

Abstract
SnoaL belongs to a family of small polyketide cyclases, which catalyse ring closure steps in the biosynthesis of polyketide antibiotics produced in Streptomyces. Several of these antibiotics are among the most used anti-cancer drugs currently in use. The crystal structure of SnoaL, involved in nogalamycin biosynthesis, with a bound product, has been determined to 1.35 Å resolution. The fold of the subunit can be described as a distorted α+β barrel, and the ligand is bound in the hydrophobic interior of the barrel. The 3D structure and site-directed mutagenesis experiments reveal that the mechanism of the intramolecular aldol condensation catalysed by SnoaL is different from that of the classical aldolases, which employ covalent Schiff base formation or a metal ion cofactor. The invariant residue Asp121 acts as an acid/base catalyst during the reaction. Stabilisation of the enol(ate) intermediate is mainly achieved by the delocalisation of the electron pair over the extended π system of the substrate. These polyketide cyclases thus form of family of enzymes with a unique catalytic strategy for aldol condensation.
Keywords: anthracycline, crystal structure, mechanism, nogalamycin, protein crystallography
Introduction
Polyketides form a large and diverse group of natural compounds produced mainly by microorganisms and plants. Among these secondary metabolites are the anthracyclines, aromatic polyketide antibiotics synthesised by Streptomyces species (Strohl, 2001). Anthracyclines are of particular medical importance, because some of the clinically most potent anti-tumour drugs are recruited from this class of compounds, for instance doxorubicin and daunorubicin (Figure 1) (Grein, 1987).
Figure 1.
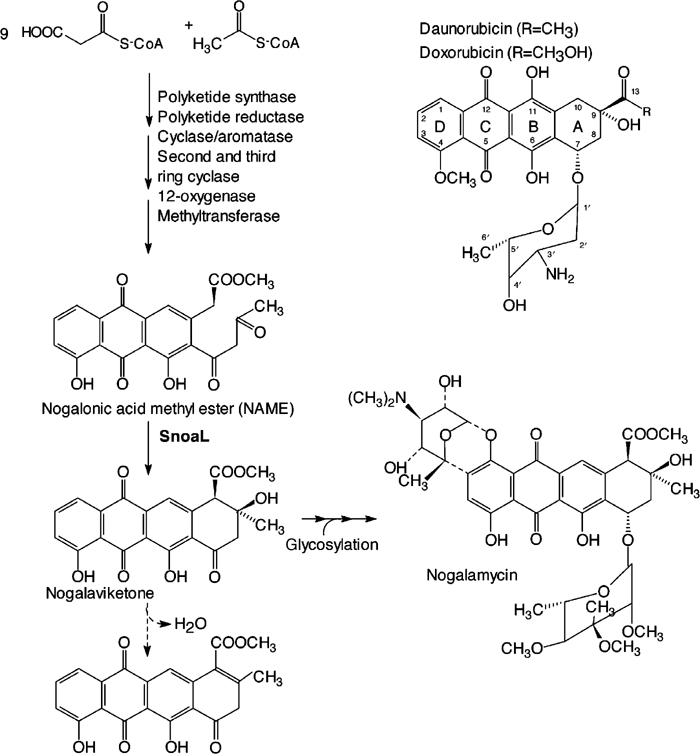
Biosynthesis of nogalamycin (left panel). The conversion of NAME into nogalaviketone is an intramolecular aldol condensation catalysed by SnoaL. The step indicated by the broken arrow, elimination of water, occurs slowly non-enzymatically. Tautomerisation at the C7 carbonyl oxygen in the dehydrated compound leads to aromatisation of the fourth ring. The structures of daunorubicin and doxorubicin are shown in the upper right part of the figure.
The basic structure of anthracyclines is a 7, 8, 9, 10- tetrahydro-5, 12-napthacene quinone. Diversity is generated by variations in the substitution patterns of the ring system and through differences in glycosylation (Strohl et al, 1989). The complexity of the chemical structure of anthracyclines makes their chemical synthesis difficult, and combinatorial biosynthesis appears to be a promising competitive route towards novel anthracyclines with improved toxicity profiles. Genetic and biochemical studies of the underlying pathways and enzymes are therefore expected to provide necessary insights into the engineering of anthracycline biosynthesis either through the hybrid antibiotic approach (Hopwood, 1981; Hopwood et al, 1985; Niemi et al, 1994; Ylihonko et al, 1996) or by redesign of biosynthetic enzymes (Hoffemeister et al, 2002).
The biosynthetic pathway of anthracyclines starts from simple carboxylic acid residues that are used to build up the polyketide backbone in a reaction catalysed by polyketide synthase (PKS) (Shen and Hutchinson, 1996; Kantola et al, 1997) (Figure 1). Acetate is the starter unit for nogalamycinone synthesis (Wiley et al, 1978) and propionate for the synthesis of aklavinone, the most common precursor of anthracyclines (Shen and Hutchinson, 1996; Kantola et al, 1997). In both cases, nine acetate residues are added to the primer carboxylic acid to yield a linear decaketide. This intermediate undergoes subsequent cyclisations by the action of aromatases and cyclases (Shen and Hutchinson, 1996; Kantola et al, 1997). The resulting multicyclic aglycone moiety is then further modified enzymatically through hydroxylation, glycosylation, methylation, reduction, decarboxylation or/and oxidation (Connors et al, 1990; Madduri et al, 1993; Dickens et al, 1997; Walczak et al, 1999).
The factors that determine the choreography of the cyclisation steps as well as their regio- and stereochemistry during the biosynthesis of the multicyclic compounds are as yet little understood. There is compelling evidence that the timing and regiospecificity of the cyclisation step is controlled by other enzymes and not by the PKS itself (Shen and Hutchinson, 1993; Kantola et al, 1997). One of the important factors for the folding of the polyketide backbone are the cyclases. The last cyclisation step, the formation of the aromatic multicyclic ring system, is catalysed by a rather small enzyme, consisting of approximately 140 amino acids. This polyketide cyclase catalyses ring closure of ring A resulting in formation of aklaviketone or nogalaviketone, aglycone precursors for doxorubicin, daunorubicin, aclacinomycin, and nogalamycin (Figure 1). Several of these cyclases, DnrD from Streptomyces peucetius (Madduri and Hutchinson, 1995; Kendrew et al, 1999), DauD from Streptomyces sp strain C5 (Dickens et al, 1995), RdmA from Streptomyces purpurascens (Niemi and Mäntsälä, 1995), AknH from Streptomyces galilaeus (Räty et al, 2002) and SnoaL from Streptomyces nogalater (Torkkell et al, 2000) have been described. While SnoaL is 60–65% identical in amino-acid sequence to other members of this family, it is unique in that the product of cyclisation differs in stereochemistry at the C9 atom from most other anthracyclines, 9S, 10R rather than 9R, 10R (Torkkell et al, 2000) (Figure 1).
In chemical terms, the reaction catalysed by these enzymes is an intramolecular aldol condensation, without the second step, the elimination of water (Figure 1). Interestingly, the cyclases do not use a Schiff base mechanism as class I aldolases, nor cofactors such as metal ions as class II aldolases (Kendrew et al, 1999) nor thiamin diphosphate as transketolases for stabilisation of the enol(ate) intermediate. They thus form a family of enzymes with a unique catalytic strategy for aldol condensations. There is no 3D structure available for any of these cyclases or of any sequence homologue. As part of our ongoing studies of the structural biology of anthracycline biosynthesis, we set out to determine the structure of a representative of this polyketide cyclase family by protein crystallography. Here, we describe the structure of nogalonic acid methyl ester cyclase, SnoaL, from S. nogalater involved in nogalamycin biosynthesis in complex with a product analogue, at atomic resolution. The structure analysis, together with site-directed mutagenesis, reveals a novel mechanism for enzymatic aldol condensations.
Results
Electron density map and quality of the model
SnoaL has been crystallised in the presence of the substrate nogalonic acid methyl ester (NAME) and the 3D structure was determined to 1.35 Å resolution using single isomorphous replacement, including anomalous scattering based on an uranium derivative as described in Materials and methods. The entire polypeptide chain, except for the N-terminal and C-terminal amino acid and residues 37–41, is well defined in electron density. Residues 37–40 are part of a 310 helix, and the side chains for these residues are not clearly defined in electron density, reflecting some degree of disorder of this part of the polypeptide chain. The N-terminal His tag, the linker peptide and the first residue of SnoaL (in total 36 residues) are disordered in the crystals of both low- and high-pH forms. There is however sufficient space in the crystal lattice to accommodate these amino-acid residues without interference with crystal packing. The stereochemistry of the model is as expected for models at this resolution, with root mean square deviations from ideal bond lengths and angles of 0.013 Å and 1.15°, respectively (Table I). The Ramachandran plot shows 89% of the amino acids in the most favoured region. One residue, Ser60, is in the disallowed region of the plot, but is well defined in the electron density map.
Table 1.
Refinement statistics
| Resolution (Å) | 1.35 |
| R values (%) | |
| Rwork (%) | 14.0 |
| Rfree (%) | 17.6 |
| Number of amino acids | 142 |
| Number of atoms | |
| Protein | 1169 |
| Ligand | 28 |
| Water | 125 |
| B-factor (Å2) | |
| Wilson plot | 16.3 |
| Protein | 18.4 |
| Main chain | 17.1 |
| Side chain | 19.8 |
| Ligand | 22.7 |
| Water | 27.3 |
| Deviations from ideals (r.m.s.d.) | |
| Bond length (Å) | 0.013 |
| Bond angle (deg) | 1.15 |
| Ramachandan plot (%) | |
| Most favoured region | 89 |
| Additional allowed region | 9.4 |
| Generously allowed region | 0.8 |
| Disallowed region | 0.8 |
The enzyme was crystallised in the presence of the substrate, and the electron density maps clearly show a bound ligand in the active site (Figure 2). The electron density is however not consistent with the structure of the substrate, but shows that cyclisation of ring A has occurred during crystallisation. There is furthermore no electron density for the hydroxyl group at the C-9 position and ring A is clearly planar. The bound ligand thus is a product derivative, formed upon elimination of water with subsequent aromatisation (Figure 1). Non-enzymatic conversion of nogalaviketone and aklaviketone into their dehydrated derivatives has been described previously (Kendrew et al, 1999; Kunnari et al, 1999) and could have occurred during the crystallisation process. The red-pink colour of the crystals, rather than the expected yellow colour due to bound substrate or product, supports the identification of the bound ligand as the planar, aromatised nogalaviketone derivative, because the latter has a red-pink colour (Kunnari et al, 1999).
Figure 2.
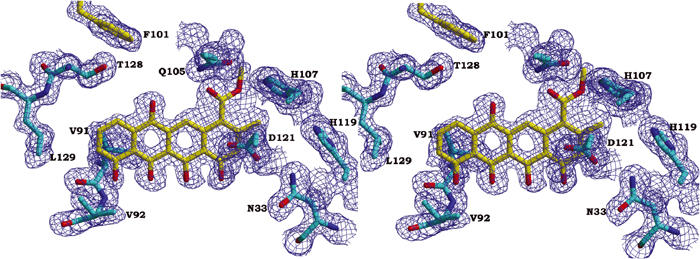
Part of a composite omit 2Fo–Fc electron density map at 1.35 Å resolution, contoured at 1.0σ. The area displayed shows the bound ligand and the surrounding amino-acid residues.
Fold of the subunit
The subunit consists of a single domain that is formed by α and β motifs, and belongs to the α+β class of structures. The core of the subunit is made up by four antiparallel β-strands (β2–β5) that are continuous in sequence. An additional β-strand (β1), considerably shorter in length, runs parallel to strand β5, giving rise to a five-stranded mixed β-sheet (Figure 3). The sheet is very curved and forms part of a barrel-like structure. Besides β1, the N-terminal half of the polypeptide chain folds into four helices H1–H4, and helices H1, H2 and H4 complete the barrel-like structure. Helices H1 and H4 are α-helices, whereas H2 and H3 are 310 helices. The C-terminal helix H5 extends β-strand 5 and thus is part of the α+β barrel. In the inside of this barrel, a large hydrophobic cavity is formed which harbours the active site. Two loops, comprising residues Asn33–Gly44 including helix H3, and Val88–Asp98 between β3 and β4, respectively, fold over the entrance of the barrel and limit access to the active site from the outer solution (Figure 3).
Figure 3.
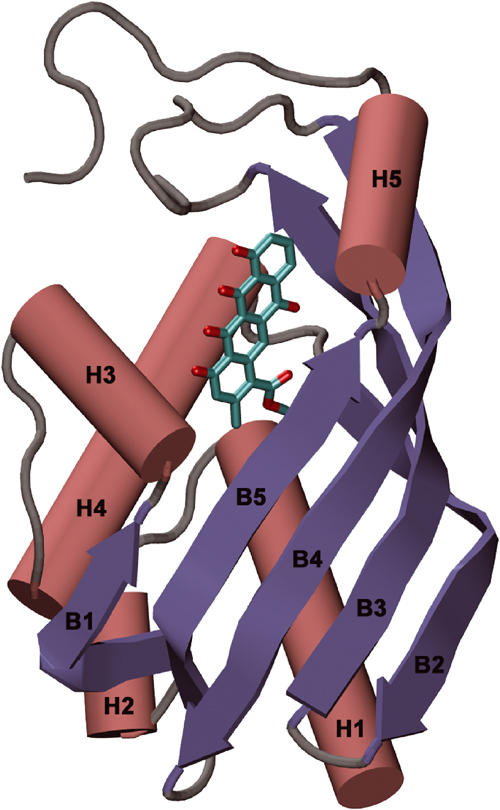
Schematic view of the subunit of SnoaL. Helices are shown in red and β-strands in blue. The location of the substrate binding site is indicated by the bound aglycone ligand, shown in cyan.
There has been some ambiguity with respect to the start of the open reading frame coding for SnoaL (Torkkell et al, 2000). The two potential start codons for the snoaL gene, ATG or a TTG located 30 bp upstream from the ATG codon, would result in a polypeptide chain of 134 or 144 amino-acid residues, respectively. The 3D structure of the enzyme, based on a construct coding for 144 amino acids, sequence alignment with related enzymes (Figure 4) and the presence of a typical ribosome binding site suggest that the start codon for the snoal gene is TTG, and that snoal codes for a polypeptide chain of 144 amino acids. The ATG start codon would result in the truncation of a large part of the N-terminal amino helix α1, which from a structural point appears less likely. Sequence alignment also shows that six out of these 10 amino acids are conserved in other members of this cyclase family, AknH, DauD and DnrD (Figure 4).
Figure 4.
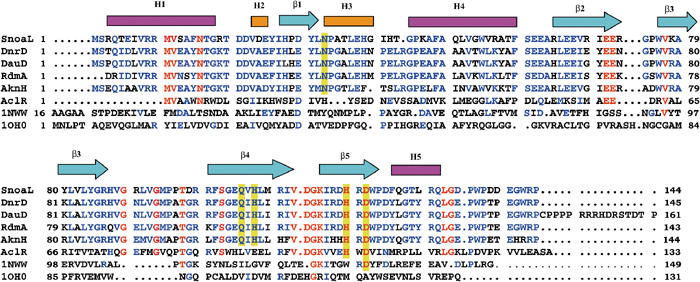
Sequence alignment of SnoaL with other polyketide cyclases and structural homologues, including the secondary structural elements of SnoaL (pink boxes indicate α-helices, yellow boxes 310 helices and arrows denote β-strands). Polyketide cyclases in anthracycline biosynthesis are DnrD (Q54808) from S. peucetius, DauD (Q55215) from Streptomyces sp strain C5, RdmA from S. purpurascens (Q54526) and AknH from S. galilaeus (Q52646). AclR is a protein related in structure, but with unknown function from S. galilaeus (Q8VWA2). A structure-based sequence alignment of the structural homologues limonene-1,2-epoxide hydrolase (1NWW; Arand et al, 2003) and Δ5-3-ketosteroid isomerase (1OHO; Kim et al, 1997) is included. Invariant residues in the polyketide cyclase family are marked in red, and highly conserved residues are shown in blue. Active site residues in polyketide cyclases are highlighted by a yellow background.
Quaternary structure
In the crystal, four SnoaL subunits are arranged as a tetramer with 222 symmetry (Figure 5). Two subunits are related by one of the crystallographic two-fold symmetry axes and form a tight interface (A–B and C–D interfaces, respectively, in Figure 5). The buried surface area between the monomers is 1730 Å2, corresponding to 21% of the total surface area in the dimer. In the crystal lattice, two of these dimers are related by another crystallographic two-fold axis. The interaction interface between the subunits across this two-fold axis (interfaces A–C and B–D, respectively, in Figure 5) is rather small, 590 Å2. Gel filtration chromatography experiments however also suggest that SnoaL forms a tetramer in solution (data not shown), consistent with the quaternary structure revealed by the crystallographic analysis.
Figure 5.
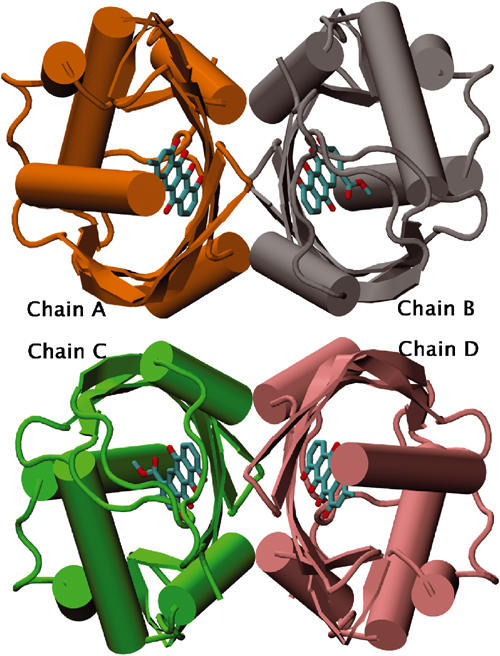
Schematic view of the SnoaL tetramer. The four subunits are shown in different colours. Bound ligands are shown in purple.
A total of 22 hydrogen bonds are formed between residues from the two subunits in the A–B (or C–D) dimer, and the amino acids participating in the hydrogen bond network are located in β1, β3, β4 and β5, helix H3 and H5 and also in the C-terminal part of the chain. There are two indirect hydrogen-bond bridges through water molecules (Trp122-W45-Leu32) and (Trp122-W82-Asp118) at the dimer interface. Most of the residues participating in these hydrogen bonds are conserved in other cyclase sequences (Figure 4). One important consequence of dimer formation is the more complete burial of the active sites. The active site and the bound product are only partly accessible in the monomer, and no atom of the bound ligand is exposed to solvent in the dimer. This on the other hand might mean that substrate binding/product release is coupled to a significant conformational change. Structural changes that would allow access to the binding pocket for substrate molecules most likely involve the loop between strands β3 and β4 (residues 90–95) and the adjacent 10 C-terminal residues after H5 (Figure 3).
The dimer–dimer interface (A–C or B–D, Figure 5) is made up mainly by β-strand β4 and H5. It contains significantly less interactions, for instance only three direct hydrogen bonds are formed. Four additional indirect hydrogen bonds involve bridging water molecules.
Structural homologues of SnoaL
A search of the PDB databank using the program DALI (Holm and Sander, 1993) revealed several structures similar to SnoaL. Limonene-1,2-epoxide hydrolase (Arand et al, 2003), Δ5-3-ketosteroid isomerase (Kim et al, 1997), the association domain of Ca2+/calmodulin-dependent kinase II (Hoelz et al, 2003), scytalone dehydratase (Lundqvist et al, 1994), a nuclear transport factor (NTF2) (Bullock et al, 1996), an NTF2-related export protein (Fribourg et al, 2001) and the β subunit of naphthalene 1,2 dioxygenase (Kauppi et al, 1998) show the same fold, with limonene-1,2-epoxide hydrolase (Figure 6) and Δ5-3-ketosteroid isomerase being most closely related to SnoaL in structure. None of these proteins shows significant overall sequence conservation to SnoaL, and the relationship of these polyketide cyclases to this fold family was not revealed until structure determination of SnoaL.
Figure 6.
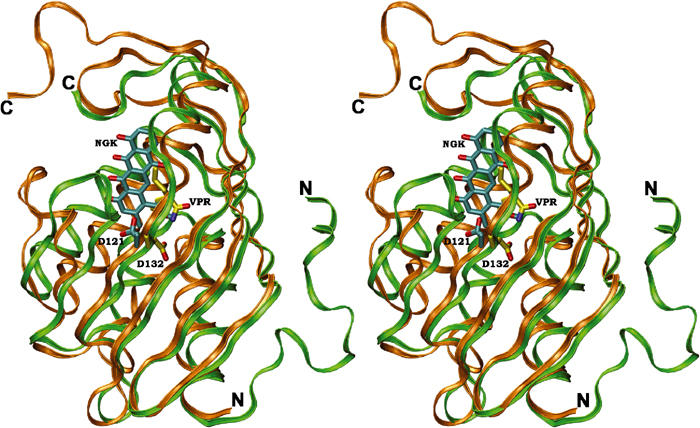
Stereoview of a superposition of the structures of SnoaL (gold) and limonene-1,2-epoxide hydrolase (green). The ligands of SnoaL (NGK) and limonene-1,2-epoxide hydrolase (VPR) are coloured in cyan and yellow, respectively. The position of the structurally conserved aspartic acid residue, involved in proton transfer steps during catalysis in both enzymes, is also shown.
With the exception of tetradecameric protein kinase II and trimeric scytalone dehydratase, most of these structural homologues of SnoaL are dimers. Since the quaternary structure of SnoaL is best described as a dimer of dimers, the packing interactions within such a dimer could be similar to those in the other dimeric structural relatives. In limonene-1,2-epoxide hydrolase, Δ5-3-ketosteroid isomerase and NTF2, the interface is formed by the faces of the β-sheet of the monomers. The orientation of the sheets varies however from roughly parallel in NTF2 to perpendicular in limonene-1,2-epoxide hydrolase, resulting in a similar, but nevertheless different, interface. In SnoaL, the dimer interface is formed between strands β4 and β5 from one subunit to helix H5 and the C-terminus of the second subunit, and vice versa, and the packing interactions are thus completely different from those seen in the structural homologues.
Ligand binding site
The ligand is bound in a predominantly hydrophobic pocket in the interior of the α+β barrel (Figure 3), and residues from most of the secondary structure elements are lining this pocket. The majority of interactions of the bound product with the enzyme are van der Waals contacts and hydrophobic interactions (Figure 7A). The aromatic ring system of the ligand packs against hydrophobic enzyme side chains, Phe15, Leu51, Trp54, Phe59, Phe125 and Leu129 located on helixes (H1, H4, H5), respectively, and Val91 and Val92 from the loop between β3 and β4. Only one direct hydrogen bond is formed between atoms of the product and the enzyme, from the side chain of Gln105 to the carbonyl oxygen atom of C14 of the bound nogalamycinone derivative.
Figure 7.
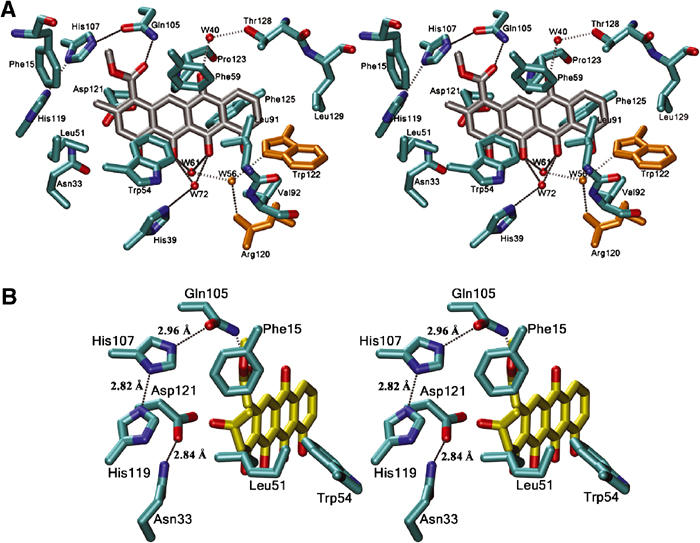
(A) Stereoview of the substrate/product binding site in SnoaL. The interactions of the bound aglycone with the surrounding protein side chains and water molecules are shown (cutoff distances: 3.2 Å for hydrogen bonds, 3.8 Å for van der Waals interactions). Residues shown in yellow are from the second subunit. (B) Modelled Michaelis complex of SnoaL with the substrate NAME. Only interactions with putative catalytic groups are shown.
Water molecules appear to be important for the binding and recognition of the aglycone moiety of the substrate, because several hydrogen bonds are formed indirectly via water molecules. The oxygen atom O18 of the aglycone is bridged via water molecule W40 to the side chain of Thr128, and atoms O20 and O21 are bridged via W72 to the side chain of His39. In addition, the latter two oxygen atoms are indirectly linked to residues from the second subunit of the dimer via a hydrogen bond to W61, which in turn is hydrogen bonded to W56. Two residues from the second subunit, Arg120 and Trp122, form a hydrogen bond to this water molecule and provide a link between the bound substrate/product in one subunit and residues from the second subunit (Figure 7A). Thus although the substrate binding sites appear to be contained within one subunit, this network of hydrogen bonds would allow cross-talk between subunits, resulting in one subunit sensing presence/absence of substrate in the other.
Active site residues
Most polar/charged residues within the binding pocket are found close to ring A of the bound product, that is, in the immediate proximity of the atoms involved in cyclisation. The side chain of the invariant residue Asp121 is in the vicinity of the C9 carbon atom of ring A (<3.5 Å distance). The natural product of the reaction catalysed by SnoaL is nogalaviketone, that is, not the product after elimination of the C9 hydroxyl group (Figure 1). Modelling of nogalaviketone into the active site is however rather straightforward. In such a model, the C9 hydroxyl group is ideally positioned to form a hydrogen bond to the side chain of Asp121. The importance of Asp121 is further emphasised by the hydrogen bond to Asn33, another residue conserved in the cyclases (Figure 4) and which may anchor the side chain of Asp121 in a position required for catalysis. Interestingly, an aspartic acid residue (Asp132) is found at the corresponding position in the structure of limonene-1,2-epoxide hydrolase (Figure 6), an enzyme that exhibits a similar fold of the subunit, but lacks significant overall sequence identity to SnoaL. In limonene-1,2-epoxide hydrolase, this residue is part of a catalytic Asp–Arg–Asp triad and activates a water molecule for nucleophilic attack on the epoxide (Arand et al, 2003).
Gln105 is another residue in contact with the substrate, which is invariant in this family of polyketide cyclases (Figure 4). The side chain of Gln105 forms a hydrogen bond to the C14 carbonyl oxygen of the bound ligand, and is linked through a hydrogen bond network to the side chains of the invariant residues His107 and His119 (Figure 7B).
Mutagenesis
The mutants Phe15Tyr, Gln105Ala and Asp121Ala were produced at levels comparable to wild-type enzyme in Escherichia coli, the other two mutants, Gln105Glu and Asp121Asn, were produced as insoluble inclusion bodies. The Asp121Ala mutant was completely inactive. The specific activity of the Gln105Ala mutant was reduced to 15% of that of the wild-type enzyme. Replacement of Phe15 by a tyrosine residue resulted in a 20-fold drop in specific activity (Table II). Activity measurements of wild-type SnoaL in the presence of 5 mM EDTA did not result in any sign of inhibition.
Table 2.
Active site mutants of SnoaL
| Enzyme variant | Specific activity (nmol/mg s) |
|---|---|
| Wild type | 21.1 |
| Phe15Tyr | 1.1 |
| Gln105Ala | 3.3 |
| Gln105Glu | Inclusion bodies |
| Asp121Ala | No detectable activity |
| Asp121Asn | Inclusion bodies |
Sequence homologues of SnoaL
A search using the program BLAST (Altschul et al, 1990) revealed a number of amino-acid sequences homologous to SnoaL, with the closest relatives (>60% sequence identity) being polyketide cyclases involved in anthracycline biosynthesis in other Streptomyces sp (Figure 4), DnrD from S. peucetius (Madduri and Hutchinson, 1995; Kendrew et al, 1999), DauD from Streptomyces sp strain C5 (Dickens et al, 1995), RdmA from S. purpurascens (Niemi and Mäntsälä, 1995) and AknH from S. galilaeus (Räty et al, 2002). The sequence alignment shows that the residues in the helical and β-sheet regions of SnoaL are well conserved and that active site residues (Asn33, Gln105, His107, Asp121, His119) are invariant in these enzymes (Figure 4). These cyclases produce an aglycone moiety with 9R configuration rather than 9S configuration found in the product of the SnoaL reaction. The high degree of sequence conservation, in particular of active site residues, suggests that the differences in stereochemical course of the reaction must be due to subtle changes in the active site topology and are not realised by completely different enzyme mechanisms.
Several other protein sequences with weak, but distinct sequence homology (20–29% sequence identity) to SnoaL were found. These sequences include hypothetical proteins from S. avermitilis (Q82GA1), Anabena sp (Q8YNM4), Gloeobacter violaceus PCC 7421 (BAC88168), Chromobacterium violaceum ATCC 12472 (AAQ57926) and Ralstonia solanacearum (Q8Y171), and these proteins most likely have the same fold. In the sequence of AclR from S. galilaeus, several active site residues (His119 and Asp121) are conserved, suggesting that this protein may act as a polyketide cyclase (Figure 4).
Discussion
Enzyme fold
SnoaL is a member of a polyketide cyclase family with hitherto unknown structure. The structure determination has revealed that this sequence family belongs to the superfamily of α+β barrel folds (Orengo and Thornton, 1993; Murzin, 1996). Sequence comparisons using BLAST (Altschul et al, 1990) show that there are homologues of these enzymes in other species than Streptomyces, for instance Anabaena sp (strain PCC 7120). Conservation of essential residues at the active site suggests that some of these enzymes may catalyse similar reactions. Other members of this sequence family share a similar fold, but it remains to be established whether or not they catalyse aldol condensation like reactions, since the key active site residues are not conserved.
Anthracycline recognition
It is striking that only one of the several possible hydrogen bonds between the product and the enzyme are formed. Recognition and specificity of binding of the aglycone moiety thus appears to be controlled by the shape and architecture of the binding site rather than by specific patterns of hydrogen bonds. A similar observation has been made with aclacinomycine methylesterase, a tailoring enzyme in rhodomycine biosynthesis (Jansson et al, 2003). This feature thus appears to be a more general characteristic of enzymes recognising the aglycone moiety of anthracyclines.
Proposed enzymatic mechanism
In enzyme-catalysed aldol condensation reactions, the key step is the abstraction of a C–H proton, that is, the generation of the carbanion/enol(ate). In most enzymes catalysing such reactions, this step is facilitated through various means of stabilising the resulting enol(ate). For instance, class I aldolases use covalent catalysis involving a Schiff base mechanism (Gefflaut et al, 1995), whereas class II aldolases contain a metal ion to stabilise the enol(ate) intermediate (Morse and Horecker, 1968). The high-resolution structure of SnoaL with bound product reveals that this polyketide cyclase does not use any of the above mechanisms. There is no lysine residue in the active site that could form a Schiff base with the substrate. We also do not observe any electron density in the vicinity of the substrate, which might represent a bound metal ion. The latter observation is consistent with the finding that the activity of the enzyme is unperturbed in the presence of 5 mM EDTA. The mechanistic differences to the classical aldolases may hold for all members of this cyclase family, because of the high degree of sequence conservation. Biochemical studies of the SnoaL homologue DnrD have shown that this enzyme is also not dependent on metal ions, and no evidence for the existence of a covalent Schiff base intermediate could be obtained (Kendrew et al, 1999). The enzymatic mechanism of DnrD remained however unresolved.
The structure presented here is that of a complex with a product analogue, and allows a reliable modelling of the Michaelis complex of SnoaL with the bound substrate NAME (Figure 7B). The arrangement of catalytic residues surrounding the substrate in this model strongly suggests that this enzyme uses only acid–base chemistry to catalyse the intramolecular aldol condensation leading to formation of the cyclised product. A mechanistic proposal consistent with the 3D structure and mutagenesis data is shown in Figure 8. The reaction is initiated by the invariant residue Asp121 abstracting the C–H proton at carbon atom C10 of the substrate. The resulting enol(ate) could then be stabilised in several ways. The hydrogen bond to the carbonyl oxygen at C14 to the invariant Gln105 may facilitate the formation of a negative charge at this atom in the enol(ate). While the mutagenesis experiments indicate that a certain contribution of this interaction to enolate stabilisation might occur, it is of only minor catalytic significance. The relatively high residual activity of the Gln105Ala mutant shows that this residue, albeit invariant in polyketide cyclase (Figure 4), is not essential for catalysis.
Figure 8.
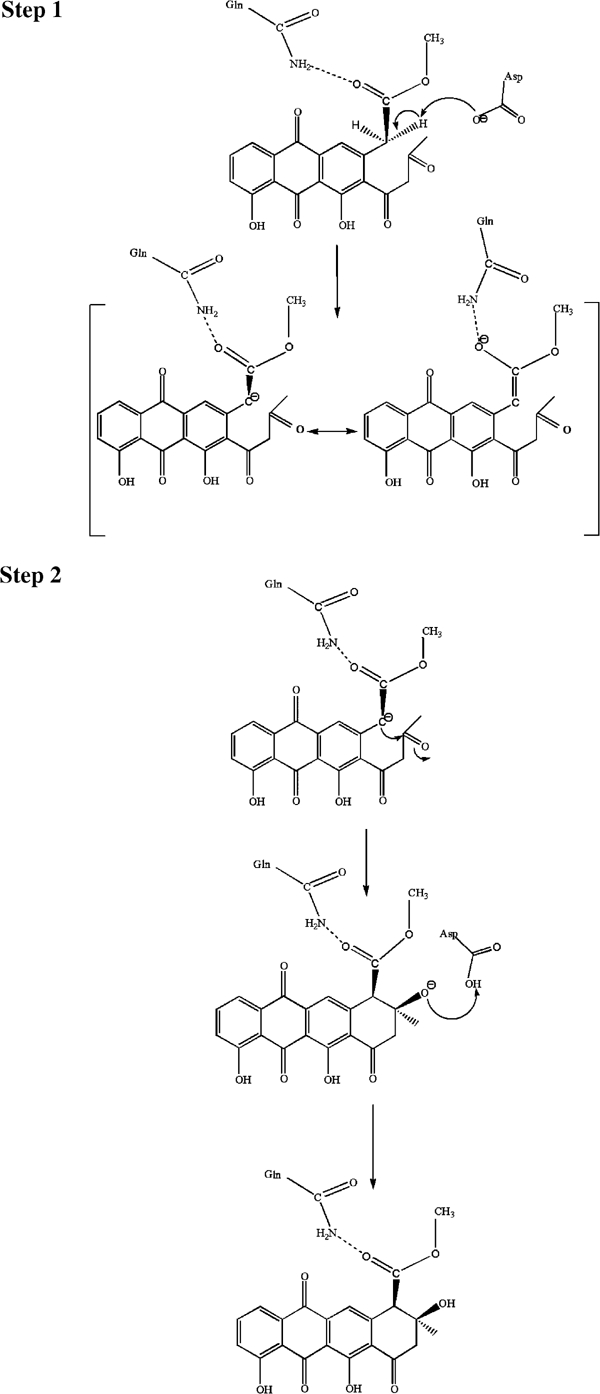
Proposed mechanism of the intramolecular aldol condensation catalysed by SnoaL. The first step of the reaction is the generation of the carbanion/enol(ate) intermediate by abstraction of a proton at carbon 10 by the conserved residue Asp121. The negative charge of the enol(ate) is stabilised by resonance over the aromatic ring system of the polyketide (not shown here). A minor stabilisation may be due to the hydrogen bond to the conserved residue Gln105. The carbanion then attacks the C9 carbonyl carbon atom resulting in ring closure. The negative charge at the carbonyl oxygen atom developing in the transition state is stabilised via a hydrogen bond to the protonated side chain of Asp121. The same residue also transfers a proton to the oxygen atom at C9 of the aglycone moiety.
A more significant contribution to the stabilisation of the enol(ate) intermediate may be due to the delocalisation of the electron pair over the extended π system of the neighbouring ring systems. This should also result in a relatively low pKa for the C–H proton compared to an aliphatic substituent, thus facilitating abstraction of this proton by a catalytic base. In fact, the stabilisation of the enol(ate) by resonance might be sufficient so that Schiff base formation or a metal ion is not needed and acid–base chemistry is utilised to carry out the necessary proton transfer steps during catalysis.
The next step of the reaction is the nucleophilic attack of the enol(ate) intermediate onto the carbonyl carbon at C9. The developing negative charge at the carbonyl oxygen in the transition state could be stabilised by the side chain of Asp121, which at this step of the catalytic cycle is in its protonated form. Transfer of the proton then leads to formation of the product nogalaviketone. The key role of this invariant residue in the mechanism of SnoaL is also buttressed by the site-directed mutagenesis experiments (Table II). Consistent with the mechanistic proposal (Figure 8), replacement of this residue by an alanine leads to an enzyme completely impaired in catalytic activity.
Stereochemistry
The model of the SnoaL–NAME Michaelis complex also provides insights into the structural basis for the stereochemical outcome of the reaction. Formation of the 9S isomer requires nucleophilic attack of the enol(ate) onto the re face of the aldehyde, and the resulting alcoholate anion then points towards the protonated side chain of Asp121. Due to the free rotation along the C9–C13 bond, the attack can in principle also occur onto the si face of the aldehyde, which produces the 9R isomer. This would however result in that the alcoholate points in the opposite direction, which is a very hydrophobic environment, and energetically is certainly disfavoured. In addition, in this orientation, there is no catalytic acid available to protonate the alcoholate anion. The topology of the active site, with a hydrophobic wall on one side and a catalytic base on the opposite side of the substrate, thus ensures the stereochemical course of the reaction, which leads to the formation of the 9S stereoisomer. The importance of the hydrophobic character of the substrate binding pocket is further emphasised by the fact that the replacement of Phe15, located within van der Waals distance to the C9 carbon atom of the product (Figure 7), by the more hydrophilic tyrosine residue results in a 20-fold drop in catalytic activity.
In the polyketide cyclases that catalyse the formation of the opposite stereoisomer, the hydrophobic character of this part of the substrate binding pocket is however changed by a substitution of Phe15 to Tyr15 (Figure 4). Modelling shows that in these enzymes the hydroxyl group of Tyr15 could be within hydrogen bonding distance to the C9 oxygen atom, when the latter has 9R configuration. This residue thus might be responsible for the stereochemistry of these cyclases. However, the Phe15Tyr mutant of SnoaL only produced the 9S stereoisomer, nogalaviketone, and no 9R isomer of the product was observed. We thus conclude that the residue at position 15 in the cyclases appears not to be the major determinant of the stereochemical outcome of the reaction. The switch in the stereochemistry of the product most likely involves additional, as yet unresolved, residue substitutions.
Conclusions
SnoaL is a member of a group of polyketide cyclases with hitherto unknown structure. The crystal structure analysis of SnoaL at 1.35 Å resolution shows that these enzymes have a α+β barrel fold, first observed in scytalone dehydratase, an enzyme not related in sequence or function. Binding of the aglycone ligand is dominated by hydrophobic interactions, and recognition and specificity appears to be controlled by the architecture of the binding pocket rather than specific hydrogen bonds. The structure of the active site suggests that the enzyme uses acid–base chemistry rather than covalent or metal ion catalysis as observed in other enzymes catalysing aldol condensations.
Materials and methods
Protein production and crystallisation
SnoaL was produced in E. coli as a fusion protein, with a 6xHis tag and a linker peptide of 29 amino acids at the N-terminal and purified as described (Sultana et al, 2004). Isomorphous crystals of the complex of SnoaL with the substrate NAME were obtained following the protocol described in Sultana et al (2004), at pH 4.6 and 8.5, respectively, with polyethylene glycol (PEG) as precipitant.
Data collection and processing
SnoaL crystals were soaked in a cryoprotectant solution consisting of crystallisation solution including 25% PEG400 for 30 s and then flash-frozen at 100 K by transfer into a gaseous nitrogen stream. The crystallisation solution consisted of 0.1 M sodium acetate, pH 4.6, 8% w/v PEG4000 (low-pH crystals) or 0.2 M sodium acetate, 0.1 M TRIS buffer, pH 8.5, and 30% w/v PEG4000, respectively. The X-ray diffraction data were collected at beamline B711, MAX Laboratory, Lund, Sweden (low-pH form) and at beamline ID14 EH-1, ESRF, Grenoble, France (high-pH form). For the preparation of a heavy atom derivative of SnoaL, crystals were soaked in mother liquor (low pH) containing 1 mM uranium acetate for 30 min, quickly transferred into the same cryoprotectant solution as above and flash-frozen at 100 K. The derivative data were collected at beamline B711, MAX Laboratory. All images were processed using MOSFLM (Collaborative Computational Project, 1994) and the data were scaled using the program SCALA from the CCP4 program suite (Collaborative Computational Project, 1994). Details of the data collection statistics are given in Table III.
Table 3.
Statistics of data collection for SnoaL
| Parameters | SnoaL (low pH) | SnoaL (high pH) | SnoaL (U derivative) |
|---|---|---|---|
| Beam line | B711 | ID14 EH-1 | B711 |
| Wavelength (Å) | 1.099 | 0.934 | 1.099 |
| Resolution (Å) | 28.63–1.87 | 36.03–1.35 | 28.65–2.64 |
| Space group | I222 | I222 | I222 |
| Unit cell parameters | |||
| a (Å) | 68.3 | 69.1 | 68.4 |
| b (Å) | 72.3 | 72.0 | 72.4 |
| c (Å) | 64.5 | 65.4 | 64.5 |
| No. of observations | 48 371 | 402 458 | 18 134 |
| No. of unique reflections | 12 194 | 35 447 | 4771 |
| Rsym (%)a | 6.6 (23.2) | 8.5 (48.5) | 5.3 (7.6) |
| Completeness (%) | 99.6 (99.6) | 98.5 (98.5) | 97.0 (97.0) |
| Multiplicity (%) | 3.8 (3.8) | 11.3 (8.3) | 3.8 (3.7) |
| Average I/σ(I) |
20.2 (4.8) |
23.3 (3.4) |
23.7 (10.9) |
| a Values in parentheses are for the highest resolution shell. | |||
Phasing and model building
For phase determination, single isomorphous replacement including anomalous scattering (SIRAS) was used, based on the uranium acetate derivative (Table IV). The positions of the heavy atoms were determined with the program SOLVE (Terwilliger and Berendzen, 1999). Inspection of the heavy metal sites after structure solution showed that the two uranium ions interacted with the side chains of Glu6 and Asp30, respectively. RESOLVE (Terwilliger, 2000) was used for density modification to 2.65 Å resolution. An initial model was built using ARP/WARP (Perrakis et al, 1999) and data from the low-pH crystal form to 1.87 Å resolution. This model contained 139 residues out of the 178 residues in the recombinant protein. Residues 37–40, not modelled by ARP/WARP, were included in the model manually using the program O (Jones et al, 1991).
Table 4.
Phasing statistics
| Parameters | Values |
|---|---|
| Derivative | Uranium acetate |
| Number of sites | 2 |
| Rder a(%) | 16.0 |
| Rcullis b,c | 0.76 |
| Phasing powerb,d | 2.05 |
| Figure of merit |
0.63 |
| a Rder=∑∣FPH−FP∣/∑∣FP∣, where FPH is the structure factor amplitude of the derivative crystal and FP that of the native. | |
| b Acentric reflections. | |
| c Rcullis=∑∣∣FPH±FP∣−∣FH(calc)∣/∑∣FPH−FP∣, where FPH and FP are defined as above and FH(calc) is the calculated heavy atom structure factor amplitude summed over centric reflections. | |
| d Phasing power=F(H)/E, the r.m.s. heavy atom structure factor amplitude divided by the lack of closure error. | |
Crystallographic refinement
The initial model was refined with the program REFMAC (Murshudov et al, 1997) using the maximum likelihood algorithm. A total of 7% of the reflections were excluded and used for Rfree calculation. The first cycles consisted of rigid body refinement, followed by positional and individual isotropic B-factor refinement. After each round, 2Fo–Fc and Fo–Fc maps were calculated and the graphics program O (Jones et al, 1991) was used for inspection of the electron density maps and manual adjustment of the model. After the Rfree had dropped to about 26%, refinement continued with the diffraction data from the isomorphous high-pH crystal form because of the considerably higher resolution (1.3 versus 1.87 Å). Water molecules were added to the model using ARP/WARP (Perrakis et al, 1999). Throughout the refinement, strong residual electron density had been observed in the active site. After inclusion of the high-resolution data, a model of a bound product analogue was fitted into this residual electron density. Refinement was continued with REFMAC (Murshudov et al, 1997), and included individual anisotropic B-factors. In the final stage, refinement with SHELX (Sheldrick and Schneider, 1997) resulted in a well-defined model with Rfac of 14.0% and Rfree of 17.6% (Table I). The final model consists of 142 amino acids (residues 2–143 of wild-type SnoaL), one bound molecule of the product analogue and 125 water molecules. A composite omit map for the model calculated using CNS (Brunger et al, 1998) was inspected for final model validation. The stereochemistry of the model was analysed with PROCHECK (Laskowski et al, 1993). A comparison of the refined low- and high-pH structures of SnoaL did not reveal any significant differences. The atomic coordinates and observed structure factor amplitudes have been deposited in the Protein Data Bank with accession code of 1sjw.
Structural comparisons
BLAST (Altschul et al, 1990) was used to search for similar protein sequences, and sequence alignments were performed with ClustalW (www2.eb.ac.uk/clustalw/) (Thompson et al, 1994). Structure comparisons were carried out with TOP (Lu, 2000), DALI (Holm and Sander, 1993) and the lsq option in O (Jones et al, 1991) using default parameters. Figures were prepared with VMD (Humphrey et al, 1996), Pov-ray and BOBSCRIPT (Esnouf, 1997).
Mutagenesis
The SnoaL Phe15Tyr, Gln105Ala, Gln105Glu, Asp121Ala and Asp121Asn mutants were generated by PCR mutagenesis with the mutagenic oligonucleotides GATGGTGAGCGCCtacAACACCGGCAGGAC, CGCTTCTCCGGTGAAgcgGTGCACCTGATG, CGGCGCTTCTCCGGTGAAgagGTGCACCTGATGCGC, GATCCGCGACCACCGGgccTGGCCCGACTTCC and AAGATCCGCGACCACCGGaacTGGCCCGACTTCCAG (mutations in lower case), and wild-type SnoaL as template. All mutations were verified by DNA sequencing, and recloned into the pBAD/His B expression plasmid. Mutant enzymes were purified using the same protocol as recombinant wild-type SnoaL. Mass spectrometric analysis resulted in the expected mass for the Phe15Tyr (20 488 Da), Gln105Ala (20 414 Da) and Asp121Ala (20 427 Da) mutant proteins, compared to the molecular mass of the wild-type enzyme, 20 471 Da.
Enzyme assay
Enzymatic activity was determined using an HPLC-based assay measuring the decrease in the concentration of the substrate NAME. The activity assay was carried out in a solution of 100 mM TES buffer, pH 7.5, containing 30 μM NAME (total volume 1 ml) and the reaction was started by the addition of enzyme. After incubation for at least 10 min, the reaction was stopped by the addition of 300 μl toluene. The substrate/product mixture was extracted in the toluene phase, dried by evaporation and resolubilised with 50 μl methanol for HPLC analysis using a LiChrosper 100 RP-18 (5 mm, 240 × 4 mm) reverse phase column. The concentration of the remaining substrate was derived from the size of the NAME peak at 420 nm in the mobile phase (65% acetonitrile, 35% 60 mM potassium phosphate, 0.7% citric acid) and a standard curve of the substrate obtained under identical conditions. Activity measurements of wild-type SnoaL were also carried out in the presence of 5 mM EDTA, under otherwise identical conditions.
The stereochemical analysis of the product of the SnoaL mutant Phe15Tyr (9S versus 9R stereoisomer) was carried out using a SnoaL/aklavinone-10-hydroxylase (RdmE) coupled enzyme assay system. RdmE exhibits stereochemical discrimination against the 9S isomer, but is able to hydroxylate only the 9R species. The reaction products can be separated using a 50:50 mobile phase and detected at 470 nm, the point of intersection of the absorption spectra of the reaction products (Sultana et al, in preparation).
Acknowledgments
We thank Dr Tero Kunnari for the samples of nogalonic acid methylester and Ms Hanna Koskiniemi for technical assistance. We acknowledge access to synchrotron radiation at beamline I711, MAX laboratory, Lund, Sweden and ID 14, ESRF, Grenoble, France. This work was supported by the Swedish Research Council, the Wallenberg Consortium North and the Academy of Finland. PK acknowledges support by the National graduate School in Informational and Structural Biology. J-SW gratefully acknowledges a fellowship from Wennergrenska Samfundet.
References
- Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ (1990) Basic local alignment search tool. J Mol Biol 215: 403–410 [DOI] [PubMed] [Google Scholar]
- Arand M, Hallberg BM, Zou J, Bergfors T, Oesch F, van der Werf MJ, de Bont JA, Jones TA, Mowbray SL (2003) Structure of Rhodococcus erythropolis limonene-1,2-epoxide hydrolase reveals a novel active site. EMBO J 22: 2583–2592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS, Read RJ, Rice LM, Simonson T, Warren GL (1998) Crystallography & NMR system: a new software suite for macromolecular structure determination. Acta Crystallogr D 54: 905–921 [DOI] [PubMed] [Google Scholar]
- Bullock TL, Clarkson WD, Kent HM, Stewart M (1996) The 1.6 angstroms resolution crystal structure of nuclear transport factor 2 (NTF2). J Mol Biol 260: 422–431 [DOI] [PubMed] [Google Scholar]
- Collaborative Computational Project No 4 (1994) The CCP4 suite: programs for protein crystallography. Acta Crystallogr D 50: 760–763 [DOI] [PubMed] [Google Scholar]
- Connors C, Bartel L, Strohl WR (1990) Biosynthesis of anthracyclines: enzyme conversion of aklanonic acid to aklavinone and e-rhodomycinone by anthracycline producing Streptomyces. J Gen Microbiol 136: 1887–1894 [Google Scholar]
- Dickens ML, Priestley ND, Strohl WR (1997) In vivo and in vitro bioconversion of epsilon-rhodomycinone glycoside to doxorubicin: functions of DauP, DauK, and DoxA. J Bacteriol 179: 2641–2650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickens ML, Ye J, Strohl WR (1995) Analysis of clustered genes encoding both early and late steps in daunomycin biosynthesis by Streptomyces sp. strain C5. J Bacteriol 177: 536–543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esnouf RM (1997) An extensively modified version of MOLSCRIPT that includes greatly enhanced coloring capabilities. J Mol Graph Model 15: 133–138 [DOI] [PubMed] [Google Scholar]
- Fribourg S, Braun IC, Izaurralde E, Conti E (2001) Structural basis for the recognition of a nucleoporin FG repeat by the NTF2-like domain of the TAP/p15 mRNA nuclear export factor. Mol Cell 8: 645–656 [DOI] [PubMed] [Google Scholar]
- Gefflaut T, Blonski C, Perie J, Willson M (1995) Class I aldolases: substrate specificity, mechanism, inhibitors and structural aspects. Prog Biophys Mol Biol 63: 301–340 [DOI] [PubMed] [Google Scholar]
- Grein A (1987) Antitumor anthracyclines produced by Streptomyces peucetius. Adv Appl Microbiol 32: 203–214 [DOI] [PubMed] [Google Scholar]
- Hoelz A, Nairn AC, Kuriyan J (2003) Crystal structure of a tetradecameric assembly of the association domain of Ca2+/calmodulin-dependent kinase II. Mol Cell 11: 1241–1251 [DOI] [PubMed] [Google Scholar]
- Hoffemeister D, Wilkinson B, Foster G, Sidebottom PJ, Ichinose K, Bechthold A (2002) Engineered urdamycin glycosyltransferases are broadened and altered in substrate specificity. Chem Biol 9: 287–295 [DOI] [PubMed] [Google Scholar]
- Holm L, Sander C (1993) Protein structure comparison by alignment of distance matrices. J Mol Biol 233: 123–138 [DOI] [PubMed] [Google Scholar]
- Hopwood DA (1981) Future possibilities for the discovery of new antibiotics by genetic engineering. In Beta-lactam Antibiotics, Salton MRJ, Shockman GD (eds) New York: Academic Press [Google Scholar]
- Hopwood DA, Malpartida F, Kieser HM, Ikeda H, Duncan J, Fujii I, Rudd BA, Floss HG, Omura S (1985) Production of ‘hybrid' antibiotics by genetic engineering. Nature 314: 642–644 [DOI] [PubMed] [Google Scholar]
- Humphrey W, Dalke A, Schulten K (1996) VMD: visual molecular dynamics. J Mol Graph 14: 33–38 [DOI] [PubMed] [Google Scholar]
- Jansson A, Niemi J, Mäntsälä P, Schneider G (2003) Crystal structure of aclacinomycin methylesterase with bound product analogues: implications for anthracycline recognition and mechanism. J Biol Chem 278: 39006–39013 [DOI] [PubMed] [Google Scholar]
- Jones TA, Zou JY, Cowan SW, Kjeldgaard M (1991) Improved methods for building protein models in electron density maps and the location of errors in these models. Acta Crystallogr A 47: 110–119 [DOI] [PubMed] [Google Scholar]
- Kantola J, Blanco G, Hautala A, Kunnari T, Hakala J, Mendez C, Ylihonko K, Mäntsälä P, Salas J (1997) Folding of the polyketide chain is not dictated by minimal polyketide synthase in the biosynthesis of mithramycin and anthracycline. Chem Biol 4: 751–755 [DOI] [PubMed] [Google Scholar]
- Kauppi B, Lee K, Carredano E, Parales RE, Gibson DT, Eklund H, Ramaswamy S (1998) Structure of an aromatic-ring-hydroxylating dioxygenase-naphthalene 1,2-dioxygenase. Structure 6: 571–586 [DOI] [PubMed] [Google Scholar]
- Kendrew SG, Katayama K, Deutsch E, Madduri K, Hutchinson CR (1999) DnrD cyclase involved in the biosynthesis of doxorubicin: purification and characterization of the recombinant enzyme. Biochemistry 38: 4794–4799 [DOI] [PubMed] [Google Scholar]
- Kim SW, Cha SS, Cho HS, Kim JS, Ha NC, Cho MJ, Joo S, Kim KK, Choi KY, Oh BH (1997) High-resolution crystal structures of Δ5-3-ketosteroid isomerase with and without a reaction intermediate analogue. Biochemistry 36: 14030–14036 [DOI] [PubMed] [Google Scholar]
- Kunnari T, Kantola, Ylihonko K, Klika KD, Mäntsälä P, Hakala J (1999) Hybrid compounds derived from the combination of anthracycline and actinorhodin biosynthetic pathways. J Chem Soc Perkin Trans 2: 1649–1652 [Google Scholar]
- Laskowski RA, Moss DS, Thornton JM (1993) Main-chain bond lengths and bond angles in protein structures. J Mol Biol 231: 1049–1067 [DOI] [PubMed] [Google Scholar]
- Lu G (2000) TOP: a new method for protein structure comparisons and similarity searches. J Appl Crystallogr 33: 176–183 [Google Scholar]
- Lundqvist T, Rice J, Hodge CN, Basarab GS, Pierce J, Lindqvist Y (1994) Crystal structure of scytalone dehydratase—a disease determinant of the rice pathogen, Magnaporthe grisea. Structure 2: 937–944 [DOI] [PubMed] [Google Scholar]
- Madduri K, Hutchinson CR (1995) Functional characterization and transcriptional analysis of a gene cluster governing early and late steps in daunorubicin biosynthesis in Streptomyces peucetius. J Bacteriol 177: 3879–3884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madduri K, Torti F, Colombo AL, Hutchinson CR (1993) Cloning and sequencing of a gene encoding carminomycin 4-O-methyltransferase from Streptomyces peucetius and its expression in Escherichia coli. J Bacteriol 175: 3900–3904 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morse DE, Horecker BL (1968) The mechanism of action of aldolases. Adv Enzymol Relat Areas Mol Biol 31: 125–181 [DOI] [PubMed] [Google Scholar]
- Murshudov G, Vagin AA, Dodson EJ (1997) Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr D 53: 240–255 [DOI] [PubMed] [Google Scholar]
- Murzin AG (1996) Structural classification of proteins: new superfamilies. Curr Opin Struct Biol 6: 386–394 [DOI] [PubMed] [Google Scholar]
- Niemi J, Mäntsälä P (1995) Nucleotide sequences and expression of genes from Streptomyces purpurascens that cause the production of new anthracyclines in Streptomyces galilaeus. J Bacteriol 177: 2942–2945 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niemi J, Ylihonko K, Hakala J, Parssinen R, Kopio A, Mäntsälä P (1994) Hybrid anthracycline antibiotics: production of new anthracyclines by cloned genes from Streptomyces purpurascens in Streptomyces galilaeus. Microbiology 140: 1351–1358 [DOI] [PubMed] [Google Scholar]
- Orengo CA, Thornton JM (1993) Alpha plus beta folds revisited: some favoured motifs. Structure 1: 105–120 [DOI] [PubMed] [Google Scholar]
- Perrakis A, Morris R, Lamzin VS (1999) Automated protein model building combined with iterative structure refinement. Nat Struct Biol 6: 458–463 [DOI] [PubMed] [Google Scholar]
- Räty K, Kantola J, Hautala A, Hakala J, Ylihonko K, Mäntsälä P (2002) Cloning and characterization of Streptomyces galilaeus aclacinomycins polyketide synthase (PKS) cluster. Gene 293: 115–122 [DOI] [PubMed] [Google Scholar]
- Sheldrick GM, Schneider TR (1997) ShelxL: High Resolution Refinement Methods in Enzymology. San Diego, CA: Academic Press [PubMed] [Google Scholar]
- Shen B, Hutchinson CR (1993) Enzymatic synthesis of a bacterial polyketide from acetyl and malonyl coenzyme A. Science 262: 1535–1540 [DOI] [PubMed] [Google Scholar]
- Shen B, Hutchinson CR (1996) Deciphering the mechanism for the assembly of aromatic polyketides by a bacterial polyketide synthase. Proc Natl Acad Sci USA 93: 6600–6604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strohl WR (2001) Biochemical engineering of natural product biosynthesis pathways. Metab Eng 3: 4–14 [DOI] [PubMed] [Google Scholar]
- Strohl WR, Bartel PL, Connors NC, Zhu C-B, Dosch DC, Beale JMJ, Floss HG, Stutzman-Engwall K, Otten SL, Hutchinson CR (1989) Biosynthesis of Natural and Hybrid Polyketides by Anthracycline Producing Streptomyces. Washington, DC: American Society of Microbiology [Google Scholar]
- Sultana A, Kallio P, Jansson A, Niemi J, Mäntsälä P, Schneider G (2004) Crystallisation and preliminary crystallographic data of SnoaL, a polyketide cyclase in nogalamycin biosynthesis. Acta Crystallogr D, in press [DOI] [PubMed] [Google Scholar]
- Terwilliger TC (2000) Maximum-likelihood density modification. Acta Crystallogr D 56: 965–972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terwilliger TC, Berendzen JB (1999) Automated MAD and MIR structure solution. Acta Crystallogr D 55: 849–861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson JD, Higgins DG, Gibson TJ (1994) CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res 22: 4673–4680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torkkell S, Kunnari T, Palmu K, Hakala J, Mäntsälä P, Ylihonko K (2000) Identification of a cyclase gene dictating the C-9 stereochemistry of anthracyclines from Streptomyces nogalater. Antimicrob Agents Chemother 44: 396–399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walczak RJ, Dickens ML, Priestley ND, Strohl WR (1999) Purification, properties, and characterization of recombinant Streptomyces sp. strain C5 DoxA, a cytochrome P-450 catalyzing multiple steps in doxorubicin biosynthesis. J Bacteriol 181: 298–304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiley PF, Elrod DW, Marshall VP (1978) Biosynthesis of the anthracycline antibiotics nogalamycin and steffimycin B. J Org Chem 43: 3457–3461 [Google Scholar]
- Ylihonko K, Hakala J, Kunnari T, Mäntsälä P (1996) Production of hybrid anthracycline antibiotics by heterologous expression of Streptomyces nogalater nogalamycin biosynthesis genes. Microbiology 142: 1965–1972 [DOI] [PubMed] [Google Scholar]