

Abstract
Tris(hydroxymethyl)aminomethane (Tris) is one of the most frequently used buffer ingredients. Among other things, it is recommended and is usually used for lectin-based affinity enrichment of glycopeptides. Here we report that sialic acid, a common ‘capping’ unit in both N- and O-linked glycans may react with this chemical, and this side reaction may compromise glycopeptide identification when ETD spectra are the only MS/MS data used in the database search. We show that the modification may alter N- as well as O-linked glycans, the Tris-derivative is still prone to fragmentation both in ‘beam-type’ CID (HCD) and ETD experiments, at the same time—since the acidic carboxyl group was ‘neutralized’—it will display a different retention time than its unmodified counterpart. We also suggest solutions that—when incorporated into existing search engines—may significantly improve the reliability of glycopeptide assignments.
Keywords: ETD, Glycopeptide, CID, HCD, Side reaction, Sialic acid
Introduction
Post-translational modifications (PTMs) may be crucial for the subcellular localization, function, and interactions of proteins. Accordingly, recent mass spectrometry-aided proteomic studies are aimed not only at large scale protein identification but also at high-throughput PTM characterization.
Although glycosylation is one of the most frequent PTMs, relatively little is known about this modification because of analytical challenges. Glycosylation cannot be predicted on a genomic basis, it is not template-based, and the glycan structures display bewildering variations [1]. Both N-linked (Asn modification) and O-linked (Ser, Thr, and Tyr modification) glycoproteins feature heterogeneity in site-occupancy as well as in glycan structure at any given modification site [1–3]. Glycosidic bonds are weaker than peptide bonds, thus, CID/HCD spectra are dominated by carbohydrate fragments and frequently do not provide sufficient information about the aminoacid sequence [2–4]. Consequently, characterization of site-specific glycosylation used to be limited to purified protein samples [5–9]. The new MS/MS techniques, electron capture dissociation (ECD, [10]) and electron transfer dissociation (ETD, [11]) opened up new possibilities in intact glycopeptide analysis [12–16], since in these processes the peptide backbone is fragmented whereas the side chains mostly stay unaltered [17]. ETD is inherently more efficient, thus, large scale glycopeptide studies use this technique. In order to shorten the duty cycle during the characterization of complex mixtures, and selectively target only glycopeptides for ETD analysis, ‘beam-type’ CID (HCD) data can be acquired on the precursor ions first, and ETD acquisition is triggered by the detection of specified diagnostic carbohydrate ions [18]. Ion trap CID (i.e., resonance activation) cannot fulfill this job since the formation of some diagnostic carbohydrate ions requires multiple bond cleavages, and most of these fragment ions feature masses that are usually lower than one-third of the glycopeptide precursor ion's m/z value (i.e., will not be detected during the analysis).
Interpretation of MS/MS data acquired from complex mixtures inevitably requires a suitable bioinformatic tool. Database search engines were developed and optimized for the reliable identification of tryptic peptides. Modified peptide identification and site assignments are a bit harder to tackle. Permitting variable modifications on frequently occurring aminoacids opens up the search space leading to increased false discovery rates, and most of the false identifications are among the modified sequences. The situation is even worse when nonspecific cleavages have to be considered, for example, in serum samples or other secreted protein mixtures where proteolytic activity is rampant. The fact that ETD spectra will not yield information on the glycan attached to the peptide is almost as big of a problem as the insufficient peptide fragmentation during CID/HCD analysis of glycopeptides, since variable modifications have to be specified prior to the database search, and permitting too many undermines the reliability of data interpretation.
We used two search engines, Byonic and Protein Prospector ‘combined’ to evaluate glycopeptide enrichment by lectin-affinity chromatography. Byonic was used to identify N-linked glycopeptides, Protein Prospector was used to find O-glycosylation. As far as we know, presently Byonic [19] is the most promising search engine for glycopeptide identification from ETD data. This search engine considers only Asn residues located in consensus sequences, NX(S/T) (where X cannot be Pro), as potential modification sites. The software is able to combine a protein database with any glycan structure pool specified/generated by the user. Thus, it is inherently efficient in N-linked glycopeptide analysis. Protein Prospector (http://prospector.ucsf.edu, [20]) cannot compete with Byonic in the N-glycosylation field for a number of reasons: (1) introducing/ creating new glycan structures is not as straightforward as in the other program; (2) all Asn residues are considered as potential modification sites. This latter feature is a definite shortcoming; however, it permits the identification of glycopeptides featuring the less frequent NXC glycosylation motif [21, 22] that Byonic cannot do without human intervention. The two search engines perform similarly in O-glycosylation analysis, since there is no consensus motif for O-glycosylation that could narrow down the number of potential modification sites to be considered. Both software handle the searches with ETD and HCD data separately; neither of them is capable combining the ‘two halves’ of the information provided by collisional activation and radical fragmentation. In addition, neither of them deciphers/interprets glycan fragmentation data, except that the newest version of Byonic indicates the sialic acid loss(es) in ETD.
Here we report a side reaction, the amidation of sialic acid that we encountered upon manual evaluation of glycopeptide ETD data, and the ‘culprit’ was Tris, the buffering agent recommended for the chromatography and widely used in proteomic experiments. Our findings demonstrate that using ETD spectra alone may lead to the misinterpretation of glycopeptide data. We also present data indicating the presence of Tris-amidated glycoforms in an earlier described large glycopeptide dataset [22], which indicates that this could be a widespread problem. These observations underline the need for improved bioinformatic tools for glycopeptide data interpretation. We propose the combined use of ETD/HCD data, obviously with utilizing the glycan fragmentation as well, and the ‘validation’ of the precursor ion cluster (i.e., the confirmation of the identity of the monoisotopic ion). In addition, incorporating chromatographic retention time information also may aid accurate structural assignments.
Experimental
Glycopeptide Enrichment by Lectin-Affinity Chromatography
A human serum tryptic digest was injected onto a 2 mm× 250 mm column packed with wheat germ agglutinin (WGA) immobilized on POROS Al resin [15]. After introducing the sample, the column was washed with WGA buffer (100 mM Tris pH 7.5, 150 mM NaCl, 2 mM MgCl2, 2 mM CaCl2, 5% acetonitrile; flow rate:125 μL/min) and a 100 μL plug of 200 mMN-acetyl-D-glucosamine (GlcNAc) in WGA buffer was injected at 12 min. Three-minute fractions were collected between 9 and 24 min. Each fraction was acidified, desalted, and dried down. Altogether, 12 mg of peptide sample was enriched in four rounds, then combined and subjected to a second round of enrichment in the same fashion as described above.
Mass Spectrometry
The isolated glycopeptide mixture was analyzed by LC-MS using a nanoflow RP-HPLC on-line coupled to a linear ion trap-Orbitrap (Orbitrap-Elite; Thermo Fisher Scientific, Bremen, Germany) mass spectrometer operating in positive ion mode.
Data acquisition was carried out in data-dependent fashion; the three most abundant, multiply charged ions were selected from each MS survey for MS/MS analysis. ‘Beam-type’ CID (HCD) data were acquired for each precursor (normalized collision energy: 35), whereas ETD data acquisition was triggered by the presence of diagnostic sugar oxonium ions 204.0867 (for N-acetylhexosamine, HexNAc) or 366.1395 (for hexosyl N-acetylhexosamine, HexNAcHex); among the 50 most abundant fragments of the HCD spectrum, the mass accuracy requirement was 15 ppm. MS and HCD spectra were acquired in the Orbitrap, and ETD spectra in the linear ion trap. For ETD experiments, fluoranthene was used as reagent and 200/z ms (z: precursor charge) activation time was applied. Supplemental activation (normalized collision energy: 15) for the ETD experiments was enabled.
Data Interpretation
Proteome Discoverer (Thermo Scientific, v1.4.0.288) was used to generate separate HCD and ETD peak lists from the raw data.
The ETD peak list was searched using the Byonic software (v1.3beta-150, Protein Metrics Inc., San Carlos, CA, USA) with the following parameter set: tryptic peptides with maximum two missed cleavages, allowing nonspecific cleavage at 1 terminus; mass accuracies within 10 ppm for precursor ions and 0.4 Da for fragment ions. Fixed modification was carbamidomethylation of Cys residues. Variable modifications were Met oxidation; cyclization of N-terminal Gln residues; and N-glycosylation. For N-glycosylation, the ‘common human’ glycan database, including ~400 glycan masses was selected. Acceptance criteria: 1% false discovery rate on the protein level.
The ETD data were also searched against the subset of proteins confidently identified by Byonic using Protein Prospector (v5.10.10) with the following parameters: trypsin with maximum two missed cleavages; mass accuracies of 5 ppm for precursor ions and 0.8 Da for ETD fragment ions; fixed modification: carbamidomethylation of Cys residues; variable modifications: acetylation of protein N-termini; Met oxidation; cyclization of N-terminal Gln residues and undefined modifications of 203–3000 Da on Ser/Thr/Asn with maximum two variable modifications per peptide. Acceptance criteria: minimum scores: 22 and 15; maximum E values: 0.01 and 0.05 for protein and peptide identifications, respectively.
Results
Unexpected O-Linked Glycan on a Human Serum Protein?
Glycopeptides were enriched from human serum using wheat germ agglutinin (WGA) lectin-affinity chromatography. The search engine Byonic was used to identify N-linked glycopeptides in the mixture (data not shown). As WGA has been shown to bind a wide variety of carbohydrate structures [22], we wanted to investigate whether unexpected N-glycan structures and any O-linked glycosylation were detected on the already identified glycoproteins. Hence, ETD data were also searched using the search engine Protein Prospector against the subset of proteins identified using Byonic, and undefined mass modifications of 203–3000 Da were permitted on Ser/Thr/Asn. Only a few O-linked glycopeptides were identified in the mixture. Interestingly, for inter-alpha-trypsin inhibitor heavy chain H1 (ITI H1, Uniprot ID: P19827) protein, the very same aminoacid sequence was identified with three different modifications (Table 1).
Table 1.
Inter-Alpha-Trypsin Inhibitor Heavy Chain H1 Peptides with Different Mass Modifications (Identification from ETD data)
| m/z | z | Peptide | RT(min) | Score | Expect | |
|---|---|---|---|---|---|---|
| Peptide 1 | 890.4311 | 3 | TFVLS(511.19)ALQPSPTHSSSNTQR | 15.617 | 34.4 | 0.0085 |
| Peptide 2 | 938.7789 | 3 | TFVLSALQPS(656.23)PTHSSSNTQR | 39.583 | 41.7 | 8.90E-05 |
| Peptide 3 | 973.1314 | 3 | TFVLSALQPS(759.29)PTHSSSNTQR | 35.433 | 29.9 | 0.0023 |
C-terminal z. fragments from z2 to z8 were observed in the ETD spectra of all three peptides suggesting that these three compounds are indeed related (Figure 1). Regarding peptide 2, the 656 Da-addition can easily be explained by a sialylated mucin core-1 structure, sialyl-galactosyl-N-acetylgalactosamine (SAGalGalNAc) that is a frequent modification on secreted proteins [1, 3]. The z. ion series along with a few c fragments confidently identify the modified sequence, and the site of modification can be restricted to Ser-10 or Thr-12 (Figure 1b). The 511 Da-modification on peptide 1 might correspond to a glycan of HexNAc1Hex1Fuc1 composition, and considering this structure the mass accuracy of the identified glycopeptide is within 0.03 ppm, well within the ±5 ppm limit afforded by the instrument during this analysis. This putative HexNAcHexFuc glycan matches two O-linked carbohydrate structures described earlier: Gal(β1,4)GlcNAc(β1,3)Fuc [23] or Fuc(α1,2)Gal(β1,3)GalNAc [1]. The presence of the former structure can be ruled out as this type of glycosylation occurs only on consensus motifs CXXGG(S/T)C of EGF domains [23]. The latter glycan sequence is a blood group antigen that may occur on secreted proteins [24]. However, the retention time of this peptide is much shorter than that of peptide 2 with the already deciphered sugar structure, and this is rather atypical for glycopeptides of the same peptide backbone with different carbohydrate structures of approximately the same size. Thus, we carefully inspected the MS/MS data of these related peptides. Upon HCD fragmentation, all three peptides yielded the HexNAc oxonium ion at m/z 204.087 and peptide 2 also featured fragments at m/z 292.102 and 274.092, characteristic for sialic acid (Figure 2). This peptide also produced extensive peptide fragmentation providing information mostly on the N-terminal half of the sequence in form of y and b fragments (Figure 2b). Peptide 1 yielded a much weaker HCD spectrum, and surprisingly enough the abundant a2-b2 ion-pair (m/z 221.129 and 249.123) was missing (Figure 2a). At the same time, most of the y fragments observed for peptide 2 were also detected in this spectrum, the HexNAc-modified y12 included. Interestingly, there is an abundant fragment ion, m/z 1910.0 in the spectrum that could be assigned as the unmodified y18. We rather assumed that this ion represents the unmodified full length peptide (i.e., we concluded that the two N-terminal amino acid residues are missing. Our conclusion was based on the relatively high abundance of this ion and its absence in the HCD of peptide 2 (Figure 2b), and on the common knowledge that O-glycopeptides produce the completely deglycosylated peptide ion upon collisional activation [25, 26]. In addition, the new a2 and b2 ions were detected albeit not very abundant (Figure 2a). Thus, the correct peptide sequence for peptide 1 is VLSALQPSPTHSSSNTQR, and the mass modification is 759 Da instead of the suggested 511 Da. The difference in the peptide structure also renders the large retention time difference for peptide 1 more agreeable. However, as far as the glycan structure is concerned, we are back to square one, except we have two peptides bearing the enigmatic 759 Da mass modification that cannot be explained by any commonly occurring human O-linked glycan structures. (The HCD spectrum of peptide 3—Figure 2c—indicates that the amino acid sequence assignment for peptide 3 was correct.)
Figure 1.

ETD spectra of (a) peptide 1, m/z 890.4311(3+); (b) peptide 2, m/z 938.7789(3+); and (c) peptide 3, m/z 973.1314(3+). Each spectrum was assigned as TFVLSALQPSPTHSSSNTQR, [642-661] of ITI H1, featuring different modifications (Table 1). The spectra are annotated according to the correct assignments that are: (a) VLSALQPS(SA(T)GalGalNAc)PTHSSSNTQR (b) TFVLSALQPS(SAGalGalNAc)PTHSSSNTQR (c) TFVLSALQPS(SA(T)GalGalNAc)PTHSSSNTQR The site of modification can be either Ser-10 shown above or Thr-12; positions are given in the full-length tryptic peptide. SA(T) indicates Tris-amidated sialic acid. ❖ Labels the precursor ions and their charged-reduced states. * Denotes the charge-reduced ions from co-eluting precursor ions of other charge state(s). Fragments labeled with asterisks are z+1 ions
Figure 2.

HCD spectra of (a) peptide 1, m/z 890.4311(3+); (b) peptide 2, m/z 938.7789(3+); and (c) peptide 3, m/z 973.1314(3+) identified with the same peptide sequence, TFVLSALQPSPTHSSSNTQR, but bearing different sugar structures (Table 1). The spectra are annotated according to the correct assignments that are: (a) VLSALQPS(SA(T)GalGalNAc)PTHSSSNTQR (b) TFVLSALQPS(SAGalGalNAc)PTHSSSNTQR (c) TFVLSALQPS(SA(T)GalGalNAc)PTHSSSNTQR; except, the site of modification can be either Ser-10 shown above or Thr-12; positions are given in the full-length tryptic peptide. SA(T) indicates Tris-amidated sialic acid. G indicates fragment ions carrying the core GalNAc. * Indicates NH3 loss from the respective sequence ion
Deciphering the 759 Da Modification
The HCD spectra of peptide 1 (Figure 2a) and also peptide 3 (Figure 2c) indicated unmodified peptide sequences. Thus, we had to assume that the modification occurred on the carbohydrate. It was reasonable to believe that something happened to the sialic acid since that is the only sugar unit featuring a differently reactive functional group. Peptide 2 with the ‘normal’ trisaccharide displayed the expected sialic acid-diagnostic fragments both in HCD (oxonium ions at m/z 292 and 274, Figure 2b) and in ETD (charge-reduced sialic acid loss at m/z 1262(2+), Figure 1b) [12]. While the sialic acid oxonium ions were not detected in the HCD spectra of the other peptides, the above-mentioned ‘neutral loss’ ion was observed in the ETD spectrum of peptide 3 (Figure 1c), and the corresponding, but obviously 2 amino acid shorter ion was detected for peptide 1 (m/z 1138(2+), Figure 1a), i.e., the residue mass of the sialic acid was implemented with 103 Da. Derivatization of the sialic acid with a 103-Da mass increase is also supported by HCD data: a series of ions (m/z 395 and 377) were detected in the MS/MS spectra of peptides 1 and 3 (Figure 2a and c) that could not be explained by peptide fragmentation and represent a 103-Da mass shift to the sialic acid oxonium ions observed in HCD of peptide 2 (m/z 292 and 274) (Figure 2b).
After careful screening of all the chemicals used during sample preparation, we concluded that the commonly used buffer ingredient Tris must be responsible for this derivatization. Mass accuracies of the precursor ions and HCD fragment ions also support our hypothesis for Tris-induced side reaction. Derivatization of the carboxylic group of sialic acids by Tris adds C4H9NO2 (monoisotopic mass: 103.0633) to the glycopeptides. The mass measurement errors of the precursor ions of peptides 1 and 3 are +2.7 and +3.3 ppm in comparison to the calculated values of the Tris-modified glycopeptides [m/z: 890.4260(3+) and 973.1314(3+), respectively] (Figure 3) and mass errors of the fragment ions attributed to the Tris-derivatized sialic acid (Figure 2a and c; theoretical m/z values: SA(T): 395.166, SA(T)-H2O: 377.155, T denoting Tris) are all within 3 ppm.
Figure 3.

The precursor ions of peptides 1 and 3 (10-scan average). The relative mass difference between the measured and calculated values (m/z: 890.4260 and 973.1314 for the shorter and full-length Tris-modified glycopeptides, respectively) are shown in the panels
Tris-Modified O-Glycopeptides Identified in Mouse Synaptosome Tryptic Digest
In order to find additional supporting evidence for the Trisderivatization of sialic acid, we have revisited our dataset acquired on glycopeptides isolated from mouse synapto-some. Glycopeptides in a mouse synaptosome tryptic digest were enriched by WGA lectin-affinity chromatography, the resulting mixture was fractionated off-line using high pH reversed phase HPLC, and then the collected fractions were analyzed by LC/MS/MS, with just ETD on an Orbitrap Velos mass spectrometer. The mass accuracy for precursors was within 5–7 ppm, and we searched the ETD data with 0.6 Da fragment mass error permitted [15].
Since the synaptosome protein mixture has been characterized during our previous experiments, it was known that numerous secreted and transmembrane proteins were present. Thus, a database search was performed against 423 such proteins, permitting unspecified modifications on Asn, Ser, and Thr residues up to 3000 Da. The resulting histogram of frequently occurring modifications identified a series of glycan structures, and this information was used for database searches with well defined structures. We published a significant part of our results [22]; however we also indicated in the paper that the mining of this dataset has not been completed.
Following our discovery of the sialic acid-Tris side reaction, we searched the above-mentioned synaptosome dataset for similar modifications, namely for 656 + 103, 947 + 103 and 947 + 2×103 Da mass modifications corresponding to the Tris-amidated sialyl- and disialyl mucin core-1 type glycans. Sixteen glycopeptides were identified, bearing a Tris-modified SAGalGalNAc structure, while seven glycopeptides were found with a disialo-GalGalNAc structure, where one of the sialic acids reacted with Tris (Table 2). Doubly modified disialo structures were not identified.
Table 2.
Tris-Amidated Sialyl- and Disialyl Mucin Core-1 Glycopeptides Identified from Mouse Synaptosome Preparation
| m/z | z | MHmes | MHcal | ppm | Sequence | Score | E |
|---|---|---|---|---|---|---|---|
| Peptides with GalNAcGalSA-TRIS-amide modification | |||||||
| 696.0357 | 3 | 2086.0915 | 2086.0900 | –1 | AEAS*IKPLVLASK | 36.3 | 0.0047 |
| 701.3181 | 4 | 2802.2489 | 2802.2476 | 0 | AGPEEVPT*AASSSHFHAGYK | 40.1 | 0.0018 |
| 1109.2417 | 3 | 3325.7095 | 3325.6924 | –5 | DLKPQPDIVLLPLPT*AYELDSTK | 32.3 | 6.20E-04 |
| 592.2780 | 4 | 2366.0885 | 2366.0915 | 1 | DLSVVPT*HGAMQHSK | 38.7 | 0.0041 |
| 670.5860 | 4 | 2679.3205 | 2679.3207 | 0 | EAGHSRLT*AQPLLEAAQK | 53.9 | 1.40E-05 |
| 765.6172 | 4 | 3059.4453 | 3059.4440 | 0 | FRPYHPEQRPT*TAAGTSLDR | 38.4 | 0.0067 |
| 760.1089 | 4 | 3037.4121 | 3037.4120 | 0 | FRPYNPEERPT*TAAGTSLDR | 43.8 | 9.70E-04 |
| 676.3311 | 3 | 2026.9777 | 2026.9802 | 1 | GKETT*FGVTLSK | 42.7 | 0.0057 |
| 725.3698 | 3 | 2174.0938 | 2174.0922 | –1 | GLT*TRPGSGLTNIK | 32.3 | 0.048 |
| 631.2930 | 3 | 1891.8634 | 1891.8655 | 1 | GPTFSAT*QAPR | 24.2 | 0.03 |
| 719.1028 | 4 | 2873.3877 | 2873.3833 | –2 | KPQAMHTGLPNPT*RPDTPR | 29.5 | 0.018 |
| 793.1401 | 4 | 3169.5369 | 3169.5383 | 0 | KQQLQEQS*APPSKPDGQLQFR | 45.0 | 1.40E-04 |
| 761.0398 | 3 | 2281.1038 | 2281.1068 | 1 | LGPAIKST*DVYTEK | 27.4 | 0.0087 |
| 938.7174 | 4 | 3751.8461 | 3751.8371 | –2 | MNHRDPLQPLLENPPLGPGVPT*AFEPR | 32.0 | 0.02 |
| 721.3731 | 3 | 2162.1037 | 2162.1074 | 2 | VRGPPAET*LLPPR | 28.1 | 0.05 |
| 704.5931 | 4 | 2815.3489 | 2815.3441 | –2 | VSEARPS*TMVVEHPEFLK | 43.2 | 2.80E-04 |
| Peptides with GalNAcGalSA,SA-TRIS-amide modification | |||||||
| 774.0908 | 4 | 3093.3397 | 3093.3430 | 1 | AGPEEVPT*AASSSHFHAGYK | 35.6 | 0.0047 |
| 700.0656 | 4 | 2797.2389 | 2797.2381 | 0 | ATTHNQPAT*VSHPETR | 34.3 | 0.015 |
| 743.3619 | 4 | 2970.4241 | 2970.4161 | –3 | EAGHSRLT*AQPLLEAAQK | 41.8 | 0.0012 |
| 838.3925 | 4 | 3350.5465 | 3350.5394 | –2 | FRPYHPEQRPT*TAAGTSLDR | 36.7 | 0.0059 |
| 773.3654 | 3 | 2318.0806 | 2318.0756 | –2 | GKETTFGVT*LSK | 32.2 | 0.0025 |
| 791.8751 | 4 | 3164.4769 | 3164.4787 | 1 | KPQAMHTGLPNPT*RPDTPR | 36.3 | 0.0021 |
| 777.3653 | 4 | 3106.4377 | 3106.4395 | 1 | VSEARPS*TMVVEHPEFLK | 39.4 | 0.002 |
All these—at least partly—‘neutralized’ sialic acid-containing glycoforms featured retention times 2–3 min shorter than the peptides bearing the corresponding underivatized glycans i.e., their chromatographic behavior was similar to that of peptide 3 (Table 1). Also, similarly to peptides 1 and 3, the amidated sialic acid remained prone to fragmentation in ETD. However, when both underivatized and modified sialic acids were present in these O-linked structures, usually only the 291 Da loss was detected (data not shown).
Tris-Modified Sialic Acid on N-Glycans
Originally very few sialic acid-containing N-linked glycopeptides were identified in this mouse synaptosome dataset [22]. However, in the process of further data evaluation utilizing the similar charge distribution profile, retention time information of the different glycoforms representing the same amino acid sequences as well as the common fragments in the ETD spectra, we managed to identify numerous additional glycoforms with more acidic structures included [22]. For example, the original 11-glycoform list for peptide VLGFKPKPPKN*ESLETYPLMMK from sodium/potassium-transporting ATPase subunit beta-1 (Uniprot ID: P14094) has been extended significantly. Among the newly discovered glycopeptides, there were neutral, mono-, di-, and trisialo-glycopeptides (data not shown), and a series of ‘mystery’ glycoforms which, based on the ETD spectra, clearly belonged to the series, but we had difficulties making sense of their masses. The ETD data of glycopeptide with precursor ion at m/z 1074.0882(5+) revealed the presence of a sialic acid, and indicated that neither of the methionines were oxidized (Figure 4b). Considering these constrains and the mass accuracy required (within 5 ppm) Glycomod in Expasy (http://web.expasy.org/glycomod/) listed only one potential N-linked glycan structure: Man3GlcNAc2 + Hex2HexNAc3Deoxyhexose3Pent2NeuAc1, contrary to our knowledge that mammalian N-linked structures do not feature pentoses [1]. However, if the sialic acid modification with Tris is considered, the modifying oligosaccharide can readily be identified as a core fucosylated, disialotriantennary complex glycan, a quite common modifier of mammalian proteins, which was also detected without derivatization (Figure 4a). The chromatographic behavior of this modified N-linked glycopeptide also reflected the ‘removal’ of an acidic group in the glycoform. The Tris-modified disialo-structure coeluted with the underivatized monosialo glycopeptides. For further illustration, Online Resource 1 shows the ETD spectra of the Tris-modified monosialo-glycoforms of this glycopeptide; while Online Resource 2 shows the HCD and ETD data of two human N-glycopeptides with Tris-modified glycan identified from our original human glycopeptide mixture.
Figure 4.

ETD spectrum of an N-linked glycopeptide and its Tris-modified counterpart. (a) VLGFKPKPPKN*ESLETYPLMMK modified with a HexNAc5Hex6FucSA2 glycan. The precursor ion was m/z 1053.4790(5+). (b) Same glycoform, except one of the sialic acids is modified by Tris. The precursor ion was m/z 1074.0882(5+). Both precursor ions are indicated with ❖. In the low mass region, the masses measured are listed in both spectra, but only the N-terminal assignments are shown in the upper panel, whereas the C-terminal fragments are indicated in the other spectrum. Both spectra feature the characteristic sialic acid losses, whereas the loss of the modified sialic acid was also detected from the respective glycan (m/z 1658.4). The entire mass range of both spectra was magnified by a factor of 2
Estimation of the Extent of the Side Reaction
Two approaches were used to estimate the extent of this side reaction. In order to estimate whether most sialoglycan structures were affected by the modification, HCD-data were ‘filtered’ and spectra representing glycopeptides, sialoglycopeptides, and Tris-sialoglycopeptides were identified relying on the presence of diagnostic fragment ions or fragment ion combinations. It was assumed that all spectra featuring the m/z 204.087 ion represent glycopeptides. Sialoglycopeptides were identified by the additional presence of m/z 274.092 and 292.103 fragments, whereas the presence of Tris-SA derivatives was indicated by m/z 377.155 and 395.166 (20 ppm mass error allowed for all fragment ions). We found that ~6% of all acidic glycopep-tides was present in Tris-amidated form.
In order to estimate what percentage of a sialoglycan was derivatized for any particular glycoform, we compared the peak areas calculated from the selected ion chromatogram of the representative ions of the ‘normal’ and modified glycoforms (Online Resource 3). It was found that the side product corresponds to approximately 10% of the unmodified structure.
Discussion
Proposed Mechanism for the Derivatization
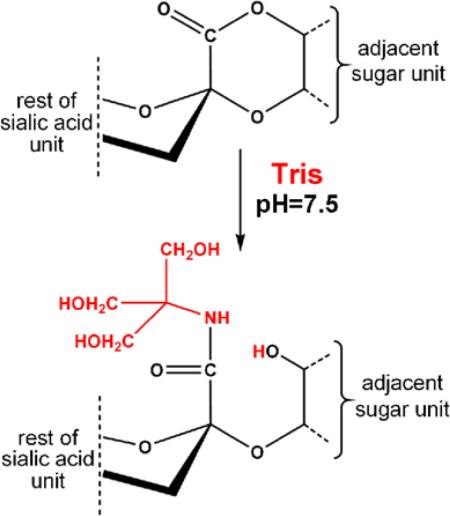
We attribute the presence of the 103 Da-‘shifted’ glycoforms to amidation of a sialic acid in the glycan by the common buffering agent Tris during sample preparation. This hypothesis is supported by the mass accuracy of the precursor ions as well as the modified sialic acid oxonium ions detected in the HCD experiments. In addition, the modified glycoforms eluted earlier than their unmodified counterparts during the LC/MS experiments (i.e., in a formic acid-containing mobile phase), while their retention time was longer in high pH reversed phase chromatography, both are clear indication for the presence of less acidic structures [2]. Conversion of free carboxylic acids into amides in dilute aqueous solutions is not expected. On the other hand, esters including lactones can readily be converted into the corresponding amides upon treatment with primary amines. Therefore, we assume that Tris amidates sialic acid lactones occurring either naturally or introduced during sample preparation (Figure 5).
Figure 5.

Proposed reaction scheme for the Tris-amidation of sialylated glycans
Lactone formation between the carboxyl group of sialic acids and adjacent hydroxyl groups may occur at physiological pH [1]. Lactonization of sialic acid-containing compounds, including oligosaccharides [27], gangliosides [28], and a synthetic glycopeptide [29], upon acid treatment has also been reported. Trace amounts of acid has been shown to be sufficient to induce the process [28]. Similar pH conditions persist during peptide extraction after in-gel digestion of proteins or C18 desalting of peptide samples.
‘Cumulative’ Difficulties in Glycopeptide Analysis
This side reaction offers a nice example to contemplate the difficulties that have to be tackled when ‘chasing’ glycopeptides. Variable site-occupancy, site-specific glycan heterogeneity, and rampant proteolytic activity in readily available body fluids including serum, which are frequently studied for glycosylation, all increase the complexity of the glycopeptide quagmire. Whatever MS/MS method is used for the glycopeptide analysis, the database search is hindered by the fact that heterogeneity is an inherent property of this PTM, and all the potential glycans have to be listed among the search parameters. Thus, either we have to have a complete glycan pool or allow the software to build the potential structures from the known ‘ingredients’—Byonic permits both options. However, since side chains usually do not fragment in ETD, glycopeptide assignment is based on the correct amino acid sequence identification and then on the mass difference between this sequence and the precursor mass measured. Unfortunately, MS/MS data frequently do not provide full sequence coverage. In addition, Pro residues prevent z. and c ion formation at their N-termini. Thus, based on ‘limited’ fragmentation, which is more of the rule than the exception, the peptide may be misidentified and, consequently, the glycan will be assigned incorrectly too.
Combining ‘Beam-Type’CID and ETD Data
The incorporation of information gained from the HCD spectra may prevent such misidentifications. The combination of ECD/CID and ETD/CID data has been proposed and utilized for more reliable sequence identification or phosphorylaton site assignments [30, 31]. Since the HCD data are rich in carbohydrate-related fragments, some information can be gained about the modifying glycan, at least the presence of certain saccharide units can be confirmed, while some structures can be excluded. For example, both sialic acid and fucose are prone to produce abundant neutral losses [32–34]; thus, the charateristic ions resulting from such fragmentation confirm their presence. In addition, as pointed out above, O-linked glycopeptides usually feature the abundant, fully deglycosylated peptide ion [26]. Similarly, N-linked glycopeptides produce an abundant ion that corresponds to the peptide with the core GlcNAc retained [2, 4]. Provided that fragment ions generated by collisional activation are measured with high resolution and high mass accuracy, a molecular mass confirmation for the amino acid sequence could be gained from the masses of the deglycosylated or GlcNAc-modified peptide ions. Obviously, whenever peptide fragmentation is also detected, that could make or break the ETD-based identification.
The Importance of Accurately Measured/Identified Monoisotopic Precursor Ions
During the characterization of complex protein mixtures, the necessity of high accuracy mass measurements on the MS1 level is inevitable. Since this mass accuracy is recently within the 5 ppm window, the question may arise why a peptide-level mass confirmation could improve glycopeptide identification. Partly because nonspecific cleavages, and fortuitous peptide- or glycan-mass altering modifications may occur, as presented here. Partly because the correct monoisotopic peak assignment still represents a challenge for the peak-picking programs whenever the monoisotopic m/z value has to be identified from an ion cluster representing a relatively large compound that is detected barely above noise level or is overlapping with some coeluting other species. Both of these situations may occur to glycopeptides somewhat more frequently than to unmodified compounds, (1) even just the N-linked core structure adds 892 Da to the peptide sequence modified; (2) the presence of numerous glycoforms makes the glycopeptide mixtures unusually rich in high mass, high charge state ions [13, 14, 16, 22]. Both Byonic and Protein Prospector allow the consideration of incorrect monoisotopic peak assigment as an option; however, this is a ‘double-edged sword.’ Unfortunately, the ‘incorrect precursor ion determination’ could also prove to be incorrect and that will result in data misinterpretation, especially with the low resolution and low mass accuracy of ion trap data. A potential solution to this problem could be a ‘feed-back’ checking mechanism, where the theoretical precursor ion cluster of the peptide tentatively identified could be compared with the precursor ion measured, and not just from a single MS survey, but perhaps from a few scans combined over a selected time range that could be determined from the selected ion extraction profile (as shown in Figure 3). Summing up MS data in such manner is already in practice, only it is used for quantification and not for improving mass accuracy, or for confirming the monoisotopic mass assignment. Thus, we suggest that such a step should be incorporated in the validation of database search results. Presently, groups that recognize this problem have to vote ‘for manual inspection of the correct monoisotopic peak’ [35].
Using All Available Information Combined
However, an accurate precursor mass alone will not solve all the problems. The precursor mass of peptide 1, computer-picked correctly from a single MS survey, fits the full tryptic sequence, TFVLSALQPSPTHSSSNTQR modified with a HexHexNAcFuc glycan within 0.3 ppm. MS/MS analysis usually clears up the matter, although, as mentioned above, ion trap ETD spectra with the low resolution and low mass accuracy have certain limitations. Obviously, with improved instrumentation the efficiency of ETD analysis can be improved and fragments can be measured in the Orbitrap with high resolution and mass accuracy. Interestingly, in our example this improvement might not solve the problem because most of the fragment ions detected fit perfectly well to the real as well as to the false structure. Thus, the HCD data were essential for solving this particular problem. In addition, we cannot forget that quite a few research groups still have to rely on lower quality, but high sensitivity ion trap data that combined with the information delivered by HCD still could yield the correct answer more unambiguously. In addition, HCD spectra could be instrumental in identifying metal-adducts, to confirm the presence of unexpected covalent modifications, and to decipher side reactions, just like the case presented here.
Our example also shows that readily available additional information, such as the chromatographic retention time may ‘flag’ some identifications as incorrect. In addition, for related structures, the changes in retention time may indicate the chemical nature of the alterations that occurred.
Conclusions
In summary, not only the functional groups of peptides but also the PTMs may participate in unforeseen chemical reactions during sample preparation, which further complicates the automated data interpretation process for high throughput proteomic analyses. Manual interrogation of the available MS/MS and chromatographic data combined permitted us to decipher a glycan modification. Dialog between investigative researchers and software developers could lead to the incorporation of similar combined strategies into search engines, which we believe would be highly beneficial for the field.
Supplementary Material
Acknowledgments
The authors gratefully acknowledge the contribution of Ralf Schoepfer and Jonathan Trinidad in the mouse synaptosome study, and thank Marshall Bern for performing the Byonic database search, Adam Kerenyi for writing the script used for HCD peak list filtering, and Zoltan Kupihar for useful discussions and technical assistance.
K.F.M. was supported by NIH grant NIGMS 8P41GM103481, and by the Howard Hughes Medical Institute (to the Bio-Organic Biomedical Mass Spectrometry Resource at UCSF, Director: A.L. Burlingame), and by the following grants: OTKA 105611 (to Z.D.), and BAROSS-DA07-DAESZK-07-2008-0036 (to the Biological Research Centre, HAS, director: P. Ormos). Z.D. was supported by the Janos Bolyai Fellowship of the HAS.
Footnotes
Electronic supplementary material The online version of this article (doi:10.1007/s13361-014-0852-9) contains supplementary material, which is available to authorized users.
References
- 1.Varki A, Cummings RD, Esko JD, Freeze HH, Stanley P, Bertozzi CR, Hart GW, Etzler ME, editors. Essentials of Glycobiology. 2nd edn. Cold Spring Harbor Laboratory Press; Cold Spring Harbor: 2009. [PubMed] [Google Scholar]
- 2.Medzihradszky KF. Characterization of protein N-glycosylation. Methods Enzymol. 2005;405:116–138. doi: 10.1016/S0076-6879(05)05006-8. [DOI] [PubMed] [Google Scholar]
- 3.Peter-Katalinić J. Methods in enzymology: O-glycosylation of proteins. Methods Enzymol. 2005;405:139–171. doi: 10.1016/S0076-6879(05)05007-X. [DOI] [PubMed] [Google Scholar]
- 4.Dodds ED. Gas-phase dissociation of glycosylated peptide ions. Mass Spectrom. Rev. 2005;31:666–682. doi: 10.1002/mas.21344. [DOI] [PubMed] [Google Scholar]
- 5.Settineri CA, Medzihradszky KF, Masiarz FR, Burlingame AL, Chu C, George-Nascimento C. Characterization of O-glycosylation sites in recombinant B-chain of platelet-derived growth factor expressed in yeast using liquid secondary ion mass spectrometry, tandem mass spectrometry and Edman sequence analysis. Biomed. Environ. Mass Spectrom. 1990;19:665–676. doi: 10.1002/bms.1200191106. [DOI] [PubMed] [Google Scholar]
- 6.Medzihradszky KF, Besman MJ, Burlingame AL. Structural characterization of site-specific N-glycosylation of recombinant human factor VIII by reversed-phase high-performance liquid chromatography-electrospray ionization mass spectrometry. Anal. Chem. 1997;69:3986–3994. doi: 10.1021/ac970372z. [DOI] [PubMed] [Google Scholar]
- 7.Stimson E, Hope J, Chong A, Burlingame AL. Site-specific characterization of the N-linked glycans of murine prion protein by high-performance liquid chromatography/electrospray mass spectrometry and exoglycosidase digestions. Biochemistry. 1999;38:4885–4895. doi: 10.1021/bi982330q. [DOI] [PubMed] [Google Scholar]
- 8.Schmitt S, Glebe D, Alving K, Tolle TK, Linder M, Geyer H, Linder D, Peter-Katalinic J, Gerlich WH, Geyer R. Analysis of the pre-S2 N- and O-linked glycans of the M surface protein from human hepatitis B virus. J. Biol. Chem. 1999;274:11945–11957. doi: 10.1074/jbc.274.17.11945. [DOI] [PubMed] [Google Scholar]
- 9.Hofsteenge J, Huwiler KG, Macek B, Hess D, Lawler J, Mosher DF, Peter-Katalinic J. C-mannosylation and O-fucosylation of the thrombospondin type 1 module. J. Biol. Chem. 2001;276:6485–6498. doi: 10.1074/jbc.M008073200. [DOI] [PubMed] [Google Scholar]
- 10.Zubarev RA, Horn DM, Fridriksson EK, Kelleher NL, Kruger NA, Lewis MA, Carpenter BK, McLafferty FW. Electron capture dissociation for structural characterization of multiply charged protein cations. Anal. Chem. 2000;72:563–573. doi: 10.1021/ac990811p. [DOI] [PubMed] [Google Scholar]
- 11.Syka JEP, Coon JJ, Schroeder MJ, Shabanowitz J, Hunt DF. Peptide and protein sequence analysis by electron transfer dissociation mass spectrometry. Proc. Natl. Acad. Sci. U. S. A. 2004;101:9528–9533. doi: 10.1073/pnas.0402700101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Darula Z, Medzihradszky KF. Affinity enrichment and characterization of mucin core–1 type glycopeptides from bovine serum. Mol. Cell Proteomics. 2009;8:2515–2526. doi: 10.1074/mcp.M900211-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Steentoft C, Vakhrushev SY, Vester-Christensen MB, Schjoldager KT, Kong Y, Bennett EP, Mandel U, Wandall H, Levery SB, Clausen H. Mining the O-glycoproteome using zinc-finger nucleaseglycoengineered SimpleCell lines. Nat. Methods. 2011;8:977–982. doi: 10.1038/nmeth.1731. [DOI] [PubMed] [Google Scholar]
- 14.Halim A, Nilsson J, Rüetschi U, Hesse C, Larson G. Human urinary glycoproteomics; attachment site specific analysis of N- and O-linked glycosylations by CID and ECD. Mol. Cell Proteomics. 2012;11(4):M111.013649. doi: 10.1074/mcp.M111.013649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Trinidad JC, Barkan DT, Gulledge BF, Thalhammer A, Sali A, Schoepfer R, Burlingame AL. Global identification and characterization of both O-GlcNAcylation and phosphorylation at the murine synapse. Mol. Cell Proteomics. 2012;11:215–229. doi: 10.1074/mcp.O112.018366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yin X, Bern M, Xing Q, Ho J, Viner R, Mayr M. Glycoproteomics analysis of the secretome of human endothelial cells. Mol. Cell Proteomics. 2013;12:956–978. doi: 10.1074/mcp.M112.024018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mikesh LM, Ueberheide B, Chi A, Coon JJ, Syka JE, Shabanowitz J, Hunt DF. The utility of ETD mass spectrometry in proteomic analysis. Biochim. Biophys. Acta. 2006;1764:1811–1822. doi: 10.1016/j.bbapap.2006.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Saba J, Dutta S, Hemenway E, Viner R. Increasing the productivity of glycopeptides analysis by using higher-energy collision dissociation-accurate mass-product-dependent electron transfer dissociation. Int. J. Proteomics. 2012;2012:560391. doi: 10.1155/2012/560391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bern M, Kil YJ, Becker C. Byonic: advanced peptide and protein identification software. Curr. Protoc. Bioinformatics. 2012 doi: 10.1002/0471250953.bi1320s40. Chapter 13:Unit13.20 doi:10.1002/0471250953.bil320s40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chalkley RJ, Baker PR, Medzihradszky KF, Lynn AJ, Burlingame AL. In-depth analysis of tandem mass spectrometry data from disparate instrument types. Mol. Cell Proteomics. 2008;7:2386–2398. doi: 10.1074/mcp.M800021-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sato C, Kim JH, Abe Y, Saito K, Yokoyama S, Kohda D. Characterization of the N-oligosaccharides attached to the atypical Asn-X-Cys sequence of recombinant human epidermal growth factor receptor. J. Biochem. 2000;127:65–72. doi: 10.1093/oxfordjournals.jbchem.a022585. [DOI] [PubMed] [Google Scholar]
- 22.Trinidad JC, Schoepfer R, Burlingame AL, Medzihradszky KF. N- and O-glycosylation in the murine synaptosome. Mol. Cell Proteomics. 2013;12:3474–3488. doi: 10.1074/mcp.M113.030007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Moloney DJ, Lin AI, Haltiwanger RS. The O-linked fucose glycosylation pathway. evidence for protein-specific elongation of O-linked fucose in Chinese hamster ovary cells. J. Biol. Chem. 1997;272:19046–19050. doi: 10.1074/jbc.272.30.19046. [DOI] [PubMed] [Google Scholar]
- 24.Rossez Y, Maes E, Lefebvre Darroman T, Gosset P, Ecobichon C, Joncquel Chevalier Curt M, Boneca IG, Michalski JC, Robbe-Masselot C. Almost all human gastric mucin O-glycans harbor blood group A; B or H antigens and are potential binding sites for Helicobacter pylori. Glycobiology. 2012;22:1193–1206. doi: 10.1093/glycob/cws072. [DOI] [PubMed] [Google Scholar]
- 25.Darula Z, Chalkley RJ, Lynn A, Baker PR, Medzihradszky KF. Improved identification of O-linked glycopeptides from ETD data with optimized scoring for different charge states and cleavage specificities. Amino Acids. 2011;41:321–328. doi: 10.1007/s00726-010-0692-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zauner G, Kozak RP, Gardner RA, Fernandes DL, Deelder AM, Wuhrer M. Protein O-glycosylation analysis. Biol. Chem. 2012;393:687–708. doi: 10.1515/hsz-2012-0144. [DOI] [PubMed] [Google Scholar]
- 27.Pan GG, Melton LD. Lactones of disialyl lactose: characterisation by NMR and mass spectra. Carbohydr. Res. 2006;341:730–737. doi: 10.1016/j.carres.2006.01.015. [DOI] [PubMed] [Google Scholar]
- 28.Bassi R, Riboni L, Sonnino S, Tettamanti G. Lactonization of GD1b ganglioside under acidic conditions. Carbohydr. Res. 1989;193(C):141–146. doi: 10.1016/0008-6215(89)85113-4. [DOI] [PubMed] [Google Scholar]
- 29.Pudelko M, Lindgren A, Tengel T, Reis CA, Elofsson M, Kihlberg J. Formation of lactones from sialylated MUC1 glycopep-tides. Org. Biomol. Chem. 2006;4:713–720. doi: 10.1039/b514918e. [DOI] [PubMed] [Google Scholar]
- 30.Nielsen ML, Savitski MM, Zubarev RA. Improving protein identification using complementary fragmentation techniques in fourier transform mass spectrometry. Mol. Cell Proteomics. 2005;4:835–845. doi: 10.1074/mcp.T400022-MCP200. [DOI] [PubMed] [Google Scholar]
- 31.Hansen TA, Sylvester M, Jensen ON, Kjeldsen F. Automated and high confidence protein phosphorylation site localization using com plementary collision-activated dissociation and electron transfer dissociation tandem mass spectrometry. Anal. Chem. 2012;84:9694–9699. doi: 10.1021/ac302364r. [DOI] [PubMed] [Google Scholar]
- 32.Darula Z, Chalkley RJ, Baker P, Burlingame AL, Medzihradszky KF. Mass spectrometric analysis; automated identification and complete annotation of O-linked glycopeptides. Eur. J. Mass Spectrom. 2010;16:421–428. doi: 10.1255/ejms.1028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hägglund P, Bunkenborg J, Elortza F, Jensen ON, Roepstorff P. A new strategy for identification of N-glycosylated proteins and unambiguous assignment of their glycosylation sites using HILIC enrichment and partial deglycosylation. J. Proteome Res. 2004;3:556–566. doi: 10.1021/pr034112b. [DOI] [PubMed] [Google Scholar]
- 34.Froesch M, Bindila L, Zamfir A, Peter-Katalinić J. Sialylation analysis of O-glycosylated sialylated peptides from urine of patients suffering from Schindler's disease by Fourier transform ion cyclotron resonance mass spectrometry and sustained off-resonance irradiation collision-induced dissociation. Rapid Commun. Mass Spectrom. 2003;17:2822–2832. doi: 10.1002/rcm.1273. [DOI] [PubMed] [Google Scholar]
- 35.Parker BL, Thaysen-Andersen M, Solis N, Scott NE, Larsen MR, Graham ME, Packer NH, Cordwell SJ. Site-specific glycan-peptide analysis for determination of N-glycoproteome heterogeneity. J. Proteome Res. 2013;12:5791–5800. doi: 10.1021/pr400783j. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.