

Abstract
We surveyed microsatellite distribution in 10 completely sequenced genomes. Using a permutation-based statistic, we assessed for all 10 genomes whether the microsatellite distribution significantly differed from expectations. Consistent with previous reports, we observed a highly significant excess of long microsatellites. Focusing on short microsatellites containing onlya few repeat units, we demonstrate that this repeat class is significantly underrepresented in most genomes. This pattern was observed across different repeat types. Computer simulations indicated that neither base substitutions nor a combination of length-dependent slippage and base substitutions could explain the observed pattern of microsatellite distribution. When we introduced one additional mutation process, a length-independent slippage (indel slippage) operating at repeats with few repetitions, our computer simulations captured the observed pattern of microsatellite distribution.
Microsatellites are short tandemly repeated sequence motifs consisting of 1-6 bp (Tautz 1993; Ellegren 2000b; Schlötterer 2000). Over the past decade, microsatellites have attracted considerable attention due to their involvement in some neurodegenerative diseases and their high polymorphism (Goldstein and Schlötterer 1999). Microsatellites gain and lose repeat units at high rates. The underlying mutation process has been termed “DNA replication slippage.” It is assumed that during DNA synthesis, the nascent strand dissociates and realigns out of register. When DNA synthesis continues, the repeat number at the microsatellite is altered at the nascent strand (Ellegren 2000b; Schlötterer 2000). Interestingly, the DNA replication slippage rate seems to be dependent on the length of the microsatellite. Alleles with a high repeat number are less stable than those with a small repeat number. This trend has been seen in pedigree studies (Brinkmann et al. 1998; Brohede et al. 2002), in vitro experiments (Shinde et al. 2003), and in surveys of population variability (Goldstein and Clark 1995; Bachtrog et al. 2000). In addition to this dependence on repeat count, microsatellite stability also depends on the repeat motif (Schlötterer and Tautz 1992; Chakraborty et al. 1997; Bachtrog et al. 2000). The major drawback of experiments aiming to characterize microsatellite mutational dynamics from direct observations of microsatellite mutations (e.g., pedigree analyses) is their limitation to microsatellites with a high mutation rate (and thus with a high repeat count). Population surveys could in principle also use shorter microsatellites, but here the observed microsatellite variability is not only a reflection of the microsatellite mutation rate, but also population history (Stumpf and Goldstein 2001) and selection (Harr et al. 2002a).
With the steadily increasing number of genomic sequences, an alternative approach to study microsatellite evolution has become feasible (Jurka and Pethiyagoda 1995; Bell and Jurka 1997; Cox and Mirkin 1997; Field and Wills 1998). Assuming that the distribution of microsatellites in the genome reflects an equilibrium state between the operating evolutionary forces, information about the underlying mutation processes could be extracted from the analysis of genomic sequences. Comparing the observed distribution of microsatellites to the expectations based on the random distribution of nucleotides conditional on their frequencies, it was noted that the distribution of microsatellites deviates from this simple Bernoulli model. The most obvious deviation from expectations was an overrepresentation of long microsatellites. Bell and Jurka (1997) used an unbiased random walk model to analyze this overrepresentation of long microsatellites. Their model incorporated two opposing forces operating on microsatellite sequences: (1) length-dependent DNA replication slippage, which results in a growth of repeats, and (2) base substitutions interrupting the repeat structure. Using the unbiased random walk model, those authors obtained a better fit for long microsatellites than the Bernoulli model.
Applying a Markov chain model of microsatellite evolution to a number of eukaryotic organisms, Kruglyak et al. (1998) were able to quantify the rates of replication slippage. Similar to Bell and Jurka (1997), they assumed that the slippage rate increases with the length of a repeat unit, and they combined this with a constant rate of base substitutions. By fitting observed microsatellite length distributions to the stationary length distribution under their model, the authors inferred a species-specific rate of replication slippage (Kruglyak et al. 1998). Although the combination of base substitutions and DNA replication slippage seemingly accounted for the lack of very long microsatellites, a refined model indicated that additional mutational forces must be incorporated to describe genomic microsatellite distributions (Calabrese et al. 2001). Evidence from yeast, D. melanogaster, and humans suggests that long microsatellite alleles have a downward mutation spectrum, which could result in a size constraint of microsatellites (Schlötterer 1998; Ellegren 2000a; Harr and Schlötterer 2000; Xu et al. 2000; Harr et al. 2002b).
The genomic distribution of long microsatellites has been studied in relation to functionally different components of the genome. Tri-nucleotide repeats are overrepresented in coding sequences, but less frequent than mono- and di-nucleotide repeats in noncoding regions (Tóth et al. 2000; Morgante et al. 2002). Furthermore, the microsatellite distribution seems to differ between intergenic and intronic sequences (Tóth et al. 2000; Morgante et al. 2002). In plants also, 5′ and 3′ UTRs differ in microsatellite density (Morgante et al. 2002). In Arabidopsis thaliana, low microsatellite densities are observed in the centromeric region containing many transposable elements (Schlötterer 2000). In D. melanogaster, the microsatellite distribution differs between X-chromosomes and autosomes (Bachtrog et al. 2000). These data suggest a significant heterogeneity in microsatellite distribution with respect to functionally different components of the genome.
Over the past years much has been learned about the complex mutational behavior of long microsatellites, but still very little is known about the mutational dynamics of short microsatellites. Early experimental results suggested that short microsatellites do not mutate by DNA replication slippage. In addition, the analysis of the complete genomic sequence of Saccaromyces cerevisiae found a close fit between the observed density of microsatellites below 8-10 bp and the density expected by the base composition (Rose and Falush 1998). Based on this close fit, the authors concluded that short microsatellites are highly stable (Rose and Falush 1998). Interestingly, another study used the same genomic sequence and concluded that even short microsatellites mutate by DNA replication slippage (Pupko and Graur 1999). Further evidence of instability of short microsatellites comes from a detailed analysis of the mutation spectrum in mice with a lacI transgene (Halangoda et al. 2001). Those authors observed an exponential increase of slippage mutations in mononucleotide runs consisting of 1-5 repeats, suggesting that short repeats mutate by DNA replication slippage. Interspecies comparisons also indicated that microsatellites with a small number of repeats also gain and lose repeat units (Schlötterer and Zangerl 1999; Webster et al. 2002). Further support for the gains and losses of repeat units has been provided in a seminal paper by Zhu et al. (2000). They analyzed the mutation spectrum in human genes resulting in a diseased phenotype. They found that new mutations were the most common events, which generated new microsatellites consisting of two repeat units. More important, however, they also observed that over 70% of the 2-4-bp insertions are duplications of adjacent sequences. Similar results were obtained from an analysis of pseudogenes. Nishizawa and Nishizawa (2002) found that between 50% and 80% of the indels are involved in a slippage-like process. Even short repeats, such as CCCC, have a 10-15-fold increased susceptibility to insertions and deletions compared to nonrepetitive sequences.
Thus, although some evidence suggests that short microsatellites are gaining/losing repeats, the current evidence is either derived from a biased sample set (Zhu et al. 2000, Nishizawa and Nishizawa 2002) or lacks statistical support (Pupko and Graur 1999). In the present study, we analyzed the genomic distribution of microsatellites and used a permutation procedure to evaluate the statistical significance of observed deviations from the expectation. Based on our analyses, we suggest that short microsatellites evolve by a slippage-like mutation process. In contrast to DNA replication slippage, this mutation process seems not to be length-dependent.
RESULTS
We determined the density of microsatellite repeats for nine different eukaryotes for which the complete genomic sequence is available. Four different microsatellite classes were analyzed, mono-, di-, tri-, and tetranucleotide repeats. Consistent with previous reports (Katti et al. 2001), we found dramatic differences among repeat classes within and between species (Table 1A, 1B, 1C, 1D). Such differences in microsatellite composition may be caused by (1) species-specific differences in the DNA synthesis and repair machinery (Harr et al. 2002b), (2) selection (Nauta and Weissing 1996), or (3) base composition (see below).
Table 1A.
Mononucleotide Repeat Microsatellite Densities (Microsatellites per 10 kb) in Different Genomes for Each Repeat Length (bp)
| Length | H. sap. | M. mus. | F. rub. | D. mel. | C. ele. | A. tha. | Rice 1a | Rice 2b | C. alb. | S. cer. |
|---|---|---|---|---|---|---|---|---|---|---|
| 2 | 1311 | 1312 | 1287 | 1357 | 1264 | 1362 | 1378 | 1397 | 1446 | 1422 |
| 3 | 436 | 425 | 404 | 426 | 409 | 414 | 374 | 381 | 435 | 401 |
| 4 | 144 | 135 | 123 | 140 | 187 | 145 | 125 | 128 | 141 | 130 |
| 5 | 50.0 | 39.9 | 39.6 | 46.7 | 85.7 | 47.0 | 39.5 | 41.0 | 50.2 | 43.1 |
| 6 | 14.5 | 9.9 | 11.0 | 13.0 | 34.7 | 14.0 | 12.7 | 12.3 | 16.0 | 14.0 |
| 7 | 5.58 | 4.00 | 5.21 | 3.88 | 13.19 | 6.38 | 6.19 | 5.94 | 6.65 | 5.81 |
| 8 | 2.02 | 1.56 | 2.55 | 1.80 | 6.55 | 2.93 | 3.58 | 3.51 | 3.52 | 2.21 |
| 9 | 1.16 | 1.08 | 1.23 | 1.23 | 4.71 | 1.85 | 1.84 | 1.86 | 2.65 | 1.04 |
| 10 | 0.754 | 0.777 | 0.817 | 0.901 | 2.237 | 1.202 | 1.018 | 0.935 | 2.342 | 0.631 |
| 11 | 0.468 | 0.456 | 0.507 | 0.541 | 0.602 | 0.589 | 0.336 | 0.322 | 1.209 | 0.401 |
| 12 | 0.364 | 0.325 | 0.351 | 0.360 | 0.249 | 0.327 | 0.186 | 0.167 | 0.789 | 0.292 |
| 13 | 0.318 | 0.252 | 0.245 | 0.233 | 0.117 | 0.207 | 0.112 | 0.090 | 0.417 | 0.201 |
| 14 | 0.283 | 0.193 | 0.174 | 0.164 | 0.068 | 0.148 | 0.067 | 0.053 | 0.280 | 0.108 |
| 15 | 0.251 | 0.148 | 0.122 | 0.122 | 0.045 | 0.115 | 0.043 | 0.031 | 0.159 | 0.073 |
| 16 | 0.215 | 0.110 | 0.085 | 0.088 | 0.030 | 0.092 | 0.026 | 0.019 | 0.106 | 0.054 |
| 17 | 0.175 | 0.077 | 0.062 | 0.053 | 0.024 | 0.067 | 0.015 | 0.012 | 0.055 | 0.044 |
| 18 | 0.140 | 0.053 | 0.046 | 0.035 | 0.018 | 0.050 | 0.012 | 0.007 | 0.039 | 0.023 |
| 19 | 0.119 | 0.041 | 0.032 | 0.022 | 0.010 | 0.038 | 0.008 | 0.005 | 0.016 | 0.029 |
| 20 | 0.101 | 0.034 | 0.021 | 0.018 | 0.008 | 0.030 | 0.005 | 0.003 | 0.015 | 0.018 |
Oryza sativa L. ssp. Indica.
Oryza sativa L. ssp. japonica.
Table 1B.
Dinucleotide Repeat Microsatellite Densities (Microsatellites per 10 kb) in Different Genomes for Each Repeat Length (bp)
| Length | H. sap. | M. mus. | F. rub. | D. mel. | C. ele. | A. tha. | Rice 1a | Rice 2b | C. alb. | S. cer. |
|---|---|---|---|---|---|---|---|---|---|---|
| 4 | 255 | 262 | 263 | 230 | 228 | 246 | 246 | 244 | 214 | 232 |
| 5 | 79.6 | 86.9 | 76.3 | 61.4 | 57.3 | 79.2 | 74.4 | 73.0 | 61.4 | 63.4 |
| 6 | 19.7 | 23.9 | 21.9 | 17.8 | 14.9 | 21.8 | 21.2 | 20.8 | 14.9 | 15.5 |
| 7 | 7.73 | 9.55 | 8.05 | 6.39 | 4.53 | 8.99 | 8.19 | 8.02 | 5.21 | 4.66 |
| 8 | 2.57 | 3.25 | 2.94 | 2.51 | 1.31 | 3.20 | 3.14 | 3.06 | 1.69 | 1.31 |
| 9 | 1.24 | 1.68 | 1.56 | 1.21 | 0.42 | 1.47 | 1.44 | 1.46 | 0.93 | 0.57 |
| 10 | 0.510 | 0.652 | 0.716 | 0.555 | 0.220 | 0.584 | 0.575 | 0.578 | 0.393 | 0.187 |
| 11 | 0.366 | 0.584 | 0.571 | 0.345 | 0.146 | 0.320 | 0.310 | 0.359 | 0.305 | 0.120 |
| 12 | 0.196 | 0.299 | 0.331 | 0.225 | 0.099 | 0.148 | 0.159 | 0.171 | 0.156 | 0.053 |
| 13 | 0.174 | 0.317 | 0.316 | 0.190 | 0.071 | 0.117 | 0.136 | 0.144 | 0.163 | 0.056 |
| 14 | 0.102 | 0.179 | 0.202 | 0.143 | 0.049 | 0.073 | 0.078 | 0.081 | 0.105 | 0.029 |
| 15 | 0.096 | 0.193 | 0.215 | 0.133 | 0.046 | 0.068 | 0.078 | 0.080 | 0.087 | 0.030 |
| 16 | 0.055 | 0.111 | 0.143 | 0.104 | 0.033 | 0.056 | 0.050 | 0.051 | 0.059 | 0.020 |
| 17 | 0.054 | 0.120 | 0.162 | 0.086 | 0.025 | 0.048 | 0.051 | 0.047 | 0.059 | 0.025 |
| 18 | 0.035 | 0.073 | 0.118 | 0.076 | 0.022 | 0.037 | 0.032 | 0.033 | 0.034 | 0.019 |
| 19 | 0.035 | 0.082 | 0.127 | 0.061 | 0.015 | 0.036 | 0.031 | 0.029 | 0.036 | 0.011 |
| 20 | 0.025 | 0.054 | 0.091 | 0.049 | 0.014 | 0.024 | 0.024 | 0.019 | 0.027 | 0.015 |
Oryza sativa L. ssp. Indica.
Oryza sativa L. ssp. japonica.
Table 1C.
Trinucleotide Repeat Microsatellite Densities (Microsatellites per 10 kb) in Different Genomes for Each Repeat Length (bp)
| Length | H. sap. | M. mus. | F. rub. | D. mel. | C. ele. | A. tha. | Rice 1a | Rice 2b | C. alb. | S. cer. |
|---|---|---|---|---|---|---|---|---|---|---|
| 6 | 91.5 | 89.7 | 102.8 | 89.9 | 88.1 | 98.8 | 104.2 | 104.2 | 110.4 | 103.7 |
| 7 | 27.8 | 27.3 | 31.8 | 28.1 | 25.5 | 33.6 | 35.8 | 36.0 | 39.7 | 32.5 |
| 8 | 8.86 | 8.98 | 11.35 | 11.30 | 9.08 | 13.06 | 14.37 | 14.57 | 17.90 | 11.74 |
| 9 | 2.82 | 2.52 | 3.71 | 3.98 | 3.02 | 4.60 | 5.08 | 5.20 | 6.62 | 3.84 |
| 10 | 1.03 | 0.95 | 1.43 | 1.71 | 1.07 | 1.97 | 2.31 | 2.40 | 3.25 | 1.43 |
| 11 | 0.485 | 0.579 | 0.781 | 1.103 | 0.613 | 1.113 | 1.274 | 1.332 | 2.314 | 0.790 |
| 12 | 0.185 | 0.215 | 0.324 | 0.474 | 0.270 | 0.505 | 0.583 | 0.608 | 1.071 | 0.265 |
| 13 | 0.114 | 0.130 | 0.208 | 0.261 | 0.155 | 0.280 | 0.374 | 0.387 | 0.721 | 0.132 |
| 14 | 0.116 | 0.174 | 0.182 | 0.235 | 0.124 | 0.226 | 0.289 | 0.280 | 0.665 | 0.115 |
| 15 | 0.048 | 0.069 | 0.084 | 0.126 | 0.078 | 0.122 | 0.175 | 0.193 | 0.395 | 0.050 |
| 16 | 0.031 | 0.053 | 0.069 | 0.084 | 0.047 | 0.086 | 0.132 | 0.129 | 0.331 | 0.049 |
| 17 | 0.050 | 0.080 | 0.077 | 0.094 | 0.036 | 0.068 | 0.112 | 0.110 | 0.361 | 0.055 |
| 18 | 0.019 | 0.031 | 0.037 | 0.053 | 0.024 | 0.046 | 0.072 | 0.071 | 0.225 | 0.023 |
| 19 | 0.014 | 0.024 | 0.034 | 0.036 | 0.016 | 0.031 | 0.052 | 0.051 | 0.180 | 0.009 |
| 20 | 0.024 | 0.041 | 0.039 | 0.045 | 0.010 | 0.032 | 0.046 | 0.042 | 0.179 | 0.029 |
Oryza sativa L. ssp. Indica.
Oryza sativa L. ssp. japonica.
Table 1D.
Tetranucleotide Repeat Microsatellite Densities (Microsatellites per 10 kb) in Different Genomes for Each Repeat Length (bp)
| Length | H. sap. | M. mus. | F. rub. | D. mel. | C. ele. | A. tha. | Rice 1a | Rice 2b | C. alb. | S. cer. |
|---|---|---|---|---|---|---|---|---|---|---|
| 8 | 27.0 | 30.0 | 26.4 | 28.1 | 22.9 | 27.1 | 29.3 | 29.1 | 27.2 | 22.4 |
| 9 | 9.00 | 9.95 | 8.23 | 9.36 | 6.91 | 9.03 | 9.33 | 9.31 | 9.10 | 6.54 |
| 10 | 3.23 | 4.17 | 2.78 | 3.59 | 2.33 | 3.23 | 3.52 | 3.49 | 3.50 | 2.19 |
| 11 | 1.57 | 2.13 | 1.45 | 1.66 | 1.03 | 1.33 | 1.53 | 1.52 | 1.62 | 0.74 |
| 12 | 0.515 | 0.795 | 0.473 | 0.608 | 0.356 | 0.467 | 0.564 | 0.563 | 0.652 | 0.197 |
| 13 | 0.277 | 0.394 | 0.234 | 0.317 | 0.172 | 0.220 | 0.261 | 0.259 | 0.338 | 0.073 |
| 14 | 0.187 | 0.281 | 0.146 | 0.202 | 0.084 | 0.111 | 0.153 | 0.150 | 0.235 | 0.038 |
| 15 | 0.297 | 0.442 | 0.191 | 0.138 | 0.052 | 0.070 | 0.114 | 0.104 | 0.181 | 0.019 |
| 16 | 0.098 | 0.154 | 0.075 | 0.069 | 0.029 | 0.031 | 0.062 | 0.056 | 0.122 | 0.013 |
| 17 | 0.071 | 0.096 | 0.050 | 0.048 | 0.022 | 0.019 | 0.037 | 0.036 | 0.079 | 0.011 |
| 18 | 0.067 | 0.087 | 0.037 | 0.036 | 0.016 | 0.013 | 0.029 | 0.028 | 0.069 | 0.010 |
| 19 | 0.120 | 0.165 | 0.069 | 0.025 | 0.007 | 0.007 | 0.027 | 0.024 | 0.059 | 0.008 |
| 20 | 0.0372 | 0.0587 | 0.0281 | 0.0201 | 0.0054 | 0.0038 | 0.0146 | 0.013 | 0.0442 | 0.0008 |
Oryza sativa L. ssp. Indica.
Oryza sativa L. ssp. japonica.
Influence of Base Composition on Microsatellite Density
We evaluated the potential influence of variation in base composition on the observed microsatellite density, by varying the GC content in a sequence between 0.2 and 0.8. Figure 1 indicates that base composition has a dramatic influence on microsatellite density. Both mono- and dinucleotide sequences have the lowest density at a balanced GC content. The expected number of microsatellites strongly increases with a more biased GC content. This effect becomes more pronounced for longer microsatellites and opposite for very short ones (data not shown). Given that the base composition differs substantially among species and also within genomes, any comparison of microsatellite densities also needs to account for local base composition.
Figure 1.

Correlation between expected microsatellite density and GC content for mononucleotide microsatellites (dashed line) and dinucleotide microsatellites (solid line). For both repeat classes, all microsatellites ≥9 bp were considered.
Statistical Evaluation of Microsatellite Densities
Assuming a random distribution of nucleotides in a given sequence (mononucleotide space), the expected number of microsatellites could be calculated analytically. The number of (CA)4 repeats in a 10-kb frequent with a balanced base composition would be  , where pa and pc are the frequency of A and C, respectively. When higher-order moments, such as a pronounced dinucleotide pattern (Gentles and Karlin 2001), are present in a sequence, the expectation for the number of microsatellites could be calculated as
, where pa and pc are the frequency of A and C, respectively. When higher-order moments, such as a pronounced dinucleotide pattern (Gentles and Karlin 2001), are present in a sequence, the expectation for the number of microsatellites could be calculated as 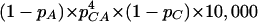 . Although both approaches have been pursued, they are somewhat unsatisfactory as they provide only an expectation for the microsatellite density, and no variance. Hence, it is not possible to determine whether an observed microsatellite density differs significantly from the expectations.
. Although both approaches have been pursued, they are somewhat unsatisfactory as they provide only an expectation for the microsatellite density, and no variance. Hence, it is not possible to determine whether an observed microsatellite density differs significantly from the expectations.
To overcome these limitations, we permuted the genomic sequences in 20-kb intervals. This strategy was chosen to account for heterogeneity in base composition within species (Lander et al. 2001). A certain repeat type was considered to deviate from expectations when the observed number of microsatellites fell outside of the 95% confidence interval determined by our permutation procedure. Irrespective of whether our permutations were based on a mononucleotide or a dinucleotide space, we observed substantial differences among repeat types and species. The different permutation schemes (permuting either single nucleotides or two adjacent bases) provided qualitatively similar results; therefore, we limit our discussion to the results obtained by those permutations assuming a mononucleotide space (Table 2, Supplemental Tables A1 and A2, available online at www.genome.org). Results based on the dinucleotide space are given in the Supplemental Tables A3 and A4.
Table 2.
Threshold of Overrepresentation a for Each Genome and Repeat Type
|
Mono
|
di
|
tri
|
||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| a/t | g/c | ac/gt | ag/ct | at/ta | cg/gc | aac/gtt | aag/ctt | aat/att | acc/ggt | acg/cgt | act/agt | agc/gct | agg/cct | atc/gat | ccg/cgg | |
| H. sap. | 3 | 9 | ≤4 | ≤4 | 8 | 13 | 11 | ≤6 | 10 | ≤6 | 15 | 16 | ≤6 | ≤6 | 9 | 11 |
| M. mus. | 7 | 10 | ≤4 | ≤4 | 12 | 12 | 13 | 13 | 14 | 11 | 14 | 14 | 12 | 11 | 13 | 12 |
| F. rub. | 3 | 5 | ≤4 | ≤4 | 10 | 13 | ≤6 | ≤6 | 8 | ≤6 | 14 | 14 | ≤6 | ≤6 | ≤6 | 7 |
| D. mel. | 3 | 6 | 5 | 9 | 6 | 13 | ≤6 | 8 | 8 | ≤6 | 9 | 14 | ≤6 | ≤6 | 10 | ≤6 |
| C. ele. | 3 | 10 | 10 | 10 | 15 | 10 | 14 | ≤6 | 16 | ≤6 | 12 | 14 | ≤6 | ≤6 | 13 | ≤6 |
| A. tha. | 3 | 11 | 7 | ≤4 | 8 | 12 | ≤6 | ≤6 | 16 | ≤6 | 12 | 14 | ≤6 | ≤6 | ≤6 | 7 |
| Rice 1b | 3 | 10 | 7 | 5 | 6 | 5 | ≤6 | ≤6 | 7 | ≤6 | 7 | 8 | ≤6 | ≤6 | ≤6 | ≤6 |
| Rice 2c | 3 | 4 | 7 | 5 | 6 | 5 | ≤6 | ≤6 | 7 | ≤6 | 7 | 8 | ≤6 | ≤6 | ≤6 | ≤6 |
| C. alb. | ≤2 d | ≤2 d | 7 | 9 | 11 | never | ≤6 | ≤6 | 10 | ≤6 | 11 | 9 | ≤6 | ≤6 | ≤6 | 11 |
| S. cer. | 3 | 11 | 12 | 13 | 13 | 11 | 13 | ≤6 | 16 | ≤6 | 12 | 14 | ≤6 | 13 | ≤6 | 11 |
Long microsatellites are overrepresented. We determined at which size a given repeat motif was more frequently detected than expected by chance (see Methods). Here we provide the number of bases at which we start to observe a significant overrepresentation.
Oryza sativa L. ssp. Indica.
Oryza sativa L. ssp. japonica.
Because our counting procedure was limited to a minimum of two repeats, we cannot make any inference about shorter repeats consisting of less than two repeats. For those cases in which we observed an overrepresentation at two repeat units, we used the “≤” symbol to indicate this uncertainty.
Overrepresentation of Microsatellites With a High Repeat Number
Previous results suggested that above a certain threshold repeat number, microsatellites are found more frequently in the genome than expected by chance (Rose and Falush 1998). Using our statistical approach, we were able to evaluate the statistical significance of such observations. Although some repeat motifs were overrepresented at all repeat numbers, most repeat types were only overrepresented beyond a certain threshold (Table 2; see Suppl. Tables A1 and A2 for more details). For some repeat types, such as (GC)n, no long microsatellites were detected in species with a small genome. In species with a larger genome, such as human and mouse, however, these repeats were present and also overrepresented.
Absolute Length in Base Pairs Versus Repeat Number
As we were comparing microsatellites with different repeat lengths (i.e., mono-, di-, tri-, and tetranucleotide repeats), we were interested whether the microsatellite distribution is better analyzed by the absolute length of the repeat structure (in bp) or by the number of repeats. Figure 2 shows the ratio of the observed and expected microsatellite density for H. sapiens (for the other genomes see Suppl. Fig. A1). Figure 2A is scaled by absolute length in base pairs, and Figure 2B is scaled by repeat number. For all species, the scaling by base pairs resulted in almost perfect parallel lines, and scaling by repeat number resulted in more pronounced differences. Thus, different microsatellites seem to show similar patterns of over- or underrepresentation when their absolute lengths in base pairs are compared. On the other hand, microsatellites with the same repeat number but different repeat lengths show drastically different patterns of over- or underrepresentation. For this reason, all of our representation plots are scaled by the size of the microsatellite stretch in base pairs.
Figure 2.

Ratio of the observed density and expected microsatellite density for H. sapiens. (A) Length scaled by absolute length (bp). (B) Length scaled by repeat number. Black lines, mononucleotide microsatellites; gray lines, dinucleotide microsatellites; (□) trinucleotide microsatellites, and (X) tetranucleotide microsatellites.
Deviations of Observed and Expected Microsatellite DensityAre Length-Dependent
Previous studies focused on microsatellites with a high repeat number, but we also analyzed microsatellites with only a small number of repeats. Although the patterns of over- and underrepresentation differed remarkably between repeat types and species, a general picture emerged (Fig. 3, Suppl. Fig. A2). In principle, three different zones could be distinguished in the representation plot. The first zone contains the very short microsatellites with the size of a few bp only. Microsatellites in this size class are often significantly underrepresented (see also Suppl. Tables A1 and A2). In the second zone, containing microsatellites of an intermediate size range (4-15 bp), the observed and expected microsatellite densities are either very similar or a slight overrepresentation is observed. Finally, the third zone is characterized by a marked overrepresentation of microsatellites. It is important to note that at the transition between zones two and three, the slope of the representation plot changes. For some repeat types, the transition to zone three is associated with an underrepresentation (Fig. 3, Suppl. Fig. A2). The overrepresentation of microsatellites in the longer size classes is consistent with previous studies and has been attributed to DNA replication slippage (Bell and Jurka 1997).
Figure 3.


Representation plots for mononucleotide microsatellites. Solid lines, observed densities; dashed lines, the expected densities. Black lines, A/T-microsatellites; gray lines with □, G/C microsatellites. (A) Homo sapiens, (B) Drosophila melanogaster, (C) Caenorhabditis elegans.
We used computer simulations to understand which genome dynamics could result in a microsatellite distribution that matches our observations in the three zones. Based on the large variation in microsatellite densities among repeat types and species, it is obvious that such computer simulations could only serve as an exploratory tool, and not as a systematic approach to estimate exact parameters. We assumed a random sequence with a balanced nucleotide composition, which experiences base substitutions (with equal transition probabilities) and slippage mutations. For computational simplicity we simulated a genome size of 2×107 bp and focused on mononucleotide repeats only, as this repeat type occurs at the highest density. The simplest model assumed only base substitutions (Table 3, Model 1). Hence, mutations could create new microsatellites and also destroy them. Figure 4A clearly indicates that this model corresponds to the expected microsatellite distribution and therefore fails to explain the observed distribution. The second evolutionary model incorporated base substitutions together with replication slippage using a length-dependent slippage rate (i.e., a linear increase with repeat number, Fig. 4B; Table 3, Model 2). This model did not match our observations in two aspects. First, the short microsatellites in the first zone were more underrepresented than in our genome survey. This result is consistent with previous publications, which also noted a large discrepancy in the observed distribution of short microsatellites (Kruglyak et al. 1998). Second, the slope of the representation graph does not change over the entire size range, whereas we observed at least one change between zone two and three. Therefore, this model does also not fully explain the observed genomic distribution of microsatellites. When this model was modified and slippage occurred only for microsatellites above a certain repeat number (Table 3, Models 3, 4, 6), a depletion of microsatellites shorter than the slippage boundary was detected (Fig. 4). One intuitive interpretation of this pattern is that microsatellites above the threshold have the tendency to grow, which results in the overrepresentation of microsatellites with a higher repeat number. At the boundary between slippage and no-slippage, this leads to an underrepresentation of this size class (as the threshold prevents the supply of shorter microsatellites). Nevertheless, this sequence evolution model still does not explain the underrepresentation of the size class of very short microsatellites. In an attempt to understand this, we introduced one additional mutation process. Rather than assuming a slippage process, the mutation rate of which is length-dependent, we introduced a process which we termed “indel slippage.” Contrary to the slippage process, we assumed indel-slippage to be not length-dependent, but to occur at a constant rate (Table 3, Model 5). This model is justified by the observation that insertions often copy the flanking sequence, which creates a short microsatellite (Zhu et al. 2000; Nishizawa and Nishizawa 2002). The representation plots for these computer simulations show the same trends observed for the genomic data (Fig. 4E). The first zone with microsatellite underrepresentation could be recognized. The second zone has an overrepresentation of microsatellites, but it is still close to the expectations, and zone three is characterized by the well described overrepresentation. Most important, the simulated data capture not only the change in slope in the representation graph, but also show an underrepresentation at the transition.
Table 3.
Simulation Parameters
| Minimum repeat number of μslip | μindel | |
|---|---|---|
| Model 1 | ∞ | 0 |
| Model 2 | 1 | 0 |
| Model 3 | 6 | 0 |
| Model 4 | 8 | 0 |
| Model 5 | 8 | 2.5 10−4 |
| Model 6 | 8 a | 0 |
Microsatellites with length 8 were not allowed to lose repeats by slipping. Thus, their slippage mutation rate was 0.5*μslip.
Figure 4.


Representation plot for simulation data. Solid lines, simulated densities; dashed lines, the corresponding “expected” densities. (A) model 1, (B) model 2, (C) model 3, (D) model 4, (E) model 5, (F) model 6 (see Table 3 for details).
Our computer simulations indicate clearly that base substitutions and DNA replication slippage alone are unlikely to explain the genomic distribution of microsatellites. More likely, other turnover mechanisms also need to be considered. Based on previous studies, we assumed an indel-slippage process and could obtain a qualitatively similar pattern. It may be needless to say that the dramatic divergence in pattern of microsatellite distribution also implies that different rates of the three processes and possibly other factors also shape the genomic distribution of microsatellites.
Microsatellite Distribution Differs Among Species
The base composition differs between species, and the number of expected microsatellites is dependent on the base composition. Thus, it is extremely difficult to compare microsatellite distributions across species. One obvious factor contributing to this difficulty is the base composition, which results in different expectations for the microsatellite density.
In an attempt to make the comparison of the mononucleotide microsatellite distribution across species more informative, we calculated for each species the difference between observed and expected microsatellite density, and we standardized this by the mean difference of all species analyzed. Figure 5 shows the relative distribution of mononucleotide repeat microsatellites between two and 20 bp. Most species show only moderate differences. C. elegans has significantly more microsatellites in the size class 3-10 bp than expected. C. albicans has a similar but less pronounced overrepresentation of microsatellites sized between 10 and 14 bp. The mononucleotide repeats longer than 14 bp were most abundant in humans. Interestingly, the microsatellite distribution in the two rice genomes was very similar and followed the same trends, but was nevertheless not identical. We also performed the same analysis for other repeat types and observed similar species-specific differences in microsatellite distribution (Suppl. Fig. A3).
Figure 5.

Relative density of mononucleotide microsatellites for all genomes plotted against repeat length. Relative densities were calculated by the comparison of a given species against the mean of all genomes (see text for details).
DISCUSSION
In our survey of microsatellite distribution in nine eukaryotic genomes, we did not discriminate between functionally different components of the genome. The reason for this is that several studies already demonstrated that the distribution of microsatellites differs substantially among exons, introns, 5′, and 3′ regions of a gene (e.g., Morgante et al. 2002). Our goal, however, was to obtain an overall pattern, rather than to distinguish between different parts of the genome.
Using our parameter-free permutation test, we showed that the pattern of microsatellite over- and underrepresentation differs among repeat types, microsatellite length classes, and species. To determine whether the observed microsatellite densities deviated significantly from expectations, we were very careful to determine the expectation correctly. We accounted for heterogeneity in base composition in a genomic sequence, by limiting our permutation test to small windows sized 20 kb. The simplest model to determine the expected number of microsatellites assumes independence of all nucleotides (Bernoulli model). Thus the expectation could be directly obtained from the frequencies of each nucleotide in the 20-kb window. In addition to this, we also considered higher-order interactions. Rather than permuting single bases, we permuted two neighboring bases. However, independently of the permutation procedure, we observed significant deviations from the expected microsatellite distribution, not only for long microsatellites, but also for the length class encompassing only two repeats. This observation suggested that other evolutionary forces must be operating, which determine the microsatellite distribution.
Consistent with previous studies, we found that long microsatellites were overrepresented. Interestingly, the length at which we observed a significant overrepresentation differed among repeat types and species. Assuming that this overrepresentation is caused by length-dependent DNA replication slippage, our observation may indicate that the rate of slippage differs among species and repeat types. Alternatively, the mutation pattern may differ among species (Harr and Schlötterer 2000; Harr et al. 2002b).
Although the slippage model assumes that long microsatellites are generated by a random walk of upward and downward mutations, which occasionally result in a high repeat number, alternative scenarios are possible, under which long repeats are generated instantaneously. An association of A-rich microsatellites with retrotransposable elements was observed (Nadir et al. 1996). Those authors suggested that A-rich microsatellites were generated by a 3′ extension of retrotranscripts, similar to mRNA polyadenylation. Similarly, the de novo generation of (GT)n microsatellites during nonhomologous repair of double-strand breaks has been described (Liang et al. 1998). To what extent such processes shape the genomic microsatellite distribution remains open to further investigation.
Highly surprising was our observation that the density of short microsatellites also deviated from expectations. Rather than a consistent overrepresentation, which may have been consistent with DNA replication slippage operating at short repeats, we observed a rather complex pattern. Some short repeats were significantly underrepresented, and others were overrepresented. Even more interestingly, several repeat type microsatellites of intermediate length either showed no or moderate overrepresentation. Building on seminal work of Zhu et al. (2000), we considered one additional mutation process to explain this pattern. Zhu et al. (2000) showed that indel-like processes tend to duplicate short sequences, and thus we termed this process indel slippage. Using computer simulations we showed that the combination of indel slippage with base substitutions and length-dependent DNA replication slippage could result in a microsatellite distribution that closely resembled our observations.
The distribution of microsatellites in protein coding regions was studied recently (Borstnik and Pumpernik 2002). For alanine codons, those authors observed that the abundance of repeats follows a curve with two different slopes. This is very similar to the pattern we observed for many repeat types. In contrast to our model, Borstnik and Pumpernik assumed only length-dependent slippage and base substitutions. Whereas we assumed complete neutrality, those authors also accounted for the higher order of the protein coding sequence. Thus, similar results regarding the distribution of microsatellite repeat types could be obtained using different assumptions. This result underpins the overall difficulty of extracting precise information about the mutation process of genomic sequences from their genomic distribution. Nevertheless, the availability of sequences from more closely related species will greatly facilitate future attempts to understand the evolution of tandemly repeated sequences. Based on such sequences, it will be possible to compare orthologous microsatellites in fully sequenced genomes (Webster et al. 2002).
METHODS
Sequences
Genome sequences were obtained in the FASTA format, and non-sequence information as well as ambiguous sequence information (e.g., N) was removed.
Arabidopsis thaliana (A. tha.) chromosomes were downloaded as pseudomolecules, which contain all available nonredundant sequences: ftp://ftp.tigr.org/pub/data/a_thaliana/ath1/PUBLICATION_RELEASE/PSEUDOMOLECULES/ (21.12.2000)
Saccharomyces cerevisiae (S. cer.), Drosophila melanogaster (D. mel.), and Caenorhabditis elegans (C. ele.) pseudomolecules were obtained from: ftp://ncbi.nlm.nih.gov/genomes/ and ftp://ncbi.nlm.nih.gov/genbank (17.03.2000)
Homo sapiens (H. sap.) pseudomolecules were downloaded from: ftp://ncbi.nlm.nih.gov/genomes/ (09.07.2001)
We obtained draft sequences from two different rice varieties, Oryza sativa L. ssp. indica (O.sat. indica or Rice 1) from http://btn.genomics.org.cn/rice (30.4.2002) and Oryza sativa L. ssp. japonica (O.sat. japonica or Rice 2) from a CD distributed by Syngenta Biotechnology (formerly the Torrey Mesa Research Institute; 3.6.2002).
The Fugu rubripes (F. rub.) unannotated draft genome assembly was obtained from: www.fugu-sg.org (25.10.2002).
Mus musculus (M. mus.) sequences were obtained from: ftp://ftp.ensembl.org/pub/current_mouse/data/fasta/dna/ (5.12.2002)
Candida albicans (C. alb.) sequence was downloaded from: ftp://ncbi.nlm.nih.gov/genomes/ (17.04.2002).
Calculation of Expected Microsatellite Density From Base Composition
The expected distribution of mononucleotide repeats could be directly obtained from the genomic frequency pa of the nucleotide forming the mononucleotide repeat:
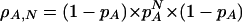 |
1 |
where N is the number of repeats.
This formula could be extended to dinucleotide repeats consisting of two bases A and C, which occur at frequency pa and pc. The expected frequency of dinucleotide repeats with a length of N base pairs could be calculated as
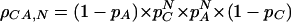 |
2 |
for even N, and
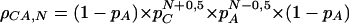 |
3 |
for odd N. (Kruglyak et al. 1998; Rose and Falush 1998).
Microsatellite Counts
We determined the number of microsatellite repeats by counting the number of bases forming a consecutive sequence stretch consisting of a single repeat type. This number was divided by the size of the repeat unit. Note that we also included noninteger repeat units (e.g., [CA]15.5). Higher-order motifs that could be decomposed into a lower-order motif were not considered (e.g., [CACA]7 was counted as [CA]14). Only those repeats consisting of a minimum of two repeats were counted. Interrupted repeat stretches were decomposed into multiple repeats without interruption. Juxtaposed microsatellite repeats consisting of two different repeat types were counted as two separate repeats. In cases in which the repeat units of such juxtaposed microsatellites share some bases, we resolved the repeat structure in the 5′ to 3′ direction (i.e., the sequence [A]10[AAT]5 would be resolved as two microsatellites [A]12 and [TAA]4.3).
Microsatellite densities are given in number of microsatellites per 10 kb. The C-code for counting microsatellite densities is available from the authors on request.
Permutation Tests
To evaluate whether the genomic microsatellite distribution deviates from the expectation based on a random genome composition, we used a permutation procedure. In nonoverlapping 20-kb windows, we permuted the original sequence and determined the microsatellite distribution. The permutation was limited to the 20-kb windows to account for heterogeneity in base composition within genomes. This procedure was chosen to account for the known local variation of the base composition over a genome (Lander et al. 2001). For each genome, 250 permutations were made. The genomic microsatellite distribution was considered to deviate significantly from randomness when an observed microsatellite density fell outside the 95% confidence interval obtained from 250 permutations. Note that for microsatellites with a low repeat count, this number of permutations is sufficient (data not shown). As microsatellites with a higher repeat count are much rarer, a higher number of permutations would be required to estimate the 95% confidence interval reliably.
The permutation procedure was modified to account for a higher-order structure in the genome (Gentles and Karlin 2001). Rather than permuting single bases, we permuted two adjacent bases.
Representation Plots
We used a representation plot to visualize the over-and underrepresentation of microsatellites over their entire size range. The expected microsatellite density and its confidence interval are obtained by permutations based on the observed mono- or dinucleotide space. Fewer than expected microsatellites fall below the diagonal, and more than expected microsatellites are located above the diagonal.
Genome Evolution Simulations
To determine the genomic microsatellite distribution when different mutational processes are operating, we analyzed the distribution of mononucleotide repeats in a sequence stretch of 20 Mb. The simulations were started with a random distribution of the four nucleotides all occurring at the same frequency. The entire genome was exposed to multiple rounds of mutation. The number of mutations occurring in each round is drawn from a Poisson distribution. The position of a mutation in the sequence is drawn from a uniform distribution. Transitions and transversions were equally likely.
DNA replication slippage mutations were added for each microsatellite length class separately. First the number of repeats (N) in each class was determined. The number of microsatellite mutations occurring in each length class was determined by a Poisson distribution with a mean of M.
 |
4 |
μslip is the per repeat unit mutation rate, N is the number of repeats, and l is the repeat number of the corresponding size class. We assumed that in one round of mutation, no more than a single slippage mutation occurs at a given microsatellite. Thus, from the total number of microsatellites in a given size class, we randomly chose one microsatellite for each microsatellite mutation. Insertion and deletions of single repeat units were selected with equal probabilities.
Indel slippage mutations were generated in a manner similar to that used for the replication slippage mutations. Only the mean of the Poisson distribution differed: M = M = μindelN, where N is the total number of microsatellites in the sequence stretch.
The simulations were discontinued after 10,000 rounds or when a stable microsatellite size distribution was reached. The C-code for these simulations is available from the authors on request.
Three different mutation processes were considered: base substitutions occurring at rate μbase (=10-4), length-dependent DNA replication slippage occurring at a rate μslip(=μslipbase * repeat number, μslipbase = 5 10-3), and indel slippage with the mutation rate μindel, which is length-independent. Slippage mutations followed the strict stepwise mutation model, which results in the gain or loss of one repeat unit with equal probability. Table 3 provides an overview of the parameters used for the simulations
Acknowledgments
We thank the members of the CS Lab and Claus Vogel for discussion and three anonymous reviewers for their helpful comments on a previous version of this manuscript. This work was supported by Fonds zur Förderung der wissenschaftlichen Forschung (FWF) grants to C.S.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
[Supplemental material is available online at www.genome.org.]
Article and publication are at http://www.genome.org/cgi/doi/10.1101/gr.1416703.
References
- Bachtrog, D., Agis, M., Imhof, M., and Schlötterer, C. 2000. Microsatellite variability differs between dinucleotide repeat motifs-evidence from Drosophila melanogaster. Mol. Biol. Evol. 17: 1277-1285. [DOI] [PubMed] [Google Scholar]
- Bell, G.I. and Jurka, J. 1997. The length distribution of perfect dimer repetitive DNA is consistent with its evolution by an unbiased single-step mutation process. J. Mol. Evol. 44: 414-421. [DOI] [PubMed] [Google Scholar]
- Borstnik, B. and Pumpernik, D. 2002. Tandem repeats in protein coding regions of primate genes. Genome Res. 12: 909-915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brinkmann, B., Klintschar, M., Neuhuber, F., Huhne, J., and Rolf, B. 1998. Mutation rate in human microsatellites: Influence of the structure and length of the tandem repeat. Am. J. Hum. Genet. 62: 1408-1415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brohede, J., Primmer, C.R., Moller, A., and Ellegren, H. 2002. Heterogeneity in the rate and pattern of germline mutation at individual microsatellite loci. Nucleic Acids Res. 30: 1997-2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calabrese, P.P., Durrett, R.T., and Aquadro, C.F. 2001. Dynamics of microsatellite divergence under stepwise mutation and proportional slippage/point mutation models. Genetics 159: 839-852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakraborty, R., Kimmel, M., Stivers, D.N., Davison, L.J., and Deka, R. 1997. Relative mutation rates at di-, tri-, and tetranucleotide microsatellite loci. Proc. Natl. Acad. Sci. 94: 1041-1046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox, R. and Mirkin, S.M. 1997. Characteristic enrichment of DNA repeats in different genomes. Proc. Natl. Acad. Sci. 94: 5237-5242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellegren, H. 2000a. Heterogeneous mutation processes in human microsatellite DNA sequences. Nat. Genet. 24: 400-402. [DOI] [PubMed] [Google Scholar]
- Ellegren, H. 2000b. Microsatellite mutations in the germline: Implications for evolutionary inference. Trends Genet. 16: 551-558. [DOI] [PubMed] [Google Scholar]
- Field, D. and Wills, C. 1998. Abundant microsatellite polymorphism in Saccharomyces cerevisiae, and the different distributions of microsatellites in eight prokaryotes and S. cerevisiae, result from strong mutation pressures and a variety of selective forces. Proc. Natl. Acad. Sci. 95: 1647-1652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gentles, A.J. and Karlin, S. 2001. Genome-scale compositional comparisons in eukaryotes. Genome Res. 11: 540-546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldstein, D. and Schlötterer, C. 1999. Microsatellites: Evolution and applications. Oxford University Press, Oxford, UK.
- Goldstein, D.B. and Clark, A.G. 1995. Microsatellite variation in North American populations of Drosophila melanogaster. Nucleic Acids Res. 23: 3882-3886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halangoda, A., Still, J.G., Hill, K.A., and Sommer, S.S. 2001. Spontaneous microdeletions and microinsertions in a transgenic mouse mutation detection system: Analysis of age, tissue, and sequence specificity. Environ. Mol. Mutagen. 37: 311-323. [DOI] [PubMed] [Google Scholar]
- Harr, B. and Schlötterer, C. 2000. Long microsatellite alleles in Drosophila melanogaster have a downward mutation bias and short persistence times, which cause their genome-wide underrepresentation. Genetics 155: 1213-1220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harr, B., Kauer, M., and Schlötterer, C. 2002a. Hitchhiking mapping—A population based fine mapping strategy for adaptive mutations in D. melanogaster. Proc. Natl. Acad. Sci. 99: 12949-12954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harr, B., Todorova, J., and Schlötterer, C. 2002b. Mismatch repair driven mutational bias in D. melanogaster. Mol. Cell 10: 199-205. [DOI] [PubMed] [Google Scholar]
- Jurka, J. and Pethiyagoda, C. 1995. Simple repetitive DNA sequences from primates: Compilation and analysis. J. Mol. Evol. 40: 120-126. [DOI] [PubMed] [Google Scholar]
- Katti, M.V., Ranjekar, P.K., and Gupta, V.S. 2001. Differential distribution of simple sequence repeats in eukaryotic genome sequences. Mol. Biol. Evol. 18: 1161-1167. [DOI] [PubMed] [Google Scholar]
- Kruglyak, S., Durret, R.T., Schug, M., and Aquadro, C.F. 1998. Equilibrium distributions of microsatellite repeat length resulting from a balance between slippage events and point mutations. Proc. Natl. Acad. Sci. 95: 10774-10778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lander, E.S., Linton, L.M., Birren, B., Nusbaum, C., Zody, M.C., Baldwin, J., Devon, K., Dewar, K., Doyle, M., FitzHugh, W., et al. 2001. Initial sequencing and analysis of the human genome. Nature 409: 860-921. [DOI] [PubMed] [Google Scholar]
- Liang, F., Han, M., Romanienko, P.J., and Jasin, M. 1998. Homology-directed repair is a major double-strand break repair pathway in mammalian cells. Proc. Natl. Acad. Sci. 95: 5172-5177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgante, M., Hanafey, M., and Powell, W. 2002. Microsatellites are preferentially associated with nonrepetitive DNA in plant genomes. Nat. Genet. 30: 194-200. [DOI] [PubMed] [Google Scholar]
- Nadir, E., Margalit, H., Gallily, T., and Ben-Sasson, S.A. 1996. Microsatellites spreading in the human genome: Evolutionary mechanisms and structural implications. Proc. Natl. Acad. Sci. 93: 6470-6475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nauta, M.J. and Weissing, F.J. 1996. Constraints on allele size at microsatellite loci: Implications for genetic differentiation. Genetics 143: 1021-1032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishizawa, N. and Nishizawa, K. 2002. A DNA sequence evolution analysis generalized by simulation and the Markov Chain Monte Carlo method implicates strand slippage in a majority of insertions and deletions. J. Mol. Evol. 55: 706-717. [DOI] [PubMed] [Google Scholar]
- Pupko, T. and Graur, D. 1999. Evolution of microsatellites in the yeast Saccharomyces cerevisiae: Role of length and number of repeated units. J. Mol. Evol. 48: 313-316. [DOI] [PubMed] [Google Scholar]
- Rose, O. and Falush, D. 1998. A threshold size for microsatellite expansion. Mol. Biol. Evol. 15: 613-615. [DOI] [PubMed] [Google Scholar]
- Schlötterer, C. 1998. Are microsatellites really simple sequences? Curr. Biol. 8: R132-R134. [DOI] [PubMed] [Google Scholar]
- Schlötterer, C. 2000. Evolutionary dynamics of microsatellite DNA. Chromosoma 109: 365-371. [DOI] [PubMed] [Google Scholar]
- Schlötterer, C. and Tautz, D. 1992. Slippage synthesis of simple sequence DNA. Nucleic Acids Res. 20: 211-215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlötterer, C. and Zangerl, B. 1999. The use of imperfect microsatellites for DNA fingerprinting and population genetics. In DNA profiling and DNA fingerprinting (eds. J.T. Epplen and T. Lubjuhn), pp. 153-165. Birkhäuser, Basel, Switzerland.
- Shinde, D., Lai, Y., Sun, F., and Arnheim, N. 2003. Taq DNA polymerase slippage mutation rates measured by PCR and quasi-likelihood analysis: (CA/GT)n and (A/T)n microsatellites. Nucleic Acids Res. 31: 974-980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stumpf, M.P. and Goldstein, D.B. 2001. Genealogical and evolutionary inference with the human Y chromosome. Science 291: 1738-1742. [DOI] [PubMed] [Google Scholar]
- Tautz, D. 1993. Notes on the definition and nomenclature of tandemly repetitive DNA sequences. In DNA fingerprinting: State of science (eds. S.D.J. Pena, R. Chakraborty, J.T. Epplen, and A.J. Jeffreys), pp. 21-28. Birkhäuser Verlag, Basel, Switzerland. [DOI] [PubMed]
- Tóth, G., Gáspári, Z., and Jurka, J. 2000. Microsatellites in different eukaryotic genomes: Survey and analysis. Genome Res. 10: 967-981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webster, M.T., Smith, N.G., and Ellegren, H. 2002. Microsatellite evolution inferred from human-chimpanzee genomic sequence alignments. Proc. Natl. Acad. Sci. 99: 8748-8753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu, X., Peng, M., and Fang, Z. 2000. The direction of microsatellite mutations is dependent upon allele length. Nat. Genet. 24: 396-399. [DOI] [PubMed] [Google Scholar]
- Zhu, Y., Strassmann, J.E., and Queller, D.C. 2000. Insertions, substitutions, and the origin of microsatellites. Genet. Res. 76: 227-236. [DOI] [PubMed] [Google Scholar]
WEB SITE REFERENCES
- ftp://ftp.tigr.org/pub/data/a_thaliana/ath1/PUBLICATION_RELEASE/PSEUDOMOLECULES/; TIGR ftp-site for A. thaliana sequence data.
- ftp://ncbi.nlm.nih.gov/genomes/; NCBI genome data Web page.
- ftp://ncbi.nlm.nih.gov/genbank; NCBI GenBank Web page.
- http://btn.genomics.org.cn/rice; Genome database of Chinese Super Hybrid Rice.
- www.fugu-sg.org; The IMCB—FUGU Genome Project Web page.
- ftp://ftp.ensembl.org/pub/current_mouse/data/fasta/dna/; The Ensembl mouse genome project, current sequences.