

Abstract
The mammalian chemokine family is segregated into four families – CC, CXC, CX3C, and XC—based on the arrangement of cysteines and the corresponding disulfides. Sequencing of the Danio rerio (zebrafish) genome has identified more than double the amount of human chemokines with the absence of the CX3C family and the presence of a new family, CX. The only other family with a single cysteine in the N-terminal region is the XC family. Human lymphotactin (XCL1) has two interconverting structures due to dynamic changes that occur in the protein. Similar to an experiment with XCL1 that identified the two structural forms, we probed for multiple forms of zCXL1 using heparin affinity. The results suggest only a single form of CXL1 is present. We used sulfur-SAD phasing to determine the three-dimensional structure CXL1. Zebrafish CXL1 (zCXL1) has three disulfides that appear to be important for a stable structure. One disulfide is common to all chemokines except those that belong to the XC family, another is similar to a subset of CC chemokines containing three disulfides, but the third disulfide is unique to the CX family. We analyzed the electrostatic potential of the zCXL1 structure and identified the likely heparin-binding site for glycosaminoglycans (GAGs). zCXL1 has a similar sequence identity with human CCL5 and CXCL12, but the structure is more related to CCL5. Our structural analysis supports the phylogenetic and genomic studies on the evolution of the CXL family.
Keywords: CX chemokine family, chemokine structure, Sulfur SAD, Danio rerio (zebrafish) chr24a2/CXL34bk3 (CXL1), heparin binding
INTRODUCTION
Chemokines are a superfamily of small, secreted proteins of 8–10 kDa that activate G-protein coupled receptors.1 These proteins are involved in embryonic development, adult homeostasis, and inflammation that direct the migration of immune cells to injured or infected tissues. Based on the arrangement of the conserved cysteines in the human superfamily, chemokines are classified into four families with 28 proteins in the CC family, 17 in the CXC family, two in the XC family, and one in the CX3C family. Chemokines in the CC or CXC families contain either no intervening residue or one intervening amino acid between the first two cysteines, respectively. These two cysteines form disulfides with two other conserved cysteines in the sequence. There are three intervening residues in the mammalian CX3C chemokine that form two disulfides. With the exception of the XC chemokines, the first and third cysteines, and the second and fourth cysteines form disulfides in chemokines that belong to the other families. The difference in the cysteine framework results, generally, in different dimeric structures.2 Only one of the two conserved cysteines is present at the N-terminal region in the XC family that forms a single disulfide with the other cysteine in the protein sequence. XCL1 has the unique property that its structure is in equilibrium between a cognate monomeric chemokine topology and a non-cognate dimer consisting of a four-stranded β-sheet without any α-helix.3 These two XCL1 structures have functional differences. The cognate chemokine activates the XCR receptor but has low affinity for heparin. In contrast, the dimeric, four-stranded β-sheet is unable to activate the receptor but binds heparin with high affinity.
Zebrafish contain the most chemokines of any species examined thus far with 82 CC chemokines, 25 CXC chemokines, and one XC chemokine.4 There are no genes in the CX3C family but there are five genes in a unique CX family that is defined by the presence of six cysteines. Although the new zebrafish CX chemokine family lacks one of the two conserved cysteine residues at the N-terminal region, it differs from the XC family in that it retains the third and the fourth cysteines common to the other families in addition to three additional cysteines that form a unique disulfide network. This new chemokine family is currently not found in any other species.4
Aside from a few homologues between zebrafish and humans, zebrafish and other teleost fish constitute an evolutionary diverse group that evolved its own set of chemokines to adapt to their environment.4 The difference in the number of chemokines and the unique species-dependent family provide a window into understanding the evolution of zebrafish. In this report, we describe the X-ray structure of zebrafish CXL1 (zCXL1; also referred as CXL-chr24a4 and CXL34bk5) solved by sulfur-SAD phasing in two different space groups. Since there are no other known zebrafish three-dimensional chemokine structures, we used the human chemokine superfamily as a surrogate for comparison of zCXL1. To validate that CXL1 has other properties that differ from XCL1,3 we compared structural and heparin-binding properties of the two chemokines. The electrostatic potential of zCXL1 identifies a potential heparin-binding site that is similar to other chemokines.6 Although the evolution of the zebrafish and human chemokine superfamily diverged, structural analysis leads to the conclusion that the zCX family is evolutionary related to the CC family, similar to the phylogenetic and genomic analysis of zebrafish chemokines.4,5
MATERIALS AND METHODS
Cloning, expression, and purification
The zCXL1 pET11d clone was a kind gift of Dr. Hisayuki Nomiyama. We expressed CXL1 from BL21 DE3 E.coli cells. Although protein expression was sufficient for structural studies, it was in the insoluble fraction, and resolubilization and re-folding proved difficult. The gene was re-cloned into Pichia pastoris plasmid pPICZαB between XbaI and XhoI restriction sites that allows the expressed protein to be secreted into the medium. The expression protocol followed the Pichia Expression Kit Manual (Invitrogen). Briefly, upon confirmation of the clone by sequencing, the plasmid was linearized with SacI and transformed into GS115 P. pastoris cells. Zeocin-resistant clones were screened for maximal expression of zCXL1. A selected clone was grown at 30°C to an OD600 of 2–6 in BMGY medium with 0.1% casamino acids. For induction, the cells were transferred to BMMY media and induced at 30°C with 1% methanol every 24 h for 3 days. The protein was secreted into the medium and purified directly using a cation-exchange SP-Sepharose column equilibrated with 25 mM Tris-HCl, pH 7.4 and eluted with a linear gradient of 0–1M NaCl. The protein was further purified on a C18 column by HPLC reverse phase chromatography. zCXL1 was eluted from the column in an acetonitrile gradient of 20–55%. The eluted protein was lyophilized and suspended in 0.1M ammonium acetate, concentrated to 13 mg/mL, and used for crystallization.
Heparin sepharose chromatography
One hundred micrograms of zCXL1 protein was loaded onto a 1 mL heparin sepharose column (GE Healthcare) in 25 mM Tris-HCl, pH 7.4 buffer and eluted with a linear gradient (0–1M NaCl in 25 mM Tris-HCl, pH 7.4) at a flow rate of 0.5 mL/min. The protein was monitored by absorbance at 280 nm, and the concentration of NaCl was determined using an in-line conductivity meter.
Crystallization, data collection and processing, phasing, and refinement
Initial screening of the crystallization conditions was carried out by the hanging-drop vapor diffusion method using the Hampton Index Formulations (Hampton Research, Laguna Hills, CA) at 293K. Approximately 2 μL of the 13 mg/mL protein sample was mixed with an equal volume of the reservoir solution. Single crystals (form A) appeared in 4 days from a condition comprised of 2.4M sodium malonate. The second form appeared in a one week from 0.1M Tris, pH 7.4 buffer with 38% PEG 3350. Unlike crystal form A, these crystals (form B) were clustered needles. An individual needle was separated for diffraction purposes and paratone oil was used as cryoprotectant for both crystal forms.
High-resolution data for P. pastoris-expressed zCXL1 were collected at Brookhaven National Laboratory for space group P21212 and Argonne National Laboratory for space group P6122 (Table IA). Molecular replacement methods using various versions of chemokine monomers (CCs, CXCs, XCs backbone or polyalanine forms with and without the flexible the N-terminal region and loops) and a variety of software (Phaser, MolRep and AMoRe) were unsuccessful for either space group. Single- or mutli-wavelength anomalous dispersion (SAD or MAD, respectively) methods using selenomethionine were not suitable because zCXL1 could not be refolded from E. coli nor is selenomethionine incorporation optimal for P. pastoris. Additional X-ray data were collected at an in-house Rigaku system at The Woodlands, TX for crystal A (P6122) to measure sulfur SAD accurately given the small Bijvoet differences. The system consisted of a MicroMax-007 rotating anode generator with a Cr anode, Osmic Confocal MaxFlux (Cr CMF) multilayer optics, a square-pyramidal helium beam path, and a large-aperture R-AXIS IV++ detector.7 The data were analyzed for anomalous signal content by comparing the anomalous signal-to-noise ratio based on the variance of F+ and F− [ΔF/σΔF]. Symmetry-related reflections were kept separate during data reduction in XSCALE to allow local scaling before calculation of anomalous FA values. The diffraction data were processed with XDS.8 Data collection and processing statistics are summarized in Table IA. Peak searches were successful in identifying seven sulfur atoms (six from cysteine and one from methionine) using Autosol.9 The initial electron density map was obtained from sulfur–SAD phasing and the structure was built automatically into the electron density. The zCXL1 structure was successfully used for molecular replacement of the high-resolution data sets from synchrotron sources. Most water molecules in the structural models were located automatically using the water-picking protocols during refinement and were manually retained or discarded by visual inspection of the electron density for each molecule. Alternating cycles of refinement with Phenix10 and model building with Coot11 were used until Rfree no longer decreased (Table IB). Structural comparisons were made with LSQKAB in ccp4i.12
Table I.
(A) Data Collection and Processing
| Beamline | Rigaku | NSLS-X25 | APS-24-ID-C |
|---|---|---|---|
| Wavelength | 2.29 | 1.0 | 0.97924 |
| Distance (cm) | 220 | 150 | |
| Resolution (Å) | 47.31-2.20(2.32-2.20) | 46.31-1.62(1.71-1.62) | 23.00-1.28 (1.35-1.28) |
| Total reflections | 64119(2330) | 129685(17865) | 165221(28067) |
| Unique reflections | 4720(593) | 9709(1370) | 21906(3203) |
| Space group | P6122 | P21212 | P6122 |
| Unit cell | |||
| a, b, c (Å) | 44.62, 44.62, 141.93 | 27.62, 46.31, 56.66 | 44.57, 44.57, 141.88 |
| α, β, γ (°) | 90, 90, 120 | 90, 90, 90 | 90, 90, 120 |
| Completeness (%) | 98.5(90.6) | 99.7(99.6) | 97.6(100) |
| Anomalous completeness (%) | 97.4(83.2) | ||
| Anomalous multiplicity | 8.0(2.2) | ||
| Redundancy | 13.6(3.9) | 13.4(13.0) | 7.5(8.8) |
| <I/σ(I)> | 29.7(2.8) | 19.7(4.9) | 11.0(3.9) |
| Rmerge | 0.049(0.405) | 0.078(0.479) | 0.084(0.369) |
| (B) Refinement Statistics | ||
|
| ||
| Beamline | NSLS-X25 | APS- 24-ID-C |
|
| ||
| No. of atoms | ||
| Proteins | 552 | 534 |
| Water | 85 | 113 |
| Rwork/Rfree(%) | 18.0/23.0 | 18.3/20.6 |
| RMS deviations | ||
| Bond angle | 0.009 | 0.005 |
| Bond length | 1.327 | 1.013 |
| Chirality | 0.099 | 0.080 |
| Dihedral | 11.422 | 8.894 |
| Ramachandran plot | ||
| Outliers (%) | 0 | 0 |
| Favored (%) | 100 | 100 |
| PDB code | 4HED | 4HCS |
Values in the parentheses are for highest resolution shell
Rmerge = Σ∣I(h,i) − < I(h) > α/ΣI(h,i), where < I(h) > is the mean intensity of reflections. Rwork and Rfree were calculated from working and test set reflections. Values for the highest resolution shells are given in parenthesis. Bijvoet pairs are merged.
RESULTS AND DISCUSSION
Structural features of zCXL1
The P. pastoris-expressed protein was secreted, purified, and crystallized. zCXL1 is one of a few dozen structures in the pdb database that used the sulfur SAD method for phasing.13 Higher resolution structures of zCXL1 using data collected at synchrotron sources were solved by molecular replacement using the sulfur SAD-phased structure. The structure in space groups P6122 and P21212 was modeled with electron density for residues 13–78 and 12–81, respectively. The lack of electron density for N-terminal residues is typical of chemokines and consistent with the flexibility of this region.2 Representative electron density for other parts of the structure from both crystal forms is shown in Figure 1(A,B). The rms difference of the overlapping Cα carbons between the structures is 0.41Å [Fig. 1(C)]. The structures are monomers with no substantial interactions typical of a bona fide dimer using crystallographic symmetry, as previously observed for CXCL8.14 The possibility of monomer-dimer equilibrium was probed by analytical and preparative size-exclusion chromatography but only one peak was visible (Supporting Information Fig. S1). The monomeric structure of zCXL1 was also confirmed by the online program PISA (Protein Interfaces, Surfaces and Assemblies) at the European Bioinformatics Institute (http://www.ebi.ac.uk/pdbe/prot_int/pistart.html).15
Figure 1.
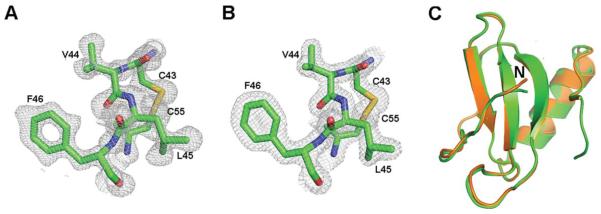
Electron density for CXL1 with one of the disulfides in space group (A) P6122 (residues 43–46, 55), (B) P21212 (residues 43–46, 55), (C) Superposition of the structures in the two space groups (4HCS, orange; 4HED, green). Structural comparisons were made with LSQKAB in ccp4i.12
The long N-terminal loop (residues 16–29), which plays an important role in receptor binding for all other chemokines, has four solvent-exposed basic residues spanning a distance of 23Å and one glutamic acid side chain that faces the helix. The loop is followed by three anti-parallel β-strands that form a β-sheet and a C-terminal α-helix [Fig. 2(A)]. The secondary structures are connected by loops commonly referred to as the 30s, 40s, and 50s loops. The topology and monomeric structure of zCXL1 is the same as the human CC, CXC, and CX3C chemokines. However, the disulfide framework that defines each chemokine family, plays an important in receptor binding specificity and biological activity16 and, in general, induces different family-dependent dimeric or higher order structures2,17,18 is unique for zCXL1. The only cysteine at the N-terminal region (Cys-15) forms a disulfide with the third cysteine present in the 30s loop (Cys-40). The second disulfide is formed between the cysteine located in the first β-sheet (Cys-31) and the sixth cysteine at the C-terminal region following the helix (Cys-77). The third and distinctive disulfide bond is between the fourth and fifth cysteines (Cys-43 and Cys-55) found in β-strands two and three, respectively [Fig. 2(B,C)].
Figure 2.

Zebrafish CXL1 structure and disulfide framework. (A) Cartoon representation of zCXL1 colored in a purple-to-red spectrum along the chain from N- to C-terminus. The structure has a typical chemokine fold with a long N-terminus (blue) followed by three anti-parallel β-strands forming a sheet, and a C-terminal helix. (B) Cartoon representation of zCXL1 highlighting the disulfide bonds as spheres are shown in yellow. (C) Sequence, secondary structure, and disulfides of zCXL1. All structural figures in the manuscripts were prepared with Pymol.29
Comparison to XCL1: heparin binding and the electrostatic potential of zCXL1
The only human chemokines that lack one of the two cysteines near the N-terminus to form only one disulfide belong to the XC family. Sequence identity between the mature human XCL1 and zebrafish CXL1 is slightly below 20% (Supporting Information Fig. S2). The lack of additional disulfides for XCL1 allows it to have a more dynamic structure that is in equilibrium under physiological conditions between a canonical chemokine monomeric structure and a non-canonical structure that replaces the C-terminal helix with a β-strand to form a four-stranded β-sheet dimer that has never been observed for any other chemokine.3 The XCL1 structures have other properties that are divided between the two but are common for other individual chemokines. For example, the canonical XCL1 chemokine binds and activates the receptor XCR1 but has weak affinity for heparin. The non-canonical, dimeric four-stranded β-sheet structure has very high affinity for heparin but does not activate the receptor.3,19 Since elution of two peaks at different NaCl concentrations from heparin affinity chromatography was one of several methods that identified the two forms of XCL1, we also used heparin chromatography to probe for other forms of zCXL1. The binding of zCXL1 to glycosaminoglycans (GAGs) was demonstrated by elution of zCXL1 from a heparin-Sepharose column at a single concentration of ~0.5M NaCl using a gradient from 0 to 1M NaCl [Fig. 3(A)], suggesting that only one zCXL1 structure is formed. Although extensive NMR studies would be required to rule out an alternative form for zCXL1, it is difficult to speculate that the chemokine fold of zCXL1 can also form a non-cognate structure for two related reasons. The interconversion of the two XCL1 structures requires global unfolding,20 which would be difficult for the zCXL1 chemokine structure stabilized by three disulfides. Second, XCL1 cysteine mutants that form a second disulfide stabilize the chemokine fold with no evidence of conversion to an alternative structure.21
Figure 3.

Heparin binding and electrostatic potential. (A) Chromatogram of zCXL1 eluted from a heparin-Sepharose column by a concentration gradient of NaCl. (B) Electrostatic potential map of CXL1 overlaid with the cartoon representation of the structure. The surface has extensive electrostatic positive potential for most of the surface, especially between the β-sheet and α-helix.
A functional role for GAG binding has been identified for a number of other chemokines,22 promoting dimerization or further aggregation that is important for chemokine function and tissue specificity.22,23 The electrostatic potential of zCXL1 shows an extensive positive region for most of the surface with islands of negative potential emanating from the helix and loops [Fig. 3(B)]. The positive potential between the β-sheet and α-helix is similar to many chemokines where the GAG binding site has been identified.6 Given the similarity in the electrostatic positive potential for GAG binding similar to other chemokines, a quaternary structure for zCXL1 cannot be dismissed.
Comparison with CC and CXC chemokines
Sequence analysis of zCXL1 with human CC and CXC chemokines reveals a maximum identity of 25% to CCL5 and 22% to CXCL12 [Fig. 4(A)]. The three-dimensional structure of CXL1 has higher similarity to CCL5 [Fig. 4(B)] than CXCL12 [Fig. 4(C)]. For this structural analysis we refer to the method of Veldkamp et al. in measuring the α-helical axis with respect to the first β-strand.24 The angle is 119° for zCXL1 and 120° for CCL5 (pdb code 1EQT) [Fig. 4(B)]. The CXCL12 dimeric X-ray structure has an angle of 90° (pdb code 2J7Z25) which does not vary much from other CXCL12 X-ray structures6,17,26,27 but varies significantly from the monomeric structure determined by NMR with an angle of 52° [Fig. 4(C)].24,28 The average rms difference for Ca’s between zCXL1 and CXCL12 or CCL5 is 2.63Å and 1.83Å, respectively. Without the α-helix in the structural alignment, the average rms difference is reduced to 2.21Å for CXCL12 with no difference for CCL5, demonstrating that a large component of the difference with zCXL1 is due to the α-helix but the rms difference for CXCL12 is still not as low as CCL5. The alignment of the helix also bears upon what type of dimer zCXL1 might make in the presence of heparin or other GAGs. Based upon the analysis of the angle of the helix relative to the first β-strand a zCXL1 dimer will resemble a CC chemokine, but this remains to be determined.
Figure 4.

Superposition of zebrafish CXL1 with human CCL5 and CXC12. (A) Sequence alignment of zCXL1, CCL5, and CXC12 chemokines. (B) Overlay of CXL1 (magenta) and CCL5 (green). (C) Superimposition of zCXL1 (magenta) with structures of CXCL12 (1SDF, yellow; 2J7Z, blue). The angle made by the C-terminal helix with respect to the second β-strand is 119° for zCXL1 (4HED), 120° for CCL5 (1EQT), 52° for the monomeric structure of CXCL12 determined by NMR (1SDF), and 90° for the X-ray structure of CXCL12 (2J7Z).
The disulfide framework was also analyzed and compared. The first CXL1 disulfide is similar to the first disulfide in chemokines from all families except those that belong to the XC family (Supporting Information Fig. S3). One of the two remaining disulfides (Cys-31-Cys-77) is similar to a disulfide from a small number of CC chemokines that also contain three disulfides but with a sequence identity less than 15% to CXL1 [Figs. 2(B), 5(A,B)]. This disulfide for human CCL1, CCL15, and CCL23, and zCXL1 is between a cysteine at the first β-strand and the sixth cysteine at the C-terminal region, constraining the movement of the α-helix (It is likely that the four other human CC chemokines with six cysteines—CCL6, CCL9, CCL21, and CCL28—have the same disulfide based upon the location of the sixth cysteine). The third disulfide is unique and defines this new chemokine family [Fig. 2(B,C), and Supporting Information Table S1).
Figure 5.

(A) Cartoon representation of human CCL15, (2HCC, cyan), CCL23 (1G91, blue), CCL1 (1ELO, green), and zCXL1 (4HCS, magenta) highlighting the three disulfide bonds as spheres shown in yellow for comparison. (B) Sequence alignment from the three known structures of human CC chemokines with three disulfides (CCL1, CCL15, CCL23) ranging in sequence similarity between 10 and 13% with zCXL1. The disulfide linkages are shown for chemokines CCL15 and CCL23 above the alignment and for zCXL1 and CCL1 below the alignment.
The evolution of zebrafish CXL based on sequence and structural analysis
To gain insight into the molecular evolution of the CX chemokine family in zebrafish, we used sequences and structures from the human chemokine superfamily even though humans do not contain CX chemokines. The rationale for using this method is there are no other zebrafish chemokine structures currently available. We first compared the zCXL1 to the XCL1 as both lacked the second cysteine at the N-terminal sequence and found there were significant differences both in the stability of the structure based on the number of disulfides and heparin-binding properties. We therefore eliminated a direct evolutionary relationship between these two proteins. Our sequence analysis to the CXC and CC family indicates zCXL1 has a similar sequence identity to human CXCL12 and CCL5, however, the structure is far more related to CCL5 based on the angle between the α-helix and the second β-strand. Part of the disulfide framework of zCXL1 is also similar to a small set of mammalian CC chemokines that contain three disulfides despite a sequence identity less than 15%. Both the structural similarity to CCL5 and the partial similarity to the disulfide framework of a subset of CC chemokines suggest that the CX chemokine family evolved from the CC family in zebrafish. This structural analysis is consistent with the phylogenetic analysis and genome chromosomal organization of the zebrafish superfamily that concluded the CX family evolved from the CC family.4 This study is an example of how structural biology can provide insight into the evolution of a protein superfamily even after the species involved in the analysis diverged in evolution.
Supplementary Material
ACKNOWLEDGMENTS
The authors thank Dr. Hisayuki Nomiyama from Kumamoto University Graduate School of Medical Sciences for the kind gift of the CXL1 clone. They also thank the Yale Keck Biotechnology Resource Laboratory for use of the mass spectrometry facility.
Grant sponsor: National Institutes of Health grant 5R01 AI082295.
Abbreviations
- MAD
multi-wavelength anomalous dispersion
- SAD
single-wavelength anomalous dispersion
Footnotes
Additional Supporting Information may be found in the online version of this article.
REFERENCES
- 1.Zlotnik A, Yoshie O. The chemokine superfamily revisited. Immunity. 2012;36:705–716. doi: 10.1016/j.immuni.2012.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Fernandez EJ, Lolis E. Structure, function, and inhibition of chemokines. Annu Rev Pharmacol Toxicol. 2002;42:469–499. doi: 10.1146/annurev.pharmtox.42.091901.115838. [DOI] [PubMed] [Google Scholar]
- 3.Tuinstra RL, Peterson FC, Kutlesa S, Elgin ES, Kron MA, Volkman BF. Interconversion between two unrelated protein folds in the lymphotactin native state. Proc Natl Acad Sci USA. 2008;105:5057–5062. doi: 10.1073/pnas.0709518105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nomiyama H, Hieshima K, Osada N, Kato-Unoki Y, Otsuka-Ono K, Takegawa S, Izawa T, Yoshizawa A, Kikuchi Y, Tanase S, Miura R, Kusuda J, Nakao M, Yoshie O. Extensive expansion and diversification of the chemokine gene family in zebrafish: identification of a novel chemokine subfamily CX. BMC Genomics. 2008;9:222. doi: 10.1186/1471-2164-9-222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nomiyama H, Osada N, Yoshie O. Systematic classification of vertebrate chemokines based on conserved synteny and evolutionary history. Genes cells. 2013;18:1–16. doi: 10.1111/gtc.12013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Murphy JW, Cho Y, Sachpatzidis A, Fan C, Hodsdon ME, Lolis E. Structural and functional basis of CXCL12 (stromal cell-derived factor-1alpha) binding to heparin. J Biol Chem. 2007;282:10018–10027. doi: 10.1074/jbc.M608796200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yang C, Pflugrath JW, Courville DA, Stence CN, Ferrara JD. Away from the edge: SAD phasing from the sulfur anomalous signal measured in-house with chromium radiation. Acta Crystallogr D. 2003;59(Pt 11):1943–1957. doi: 10.1107/s0907444903018547. [DOI] [PubMed] [Google Scholar]
- 8.Kabsch W. XDS. Acta Crystallogr D. 2010;66(Pt 2):125–132. doi: 10.1107/S0907444909047337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Terwilliger TC, Adams PD, Read RJ, McCoy AJ, Moriarty NW, Grosse-Kunstleve RW, Afonine PV, Zwart PH, Hung LW. Decision-making in structure solution using Bayesian estimates of map quality: the PHENIX AutoSol wizard. Acta Crystallogr D. 2009;65(Pt 6):582–601. doi: 10.1107/S0907444909012098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Terwilliger TC, Grosse-Kunstleve RW, Afonine PV, Moriarty NW, Zwart PH, Hung LW, Read RJ, Adams PD. Iterative model building, structure refinement and density modification with the PHENIX AutoBuild wizard. Acta Crystallogr D. 2008;64(Pt 1):61–69. doi: 10.1107/S090744490705024X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of Coot. Acta Crystallogr D. 2010;66(Pt 4):486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Winn MD, Ballard CC, Cowtan KD, Dodson EJ, Emsley P, Evans PR, Keegan RM, Krissinel EB, Leslie AG, McCoy A, McNicholas SJ, Murshudov GN, Pannu NS, Potterton EA, Powell HR, Read RJ, Vagin A, Wilson KS. Overview of the CCP4 suite and current developments. Acta Crystallogr D. 2011;67(Pt 4):235–242. doi: 10.1107/S0907444910045749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Liu Q, Dahmane T, Zhang Z, Assur Z, Brasch J, Shapiro L, Mancia F, Hendrickson WA. Structures from anomalous diffraction of native biological macromolecules. Science. 2012;336:1033–1037. doi: 10.1126/science.1218753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Baldwin ET, Weber IT, St Charles R, Xuan JC, Appella E, Yamada M, Matsushima K, Edwards BF, Clore GM, Gronenborn AM, Wlodawer A. Crystal structure of interleukin 8: symbiosis of NMR and crystallography. Proc Natl Acad Sci USA. 1991;88:502–506. doi: 10.1073/pnas.88.2.502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Krissinel E, Henrick K. Inference of Macromolecular Assemblies from Crystalline State. J Mol Biol. 2007;372:774–797. doi: 10.1016/j.jmb.2007.05.022. [DOI] [PubMed] [Google Scholar]
- 16.Rajarathnam K, Sykes BD, Dewald B, Baggiolini M, Clark-Lewis I. Disulfide bridges in interleukin-8 probed using non-natural disulfide analogues: dissociation of roles in structure from function. Biochemistry. 1999;38:7653–7658. doi: 10.1021/bi990033v. [DOI] [PubMed] [Google Scholar]
- 17.Murphy JW, Yuan H, Kong Y, Xiong Y, Lolis EJ. Heterologous quaternary structure of CXCL12 and its relationship to the CC chemokine family. Proteins. 2010;78:1331–1337. doi: 10.1002/prot.22666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang X, Watson C, Sharp Joshua S, Handel Tracy M, Prestegard James H. Oligomeric Structure of the Chemokine CCL5/RANTES from NMR, MS, and SAXS Data. Structure. 2011;19:1138–1148. doi: 10.1016/j.str.2011.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Peterson FC, Elgin ES, Nelson TJ, Zhang F, Hoeger TJ, Linhardt RJ, Volkman BF. Identification and characterization of a glycosaminoglycan recognition element of the C chemokine lymphotactin. J Biol Chem. 2004;279:12598–12604. doi: 10.1074/jbc.M311633200. [DOI] [PubMed] [Google Scholar]
- 20.Tyler RC, Murray NJ, Peterson FC, Volkman BF. Native-state inter-conversion of a metamorphic protein requires global unfolding. Biochemistry. 2011;50:7077–7079. doi: 10.1021/bi200750k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tuinstra RL, Peterson FC, Elgin ES, Pelzek AJ, Volkman BF. An engineered second disulfide bond restricts lymphotactin/XCL1 to a chemokine-like conformation with XCR1 agonist activity. Biochemistry. 2007;46:2564–2573. doi: 10.1021/bi602365d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Handel TM, Johnson Z, Crown SE, Lau EK, Sweeney M, Proudfoot AE. Regulation of protein function by glycosoaminoglycans–as exemplified by chemokines. Annu Rev Biochem. 2005;74:385–410. doi: 10.1146/annurev.biochem.72.121801.161747. [DOI] [PubMed] [Google Scholar]
- 23.Rajasekaran D, Keeler C, Syed MA, Jones MC, Harrison JK, Wu D, Bhandari V, Hodsdon ME, Lolis EJ. A Model of GAG/MIP-2/CXCR2 Interfaces and Its Functional Effects. Biochemistry. 2012;51:5642–5654. doi: 10.1021/bi3001566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Veldkamp CT, Ziarek JJ, Su J, Basnet H, Lennertz R, Weiner JJ, Peterson FC, Baker JE, Volkman BF. Monomeric structure of the cardioprotective chemokine SDF-1/CXCL12. Prot Sci. 2009;18:1359–1369. doi: 10.1002/pro.167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ryu EK, Kim TG, Kwon TH, Jung ID, Ryu D, Park Y-M, Kim J, Ahn KH, Ban C. Crystal structure of recombinant human stromal cell-derived factor-1a. Proteins. 2007;67:1193–1197. doi: 10.1002/prot.21350. [DOI] [PubMed] [Google Scholar]
- 26.#Dealwis C, Fernandez EJ, Thompson DA, Simon RJ, Siani MA, Lolis E. Crystal structure of chemically synthesized [N33A] stromal cell-derived factor 1alpha, a potent ligand for the HIV-1 “fusin” coreceptor. Proc Natl Acad Sci USA. 1998;95:6941–6946. doi: 10.1073/pnas.95.12.6941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ohnishi Y, Senda T, Nandhagopal N, Sugimoto K, Shioda T, Nagal Y, Mitsui Y. Crystal structure of recombinant native SDF-1alpha with additional mutagenesis studies: an attempt at a more comprehensive interpretation of accumulated structure-activity relationship data. J Interferon Cytokine Res. 2000;20:691–700. doi: 10.1089/10799900050116390. [DOI] [PubMed] [Google Scholar]
- 28.Crump MP, Gong JH, Loetscher P, Rajarathnam K, Amara A, Arenzana-Seisdedos F, Virelizier JL, Baggiolini M, Sykes BD, Clark-Lewis I. Solution structure and basis for functional activity of stromal cell- derived factor-1; dissociation of CXCR4 activation from binding and inhibition of HIV-1. EMBO J. 1997;16:6996–7007. doi: 10.1093/emboj/16.23.6996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.DeLano WL. The PyMOL molecular graphics system. DeLano Scientific; Palo Alto, CA: 2002. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.