

Abstract
The human pathogen Tropheryma whipplei is the only known reduced genome species (<1 Mb) within the Actinobacteria [high G+C Gram-positive bacteria]. We present the sequence of the 927,303-bp circular genome of T. whipplei Twist strain, encoding 808 predicted protein-coding genes. Specific genome features include deficiencies in amino acid metabolisms, the lack of clear thioredoxin and thioredoxin reductase homologs, and a mutation in DNA gyrase predicting a resistance to quinolone antibiotics. Moreover, the alignment of the two available T. whipplei genome sequences (Twist vs. TW08/27) revealed a large chromosomal inversion the extremities of which are located within two paralogous genes. These genes belong to a large cell-surface protein family defined by the presence of a common repeat highly conserved at the nucleotide level. The repeats appear to trigger frequent genome rearrangements in T. whipplei, potentially resulting in the expression of different subsets of cell surface proteins. This might represent a new mechanism for evading host defenses. The T. whipplei genome sequence was also compared to other reduced bacterial genomes to examine the generality of previously detected features. The analysis of the genome sequence of this previously largely unknown human pathogen is now guiding the development of molecular diagnostic tools and more convenient culture conditions.
Tropheryma whipplei is the bacterial agent of Whipple's disease, a spectacular chronic disease described in 1907 by Nobel laureate George Whipple (Whipple 1907). Whipple's disease is characterized by intestinal malabsorption leading to cachexia and death if appropriate antibiotic treatment is not given. T. whipplei resisted reproducible culture until grown in human fibroblasts in 2000 (Raoult et al. 2000). The bacterium is small (0.3 × 1.5 mm) and Gram-negative on staining (La Scola 2001). It possesses an atypical envelope and a thick cell wall. In culture, it exhibits giant rope-like structures similar to Mycobacterium tuberculosis. Very little is known of its physiology (La Scola 2001).
At the time that genome sequencing of T. whipplei was initiated, only five genes were identified: 16S rRNA, 5S rRNA, 23S rRNA, groEL, and rpoB (Wilson et al. 1991; Drancourt and Raoult 1999; Hinrikson et al. 2000; Maiwald et al. 2000). Phylogenetic analyses classified T. whipplei within the high G+C content Gram-positive bacteria class (Actinobacteria, including Mycobacterium tuberculosis and Mycobacterium leprae), albeit not closely related to any known genus (Drancourt et al. 2001; Marth and Raoult 2003). The T. whipplei reservoir is suspected to be environmental, as previous studies using PCR experiments revealed its presence in sewage water (Maiwald et al. 1999). In human beings, the bacterium is mostly seen within cells, but its strictly intracellular niche is still debated. T. whipplei has been observed within intestinal macrophages and circulating monocytes (Raoult et al. 2001a,b), while Fredricks and Relman (2001) reported extracellular metabolically active bacteria in the intestinal lumen. T. whipplei is able to multiply in acidic lysosome-like vacuoles in HeLa cells (Ghigo et al. 2002). In the laboratory, T. whipplei has only been cultured within eukaryotic cells. The first subcultures exhibited a slow doubling time of 17 d (Raoult et al. 2000), comparable to 14 d for M. leprae (Cole et al. 2001).
Genomes of intracellular/parasitic bacteria undergo massive reduction compared to their free-living relatives. Examples include Mycoplasma (Fraser et al. 1995) for Firmicutes (the low G+C content Gram-positive), Rickettsia (Andersson et al. 1998) for alpha proteobacteria, and Wigglesworthia (Akman et al. 2002) and Buchnera (Shigenobu et al. 2000) for gamma proteobacteria. With a genome size of less than 1 Mb, T. whipplei offers the prime example of genome reduction among Actinobacteria.
The sequencing of T. whipplei genome was undertaken as an efficient way to learn more about this largely unknown fastidious human pathogen, to guide the development of molecular diagnostic tools, and eventually suggest improved culture conditions. These new data also offer an opportunity to reevaluate the generality of features previously proposed to characterize reduced genomes.
RESULTS AND DISCUSSION
Genome Sequence and Predicted Functions
General Features
The 927,303-base pair (bp) circular genome of T. whipplei Twist exhibits 808 predicted protein coding genes and 54 RNA genes (Fig. 1A,B; Table 1). The average G+C content is 46%, by far the lowest among the genome sequences available for other high G+C content Gram-positive bacteria. Coding content is 85.6%. On a global scale, T. whipplei exhibits no detectable colinearity with any of its close relatives with much larger genomes such as M. leprae (1605 open reading frames, ORFs), Corynebacterium glutamicum (3040 ORFs), M. tuberculosis (3927 ORFs), and Streptomyces coelicolor (7897 ORFs; Supplementary Fig. S1, available online at www.genome.org). Predicted gene functions (Fig. 2) indicate that T. whipplei is relatively well equipped, with different biological functions compared to other bacteria with reduced genomes (<1 Mb).
Figure 1.


(A) Circular representation of the T. whipplei Twist genome (upper panel), and the alignment of the Twist and TW08/27 genomes (lower panel). The origin of replication was predicted to be near the dnaA gene, based on the conservation of dnaA-dnaN-recF gene cluster and the change of AT-skew signal. One leading strand displays a pronounced excess in T vs. A and a slight excess of G vs. C. The location of the oriC was verified by Southern-blot hybridization with a digoxigenin-labeled oriC probe onto the Not I restriction profile. In the upper panel, the outermost (1st) circle indicates the nucleotide positions. 2nd circle: The two chromosomal segments, one of which exhibits an inversion (see text). 3rd and 4th circles: The ORF locations on the plus and minus strands, respectively. Functional categories are color-coded (see Fig. 1B). 5th and 6th circles: tRNAs. 7th circle: The locations of three rRNAs are indicated by black arrows. 8th circle: The repeat locations for the two largest families. The most internal circle shows the AT skew (A-T/A+T) computed with a sliding window size of 10 kb. The lower panel shows the detailed structure of the chromosomal inversion (red arrows), flanked by repeated sequences coding for WND-domains (green arrows). Black arrows: WiSP protein-coding genes at the extremities of the inverted segments. (B) Linear view of the complete circular genome of T. whipplei. Arrows = genes, with color-coded functional categories; small red flags = TRNAs; open arrows = other RNAs; open boxes = repeats belonging to the two major categories.
Table 1.
General Features of the T. whipplei Genome
| Genome size | 927,303 bp |
| G + C content | 46.3% |
| Coding content | 85.6% |
| ORFs | 808 |
| Median size | 287 aa |
| Annotated | 535 (66%) |
| Best database match | 678 (84%) |
| High G + C gram-positive bacteria | 552 |
| Streptomyces coelicolor | 292 |
| Mycobacterium tuberculosis | 95 |
| Corynebacterium glutamicum | 72 |
| Mycobacterium leprae | 36 |
| Other species | 57 |
| Low G + C Gram-positive bacteria | 39 |
| Proteobacteria | 47 |
| Other bacteria | 19 |
| Archaca | 13 |
| Eucarya | 7 |
| Viruses (Equine herpesvirus 1) | 1 |
| ORFans | 130 (16%) |
| RNA | 54 |
| rRNA | 3 (1 set) |
| tRNA | 49 (20 species) |
| tmRNA | 1 |
| M1 RNA (RNase P) | 1 |
Figure 2.

Comparative analysis of the number of genes present in each functional category as defined by the COG database. For the sake of comparing bacterial genomes of largely different sizes, a percentage graph is provided in Suppl. Fig. S2.
Information Processing
T. whipplei exhibits a complement of information processing genes comparable to that found in other small parasitic bacteria (Fig. 2). The DNA polymerase III complex—the primary replication machinery—is composed of the alpha (dnaE), beta (dnaN), gamma-tau (dnaZX), and putative delta subunits, as found in Mycoplasma genitalium. T. whipplei lacks homologs for the DNA polymerase polC, the second and essential polymerase in Bacillus subtilis (Dervyn et al. 2001). In contrast, T. whipplei appears to have two DNA gyrases, each made of two subunits, as found in S. coelicolor, another Actinobacteria. T. whipplei's TWT006/TWT005 gene pair appears to be the orthologs of the gyrA/gyrB subunits common to Actinobacteria; their role is to eliminate positive supercoils at the replication fork. The second gyrase-like copy, TWT491/TWT494, are orthologous to S. coelicolor SCO5836/SCO5822 genes that probably encode topoisomerase IV (ParC/ParE); this system is involved in chromosome segregation. Interestingly, an alanine residue was found at position 81 of GyrA and at position 96 of ParC, at which serine residues are usually found. In Escherichia coli, mutations at these positions are associated with the acquisition of resistance to quinolone antibiotics (Drlica and Zhao 1997; Drlica 1999). Based on these data, T. whipplei is predicted to be resistant to quinolones. This was recently confirmed experimentally (Masselot et al. 2003).
T. whipplei contains two paralogous genes for chromosome partitioning protein ParA. One (parA) is apparently orthologous to those of Mycobacterium and Streptomyces. The other (parA2) is rather similar to plasmid-encoded parA in the actinomycete Rhodococcus erythropolis. In addition, T. whipplei exhibits two replicative DNA helicases, dnaC and pcrA; the latter is involved in plasmid rolling-circle replication and ultraviolet-induced damage repair in B. subtilis (Petit et al. 1998). T. whipplei contains three competence-related genes homologous to B. subtilis comEA, comEC, and comFC. T. whipplei might thus naturally take up DNA from its environment, although this remains to be confirmed. In addition, we identified two putative site-specific integrase/recombinase genes (xerC and xerD) that may be involved in the genome rearrangements discussed below.
Four RNA polymerase subunit genes were identified: alpha (rpoA), beta (rpoB), beta′ (rpoC), and omega (rpoZ). As for the translation apparatus, T. whipplei exhibits 53 ribosomal protein genes and 20 genes for aminoacyl-tRNA synthetases (aaRSs). All amino acids are represented among these aaRSs, except for glutamine and asparagine. The gatCAB operon for the glutamyl-tRNAGln amidotransferase found in the genome probably compensates for the lack of glutaminyl-tRNA synthetase (GlnRS) and asparaginyl-tRNA synthetase (AsnRS), as in Chlamydia trachomatis (Raczniak et al. 2001).
Energy and Small-Molecule Metabolisms
T. whipplei shows a limited complement of genes related to energy metabolisms. T. whipplei appears capable of producing energy by glycolysis, the pentose-phosphate cycle, and oxidative phosphorylation. All glycolysis genes were identified except for those encoding 6-phosphofructokinase (Pfk) and fructose-bisphosphate aldolase (Fba). In contrast, none of the genes corresponding to the tricarboxylic acid cycle was found. The lack of the two glycolysis enzymes is compensated for by the complete pentose phosphate pathway, which on one hand generates NADPH+H+, required for the biosynthesis of fatty acids, and on the other hand, the ribose-5-phosphate required for nucleic acids biosynthesis. The gene complement for oxidative phosphorylation is limited. We identified 21 genes corresponding to the components of complex III, IV, V, plus one NADH dehydrogenase gene (ndh). T. whipplei has a putative transport system for L-arabinose (and/or D-xylose). Given the abundance of these sugars in plants, this suggests a possible environmental origin for T. whipplei. Overall, these predicted metabolic pathways indicate that T. whipplei could use glucose, fructose, maltose, and L-arabinose as primary energy sources.
Among parasitic bacteria with reduced genomes, T. whipplei has the most complete biosynthetic pathways for purine and pyrimidine nucleotides, fatty acids, several cofactors, and other small molecules (Fig. 2). In contrast, a detailed comparison of T. whipplei with the M. tuberculosis metabolic pathways in the KEGG database indicates rather important deficiencies in amino acid metabolisms (Fig. 3). Biosynthetic pathways appear to be lacking nine amino acids (histidine, tryptophan, leucine, arginine, proline, lysine, methionine, cysteine, and asparagine). In addition, partial deficiencies are predicted for seven other amino acids (glutamate, glutamine, aspartate, threonine, valine, isoleucine, and phenylalanine). The lack of asparagine synthetase (AsnB) might not affect protein synthesis, because of the presence of the amidotransferase (GatCAB) as described above. To compensate for the other defective biosynthetic pathways, the missing amino acids must be obtained from the environment or the host. This might involve membrane transport systems such as ABC transporters for amino acids and peptides, two of which were identified in the T. whipplei genome sequence. This suggests that enriching the medium in amino acids might improve the growth of T. whipplei in laboratory culture (P. Renesto, N. Crapoulet, H. Ogata, B. La Scola, G. Vestris, J.-M. Claverie, and D. Raoult, in prep.).
Figure 3.
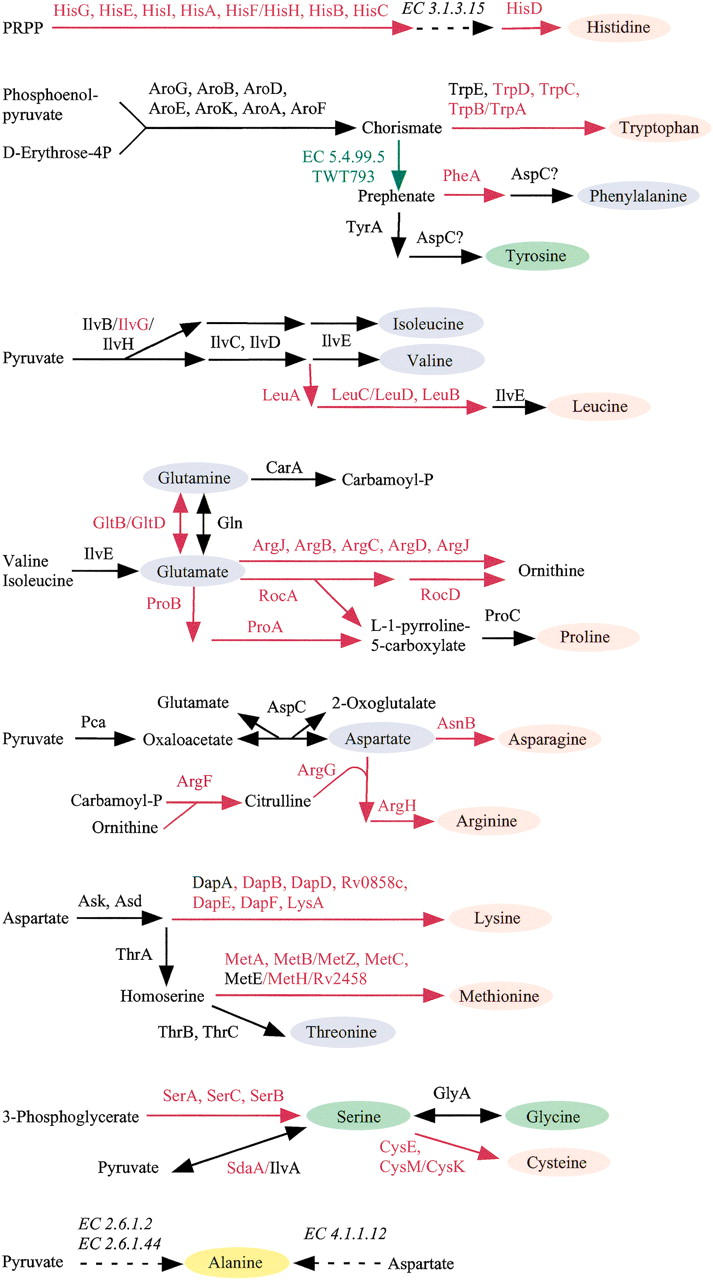
Predicted amino acid metabolisms of T. whipplei based on the M. tuberculosis metabolisms described in KEGG. Metabolic pathways were predicted to be lost for nine amino acids (pink ovals). Additional deficiencies were predicted for other seven amino acids (blue ovals). Metabolic pathways for three amino acids (green ovals) were retained. Enzymes for the alanine (yellow oval) biosynthesis were not identified in both M. tuberculosis and T. whipplei. Enzymatic steps predicted to be present in both M. tuberculosis and T. whipplei are shown in black. Enzymatic steps present only in M. tuberculosis or in T. whipplei are shown in red and green, respectively. Enzymatic steps absent in both M. tuberculosis and T. whipplei are shown with dashed black lines.
Both T. whipplei and Buchnera (Shigenobu et al. 2000) have retained approximately half of the amino-acid biosynthetic pathways. It is thought that Buchnera species specifically retained the pathways corresponding to amino acids essential for their insect hosts. As no such symbiotic association is known for T. whipplei, its residual biosynthetic capacity might point out the amino acids that are in the most limited supply from its environment or host.
T. whipplei lacks clear orthologs for thioredoxins (Trx) and thioredoxin reductase (TrxR) of M. tuberculosis, whose genome encodes three Trx (Rv3914, Rv1470, Rv1471) and one TrxR (Rv3913) in two operons. TrxR is a ubiquitous enzyme that reduces Trx, which in turn acts as electron donor in various essential redox reactions in the cell. TrxR- and Trx-encoding genes have been found in all bacterial genomes sequenced so far (Fig. 4). This includes Coxiella burnetii, which shares the same intracellular acidic vacuoles niche as T. whipplei. Because this would represent the first case of a bacterium without a functional thioredoxin system, this matter was investigated in greater detail. Protein motif searches using the PROSITE database entries (PS00194 for Trx; PS00573 for TrxR) failed to identify any ORFs with the sequence signatures in the T. whipplei genome. Another search using the TIGRFAM database (TIGR01068 for Trx; TIGR01292 for TrxR) again failed to identify any significantly similar sequences (above the noise cutoff). However, we further analyzed some candidates listed below this threshold. One was ORF TWT756, similar to B. subtilis thioredoxin-like protein gene resA. Another was ORF TWT210, exhibiting a significant sequence similarity to proteins of the pyridine nucleotide-disulfide oxidoreductases class II family. TWT210 might thus encode the thioredoxin reductase function, despite its weak similarity to known thioredoxin reductases. The glutaredoxin system serves similar roles as the thioredoxin system. M. tuberculosis has one copy of glutathione reductase gene (Rv2855), but the glutaredoxin system appears incomplete because of the lack of glutaredoxin genes. T. whipplei exhibits a distant homolog TWT629 to glutathione reductase, but shows no evidence of glutaredoxin genes. In conclusion, experiments are necessary to confirm that T. whipplei might be the first example of a bacterium without the—usually—essential thioredoxin pathway.
Figure 4.

Thioredoxin and glutaredoxin systems. Proteins in pink rectangles are predicted to be present in M. tuberculosis, for which only remote homologs are detected in T. whipplei. Glutaredoxin gene is absent in both T. whipplei and M. tuberculosis (see text). 2G-SH and 2G-S represent reduced and oxidized forms of glutathione, respectively.
Interaction With the Environment
We identified 40 genes for transporters, which probably comprise 10 to 15 different transport systems. These include two systems for amino acids transport, one for L-arabinose, two for iron, and one for phosphate. We identified two putative membrane proteins related to drug efflux proteins that may play a role in modulating antibiotic susceptibility. T. whipplei also exhibits a gene for dimethyladenosine transferase (ksgA), known to be related to kasugamycin resistance in E. coli. Those genes may help T. whipplei resist various antibiotics. A cold-shock protein gene capB was found, suggesting that the lifestyle of T. whipplei may include some cold periods and arguing against obligate human parasitism. The genes for two sensor histidine kinase/response regulator systems were also identified. Two protein translocation gene sets were identified, a Sec system and a twin-arginine translocase (TAT).
Finally, T. whipplei possesses whiA and whiB, two regulatory (possibly transcriptional) factors essential for the sporulation of S. coelicolor (Molle et al. 2000), and another whiB homolog (wblB). Spores have not yet been observed for T. whipplei, but the presence of these genes suggests that they may arise in environments not yet mimicked in laboratory culture conditions.
Gene Families
We identified 38 families of predicted gene paralogs (Suppl. Table S1). Of these, eight are lineage-specific families (exhibiting higher similarities within the family than to any other genes from other bacteria). They include WiSP membrane proteins (Bentley et al. 2003), other predicted membrane proteins, exodeoxyribonuclease III, inorganic pyrophosphatases, and iron ABC transporters.
Comparative Genome Analysis
The genome sequence of another T. whipplei isolate (strain TW08/27; GenBank: BX072543) recently became available (Bentley et al. 2003). The two genomic sequences of the different T. whipplei strains are mostly identical (>99% identity at the nucleotide sequence level), and encode quasi-identical gene complements. However, the Twist and TW08/27 genome organizations differ by the inversion of a large chromosomal segment (Twist coordinates: 182333–713982; Fig. 1A). Such inversions, approximately symmetrical to the origin of replication, are frequently observed in interspecific bacterial genome comparisons (Eisen et al. 2000; Hughes 2000; Makino and Suzuki 2001). However, such a difference between otherwise almost identical strains is an indication of a very active genome rearrangement process in T. whipplei. As already suggested for several Gram-negative species (Salmonella, Neisseira, Pseudomonas, and Bordetella), such genome rearrangement might be the consequence of the host-bacteria interaction (Hughes 2000). Interestingly, the boundaries of T. whipplei genome inversion are within the coding regions of two genes of the WiSP membrane protein family. More precisely, they involve two virtually identical nucleotide sequences, corresponding to the N-terminal WND-domains of the WiSP proteins. In the re combination process, the WiSP genes in the TW08/27 isolate (TW157, TW625) were significantly altered in the Twist isolate. The TW08/27 genome exhibits eight copies of WND-domain sequences that are up to 99% identical over an 800-nucleotide span. In contrast, the rest of these WiSP genes are all quite different. This strongly suggests that WND-domain sequences act both as coding regions and as DNA repeats promoting genome recombination. PCR experiments were performed to validate the genome organization of our Twist isolate, using primers precisely defining the boundaries of the inversion. While the experiments confirmed the Twist genome organization for both extremities of the inverted segment, a PCR product compatible with one extremity of the TW08/27-like arrangement was also observed. This intra-isolate variation, as well as the differences between the Twist and TW08/27 genomes, suggests that T. whipplei exhibits a large genomic plasticity mediated by the highly conserved WND-domain repeats. We also found a paralogous gene family for another type of predicted membrane spanning protein (TWT569, TWT580, TWT653) that exhibits highly conserved bipartite segments separated by a variable sequence.
It is tempting to speculate that the frequent genome rearrangements mediated by such “coding” repeats lead to significant changes in the set of proteins exposed at the surface of the bacteria, and might constitute an adaptive response to the host defense or various environmental conditions.
T. whipplei gene content was classified into functional categories (Fig. 2) and compared with the other bacteria with reduced (<1Mb) genomes (Mycoplasma species, Ureaplasma, Buchnera, and Wiggleworthia). Overall, T. whipplei exhibits a larger complement of genes for most functional categories compared to these other bacteria. However, Wiggleworthia exhibits more genes relevant to lipid metabolism, cell envelope biogenesis, the outer membrane, and coenzyme metabolism. For the categories of carbohydrate transport and metabolism and DNA replication and repair, Mycoplasma pulmonis is better equipped. Finally, Buchnera exhibits a larger gene complement for energy production and conversion. This variability indicates that small bacterial genomes are not the result of a universal and unique reductive evolution pathway.
Genomic Features of Reduced Genomes
A certain number of genome reduction “rules” have been enunciated, following the analysis of the available genomes of parasitic/intracellular bacteria. Characteristic features of reduced genomes may be associated with the adaptive strategies linked to their strict or facultative association with hosts (Koonin et al. 2001; Doolittle 2002). The genome sequence of T. whipplei provides the first opportunity to examine these rules for a high G+C Gram-positive bacteria.
G+C Content
Within each clade, reduced genome bacteria tend to exhibit the lowest G+C content. Rickettsia, Buchnera, Wigglesworthia, Mycoplasma, and Ureaplasma species are all in the range of 22%–33% except for Mycoplasma pneumoniae (40%). At 46%, the G+C content of T. whipplei is by far the lowest of the Actinobacteria, including M. leprae (58%), M. tuberculosis (66%), and S. coelicolor (72%). The cause for this general trend remains unknown, although it has been linked to a mutational bias due to the loss of repair and recombination functions (Moran 1996; Moran and Wernegreen 2000). Consistently, the genome of T. whipplei is lacking most of the base excision repair genes found in M. tuberculosis. We also noticed that the genes most conserved (ubiquitous) across distant clades tend to resist this trend. For example, the average G+C content of the T. whipplei ORFs for 53 ribosomal proteins is 50%, whereas that for all of the ORFs is 47%.
Horizontal Gene Transfer
Gene acquisition by horizontal transfer appears less frequent for small genome parasitic bacteria such as M. genitalium and Chlamidia (Ochman et al. 2000; Brinkman et al. 2002) than for free-living bacteria. One trivial explanation might be found in their less promiscuous lifestyles, offering much fewer opportunities to exchange DNA with other microorganisms. The analysis of two Rickettsia genomes (Ogata et al. 2001) confirmed this tendency by revealing the lack of horizontal gene transfer over a period of 40–80 million years. The reduced T. whipplei genome appears to follow the same rule. Potentially foreign genes can be identified by their atypical G+C content (Lawrence and Ochman 1998). In the T. whipplei genome, only two such ORFs are identified (TWT513 and TWT613). Indeed, the G+C content of the T. whipplei ORFs varies little (the standard deviation is 3%), as in other obligate intracellular bacteria (Brinkman et al. 2002). Horizontally transferred genes are also pointed out by inconsistencies in phylogenetic trees. This approach identified nine candidate genes likely to have been acquired by horizontal gene transfer (Suppl. Table S2), thus amounting to about 1% of the genome. Of these nine, five are aminoacyl-tRNA synthetases, including a valyl-tRNA synthetase of the archaeal-type, also observed in Rickettsia (Woese et al. 2000). Four of these anomalous phylogenies with different topologies are shown in Figure 5, for two aminoacyl-tRNA synthetases (AspRS, ValRS), and two enzymes (PurB, PyrB) of the nucleotide metabolism. It should be noted that some of those anomalous phylogenies might also originate from ancient gene duplication followed by lineage-specific loss of paralogs.
Figure 5.

Typical examples of anomalous phylogenies exhibited by T. whipplei genes. Trees were constructed using CLUSTAL W alignments and the neighbor-joining method (Thompson et al. 1994). Bootstrap values are indicated when larger than 80%. Other examples are provided in Supplemental Figure S3.
Repeated Sequences
Another proposed rule is that repeated sequences should be less frequent in the reduced genomes of parasitic bacteria (Frank et al. 2002). A possible cause is again that the sequestered lifestyle of these bacteria diminishes their exposure to foreign selfish DNA elements (bacteriophages and transposons). Concurrently, eventual repeated sequences are eliminated from these genomes by recombination. However, there are already some strong exceptions to that rule. M. pneumoniae exhibits a large number of repeats (Rocha et al. 1999). We observed many repeated sequences in an obligate intracellular parasite Rickettsia conorii (Ogata et al. 2002; Claverie and Ogata 2003). T. whipplei is another counter-example, with a high frequency of repeats. Thirty-eight repeat families were identified representing 6% of the T. whipplei genome. The two largest families account for 29,850 nt (3.22%) and 11,137 nt (1.20%) respectively (Fig. 1A,B). The family of the longest repeat consists of a pair of nearly identical sequences (99.91% identity over 4692 nucleotides). The positions of the repeats frequently correspond to abrupt changes in the AT-skew profile (Fig. 1A), suggesting their involvement in frequent local genome rearrangements (Petes and Hill 1988).
Genome Degradation
Finally, “on-going genome degradation” (Andersson and Andersson 2001; Lawrence et al. 2001) has been proposed to be induced by the sheltered and isolated lifestyle of parasitic bacteria. Accordingly, R. prowazekii (Andersson et al. 1998) and M. leprae (Cole et al. 2001) both exhibit a number of pseudogenes in their genomes, which appear under the process of elimination. However, this tendency is not so clear for other reduced genome bacteria such as Chlamydia. Moreover, two closely related Rickettsia exhibit very different levels of genome degradation (Ogata et al. 2001). Here, the T. whipplei genome exhibits a coding content (86%) comparable to the one of free-living bacteria, and shows few pseudogenes and little sign of on-going degradation.
Conclusion
An important finding derived from T. whipplei Twist (this work) and TW08/27 (Bentley et al. 2003) genome sequences is the existence of frequent genomic instability mediated by protein-coding repeats within genes of membrane proteins. This active recombination process probably causes the bacteria to expose different sets of proteins at their surface, in response to the host defense or various environmental conditions. At the same time, frequent recombinations homogenize the repeat sequences and maintain or amplify their capacity to be involved in forthcoming genome rearrangements.
A related phenomenon has been reported to explain the variation of the MSP2 outer membrane protein of Anaplasma marginale, a rickettsial pathogen. Gene conversions between functional msp2 genes and their pseudogenes (Brayton et al. 2001) have been invoked as the most likely mechanism. The msp2 genes also exhibit highly conserved DNA segments, as found in T. whipplei WiSP genes. UV-induced genomic inversions mediated by highly conserved lipoprotein-like ORF sequences were also recently reported (Uchida et al. 2003) in Streptomyces griseus, an environmental actinobacterium. Intra-ORF genome inversions might thus be a process generally used by Actinobacteria to modulate the expression of their surface proteins in response to their environment.
The T. whipplei genome sequence now provides important and practical information on a poorly characterized bacterium, isolated only three years ago. In an attempt to improve the current molecular diagnosis for Whipple's disease, new PCR primers were designed according to the sequence of the highly conserved WND-domain repeats. The primers (Tw53–3F: 5′- TGT GTC TGT GGT TGG GGT AA-3′ / Tw53–3R: 5′-CCT CCT GCT CTA TCC CTC CT-3′) were tested against diluted T. whipplei cultures, and detected 10 to 100 more cells than rpoB-based primers (Drancourt et al. 2001).
The prediction of the T. whipplei resistance to quinolone antibiotics, which was later confirmed (Masselot et al. 2003), suggested the avoidance of the use of these compounds in the treatment of Whipple's disease.
Finally, the detailed analysis of the predicted metabolism of T. whipplei suggests useful clues on how to rationally modify the current culture conditions and improve our capacity to grow and study this extremely fastidious bacterium in the laboratory (P. Renesto, N. Crapoulet, H. Ogata, B. La Scola, G. Vestris, J.-M. Claverie, and D. Raoult, in prep.).
METHODS
Source and Preparation of DNA
T. whipplei Twist strain, a type 2A, was cocultivated with HEL cells in 150-cm2 flasks as described (Raoult et al. 2000) to the 20th passage. Purified bacteria (analyzed using electron microscopy) were submitted to pressure shock using a French-pressure device (Bioritech), and lysed bacteria were mixed with 1% low melting point agarose (FMC Bio Products) in TE to form plugs in a mold at 37°C. DNA purity was assessed after logarithmic dilutions of purified T. whipplei DNA were subjected to a competitive PCR, targeting both human β-globin and a portion of the rpoB gene encoding the β-subunit of the RNA polymerase of T. whipplei. There was a 10:6 ratio between the concentrations of DNA of T. whipplei and that of the cells in which the organism was cultured. Digestion was performed by incubation of plugs in 25 mL of Tris-Sodium EDTA (6 mM Tris-HC1, pH 7.5, 1 M NaCl, 0.2 M EDTA, pH 8.0) and 1.25 mL of 10% sodium lauryl sarcosine solution (Sigma) to which 30 mg lysozyme (Boehringer Mannheim) was added and incubated at 37°C for 16 h on a roller. After two gentle washings in TE, the plugs were incubated overnight in a solution of EDTA-sarcosine-proteinase (30 mL 0.5 M EDTA, pH 8.5, 1.5 mL 10% sodium lauryl sarcosine, 60 mg proteinase K [Euromedex]) at 50°C. This operation was repeated three times, and the plugs were finally washed gently three times. Digested plugs were stored in EDTA 0.2 M, pH 8.0 at 4°C until used for cloning.
Genome Sequence Analysis
A first library A (5-kb inserts obtained by mechanic shearing, cloned in pcDNA 2.1 with Bst XI adaptors) was constructed, and a preliminary analysis of 96 clones disclosed <10-4 HEL cell DNA contamination. Plasmid clones were sequenced at both ends of the insert with flanking vector sequences as primers. Dye primer reactions were analyzed on an L1-COR 4200L, and six genomic equivalents were sequenced. We generated a second library B by cloning 20-kb fragments in pCNS with BstXI adaptors. Dye primer reactions were analyzed on a capillary ABI3700. The whole-genome assembly was performed by means of the PHRED, PHRAP, and CONSED 11.0 software packages (Ewing and Green 1998; Ewing et al. 1998; Gordon et al. 1998). A total of 6340 (6.34 X) and 5286 (2.93 X) end sequences (X's indicate genome equivalents) respectively, from libraries A and B were incorporated into contigs. Cloning gaps and several sensitive regions were resolved and confirmed by sequencing-duplicated PCR products. In addition, regions with lower quality were optimized using dye terminator reaction analyzed on a capillary ABI3100. The final sequence includes 99.9% positions with PHRED scores over 40. The coherence of the assembly was verified by comparison of the Spe I and Not I restriction site pattern previously obtained by pulsed-field gel electrophoresis. Also, the assembly was confirmed by sequencing PCR products obtained by incorporating primers designed on the basis of the scaffold of the molecule.
Informatics
Nucleotides in the T. whipplei genome were numbered according to the M. tuberculosis genome, where position one corresponds to the predicted origin of replication. Genes for tRNA were identified with the use of tRNAscan-SE (Lowe and Eddy 1997). Other RNAs were identified using BLAST (Altschul et al. 1997). ORFs at least 50 amino acid residues-long were predicted using SelfID (Audic and Claverie 1998). Additional ORFs shorter than 50 aa residues were identified by homology searches. ORFs overlapping with other ORFs were removed retrospectively. Repeated sequences were delineated using RepeatFinder (Volfovsky et al. 2001). ORF functions were predicted based on the homology searches by BLAST against the SWISS-PROT and TrEMBL sequence databases (Boeckmann et al. 2003). Metabolic pathways were analyzed using KEGG (Kanehisa et al. 2000). Preliminary sequence data for C. burnetii were obtained from The Institute for Genomic Research (http://www.tigr.org/). The functional classifications of the genes were performed in reference to the COG database (Tatusov et al. 2001) except for T. whipplei. COG categories for T. whipplei were determined by homology to COG database sequences.
Acknowledgments
We thank J. Weissenbach and the Genoscope team for shotgun sequencing and B. La Scola for helpful discussion. We thank C. Corona for her help in the preparation of the manuscript. This work was supported in part by a French Ministry for Health Grant (Programme Hospitalier de Recherche Clinique).
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Article and publication are at http://www.genome.org/cgi/doi/10.1101/gr.1474603.
Footnotes
[Supplemental material is available online at www.genome.org. The sequence data from this study have been submitted to GenBank under accession no. AE014184. Genome sequence and annotation are also available at http://igs-server.cnrs-mrs.fr/.]
References
- Akman, L., Yamashita, A., Watanabe, H., Oshima, K., Shiba, T., Hattori, M., and Aksoy, S. 2002. Genome sequence of the endocellular obligate symbiot of tsetse flies, Wiggleworthia glossinidia. Nat. Genet. 32: 402-407. [DOI] [PubMed] [Google Scholar]
- Altschul, S.F., Madden, T.L., Schaffer, A.A., Zhang, J., Zhang, Z., Miller, W., and Lipman, D.J. 1997. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 25: 3389-3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersson, J.O. and Andersson, S.G.E. 2001. Pseudogenes, junk DNA, and the dynamics of Rickettsia genomes. Mol. Biol. Evol. 18: 829-839. [DOI] [PubMed] [Google Scholar]
- Andersson, S.G., Zomorodipour, A., Andersson, J.O., Sicheritz-Ponten, T., Alsmark, U.C., Podowski, R.M., Naslund, A.K., Eriksson, A.S., Winkler, H.H., and Kurland, C.G. 1998. The genome sequence of Rickettsia prowazekii and the origin of mitochondria. Nature 396: 133-140. [DOI] [PubMed] [Google Scholar]
- Audic, S. and Claverie, J.M. 1998. Self-identification of protein-coding regions in microbial genomes. Proc. Natl. Acad. Sci. 95: 10026-10031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bentley, S.D., Maiwald, M., Murphy, L.D., Pallen, M.J., Yeats, C.A., Dover, L.G., Norbertczak, H.T., Besra, G.S., Quail, M.A., Harris, D.E., et al. 2003. Sequencing and analysis of the genome of the Whipple's disease bacterium Tropheryma whipplei. Lancet 361: 637-644. [DOI] [PubMed] [Google Scholar]
- Boeckmann, B., Bairoch, A., Apweiler, R., Blatter, M.C., Estreicher, A., Gasteiger, E., Martin, M.J., Michoud, K., O'Donovan, C., Phan, I., et al. 2003. The SWISS-PROT protein knowledgebase and its supplement TrEMBL in 2003. Nucleic Acids Res. 31: 365-370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brayton, K.A., Knowles, D.P., McGuire, T.C., and Palmer, G.H. 2001. Efficient use of a small genome to generate antigenic diversity in tick-borne ehrlichial pathogens. Proc. Natl. Acad. Sci. 98: 4130-4135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brinkman, F.S., Blanchard, J.L., Cherkasov, A., Av-Gay, Y., Brunham, R.C., Fernandez, R.C., Finlay, B.B., Otto, S.P., Ouellette, B.F., Keeling, P.J., et al. 2002. Evidence that plant-like genes in Chlamydia species reflect an ancestral relationship between Chlamydiaceae, cyanobacteria, and the chloroplast. Genome Res. 12: 1159-1167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Claverie, J.M. and Ogata, H. 2003. The insertion of palindromic repeats in the evolution of proteins. Trends Biochem. Sci. 28: 75-80. [DOI] [PubMed] [Google Scholar]
- Cole, S.T., Eiglmeier, K., Parkhill, J., James, K.D., Thomson, N.R., Wheeler, P.R., Honore, N., Garnier, T., Churcher, C., Harris, D., et al. 2001. Massive gene decay in the leprosy bacillus. Nature 409: 1007-1011. [DOI] [PubMed] [Google Scholar]
- Dervyn, E., Suski, C., Daniel, R., Bruand, C., Chapuis, J., Errington, J., Janniere, L., and Ehrlich, S.D. 2001. Two essential DNA polymerases at the bacterial replication fork. Science 294: 1716-1719. [DOI] [PubMed] [Google Scholar]
- Doolittle, R.F. 2002. Microbial genomes multiply. Nature 416: 697-700. [DOI] [PubMed] [Google Scholar]
- Drancourt, M., and Raoult, D. 1999. Characterization of mutations in the rpoB gene in naturally rifampin-resistant Rickettsia species. Antimicrob. Agents Chemother. 43: 2400-2403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drancourt, M., Carlioz, A., and Raoult, D. 2001. rpoB sequence analysis of cultured Tropheryma whippelii. J. Clin. Microbiol. 39: 2425-2430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drlica, K. 1999. Mechanism of fluoroquinolone action. Curr. Opin. Microbiol. 2: 504-508. [DOI] [PubMed] [Google Scholar]
- Drlica, K. and Zhao, X. 1997. DNA gyrase, topoisomerase IV, and the 4-quinolones. Microbiol. Mol. Biol. Rev. 61: 377-392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisen, J.A., Heidelberg, J.F., White, O., and Salzberg, S.L. 2000. Evidence for symmetric chromosomal inversions around the replication origin in bacteria. Genome Biol. 1: research0011. [DOI] [PMC free article] [PubMed]
- Ewing, B.G. and Green, P. 1998. Base-calling of automated sequencer traces using phred. II. Error probabilities. Genome Res. 8: 186-194. [PubMed] [Google Scholar]
- Ewing, B.G., Hillier, L., Wendl, M.C., and Green, P. 1998. Base calling of automated sequencer traces using phred. I. Genome Res. 8: 175-185. [DOI] [PubMed] [Google Scholar]
- Frank, A.C., Amiri, H., and Andersson, S.G. 2002. Genome deterioration: Loss of repeated sequences and accumulation of junk DNA. Genetica 115: 1-12. [DOI] [PubMed] [Google Scholar]
- Fraser, C.M., Gocayne, J.D., White, O., Adams, M.D., Clayton, R.A., Fleischmann, R.D., Bult, C.J., Kerlavage, A.R., Sutton, G., Kelley, J.M., et al. 1995. The minimal gene complement of Mycoplasma genitalium. Science 270: 397-403. [DOI] [PubMed] [Google Scholar]
- Fredricks, D.N. and Relman, D.A. 2001. Localization of Tropheryma whippelii rRNA in tissues from patients with Whipple's disease. J. Infect. Dis. 183: 1229-1237. [DOI] [PubMed] [Google Scholar]
- Ghigo, E., Capo, C., Aurouze, M., Tung, C.H., Gorvel, J.P., Raoult, D., and Mege, J.L. 2002. Survival of Tropheryma whipplei, the agent of Whipple's disease, requires phagosome acidification. Infect. Immun. 70: 1501-1506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon, D., Abajian, C., and Green, P. 1998. Consed: A graphical tool for sequence finishing. Genome Res. 8: 195-202. [DOI] [PubMed] [Google Scholar]
- Hinrikson, H.P., Dutly, F., and Altwegg, M. 2000. Evaluation of a specific nested PCR targeting domain III of the 23S rRNA gene of “Tropheryma whippelii” and proposal of a classification system for its molecular variants. J. Clin. Microbiol. 38: 595-599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes, D. 2000. Evaluating genome dynamics: The constraints on rearrangements within bacterial genomes. Genome Biol. 1: reviews0006. [DOI] [PMC free article] [PubMed]
- Kanehisa, M., Goto, S., Kawashima, S., and Nakaya, A. 2002. The KEGG databases at GenomeNet. Nucleic Acids Res. 30: 42-46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koonin, E.V., Makarova, K.S., and Aravind, L. 2001. Horizontal gene transfer in prokaryotes: Quantification and classification. Annu. Rev. Microbiol. 55: 709-742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- La Scola, B., Fenollar, F., Fournier, P.E., Altwegg, M., Mallet, M.N., and Raoult, D. 2001. Description of Tropheryma whipplei gen.nov., sp.nov., the Whipple's disease bacillus. Int. J. Syst. Evol. Microbiol. 51: 1471-1479. [DOI] [PubMed] [Google Scholar]
- Lawrence, J.G. and Ochman, H. 1998. Molecular archaeology of the Escherichia coli genome. Proc. Natl. Acad. Sci. 95: 9413-9417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawrence, J.G., Hendrix, R.W., and Casjens, S. 2001. Where are the pseudogenes in bacterial genomes? Trends Microbiol. 9: 535-540. [DOI] [PubMed] [Google Scholar]
- Lowe, T.M. and Eddy, S.R. 1997. t-RNAscan-SE: A program for improved detection of transfer RNA gene in genomic sequence. Nucleic Acids Res. 25: 955-964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maiwald, M., Schuhmacher, F., Ditton, H.J., and Von Herbay, A. 1999. Environmental occurrence of the Whipple's disease bacterium (Tropheryma whippelii). Appl. Environ. Microbiol. 64: 760-762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maiwald, M., Von Herbay, A., Lepp, P.W., and Relman, D.A. 2000. Organization, structure, and variability of the rRNA operon of the Whipple's disease bacterium (Tropheryma whippelii). J. Bacteriol. 182: 3292-3297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makino, S. and Suzuki, M. 2001. Bacterial genomic reorganization upon DNA replication. Science 292: 803. [DOI] [PubMed] [Google Scholar]
- Marth, T. and Raoult, D. 2003. Whipple's disease. Lancet 361: 239-246. [DOI] [PubMed] [Google Scholar]
- Masselot, F., Boulos, A., Maurin, M., Rolain, J.M., and Raoult, D. 2003. Molecular evaluation of antibiotic susceptibility: Tropheryma whipplei paradigm. Antimicrob. Agents Chemother. 47: 1658-1664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molle, V., Palframan, W.J., Findlay, K.C., and Buttner, M.J. 2000. WhiD and WhiB, homologous proteins required for different stages of sporulation in Streptomyces coelicolor A3(2). J. Bacteriol. 182: 1286-1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moran, N.A. 1996. Accelerated evolution and Muller's rachet in endosymbiotic bacteria. Proc. Natl. Acad. Sci. 93: 2873-2878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moran, N.A. and Wernegreen, J.J. 2000. Lifestyle evolution in symbiotic bacteria: Insights from genomics. Trends Ecol. Evol. 15: 321-326. [DOI] [PubMed] [Google Scholar]
- Ochman, H., Lawrence, J.G., and Groisman, E.A. 2000. Lateral gene transfer and the nature of bacterial innovation. Nature 405: 299-304. [DOI] [PubMed] [Google Scholar]
- Ogata, H., Audic, S., Renesto-Audiffren, P., Fournier, P.E., Barbe, V., Samson, D., Roux, V., Cossart, P., Weissenbach, J., Claverie, J.M., et al. 2001. Mechanisms of evolution in Rickettsia conorii and R. prowazekii. Science 293: 2093-2098. [DOI] [PubMed] [Google Scholar]
- Ogata, H., Audic, S., Abergel, C., Fournier, P.E., and Claverie, J.M. 2002. Protein coding palindromes are a unique but recurrent feature in Rickettsia. Genome Res. 12: 808-816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petes, T.D. and Hill, C.W. 1988. Recombination between repeated genes in microorganisms. Annu. Rev. Genet. 22: 147-168. [DOI] [PubMed] [Google Scholar]
- Petit, M.A., Dervyn, E., Rose, M., Entian, K.D., McGovern, S., Ehrlich, S.D., and Bruand, C. 1998. PcrA is an essential DNA helicase of Bacillus subtilis fulfilling functions both in repair and rolling-circle replication. Mol. Microbiol. 29: 261-273. [DOI] [PubMed] [Google Scholar]
- Raczniak, G., Becker, H.D., Min, B., and Soll, D. 2001. A single amidotransferase forms asparaginyl-tRNA and glutaminyl-tRNA in Chlamydia trachomatis. J. Biol. Chem. 276: 45862-45867. [DOI] [PubMed] [Google Scholar]
- Raoult, D., Birg, M.L., La Scola, B., Fournier, P.E., Enea, M., Lepidi, H., Roux, V., Piette, J.C., Vandenesch, F., Vital-Durand D., et al. 2000. Cultivation of the bacillus of Whipple's disease. N. Engl. J. Med. 342: 620-625. [DOI] [PubMed] [Google Scholar]
- Raoult, D., La Scola, B., Lecocq, P., Lepidi, H., and Fournier, P.E. 2001a. Culture and immunological detection of Tropheryma whippelii from the duodenum of a patient with Whipple disease. JAMA 285: 1039-1043. [DOI] [PubMed] [Google Scholar]
- Raoult, D., Lepidi, H., and Harle, J.R. 2001b. Tropheryma whipplei circulating in blood monocytes. N. Engl. J. Med. 345: 548-548. [DOI] [PubMed] [Google Scholar]
- Rocha, E.P., Danchin, A., and Viari, A. 1999. Functional and evolutionary roles of long repeats in prokaryotes. Res. Microbiol. 150: 725-733. [DOI] [PubMed] [Google Scholar]
- Shigenobu, S., Watanabe, H., Hattori, M., Sakaki, Y., and Ishikawa, H. 2000. Genome sequence of the endocellular bacterial symbiont of aphids Buchnera sp. APS. Nature 407: 81-86. [DOI] [PubMed] [Google Scholar]
- Tatusov, R.L., Natale, D.A., Garkavtsev, I.V., Tatusova, T.A., Shankavaram, U.T., Rao, B.S., Kiryutin, B., Galperin, M.Y., Fedorova, N.D., and Koonin, E.V. 2001. The COG database: New developments in phylogenetic classification of proteins from complete genomes. Nucleic Acids Res. 29: 22-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson, J.D., Higgins, D.G., and Gibson, T.J. 1994. CLUSTAL W: Improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 22: 4673-4680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uchida, T., Miyawaki, M., and Kinashi, H. 2003. Chromosomal arm replacement in Streptomyces griseus. J. Bact. 185: 1120-1124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volfovsky, N., Haas, B.J., and Salzberg, S.L. 2001. A clustering method for repeat analysis in DNA sequences. Genome Biol. 2: RESEARCH0027. [DOI] [PMC free article] [PubMed]
- Whipple, G.H. 1907. A hitherto undescribed disease characterized anatomically by deposits of fat and fatty acids in the intestinal and mesenteric lymphatic tissues. Bull. Johns Hopkins Hosp. 18: 382-393. [Google Scholar]
- Wilson, K.H., Blitchington, R., Frothingham, R., and Wilson, J.A. 1991. Phylogeny of the WhippIe's-disease-associated bacterium. Lancet 338: 474-475. [DOI] [PubMed] [Google Scholar]
- Woese, C.R., Olsen, G.J., Ibba, M., and Söll, D. 2000. Aminoacyl-tRNA synthetases, the genetic code, and the evolutionary process. Microbiol. Mol. Biol. Rev. 64: 202-236. [DOI] [PMC free article] [PubMed] [Google Scholar]