

ABSTRACT
The phosphatidylinositol 3-kinase (PI3K)/Akt signaling pathway plays key roles in diverse cellular activities and promotes cell growth and survival. It is therefore unsurprising that most viruses modify this pathway in order to facilitate their replication and spread. Previous work has suggested that the herpes simplex virus 1 (HSV-1) tegument proteins VP11/12 and US3 protein kinase modulate the PI3K/Akt pathway, albeit in opposing ways: VP11/12 binds and activates Src family kinases (SFKs), is tyrosine phosphorylated, recruits PI3K in an SFK-dependent fashion, and is required for HSV-induced phosphorylation of Akt on its activating residues; in contrast, US3 inhibits Akt activation and directly phosphorylates downstream Akt targets. We examined if US3 negatively regulates Akt by dampening the signaling activity of VP11/12. Consistent with this hypothesis, the enhanced Akt activation that occurs during US3-null infection requires VP11/12 and correlates with an increase in SFK-dependent VP11/12 tyrosine phosphorylation. In addition, deleting US3 leads to a striking increase in the relative abundances of several VP11/12 species that migrate with reduced mobility during SDS-PAGE. These forms arise through phosphorylation, strictly require the viral UL13 protein kinase, and are excluded from virions. Taken in combination, these data indicate that US3 dampens SFK-dependent tyrosine and UL13-dependent serine/threonine phosphorylation of VP11/12, thereby inhibiting VP11/12 signaling and promoting virion packaging of VP11/12. These results illustrate that protein phosphorylation events mediated by viral protein kinases serve to coordinate the roles of VP11/12 as a virion component and intracellular signaling molecule.
IMPORTANCE Herpesvirus tegument proteins play dual roles during the viral life cycle, serving both as structural components of the virus particle and as modulators of cellular and viral functions in infected cells. How these two roles are coordinated during infection and virion assembly is a fundamental and largely unanswered question. Here we addressed this issue with herpes simplex virus VP11/12, a tegument protein that activates the cellular PI3K/Akt signaling pathway. We showed that protein phosphorylation mediated by the viral US3 and UL13 kinases serves to orchestrate its functions: UL13 appears to inhibit VP11/12 virion packaging, while US3 antagonizes UL13 action and independently dampens VP11/12 signaling activity.
INTRODUCTION
Most if not all viruses modulate intracellular signaling pathways in order to promote virus replication and dampen host innate and adaptive immune responses. One commonly targeted pathway is the phosphatidylinositol-3-kinase (PI3K)/Akt axis, which plays critical roles in cell survival, growth, proliferation, gene expression, and metabolism (reviewed in references 1 and 2). The Akt signaling cascade is typically triggered by activation of cell surface receptors (including receptor tyrosine kinases, B and T cell receptors, and G protein-coupled receptors) by extracellular ligands such as growth factors, leading to membrane recruitment and activation of PI3K through interactions between the p85 regulatory subunit of PI3K and membrane-associated proteins. Once proximal to its substrate, PI3K converts phosphatidylinositol 4,5-biphosphate (PIP2) to phosphatidylinositol 3,4,5-triphosphate (PIP3). PIP3 then binds Akt and its activator phosphoinositide-dependent kinase 1 (PDK1) through their pleckstrin homology domains. Colocalization of Akt and PDK1, along with a conformational change in Akt induced by PIP3, results in phosphorylation of Akt by PDK1 at threonine 308 (T308) in the activation loop (3, 4). Full activation of Akt requires a second phosphorylation event at serine 473 (S473) in the hydrophobic motif by mammalian target of rapamycin complex 2 (mTORC2) (3–5). These phosphorylation events stabilize the active conformation of Akt and potentiate its kinase activity. Akt then phosphorylates numerous downstream targets, including those involved in apoptosis (NF-κB, FOXO1, and Bad), cell cycle control (MDM2), glucose metabolism (GSK3β), and protein synthesis (mTORC1, 4EBP1, and p70S6K) (1, 6).
The PI3K/Akt pathway is exploited by viruses to manipulate a variety of processes, including virus entry, transcription, translation, and cell survival (reviewed in references 7 and 8). Virtually all viruses regulate this pathway either positively or negatively in order to create a favorable environment for their survival and spread. To these ends, viruses use diverse strategies, including producing proteins that mimic an activated cell surface receptor (polyoma middle T antigen [9]), directly interact with PI3K (NSP1 of rotavirus [10]) or Akt (M-T5 of myxoma virus [11]), inactivate Akt (vesicular stomatitis virus matrix protein [12]), or mimic Akt (vAKT from mammalian AKT8 retrovirus [13]).
Herpes simplex virus 1 (HSV-1) is a large double-stranded DNA virus (152 kb) and a significant human pathogen. The HSV-1 virion is composed of a DNA-containing icosahedral nucleocapsid enclosed by the tegument, which is in turn surrounded by a host-derived lipid envelope containing virally encoded glycoproteins. The tegument is a proteinaceous layer containing at least 20 viral proteins (14, 15). Tegument proteins are released shortly after viral entry directly into the cytoplasm of infected cells, allowing the virus to regulate cellular activities prior to immediate early gene expression. Because tegument proteins are among the first viral proteins encountered by the host cell, many play key roles in disabling host cell defenses and regulating host and virus gene expression.
Two tegument proteins have been shown to modulate PI3K/Akt activity during HSV-1 infection: the US3 serine/threonine protein kinase (16) and the VP11/12 protein, encoded by the gene UL46 (17, 18). Benetti and Roizman showed that HSV-1 infection leads to activation of Akt in a PI3K-dependent fashion (16), and Wagner and Smiley later showed that this effect strictly requires VP11/12 (18), a highly abundant (between 1,000 and 2,000 copies) tegument phosphoprotein (19, 20). Current evidence suggests that VP11/12 activates PI3K and Akt by mimicking an activated growth factor receptor (17): it associates with the plasma membrane in infected cells (21, 22), binds and activates Src family tyrosine kinases (SFKs) (23), becomes phosphorylated on several tyrosine-based motifs (17), and recruits the p85 subunit of PI3K in an SFK-dependent manner (17, 18). The varicella-zoster virus VP11/12 orthologue also activates PI3K/Akt signaling (24), but the mechanisms involved remain unknown.
Several downstream targets of Akt are phosphorylated during HSV-1 infection (18, 25); however, VP11/12 is not required for this effect (18). This surprising observation can likely be explained by the finding of Chuluunbaatar et al. that the viral kinase US3 directly phosphorylates at least some Akt targets (including TSC2, FOXO1, and GSK3) on the same residues as Akt, thus serving as an Akt mimic (25). These data raise the possibility that HSV-1 exerts redundant control over the Akt pathway. US3 is a multifunctional protein that plays diverse roles in virus replication and pathogenesis (26, 27). It acts by phosphorylating a number of viral and cellular substrates, including UL47 (28), UL34 (29), histone deacetylases 1 and 2 (30–32), lamin A/C (33), and Bad (34). Functions ascribed to US3 include preventing apoptosis (35–41), enhancing viral gene expression by inhibiting histone deacetylation (32, 42, 43), promoting nuclear egress of progeny nucleocapsids (44–48), stimulating viral cell-to-cell spread (49–51), evading the immune system (52–55), modulating membrane biosynthesis (56), and downregulating extracellular signal-regulated kinase (ERK) activity (57). Relevant to the present report, Akt activation is enhanced during US3-null infection (16), suggesting that US3 also negatively regulates Akt during HSV-1 infection. The mechanism of this downregulation has yet to be established.
The results outlined above suggest that the PI3K/Akt pathway is subject to complex control during HSV-1 infection and raise the possibility that the functions of US3 and VP11/12 are intertwined. Indeed, Matsuzaki et al. have shown that HSV-2 US3 alters the electrophoretic mobility of VP11/12 and enhances its stability and virion packaging during HSV-2 infection and that US3 phosphorylates VP11/12 in vitro (58). We therefore examined the effect of inactivating HSV-1 US3 on VP11/12-dependent signaling, posttranslational modifications, and virion packaging. We present evidence that HSV-1 US3 inhibits phosphorylation of VP11/12 by both SFKs and HSV-1 UL13, dampens VP11/12 signaling activity, and promotes its virion packaging.
MATERIALS AND METHODS
Cells and viruses.
Vero and Cre-Vero cells (ATCC) were maintained in Dulbecco's modified Eagle medium (DMEM) (Gibco) supplemented with 5% heat-inactivated fetal bovine serum (FBS), 100 U/ml penicillin, and 100 μg/ml streptomycin. Cre-Vero cells were supplemented with 400 μg/ml hygromycin B (Invitrogen) every fourth passage to maintain the transgene. Telomerase-immortalized human foreskin fibroblasts (HFF-Tel12 [59]) were propagated in DMEM supplemented with 10% heat-inactivated FBS, 1 mM sodium pyruvate, 100 U/ml penicillin, and 100 μg/ml streptomycin. Cell lysates being examined for total and phospho-Akt (S473) were serum starved (DMEM only) 24 h prior to and during infection. HSV-1 KOS37 was derived from the KOS37 bacmid (60). The ΔUL46 (VP11/12−) and ΔUL46R (VP11/12 repaired) KOS37-derived mutants have been described previously (61). The ΔUS3 (US3−), ΔUS3R (US3 repaired), ΔUL46ΔUS3 (VP11/12− US3−), and ΔUL46ΔUS3R (US3 repaired) mutants were constructed as described below. All virus stocks were prepared from infected Vero cells. Infections of Vero and HFF cells were carried out at 37°C at a multiplicity of infection (MOI) of 10 PFU per cell.
Construction of ΔUS3 and ΔUL46ΔUS3 mutants.
HSV-1 mutants lacking US3 or both US3 and UL46 were generated by bacteriophage λ Red-mediated homologous recombination in Escherichia coli, reviewed in reference 62, using the E. coli galK gene as the positive and negative selection marker (63), as described previously (61). All synthetic oligonucleotides were from Integrated DNA Technologies (Coralville, IA, USA). To generate ΔUS3, we deleted nucleotides 209 to 1068 from the US3 open reading frame, exactly the same sequences that were removed to construct the HSV-1 F strain US3 mutant R7041 (64). To this end, the galK gene flanked by 50 nucleotides (nt) of the US3 sequences upstream and downstream of the desired deletion endpoints was amplified by PCR using the primers ΔUS3galK-F and ΔUS3galK-R (Table 1). The amplified galK cassette was first incorporated into the KOS37 bacmid and then was precisely removed to generate the desired markerless deletion bacmid KOS37ΔUS3, using the primers ΔUS3ΔgalK-F and ΔUS3ΔgalK-R (Table 1). As controls, the US3 loci of KOS37ΔUS3 and KOS37ΔUL46ΔUS3 were repaired by reinserting the galk cassette across the US3 deletion, followed by excision using a US3 amplimer generated using the primers ΔUS3Repair-F and ΔUS3Repair-R (Table 1).
TABLE 1.
Primers used for generating virusesa
| Primer name | Sequence (5′–3′) |
|---|---|
| ΔUs3galK-F | CAAACCTTCCCACACCACACCACCCAGCGAGGCCGAGCGCCTGTGTCATCcctgttgacaattaatcatcggca |
| ΔUs3galK-R | AAGATCACCAGACCGGCGCTCCAAATGTCGACGGTCGTGGTATACGGATCtcagcactgtcctgctcct |
| ΔUs3ΔgalK-F | CAAACCTTCCCACACCACACCACCCGGCGATGCCGAGCGCCTGTGTCATC/GATCCGTATACCACGACCGTCGACATTTGGAGCGCCGGTCTGGTGATCTT |
| ΔUs3ΔgalK-R | AAGATCACCAGACCGGCGCTCCAAATGTCGACGGTCGTGGTATACGGATC/GATGACACAGGCGCTCGGCATCGCCGGGTGGTGTGGTGTGGGAAGGTTTG |
| ΔUs3Repair-F | CGGGGCCCGTCGTTCGG |
| ΔUs3Repair-R | TCGGGGTCTTTTTGTGCCAACCCG |
| ΔUs3-EP-F | CACACCACACCACCCAGCGAGGCCGAGCGCCTGTGTCATCGATCCGTATACCACGACCGTTAGGGATAACAGGGTAATCGATTT |
| ΔUs3-EP-R | GACCGGCGCTCCAAATGTCGACGGTCGTGGTATACGGATCGATGACACAGGCGCTCGGCCGCCAGTGTTACAACCAATTAACC |
| KDUL13-EP-F | ATGAGCTCAACGGCAAACCACTCCTTTTCCTTTATGGTCATAACGGCAAGCTTATGTTCGCTAGGGATAACAGGGTAATCGATTT |
| KDUL13-EP-R | GACGTCCAACTGATTCGCGAACATAAGCTTGCCGTTATGACCATAAAGGAAAAGGAGTGGCCAGTGTTACAACCAATTAACC |
HSV-1 sequences are in capitals, galK sequences are in lowercase, I-SceI-aphAI sequences are in italics, mutations are in bold, and locations of deletions are indicated with a slash (“/”).
Virus stocks were generated from bacmids following transfection of Cre-Vero cells using Lipofectamine 2000 (Invitrogen), followed by three rounds of plaque purification on Cre-Vero cells (60).
Construction of KDUL13 and KDUL13ΔUS3 mutants.
En passant mutagenesis was used to create a kinase-dead UL13 mutant virus (KDUL13) and a kinase-dead UL13/US3-null (KDUL13ΔUS3) virus from the KOS37 bacterial artificial chromosome (BAC) (65). Briefly, the selection cassette I-SceI-aphAI was amplified from the plasmid pEPkan-S (66) using primers containing sequences surrounding the desired mutation or deletion. To eliminate kinase activity of UL13, lysine 176 of the UL13 gene was converted to methionine (primers KDUL13-EP-F and KDUL13-EP-R; Table 1 [67–71]). US3 nt 209 to 1068 were then deleted to construct the US3 null derivative of the UL13 kinase-dead mutant (primers ΔUS3-EP-F and ΔUS3-EP-R; Table 1). The linear DNA containing the gene-specific sequence, a sequence duplication, I-SceI site, and aphAI was targeted into the KOS37 BAC. Induction of I-SceI expression resulted in cleavage of the I-SceI site, allowing recombination between the duplicated sequences. This recombination event released the aphAI cassette, leaving behind only the desired sequence.
Antibodies.
Antibodies used for primary detection in Western blotting were as follows: phospho-AKT (S473, 1/1,000; Cell Signaling), total AKT (1/1,000; Cell Signaling), β-actin (1/5,000; Sigma), HSV-2 US3 (1/500; kindly provided by Bruce Banfield), HSV-2 UL46 (1/15,000; kindly provided by Yukihiro Nishiyama), phosphotyrosine (4G10, 1/10,000; Upstate), HSV1 VP5 (1/50; Santa Cruz), HSV-1 ICP27 (1/3,000; Virusys Corporation), HSV-1 TK (1/500; kindly provided by William C. Summer), HSV-1 gC (1/2,000; Virusys Corporation), and HSV-1 VP16 (LP1, 1/50; kindly provided by Tony Minson). Primary antibodies were detected using the following secondary antibodies: Alexa Fluor 680 goat anti-mouse IgG (1/10,000; Invitrogen), goat anti-rabbit Alexa Fluor 680 (1/10,000; Invitrogen), IRDye 800-conjugated goat anti-mouse IgG (1/10,000; Rockland), IRDye 800-conjugated goat anti-rabbit IgG (1/10,000; Rockland), and rabbit anti-rat horseradish peroxidase (1/40,000; Sigma).
Drug treatments.
PP2 (4-amino-5-(4-chlorophenyl)-7-(t-butyl) pyrazolo[3,4-d]pyramidine) and its negative control, PP3 (4-amino-7-phenylpyrazolo[3,4-d]pyramidine), were added to cells 2 h postinfection and were left on for the remainder of the infection. PP3 was added at a concentration of 30 μM and PP2 at 20 μM.
Western blotting.
Infected cells were washed one time with phosphate-buffered saline (PBS) and lysed for 20 min in RIPA buffer (150 mM NaCl, 1 mM EGTA, 50 mM Tris-HCl [pH 7.4], 1% NP-40, 0.25% sodium deoxycholate, cOmplete [EDTA-free] protease inhibitor cocktail [Roche], and PhosSTOP phosphatase inhibitor cocktail [Roche]) at 4°C. Following lysis, appropriate amounts of 6× loading buffer (375 mM Tris-HCl [pH 6.8], 30% glycerol, 10% sodium dodecyl sulfate, 1.43 M β-mercaptoethanol, and 0.03% bromophenol blue) were added to the samples, and the samples were boiled for 5 min. Lysates were then separated electrophoretically by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) (8 to 10%) and were transferred to a nitrocellulose membrane (Hybond ECL; GE Healthcare). Membranes were blocked for 1 h at room temperature, and primary antibodies were added overnight at 4°C. Secondary antibodies were added at room temperature for 1 h. Membranes imaged using chemiluminescence detection were blocked in 3% bovine serum albumin (BSA) in TBST (50 mM Tris, 150 mM NaCl, and 0.05% Tween 20). Antibodies were incubated in 1% BSA in TBST, and proteins were visualized using the ECL Plus reagent (GE Healthcare). Membranes imaged using infrared fluorescent detection were blocked in 1:1 Odyssey blocking buffer (Li-Cor)-TBST. Antibodies were incubated in 1:1 Odyssey blocking buffer-TBST, and proteins were detected using the Odyssey Infrared Imaging System (Li-Cor).
Phosphatase treatment.
HFF cells infected for 15 h were washed once in PBS and lysed for 20 min at 4°C in RIPA buffer, including protease inhibitors or both phosphatase and protease inhibitors. Samples lysed in RIPA buffer with protease inhibitors only were left untreated or were treated with 2 U of calf intestinal alkaline phosphatase (CIAP) (Invitrogen). The phosphatase reaction mixture was incubated at 37°C for 30 min. Proteins were then solubilized in loading buffer and visualized using Western blotting.
Partial purification of virions.
Vero cells grown to confluence in a T150 flask were infected for 18 h with KOS37, ΔUS3, and ΔUS3R viruses at an MOI of 10. Following infection, medium was collected and cellular debris was removed from the medium by low-speed centrifugation (1,500 × g) for 10 min at 4°C. The supernatant was then spun through a 30% (wt/vol) sucrose cushion in TNE buffer (50 mm Tris–HCl [pH 7.4], 100 mm NaCl, and 0.1 mm EDTA) for 60 min at 26,000 rpm in a Beckman SW40Ti rotor to pellet extracellular virions. The resulting pellet was resuspended in 70 μl TNE buffer, and samples were analyzed by Western blotting.
RESULTS AND DISCUSSION
Deleting US3 drastically alters the electrophoretic mobility of VP11/12.
In order to investigate potential functional links between US3 and VP11/12 during HSV-1 infection, we examined the effect of deleting US3 on the electrophoretic mobility, signaling activity, and virion packaging of VP11/12. To this end, we constructed US3-null and UL46-null/US3-null viruses to use alongside previously constructed UL46-null (KOS37 ΔUL46) and UL46 repair (KOS37 ΔUL46R) viruses (61). The US3-null and UL46-null/US3-null viruses were constructed using bacteriophage λ Red-mediated targeted homologous recombination of an infectious HSV-1 KOS37 BAC (63). The US3-null mutation (ΔUS3) removes nt 209 to 1068 from the US3 open reading frame, the same residues that were deleted to generate the HSV-1 strain F US3 mutant R7041 (64). Repair viruses in which the US3 gene was restored were also created (KOS37 ΔUS3R and KOS37 ΔUL46ΔUS3R). All mutations were confirmed by DNA sequencing.
We first examined the effects of deleting US3 on the levels and electrophoretic mobility of VP11/12. HFF cells were mock infected or infected with wild-type KOS37 and the ΔUL46, ΔUL46R, ΔUS3, ΔUS3R, ΔUL46ΔUS3, and ΔUL46ΔUS3R mutants for 15 h. Cells were then harvested and examined by Western blotting with polyclonal antibodies to VP11/12 and US3 (Fig. 1). The VP11/12 antibody detected a prominent band of ca. 85 kDa and several less-abundant more slowly migrating species in cells infected with wild-type KOS37, while US3 migrated as a tight doublet of ca. 68 kDa. As expected, VP11/12 and US3 were not detected in samples infected with ΔUL46 or ΔUS3 virus, respectively, or with the ΔUL46ΔUS3 double mutant, and expression was restored in the corresponding repaired viruses (ΔUL46R, ΔUS3R, and ΔUL46ΔUS3R viruses) (Fig. 1). Consistent with the observations made with HSV-2 (58), cells infected with ΔUS3 virus displayed a striking increase in the relative abundance of the more slowly migrating forms of VP11/12 compared to results with wild-type infection (Fig. 1), an effect that was eliminated in the ΔUS3 repair virus (ΔUS3R). However, in contrast to the results of Matsuzaki et al. (58), the overall abundance of VP11/12 was not reduced in the absence of US3. These data suggest that HSV-1 US3 influences one or more posttranslational modifications of VP11/12 without greatly altering its stability.
FIG 1.
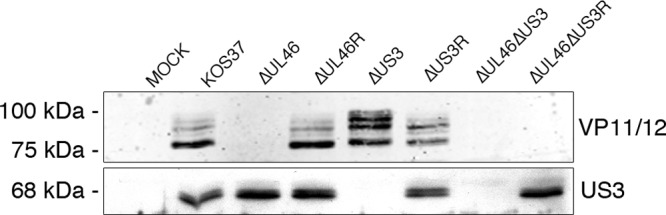
Deletion of US3 induces an electrophoretic mobility shift in VP11/12. HFF cells were mock infected or infected with the indicated viruses for 15 h. Cell extracts were prepared and subjected to Western blot analysis using antibodies directed against VP11/12 and US3.
The slowly migrating forms of VP11/12 arise through phosphorylation.
VP11/12 is highly phosphorylated (19, 72, 73) on serine (74), tyrosine (61), and perhaps threonine residues, and it therefore seemed possible that the more slowly migrating species observed in the absence of US3 might arise through differential phosphorylation. Indeed, Murphy et al. reported that the more slowly migrating forms of VP11/12 present in wild-type-infected cells are converted to the most rapidly migrating form by in vitro phosphatase treatment (21). To test this possibility, HFF cells were mock infected or infected with wild-type KOS37, ΔUS3, and ΔUS3R virus. At 15 h postinfection, cells were harvested and lysates were incubated in the presence or absence of phosphatase inhibitors or treated with calf intestinal alkaline phosphatase (CIAP), and the mobility of VP11/12 was assessed by Western blotting (Fig. 2A). In the extracts prepared from cells infected with ΔUS3 virus, the abundance of the slowly migrating forms of VP11/12 was severely reduced after incubation in the absence of phosphatase inhibitors and was further reduced by the addition of CIAP, while the relative abundance of the most rapidly migrating species was increased by these treatments. Similar reductions were also observed for the (much less abundant) slowly migrating species present in cells infected with KOS37 and ΔUS3R virus, in agreement with the findings of Murphy et al. (21). Comparable results were obtained with extracts prepared from infected Vero cells (data not shown). These data suggest that the more slowly migrating forms of VP11/12 that accumulate in the absence of US3 arise through phosphorylation. Additional evidence supporting this conclusion is presented below.
FIG 2.
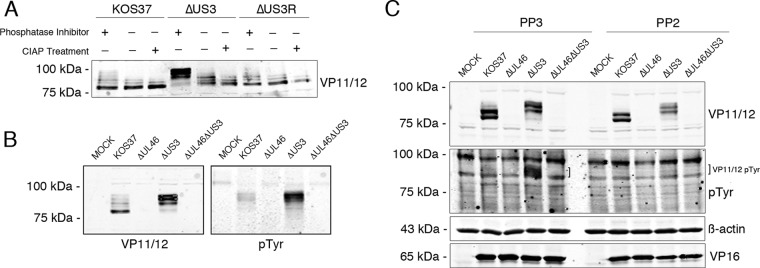
US3 inhibits phosphorylation of VP11/12. (A) The electrophoretic mobility shift is due to phosphorylation. HFF cells were infected with the indicated viruses at an MOI of 10. At 15 h postinfection, cells were harvested in lysis buffer containing protease and phosphatase inhibitors, or lysis buffer containing protease inhibitors with or without subsequent CIAP treatment. Cell lysates were subjected to SDS-PAGE and Western blotting with anti-VP11/12 antibodies. (B) HFF cells were infected for 15 h, and lysates were then assessed for total VP11/12 and tyrosine phosphorylation by Western blotting. (C) Vero cells were infected as for panel A. At 2 h postinfection, the SFK inhibitor PP2 was added to cells at a concentration of 20 μM and the PP2 negative control PP3 was added at a concentration of 30 μM. Total protein was isolated 8 h later and analyzed by immunoblotting with the indicated antisera. β-Actin and VP16 were used as controls for loading and viral infection, respectively.
Increased tyrosine phosphorylation of VP11/12 in the absence of US3.
In order to determine whether US3 affects tyrosine phosphorylation of VP11/12, mock-infected and infected HFF cells were harvested as described above and analyzed by Western blotting using antibodies directed against VP11/12 and phosphotyrosine (pTyr) (Fig. 2B, left and right panels, respectively). As described above, deletion of US3 caused a marked increase in the abundance of the more slowly migrating forms of VP11/12 relative to results with KOS37 infection (Fig. 2B, left panel). Strikingly, the antiphosphotyrosine antibody reacted predominately with a cluster of bands that comigrate with these slowly migrating species in extracts of both KOS37- and ΔUS3 virus-infected cells (Fig. 2B, right panel). Previous work has shown that these tyrosine-phosphorylated species correspond to VP11/12 (17, 18, 61). In addition, the ΔUS3 sample produced a stronger VP11/12 pTyr signal than did the KOS37 sample. Similar results were obtained with Vero cells (data not shown). These data indicate that most of the tyrosine-phosphorylated VP11/12 molecules migrate more slowly than the bulk of VP11/12. In addition, the tyrosine-phosphorylated forms increase in abundance in the absence of US3. It is important to note that although these results show that most of the tyrosine-phosphorylated VP11/12 molecules display reduced electrophoretic mobility, they do not establish whether tyrosine phosphorylation itself causes the mobility shift. This issue is addressed below.
The VP11/12 mobility shift is not due to tyrosine phosphorylation.
SFKs are membrane-associated nonreceptor tyrosine kinases that regulate a large number of intracellular signaling pathways, including the PI3K-Akt pathway (reviewed in references 75 and 76). VP11/12 binds and activates SFKs, and SFK activation is required for efficient VP11/12 tyrosine phosphorylation and downstream PI3K/Akt activation (17, 18, 23, 61). In order to determine whether SFK activity is required for the VP11/12 mobility shift, Vero cells were mock infected or infected with KOS37 or the ΔUL46, ΔUS3, or ΔUL46ΔUS3 virus. Two hours postinfection, the cells were treated with the SFK inhibitor PP2 or the inactive analog PP3 as a negative control. Cells were harvested 8 h later and analyzed by immunoblotting using antibodies directed against VP11/12 and phosphotyrosine. VP16 served as a control for virus infection, and β-actin was used as a loading control (Fig. 2C). PP2 had only a relatively minor effect on the overall levels of VP11/12 in both KOS37 and ΔUS3 virus-infected cells and did not obviously alter the mobility shift observed in the absence of US3 (Fig. 2C, upper panel). In contrast, the drug essentially eliminated the tyrosine-phosphorylated forms of VP11/12 (Fig. 2C, pTyr), as previously described (17, 18, 61). These data indicate that the mobility shift is not due to tyrosine phosphorylation and raise the possibility that phosphorylation by one or more cellular or viral serine/threonine kinases is involved. Additional evidence supporting this conclusion is presented below.
US3 inhibits VP11/12-dependent Akt activation.
As described in the introduction, US3 and VP11/12 appear to play opposing roles in modulating the host Akt signaling pathway during HSV-1 infection. Benetti and Roizman (16) showed that Akt is phosphorylated early during wild-type HSV-1 infection and that the levels of phosphorylated Akt diminish at later times. In contrast, high levels of phosphorylated Akt are observed throughout the course of infection with a US3 null mutant, indicating that US3 negatively regulates Akt phosphorylation (16). Because VP11/12 is required for Akt activation by wild-type HSV-1 in human fibroblasts (18), we asked whether US3 negatively regulates the activity of VP11/12. If this is indeed the case, then one predicts that enhanced Akt phosphorylation during US3-null infection would be eliminated by deleting VP11/12. To test this prediction, serum-starved HFF and Vero cells were mock infected or infected with KOS37, ΔUL46, ΔUS3, and ΔUL46ΔUS3 virus. Samples were then subjected to Western blot analysis using antibodies directed against phosphorylated (S473) and total Akt (Fig. 3). The signals obtained with the two antibodies were quantified using an infrared imager, and the signal obtained for Akt phosphorylated on S473 was normalized to the total Akt levels and adjusted to β-actin levels (not shown). The results were then expressed as fold increases over the mock-infected values (Fig. 3). The leaky late protein VP16 was used as a control for viral infection.
FIG 3.

US3 negatively regulates VP11/12-induced Akt activation. Serum-deprived HFF and Vero cells were infected as in Fig. 1. Cells were then harvested, solubilized, and subjected to Western blot analysis using antibodies against total and phospho-AKT (S473). (A) HFF cells were harvested at 3-h intervals for a total of 15 h. (B) Vero cells were harvested at 2-h intervals for a total infection time of 12 h. Fold increase: the infrared signals acquired from the phospho-Akt antibody were divided by the infrared signal from the total Akt antibody and normalized to β-actin. The results are expressed relative to those for mock-infected cells.
Consistent with the observations of Benetti and Roizman (16), HFF cells infected with ΔUS3 virus displayed enhanced Akt phosphorylation relative to those infected with KOS37 at all time points, with the difference increasing at later times (Fig. 3A). As expected (18), ΔUL46 virus did not stimulate Akt phosphorylation, and the enhanced Akt phosphorylation observed during ΔUS3 virus infection was abrogated for the ΔUL46 ΔUS3 double mutant (Fig. 3A). These data indicate that VP11/12 is required for HSV-1-induced Akt phosphorylation in HFF cells in both the presence and absence of US3.
Broadly similar results were obtained with Vero cells: ΔUS3 virus displayed elevated levels of Akt phosphorylation relative to KOS37 over the course of the infection (Fig. 3B), and deleting VP11/12 reduced Akt phosphorylation at all time points in both the presence and absence of US3. However, in contrast to the results obtained with HFF cells, Akt phosphorylation peaked around 6 h postinfection and declined somewhat later for both KOS37 and ΔUS3 virus. In addition, deleting VP11/12 did not entirely eliminate Akt activation at the 4- and 6-h time points, raising the possibility that an HSV-1 function other than VP11/12 may contribute to Akt activation at early times postinfection in Vero cells. Further studies will be required to test this hypothesis and identify the relevant gene product(s).
Taken in combination, the data presented in this section show that VP11/12 is required for the enhanced Akt phosphorylation observed during US3-null infection, supporting the idea that US3 inhibits Akt activation by antagonizing the activity of VP11/12. This hypothesis is fully consistent with the observation that US3 also antagonizes tyrosine phosphorylation of VP11/12 (Fig. 2B).
The HSV-1 UL13 protein kinase is essential for the production of the slowly migrating forms of VP11/12.
Matsuzaki et al. (58) and Murphy et al. (21) found that VP11/12 expressed in the absence of other HSV gene products migrates as a single band during SDS-PAGE, while more slowly migrating forms accumulate following HSV superinfection. These observations suggest that one or more HSV gene products other than US3 induce the mobility shift. Given that the mobility shift is due to phosphorylation (21), we examined whether the HSV-1 UL13 protein kinase plays a role. UL13 is a serine/threonine kinase that is packaged within the tegument and phosphorylates diverse cellular and viral targets during infection, including US3 (70, 77–86). We used two UL13 mutants to examine the role of UL13: d13LacZ (82), an HSV-1 strain KOS1.1 mutant bearing a lacZ cassette inserted across a 780-nt deletion of UL13 coding sequences, and KDUL13 (see Materials and Methods), a KOS37-derived kinase-dead UL13 mutant bearing a mutation that converts UL13 amino acid residue lysine 176 to methionine (67–70). We also constructed and analyzed a KOS37-derived UL13 kinase-dead/US3− null doubly mutant virus (KDUL13ΔUS3; see Materials and Methods).
Serum-starved Vero cells were mock infected or infected with KOS37, ΔUL46, ΔUS3, ΔUL46ΔUS3, d13lacZ, KDUL13, and KDUL13ΔUS3 viruses for 10 h, and then cell lysates were analyzed by immunoblotting using antibodies against VP11/12, phosphotyrosine, and phospho-AKT (S473) (Fig. 4). HSV ICP27, TK, VP16, and gC were also examined to assess possible defects in viral gene expression. Strikingly, cells infected with both UL13 mutants displayed a single VP11/12 band, which comigrated with the most rapidly migrating species produced during wild-type infection (Fig. 4). Similar results were obtained when UL13 was inactivated in the US3-null background (Fig. 4, compare the ΔUS3 and KDUL13ΔUS3 samples). Overall, these data demonstrate that that UL13 is required for the mobility shift and imply that US3 inhibits UL13-dependent phosphorylation of VP11/12 in addition to dampening its SFK-dependent tyrosine phosphorylation. Further studies are required to determine if VP11/12 is a direct substrate of UL13. It is important to note that these findings provide no information about whether UL13 is required for tyrosine phosphorylation of VP11/12, a question that is addressed below.
FIG 4.
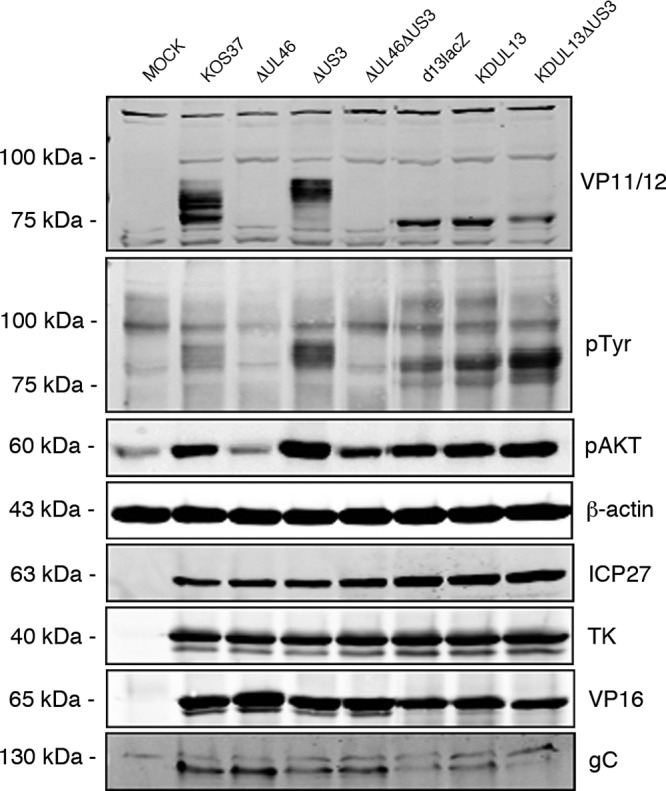
The VP11/12 electrophoretic mobility shift requires the UL13 protein kinase. Serum-starved Vero cells were mock infected or infected with wild-type KOS37 or ΔUL46, ΔUS3, or ΔUL46ΔUS3 virus, the UL13 deletion virus d13lacZ, the kinase-dead UL13 virus KDUL13, or the kinase-dead UL13, US3 knockout virus KDUL13ΔUS3. Ten hours postinfection VP11/12 expression, tyrosine phosphorylation, and Akt activation were assessed by Western blotting using the indicated antibodies. Cell lysates were analyzed for viral gene expression using antibodies against the HSV-1 proteins ICP27, TK, VP16, and gC. β-Actin was used as a loading control.
UL13 is not required for VP11/12 tyrosine phosphorylation or Akt activation.
As shown in Fig. 4, cells infected with d13lacZ and KDUL13 both displayed a prominent pTyr signal that migrated between the most rapidly migrating form of VP11/12 and the UL13-dependent species detected with the VP11/12 antibody. We interpret this species as arising from tyrosine-phosphorylated VP11/12 molecules that have not been subjected to the UL13-dependent serine/threonine phosphorylation events that lead to the major mobility shift. Consistent with this interpretation, this pTyr signal was significantly enhanced in cells infected with KDUL13ΔUS3, although the total amount of VP11/12 was if anything reduced relative to that with d13lacZ and KDUL13. These data indicate that VP11/12 is tyrosine phosphorylated in both the presence and absence of UL13 and that in both cases such tyrosine phosphorylation is increased in the absence of US3. Consistent with these observations, both UL13 mutants activated Akt to the same extent as wild-type KOS37 (Fig. 4), indicating that UL13 plays little if any role in downstream signaling by VP11/12.
US3 enhances virion packaging of HSV-1 VP11/12.
Murphy et al. (21) showed that only the most rapidly migrating form of VP11/12 is packaged into virions, and similar data were reported by Matsuzaki et al. for HSV-2 (58). Matsuzaki et al. also documented that deleting US3 dramatically reduces the amount of VP11/12 packaged into HSV-2 virions (58). To determine if this is also the case for HSV-1, Vero cells were mock infected or infected with wild-type KOS37, ΔUS3, and ΔUS3R virus. Eighteen hours postinfection, the medium was collected and cleared of debris, and extracellular virions were pelleted through a 30% sucrose cushion. The presence of VP11/12 was then determined by Western blotting (Fig. 5). The data confirmed that only the most rapidly migrating form of VP11/12 is packaged into extracellular wild-type KOS37 virions and demonstrated that this is also the case for ΔUS3 virus (Fig. 5A). Moreover, the amount of VP11/12 present in ΔUS3 virions was substantially reduced relative to that with KOS37 and ΔUS3R virus, although similar levels of the major capsid protein VP5 (UL19) were detected (Fig. 5B). Comparable results were obtained with HFF cells (data not shown). Thus, US3 is required for optimal packaging of VP11/12 into HSV-1 virus particles, consistent with the previous observations made with HSV-2 (58). As discussed further below, an interesting possibility is that US3 promotes virion incorporation of VP11/12 by dampening its UL13-dependent phosphorylation.
FIG 5.
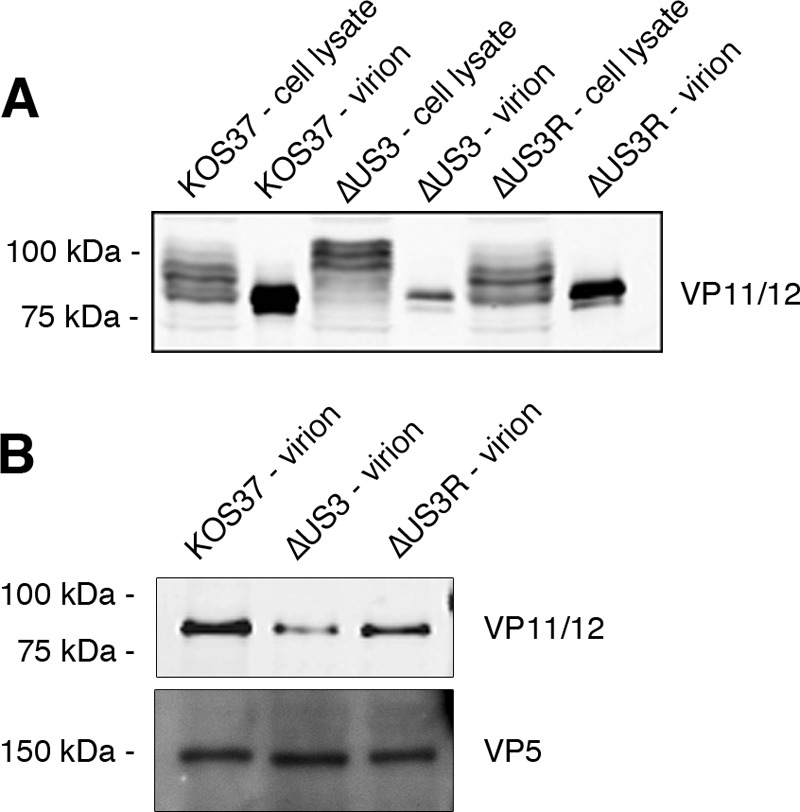
VP11/12 incorporation into virions is enhanced by US3. (A) Vero cells were mock infected or infected with KOS37, ΔUS3, or ΔUS3R virus at an MOI of 10. At 18 h postinfection, medium was collected and cleared of debris, and virions were pelleted through a 30% sucrose cushion. Cell lysates and virions were then analyzed by Western blotting with an anti-VP11/12 antibody. (B) Aliquots of virion preparations shown in panel A, containing approximately equivalent amounts of the capsid protein VP5, were analyzed by Western blotting for VP11/12 and VP5.
Conclusions and potential mechanisms.
Herpesvirus tegument proteins play dual roles during the viral life cycle, serving both as structural components of the virus particle and as modulators of cellular and viral functions in the infected cells. How these two roles are coordinated during infection and virion assembly remains a fundamental and largely unanswered question. Our results illustrate that in the case of HSV-1 VP11/12, protein phosphorylation mediated by the viral US3 and UL13 kinases serve to coordinate these functions.
We have reached five major conclusions based on our results and previously published data. First, VP11/12 is subject to at least two distinct sets of phosphorylation events during HSV-1 infection: SFK-dependent phosphorylation of tyrosine residues, leading to downstream PI3K/Akt signaling, and UL13-dependent serine/threonine phosphorylation, producing the major electrophoretic mobility shift. Second, these two classes of phosphorylation events can be functionally uncoupled, since SFK activity is not required for the UL13-dependent mobility shift (Fig. 2C) and UL13 is not required for VP11/12 tyrosine phosphorylation (Fig. 4). Nevertheless, they appear to be at least partially correlated, since most of the tyrosine-phosphorylated VP11/12 molecules present in infected cells display the UL13-dependent mobility shift (Fig. 2B). Third, US3 inhibits both SFK- and UL13-dependent phosphorylation of VP11/12. Fourth, US3 is required for efficient packaging of VP11/12 into HSV-1 virions (Fig. 5), as previously observed for HSV-2 (58). Fifth, the UL13-modified forms of VP11/12 are excluded from virions (Fig. 5).
The finding that UL13 is essential for the accumulation of detectable levels of the slowly migrating forms of VP11/12 raises the possibility that VP11/12 is a direct target of UL13. Alternatively, UL13 could indirectly promote the phosphorylation events leading to the electrophoretic mobility shift by stimulating the activity of a cellular protein kinase. The finding that US3 suppresses these UL13-dependent events implies that US3 directly or indirectly inhibits the activity of UL13 or downstream kinase or alters the conformation and/or subcellular localization of VP11/12. In order to help distinguish between these and other possibilities, it will be essential to determine if VP11/12 is a direct target of UL13 and if US3 also inhibits phosphorylation of other direct UL13 targets, such as ICP22, UL41, and UL49 (87). In addition, US3 is a known substrate of UL13 (86), and it will therefore be important to determine if UL13 also regulates US3 activity.
The slowly migrating UL13-dependent forms of VP11/12 are completely absent from virions (Fig. 5), even when these represent the majority of the VP11/12 molecules present in the infected cell (as during US3-null infection). This striking finding carries three implications. First, it strongly suggests that UL13-dependent phosphorylation prevents incorporation of VP11/12 into virions. Perhaps UL13-dependent phosphorylation disrupts some of the protein-protein interactions that are used to assemble the tegument. Consistent with this hypothesis, UL13 has been previously implicated in the dissolution of the tegument in vitro (88). It will therefore be important to determine if the UL13-modified forms of VP11/12 retain the ability to interact with its known tegument binding partners VP16 and VP22 (89). Alternatively, it is possible that the UL13-dependent modifications divert VP11/12 from the virion assembly pathway by altering its subcellular trafficking. Second, VP11/12 and UL13 are both components of the tegument. Therefore, if VP11/12 is indeed a direct UL13 substrate, then one or more mechanisms likely exist to restrain UL13 action on VP11/12 during virion assembly. Third, the finding suggests that tyrosine-phosphorylated forms of VP11/12 are also largely absent from virions, since these molecules display the UL13-dependent mobility shift.
If, as seems likely, the UL13-dependent modifications inhibit virion packaging of VP11/12, then the ability of US3 to antagonize such modification could provide a simple explanation of how US3 enhances packaging. According to this view, the reduced levels of VP11/12 packaging during US3-null infection stem from the overall depletion of the most rapidly migrating form due to exaggerated UL13 activity (Fig. 4). However, it is possible that US3 also plays a positive role in enhancing VP11/12 packaging, for example, by binding VP11/12 or indirectly altering its subcellular trafficking.
It will be important to learn how US3 inhibits SFK-dependent tyrosine phosphorylation of VP11/12 and downstream Akt activation. Possible mechanisms include directly or indirectly inhibiting SFK activity, modulating the conformation and/or subcellular localization of VP11/12, direct binding of US3 to VP11/12, or channeling VP11/12 into the virion assembly pathway. It is also possible that US3 modulates one or more cellular components of the PI3K/Akt axis downstream of VP11/12, such as the PH domain leucine-rich repeat protein phosphatase (PHLLP) or the lipid phosphatase PTEN (90, 91), in addition to its inhibitory effects on VP11/12. Further studies are required to distinguish between these possibilities, which are not mutually exclusive.
The finding that US3 downregulates tyrosine phosphorylation of VP11/12 and dampens VP11/12-dependent Akt activation is very intriguing, given that US3 phosphorylates certain cellular Akt targets on the same residues as Akt and thus serves as an Akt mimic (25, 92). These observations suggest that VP11/12-dependent PI3K/Akt signaling may assume particular importance in situations where US3 activity is limiting. Future studies are required to delineate the relative importance of VP11/12- and US3-dependent manipulation of Akt targets in a wide range of cell types and in vivo situations.
ACKNOWLEDGMENTS
We thank Steve Rice for d13lacZ and Bruce Banfield, Tony Minson, Yukihiro Nishiyama, and Bill Summers for the antibodies to US3, VP16, VP11/12, and TK, respectively.
This work was supported by an operating grant from the Canadian Institutes for Health Research (FRN 12172). J.R.S. is a Tier I Canada Research Chair in Molecular Virology, and H.H.E. held a postdoctoral fellowship from Alberta Innovates—Health Solutions.
Footnotes
Published ahead of print 16 April 2014
REFERENCES
- 1.Vivanco I, Sawyers CL. 2002. The phosphatidylinositol 3-kinase AKT pathway in human cancer. Nat. Rev. Cancer 2:489–501. 10.1038/nrc839 [DOI] [PubMed] [Google Scholar]
- 2.Liu P, Cheng H, Roberts TM, Zhao JJ. 2009. Targeting the phosphoinositide 3-kinase pathway in cancer. Nat. Rev. Drug Discov. 8:627–644. 10.1038/nrd2926 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Alessi DR, Deak M, Casamayor A, Caudwell FB, Morrice N, Norman DG, Gaffney P, Reese CB, MacDougall CN, Harbison D, Ashworth A, Bownes M. 1997. 3-Phosphoinositide-dependent protein kinase-1 (PDK1): structural and functional homology with the Drosophila DSTPK61 kinase. Curr. Biol. 7:776–789. 10.1016/S0960-9822(06)00336-8 [DOI] [PubMed] [Google Scholar]
- 4.Stephens L, Anderson K, Stokoe D, Erdjument-Bromage H, Painter GF, Holmes AB, Gaffney PR, Reese CB, McCormick F, Tempst P, Coadwell J, Hawkins PT. 1998. Protein kinase B kinases that mediate phosphatidylinositol 3,4,5-trisphosphate-dependent activation of protein kinase B. Science 279:710–714. 10.1126/science.279.5351.710 [DOI] [PubMed] [Google Scholar]
- 5.Sarbassov DD, Guertin DA, Ali SM, Sabatini DM. 2005. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science 307:1098–1101. 10.1126/science.1106148 [DOI] [PubMed] [Google Scholar]
- 6.Manning BD, Cantley LC. 2007. AKT/PKB signaling: navigating downstream. Cell 129:1261–1274. 10.1016/j.cell.2007.06.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cooray S. 2004. The pivotal role of phosphatidylinositol 3-kinase-Akt signal transduction in virus survival. J. Gen. Virol. 85:1065–1076. 10.1099/vir.0.19771-0 [DOI] [PubMed] [Google Scholar]
- 8.Buchkovich NJ, Yu Y, Zampieri CA, Alwine JC. 2008. The TORrid affairs of viruses: effects of mammalian DNA viruses on the PI3K-Akt-mTOR signalling pathway. Nat. Rev. Microbiol. 6:266–275. 10.1038/nrmicro1855 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schaffhausen BS, Roberts TM. 2009. Lessons from polyoma middle T antigen on signaling and transformation: A DNA tumor virus contribution to the war on cancer. Virology 384:304–316. 10.1016/j.virol.2008.09.042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bagchi P, Dutta D, Chattopadhyay S, Mukherjee A, Halder UC, Sarkar S, Kobayashi N, Komoto S, Taniguchi K, Chawla-Sarkar M. 2010. Rotavirus nonstructural protein 1 suppresses virus-induced cellular apoptosis to facilitate viral growth by activating the cell survival pathways during early stages of infection. J. Virol. 84:6834–6845. 10.1128/JVI.00225-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang G, Barrett JW, Stanford M, Werden SJ, Johnston JB, Gao X, Sun M, Cheng JQ, McFadden G. 2006. Infection of human cancer cells with myxoma virus requires Akt activation via interaction with a viral ankyrin-repeat host range factor. Proc. Natl. Acad. Sci. U. S. A. 103:4640–4645. 10.1073/pnas.0509341103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dunn EF, Connor JH. 2011. Dominant inhibition of Akt/protein kinase B signaling by the matrix protein of a negative-strand RNA virus. J. Virol. 85:422–431. 10.1128/JVI.01671-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bellacosa A, Testa JR, Staal SP, Tsichlis PN. 1991. A retroviral oncogene, akt, encoding a serine-threonine kinase containing an SH2-like region. Science 254:274–277. 10.1126/science.1833819 [DOI] [PubMed] [Google Scholar]
- 14.Loret S, Guay G, Lippe R. 2008. Comprehensive characterization of extracellular herpes simplex virus type 1 virions. J. Virol. 82:8605–8618. 10.1128/JVI.00904-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mettenleiter TC. 2004. Budding events in herpesvirus morphogenesis. Virus Res. 106:167–180. 10.1016/j.virusres.2004.08.013 [DOI] [PubMed] [Google Scholar]
- 16.Benetti L, Roizman B. 2006. Protein kinase B/Akt is present in activated form throughout the entire replicative cycle of deltaU(S)3 mutant virus but only at early times after infection with wild-type herpes simplex virus 1. J. Virol. 80:3341–3348. 10.1128/JVI.80.7.3341-3348.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Strunk U, Saffran HA, Wu FW, Smiley JR. 2013. Role of herpes simplex virus VP11/12 tyrosine-based motifs in binding and activation of the Src family kinase Lck and recruitment of p85, Grb2, and Shc. J. Virol. 87:11276–11286. 10.1128/JVI.01702-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wagner MJ, Smiley JR. 2011. Herpes simplex virus requires VP11/12 to activate Src family kinase-phosphoinositide 3-kinase-Akt signaling. J. Virol. 85:2803–2812. 10.1128/JVI.01877-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhang Y, McKnight JL. 1993. Herpes simplex virus type 1 UL46 and UL47 deletion mutants lack VP11 and VP12 or VP13 and VP14, respectively, and exhibit altered viral thymidine kinase expression. J. Virol. 67:1482–1492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Heine JW, Honess RW, Cassai E, Roizman B. 1974. Proteins specified by herpes simplex virus. XII. The virion polypeptides of type 1 strains. J. Virol. 14:640–651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Murphy MA, Bucks MA, O'Regan KJ, Courtney RJ. 2008. The HSV-1 tegument protein pUL46 associates with cellular membranes and viral capsids. Virology 376:279–289. 10.1016/j.virol.2008.03.018 [DOI] [PubMed] [Google Scholar]
- 22.Willard M. 2002. Rapid directional translocations in virus replication. J. Virol. 76:5220–5232. 10.1128/JVI.76.10.5220-5232.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wagner MJ, Smiley JR. 2009. Herpes simplex virus requires VP11/12 to induce phosphorylation of the activation loop tyrosine (Y394) of the Src family kinase Lck in T lymphocytes. J. Virol. 83:12452–12461. 10.1128/JVI.01364-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Liu X, Cohen JI. 2013. Varicella-zoster virus ORF12 protein activates the phosphatidylinositol 3-kinase/Akt pathway to regulate cell cycle progression. J. Virol. 87:1842–1848. 10.1128/JVI.02395-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chuluunbaatar U, Roller R, Feldman ME, Brown S, Shokat KM, Mohr I. 2010. Constitutive mTORC1 activation by a herpesvirus Akt surrogate stimulates mRNA translation and viral replication. Genes Dev. 24:2627–2639. 10.1101/gad.1978310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kurachi R, Daikoku T, Tsurumi T, Maeno K, Nishiyama Y, Kurata T. 1993. The pathogenicity of a US3 protein kinase-deficient mutant of herpes simplex virus type 2 in mice. Arch. Virol. 133:259–273. 10.1007/BF01313767 [DOI] [PubMed] [Google Scholar]
- 27.Nishiyama Y, Yamada Y, Kurachi R, Daikoku T. 1992. Construction of a US3 lacZ insertion mutant of herpes simplex virus type 2 and characterization of its phenotype in vitro and in vivo. Virology 190:256–268. 10.1016/0042-6822(92)91212-D [DOI] [PubMed] [Google Scholar]
- 28.Kato A, Liu Z, Minowa A, Imai T, Tanaka M, Sugimoto K, Nishiyama Y, Arii J, Kawaguchi Y. 2011. Herpes simplex virus 1 protein kinase Us3 and major tegument protein UL47 reciprocally regulate their subcellular localization in infected cells. J. Virol. 85:9599–9613. 10.1128/JVI.00845-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Purves FC, Spector D, Roizman B. 1991. The herpes simplex virus 1 protein kinase encoded by the US3 gene mediates posttranslational modification of the phosphoprotein encoded by the UL34 gene. J. Virol. 65:5757–5764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kato A, Tanaka M, Yamamoto M, Asai R, Sata T, Nishiyama Y, Kawaguchi Y. 2008. Identification of a physiological phosphorylation site of the herpes simplex virus 1-encoded protein kinase Us3 which regulates its optimal catalytic activity in vitro and influences its function in infected cells. J. Virol. 82:6172–6189. 10.1128/JVI.00044-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Morimoto T, Arii J, Tanaka M, Sata T, Akashi H, Yamada M, Nishiyama Y, Uema M, Kawaguchi Y. 2009. Differences in the regulatory and functional effects of the Us3 protein kinase activities of herpes simplex virus 1 and 2. J. Virol. 83:11624–11634. 10.1128/JVI.00993-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Poon AP, Liang Y, Roizman B. 2003. Herpes simplex virus 1 gene expression is accelerated by inhibitors of histone deacetylases in rabbit skin cells infected with a mutant carrying a cDNA copy of the infected-cell protein no. 0. J. Virol. 77:12671–12678. 10.1128/JVI.77.23.12671-12678.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mou F, Forest T, Baines JD. 2007. US3 of herpes simplex virus type 1 encodes a promiscuous protein kinase that phosphorylates and alters localization of lamin A/C in infected cells. J. Virol. 81:6459–6470. 10.1128/JVI.00380-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Munger J, Roizman B. 2001. The US3 protein kinase of herpes simplex virus 1 mediates the posttranslational modification of BAD and prevents BAD-induced programmed cell death in the absence of other viral proteins. Proc. Natl. Acad. Sci. U. S. A. 98:10410–10415. 10.1073/pnas.181344498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Leopardi R, Van Sant C, Roizman B. 1997. The herpes simplex virus 1 protein kinase US3 is required for protection from apoptosis induced by the virus. Proc. Natl. Acad. Sci. U. S. A. 94:7891–7896. 10.1073/pnas.94.15.7891 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Munger J, Chee AV, Roizman B. 2001. The U(S)3 protein kinase blocks apoptosis induced by the d120 mutant of herpes simplex virus 1 at a premitochondrial stage. J. Virol. 75:5491–5497. 10.1128/JVI.75.12.5491-5497.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ogg PD, McDonell PJ, Ryckman BJ, Knudson CM, Roller RJ. 2004. The HSV-1 Us3 protein kinase is sufficient to block apoptosis induced by overexpression of a variety of Bcl-2 family members. Virology 319:212–224. 10.1016/j.virol.2003.10.019 [DOI] [PubMed] [Google Scholar]
- 38.Jerome KR, Fox R, Chen Z, Sears AE, Lee H, Corey L. 1999. Herpes simplex virus inhibits apoptosis through the action of two genes, Us5 and Us3. J. Virol. 73:8950–8957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Galvan V, Roizman B. 1998. Herpes simplex virus 1 induces and blocks apoptosis at multiple steps during infection and protects cells from exogenous inducers in a cell-type-dependent manner. Proc. Natl. Acad. Sci. U. S. A. 95:3931–3936. 10.1073/pnas.95.7.3931 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Benetti L, Munger J, Roizman B. 2003. The herpes simplex virus 1 US3 protein kinase blocks caspase-dependent double cleavage and activation of the proapoptotic protein BAD. J. Virol. 77:6567–6573. 10.1128/JVI.77.11.6567-6573.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Benetti L, Roizman B. 2004. Herpes simplex virus protein kinase US3 activates and functionally overlaps protein kinase A to block apoptosis. Proc. Natl. Acad. Sci. U. S. A. 101:9411–9416. 10.1073/pnas.0403160101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Poon AP, Gu H, Roizman B. 2006. ICP0 and the US3 protein kinase of herpes simplex virus 1 independently block histone deacetylation to enable gene expression. Proc. Natl. Acad. Sci. U. S. A. 103:9993–9998. 10.1073/pnas.0604142103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Walters MS, Kinchington PR, Banfield BW, Silverstein S. 2010. Hyperphosphorylation of histone deacetylase 2 by alphaherpesvirus US3 kinases. J. Virol. 84:9666–9676. 10.1128/JVI.00981-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mou F, Wills E, Baines JD. 2009. Phosphorylation of the U(L)31 protein of herpes simplex virus 1 by the U(S)3-encoded kinase regulates localization of the nuclear envelopment complex and egress of nucleocapsids. J. Virol. 83:5181–5191. 10.1128/JVI.00090-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Reynolds AE, Wills EG, Roller RJ, Ryckman BJ, Baines JD. 2002. Ultrastructural localization of the herpes simplex virus type 1 UL31, UL34, and US3 proteins suggests specific roles in primary envelopment and egress of nucleocapsids. J. Virol. 76:8939–8952. 10.1128/JVI.76.17.8939-8952.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ryckman BJ, Roller RJ. 2004. Herpes simplex virus type 1 primary envelopment: UL34 protein modification and the US3-UL34 catalytic relationship. J. Virol. 78:399–412. 10.1128/JVI.78.1.399-412.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wisner TW, Wright CC, Kato A, Kawaguchi Y, Mou F, Baines JD, Roller RJ, Johnson DC. 2009. Herpesvirus gB-induced fusion between the virion envelope and outer nuclear membrane during virus egress is regulated by the viral US3 kinase. J. Virol. 83:3115–3126. 10.1128/JVI.01462-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wagenaar F, Pol JM, Peeters B, Gielkens AL, de Wind N, Kimman TG. 1995. The US3-encoded protein kinase from pseudorabies virus affects egress of virions from the nucleus. J. Gen. Virol. 76(Part 7):1851–1859. 10.1099/0022-1317-76-7-1851 [DOI] [PubMed] [Google Scholar]
- 49.Van den Broeke C, Radu M, Deruelle M, Nauwynck H, Hofmann C, Jaffer ZM, Chernoff J, Favoreel HW. 2009. Alphaherpesvirus US3-mediated reorganization of the actin cytoskeleton is mediated by group A p21-activated kinases. Proc. Natl. Acad. Sci. U. S. A. 106:8707–8712. 10.1073/pnas.0900436106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Finnen RL, Roy BB, Zhang H, Banfield BW. 2010. Analysis of filamentous process induction and nuclear localization properties of the HSV-2 serine/threonine kinase Us3. Virology 397:23–33. 10.1016/j.virol.2009.11.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Favoreel HW, Van Minnebruggen G, Adriaensen D, Nauwynck HJ. 2005. Cytoskeletal rearrangements and cell extensions induced by the US3 kinase of an alphaherpesvirus are associated with enhanced spread. Proc. Natl. Acad. Sci. U. S. A. 102:8990–8995. 10.1073/pnas.0409099102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wang S, Wang K, Lin R, Zheng C. 2013. Herpes simplex virus 1 serine/threonine kinase US3 hyperphosphorylates IRF3 and inhibits beta interferon production. J. Virol. 87:12814–12827. 10.1128/JVI.02355-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Piroozmand A, Koyama AH, Shimada Y, Fujita M, Arakawa T, Adachi A. 2004. Role of Us3 gene of herpes simplex virus type 1 for resistance to interferon. Int. J. Mol. Med. 14:641–645. 10.3892/ijmm.14.4.641 [DOI] [PubMed] [Google Scholar]
- 54.Liang L, Roizman B. 2008. Expression of gamma interferon-dependent genes is blocked independently by virion host shutoff RNase and by US3 protein kinase. J. Virol. 82:4688–4696. 10.1128/JVI.02763-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Peri P, Mattila RK, Kantola H, Broberg E, Karttunen HS, Waris M, Vuorinen T, Hukkanen V. 2008. Herpes simplex virus type 1 Us3 gene deletion influences toll-like receptor responses in cultured monocytic cells. Virol. J. 5:140. 10.1186/1743-422X-5-140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wild P, de Oliveira AP, Sonda S, Schraner EM, Ackermann M, Tobler K. 2012. The herpes simplex virus 1 U(S)3 regulates phospholipid synthesis. Virology 432:353–360. 10.1016/j.virol.2012.06.020 [DOI] [PubMed] [Google Scholar]
- 57.Chuluunbaatar U, Roller R, Mohr I. 2012. Suppression of extracellular signal-regulated kinase activity in herpes simplex virus 1-infected cells by the Us3 protein kinase. J. Virol. 86:7771–7776. 10.1128/JVI.00622-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Matsuzaki A, Yamauchi Y, Kato A, Goshima F, Kawaguchi Y, Yoshikawa T, Nishiyama Y. 2005. US3 protein kinase of herpes simplex virus type 2 is required for the stability of the UL46-encoded tegument protein and its association with virus particles. J. Gen. Virol. 86:1979–1985. 10.1099/vir.0.80949-0 [DOI] [PubMed] [Google Scholar]
- 59.Bresnahan WA, Hultman GE, Shenk T. 2000. Replication of wild-type and mutant human cytomegalovirus in life-extended human diploid fibroblasts. J. Virol. 74:10816–10818. 10.1128/JVI.74.22.10816-10818.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Gierasch WW, Zimmerman DL, Ward SL, Vanheyningen TK, Romine JD, Leib DA. 2006. Construction and characterization of bacterial artificial chromosomes containing HSV-1 strains 17 and KOS. J. Virol. Methods 135:197–206. 10.1016/j.jviromet.2006.03.014 [DOI] [PubMed] [Google Scholar]
- 61.Zahariadis G, Wagner MJ, Doepker RC, Maciejko JM, Crider CM, Jerome KR, Smiley JR. 2008. Cell-type-specific tyrosine phosphorylation of the herpes simplex virus tegument protein VP11/12 encoded by gene UL46. J. Virol. 82:6098–6108. 10.1128/JVI.02121-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Court DL, Sawitzke JA, Thomason LC. 2002. Genetic engineering using homologous recombination. Annu. Rev. Genet. 36:361–388. 10.1146/annurev.genet.36.061102.093104 [DOI] [PubMed] [Google Scholar]
- 63.Warming S, Costantino N, Court DL, Jenkins NA, Copeland NG. 2005. Simple and highly efficient BAC recombineering using galK selection. Nucleic Acids Res. 33:e36. 10.1093/nar/gni035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Purves FC, Longnecker RM, Leader DP, Roizman B. 1987. Herpes simplex virus 1 protein kinase is encoded by open reading frame US3 which is not essential for virus growth in cell culture. J. Virol. 61:2896–2901 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Tischer BK, Smith GA, Osterrieder N. 2010. En passant mutagenesis: a two step markerless red recombination system. Methods Mol. Biol. 634:421–430. 10.1007/978-1-60761-652-8_30 [DOI] [PubMed] [Google Scholar]
- 66.Tischer BK, von Einem J, Kaufer B, Osterrieder N. 2006. Two-step red-mediated recombination for versatile high-efficiency markerless DNA manipulation in Escherichia coli. Biotechniques 40:191–197. 10.2144/000112096 [DOI] [PubMed] [Google Scholar]
- 67.Hanks SK, Quinn AM, Hunter T. 1988. The protein kinase family: conserved features and deduced phylogeny of the catalytic domains. Science 241:42–52. 10.1126/science.3291115 [DOI] [PubMed] [Google Scholar]
- 68.Iyer GH, Garrod S, Woods VL, Jr, Taylor SS. 2005. Catalytic independent functions of a protein kinase as revealed by a kinase-dead mutant: study of the Lys72His mutant of cAMP-dependent kinase. J. Mol. Biol. 351:1110–1122. 10.1016/j.jmb.2005.06.011 [DOI] [PubMed] [Google Scholar]
- 69.He Z, He YS, Kim Y, Chu L, Ohmstede C, Biron KK, Coen DM. 1997. The human cytomegalovirus UL97 protein is a protein kinase that autophosphorylates on serines and threonines. J. Virol. 71:405–411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Kawaguchi Y, Kato K, Tanaka M, Kanamori M, Nishiyama Y, Yamanashi Y. 2003. Conserved protein kinases encoded by herpesviruses and cellular protein kinase cdc2 target the same phosphorylation site in eukaryotic elongation factor 1delta. J. Virol. 77:2359–2368. 10.1128/JVI.77.4.2359-2368.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kenyon TK, Lynch J, Hay J, Ruyechan W, Grose C. 2001. Varicella-zoster virus ORF47 protein serine kinase: characterization of a cloned, biologically active phosphotransferase and two viral substrates, ORF62 and ORF63. J. Virol. 75:8854–8858. 10.1128/JVI.75.18.8854-8858.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Lemaster S, Roizman B. 1980. Herpes simplex virus phosphoproteins. II. Characterization of the virion protein kinase and of the polypeptides phosphorylated in the virion. J. Virol. 35:798–811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Stevely WS, Katan M, Stirling V, Smith G, Leader DP. 1985. Protein kinase activities associated with the virions of pseudorabies and herpes simplex virus. J. Gen. Virol. 66(Part 4):661–673 [DOI] [PubMed] [Google Scholar]
- 74.Bell C, Desjardins M, Thibault P, Radtke K. 2013. Proteomics analysis of herpes simplex virus type 1-infected cells reveals dynamic changes of viral protein expression, ubiquitylation, and phosphorylation. J. Proteome Res. 12:1820–1829. 10.1021/pr301157j [DOI] [PubMed] [Google Scholar]
- 75.Parsons SJ, Parsons JT. 2004. Src family kinases, key regulators of signal transduction. Oncogene 23:7906–7909. 10.1038/sj.onc.1208160 [DOI] [PubMed] [Google Scholar]
- 76.Lowell CA. 2004. Src-family kinases: rheostats of immune cell signaling. Mol. Immunol. 41:631–643. 10.1016/j.molimm.2004.04.010 [DOI] [PubMed] [Google Scholar]
- 77.Daikoku T, Yamashita Y, Tsurumi T, Maeno K, Nishiyama Y. 1993. Purification and biochemical characterization of the protein kinase encoded by the US3 gene of herpes simplex virus type 2. Virology 197:685–694. 10.1006/viro.1993.1644 [DOI] [PubMed] [Google Scholar]
- 78.Smith RF, Smith TF. 1989. Identification of new protein kinase-related genes in three herpesviruses, herpes simplex virus, varicella-zoster virus, and Epstein-Barr virus. J. Virol. 63:450–455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Overton HA, McMillan DJ, Klavinskis LS, Hope L, Ritchie AJ, Wong-kai-in P. 1992. Herpes simplex virus type 1 gene UL13 encodes a phosphoprotein that is a component of the virion. Virology 190:184–192. 10.1016/0042-6822(92)91204-8 [DOI] [PubMed] [Google Scholar]
- 80.Coulter LJ, Moss HW, Lang J, McGeoch DJ. 1993. A mutant of herpes simplex virus type 1 in which the UL13 protein kinase gene is disrupted. J. Gen. Virol. 74(Part 3):387–395. 10.1099/0022-1317-74-3-387 [DOI] [PubMed] [Google Scholar]
- 81.Kawaguchi Y, Van Sant C, Roizman B. 1998. Eukaryotic elongation factor 1delta is hyperphosphorylated by the protein kinase encoded by the U(L)13 gene of herpes simplex virus 1. J. Virol. 72:1731–1736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Long MC, Leong V, Schaffer PA, Spencer CA, Rice SA. 1999. ICP22 and the UL13 protein kinase are both required for herpes simplex virus-induced modification of the large subunit of RNA polymerase II. J. Virol. 73:5593–5604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Ng TI, Ogle WO, Roizman B. 1998. UL13 protein kinase of herpes simplex virus 1 complexes with glycoprotein E and mediates the phosphorylation of the viral Fc receptor: glycoproteins E and I. Virology 241:37–48. 10.1006/viro.1997.8963 [DOI] [PubMed] [Google Scholar]
- 84.Ogle WO, Ng TI, Carter KL, Roizman B. 1997. The UL13 protein kinase and the infected cell type are determinants of posttranslational modification of ICP0. Virology 235:406–413. 10.1006/viro.1997.8710 [DOI] [PubMed] [Google Scholar]
- 85.Purves FC, Roizman B. 1992. The UL13 gene of herpes simplex virus 1 encodes the functions for posttranslational processing associated with phosphorylation of the regulatory protein alpha 22. Proc. Natl. Acad. Sci. U. S. A. 89:7310–7314. 10.1073/pnas.89.16.7310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Kato A, Yamamoto M, Ohno T, Tanaka M, Sata T, Nishiyama Y, Kawaguchi Y. 2006. Herpes simplex virus 1-encoded protein kinase UL13 phosphorylates viral Us3 protein kinase and regulates nuclear localization of viral envelopment factors UL34 and UL31. J. Virol. 80:1476–1486. 10.1128/JVI.80.3.1476-1486.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Asai R, Ohno T, Kato A, Kawaguchi Y. 2007. Identification of proteins directly phosphorylated by UL13 protein kinase from herpes simplex virus 1. Microbes Infect. 9:1434–1438. 10.1016/j.micinf.2007.07.008 [DOI] [PubMed] [Google Scholar]
- 88.Morrison EE, Wang YF, Meredith DM. 1998. Phosphorylation of structural components promotes dissociation of the herpes simplex virus type 1 tegument. J. Virol. 72:7108–7114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Lee JH, Vittone V, Diefenbach E, Cunningham AL, Diefenbach RJ. 2008. Identification of structural protein-protein interactions of herpes simplex virus type 1. Virology 378:347–354. 10.1016/j.virol.2008.05.035 [DOI] [PubMed] [Google Scholar]
- 90.Cantley LC, Neel BG. 1999. New insights into tumor suppression: PTEN suppresses tumor formation by restraining the phosphoinositide 3-kinase/AKT pathway. Proc. Natl. Acad. Sci. U. S. A. 96:4240–4245. 10.1073/pnas.96.8.4240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Cully M, You H, Levine AJ, Mak TW. 2006. Beyond PTEN mutations: the PI3K pathway as an integrator of multiple inputs during tumorigenesis. Nat. Rev. Cancer. 6:184–192. 10.1038/nrc1819 [DOI] [PubMed] [Google Scholar]
- 92.Chuluunbaatar U, Mohr I. 2011. A herpesvirus kinase that masquerades as Akt: you don't have to look like Akt, to act like it. Cell Cycle 10:2064–2068. 10.4161/cc.10.13.16242 [DOI] [PMC free article] [PubMed] [Google Scholar]