

Abstract
Background
Plant-microbe interactions feature complex signal interplay between pathogens and their hosts. Phytophthora species comprise a destructive group of fungus-like plant pathogens, collectively affecting a wide range of plants important to agriculture and natural ecosystems. Despite the availability of genome sequences of both hosts and microbes, little is known about the signal interplay between them during infection. In particular, accurate descriptions of coordinate relationships between host and microbe transcriptional programs are lacking.
Results
Here, we explore the molecular interaction between the hemi-biotrophic broad host range pathogen Phytophthora capsici and tomato. Infection assays and use of a composite microarray allowed us to unveil distinct changes in both P. capsici and tomato transcriptomes, associated with biotrophy and the subsequent switch to necrotrophy. These included two distinct transcriptional changes associated with early infection and the biotrophy to necrotrophy transition that may contribute to infection and completion of the P. capsici lifecycle
Conclusions
Our results suggest dynamic but highly regulated transcriptional programming in both host and pathogen that underpin P. capsici disease and hemi-biotrophy. Dynamic expression changes of both effector-coding genes and host factors involved in immunity, suggests modulation of host immune signaling by both host and pathogen. With new unprecedented detail on transcriptional reprogramming, we can now explore the coordinate relationships that drive host-microbe interactions and the basic processes that underpin pathogen lifestyles. Deliberate alteration of lifestyle-associated transcriptional changes may allow prevention or perhaps disruption of hemi-biotrophic disease cycles and limit damage caused by epidemics.
Background
Plant-pathogen interactions exhibit a dynamic interplay between host defense mechanisms and specialized pathogen structures that aim to subvert immunity. Although plants lack an adaptive immune system, they carry pattern recognition receptors (PRRs) that recognize microbe or pathogen-associated molecular patterns (MAMPs or PAMPs) and initiate effective defense responses. This form of innate immunity, termed PAMP-triggered immunity (PTI), ensures an early response to a broad range of potential pathogens, and generates systemic signals that travel to healthy tissues and prime defense signaling networks [1]. To counter plant defenses, pathogens deploy repertoires of secreted molecules (effectors) that, upon delivery into the host apoplast (extracellular effectors) or cell cytoplasm (intracellular effectors), modify cellular targets to suppress PTI and enable parasitic infection and reproduction [2-4]. In addition, pathogens may also secrete classes of effectors that provoke execution of host cellular processes required for disease development. Consequently, both host and microbe tightly control transcriptional programs that drive responses to external signals.
Phytophthora species are a destructive group of filamentous plant pathogens, which have a global distribution and devastating effect on a wide range of plants important to agriculture and natural ecosystems [5]. For example, Phytophthora infestans, the causal agent of late blight in potato and tomato crops, and Phytophthora capsici, an important pathogen of tomato, other solanaceous, and cucurbit plants, cause multi-billion dollar losses in crop production annually [6,7]. The economic impact of this group of pathogens remains the principal driving force behind the need to understand Phytophthora parasitism and epidemics. Phytophthora spp. are hemi-biotrophic pathogens, having a lifestyle that features a biotrophic phase, followed by a switch to necrotrophy [8-10]. This lifestyle is also common to other detrimental filamentous plant pathogens such as fungi that fall into Magnaporthe, Colletotrichum, and Mycosphaerella genera [11]. In the early biotrophic phase, specialized infection structures, termed haustoria, are formed to breach the plant cell walls and interface with the host membrane [12,13]. The initial biotrophic phase is crucial for infection and disease establishment, after which rapid intercellular growth and colonization occurs, ultimately leading to host cell death, sporulation, and initiation of a new infection cycle. Despite the availability of multiple Phytophthora genome sequences [7,14,15], little is known about the signal interplay between organisms that occurs during infection, or which processes in the plant contribute to the Phytophthora hemi-biotrophic lifestyle.
Throughout the infection cycle, Phytophthora secretes effectors into its host with the aim to promote pathogen growth and reproduction [16-18]. The Phytophthora effector repertoire consists of extracellular proteins (apoplastic effectors) that inhibit or counter defense-associated compounds and lytic enzymes, as well as classes of secreted proteins that traverse the host membrane and target intracellular processes (intracellular effectors) [19]. Of the intracellular effectors, the RXLRs, named after their RXLR-dEER amino acid motif, translocate across the haustorial host-pathogen interface, where they are thought to perturb host cellular signaling and suppress immunity [13,20,21]. Besides the RXLRs, Phytophthora genomes encode another large class of intracellular effectors, termed crinklers (CRN), which feature a conserved LFLAK motif that is required for effector translocation. These effectors exclusively target the host nucleus upon delivery [14,22,23].
Owing to the tremendous economic impact Phytophthora-host associations have on crop production, it is crucial to understand how this group of pathogens manipulates their host and promote damage. Current breeding strategies rely on introgression of resistance genes and identifying new R-gene variants [24-26], which are often rapidly overcome by the pathogen [27-29]. Therefore, increasing interest lies in the mechanisms underpinning infection, disease establishment, and epidemics. One strategy for studying these processes is to examine host and pathogen gene-expression patterns during the course of infection. This will enable the identification of essential transcriptional changes that occur in the pathogen during infection, its establishment, and the transition from biotrophy to necrotrophy. Equally important for understanding disease is identification of those host processes and signaling pathways that are perturbed by the pathogen while it progresses through its specific life stages.
In the current study, we explored the association between the broad host range pathogen Phytophthora capsici and tomato. Infection time-course assays reveal a distinct hemi-biotrophic infection cycle, featuring haustoria formation early in, infection, followed by necrotrophy in the late disease stages. We exploited the availability of genome sequences for P. capsici and tomato to measure gene-expression changes in both P. capsici and tomato simultaneously during the course of infection. Microarray analyses, using a custom-designed combined pathogen and host whole-genome array helped define transcriptional changes in Phytophthora that were linked to (disease) development, and identified distinct transcriptional responses in tomato, associated with pathogen lifestyle. These unveil a requirement for Phytophthora to have enhanced protein production and metabolism in biotrophy, catabolism during its transition to necrotrophy, and induction of signaling and developmental processes upon sporulation. P. capsici infection of tomato results in two dramatic changes in the host transcriptome, favoring defense signaling and metabolism early after infection, whereas during the transition from biotrophy to necrotrophy, genes required for (a)biotic stress, signaling, and regulation are activated. We hypothesize that these changes are driven by differentially regulated and stage-specific effector genes identified in our study.
Our results provide unique detail about the coordinated transcriptional reprogramming in both host and pathogen during infection, and lay a foundation for future studies on transcriptional programs that drive parasitic lifestyles. This work opens the door towards comparative transcriptomics studies that should help unravel pathogen infection strategies and exploit host basal defense responses.
Results
P. capsici-tomato interactions feature an early biotrophic and late necrotrophic phase.
We investigated the interaction between P. capsici strain LT1534 and tomato (Solanum lycopersicum 'Moneymaker') in time-course experiments (Figure 1). Inoculations followed by phenotypic analyses across time points suggested that in the early stages of infection (up to 24 hours post-infection (hpi)), P. capsici ingress features a biotrophic phase during which host tissues appear healthy and unaffected, followed by a necrotrophic phase (>24 hpi), marked by tissue collapse (Figure 1A). Multiple inoculation experiments showed distinct phenotypic changes in the later stages of infection, which included host tissue water-soaking, cell death, and tissue collapse (Figure 1A).
Figure 1.
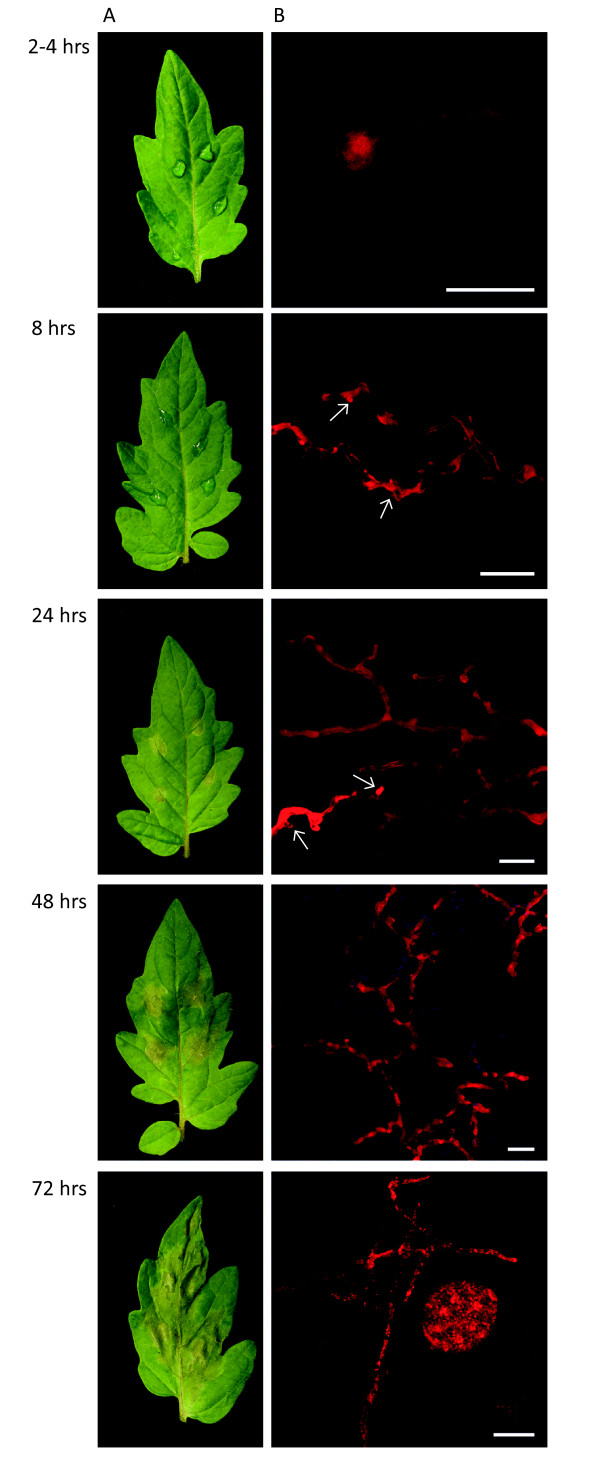
Phytophthora capsici infection of tomato features a hemi-biotrophic lifecycle. (A) Tomato leaves infected with zoospore suspensions of P. capsici at 2-4, 8, 24, 48, and 72 hours post-infection (hpi). (B) Confocal microscopy images of tomato leaves infected with a transgenic P. capsici strain expressing the fluorescent protein TdTomato (red). Infection featured rapid germination of cysts and infection at 8 hpi, formation of biotrophy associated haustoria (arrowheads) visible up to 48 hpi after infection, and rapid growth and sporulation at 48 and 72 hpi respectively. Scale bar = 20 μm.
Based on these observations, we investigated whether P. capsici forms haustoria in planta and inoculated tomato plants. Zoospores derived from a transgenic P. capsici strain were used, which expressed the red fluorescent protein-coding gene tdTomato, and infection was monitored through confocal microscopy (Figure 1B). We observed germinating cysts as early as 1 hpi, and found germinating cysts with hyphae penetrating into the plant cells at 8 hpi (Figure 1B). Infection and subsequent colonization of leaf tissue was evidenced by growth of red fluorescent P. capsici mycelia in inoculated host tissues, and the formation of distinct haustorial structures at the early time points (Figure 1B). Confocal microscopy of leaf tissues in the late infection stages showed significant colonization of tissues with formation of sporangia at 72 hpi (Figure 1B). To assess whether cells were viable, we also inoculated transgenic Nicotiana benthamiana plants expressing ER-eGFP (associated with the endoplasmic reticulum; ER-eGFP) and assessed cell viability at relevant time points at drop inoculation sites (see Additional file 10: Figure S1). These analyses identified haustoria in living cells and low levels of cell death in the early phase (0 to 16 hpi), with increasing numbers of dead cells at 24 and 48 hpi (see Additional file 10: Figure S1). After tissue collapse at inoculation sites, living haustoriated cells were commonly seen at lesion edges, suggesting a dynamic infection cycle in which phase transition is separated spatially. These results are consistent with a hemi-biotrophic infection cycle and further confirm the presence of distinct developmental stages accompanying tomato infection.
A composite host-pathogen microarray approach to simultaneously profile transcriptional changes during the P. capsici-tomato interaction
Considering that PTI features a shift in gene expression and cellular activity towards defense, and that pathogen effectors act to modulate defense gene induction, simultaneous profiling of the gene expression of both pathogen and host should help deduce coordinated relationships between transcriptional programs in host and pathogen. To understand the processes underpinning both P. capsici infection and disease progression in tomato, we designed a custom microarray (Agilent Technologies, Inc., Santa Clara, CA, USA) with 60-mer oligonucleotide probes to all gene models of P. capsici and S. lycopersicum [23], and measured gene-expression changes in both organisms across the same time-course infection experiment. Sites on detached tomato leaves that had been drop-inoculated with P. capsici strain LT1534 were harvested at 0, 8, 16, 24, 48 and 72 hpi (Figure 2). In addition to the infectious stages, samples were taken from tomato leaves that had been mock-inoculated with water (designated 'Non-infected tissue': Ni) and harvested at 0 hpi. Further sporangia and zoospores (taken at 0 hpi), germinating cysts (taken at 16 hpi), and mycelia grown in vitro (harvested at 48 hpi), were collected directly from the inoculum after the various incubation times. This experiment was repeated two times to generate three fully independent biological replicates for analysis.
Figure 2.
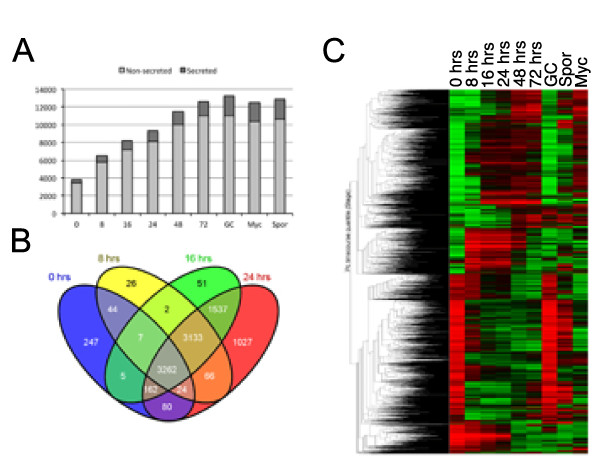
Expression of Phytophthora capsici gene complement during infection and disease progression. (A) Overview of genes that were expressed as detected on the P. capsici-tomato two-genome microarray. The proportion of genes encoding putative secreted proteins (effectors) are indicated by dark grey. (B) Assessment of overlap of genes expressed in infectious stages and (C) overall assessment of differentially expressed P. capsici genes, determined by ANOVA as described in the text. Red and green represent upregulated and downregulated genes respectively. The y-axis shows average linkage of Pearson correlations of gene-expression profiles. The Venn diagram was generated using Venny [49].
P. capsici gene-expression analyses showed that of the 20,530 gene models represented on the array, 15,430 (75%) were expressed in at least one of the 6 infection and 3 in vitro stages sampled (Figure 2A,B). In each of the stages, a significant fraction of expressed genes encoded secreted proteins, ranging from 10.4% to 12.6% during all stages of infection and up to 17.4 % in the in vitro stages (Figure 2A; see Additional file 1: Table S1). Given the dynamic nature of pathogen infection and development, we assessed the expression patterns of P. capsici genes, and found large suites of genes that are specifically expressed in early stages of infection and throughout the infection process (Figure 2B). Differences in expression patterns were not solely due to low levels of detection in the early stages of infection, as distinct sets of genes, expressed only in these stages, were identified (Figure 2B). Subsequent statistical analyses identified 3,691 differentially expressed genes (one-way ANOVA, using Benjamini and Hochberg multiple testing correction, P≤0.005), suggesting dramatic transcriptional changes throughout the infection and growth process of P. capsici (Figure 2C).
P. capsici shows defined shifts in gene expression during specific life stages
To understand the infection process in more detail, we explored the P. capsici gene model set. We used pre-existing information to identify genes that mark specific infection stages in Phytophthora, and assessed their gene-expression profiles across our time-course experiment. Expression of PcHmp1 (P. capsici ortholog of the P. infestans Haustorial membrane protein 1, PiHmp1) [30], PcNpp1 (Nep1-Iike Protein 1) [31,32], and PcCdc14 [33], markers for biotrophy, necrotrophy, and sporulation respectively, showed distinct expression patterns in both the microarray (Figure 3A) and reverse transcription (RT)-PCR (see Additional file 11: Figure S2) data, and are consistent with stage-specific gene expression reported in other Phytophthora species [34]. Our results also agreed with the phenotypic changes and disease progression seen in our infection assays on tomato leaves (Figure 1A,B; see Additional file 10: Figure S1).
Figure 3.
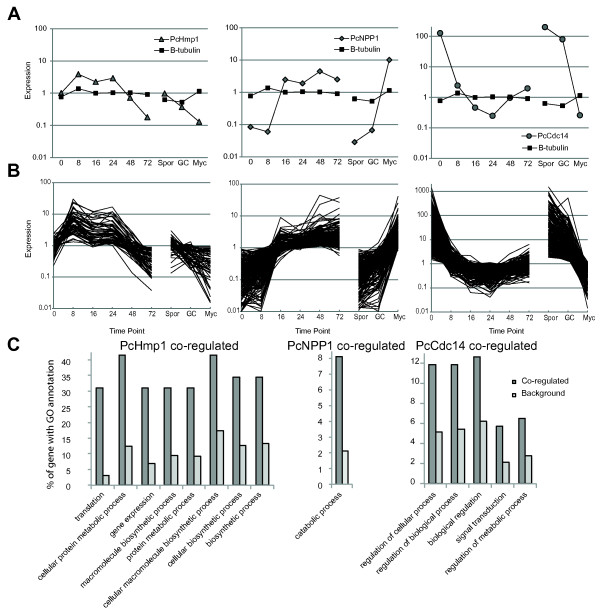
Marker gene-assisted identification of stage-specific processes in P. capsici. (A) Expression of PcHmp1 (left panel), PcNpp1 (middle panel), and PcCdc14 (right panel) as determined by whole-genome microarray analyses and compared with the constitutive control gene β-tubulin. (B) Marker genes were used in cluster analyses to identify genes that were coregulated. The y-axis represents fold change in expression values, determined by calculating fold changes over mean expression values across all treatments. (C) Overview of significantly enriched ontologies present in marker coregulated genes. Dark bar shows the percentage of genes in the coregulated fraction compared with the background fraction (light grey). All ontologies shown were significantly enriched (P<0.05, false discovery rate <0.05).
To refine our view of transcriptional changes associated with infectious stages in P. capsici, we further explored the differentially expressed gene model set and used microarray-derived expression values to identify P. capsici genes that are coregulated with PcHmp1, PcNpp1, and PcCdc14 (Figure 3B). Based on expression patterns, we were able to group 57 genes coregulated with PcHmp1, 209 genes with PcNpp1, and 533 genes coregulated with PcCdc14 (Figure 3B; see Additional file 2, Table S2). We then classified these coregulated genes based on available gene annotations and their corresponding proposed biological processes, and assessed enrichment for specific terms (Figure 3C; see Additional file 3, Table S3). These analyses showed that for the PcHmp1 coregulated genes, annotation terms were significantly enriched (P<0.05) for protein metabolism, (Gene Ontology (GO) number GO:0044267), gene expression (GO:0010467), and biosynthetic processes (GO:0034645) (Table S3). These results suggest activation of cellular machineries required for gene expression and translation. Transcriptional reprogramming would allow an increase in the production and processing of protein factors required for initiation and maintenance of biotrophy. Consistent with an association of Hmp1 with biotrophy, we also found candidate effector genes that are coregulated with Hmp1 (see Additional file 2, Table S2), suggesting effector-mediated dampening of host immune responses in biotrophy.
We consistently found a biotrophic phase in the first 24 hours after infection, followed by host tissue collapse and necrosis in our inoculation experiments, suggesting a distinct transition to necrotrophy. We thus determined expression of PcNpp1, a marker for this transition, and identified 209 genes that are coregulated in P. capsici (Table S2). Annotation-term enrichment analyses of this gene set showed specific enrichment for catabolic processes (GO:0009056) (Table S3). Within this gene set, a large number of peptidases and proteasomal subunits were present, suggesting active involvement of proteosomal degradation of pathogen proteins during the transition from biotrophy to necrotrophy (Table S2). Although the mechanisms of this proteasomal machinery and its targets need to be characterized, our results suggest dramatic shifts in protein modification and degradation processes, which may represent a committed step in disease development.
In the late infection stages, Phytophthora spp. form sporangia that emerge from necrotic tissues, a process that in P. infestans features upregulation of Cdc14. We identified Cdc14 coexpressed genes that were again enriched for signal transduction (GO:0007165) and metabolic processes (GO:0019222) (Table S3), which could be required for extensive cellular reprogramming underpinning spore formation. Altogether, these results are consistent with the view that Phytophthora infection features stage-specific transcriptional programs [34].
P. capsici infection features dynamic transcriptional regulation of effector-coding genes
To learn about expression of known effector genes in P. capsici, we extracted expression profiles for RXLR coding genes identified in the P. capsici genome [7] (see Additional file 4, Table S4). We detected expression of 346 RXLR-encoding genes (73%) in all tested stages and treatments, of which 73 were differentially expressed (Figure 4A; see Additional file 4, Table S4) during infection. We grouped the RXLR genes based on differential expression patterns, and defined four classes using cluster analyses (Figure 4B-E). These analyses identified 26 genes upregulated during biotrophy (8 to 24 hpi), and 13 RXLRs that were expressed in the early infection stages (0 to 16 hpi) but showed lower expression at only one biotrophic time point (24 hpi) and during necrotrophy. We also identified 9 RXLR genes that were expressed only in the sporulation stages and 13 genes that were specifically expressed in the late infection stages. In addition, 273 RXLR protein-encoding genes were expressed without any significant changes in transcript levels (see Additional file 4, Table S4). This shows that regulation of the expression of P. capsici genes takes place both before and during infection.
Figure 4.
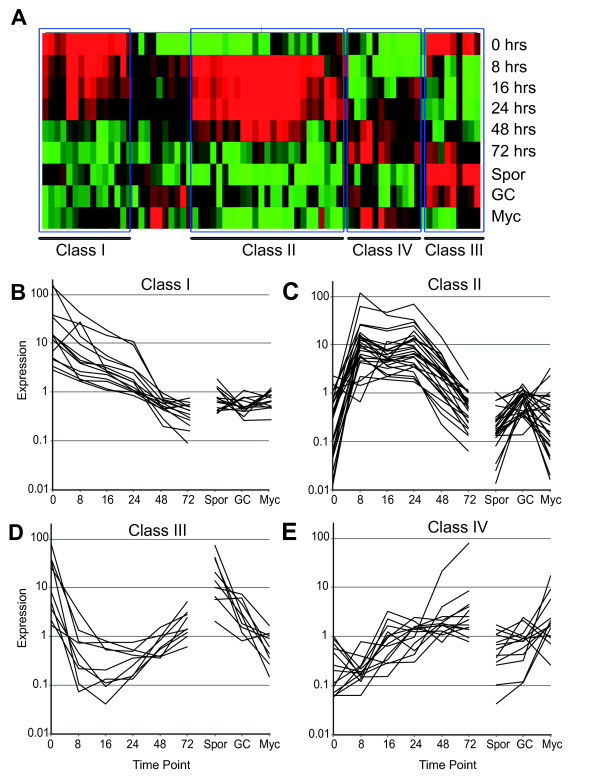
Identification of classes of differentially expressed RXLR genes in Phytophthora capsici. (A) Cluster analyses of putative RXLR genes that were found to be differentially expressed across infectious and developmental stages identified four different groups of genes. (B-E) Overview of expression patterns corresponding to the groups shown in (A). Values on the y-axis represents fold change over mean expression as determined across all treatments.
These results suggest an active involvement of pathogen effector proteins in the initiation and progression of disease, facilitated by modification of host cellular processes.
Host transcriptional changes associated with P. capsici infection
To learn more about P. capsici-mediated changes in host gene expression, we simultaneously measured host gene expression during infection with P. capsici. Measurements of transcript levels for 34,727 gene models (ITAG, version 2.3 [35]) revealed detectable expression of 24,390 genes at a minimum of one time point, representing 65% of the predicted tomato transcriptome. We aimed to identify genes that were differentially expressed in the time-course experiment, and identified 12,883 genes for which significant changes in gene expression were measured (one-way ANOVA, using Benjamini and Hochberg multiple testing correction, P≤0.005). Given that a significant change may occur between two unrelated treatments or time points (for example 8 versus 72 hpi), we also used pairwise comparisons (Student's t-test) to identify genes that are differentially regulated between adjacent time points (Figure 5). These analyses identified a set of 7,314 non-redundant tomato genes, suggesting dynamic transcriptional changes in tomato gene expression over the course of infection (see Additional file 5, Table S5). We determined the number of differentially expressed genes per comparison, and noted large differences in the numbers of genes that are either upregulated or downregulated between time points (Figure 5A). Our analyses suggest a major shift in gene expression (3,720 genes) between the 0 and 8 hour time points (Figure 5A; see Additional file 6, Table S6) suggesting drastic transcriptional reprogramming associated with P. capsici ingress and disease establishment. The water-inoculated Ni tissue was used as a control sample, and showed no significant upregulation of genes. Comparisons between the later infection stages revealed a further major shift in gene expression between the 24 and 48 hour time points (Figure 5A; see Additional file 6, Table S6), coinciding with the transition from biotrophy to necrotrophy observed during infection (Figure 1, Figure 3).
Figure 5.
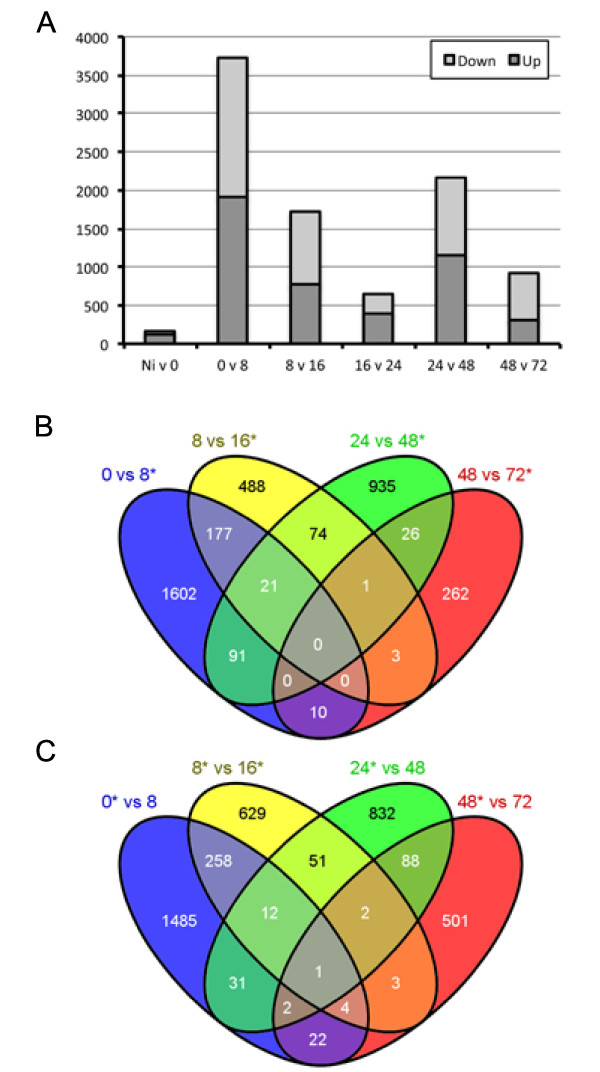
Phytophthora capsici infection of tomato results in two distinct transcriptional responses. (A) Overview of the number of significantly upregulated (dark grey) or downregulated (light grey) between adjacent timepoints. Differences in the number of differentially expressed genes can be seen between specific early (0 versus 8 hpi) and late (24 versus 48 hpi) time-point comparisons. The non-infected (Ni) tissue was a water-inoculated control sample. Comparisons between gene lists generated in pairwise comparisons revealed limited overlap in both (B) upregulated and (C) downregulated gene sets. Diagrams were generated using Venny [49].
Given the dramatic changes in gene expression, we determined the level of overlap of differentially expressed genes between sampled time points (Figure 5B,C). These analyses revealed only a limited number of genes that were upregulated or downregulated at multiple time points, and large suites of genes that were uniquely regulated between 0 versus 8 (2,087), 8 versus 16 (1,117), and 24 versus 48 (1,757) hpi (Figure 5B,C). Crucially, little overlap was found between differentially expressed gene sets from the 0 versus 8 hpi and 24 versus 48 hpi comparisons (Figure 5B,C). These results suggest two major but distinct transcriptome changes in the host occurring at initial infection (0 to 8 hpi) and the transition from biotrophy to necrotrophy (24 to 48 hpi).
P. capsici infection leads to two distinct transcriptional responses in tomato
To identify the biological processes affected by those two distinct responses, we assessed relative enrichment of annotation categories from genes that were present in both our ANOVA and pairwise comparisons sets. As expected, we found no enrichment of processes in the non-infected versus 0 hpi time points, partly owing to a relatively small number (n = 171) of genes that were differentially expressed between these treatments. However, further assessment of sets emerging from other comparisons revealed significant enrichment for specific processes at the 0 versus 8 hpi and 24 versus 48 hpi time points (Figure 6; see Additional file 7, Table S7). Processes associated with (primary) metabolism (GO:0008152, GO:0044238) were significantly enriched in the fraction upregulated at 0 hpi, suggesting a drop in core metabolic genes after infection (Figure 6; see Additional file 7, Table S7). Both catabolic processes (GO:0009056) and specific metabolic processes were enriched in the fraction upregulated at 8 hpi, suggesting major metabolic reprogramming in early infection (Figure 6,; see Additional file 7, Table S7).
Figure 6.
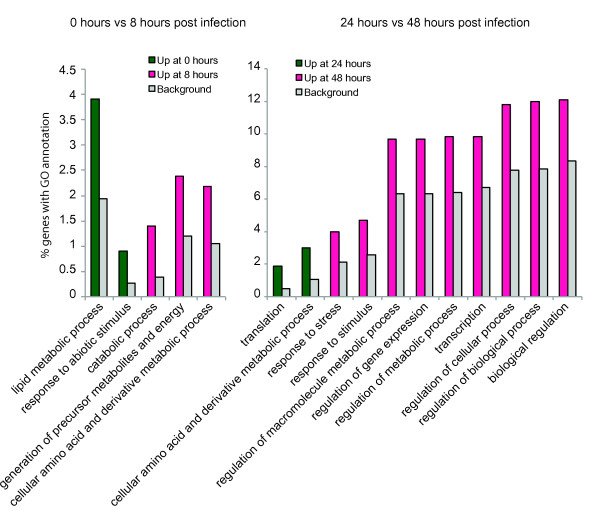
Gene ontology (GO) enrichment analyses of tomato genes identified in the early (0 versus 8) and late (24 versus 48) transcriptional response. Percentage of genes with significantly enriched GO terms that were specifically expressed in either of the time points (magenta/green) in our pairwise comparisons, compared with the background (grey). The y-axis shows the percentage of genes falling within each given GO annotation class.
The switch from 24 to 48 hpi showed drastic re-regulation of metabolic and biosynthetic processes. Interestingly, the genes specifically upregulated at 48 hpi showed enrichment for a relatively large number of ontologies, including response to stimulus (GO:0006950) and response to stress (GO:0050896), and a number of gene regulation-related ontologies (Figure 6; see Additional file 7, Table S7). These results suggest an active response of the host that accompanies the initiation of necrotrophy by P. capsici, suggesting a pathogen-derived cue that causes host cell death. If true, perturbation of this process may limit initiation of pathogen necrotrophy, which in turn could lead to reduced pathogen growth and sporulation.
P. capsici infection features differential expression of candidate PAMP perception and signaling genes in tomato
We noted a vast transcriptional shift in tomato between the 0 and 8 hpi time points, and hypothesized that these changes are due either to an initial PAMP or effector induced-response upon pathogen ingress. We also hypothesized that upon infection, the PTI response is dampened by effectors that are expressed and delivered during infection and biotrophy (Figure 4). If true, immune signaling gene candidates that help determine interaction outcomes could be identified. We thus investigated transcriptional changes in gene classes involved in pathogen perception and signaling.
We exploited available annotations for tomato gene models in MAPMAN, and identified 202 signaling genes in our differentially regulated dataset (Figure 7; see Additional file 8, Table S8). Subsequent cluster analyses and classification revealed a set of 84 genes that are induced between 0 and 8 hpi but that decrease in expression in the later stages (group A) (see Additional file 8, Table S8) and 61 genes that appear to be specifically suppressed in biotrophy (group B) (see Additional file 8, Table S8). Another group of 57 genes (group C) (see Additional file 8, Table S8) were found to be transcriptionally activated throughout the time course after pathogen ingress, possibly reflecting activation of signaling networks that allow pathogen growth. Notably, in our differentially regulated set, we identified large suites of receptor-like kinases (RLKs) that are downregulated in biotrophy (group B), suggesting that they may be targeted by P. capsici effectors and their downregulation may enhance virulence. We noted that a large proportion of differentially regulated receptor-like genes are annotated as receptors involved in nodulation, suggesting overlap between symbiotic and pathogenic associations with host plants. Importantly, in our set of RLKs, we identified a homolog of AtPepR1 [36] that is suppressed during disease progression (group B), suggesting that immune suppression is important in biotrophy. Approaches that aim to maintain or enhance their expression during biotrophy may limit disease progression.
Figure 7.
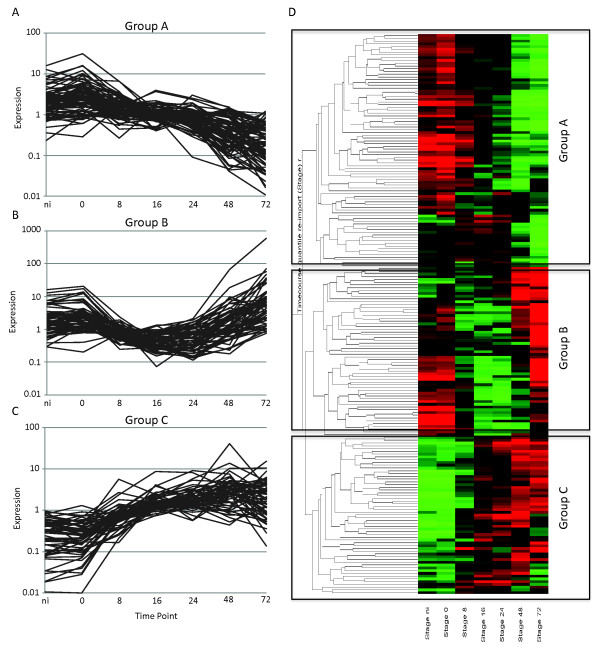
Differentially expressed immune signaling candidate genes identified in microarray analyses. Overview of differentially expressed immune signaling candidate genes identified in pairwise comparisons between time points (Student's t-test) and ANOVA (P = 0.005) analyses. (A-C) Expression profiles are presented for class A, B, and C genes, identified by (D) cluster analyses in Genespring. Red and green represent upregulated and downregulated genes respectively. The y-axis shows average linkage of Pearson correlations of gene-expression profiles. The non-infected (Ni) tissue was a water-inoculated control sample.
Differential regulation of host transcription factors underpins transcriptional responses to P. capsici infection in tomato
The observed dramatic changes in gene expression, both early in infection and during the transition from biotrophy to necrotrophy, prompted us to extract expression profiles for known and differentially regulated transcription factors (Figure 8). Clustering and subsequent grouping of these transcription factors based on expression patterns revealed the presence of distinct transcriptional profiles, consistent with wholesale transcriptional changes in tomato during infection. A large group of transcription factors was found to be induced upon infection (class A) (Figure 8; see Additional file 8, Table S8), whereas others were either repressed during biotrophy (class B) (Figure 8; see Additional file 8, Table S8) or were expressed throughout infection but specifically downregulated during necrotrophy (class C) (Figure 8; see Additional file 8, Table S8). We assessed transcription-factor family membership for each of these expression classes, and found that class A contained a relatively large fraction of the WRKY-type transcription-factor families (see Additional file 8, Table S8).
Figure 8.
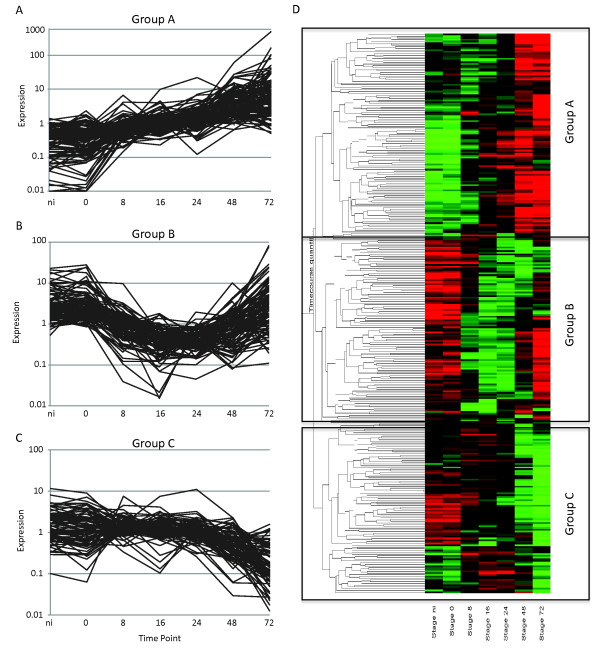
Phytophthora capsici infection leads to dynamic changes in host transcription-factor genes. Overview of differentially expressed candidate transcription-factor genes, identified in pairwise comparisons between time points (Student's t-test) and ANOVA (P = 0.005) analyses. (A-C) Expression profiles are presented for class A, B, and C genes, identified by (D) cluster analyses in Genespring, showing distinct expression changes during infection. Red and green represent upregulated and downregulated genes respectively. The y-axis shows average linkage of Pearson correlations of gene-expression profiles. The non-infected (Ni) tissue was a water-inoculated control sample.
These results are consistent with the activation of genes involved in (biotic) stress responses, and suggest execution of specific transcriptional programs possibly leading to tissue necrosis. Our results also suggest involvement of the phytohormone ethylene and its responsive transcription factors in disease development, as a sizeable fraction was found to fall into classes A and B (Figure 8; see Additional file 8, Table S8). These results suggest repression of specific transcriptional regulators by P. capsici during early and biotrophic infection stages. Taken together, our results lead us to suggest that further investigation into and alteration of specific transcriptional changes leading to necrosis in hosts may prevent or limit progression of P. capsici infection beyond biotrophy and limit sporulation.
Discussion
A composite whole-genome microarray approach to study plant-microbe interactions in new detail
In this paper, we report on a genome-wide analysis of transcriptional changes that take place in tomato and its pathogen P. capsici. By utilizing their full genomes, our work provides the first detailed simultaneous overview of gene-expression changes during the course of infection in both a pathogen and its plant host. This approach allows an unprecedented view in great detail of the processes that underpin infection, disease progression, and lifestyle transitions. Given the immense damage Phytophthora species continue to cause in important crops, these analyses will thus provide new means and exciting opportunities to investigate complex yet important plant-microbe interactions, in which extensive signal interplay is known to occur. Our approach sets the stage for standardized experiments that can compare the effect of pathogen infection strategies on a given host, or the importance of host factors on pathogen transcriptional programs.
P. capsici infection features a hemi-biotrophic lifecycle
Using confocal microscopy and microarray analyses, we found evidence of a distinct biotrophic phase, followed by transition to necrotrophy after 24 hpi and sporulation at 72 hpi on susceptible tomato (Figure 1). Biotrophy is marked by the formation of distinct haustorial structures that invaginate living cells, an important feature we were able to show on N. benthamiana plants expressing ER-eGFP (see Additional file 10: Figure S1). These results are similar to observations made in other Phytophthora species [30,37,38], although we note that P. capsici has a relatively short infection cycle compared with related organisms such as P. infestans [27]. We took advantage of the availability of genome sequences for both P. capsici and tomato, and used this information to design a custom two-genome array and measure gene expression in both organisms in a detailed time-course experiment. Using this approach, we were able to measure and demonstrate the expression of 20,530 P. capsici and 24,390 tomato genes in a replicated time-course experiment. We determined transcriptional programs associated with distinct stages of pathogen infection.
The P. capsici lifecycle is marked by activation of stage-specific processes
Using the existing literature, we identified and selected three Phytophthora marker genes that could provide information on disease progression and development after infection. Assessment of expression for PcHmp1, PcNpp1, and PcCdc14 during P. capsici infection confirmed the presence of a hemi-biotrophic lifecyle that features biotrophy in the first 24 hpi, a switch to necrotrophy between 24 and 48 hpi, and sporulation at 72 hpi (Figure 2). Identification of coregulated genes followed by GO term enrichment analyses revealed that genes associated with expression and translation of genes and with protein metabolism were over-represented in biotrophy (Figure 3). These analyses suggest that the de novo expression, production and modification of proteins are crucial requirements for initiation and maintenance of biotrophy, and that P. capsici does not take amino acids directly from the host. This is consistent with previous work showing that haustoriating P. infestans cysts show relatively high expression of amino acid biosynthesis genes [39].
Given that the plant-haustorial membrane interface is a crucial site where effector proteins are secreted and delivered into host tissues and cells, it is plausible that Hmp1-coregulated genes are required for haustorial development and enhancement of effector protein production and delivery. The identification of stage-specific genes, encoding secreted proteins of unknown function or cellular destination, may help identify novel effector (classes) and help determine their roles in virulence.
The NEP1-like protein superfamily forms an important class of necrosis-inducing peptides with proposed roles in pathogen virulence. In this study, we found that, together with PcNPP1, a significant group of genes is induced during the transition from biotrophy to necrotrophy, suggesting a committed transcriptional shift between stages. Enrichment analyses revealed a significant gene complement associated with catabolism and degradation, suggesting that transcriptional shifts may result in cellular reprogramming of Phytophthora hyphae or accommodate the breakdown of compounds released during host cell death. These results illustrate a dynamic transcriptional program used by P. capsici to drive differentiation and adaptation.
Successful Phytophthora infection must lead to the formation of sporangia, an essential developmental process required for propagation of the Phytophthora disease cycle. Given the devastation caused by often explosive Phytophthora epidemics, there is considerable interest in the mechanisms governing sporulation and dissemination. In this study, we assessed genes coregulated with the sporulation marker Cdc14 and identified genes required for signaling, regulation, and expression. These results are consistent with the idea of extensive signaling cascades that drive the formation and differentiation of sporangia from a hyphal stage. Although the exact cascades driving sporulation still require elucidation, our work, together with gene-expression studies on other Phytophthora spp., should allow identification of common genes associated with spore formation, which in turn could inform strategies that stop pathogen dissemination and limit epidemics on crops. Interestingly, a relatively small percentage (22%) of differentially expressed genes fall into one of the coregulated gene sets. Although this could be due to high levels of stringency during our analyses, it could also point to the existence of other coregulated gene classes, driving as yet unknown processes required for disease development. More detailed cluster analyses and investigation of candidate gene function may thus give rise to additional sets of marker genes, suited to study Phytophthora infection.
P. capsici effector gene expression is regulated by developmental and plant signaling cues
We assessed gene-expression patterns for the RXLR class of effectors, and detected expression for a relatively high proportion of RXLR-coding genes (73%). These results could be due to the high level of sensitivity provided by the Agilent platform (as evidenced by the large number of P. capsici genes detected at 0 hpi) or the number of time points and stages assayed in our microarray experiments (9), or might reflect the biology of a pathogen that has a broad host range. We found that, based on expression changes, RXLR genes can be grouped into four distinct classes. Class I and III RXLRs were highly expressed in the early phase of infection, and showed either low (class I) or high (class III) expression levels in germinating cysts. These results suggest the presence of both a developmental program and specific plant signals that drive RXLR gene induction. Given their expression early in infection, these genes are likely to play roles in prevention or suppression of initial immune responses. Besides genes expressed in the early infection stages, we also found RXLR-coding genes that were upregulated in biotrophy (classes II and IV), most of which were downregulated in necrotrophy (class II), and some that remained highly expressed in the late stages (class III). Given the observation that biotrophy features suppression of defense responses, we suggest that secretion and delivery of effectors are required for the maintenance of biotrophy. Our results also suggest continuous reprogramming of host cells in favor of pathogen growth. Effector genes expressed in the late stages could stimulate cell death in the necrotrophic phase or modulate host metabolism.
P. capsici infection and disease progression induces two distinct responses in tomato
By characterizing host gene expression during P. capsici infection, we identified processes associated with pathogen infection and lifestyle. Pairwise comparisons between time points identified two distinct transcriptional changes in tomato, coinciding with initial infection (0 versus 8 hpi) and the transition from biotrophy to necrotrophy (24 versus 48 hpi). Characterization of the early response revealed downregulation of genes required for primary metabolism, whereas genes falling into secondary metabolism categories were induced as part of early responses to infection. These results are in line with previously reported observations, and could reflect production of antimicrobial compounds upon initial Phytophthora ingress.
Phytophthora infection results in the induction and suppression of PAMP-like responses
Whereas some Phytophthora-host interactions feature suppression of initial host defense responses [40], we found evidence suggesting defense responses occurring early in infection. These included differential regulation of genes encoding RLKs, including the PEPR1 receptor and classes with similarity to Nod factor receptors. These results suggest activation of PAMP or effector-triggered immune responses that may overlap with pathways that are regulated by Nod receptor-like genes in plants. These results may indicate co-opting of signaling pathways normally activated in symbiosis, and would give weight to recent observations made in Lotus japonicus [41]. Crucially, in a set of differentially expressed RLK-coding genes, we identified a subset of candidate receptors whose expression was specifically repressed in biotrophy. These results, together with the identification of effectors induced in the early stages of infection, lead us to suggest that, consistent with current models describing plant-microbe interactions, P. capsici secretes and delivers effectors into host tissues to limit PAMP perception, inhibit immune signaling, and promote virulence. With both effector and immune signaling genes now characterized in P. capsici and tomato respectively, it is now possible to investigate the mechanisms driving P. capsici virulence and host immune signaling. Over-expression of host RLKs normally downregulated in biotrophy may lead to enhanced PTI responses that limit pathogen growth and disease development.
Conclusions
Our results suggest dynamic but highly regulated transcriptional programming in both host and pathogen that underpin P. capsici disease and hemi-biotrophy. We found expression changes in both effector-coding genes and host factors involved in immunity, suggesting distinct roles for effectors towards susceptibility by modulating host processes. With new unprecedented detail on transcriptional reprogramming during infection in both host and pathogen, the coordinate relationships that drive host-microbe interactions and the basic processes that drive hemi-biotrophy can now be explored. Importantly, and with the availability of genome sequences for both hosts and distinct classes of pathogens that share parasitic lifestyles, it is possible to identify and study the processes that underpin pathogen lifestyles. Given that major transcriptional switches can be observed in both Phytophthora and tomato during infection, deliberate alteration of lifestyle-associated transcriptional changes may allow prevention or perhaps disruption of hemi-biotrophic disease cycles, and limit damage caused by epidemics.
Methods
Plant material
S. lycopersicum 'Moneymaker' plants were grown in controlled growth chambers at 22°C, with a photoperiod of 16 hours, supplemented by artificial light. The third leaf from the top of every plant was detached and placed upside-down in humid transparent plastic trays in a controlled incubator with the same settings as in the growth chamber. Leaf discs centered on mock-inoculated tissue and infected lesion tissue were harvested using a cork borer (diameter 7 mm), and frozen in liquid nitrogen before RNA extraction.
P. capsici inoculation and in vitro samples
P. capsici wild-type strain LT1534 was grown in petri dishes on V8 agar medium in a dark climate chamber at 25°C for 4 days and under standard light at 22°C for 3 days. To induce zoospore release, plates were flooded with ice-cold distilled water, and spores were harvested from sporulating mycelia by dislodging the sporangia with a sterile glass spreader. Sporangial suspensions were collected and incubated at room temperature under bright light conditions. Release of zoospores was monitored, and their numbers counted in a hemocytometer under a microscope, and adjusted to 1 × 105/ml. The detached leaves were inoculated with four 20 µl droplets of the zoospore solution. In addition to samples taken during the infectious stages, three in vitro samples were taken: sporangia/zoospores (Spor), germinating cysts (GC), and mycelia (Myc) grown in vitro. Spor (taken at 0 hpi) and GC (taken at 16 hpi) were sampled from the same inoculum/sporangial suspension, differing only in harvesting times. They were collected from 10 ml of sporangial suspension after an incubation time of 1 h (Spor) and 16 h (GC) at 22 °C. The mycelia were grown in 1 ml pea broth, infected with 20 µl of inoculum at 22°C, and harvested 48 hpi by collecting the mycelial mat into 10 ml tubes. All samples were placed in the controlled incubator with the same settings and conditions as the leaf samples, and harvested after centrifugation for 2 minutes at 1,200 × g. After the supernatant was removed, the pellets were collected and frozen in liquid nitrogen.
RNA extractions and cDNA synthesis
RNA was isolated from frozen leaf tissue (RNeasy Plant Mini Kit; Qiagen Inc., Valencia, CA, USA) and treated afterwards with DNAse (Ambion, Foster City, CA, USA) to remove genomic DNA contamination, in accordance with the instructions of the manufacturers. To test for genomic DNA contamination, PCR using primers specific for PcTubulin (Table 1), was performed on the extracted RNA. cDNA was synthesized using 500 ng of total RNA, using a commercial kit and primer (SuperScript™ II cDNA synthesis kit and Oligo dT primer; Invitrogen Corp., Carlsbad, CA, USA) following the manufacturer's instructions.
Table 1.
Primers used in study.
| Primer | Direction | Sequence |
|---|---|---|
| PcTubulin | Forward | GACTCGGTGCTTGATGTTGTC |
| Reverse | CCATCTCATCCATACCCTCGCCAG | |
| PcHmp1-F | Forward | CATGATGGCAGTCATGGTCGGTGAAG |
| PcHmp1-R | Reverse | TTAGCTAACATTGAGGCGGGCATGCAG |
| PcNPP1-F: | Forward | CAGCTCCACATCACCAACGGct |
| PcNPP1-R | Reverse | CTCTTCCCGTTCAAATAGTTC |
| PcCDC14-F | Forward | GGAAGCGATTGAGTTCTTGC |
| PcCDC14-R | Reverse | TTCTCCACACGCTCAAAGTG |
Microarray design and analysis
A custom 60-mer oligonucleotide microarray was designed from predicted transcripts of the P. capsici (LT1534 v11.0 [7]), and S. lycopersicum (ITAG 2.3, [35]) genomes using eArray software (Agilent Technologies, Inc., Santa Clara, CA, USA). The P. capsici predicted transcriptome (Phyca11_No.) was supplemented with separately predicted CRN effectors (Scaffold_No.) as described by Stam et al. [23], and RXLR effectors (PhycaSCAFFOLD_No.) as described below. The design and sequences are available at ArrayExpress (accession A-MEXP-2253), and represent 20,530 transcripts for P. capsici and 34,510 transcripts for S. lycopersicum. RNA labeling and microarray hybridization procedures were performed (Genome Technology, The James Hutton Institute, Dundee, UK) as described previously. In short, fluorescent one-color labeling of the RNA and hybridization was performed as recommended (Agilent One-Color Microarray-Based Gene Expression Analysis (Low Input Quick Amp Labeling) version 6.5; Agilent Technologies, Inc., Santa Clara, CA, USA) using 8 × 60 k format slides.
The microarray experimental design, along with raw datasets, is available at ArrayExpress (Accession A-MEXP-2253). The extracted dataset was separated for each array into P. capsici and S. lycopersicum data to allow independent processing of each dataset. Datasets were each independently quality filtered using flag values (present or marginal in two-thirds of replicates) and then quantile-normalized with Genomics Suite software (Partek Inc,. St Louis, MO, USA), before being loaded into Genespring (version 7.3; Agilent Technologies) software for analysis. Statistical tests were performed using one-way ANOVA (Benjamini and Hochberg multiple testing correction, P≤0.005) to identify significantly changed genes across the in planta time course. For grouping genes that are coregulated with markers of Phytophthora infection stages, a minimum Pearson correlation of at least 85% was used to define clusters. Candidate secreted proteins were identified by using SignalP (version 3) analyses on the predicted P. capsici proteome [42], applying a hidden Markov model (HMM) cut-off score of less than 0.5. Predicted membrane proteins were identified using TMHMM [43], and removed from the secreted protein set as described previously [23]. This set was then augmented with predicted RXLRs (this study) and previously described CRN gene models [23].
Marker gene sequences
For all marker genes, the original P. infestans sequences were retrieved from NCBI using the published accession numbers (PiHmp1: EU680858.1; PiNPP1: AF356840.1; PiCDC14: AY204881.1). The sequences were then used in a tBLASTn and BLASTp [44] search against the P. capsici genome version 11 [7] to obtain the corresponding P. capsici homologous sequences, for which primer pairs (Table 1) were designed. Reverse transcription PCR was performed in 25-µl reaction volumes with 1 µl of cDNA (1:5 dilution) as template. Thermocycling conditions of the PCR were: 94°C for 2 minutes, followed by 30 cycles at 94°C for 30 seconds, 57 C for 30 seconds and 72°C for 2.5 minutes. Extension was finalized at 72°C for 10 minutes to allow trimming of incomplete polymerizations. Amplicons from cDNA were Sanger-sequenced, and derived sequences were aligned to P. infestans reference sequences using the program ClustalW Multiple Sequence Alignment [45] to investigate the levels of sequence similarity. P. capsici marker genes were highly similar to those identified in P. infestans, with the similarity of the protein sequences being 77% for HMP1 and NPP1, and 88% for CDC14. [We refer here to proteins]
Identification of PcRXLR complement
Analysis of previously published P. capsici RXLRs [7] indicated that the present database was incomplete. We therefore implemented a new identification strategy, in which RXLRs were sought using previously published methods [13,46,47]. All output was collated and compared with previously predicted P. capsici RXLR component using BLASTN. Redundancies were removed, and in cases of difference in predicted open reading frame length, sequences were compared with known PiRXLR sequences and manually curated. This yielded a set of 516 RXLR candidates, of which 471 were represented on the array (see Additional file 9, Table S9).
Confocal imaging
Zoospores (5 × 105/ml, generated as described above) of transformed P. capsici LT1534:tdTomato were inoculated in 20 µl droplets onto leaves of S. lycopersicum 'Moneymaker' or N. benthamiana (Line 16c) plants. Plants were incubated in a small climate chamber to maintain humidity and kept at 20°C for a maximum of 72 hours to allow P. capsici to infect leaves, form haustoria, and colonize host tissues. Imaging was conducted on a confocal microscope (LSM 710; Zeiss, Jena, Germany) using a water dipping lens (W Plan-Apochromat 40x/1.0 DIC M27; Zeiss) and the following settings: tdTomato (561 nm excitation and 573 to 612 nm emission) and chlorophyll (488 nm excitation and 650 to 700 nm emission). Haustoria are indicated with white arrows. The scale bars shown are 20 μm.
GO enrichment analysis
To investigate enrichment of specific gene ontologies in either P. capsici marker coregulated genes or S. lycopesicum genes in our pairwise analysis, we used a singular enrichment analysis (SEA) strategy. All genes with no GO annotations were filtered from the set, and compared with a customized background set containing all genes on the array with known ontologies for P. capsici or tomato, respectively. SEA was done using AgriGO. Significance was tested using Fisher's exact test, results were reported for P<0.05 after correction for false discovery rate [48]. P. capsici results were reported using GO Slim annotations, and tomato results with GO plant Slim.
Authors' contributions
JJ carried out time-course experiments and microarray sample preparation, and JJ, PH, RS, and EH designed the custom array. The technical part of the microarray was carried out by JM and JJ. Quantile normalization was performed by RZ and PH. P. capsici analysis was carried out by JJ, PH and RS and tomato analysis by RS, EH, and PH. Confocal microscopy was performed by AH. EH, JJ, RS, and AH wrote the manuscript. All authors read and approved this manuscript.
Supplementary Material
Table S1. Phytophthora capsici genes encoding putative secreted proteins found expressed in microarray experiments. Lists of P. capsici genes encoding candidate secreted proteins are given. These represents gene for which expression was detected in the infectious and in vitro stages.
Table S2. PcHmp1, PcNPP1, and PcCdc14 coregulated genes Overview of genes found to be coregulated with marker genes in Phytophthora capsici. Gene identifier, probe ID number, and normalized expression value (fold change over mean expression) are given for each gene and at each time point. Gene Ontology (GO) annotations are also given for each gene where available.
Table S3. Significantly enriched ontologies coregulated with marker genes, Overview of gene ontologies that are significantly enriched in the fractions that are specifically coregulated with P. capsici marker genes as shown in Figure 3C. Gene Ontology (GO) terms, P-values, false discovery rates (FDRs), and query and reference sample sizes are given.
Table S4. Phytophthora capsici candidate RXLR genes expressed in microarray experiments. Overview of RXLR effector genes found to be upregulated during specific lifecycle stages in P. capsici. Gene identifier, probe ID number, and normalized expression value (fold change over mean expression) are given for each gene and at each time point. Genes are listed per class as shown in Figure 4.
Table S5. Annotation and expression of tomato genes differentially expressed in pairwise comparisons. Overview of genes found to be differentially expressed between timepoints during Phytophthora capsici infection. Gene identifiers, normalized expression values, and Gene Ontology (GO) annotations are given.
Table S6. Tomato genes differentially expressed in each pairwise comparison. Overview of genes found to be differentially expressed during Phytophthora capsici infection in pairwise comparisons between two timepoints. Gene identifiers and normalized expression values are given.
Table S7. Genes corresponding to enriched gene ontologies (GOs) in pairwise comparisons. Overview of GOs that were significantly enriched in the fractions that were specifically upregulated or downregulated between two time points as shown in Figure 6. GOs, P-values, false discovery rates (FDRs), and query and reference sample sizes are given.
Table S8. Tomato genes with possible roles in pathogen-associated molecular pattern (PAMP) perception and signaling, differentially expressed during Phytophthora capsici infection. Overview of candidate PAMP signaling and transcription-factor genes found to be differentially expressed during P. capsici infection. Gene identifiers, normalized expression values, and annotations are given. Genes are grouped based on their expression patterns as shown in Figure 7 and 8 respectively.
Table S9. Putative RXLR effectors used in this study. Overview of putative RXLR effectors. Gene name, probe ID, and nucleotide sequence are given for each gene.
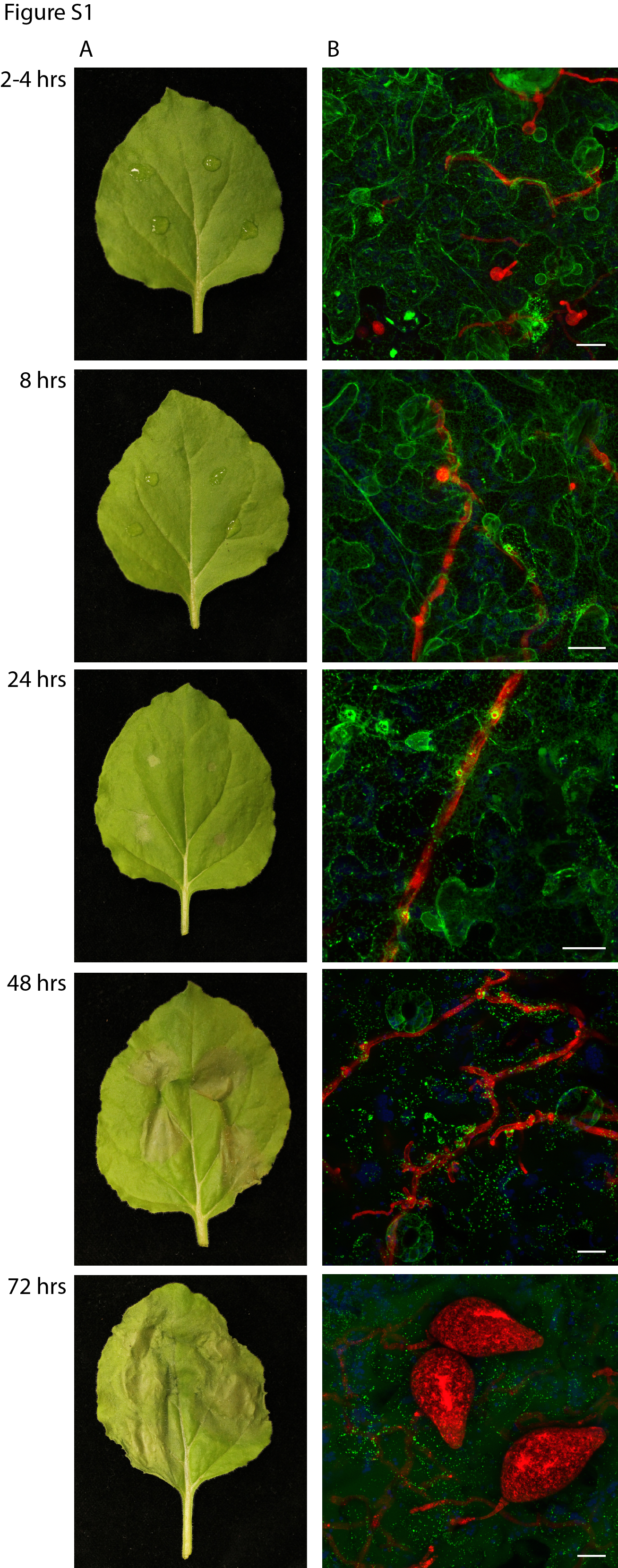
Figure S1. Assessment of cell viability during Phytophthora capsici infection. Transgenic Nicotiana benthamiana plants constitutively expressing ER-eGFP, a green fluorescent protein (GFP) localized to the endoplasmic reticulum (ER) were used to assess whether host cells were alive during the course of infection. (A) Photographs of N. benthamiana leaves infected with zoospore suspensions of P. capsici at 0, 8, 24, 48, and 72 hpi. (B) Confocal microscopy images of N. benthamiana leaves infected with a transgenic P. capsici strain expressing the fluorescent protein TdTomato. Within the first 24 hours, the host ER was largely intact despite the presence of P. capsici, and haustoria were often seen to invaginate living cells. After 24 hours, the ER network was disrupted as shown by the unstructured distribution of GFP, suggesting dead or dying cells. Bar = 20 μm.
{kind=link}
Figure S2. Reverse transcription PCR verification of marker-gene expression during infection. Expression of the marker genes PcHmp1, PcNpp1, PcCdc14, and PcTub (constitutive control) was tested by semi-quantitative PCR on cDNA derived from a time-course infection series used for the microarrays. Amplification of genes on cDNA derived from water-inoculated control (non-infected; ni) and tomato harvested 0, 8, 16, 24, 48, and 72 hpi with Phytophthora capsici.
Contributor Information
Julietta Jupe, Email: julietta.jupe@hutton.ac.uk.
Remco Stam, Email: remco.stam@hutton.ac.uk.
Andrew JM Howden, Email: andrew.howden@hutton.ac.uk.
Jenny A Morris, Email: Jenny.Morris@hutton.ac.uk.
Runxuan Zhang, Email: runxuan.zhang@hutton.ac.uk.
Pete E Hedley, Email: Pete.Hedley@hutton.ac.uk.
Edgar Huitema, Email: e.huitema@dundee.ac.uk.
Acknowledgements
We thank Dr Ariane Kemen and Professor Jonathan D.G. Jones (TSL) for providing P. capsici strain LT1534pTORtdTomato, Professor David Baulcombe for making available N. benthamiana line 16c (ER-eGFP), and members of the Dundee Effector Consortium for support and advice. This work was funded in part by the Royal Society of Edinburgh (cofunded by Marie-Curie) and the Biological Sciences Research Council (BBSRC grant BB/I00386X/1).
References
- Jones JDG, Dangl JL. The plant immune system. Nature. 2006;14:323–329. doi: 10.1038/nature05286. [DOI] [PubMed] [Google Scholar]
- Birch PRJ, Rehmany AP, Pritchard L, Kamoun S, Beynon JL. Trafficking arms: oomycete effectors enter host plant cells. Trends Microbiol. 2006;14:8–11. doi: 10.1016/j.tim.2005.11.007. [DOI] [PubMed] [Google Scholar]
- Chisholm ST, Coaker G, Day B, Staskawicz BJ. Host-microbe interactions: shaping the evolution of the plant immune response. Cell. 2006;14:803–814. doi: 10.1016/j.cell.2006.02.008. [DOI] [PubMed] [Google Scholar]
- Kamoun S. A catalogue of the effector secretome of plant pathogenic oomycetes. Annu Rev Phytopathol. 2006;14:41–60. doi: 10.1146/annurev.phyto.44.070505.143436. [DOI] [PubMed] [Google Scholar]
- Fisher MC, Henk DA, Briggs CJ, Brownstein JS, Madoff LC, McCraw SL, Gurr SJ. Emerging fungal threats to animal, plant and ecosystem health. Nature. 2012;14:186–194. doi: 10.1038/nature10947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamour KH, Stam R, Jupe J, Huitema E. The oomycete broad-host-range pathogen Phytophthora capsici. Mol Plant Pathol. 2012;14:329–337. doi: 10.1111/j.1364-3703.2011.00754.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamour KH, Mudge J, Gobena D, Hurtado-Gonzales OP, Schmutz J, Kuo A, Miller NA, Rice BJ, Raffaele S, Cano L, Bharti AK, Donahoo RS, Finley SL, Huitema E, Hulvey J, Platt D, Salamov A, Savidor A, Sharma R, Stam R, Storey D, Thines M, Win J, Haas B, Dinwiddie D, Jenkins J, Knight J, Affourtit J, Han CS, Chertkov O. et al. Genome sequencing and mapping reveal loss of heterozygosity as a mechanism for rapid adaptation in the vegetable pathogen Phytophthora capsici. Mol Plant Microbe Interact. 2012;14(10):1350–1360. doi: 10.1094/MPMI-02-12-0028-R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Judelson HS, Blanco FA. The spores of Phytophthora: weapons of the plant destroyer. Nat Rev Microbiol. 2005;14:47–58. doi: 10.1038/nrmicro1064. [DOI] [PubMed] [Google Scholar]
- Horbach R, Navarro-Quesada AR, Knogge W, Deising HB. When and how to kill a plant cell: infection strategies of plant pathogenic fungi. Journal Plant Physiol. 2011;14:51–62. doi: 10.1016/j.jplph.2010.06.014. [DOI] [PubMed] [Google Scholar]
- Koeck M, Hardham AR, Dodds PN. The role of effectors of biotrophic and hemibiotrophic fungi in infection. Cell Microbiol. 2011;14:1849–1857. doi: 10.1111/j.1462-5822.2011.01665.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Infection structures of biotrophic and hemibiotrophic fungal plant pathogens. Mol Plant Pathol. 2001;14:101–108. doi: 10.1046/j.1364-3703.2001.00055.x. [DOI] [PubMed] [Google Scholar]
- van West P, Shepherd SJ, Walker CA, Li S, Appiah AA, Grenville-Briggs LJ, Govers F, Gow NAR. Internuclear gene silencing in Phytophthora infestans is established through chromatin remodelling. Microbiology. 2008;14:1482–1490. doi: 10.1099/mic.0.2007/015545-0. [DOI] [PubMed] [Google Scholar]
- Whisson SC, Boevink PC, Moleleki L, Avrova AO, Morales JG, Gilroy EM, Armstrong MR, Grouffaud S, van West P, Chapman S, Hein I, Toth IK, Pritchard L, Birch PR. A translocation signal for delivery of oomycete effector proteins into host plant cells. Nature. 2007;14:115–118. doi: 10.1038/nature06203. [DOI] [PubMed] [Google Scholar]
- Haas BJ, Kamoun S, Zody MC, Jiang RHY, Handsaker RE, Cano LM, Grabherr M, Kodira CD, Raffaele S, Torto-Alalibo T, Bozkurt TO, Ah-Fong AMV, Alvarado L, Anderson VL, Armstrong MR, Avrova A, Baxter L, Beynon J, Boevink PC, Bollmann SR, Bos JIB, Bulone V, Cai G, Cakir C, Carrington JC, Chawner M, Conti L, Costanzo S, Ewan R, Fahlgren N. et al. Genome sequence and analysis of the Irish potato famine pathogen Phytophthora infestans. Nature. 2009;14:393–398. doi: 10.1038/nature08358. [DOI] [PubMed] [Google Scholar]
- Tyler BM, Tripathy S, Zhang X, Dehal P, Jiang RH, Aerts A, Arredondo FD, Baxter L, Bensasson D, Beynon JL, Chapman J, Damasceno CM, Dorrance AE, Dou D, Dickerman AW, Dubchak IL, Garbelotto M, Gijzen M, Gordon SG, Govers F, Grunwald NJ, Huang W, Ivors KL, Jones RW, Kamoun S, Krampis K, Lamour KH, Lee MK, McDonald WH, Medina M. et al. Phytophthora genome sequences uncover evolutionary origins and mechanisms of pathogenesis. Science. 2006;14:1261–1266. doi: 10.1126/science.1128796. [DOI] [PubMed] [Google Scholar]
- Bozkurt TO, Schornack S, Win J, Shindo T, Ilyas M, Oliva R, Cano LM, Jones AME, Huitema E, van der Hoorn RAL, Kamoun S. Phytophthora infestans effector AVRblb2 prevents secretion of a plant immune protease at the haustorial interface. Proc Natl Acad Sci USA. 2011;14:20832–20837. doi: 10.1073/pnas.1112708109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saunders DGO, Win J, Cano LM, Szabo LJ, Kamoun S, Raffaele S. Using hierarchical clustering of secreted protein families to classify and rank candidate effectors of rust fungi. PLoS ONE. 2012;14:e29847. doi: 10.1371/journal.pone.0029847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bozkurt TO, Schornack S, Banfield MJ, Kamoun S. Oomycetes, effectors, and all that jazz. Current Opinion in Plant Biology. 2012;14:9. doi: 10.1016/j.pbi.2012.03.008. [DOI] [PubMed] [Google Scholar]
- Oliva R, Win J, Raffaele S, Boutemy L, Bozkurt TO, Chaparro-Garcia A, Segretin ME, Stam R, Schornack S, Cano LM, Damme Mv, Huitema E, Thines M, Banfield MJ, Kamoun S. Recent developments in effector biology of filamentous plant pathogens. Cell Microbiol. 2010;14:705–715. doi: 10.1111/j.1462-5822.2010.01471.x. [DOI] [PubMed] [Google Scholar]
- Birch PR, Boevink PC, Gilroy EM, Hein I, Pritchard L, Whisson SC. Oomycete RXLR effectors: delivery, functional redundancy and durable disease resistance. Curr Opin Plant Biol. 2008;14:373–379. doi: 10.1016/j.pbi.2008.04.005. [DOI] [PubMed] [Google Scholar]
- Morgan W, Kamoun S. RXLR effectors of plant pathogenic oomycetes. Curr Opin Microbiol. 2007;14:332–338. doi: 10.1016/j.mib.2007.04.005. [DOI] [PubMed] [Google Scholar]
- Schornack S, van Damme M, Bozkurt TO, Cano LM, Smoker M, Thines M, Gaulin E, Kamoun S, Huitema E. Ancient class of translocated oomycete effectors targets the host nucleus. Proc Natl Acad Sci USA. 2010;14:17421–17426. doi: 10.1073/pnas.1008491107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stam R, Jupe J, Howden AJM, Morris JA, Boevink PC, Hedley PE, Huitema E. Identification and characterisation of CRN effectors in Phytophthora capsici shows modularity and functional diversity. PLOS ONE. 2013;14:e59517. doi: 10.1371/journal.pone.0059517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan MYA, Hutten RB, Visser RF, Eck H. The effect of pyramiding Phytophthora infestans resistance genes R Pi-mcd1 and R Pi-ber in potato. Theor Appl Genet. 2010;14:117–125. doi: 10.1007/s00122-010-1295-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu S, Li Y, Vossen J, Visser RF, Jacobsen E. Functional stacking of three resistance genes against Phytophthora infestans in potato. Transgenic Res. 2012;14:89–99. doi: 10.1007/s11248-011-9510-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jupe F, Pritchard L, Etherington G, MacKenzie K, Cock P, Wright F, Sharma SK, Bolser D, Bryan G, Jones J, Hein I. Identification and localisation of the NB-LRR gene family within the potato genome. BMC Genomics. 2012;14:75. doi: 10.1186/1471-2164-13-75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fry WE. Phytophthora infestans: the plant (and R gene) destroyer. Mol Plant Pathol. 2008;14:385–402. doi: 10.1111/j.1364-3703.2007.00465.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballvora A, Ercolano MR, Weiß J, Meksem K, Bormann CA, Oberhagemann P, Salamini F, Gebhardt C. The R1 gene for potato resistance to late blight (Phytophthora infestans) belongs to the leucine zipper/NBS/LRR class of plant resistance genes. Plant J. 2002;14:361–371. doi: 10.1046/j.1365-313X.2001.01292.x. [DOI] [PubMed] [Google Scholar]
- Champouret N, Bouwmeester K, Rietman H, van der Lee T, Maliepaard C, Heupink A, van de Vondervoort PJI, Jacobsen E, Visser RGF, van der Vossen EAG, Govers F, Vleeshouwers VGAA. Phytophthora infestans isolates lacking class I ipiO Variants are virulent on Rpi-blb1 potato. Mol Plant Microbe Interact. 2009;14:1535–1545. doi: 10.1094/MPMI-22-12-1535. [DOI] [PubMed] [Google Scholar]
- Avrova AO, Boevink PC, Young V, Grenville-Briggs LJ, van West P, Birch PR, Whisson SC. A novel Phytophthora infestans haustorium-specific membrane protein is required for infection of potato. Cell Microbiol. 2008;14:2271–2284. doi: 10.1111/j.1462-5822.2008.01206.x. [DOI] [PubMed] [Google Scholar]
- Kanneganti TD, Huitema E, Cakir C, Kamoun S. Synergistic interactions of the plant cell death pathways induced by Phytophthora infestans Nep1-like protein PiNPP1.1 and INF1 elicitin. Mol Plant Microbe Interact. 2006;14:854–863. doi: 10.1094/MPMI-19-0854. [DOI] [PubMed] [Google Scholar]
- Qutob D, Kamoun S, Gijzen M. Expression of a Phytophthora sojae necrosis-inducing protein occurs during transition from biotrophy to necrotrophy. Plant J. 2002;14:361–373. doi: 10.1046/j.1365-313X.2002.01439.x. [DOI] [PubMed] [Google Scholar]
- Ah Fong AM, Judelson HS. Cell cycle regulator Cdc14 is expressed during sporulation but not hyphal growth in the fungus-like oomycete Phytophthora infestans. Mol Microbiol. 2003;14:487–494. doi: 10.1046/j.1365-2958.2003.03735.x. [DOI] [PubMed] [Google Scholar]
- Randall TA, Dwyer RA, Huitema E, Beyer K, Cvitanich C, Kelkar H, Ah Fong AMV, Gates K, Roberts S, Yatzkan E, Gaffney T, Law M, Testa A, Torto T, Zhang M, Zheng L, Mueller E, Windass J, Binder A, Birch PRJ, Gisi U, Govers F, Gow N, Mauch F, van West P, Waugh M, Yu J, Boller T, Kamoun S, Lam ST, Judelson HS. Large-scale gene discovery in the oomycete Phytophthora infestans reveals likely components of phytopathogenicity shared with true fungi. Mol Plant Microbe Interact. 2005;14:229–243. doi: 10.1094/MPMI-18-0229. [DOI] [PubMed] [Google Scholar]
- consortium sg. The tomato genome sequence provides insights into fleshy fruit evolution. Nature. 2012;14:635–641. doi: 10.1038/nature11119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamaguchi Y, Pearce G, Ryan CA. The cell surface leucine-rich repeat receptor for AtPep1, an endogenous peptide elicitor in Arabidopsis, is functional in transgenic tobacco cells. Proc Natl Acad Sci USA. 2006;14:10104–10109. doi: 10.1073/pnas.0603729103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moy P, Qutob D, Chapman BP, Atkinson I, Gijzen M. Patterns of gene expression upon infection of soybean plants by Phytophthora sojae. Mol Plant Microbe Interact. 2004;14:1051–1062. doi: 10.1094/MPMI.2004.17.10.1051. [DOI] [PubMed] [Google Scholar]
- Qutob D, Kemmerling B, Brunner F, Kufner I, Engelhardt S, Gust AA, Luberacki B, Seitz HU, Stahl D, Rauhut T, Glawischnig E, Schween G, Lacombe B, Watanabe N, Lam E, Schlichting R, Scheel D, Nau K, Dodt G, Hubert D, Gijzen M, Nurnberger T. Phytotoxicity and innate immune responses induced by NEP1-like proteins. Plant Cell. 2006;14:3721–3744. doi: 10.1105/tpc.106.044180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grenville-Briggs LJ, Avrova AO, Bruce CR, Williams A, Whisson SC, Birch PR, van West P. Elevated amino acid biosynthesis in Phytophthora infestans during appressorium formation and potato infection. Fungal Genet Biol. 2005;14:244–256. doi: 10.1016/j.fgb.2004.11.009. [DOI] [PubMed] [Google Scholar]
- Schlink K. Down-regulation of defense genes and resource allocation into infected roots as factors for compatibility between Fagus sylvatica and Phytophthora citricola. Funct Integr Genomics. 2009;14:253–264. doi: 10.1007/s10142-009-0143-x. [DOI] [PubMed] [Google Scholar]
- Lopez-Gomez M, Sandal N, Stougaard J, Boller T. Interplay of flg22-induced defence responses and nodulation in Lotus japonicus. J Exp Bot. 2012;14:393–401. doi: 10.1093/jxb/err291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dyrløv Bendtsen J, Nielsen H, von Heijne G, Brunak S. Improved Prediction of Signal Peptides: SignalP 3.0. J Mol Biol. 2004;14:783–795. doi: 10.1016/j.jmb.2004.05.028. [DOI] [PubMed] [Google Scholar]
- Krogh A, Larsson Br, von Heijne G, Sonnhammer ELL. Predicting transmembrane protein topology with a hidden markov model: application to complete genomes. J Mol Biol. 2001;14:567–580. doi: 10.1006/jmbi.2000.4315. [DOI] [PubMed] [Google Scholar]
- Altschul SF, Gish W, W M, EW M, DJ L. Basic local alignment search tool. J Mol Biol. 1990;14:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- Thompson JD, Higgins DG, Gibson TJ. Improved sensitivity of profile searches through the use of sequence weights and gap excision. Comput appl biosci: CABIOS. 1994;14:19–29. doi: 10.1093/bioinformatics/10.1.19. [DOI] [PubMed] [Google Scholar]
- Win J, Morgan W, Bos J, Krasileva KV, Cano LM, Chaparro-Garcia A, Ammar R, Staskawicz BJ, Kamoun S. Adaptive evolution has targeted the C-terminal domain of the RXLR effectors of plant pathogenic oomycetes. Plant Cell. 2007;14:2349–2369. doi: 10.1105/tpc.107.051037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhattacharjee S, Hiller NL, Liolios K, Win J, Kanneganti TD, Young C, Kamoun S, Haldar K. The malarial host-targeting signal is conserved in the Irish potato famine pathogen. PLoS Pathog. 2006;14:e50. doi: 10.1371/journal.ppat.0020050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du Z, Zhou X, Ling Y, Zhang Z, Su Z. agriGO: a GO analysis toolkit for the agricultural community. Nucleic Acids Res. 2010;14:W64–W70. doi: 10.1093/nar/gkq310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VENNY. An interactive tool for comparing lists with Venn Diagrams. http://bioinfogp.cnb.csic.es/tools/venny/index.html
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Phytophthora capsici genes encoding putative secreted proteins found expressed in microarray experiments. Lists of P. capsici genes encoding candidate secreted proteins are given. These represents gene for which expression was detected in the infectious and in vitro stages.
Table S2. PcHmp1, PcNPP1, and PcCdc14 coregulated genes Overview of genes found to be coregulated with marker genes in Phytophthora capsici. Gene identifier, probe ID number, and normalized expression value (fold change over mean expression) are given for each gene and at each time point. Gene Ontology (GO) annotations are also given for each gene where available.
Table S3. Significantly enriched ontologies coregulated with marker genes, Overview of gene ontologies that are significantly enriched in the fractions that are specifically coregulated with P. capsici marker genes as shown in Figure 3C. Gene Ontology (GO) terms, P-values, false discovery rates (FDRs), and query and reference sample sizes are given.
Table S4. Phytophthora capsici candidate RXLR genes expressed in microarray experiments. Overview of RXLR effector genes found to be upregulated during specific lifecycle stages in P. capsici. Gene identifier, probe ID number, and normalized expression value (fold change over mean expression) are given for each gene and at each time point. Genes are listed per class as shown in Figure 4.
Table S5. Annotation and expression of tomato genes differentially expressed in pairwise comparisons. Overview of genes found to be differentially expressed between timepoints during Phytophthora capsici infection. Gene identifiers, normalized expression values, and Gene Ontology (GO) annotations are given.
Table S6. Tomato genes differentially expressed in each pairwise comparison. Overview of genes found to be differentially expressed during Phytophthora capsici infection in pairwise comparisons between two timepoints. Gene identifiers and normalized expression values are given.
Table S7. Genes corresponding to enriched gene ontologies (GOs) in pairwise comparisons. Overview of GOs that were significantly enriched in the fractions that were specifically upregulated or downregulated between two time points as shown in Figure 6. GOs, P-values, false discovery rates (FDRs), and query and reference sample sizes are given.
Table S8. Tomato genes with possible roles in pathogen-associated molecular pattern (PAMP) perception and signaling, differentially expressed during Phytophthora capsici infection. Overview of candidate PAMP signaling and transcription-factor genes found to be differentially expressed during P. capsici infection. Gene identifiers, normalized expression values, and annotations are given. Genes are grouped based on their expression patterns as shown in Figure 7 and 8 respectively.
Table S9. Putative RXLR effectors used in this study. Overview of putative RXLR effectors. Gene name, probe ID, and nucleotide sequence are given for each gene.
Figure S1. Assessment of cell viability during Phytophthora capsici infection. Transgenic Nicotiana benthamiana plants constitutively expressing ER-eGFP, a green fluorescent protein (GFP) localized to the endoplasmic reticulum (ER) were used to assess whether host cells were alive during the course of infection. (A) Photographs of N. benthamiana leaves infected with zoospore suspensions of P. capsici at 0, 8, 24, 48, and 72 hpi. (B) Confocal microscopy images of N. benthamiana leaves infected with a transgenic P. capsici strain expressing the fluorescent protein TdTomato. Within the first 24 hours, the host ER was largely intact despite the presence of P. capsici, and haustoria were often seen to invaginate living cells. After 24 hours, the ER network was disrupted as shown by the unstructured distribution of GFP, suggesting dead or dying cells. Bar = 20 μm.
Figure S2. Reverse transcription PCR verification of marker-gene expression during infection. Expression of the marker genes PcHmp1, PcNpp1, PcCdc14, and PcTub (constitutive control) was tested by semi-quantitative PCR on cDNA derived from a time-course infection series used for the microarrays. Amplification of genes on cDNA derived from water-inoculated control (non-infected; ni) and tomato harvested 0, 8, 16, 24, 48, and 72 hpi with Phytophthora capsici.