochem. 62, 685–713. [DOI] [PubMed] [Google Scholar]
- Joyce C. M.; Steitz T. A. (1994) Function and structure relationships in DNA polymerases. Annu. Rev. Biochem. 63, 777–822. [DOI] [PubMed] [Google Scholar]
- Young M. C.; Weitzel S. E.; von Hippel P. H. (1996) The kinetic mechanism of formation of the bacteriophage T4 DNA polymerase sliding clamp. J. Mol. Biol. 264, 440–452. [DOI] [PubMed] [Google Scholar]
- Kunkel T. A.; Bebenek K. (2000) DNA replication fidelity. Annu. Rev. Biochem. 69, 497–529. [DOI] [PubMed] [Google Scholar]
- Hubscher U.; Maga G.; Spadari S. (2002) Eukaryotic DNA polymerases. Annu. Rev. Biochem. 71, 133–163. [DOI] [PubMed] [Google Scholar]
- Beard W.; Wilson S. (2014) Structure and Mechanism of DNA Polymerase β. Biochemistry 53, DOI: 10.1021/bi500139h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bebenek K.; Pedersen L. C.; Kunkel T. A. (2014) Structure–Function Studies of DNA Polymerase λ. Biochemistry 53, DOI: 10.1021/bi4017236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maxwell B. A.; Suo Z. (2014) Recent Insight into the Kinetic Mechanisms and Conformational Dynamics of Y-Family DNA Polymerases. Biochemistry 53, DOI: 10.1021/bi5000405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang W. (2014) A Summary of Y-Family DNA Polymerases and a Case Study of Human DNA Polymerase η. Biochemistry 53, DOI: 10.1021/bi500019s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C. C.; Yeh L. S.; Karam J. D. (1995) Modular organization of T4 DNA polymerase. Evidence from phylogenetics. J. Biol. Chem. 270, 26558–26564. [DOI] [PubMed] [Google Scholar]
- Wang J.; Sattar A. K.; Wang C. C.; Karam J. D.; Konigsberg W. H.; Steitz T. A. (1997) Crystal structure of a pol α family replication DNA polymerase from bacteriophage RB69. Cell 89, 1087–1099. [DOI] [PubMed] [Google Scholar]
- Wang M.; Xia S.; Blaha G.; Steitz T. A.; Konigsberg W. H.; Wang J. (2011) Insights into base selectivity from the 1.8 Å resolution structure of an RB69 DNA polymerase ternary complex. Biochemistry 50, 581–590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burd C. G.; Dreyfuss G. (1994) Conserved structures and diversity of functions of RNA-binding proteins. Science 265, 615–621. [DOI] [PubMed] [Google Scholar]
- Liu S.; Knafels J. D.; Chang J. S.; Waszak G. A.; Baldwin E. T.; Deibel M. R. Jr.; Thomsen D. R.; Homa F. L.; Wells P. A.; Tory M. C.; Poorman R. A.; Gao H.; Qiu X.; Seddon A. P. (2006) Crystal structure of the herpes simplex virus 1 DNA polymerase. J. Biol. Chem. 281, 18193–18200. [DOI] [PubMed] [Google Scholar]
- Tuerk C.; Eddy S.; Parma D.; Gold L. (1990) Autogenous translational operator recognized by bacteriophage T4 DNA polymerase. J. Mol. Biol. 213, 749–761. [DOI] [PubMed] [Google Scholar]
- Pavlov A. R.; Karam J. D. (1994) Binding specificity of T4 DNA polymerase to RNA. J. Biol. Chem. 269, 12968–12972. [PubMed] [Google Scholar]
- Wang C. C.; Pavlov A.; Karam J. D. (1997) Evolution of RNA-binding specificity in T4 DNA polymerase. J. Biol. Chem. 272, 17703–17710. [DOI] [PubMed] [Google Scholar]
- Reha-Krantz L. J.; Nonay R. L. (1993) Genetic and biochemical studies of bacteriophage T4 DNA polymerase 3′→5′-exonuclease activity. J. Biol. Chem. 268, 27100–27108. [PubMed] [Google Scholar]
- Steitz T. A. (1999) DNA polymerases: Structural diversity and common mechanisms. J. Biol. Chem. 274, 17395–17398. [DOI] [PubMed] [Google Scholar]
- Derbyshire V.; Freemont P. S.; Sanderson M. R.; Beese L.; Friedman J. M.; Joyce C. M.; Steitz T. A. (1988) Genetic and crystallographic studies of the 3′,5′-exonucleolytic site of DNA polymerase I. Science 240, 199–201. [DOI] [PubMed] [Google Scholar]
- Abdus Sattar A. K.; Lin T. C.; Jones C.; Konigsberg W. H. (1996) Functional consequences and exonuclease kinetic parameters of point mutations in bacteriophage T4 DNA polymerase. Biochemistry 35, 16621–16629. [DOI] [PubMed] [Google Scholar]
- Wang C. X.; Zakharova E.; Li J.; Joyce C. M.; Wang J.; Konigsberg W. (2004) Pre-steady-state kinetics of RB69 DNA polymerase and its exo domain mutants: Effect of pH and thiophosphoryl linkages on 3′-5′ exonuclease activity. Biochemistry 43, 3853–3861. [DOI] [PubMed] [Google Scholar]
- Frey M. W.; Nossal N. G.; Capson T. L.; Benkovic S. J. (1993) Construction and characterization of a bacteriophage T4 DNA polymerase deficient in 3′→5′ exonuclease activity. Proc. Natl. Acad. Sci. U.S.A. 90, 2579–2583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang G.; Lin T.; Karam J.; Konigsberg W. H. (1999) Steady-state kinetic characterization of RB69 DNA polymerase mutants that affect dNTP incorporation. Biochemistry 38, 8094–8101. [DOI] [PubMed] [Google Scholar]
- a Santos E.; Lazaro J. M.; Perez-Arnaiz R.; Salas M.; deVega M. (2014) Role of Lex E motif protein primed DNA polymerases in interaction with the incoming nucleotide. J. Biol. Chem. 289, 2888–98. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Perumal S. K.; Ren W.; Lee T. H.; Benkovic S. J. (2013) How a holoenzyme for DNA replication is formed. Proc. Natl. Acad. Sci. U.S.A. 110, 99–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beese L. S.; Derbyshire V.; Steitz T. A. (1993) Structure of DNA polymerase I Klenow fragment bound to duplex DNA. Science 260, 352–355. [DOI] [PubMed] [Google Scholar]
- Dong Q.; Copeland W. C.; Wang T. S. (1993) Mutational studies of human DNA polymerase α. Identification of residues critical for deoxynucleotide binding and misinsertion fidelity of DNA synthesis. J. Biol. Chem. 268, 24163–24174. [PubMed] [Google Scholar]
- Astatke M.; Grindley N. D.; Joyce C. M. (1995) Deoxynucleoside triphosphate and pyrophosphate binding sites in the catalytically competent ternary complex for the polymerase reaction catalyzed by DNA polymerase I (Klenow fragment). J. Biol. Chem. 270, 1945–1954. [DOI] [PubMed] [Google Scholar]
- Bell J. B.; Eckert K. A.; Joyce C. M.; Kunkel T. A. (1997) Base miscoding and strand misalignment errors by mutator Klenow polymerases with amino acid substitutions at tyrosine 766 in the O helix of the fingers subdomain. J. Biol. Chem. 272, 7345–7351. [DOI] [PubMed] [Google Scholar]
- Xia S.; Wang M.; Blaha G.; Konigsberg W. H.; Wang J. (2011) Structural Insights into Complete Metal Ion Coordination from Ternary Complexes of B Family RB69 DNA Polymerase. Biochemistry 50, 9114–9124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Latham G. J.; Pietroni P.; Dong F.; Young M. C.; von Hippel P. H. (1996) Fluorescence monitoring of T4 polymerase holoenzyme accessory protein interactions during loading of the sliding clamp onto the template-primer junction. J. Mol. Biol. 264, 426–439. [DOI] [PubMed] [Google Scholar]
- Shamoo Y.; Steitz T. A. (1999) Building a replisome from interacting pieces: Sliding clamp complexed to a peptide from DNA polymerase and a polymerase editing complex. Cell 99, 155–166. [DOI] [PubMed] [Google Scholar]
- Zakharova E.; Wang J.; Konigsberg W. (2004) The activity of selected RB69 DNA polymerase mutants can be restored by manganese ions: The existence of alternative metal ion ligands used during the polymerization cycle. Biochemistry 43, 6587–6595. [DOI] [PubMed] [Google Scholar]
- Yang G.; Franklin M.; Li J.; Lin T. C.; Konigsberg W. (2002) A conserved Tyr residue is required for sugar selectivity in a Pol α DNA polymerase. Biochemistry 41, 10256–10261. [DOI] [PubMed] [Google Scholar]
- Brown J. A.; Suo Z. (2011) Unlocking the sugar “steric gate” of DNA polymerases. Biochemistry 50, 1135–1142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joyce C. M. (1997) Choosing the right sugar: How polymerases select a nucleotide substrate. Proc. Natl. Acad. Sci. U.S.A. 94, 1619–1622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Astatke M.; Ng K.; Grindley N. D.; Joyce C. M. (1998) A single side chain prevents Escherichia coli DNA polymerase I (Klenow fragment) from incorporating ribonucleotides. Proc. Natl. Acad. Sci. U.S.A. 95, 3402–3407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang G.; Franklin M.; Li J.; Lin T. C.; Konigsberg W. (2002) Correlation of the kinetics of finger domain mutants in RB69 DNA polymerase with its structure. Biochemistry 41, 2526–2534. [DOI] [PubMed] [Google Scholar]
- Castro C.; Smidansky E.; Maksimchuk K. R.; Arnold J. J.; Korneeva V. S.; Gotte M.; Konigsberg W.; Cameron C. E. (2007) Two proton transfers in the transition state for nucleotidyl transfer catalyzed by RNA- and DNA-dependent RNA and DNA polymerases. Proc. Natl. Acad. Sci. U.S.A. 104, 4267–4272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang G.; Wang J.; Konigsberg W. (2005) Base selectivity is impaired by mutants that perturb hydrogen bonding networks in the RB69 DNA polymerase active site. Biochemistry 44, 3338–3346. [DOI] [PubMed] [Google Scholar]
- Zhang H.; Rhee C.; Bebenek A.; Drake J. W.; Wang J.; Konigsberg W. (2006) The L561A substitution in the nascent base-pair binding pocket of RB69 DNA polymerase reduces base discrimination. Biochemistry 45, 2211–2220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H.; Beckman J.; Wang J.; Konigsberg W. (2009) RB69 DNA polymerase mutants with expanded nascent base-pair-binding pockets are highly efficient but have reduced base selectivity. Biochemistry 48, 6940–6950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia S.; Wang J.; Konigsberg W. H. (2013) DNA mismatch synthesis complexes provide insights into base selectivity of a B family DNA polymerase. J. Am. Chem. Soc. 135, 193–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watson J. D.; Crick F. H. (1953) Genetical implications of the structure of deoxyribonucleic acid. Nature 171, 964–967. [DOI] [PubMed] [Google Scholar]
- Showalter A. K.; Tsai M. D. (2002) A reexamination of the nucleotide incorporation fidelity of DNA polymerases. Biochemistry 41, 10571–10576. [DOI] [PubMed] [Google Scholar]
- Arndt J. W.; Gong W.; Zhong X.; Showalter A. K.; Liu J.; Dunlap C. A.; Lin Z.; Paxson C.; Tsai M. D.; Chan M. K. (2001) Insight into the catalytic mechanism of DNA polymerase β: Structures of intermediate complexes. Biochemistry 40, 5368–5375. [DOI] [PubMed] [Google Scholar]
- Rothwell P. J.; Mitaksov V.; Waksman G. (2005) Motions of the fingers subdomain of klentaq1 are fast and not rate limiting: Implications for the molecular basis of fidelity in DNA polymerases. Mol. Cell 19, 345–355. [DOI] [PubMed] [Google Scholar]
- Luo G.; Wang M.; Konigsberg W. H.; Xie X. S. (2007) Single-molecule and ensemble fluorescence assays for a functionally important conformational change in T7 DNA polymerase. Proc. Natl. Acad. Sci. U.S.A. 104, 12610–12615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson K. A. (2010) The kinetic and chemical mechanism of high-fidelity DNA polymerases. Biochim. Biophys. Acta 1804, 1041–1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai Y. C.; Johnson K. A. (2006) A new paradigm for DNA polymerase specificity. Biochemistry 45, 9675–9687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hariharan C.; Bloom L. B.; Helquist S. A.; Kool E. T.; Reha-Krantz L. J. (2006) Dynamics of nucleotide incorporation: Snapshots revealed by 2-aminopurine fluorescence studies. Biochemistry 45, 2836–2844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beckman J.; Wang M.; Blaha G.; Wang J.; Konigsberg W. H. (2010) Substitution of Ala for Tyr567 in RB69 DNA polymerase allows dAMP to be inserted opposite 7,8-dihydro-8-oxoguanine. Biochemistry 49, 4116–4125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beckman J.; Wang M.; Blaha G.; Wang J.; Konigsberg W. H. (2010) Substitution of Ala for Tyr567 in RB69 DNA polymerase allows dAMP and dGMP to be inserted opposite guanidinohydantoin. Biochemistry 49, 8554–8563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reha-Krantz L. J.; Hariharan C.; Subuddhi U.; Xia S.; Zhao C.; Beckman J.; Christian T.; Konigsberg W. (2011) Structure of the 2-aminopurine-cytosine base pair formed in the polymerase active site of the RB69 Y567A-DNA polymerase. Biochemistry 50, 10136–10149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia S.; Konigsberg W. H.; Wang J. (2011) Hydrogen-bonding capability of a templating difluorotoluene nucleotide residue in an RB69 DNA polymerase ternary complex. J. Am. Chem. Soc. 133, 10003–10005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia S.; Wang M.; Lee H. R.; Sinha A.; Blaha G.; Christian T.; Wang J.; Konigsberg W. (2011) Variation in mutation rates caused by RB69pol fidelity mutants can be rationalized on the basis of their kinetic behavior and crystal structures. J. Mol. Biol. 406, 558–570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia S.; Beckman J.; Wang J.; Konigsberg W. H. (2012) Using a Fluorescent Cytosine Analogue tC° To Probe the Effect of the Y567 to Ala Substitution on the Preinsertion Steps of dNMP Incorporation by RB69 DNA Polymerase. Biochemistry 51, 4609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia S.; Christian T. D.; Wang J.; Konigsberg W. H. (2012) Probing minor groove hydrogen bonding interactions between RB69 DNA polymerase and DNA. Biochemistry 51, 4343–4353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia S.; Eom S. H.; Konigsberg W. H.; Wang J. (2012) Structural Basis for Differential Insertion Kinetics of dNMPs Opposite a Difluorotoluene Nucleotide Residue. Biochemistry 51, 1476–1485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia S.; Eom S. H.; Konigsberg W. H.; Wang J. (2012) Bidentate and tridentate metal-ion coordination states within ternary complexes of RB69 DNA polymerase. Protein Sci. 21, 447–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia S.; Vashishtha A.; Bulkley D.; Eom S. H.; Wang J.; Konigsberg W. H. (2012) Contribution of Partial Charge Interactions and Base Stacking to the Efficiency of Primer Extension at and beyond Abasic Sites in DNA. Biochemistry 51, 4922–4931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia S.; Wood M.; Bradley M. J.; De La Cruz E. M.; Konigsberg W. H. (2013) Alteration in the cavity size adjacent to the active site of RB69 DNA polymerase changes its conformational dynamics. Nucleic Acids Res. 41, 9077–9089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee H. R.; Wang M.; Konigsberg W. (2009) The reopening rate of the fingers domain is a determinant of base selectivity for RB69 DNA polymerase. Biochemistry 48, 2087–2098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Florian J.; Goodman M. F.; Warshel A. (2003) Computer simulation studies of the fidelity of DNA polymerases. Biopolymers 68, 286–299. [DOI] [PubMed] [Google Scholar]
- Kirmizialtin S.; Nguyen V.; Johnson K. A.; Elber R. (2012) How conformational dynamics of DNA polymerase select correct substrates: Experiments and simulations. Structure 20, 618–627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kool E. T.; Sintim H. O. (2006) The difluorotoluene debate: A decade later. Chem. Commun. 3665–3675. [DOI] [PubMed] [Google Scholar]
- Moore C. L.; Zivkovic A.; Engels J. W.; Kuchta R. D. (2004) Human DNA primase uses Watson-Crick hydrogen bonds to distinguish between correct and incorrect nucleoside triphosphates. Biochemistry 43, 12367–12374. [DOI] [PubMed] [Google Scholar]
- Beckman J.; Kincaid K.; Hocek M.; Spratt T.; Engels J.; Cosstick R.; Kuchta R. D. (2007) Human DNA polymerase α uses a combination of positive and negative selectivity to polymerize purine dNTPs with high fidelity. Biochemistry 46, 448–460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seo Y. J.; Hwang G. T.; Ordoukhanian P.; Romesberg F. E. (2009) Optimization of an unnatural base pair toward natural-like replication. J. Am. Chem. Soc. 131, 3246–3252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuchta R. D. (2010) Nucleotide Analogues as Probes for DNA and RNA Polymerases. Curr. Protoc. Chem. Biol. 2, 111–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Motea E. A.; Berdis A. J. (2010) Terminal deoxynucleotidyl transferase: The story of a misguided DNA polymerase. Biochim. Biophys. Acta 1804, 1151–1166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Betz K.; Malyshev D. A.; Lavergne T.; Welte W.; Diederichs K.; Dwyer T. J.; Ordoukhanian P.; Romesberg F. E.; Marx A. (2012) KlenTaq polymerase replicates unnatural base pairs by inducing a Watson-Crick geometry. Nat. Chem. Biol. 8, 612–614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Betz K.; Malyshev D. A.; Lavergne T.; Welte W.; Diederichs K.; Romesberg F. E.; Marx A. (2013) Structural insights into DNA replication without hydrogen bonds. J. Am. Chem. Soc. 135, 18637–18643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Motea E. A.; Lee I.; Berdis A. J. (2013) Insights into the roles of desolvation and π-electron interactions during DNA polymerization. ChemBioChem 14, 489–498. [DOI] [PubMed] [Google Scholar]
- Schweitzer B. A.; Kool E. T. (1994) Aromatic Nonpolar Nucleosides as Hydrophobic Isosteres of Pyrimidine and Purine Nucleosides. J. Org. Chem. 59, 7238–7242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schweitzer B. A.; Kool E. T. (1995) Hydrophobic, Non-Hydrogen-Bonding Bases and Base Pairs in DNA. J. Am. Chem. Soc. 117, 1863–1872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kool E. T. (2002) Active site tightness and substrate fit in DNA replication. Annu. Rev. Biochem. 71, 191–219. [DOI] [PubMed] [Google Scholar]
- Moran S.; Ren R. X.; Kool E. T. (1997) A thymidine triphosphate shape analog lacking Watson-Crick pairing ability is replicated with high sequence selectivity. Proc. Natl. Acad. Sci. U.S.A. 94, 10506–10511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moran S.; Ren R. X.; Rumney S.; Kool E. T. (1997) Difluorotoluene, a Nonpolar Isostere for Thymine, Codes Specifically and Efficiently for Adenine in DNA Replication. J. Am. Chem. Soc. 119, 2056–2057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kool E. T. (2001) Hydrogen bonding, base stacking, and steric effects in DNA replication. Annu. Rev. Biophys. Biomol. Struct. 30, 1–22. [DOI] [PubMed] [Google Scholar]
- Kim T. W.; Delaney J. C.; Essigmann J. M.; Kool E. T. (2005) Probing the active site tightness of DNA polymerase in subangstrom increments. Proc. Natl. Acad. Sci. U.S.A. 102, 15803–15808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaudhari S. R.; Mogurampelly S.; Suryaprakash N. (2013) Engagement of CF3 group in N-H···F-C hydrogen bond in the solution state: NMR spectroscopy and MD simulation studies. J. Phys. Chem. B 117, 1123–1129. [DOI] [PubMed] [Google Scholar]
- Lee H. R.; Helquist S. A.; Kool E. T.; Johnson K. A. (2008) Importance of hydrogen bonding for efficiency and specificity of the human mitochondrial DNA polymerase. J. Biol. Chem. 283, 14402–14410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khakshoor O.; Wheeler S. E.; Houk K. N.; Kool E. T. (2012) Measurement and theory of hydrogen bonding contribution to isosteric DNA base pairs. J. Am. Chem. Soc. 134, 3154–3163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doublie S.; Tabor S.; Long A. M.; Richardson C. C.; Ellenberger T. (1998) Crystal structure of a bacteriophage T7 DNA replication complex at 2.2 Å resolution. Nature 391, 251–258. [DOI] [PubMed] [Google Scholar]
- Johnson S. J.; Beese L. S. (2004) Structures of mismatch replication errors observed in a DNA polymerase. Cell 116, 803–816. [DOI] [PubMed] [Google Scholar]
- Shibutani S.; Takeshita M.; Grollman A. P. (1997) Translesional synthesis on DNA templates containing a single abasic site. A mechanistic study of the “A rule”. J. Biol. Chem. 272, 13916–13922. [DOI] [PubMed] [Google Scholar]
- Greenberg M. M.; Weledji Y. N.; Kroeger K. M.; Kim J. (2004) In vitro replication and repair of DNA containing a C2′-oxidized abasic site. Biochemistry 43, 15217–15222. [DOI] [PubMed] [Google Scholar]
- Sheriff A.; Motea E.; Lee I.; Berdis A. J. (2008) Mechanism and dynamics of translesion DNA synthesis catalyzed by the Escherichia coli Klenow fragment. Biochemistry 47, 8527–8537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabouri N.; Johansson E. (2009) Translesion synthesis of abasic sites by yeast DNA polymerase epsilon. J. Biol. Chem. 284, 31555–31563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zahn K. E.; Wallace S. S.; Doublie S. (2011) DNA polymerases provide a canon of strategies for translesion synthesis past oxidatively generated lesions. Curr. Opin. Struct. Biol. 21, 358–369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loeb L. A.; Preston B. D. (1986) Mutagenesis by apurinic/apyrimidinic sites. Annu. Rev. Genet. 20, 201–230. [DOI] [PubMed] [Google Scholar]
- Randall S. K.; Eritja R.; Kaplan B. E.; Petruska J.; Goodman M. F. (1987) Nucleotide insertion kinetics opposite abasic lesions in DNA. J. Biol. Chem. 262, 6864–6870. [PubMed] [Google Scholar]
- Mozzherin D. J.; Shibutani S.; Tan C. K.; Downey K. M.; Fisher P. A. (1997) Proliferating cell nuclear antigen promotes DNA synthesis past template lesions by mammalian DNA polymerase δ. Proc. Natl. Acad. Sci. U.S.A. 94, 6126–6131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strauss B. S. (2002) The “A” rule revisited: Polymerases as determinants of mutational specificity. DNA Repair 1, 125–135. [DOI] [PubMed] [Google Scholar]
- Beard W. A.; Shock D. D.; Batra V. K.; Pedersen L. C.; Wilson S. H. (2009) DNA polymerase β substrate specificity: Side chain modulation of the “A-rule”. J. Biol. Chem. 284, 31680–31689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan K.; Resnick M. A.; Gordenin D. A. (2013) The choice of nucleotide inserted opposite abasic sites formed within chromosomal DNA reveals the polymerase activities participating in translesion DNA synthesis. DNA Repair 12, 878–889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trincao J.; Johnson R. E.; Wolfle W. T.; Escalante C. R.; Prakash S.; Prakash L.; Aggarwal A. K. (2004) Dpo4 is hindered in extending a G·T mismatch by a reverse wobble. Nat. Struct. Mol. Biol. 11, 457–462. [DOI] [PubMed] [Google Scholar]
- Wilson R. C.; Pata J. D. (2008) Structural insights into the generation of single-base deletions by the Y family DNA polymerase dbh. Mol. Cell 29, 767–779. [DOI] [PubMed] [Google Scholar]
- Wang M.; Lee H. R.; Konigsberg W. (2009) Effect of A and B metal ion site occupancy on conformational changes in an RB69 DNA polymerase ternary complex. Biochemistry 48, 2075–2086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freudenthal B. D.; Beard W. A.; Shock D. D.; Wilson S. H. (2013) Observing a DNA polymerase choose right from wrong. Cell 154, 157–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakhtina M.; Lee S.; Wang Y.; Dunlap C.; Lamarche B.; Tsai M. D. (2005) Use of viscogens, dNTPαS, and rhodium(III) as probes in stopped-flow experiments to obtain new evidence for the mechanism of catalysis by DNA polymerase β. Biochemistry 44, 5177–5187. [DOI] [PubMed] [Google Scholar]
- Brunori M.; Vallone B.; Cutruzzola F.; Travaglini-Allocatelli C.; Berendzen J.; Chu K.; Sweet R. M.; Schlichting I. (2000) The role of cavities in protein dynamics: Crystal structure of a photolytic intermediate of a mutant myoglobin. Proc. Natl. Acad. Sci. U.S.A. 97, 2058–2063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonavane S.; Chakrabarti P. (2008) Cavities and atomic packing in protein structures and interfaces. PLoS Comput. Biol. 4, e1000188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matthews B. W.; Liu L. (2009) A review about nothing: Are apolar cavities in proteins really empty?. Protein Sci. 18, 494–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merski M.; Shoichet B. K. (2012) Engineering a model protein cavity to catalyze the Kemp elimination. Proc. Natl. Acad. Sci. U.S.A. 109, 16179–16183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura T.; Zhao Y.; Yamagata Y.; Hua Y. J.; Yang W. (2012) Watching DNA polymerase η make a phosphodiester bond. Nature 487, 196–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Espinoza-Herrera S. J.; Gaur V.; Suo Z.; Carey P. R. (2013) Following DNA Chain Extension and Protein Conformational Changes in Crystals of a Y-Family DNA Polymerase via Raman Crystallography. Biochemistry 52, 4881–4890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu E. Y.; Beese L. S. (2011) The structure of a high fidelity DNA polymerase bound to a mismatched nucleotide reveals an “ajar” intermediate conformation in the nucleotide selection mechanism. J. Biol. Chem. 286, 19758–19767. [DOI] [PMC free article] [PubMed] [Google Scholar]