

Abstract
Heat-shock protein 104 (Hsp104p) is a protein-remodeling factor that promotes survival after extreme stress by disassembling aggregated proteins and can either promote or prevent the propagation of prions (protein-based genetic elements). Hsp104p can be greatly overexpressed without slowing growth, suggesting tight control of its powerful protein-remodeling activities. We isolated point mutations in Hsp104p that interfere with this control and block cell growth. Each mutant contained alterations in the middle region (MR). Each of the three MR point mutations analyzed in detail had distinct phenotypes. In combination with nucleotide binding site mutations, Hsp104pT499I altered bud morphology and caused septin mislocalization, colocalizing with the misplaced septins. Point mutations in the septin Cdc12p suppressed this phenotype, suggesting that it is due to direct Hsp104p–septin interactions. Hsp104pA503V did not perturb morphology but stopped cell growth. Remarkably, when expressed transiently, the mutant protein promoted survival after extreme stress as effectively as did wild-type Hsp104p. Hsp104pA509D had no deleterious effects on growth or morphology but had a greatly reduced ability to promote thermotolerance. That mutations in an 11-amino acid stretch of the MR have such profound and diverse effects suggests the MR plays a central role in regulating Hsp104p function.
INTRODUCTION
The AAA+ proteins are ATPases associated with various cellular activities. They are important proteins with a great diversity of functions, including protein folding, membrane trafficking, organelle biogenesis, proteolysis, intracellular motility, and DNA replication (Neuwald et al., 1999; Vale, 2000; Ogura and Wilkinson, 2001). Little is known about how most AAA+ proteins recognize substrates and use ATP binding and hydrolysis to remodel them. The intrinsic complexity and multiplicity of conformational states of the AAA+ proteins make them difficult to study. The Clp/HSP100 proteins are members of the AAA+ superfamily and have been the subject of intense biochemical analysis in vitro and genetic analysis in vivo (Wickner et al., 1999; Glover and Tkach, 2001). The functional unit of the yeast HSP100 heat-shock protein 104 (Hsp104p) is composed of six monomers, each with two ATP binding sites (nucleotide binding domains one and two; NBD1 and NBD2) flanked by amino-terminal, middle, and carboxy-terminal regions (Figure 1).
Figure 1.

Mutations recovered in the screen for dominant lethal HSP104 mutants. Hsp104p (908 amino acids) has two highly conserved but distinct NBDs (NBD1 and NBD2) flanked by less conserved N-terminal and C-terminal regions. The MR is an insertion within NBD1. The boundaries of NBD1 (according to an alignment of AAA+ protein sequences; Neuwald et al., 1999) are from residue 180 to residue 400 and again from residue 549 to residue 586 for the sensor 2 domain. The MR is the area bounded by these regions (residue 401 to residue 548). Areas of greater conservation are denoted by a thicker line (Schirmer et al., 1996), and signature sequences of the NBDs are denoted: A and B, the Walker-type P-loop motifs; S1 and S2, the two sensor regions. Asterisks indicate the positions of the mutations in sequenced mutants, and the amino acid substitutions are specified on the left. Conserved sequence elements where mutations tended to cluster are further marked by open boxes. The mutants are grouped by phenotype.
Hsp104p has remarkable functions, one of which is to allow survival after extreme stress. For example, yeast cells expressing Hsp104p are 1000 times more viable after exposure to temperatures ≥50°C or to an ethanol concentration of 20% than cells carrying deletions of HSP104 (Sanchez and Lindquist, 1990; Sanchez et al., 1992). This survival capacity is directly attributable to Hsp104p's ability to resolubilize protein aggregates and, together with Hsp70p and Hsp40p, return them to their folded and active states (Parsell et al., 1994; Glover and Lindquist, 1998; Goloubinoff et al., 1999). This is in contrast to other heat shock proteins, which generally act by preventing aggregate accumulation or by promoting the degradation of misfolded proteins (Schirmer et al., 1996; Zolkiewski, 1999). During times of severe stress, the rate of protein aggregation exceeds the capacity of other heat shock proteins to prevent aggregate accumulation, and Hsp104p becomes critical to survival. This explains the observation that Hsp104p is not required for normal growth, or even growth at high temperatures, but is vital for surviving extreme conditions. The relationship between Hsp104p and thermotolerance is simple and direct: the more Hsp104p present, the higher the level of thermotolerance.
Another remarkable activity of Hsp104p is prion maintenance (Chernoff et al., 1995; Patino et al., 1996; Paushkin et al., 1996; DebBurman et al., 1997; Moriyama et al., 2000). Prions are proteinaceous genetic elements that have the ability to undergo heritable, self-perpetuating changes in conformation. As with stress survival, prion maintenance is dependent on Hsp104p's control of protein aggregation, but in this case the relationship is more complex. For maintenance of the [PSI+] prion, intermediate levels of Hsp104p are necessary: either deletion or overexpression of HSP104 eliminates [PSI+].
The importance of the ATP hydrolysis sites in the Hsp104p NBD regions is apparent from the debilitating effects of mutations in either NBD1 or NBD2 on thermotolerance and prion maintenance (Parsell et al., 1991; Chernoff et al., 1995; Patino et al., 1996; Schirmer et al., 2001) and from the well documented importance of these domains in other AAA+ proteins (Ogura and Wilkinson, 2001; Lupas and Martin, 2002). The function of Hsp104p's other domains, which are much more variable between different members of the AAA+ superfamily, and how they cooperate with the NBDs to accomplish varied protein-remodeling actions, remain mysterious.
As an alternative to using site-directed mutagenesis to investigate the functions of domains of predefined importance, here we undertook the first genetic screen to identify other critical residues. The mutations recovered, and their analysis, establish that the middle region (MR) has crucial roles in Hsp104p function.
MATERIALS AND METHODS
Yeast Strains and Media
Plasmids (see below) were introduced into Saccharomyces cerevisiae strain LP112, W303a (an isogenic haploid), or SL304A (a W303a derivative with HSP104 codons 1–321 replaced by LEU2) (Table 1). To allow use of LEU2 as an auxotrophic marker in suppressor screens, a new Δhsp104 strain, A3224, was created in which HSP104 codons 18–892 in W303a were replaced by kanr by using pFA6a-kanMX4 (Wach et al., 1994) as template to generate the desired polymerase chain reaction (PCR) product; the screen itself was conducted in a modified variant, A3330, with a galactose-inducible LacZ reporter integrated at the HIS3 locus. The pRS303-LacZ reporter was created for this study by transferring LacZ (BamHI/XbaI) from pCM171 (Gari et al., 1997) to pRS303:GAL1–10. Strains A3685 and A3686 were derived from the originally isolated suppressor strains by mating each to W303α, sporulating, and selecting strains that maintained the suppressor phenotype but had lost the transposon insertion and LacZ reporter.
Table 1.
Strains used in this study
| Strain | Genotype | Source |
|---|---|---|
| E. coli | ||
| KC8 | r-m+ leu B600 trpC9830 ΔLacX74 strA galUK pyrF::Tn5 his B463 | M. Casadaban |
| DH10B | F-mcrA Δ(mrr-hsdRMS-mcrBC) φ80dlacZΔM15 ΔlacX74 deoR recA1 endA1 araΔ139 D(ara, leu)7697 galU galK λ-rpsL nupG | Invitrogen |
| S. cerevisiae | ||
| LP112 | MAT a/α leu2-3,112/leu2-3,112 trp1-1/trp1-1 ura3-1/ura3-1 ade2-1/ade2-1 his3-11,15/his3-11,15 lys2Δ/lys2Δ can1-100/can1-100 | R. Rothstein |
| W303a | MAT a leu2-3,112 trp1-1 ura3-1 ade2-1 his3-11,15 lys2Δ can1-100 | R. Rothstein |
| W303α | MAT α leu2-3,112 trp1-1 ura3-1 ade2-1 his3-11,15 lys2Δ can1-100 | R. Rothstein |
| SL304A | MAT a leu2-3,112 trp1-1 ura3-1 ade2-1 his3-11,15 lys2Δ can1-100 hsp104::LEU2 | Sanchez and Lindquist, 1990 |
| A3224 | MAT a leu2-3,112 trp1-1 ura3-1 ade2-1 his3-11,15 lys2Δ can1-100 hsp104::KanMX4 | See text |
| A3330 | MAT a leu2-3,112 trp1-1 ura3-1 ade2-1 his3-11,15::HIS3 (GAL1-LACZ) lys2Δ can1-100 hsp104::KanMX4 | See text |
| A3685 | MAT a leu2-3,112 trp1-1 ura3-1 ade2-1 his3-11,15 lys2Δ can1-100 hsp104::KanMX4 cdc12K351N | See text |
| A3686 | MAT a leu2-3,112 trp1-1 ura3-1 ade2-1 his3-11,15 lys2Δ can1-100 hsp104::KanMX4 cdc12E368Q | See text |
Transformants were maintained on synthetic dextrose medium (SD) (Schirmer et al., 1994). Raffinose (SR) or galactose (SG) replaced dextrose in this medium to preadapt and induce GAL1-regulated plasmids, respectively. (Raffinose relieves glucose repression, allowing more rapid induction by galactose.) In strains that also carried plasmid GAL4.ER.VP16 (encoding a chimeric transcriptional activator), galactose-regulated 104b-U1 plasmids were induced in SD by 10 nM β-estradiol. MET14-regulated genes were induced by washing and incubation in SD lacking methionine.
Plasmids
A URA3-selected plasmid carrying GAL1-regulated HSP104 was used for the mutagenesis screen (Table 2, pYS-GAL104). Subsequent experiments used a modified HSP104 plasmid that had three guanosine nucleotides inserted before the ATG to lower basal expression in both yeast and Escherichia coli. This plasmid also had additional unique restriction endonuclease sites every ∼500 base pairs that facilitated insertion of directed mutations but did not change the protein sequence (HSP104R; Schirmer and Lindquist, 1998). The GAL1:10 promoter was inserted between the BamHI and EcoRI sites of the polylinker of the pRS300 series vectors (Sikorski and Hieter, 1989), converting pRS303 into pRS303:GAL1–10, pRS313 into pLA1, pRS315 into pRS315:GAL1-10, and pRS316 into pRS316:GAL1-10. HSP104R (BamHI to the SacI site after the poly A) was then inserted behind the GAL1 promoter in the modified vectors, converting pLA1 into pGALSc104, pRS315:GAL1-10 into HSP104-b/Leu, and pRS316:GAL1-10 into 104b-U1.
Table 2.
Plasmids used
| Name | Genea | Promoter | Featuresb | Use | Source |
|---|---|---|---|---|---|
| pYS-GAL104 | HSP104 | GAL1 | CEN, URA3 | Original screen | Sanchez and Lindquist 1990 |
| pLA1 | none | GAL1 | CEN, HIS3 | Vector control for pGALSc104 | L. Arwood and S. Lindquist |
| pGALSc104 | HSP104/mutants | GAL1 | CEN, HIS3 | Directed mutants | See text |
| pRS316:GAL1-10 | none | GAL1 | CEN, URA3 | Vector control for 104b-U1 | J. Taulien and S. Lindquist |
| I04b-U1 | HSP104/mutants | GAL1 | CEN, URA3 | Directed mutants | See text |
| pRS315:GAL1-10 | none | GAL1 | CEN, LEU2 | Vector control for HSP104-b/Leu | J. Taulien and S. Lindquist |
| HSP104-b/Leu | HSP104/mutants | GAL1 | CEN, LEU2 | Suppressor screen | Ter-Avanesyan et al., 1993 |
| TM104b | HSP104/mutants | MET14 | CEN, TRP1 | Alternative induction system | E. Schirmer and S. Lindquist |
| GAL4.ER.VP16 | GAL4/ER/VP16 | ADH | CEN, HIS3 | Estradiol induction of GAL1 plasmids | Louvion et al., 1993 |
| pEMBL-Δ3AUG | SUP35 C-terminus | SUP35 | 2μm, URA3 | Test suppression | Ter-Avanesyan et al., 1993 |
| pRS303-LacZ | LacZ | GAL1 | none, HIS3 | Integration into suppressor strain | See text |
| CDC12-WT | CDC12/mutants | CDC12 | CEN, URA3 | Suppressor screen | See text |
| GAL-CDC12 | CDC12/mutants | GAL1 | CEN, URA3 | Suppressor screen | See text |
| GFP-CDC3 | GFP-CDC3 | CDC3 | CEN, URA3 | Septin visualization | M. Longtine |
| GFP-CDC12 | GFP-CDC12 | CDC12 | CEN, URA3 | Septin visualization | See text |
Plasmids containing directed mutants are indicated in the text by the plasmid name with the mutation in parentheses, e.g., pGALSc104 carrying a mutant G217S/T499I allele is designated pGALSc104(G217S/T449I).
Features listed are yeast replication (CEN is low copy number and 2μm is high copy number) and auxotrophic selection.
Individual substitutions were introduced into each ∼500-base pair segment from HSP104R in pBluescript KS+ by using the Muta-Gene M13 in vitro mutagenesis kit (Bio-Rad, Hercules, CA). The segments were completely sequenced and then replaced into the corresponding region of pGALSc104, HSP104-b/Leu, or 104b-U1.
The wild-type CDC12 and suppressor cdc12K351N and cdc12E368Q genes were amplified with and without 266 base pairs of upstream sequence (this 266 base pairs contains at least the partial endogenous promoter) from strains A3330, A3685, and A3686, respectively (Table 1). Using primers that added BamHI and XbaI sites, the amplified genes were cloned into pRS316 (266 base pairs endogenous promoter; plasmid CDC12-WT) and pRS316:GAL1–10 (exogenous promoter; plasmid GAL-CDC12). GFP-CDC12 was constructed by introducing a silent mutation into codon 2 of CDC12 (AGT → AGC) in plasmid CDC12-WT to create an Eco47 III site into which green fluorescent protein (GFP) (amplified from plasmid GFP-CDC3) was blunt-end cloned.
Mutagenesis Screen
pYS-GAL104 (10 μg of DNA/500-μl reaction) was mutagenized by incubation with 1 M hydroxylamine (pH 5.5–6.5) at 75°C for 45–90 min (Busby et al., 1982). After phenol-chloroform purification, 1/100 of each preparation was transformed into KC8 bacteria (Table 1), and colonies were replica plated to +/– uracil plates to assess the mutagenesis frequency in the URA3 marker. (URA3 rescues the bacterial pyrF mutation, similar to URA3 auxotrophic selection in yeast.) The remainders of two preparations, with only small (1–3%) losses of URA3 function, were transformed into yeast strain LP112, and it was predicted from the URA3 mutation rate that only single substitutions would be isolated in HSP104 (Sikorski and Boeke, 1991). Transformants were selected on –Ura glucose plates and replica plated onto two –Ura glucose (for HSP104 repression) and two –Ura galactose (for HSP104 induction) plates. Cells were incubated at 25 or 37°C for 3–5 d, and strains exhibiting a significant reduction in growth on galactose compared with strains containing unmutagenized plasmid were retested quantitatively by growing in liquid SD to mid-log phase, equalizing cell densities, and spotting 5 μl of fivefold serial dilutions onto –Ura glucose and –Ura galactose plates.
To eliminate one class of false positives (growth inhibition on galactose due to general respiratory deficiency), colonies on glucose plates were overlaid with 20 ml of 0.75% Bacto agar in 0.067 M phosphate buffer, pH 7.0, containing 0.1% 2,3,5-triphenyltetrazolium chloride (Colson et al., 1974). After 3 h, colonies were scored as respiration positive (red) or negative (white). A second class of false positives (carrying extraneous growth-inhibiting genomic mutations) was eliminated by testing to ensure that the growth-inhibition phenotype depended upon maintenance of the HSP104 plasmid, using 5-fluoro-orotic acid to select for plasmid loss (Boeke et al., 1984).
To determine the DNA sequences of the mutant plasmids, two overlapping fragments were amplified from yeast by using HSP104 and vector primer combinations. PCR products were completely sequenced on both strands, and regions containing mutations were resequenced from independent amplifications.
Suppressor Screens
Two different suppressor screens were conducted in strain A3330 (Table 1) carrying plasmid HSP104(G217S/T499I)-b/Leu. For the first screen, ∼4 × 109 cells were transformed with 200 μg of a galactose-inducible genomic library (ATCC 87311; Ramer et al., 1992), plated on –Leu glucose and then replicaplated to –Leu galactose. Screening of ∼106 colonies yielded 534 suppressor candidates. None passed a secondary screen for restoration of the Hsp104pG217S/T499I phenotype upon loss of the library plasmid. For the second screen, ∼108 cells were transformed with the mTN-3 × HA/lacZ library (provided by M. Snyder; Ross-Macdonald et al., 1997), plated on –Ura –Leu glucose, and then replica plated to –Ura –Leu galactose. Screening ∼105 transformants yielded 90 strong candidate suppressors. Only nine of these strains retained expression of the mutant Hsp104p, and seven could not suppress a freshly introduced HSP104(G217S/T499I)-b/Leu plasmid.
The two remaining bona fide suppressors were due to random genomic mutations and were not dependent on an integrated transposon. To isolate these, a library was constructed from suppressor strain A3685 to identify the dominant suppressor. Genomic DNA was partially digested with Sau 3AI, and fragments of 4–10 kb were purified and ligated to BamHI-cut YCp50 (Rose et al., 1987). The resulting plasmids were amplified in Electromax DH10B cells (Invitrogen, Carlsbad, CA) and transformed into A3224 cells carrying HSP104(G217S/T499I)-b/Leu, and ∼105 colonies were screened for suppression of the Hsp104pG217S/T499I growth phenotype.
Thermotolerance and ATPase Assays
Heat-stress treatments were conducted as described previously (Schirmer et al., 1994). Briefly, mutant and wild-type proteins encoded in the pGALSc104 plasmid were induced in strain SL304A for 4 h in mid-log phase, and cells were then heat shocked at 50°C with or without a preconditioning treatment at 37°C, fivefold serially diluted, and spotted onto SD plates at 25°C to repress further expression of the mutant Hsp104p. Induced cells that did not receive a heat stress were plated onto SD and SG to confirm the growth-inhibition phenotype of the plasmid they contained. Experiments were repeated at least three times with strains isolated from independent transformations, yielding similar results in all cases.
Proteins for ATPase assays were purified from E. coli as described by Schirmer and Lindquist (1998) by using the pET28a expression vector (Novagen, Madison, WI), and the assays were performed as described by Schirmer et al. (1998).
Microscopy
SL304A cells carrying pGALSc104, its mutant variants, or the parent vector were induced from mid-log phase for 7–13 h in SG. Cells were fixed for 1 h in 3.7% formaldehyde and permeabilized after cell wall removal with Triton X-100, as described by Pringle et al. (1991). Next, the cells were incubated with anti-Hsp104p antibody 8–1 (polyclonal to the C-terminal 15 residues; Parsell and Lindquist, unpublished data), 4G-10 (monoclonal to the MR; Jison, Ramakrishnan, and Lindquist, unpublished data), or anti-septin antibodies (affinity purified, polyclonal) specific for Cdc11p (Ford and Pringle, 1991) or Cdc3p (Kim et al., 1991). After incubation with the appropriate secondary antibodies (Organon-Teknika, Durham, NC), images were obtained either on an Olympus epifluorescence microscope and scanned from 35-mm slides, or on an Axioplan 2 microscope interfaced with a LSM410 confocal module (Carl Zeiss, Thornwood, NY) microscope with Openlab 2.25 software.
The same strain and plasmids used for the light/fluorescence microscopy were similarly induced before fixation for electron microscopy. The electron microscopy was carried out as described previously (Parsell et al., 1994).
Wild-type (A3224) and suppressor (A3685, A3686) strains carrying the GFP-CDC3 and HSP104(G217S/T499I)-b/Leu plasmids were analyzed live after a 12-h induction by using the Lab-Tek II chambered #1.5 German coverglass system (Nalge Nunc International, Naperville, IL). Images were obtained using an Axiovert S100TV microscope (Carl Zeiss) interfaced to a Bio-Rad MRC1024 confocal system. All images were processed using Photoshop 6.
RESULTS
Screen for Mutations That Perturb the Regulated Function of Hsp104p
Our approach was based on the observation that although Hsp104p has powerful protein-remodeling functions, it can be overexpressed at high levels without affecting growth (Lindquist and Kim, 1996); thus, its broad capacity to interact with varied substrates and remodel them must normally be tightly controlled. Plasmids carrying HSP104 were randomly mutagenized to determine whether Hsp104p is under tight regulation, and if so, to identify critical regulatory regions and residues. The mutants were screened for the capacity to inhibit growth at normal temperatures. Such a phenotype would reflect disruption of the normally tight controls on Hsp104p activities.
Pools of plasmid carrying HSP104 under the control of a galactose-inducible promoter (GAL1) were chemically mutagenized to various extents (see MATERIALS AND METHODS). To favor the recovery of single point mutations, the frequency of mutations in a marker on the same plasmid was used to select a pool with a relatively low mutation rate. Wild-type cells transformed with these plasmids were screened for the ability to grow on glucose (which represses GAL1 expression) but not on galactose (which induces expression). We used a diploid strain to reduce the recovery of extraneous mutations that simply perturb galactose metabolism; most such mutations are recessive. Of ∼3100 transformants screened, 35 had strong growth-inhibition phenotypes on galactose. Each was put through several secondary screens to eliminate false positives and to eliminate mutations that had complex interactions with spontaneous genomic mutations (see MATERIALS AND METHODS for details). Of the 22 mutants passing these secondary screens, five were inhibited for growth on galactose at either 25 or 37°C, whereas 17 were inhibited for growth at 37°C but not at 25°C.
Positions of Mutations
Unexpectedly, although wild-type HSP104 plasmids are readily recovered from E. coli (Schirmer et al., 1994), the mutant plasmids were not. Further experiments indicated that this was due to toxicity of the mutant proteins in E. coli. The GAL1 promoter is generally silent in this organism, but we found that sequences in the leader region of HSP104 promoted transcription and, hence, protein expression (Schirmer and Lindquist, unpublished data). This was innocuous with the wild-type protein, but lethal with the mutants. To circumvent this problem, the mutant genes were recovered directly from yeast (where tight regulation of the galactose promoter kept them silent) by PCR (see MATERIALS AND METHODS). We focused on analysis of six mutants, representing the two growth phenotypes (five that inhibited growth at 25 or 37°C and one that inhibited growth at 37°C but not 25°C). These six mutants contained a total of 30 nucleotide changes, 22 of which created amino acid substitutions (Figure 1). Among them, substitutions at three residues in the 908-amino acid protein were recovered more than once, suggesting that they are particularly important in Hsp104p function. Notably, despite the low rate of mutagenesis observed within the marker gene in the same plasmid, every sequenced Hsp104p mutant contained multiple substitutions. This suggested that more than one mutation might generally be required to produce a dominant growth-inhibition phenotype for Hsp104p, a suggestion confirmed by further analysis (see below).
Ten of the 22 amino acid altering mutations occurred in the first nucleotide-binding domain (NBD1); four of these were in or very near the Walker A and B consensus sequences of the highly conserved phosphate-binding loop (P-loop; Walker et al., 1982; Saraste et al., 1990; Leipe et al., 2002). Two occurred in the putative sensor 2 consensus of NBD1, which is thought to be located after the MR (Neuwald et al., 1999). Unexpectedly, seven of the remaining 12 substitutions clustered in the highly variable MR. In fact, every mutant recovered in the screen for disruption of Hsp104p regulation had at least one MR mutation.
Although the amino acid sequence of the MR is highly variable, one small segment of 11 residues (Figure 2) exhibits moderate conservation among those members of the HSP100 family that function in thermotolerance (including plant, bacterial, and fungal members; Gottesman et al., 1990; Schirmer et al., 1996). Five of the seven MR mutations recovered in our screen were located in this small conserved segment (Figures 1, open box under MR; and 2).
Figure 2.

Mutations in the conserved sequence element within the MR. The core of this element, KAAXLR, occurs in both the B and C subfamilies of HSP100/Clp proteins; however, each has its own variation. h, a hydrophobic residue.
All mutant proteins that affected growth at both temperatures carried at least one NBD mutation in addition to the MR mutation. The mutant that affected growth only at 37°C did not contain an NBD substitution. We chose one mutant from each of the two growth-inhibition categories for further detailed analysis. G217S/T499I was selected because it was independently recovered twice in the screen (the second time with an additional mutation; Figure 1) and had a relatively small number of substitutions. A503V/A509D was selected because its ability to block growth was temperature dependent.
General Characterization of Mutants
We first confirmed that the Hsp104p mutations were solely responsible for the selected phenotypes by recreating the mutant alleles through directed mutagenesis in fresh plasmids and fresh strains (our unpublished data). Next, we asked whether the growth-inhibition phenotype was dependent on either the high levels of expression characteristic of the GAL1 promoter or some feature of galactose metabolism or galactose-regulated gene expression. The mutants were transferred to galactose expression vectors with different selectable markers (plasmids pGALSc104, 104b-U1, and HSP104-b/Leu; Table 2). A hormone-responsive promoter (Louvion et al., 1993) and a methionine-regulated promoter (Korch et al., 1991) were tested as alternative induction systems (see MATERIALS AND METHODS). The phenotypes of the two mutants were reproduced with all induction systems, which varied >20-fold in the levels of protein they produced (our unpublished data), and with different selectable markers. Thus, the mutant phenotypes do not require high levels of protein expression and are not specific to any particular nutritional state.
Third, we addressed the possibility that the phenotypes were not due to the activities of the mutant proteins themselves, but rather to their ability to form mixed complexes with wild-type Hsp104p and perturb its function. Because Hsp104p functions in a hexameric complex, and the strain used in the screen expressed wild-type Hsp104p, mixed hexamers were probably formed. We compared plasmid-dependent phenotypes in isogenic wild-type and hsp104 deletion strains (Figure 3A). Each of the phenotypes was recapitulated in both genetic backgrounds: Hsp104pG217S/T499I inhibited growth at 25 or 37°C, and Hsp104pA503V/A509D inhibited growth only at 37°C. However, both growth defects were more severe in the Δhsp104 background (Figure 3A, 37°C; our unpublished data). We conclude that the loss-of-growth phenotypes were not dependent upon interaction with wild-type Hsp104p, and, indeed, were partially attenuated by its presence.
Figure 3.

Dominant lethal phenotypes of mutants. (A) HSP104 mutants inhibited growth. Wild-type (W303a) and Δhsp104 mutant cells (SL304A) carrying pGALSc104, its mutant variants, or the pLA1 vector control plasmid were first grown in noninducing liquid medium. Equal numbers of cells were then spotted onto galactose plates in fivefold serial dilutions (shown) to induce expression of the mutant proteins, or onto glucose plates to repress synthesis of the mutant proteins. No inhibition of growth was seen on the glucose plates (not shown). (B) Sorbitol suppressed the Hsp104pA503V/A509D mutant but not the Hsp104pG217S/T499I mutant. SL304A (Δhsp104) cells carrying the same plasmids and grown as in A were plated on galactose medium with or without sorbitol.
Because the mutant proteins produce dominant gain-of-function phenotypes, we asked whether their effects were due to an alteration in their interaction with Sup35p, the only known essential substrate of Hsp104p. Sup35p, a translation-termination factor, is the protein determinant of the yeast prion [PSI+] (reviewed in Serio and Lindquist, 2000). The C-terminal region of Sup35p contains the essential translation activity (Ter-Avanesyan et al., 1993), and the N-terminal region confers upon Sup35p the capacity to assume distinct prion and nonprion conformations. The change between these states is regulated by interactions between the N-terminal domain and Hsp104p (Patino et al., 1996; Schirmer and Lindquist, 1997; Cashikar et al., 2002). When Sup35p prion conversion is too efficient it can be toxic (Ter-Avanesyan et al., 1993; Derkatch et al., 1996; Li and Lindquist, 2000), because the essential translation-termination activity of the C-terminal domain is inhibited when the protein is in the [PSI+] state. Thus, it is possible that the gain-of-function mutant HSP104 phenotypes were due to excess conversion of Sup35p to the prion state. To examine this possibility, we introduced a high-copy plasmid expressing the Sup35p C-terminal domain (Table 2), which does not enter the prion state, into the mutant cells. Expression of the C terminus alone did not mitigate the phenotype of either Hsp104p mutant (our unpublished data). Therefore, the growth inhibition caused by the Hsp104p mutants is not due to an enhancement of Sup35p prion conversion.
Finally, we explored the basis of the temperature sensitivity of the A503V/A509D allele. When Δhsp104 cells carrying the A503V/A509D mutant plasmid were grown at 37°C on medium containing 0.5 M sorbitol, growth was restored (Figure 3B). Because sorbitol stabilizes protein structure (Gekko and Ito, 1990), this suggested that the mutant phenotype was caused by an effect of temperature on the structural state of the protein. Sorbitol can also stabilize cells against cell lysis defects, but mutant cells had no such defects. Further suggesting that the temperature sensitivity of the Hsp104pA503V/A509D phenotype involves a protein-folding problem, it was partially suppressed by overexpression of the chaperones Hsp90p and Sis1p (an HSP40 member; our unpublished data). In contrast, neither sorbitol nor the chaperone proteins alleviated the non-temperature-sensitive phenotype of Hsp104pG217S/T499I.
Effects of Individual Substitutions on Growth
The fact that the mutants recovered in our original screen contained more substitutions than expected (see MATERIALS AND METHODS) suggested that more than one mutation may generally be required to produce a dominant growth-inhibiting form of Hsp104p. To investigate this, we recreated the individual amino acid substitutions in two plasmids, pGALSc104 and 104b-U1 and compared the phenotypes they produced with those of the original double mutants in both wild-type (our unpublished data) and Δhsp104 (Figure 4) strain backgrounds.
Figure 4.

Phenotypes of individual mutations. Left, lethal phenotypes. pGALSc104 plasmids carrying the individual or double substitutions were tested in strain SL304A (Δhsp104) on inducing medium. Cells were grown at 25°C (top) or 37°C (bottom). Right, thermotolerance phenotypes. Using the same strain and plasmids as for the growth assessments, mutant proteins were first induced for 4 h. Cells were then pretreated at 37°C for 30 min (to induce other tolerance factors) before an extreme heat stress of 50°C for 30 min. To determine survival rates, cells were plated at 25°C on glucose medium (which repressed further synthesis of the mutant protein). All were plated in fivefold serial dilutions.
Neither G217S nor T499I alone produced a growth inhibition at either 25°C (Figure 4, top left) or at 37°C (our unpublished data). The single mutation A503V was sufficient to recapitulate the growth-inhibition phenotype at 37°C. A509D alone produced no significant growth defect (Figure 4, bottom left). In some experiments, with longer incubations or higher cell densities, the A509D mutation enhanced the severe growth defect caused by A503V (our unpublished data).
We also tested individual substitutions from other mutants recovered in the screen (A392T, A551T, P679L, and T726I). None produced a growth-inhibition phenotype on its own at either 25 or 37°C (our unpublished data). This suggests that with rare exceptions, such as the A503V mutation at high temperatures, gain-of-function growth-inhibiting phenotypes require more than one mutation in Hsp104p.
Effects of Mutations on Thermotolerance Function
Next, we asked what effects the mutations had on Hsp104p's ability to perform its normal functions. Preliminary results indicated that the mutations affected propagation of the prion [PSI+] (Chernoff, Schirmer, and Lindquist, unpublished data). However, defining the molecular nature of these effects was complicated by the fact that prion maintenance is disrupted by either too much or too little Hsp104p function. The relationship between Hsp104p and thermotolerance is simpler: the higher the concentration of Hsp104p, the greater the thermotolerance (Lindquist and Kim, 1996).
Thermotolerance assays for Hsp104 function were possible with the mutant proteins because such assays use short-term inductions, whereas growth-inhibition phenotypes require many hours of induction. Accordingly, mutant proteins were induced with galactose to a level comparable with that obtained with wild-type Hsp104p after a strong tolerance-inducing preheat treatment. Cells were then exposed to a severe heat shock at 50°C and plated on glucose medium at normal temperatures to repress further expression of the mutant proteins and to determine whether they had been able to increase the number of cells surviving the heat shock.
Neither mutant protein had any effect on the thermotolerance function of the endogenous Hsp104p (our unpublished data). In a Δhsp104 background, neither Hsp104pG217S/T499I nor Hsp104pA503V/A509D was capable of providing thermotolerance (Figure 4, right). Surprisingly, the individual constituent mutations had very different effects (Figure 4, right). Protein containing only the T499I mutation had thermotolerance function, but protein containing only the G217S mutation did not. Protein containing only the A503V substitution, which severely inhibits growth when continuously expressed at 37°C (Figure 4, left), conferred wild-type levels, or greater, of thermotolerance (Figure 4, right). In contrast, protein containing only the A509D substitution, which did not inhibit growth, had a greater than 50-fold reduction in thermotolerance function relative to wild-type Hsp104p. Similar results were obtained with (Figure 4, right) and without (our unpublished data) a preconditioning treatment at 37°C, that is, with or without other heat-inducible factors.
MR Mutations Alter ATP Hydrolysis
Because Hsp104p function requires ATP hydrolysis (Parsell et al., 1991; Schirmer et al., 1998), we examined the effects of the mutations on ATPase activity. Due to difficulties in recovering the G217S/T499I double-mutant protein from bacteria and yeast cells, we confined the analysis to the individual mutations.
The G217S substitution drastically lowered activity (our unpublished data); this was not unexpected because a G217V substitution (produced by site-directed mutagenesis, Schirmer et al., 1998) lowers the ATPase activity of Hsp104p to undetectable levels. Hsp104pT499I exhibited reduced ATP hydrolysis (less than one-half that of wild-type; Figure 5A), whereas Hsp104pA503V exhibited significantly elevated basal ATPase activity at 37°C (Figure 5A; Cashikar et al., 2002). Hsp104pA509D hydrolysis rates were indistinguishable from those of wild-type protein (Figure 5, A and B). Thus, the distinct phenotypes of the MR substitutions are associated with distinct biochemical characteristics.
Figure 5.

ATPase activities of proteins carrying single substitutions. Wild-type Hsp104p, Hsp104pT499I, Hsp104pA503V, and Hsp104pA509D proteins were purified from E. coli. (A) Basal ATP hydrolysis rates of each protein were assayed in a physiological salt buffer by release of Pi from ATP. (B) To establish that the similarity in ATP hydrolysis rates between Hsp104pA509D and wild-type Hsp104p is not restricted to early time points, they were compared over a range of time.
Aberrant Morphology Produced by Hsp104pG217S/T499I but Not by Hsp104pA503V/A509D
Hsp104pG217S/T499I produced a striking morphological abnormality: cells arrested with elongated, often grossly distended buds in both wild-type and Δhsp104 backgrounds (Figure 6A; our unpublished data). Electron microscopy revealed abundant, abnormally placed cell wall material (Figure 6B) similar to that observed in cdc3 mutants (Slater et al., 1985). In contrast, Hsp104pA503V/A509D produced no morphological changes that were obvious, by either light or electron microscopy (our unpublished data).
Figure 6.

Cells that express proteins carrying the T499I substitution in combination with NBD substitutions exhibit elongated buds with misplaced cell-wall deposition. (A) Differential interference contrast micrographs of Δhsp104 cells (strain SL304A) carrying the parent vector or expressing Hsp104p, Hsp104pG217S/T499I, or Hsp104pA392T/T499I/P679L. Elongated buds began to occur after 5–6 h of mutant protein expression. Cells were fixed at ∼10 h. Bars, 10 μm. (B) Electron microscopy of the hsp104G217S/T499I mutant cells, grown as described in A. Material resembling cell wall was deposited in strands or sheets within the cell and between the mother and daughter cells. Bars, 2 μm.
The similarity of the Hsp104pG217S/T499I phenotype to that produced by mutant septin proteins (Hartwell, 1971; Slater et al., 1985; Longtine et al., 1996; Gladfelter et al., 2001) led us to investigate the effect of the mutant Hsp104p on septin localization. To do so, we used antibodies specific to two primary structural components of the septin ring, Cdc11p (Figure 7) and Cdc3p (our unpublished data) (Ford and Pringle, 1991; Kim et al., 1991). In cells arrested by Hsp104pG217S/T499I, the septins often failed to localize to the bud neck and sometimes formed linear strands or rings in aberrant locations rather than normal rings at the neck (Figure 7, A and B).
Figure 7.
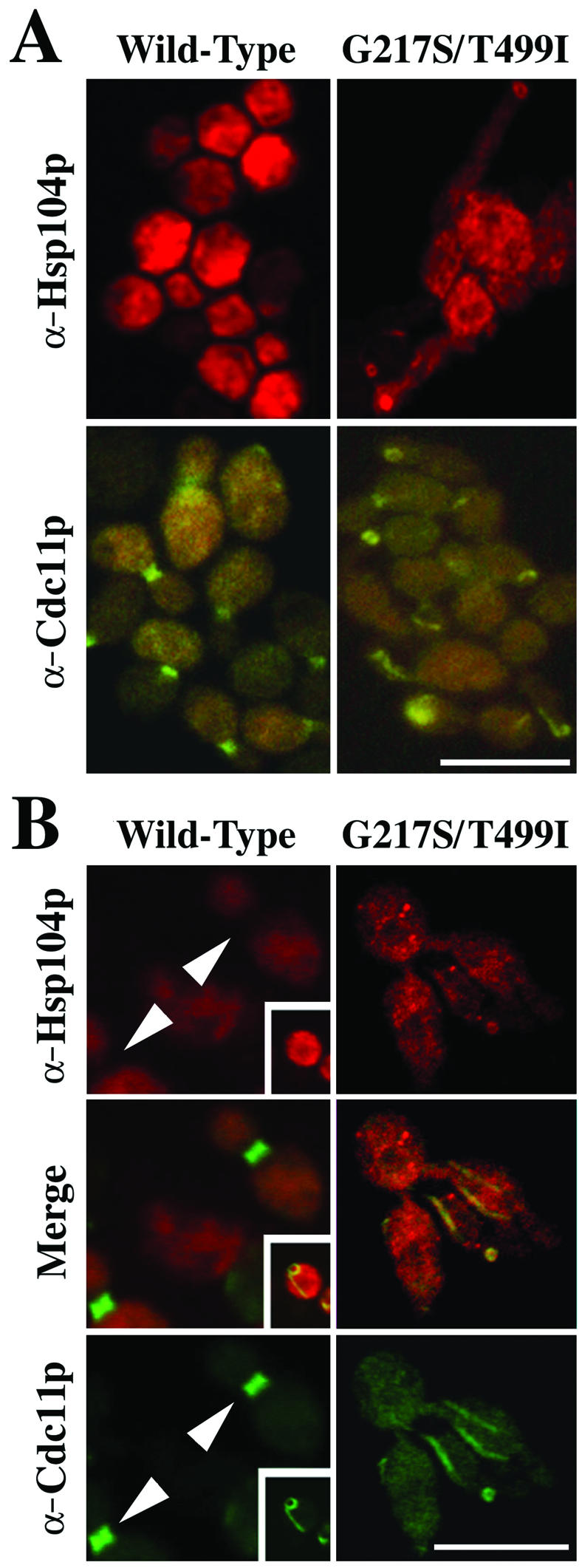
Abnormal septin organization and colocalization of Hsp104pG217S/T499I with the septins in cells expressing the mutant, but not the wild-type, Hsp104p. SL304A (Δhsp104) cells expressing wild-type Hsp104p (left) or Hsp104pG217S/T499I (right) were examined. (A) Cells were stained with polyclonal antibodies that recognize either the C terminus of Hsp104p (top) or the Cdc11p septin (bottom). Cells were fixed after ∼9 h of expression of wild-type or mutant protein. No difference was observed between cells processed with formaldehyde/Triton or with MeOH/acetone. Images were obtained with an Olympus epifluorescence microscope. (B) Cells were stained with an anti-Hsp104p monoclonal antibody against the Hsp104p MR region and the anti-Cdc11p affinity-purified polyclonal antibody. Arrowheads, regions of septin rings, showing little staining for Hsp104p. Inset, example of rare cells with mislocalized septins in a cell overexpressing wild-type Hsp104p. Cells were fixed after ∼12 h of expression. Photomicrographs are confocal sections. Bars, 10 μm.
To determine whether Hsp104pG217S/T499I colocalizes with the misplaced septins, cells were stained with antibodies specific to either the C terminus or the middle region of Hsp104p (Cashikar et al., 2002). Both antibodies gave the same pattern: cells expressing Hsp104pG217S/T499I often had strong staining of aberrant ring structures superimposed on diffuse general staining (Figure 7, A and B). These rings resembled (Figure 7A) and seemed to colocalize with (Figure 7B) the aberrant septin rings found in the same cells. In contrast, cells expressing either Hsp104p or Hsp104pA503V/A509D exhibited only diffuse Hsp104p staining (Figure 7; our unpublished data). The diffuse staining of wild-type Hsp104p was absent at the septin rings (Figure 7B, arrowheads). However, overexpression of wild-type Hsp104p did cause septin mislocalization in some (<5%) cells (Figure 7B, insets), suggesting that Hsp104p does have a very weak intrinsic potential to interact with the septin ring.
Mutations in CDC12 Partially Suppress the G217S/T499I Phenotype
To investigate the nature of the interactions of Hsp104pG217S/T499I with the septins, we conducted two types of screens for suppressors of the Hsp104pG217S/T499I-induced growth inhibition. First, we screened for proteins that would suppress the mutant phenotype when overexpressed. In a screen of approximately 1 million transformants from a galactose-inducible genomic expression library, no suppressors were isolated (see MATERIALS AND METHODS). Second, using a mini-transposon insertion library, we searched for proteins whose deletion or truncation would suppress the mutant phenotype. Screening of ∼105 transformants yielded two strains in which growth was restored (see MATERIALS AND METHODS).
In both cases, suppression of the Hsp104pG217S/T499I-induced growth defect was dominant when the haploid suppressor strain was mated to a wild-type strain, and each diploid yielded 2:2 segregation of the suppressor in all six tetrads dissected. Surprisingly, suppression was unlinked to the transposon insertion. Thus, the suppression must have been due to spontaneous genomic mutations that were unrelated to the insertional mutagenesis. When the two suppressor strains were mated and sporulated, all progeny exhibited the suppressor phenotype, suggesting that the two independently isolated suppressors mapped to the same locus.
To identify this locus, we constructed a genomic library from one of the suppressor strains and transformed it into cells expressing Hsp104pG217S/T499I (see MATERIALS AND METHODS). Transformants were screened for suppression of the Hsp104pG217S/T499I loss-of-growth phenotype on galactose medium. Three plasmids that conferred suppression were sequenced. All carried a mutant allele of the septin gene CDC12 (K351N) in which the lysine was changed to asparagine at residue 351, adjacent to the predicted coiled-coil domain of Cdc12p (amino acids 356–407 as defined by BioKnowledge Library [Incyte Palo Alto, CA]; amino acids 370–407, as defined using the algorithm of Wolf et al., 1997). Sequencing the CDC12 locus of the second independently obtained suppressor strain revealed a mutation in the same region, E368Q, that changed glutamate to glutamine at position 368. Notably, Cdc12p is a component of the septin ring at the mother-bud neck (Haarer and Pringle, 1987).
The suppressor strains expressing Cdc12pK351N or Cdc12pE368Q grew well in the presence of Hsp104pG217S/T499I (Figure 8A). Although some cells still exhibited the elongated bud morphology, this was much less frequent and less severe than in strains expressing Hsp104pG217S/T499I without the mutant Cdc12p (Figures 6A and 8B). The ability of each CDC12 mutant to suppress the Hsp104pG217S/T499I phenotype was confirmed by subcloning the mutant CDC12 open reading frames into plasmids and retesting them for suppression. Each mutant allele, but not the wild-type CDC12 allele, suppressed the growth defect of Hsp104pG217S/T499I when expressed using either a galactose-inducible promoter or the upstream region of CDC12 itself (our unpublished data).
Figure 8.

Suppression of the G217S/T499I mutant by mutations in CDC12. (A) hsp104Δ strains A3224 (CDC12), A3685 (CDC12K351N), and A3686 (CDC12E368Q) carrying plasmid HSP104(G217S/T499I)-b/Leu were grown to mid-log phase in SD and plated on SG inducing medium at 30°C in fivefold serial dilutions. (B) Strains used in A were transformed with the additional plasmid GFP-CDC3, grown to late log phase in SD, washed, and induced in SG for ∼13 h. Cells were viewed live by confocal microscopy. Pictures are merged differential interference contrast and GFP images. Bar, 10 μm. Insets, cells at higher magnification with arrows denoting Cdc3p septin fluorescence. Linear septin strands were only observed in the presence of wild-type (WT) Cdc12p; mislocalized septin rings were observed with WT and occasionally with the mutant forms of Cdc12p.
To determine whether the suppressors restored normal septin localization in cells expressing Hsp104pG217S/T499I, we used GFP fused at or near the N terminus of either wild-type Cdc3p (Figure 8B) or wild-type Cdc12p (our unpublished data). Both proteins localized normally in wild-type and CDC12 suppressor strain backgrounds in either the presence or absence of wild-type Hsp104p (our unpublished data). However, in cells expressing Hsp104pG217S/T499I, the GFP-septins were mislocalized in virtually all cells expressing wild-type Cdc12p, but in only a fraction of those expressing the suppressor mutants Cdc12pK351N or Cdc12pE368Q (Figure 8B).
The identification of mutant septin alleles as efficient suppressors of Hsp104pG217S/T499I indicates that the toxic effect of Hsp104pG217S/T499I is directly linked to the septin defects observed after its induction. The lack of a visible septin defect in strains expressing Hsp104pA503V/A509D suggested that this mutant protein has a different mode of toxicity. Indeed, the Cdc12p mutations (K351N and E368Q) did not suppress the loss-of-growth phenotype of Hsp104pA503V/A509D (our unpublished data).
Specificity of the Cell Morphology Phenotype
Neither of the CDC12 mutations produced a mutant phenotype on its own. Either in the presence of wild-type Hsp104p or in its absence (a Δhsp104 strain), the cells grew at normal rates and seemed normal by light microscopy and septin staining (our unpublished data). Thus, the Cdc12p mutations did not perturb septin function. Rather, they specifically and strongly suppressed the ability of Hsp104pG217S/T499I to interact with the septin complex and perturb its function.
To determine whether the specificity of the bud morphology defect observed with Hsp104pG217S/T499I was due to the MR substitution (T499I) or to the NBD substitution (G217S), we asked whether T499I produced a similar defect when combined with other NBD mutations. The substitutions A392T and P679L (originally isolated in combination with other mutations; Figure 1) were chosen because they were located in different structural elements than G217S (found in the Walker A sequence of NBD1); A392 is located in box VII″ of NBD1 (a highly conserved sequence motif; Neuwald et al., 1999), whereas P679 occurs at the edge of the Walker B sequence of NBD2. Neither A392T nor P679L produced a thermotolerance defect on its own. In combination with each other, they produced only a partial defect (our unpublished data). More importantly, expression of these NBD mutants, like that of the G217S mutant, produced no growth defects, either alone or in combination (our unpublished data).
When the individual NBD mutations A392T or P679L were combined with the T499I substitution (Hsp104pA392T/T499I or Hsp104pT499I/P679L), no inhibition of growth was observed. However, when both mutations were combined with T499I (Hsp104pA392T/T499I/P679L), the triple mutant had the same phenotype as Hsp104pG217S/T499I: growth inhibition was observed that was not suppressed by sorbitol, elongated buds were produced with misplaced septin rings, the mutant Hsp104p colocalized with the septins, and the phenotype was suppressed by both Cdc12pK351N and Cdc12pE368Q (Figure 6A, right; our unpublished data). Thus, the specificity of this septin-assembly phenotype depended upon the MR substitution, but its manifestation also required a perturbation of NBD function.
DISCUSSION
Critical Role of the Middle Region
Using random mutagenesis, we have conducted a screen for mutations that disrupt the normally tight regulation of Hsp104p activity. Specifically, we looked for mutations that created a dominant gain-of-function effect that blocks cell growth. To our knowledge, this is the first such screen conducted with any member of the large superfamily of AAA+ proteins. It has provided important and unexpected insights, suggesting a promising strategy for study of other members of this broad family. Every mutant we isolated depended upon a substitution in the MR of Hsp104p to exert its effect. This unequivocally demonstrates the importance of the MR, a region little studied and, until recently, thought to be of little significance.
Beyond establishing the MR as an important region, our work pinpoints a small, moderately conserved sequence of 11 amino acids (residues 499–509) as being particularly critical for Hsp104p function. Remarkably, three single amino acid substitutions found in this region caused extremely diverse alterations of Hsp104p function. Hsp104pA503V strongly inhibited growth at 37°C, yet it could promote survival after extreme stress as effectively as wild-type Hsp104p. Conversely, the MR mutant Hsp104pA509D had no deleterious effect on growth but strongly impaired thermotolerance function. (It is the first Hsp104p mutation to impair thermotolerance without also reducing basal ATP hydrolysis.) The third MR mutant, Hsp104pT499I, had reduced ATP hydrolysis but was able to confer thermotolerance and did not affect growth. Strikingly, the combination of T499I with very different mutations in the NBDs of Hsp104p (G217S or A392T plus P679L) caused mislocalization of septins and associated defects in bud morphogenesis and cell wall deposition.
The importance of the MR in Hsp104p function was unexpected due to its extreme variability: it is the most diverse region among class 1 HSP100/Clp proteins, in both length and sequence (Gottesman et al., 1990; Schirmer et al., 1996). In fact, it has previously been termed, very naturally, the “spacer” region (Gottesman et al., 1990). It is also highly variable even among members of the B-type HSP100 proteins from plants, bacteria, and fungi (Schirmer et al., 1996), all of which function in stress tolerance (Figure 2). While this manuscript was in preparation, a complete deletion of the MR in the E. coli HSP100 protein ClpB was found to block the protein's chaperone activity without affecting its stability or expression levels (Mogk et al., 2003). This strongly suggests that despite variation in its sequence, the MR will prove to have important functions in all members of this large family of protein-remodeling factors.
Further biochemical analysis of the A503V substitution in our laboratory (Cashikar et al., 2002) has suggested that it disrupts the mechanism of communication between Hsp104p's two NBDs. When poly-lysine binds the C-terminal region of wild-type Hsp104p, it triggers nucleotide hydrolysis in NBD2, which in turn causes a conformational change in the MR and stimulates hydrolysis at NBD1. Protein carrying the A503V substitution binds poly-lysine but does not respond with elevated hydrolysis. The unpublished crystal structure of the Thermus thermophilus ClpB protein indicates that the MR forms a large coiled-coil domain in a position where it might influence communication between the two NBDs (Lee and Tsai, personal communication). The 11-amino acid sequence that we have identified by genetic analysis as critical for Hsp104p function lies at one apex of the coiled-coil domain. Together, genetic, structural, and biochemical data from several laboratories are beginning to produce a picture of how interdomain communication might be involved in driving the complex conformational changes that are the heart of Hsp104p function.
Molecular Explanation for a Gain-of-Function Phenotype
In addition to establishing that changes in the MR can produce dominant gain-of-function mutations in Hsp104p, we have used cell biological and genetic methods to provide a specific molecular explanation for one of the phenotypes. Hsp104pG217S/T499I affects cell morphology by interacting with the septins. This effect on morphology was unexpected for several reasons: 1) no other Hsp104 mutant analyzed to date (in this or several other studies) affects morphology; 2) deletion of HSP104 has no effect on septin localization or cell morphology, either at 25°C or at higher temperatures; and 3) even very strong overexpression of wild-type Hsp104p causes septin mislocalization in only a few cells. Either wild-type Hsp104p does not interact with septins, or Hsp104p has a weak ability to interact with septins, but this interaction is transient and unimportant under laboratory conditions. It might facilitate assembly or disassembly of the septin ring in a manner that matters under some circumstances in the wild. Regardless, the phenotype is a novel gain of function, in which a potentially very weak intrinsic capacity to interact with the septins is strongly enhanced by the T499I mutation in the MR.
An extensive search for suppressors of the Hsp104pG217S/T499I growth defect failed to uncover any high-copy suppressors or deletion mutant suppressors. Instead, two spontaneous genomic suppressor mutations were found, each residing in CDC12, which encodes a component of the septin ring (Haarer and Pringle, 1987; Longtine et al., 1996) and of the septin complex that can assemble into filaments in vitro (Frazier et al., 1998). Interestingly, Cdc12p resembles the best characterized substrate of Hsp104p, the NM domain of the Sup35p prion protein, which forms filaments and has a domain enriched in lysines (Glover et al., 1997) that influences its interaction with Hsp104p (Liu et al., 2002). The two independently isolated Cdc12p suppressor mutants, Cdc12pK351N and Cdc12pE368Q, each contained a substitution in a lysine-rich region.
Although specificity of the Hsp104pG217S/T499I mutation for interaction with the septins is dictated by the T499I substitution, the associated phenotype requires an additional impairment of Hsp104p NBD function. The fact that perturbation of septin structure by either Hsp104pG217S/T499I or Hsp104pA392T/T499I/P679L is accompanied by colocalization of the protein with the septins suggests that impairment of ATPase activity inhibits the release of binding with Cdc12p. Thus, the Hsp104pG217S/T499I:Cdc12p interaction may represent a snapshot of the normally very transient interaction between this AAA+ protein and its substrates, which has been difficult to capture with wild-type protein. In conjunction with the Cdc12p suppressors, the Hsp104pG217S/T499I mutant could serve as a valuable tool for further exploration of the determinants of Hsp104p substrate recognition.
The Evolution of AAA+ Protein Functions
A common theme in molecular evolution is the radiation of proteins with broad functions into a class of proteins that fill specific niches through alterations in their regulation and substrate selection. In some cases, such as the acquisition of a new domain, the process leading to the acquisition of a novel function is clearly defined (Patthy, 2003). It remains a major puzzle, however, for the broad superfamily of AAA+ proteins, which couple the function of their ATP-hydrolysis domains to the remodeling of a bewildering variety of substrates. Our gain-of-function mutants provide an enticing picture of how such radiations might occur. Single amino acid changes in one small region of the protein can create a variety of functionally distinct mutants. Thus, it becomes possible to imagine how subtle changes in Hsp104p MR sequence and NBD function could serve to modulate substrate recognition and the duration of substrate interactions, as we have observed in our Hsp104pG217S/T499I mutant. Hence, the apparent paradox we have uncovered, that the most variable region in the broad family of proteins to which Hsp104p belongs plays such a critical role in Hsp104p function, can be resolved. Some MR residues may vary due to low sequence constraints, but others may vary precisely because they are so important that they provide an opportunity to rapidly modulate the function and specificity of the protein during evolution.
Acknowledgments
We thank J. Pringle and M. Longtine for the generous gift of affinity-purified antibodies to Cdc11p, antiserum to Cdc3p, and the GFP-CDC3 expression construct. We also thank D. Picard and Y. Chernoff for plasmids, and D. Kim for advice on library construction. We thank D. Hattendorf, A. Cashikar, J. Feder, and in particular J. Pringle for helpful comments on the manuscript. E.C.S. was supported by Public Health Service grant 6 T32 GM-07183-19, and O.R.H. was supported by National Science Foundation Graduate Fellowship Award DGE 9616042. This work was supported by the Howard Hughes Medical Institute and the National Institutes of Health (GM-25874).
Article published online ahead of print. Mol. Biol. Cell 10.1091/mbc.E02–08–0502. Article and publication date are available at www.molbiolcell.org/cgi/doi/10.1091/mbc.E02–08–0502.
References
- Boeke, J.D., Lacroute, F., and Fink, G.R. (1984). A positive selection for mutants lacking orotidine-5′-phosphate decarboxylase activity in yeast: 5-fluoro-orotic acid resistance. Mol. Gen. Genet. 197, 345–346. [DOI] [PubMed] [Google Scholar]
- Busby, S., Irani, M., and DeCrombrugghe, B. (1982). Isolation of mutant promoters in the Escherichia coli galactose operon using local mutagenesis on cloned DNA fragments. J. Mol. Biol. 154, 197–209. [DOI] [PubMed] [Google Scholar]
- Cashikar, A.G., Schirmer, E.C., Hattendorf, D.A., Glover, J.R., Ramakrishnan, M.S., Ware, D.M., and Lindquist, S.L. (2002). Defining a pathway of communication from the C-terminal peptide binding domain to the N-terminal ATPase domain in a AAA protein. Mol. Cell 9, 751–760. [DOI] [PubMed] [Google Scholar]
- Chernoff, Y.O., Lindquist, S.L., Ono, B-i., Inge-Vechtomov, S.G., and Liebman, S.W. (1995). Role of the chaperone protein Hsp104 in propagation of the yeast prion-like factor [psi+]. Science 268, 880–883. [DOI] [PubMed] [Google Scholar]
- Colson, A.-M., Colson, C., and Goffeau, A. (1974). Systems for membrane alteration: genetic perturbations of mitochondria in a “petite-negative” yeast. Methods Enzymol. 32, 838–843. [PubMed] [Google Scholar]
- DebBurman, S.K., Raymond, G.J., Caughey, B., and Lindquist, S. (1997). Chaperone-supervised conversion of prion protein to its protease-resistant form. Proc. Natl. Acad. Sci. USA 94, 13938–13943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derkatch, I.L., Chernoff, Y.O., Kushnirov, V.V., Inge-Vechtomov, S.G., and Liebman, S.W. (1996). Genesis and variability of [PSI] prion factors in Saccharomyces cerevisiae. Genetics 144, 1375–1386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ford, S.K., and Pringle, J.R. (1991). Cellular morphogenesis in the Saccharomyces cerevisiae cell cycle: localization of the CDC11 gene product and the timing of events at the budding site. Dev. Genet. 12, 281–292. [DOI] [PubMed] [Google Scholar]
- Frazier, J.A., Wong, M.L., Longtine, M.S., Pringle, J.R., Mann, M., Mitchison, T.J., and Field, C. (1998). Polymerization of purified yeast septins: evidence that organized filament arrays may not be required for septin function. J. Cell Biol. 143, 737–749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gari, E., Piedrafita, L., Aldea, M., and Herrero, E. (1997). A set of vectors with a tetracycline-regulatable promoter system for modulated gene expression in Saccharomyces cerevisiae. Yeast 13, 837–848. [DOI] [PubMed] [Google Scholar]
- Gekko, K., and Ito, H. (1990). Competing solvent effects of polyols and guanidine hydrochloride on protein stability. J. Biochem. 107, 572–577. [DOI] [PubMed] [Google Scholar]
- Gladfelter, A.S., Pringle, J.R., and Lew, D.J. (2001). The septin cortex at the yeast mother-bud neck. Curr. Opin. Microbiol. 4, 681–689. [DOI] [PubMed] [Google Scholar]
- Glover, J.R., Kowal, A.S., Schirmer, E.C., Patino, M.M., Liu, J.-J., and Lindquist, S. (1997). Self-seeded fibers formed by Sup35, the protein determinant of [PSI+], a heritable prion-like factor of S. cerevisiae. Cell 89, 811–819. [DOI] [PubMed] [Google Scholar]
- Glover, J.R., and Lindquist, S. (1998). Hsp104, Hsp70, and Hsp 40, a novel chaperone system that rescues previously aggregated proteins. Cell 94, 73–82. [DOI] [PubMed] [Google Scholar]
- Glover, J.R., and Tkach, J.M. (2001). Crowbars and ratchets: hsp100 chaperones as tools in reversing protein aggregation. Biochem. Cell Biol. 79, 557–568. [PubMed] [Google Scholar]
- Goloubinoff, P., Mogk, A., Zvi, A.P.B., Tomoyasu, T., and Bukau, B. (1999). Sequential mechanism of solubilization and refolding of stable protein aggregates by a bichaperone network. Proc. Natl. Acad. Sci. USA 96, 13732–13737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gottesman, S., Squires, C., Pichersky, E., Carrington, M., Hobbs, M., Mattick, J.S., Dalrymple, B., Kuramitsu, H., Shiroza, T., and Foster, T. (1990). Conservation of the regulatory subunit for the Clp ATP-dependent protease in prokaryotes and eukaryotes. Proc. Natl. Acad. Sci. USA 87, 3513–3517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haarer, B.K., and Pringle, J.R. (1987). Immunofluorescence localization of the Saccharomyces cerevisiae CDC12 gene product to the vicinity of the 10-nm filaments in the mother-bud neck. Mol. Cell. Biol. 7, 3678–3687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartwell, L.H. (1971). Genetic control of the cell division cycle in yeast. IV. Genes controlling bud emergence and cytokinesis. Exp. Cell Res. 69, 265–276. [DOI] [PubMed] [Google Scholar]
- Kim, H.B., Haarer, B.K., and Pringle, J.R. (1991). Cellular morphogenesis in the Saccharomyces cerevisiae cell cycle: localization of the CDC3 gene product and the timing of events at the budding site. J. Cell Biol. 112, 535–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korch, C., Mountain, H.A., and Bystrom, A.S. (1991). Cloning, nucleotide sequence, and regulation of MET14, the gene encoding the APS kinase of Saccharomyces cerevisiae. Mol. Gen. Genet. 229, 96–108. [DOI] [PubMed] [Google Scholar]
- Leipe, D.D., Wolf, Y.I., Koonin, E.V., and Aravind, L. (2002). Classification and evolution of P-loop GTPases and related ATPases. J. Mol. Biol. 317, 41–72. [DOI] [PubMed] [Google Scholar]
- Li, L., and Lindquist, S. (2000). Creating a protein-based element of inheritance. Science 287, 661–664. [DOI] [PubMed] [Google Scholar]
- Lindquist, S., and Kim, G. (1996). Heat-shock protein 104 expression is sufficient for thermotolerance in yeast. Proc. Natl. Acad. Sci. USA 93, 5301–5306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, J.-J., Sondheimer, N., and Lindquist, S.L. (2002). Changes in the middle region of Sup35 profoundly alter the nature of epigenetic inheritance for the yeast prion [PSI+]. Proc. Natl. Acad. Sci. USA 99 (suppl 4) 16446–16453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Longtine, M.S., DeMarini, D.J., Valencik, M.L., Al-Awar, O.S., Fares, H., De Virgilio, C., and Pringle, J.R. (1996). The septins: roles in cytokinesis and other processes. Curr. Opin. Cell Biol. 8, 106–119. [DOI] [PubMed] [Google Scholar]
- Louvion, J.-F., Havaux-Copf, B., and Picard, D. (1993). Fusion of GAL4-VP16 to a steroid-binding domain provides a tool for gratuitous induction of galactose-responsive genes in yeast. Gene 131, 129–134. [DOI] [PubMed] [Google Scholar]
- Lupas, A.N., and Martin, J. (2002). AAA proteins. Curr. Opin. Struct. Biol. 12, 746–753. [DOI] [PubMed] [Google Scholar]
- Mogk, A., Schlieker, C., Strub, C., Rist, W., Weibezahn, J., and Bukau, B. (2003). Roles of individual domains and conserved motifs of the AAA+ chaperone ClpB in oligomerization, ATP hydrolysis, and chaperone activity. J. Biol. Chem. 278, 17615–17624. [DOI] [PubMed] [Google Scholar]
- Moriyama, H., Edskes, H.K., and Wickner, R.B. (2000). [URE3] prion propagation in Saccharomyces cerevisiae: requirement for chaperone Hsp104 and curing by overexpressed chaperone Ydj1p. Mol. Cell. Biol. 20, 8916–8922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neuwald, A.F., Aravind, L., Spouge, J.L., and Koonin, E.V. (1999). AAA+: a class of chaperone-like ATPases associated with the assembly, operation, and disassembly of protein complexes. Genome Res. 9, 27–43. [PubMed] [Google Scholar]
- Ogura, T., and Wilkinson, A.J. (2001). AAA+ superfamily ATPases: common structure–diverse function. Genes Cells 6, 575–597. [DOI] [PubMed] [Google Scholar]
- Parsell, D.A., Kowal, A.S., Singer, M.A., and Lindquist, S. (1994). Protein disaggregation mediated by heat-shock protein Hsp104. Nature 372, 475–478. [DOI] [PubMed] [Google Scholar]
- Parsell, D.A., Sanchez, Y., Stitzel, J.D., and Lindquist, S. (1991). Hsp104 is a highly conserved protein with two essential nucleotide-binding sites. Nature 353, 270–273. [DOI] [PubMed] [Google Scholar]
- Patino, M.M., Liu, J.-J., Glover, J.R., and Lindquist, S. (1996). Support for the prion hypothesis for inheritance of a phenotypic trait in yeast. Science 273, 622–626. [DOI] [PubMed] [Google Scholar]
- Patthy, L. (2003). Modular assembly of genes and the evolution of new functions. Genetica 118, 217–231. [PubMed] [Google Scholar]
- Paushkin, S.V., Kushnirov, V.V., Smirnov, V.N., and Ter-Avanesyan, M.D. (1996). Propagation of the yeast prion-like [psi+] determinant is mediated by oligomerization of the SUP35-encoded polypeptide chain release factor. EMBO J. 15, 3127–3134. [PMC free article] [PubMed] [Google Scholar]
- Pringle, J.R., Adams, A.E.M., Drubin, D.G., and Haarer, B.K. (1991). Immunofluorescence methods for yeast. Methods Enzymol. 194, 565–602. [DOI] [PubMed] [Google Scholar]
- Ramer, S.W., Elledge, S.J., and Davis, R.W. (1992). Dominant genetics using a yeast genomic library under the control of a strong inducible promoter. Proc. Natl. Acad. Sci. USA 89, 11589–11593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rose, M.D., Novick, P., Thomas, J.H., Botstein, D., and Fink, G.R. (1987). A Saccharomyces cerevisiae genomic plasmid bank based on a centromere-containing shuttle vector. Gene 60, 237–243. [DOI] [PubMed] [Google Scholar]
- Ross-Macdonald, P., Sheehan, A., Roeder, G.S., and Snyder, M. (1997). A multipurpose transposon system for analyzing protein production, localization, and function in Saccharomyces cerevisiae. Proc. Natl. Acad. Sci. USA 94, 190–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez, Y., and Lindquist, S.L. (1990). HSP104 required for induced thermotolerance. Science 248, 1112–1115. [DOI] [PubMed] [Google Scholar]
- Sanchez, Y., Taulien, J., Borkovich, K.A., and Lindquist, S. (1992). Hsp104 is required for tolerance to many forms of stress. EMBO J. 11, 2357–2364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saraste, M., Sibbald, P.R., and Wittinghofer, A. (1990). The P-loop–a common motif in ATP- and GTP-binding proteins. Trends Biochem. Sci. 15, 430–434. [DOI] [PubMed] [Google Scholar]
- Schirmer, E.C., Glover, J.R., Singer, M.A., and Lindquist, S. (1996). HSP100/Clp proteins: a common mechanism explains diverse functions. Trends Biochem. Sci. 21, 289–296. [PubMed] [Google Scholar]
- Schirmer, E.C., and Lindquist, S. (1997). Interactions of the chaperone Hsp104 with yeast Sup35 and mammalian PrP. Proc. Natl. Acad. Sci. USA 94, 13932–13937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schirmer, E.C., and Lindquist, S. (1998). Purification and properties of Hsp104 from yeast. Methods Enzymol. 290, 430–444. [DOI] [PubMed] [Google Scholar]
- Schirmer, E.C., Lindquist, S., and Vierling, E. (1994). An Arabidopsis heat shock protein complements a thermotolerance defect in yeast. Plant Cell 6, 1899–1909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schirmer, E.C., Queitsch, C., Kowal, A.S., Parsell, D.A., and Lindquist, S. (1998). The ATPase activity of Hsp 104, effects of environmental conditions and mutations. J. Biol. Chem. 273, 15546–15552. [DOI] [PubMed] [Google Scholar]
- Schirmer, E.C., Ware, D.M., Queitsch, C., Kowal, A.S., and Lindquist, S.L. (2001). Subunit interactions influence the biochemical and biological properties of Hsp104. Proc. Natl. Acad. Sci. USA 98, 914–919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serio, T.R., and Lindquist, S.L. (2000). Protein-only inheritance in yeast: something to get [PSI+]-ched about. Trends Cell Biol. 10, 98–105. [DOI] [PubMed] [Google Scholar]
- Sikorski, R.S., and Boeke, J.D. (1991). In vitro mutagenesis and plasmid shuffling: from cloned gene to mutant yeast. Methods Enzymol. 194, 302–318. [DOI] [PubMed] [Google Scholar]
- Sikorski, R.S., and Hieter, P. (1989). A system of shuttle vectors and yeast host strains designed for efficient manipulation of DNA in Saccharomyces cerevisiae. Genetics 122, 19–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slater, M.L., Bowers, B., and Cabib, E. (1985). Formation of septum-like structures at locations remote from the budding sites in cytokinesis-defective mutants of Saccharomyces cerevisiae. J. Bacteriol. 162, 763–767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ter-Avanesyan, M.D., Kushnirov, V.V., Dagkesamanskaya, A.R., Didichenko, S.A., Chernoff, Y.O., Inge-Vechtomov, S.G., and Smirnov, V.N. (1993). Deletion analysis of the SUP35 gene of the yeast Saccharomyces cerevisiae reveals two non-overlapping functional regions in the encoded protein. Mol. Microbiol. 7, 683–692. [DOI] [PubMed] [Google Scholar]
- Vale, R.D. (2000). AAA proteins. Lords of the ring. J. Cell Biol. 150, F13–F19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wach, A., Brachat, A., Pohlmann, R., and Philippsen, P. (1994). New heterologous modules for classical or PCR-based gene disruptions in Saccharomyces cerevisiae. Yeast 10, 1793–1808. [DOI] [PubMed] [Google Scholar]
- Walker, J.E., Saraste, M., Runswick, M.J., and Gay, N.J. (1982). Distantly related sequences in the a- and b-subunits of ATP synthase, myosin, kinases and other ATP-requiring enzymes and a common nucleotide binding fold. EMBO J. 1, 945–951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wickner, S., Maurizi, M.R., and Gottesman, S. (1999). Posttranslational quality control: folding, refolding, and degrading proteins. Science 286, 1888–1893. [DOI] [PubMed] [Google Scholar]
- Wolf, E., Kim, P.S., and Berger, B. (1997). MultiCoil: a program for predicting two- and three-stranded coiled coils. Protein Sci. 6, 1179–1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zolkiewski, M. (1999). ClpB cooperates with DnaK, DnaJ, and GrpE in suppressing protein aggregation. A novel multi-chaperone system from Escherichia coli. J. Biol. Chem. 274, 28083–28086. [DOI] [PubMed] [Google Scholar]