

Abstract
Determining RNA secondary structure is important for understanding structure-function relationships and identifying potential drug targets. This paper reports the use of microarrays with heptamer 2′-O-methyl oligoribonucleotides to probe the secondary structure of an RNA and thereby improve prediction of that secondary structure. When experimental constraints from hybridization results are added to a free energy minimization algorithm, the prediction of the secondary structure of E. coli 5S rRNA improves from 27% to 92% of the known canonical base pairs. Optimization of buffer conditions for hybridization and application of 2′-O-methyl-2-thiouridine to enhance binding and improve discrimination between AU and GU pairs are also described. The results suggest that probing RNA with oligonucleotide microarrays can facilitate determination of secondary structure.
The rapid increase in the size of the genome sequence database is making computational analysis of RNA increasingly important for revealing structure-function relationships and potential drug targets. While NMR and x-ray crystallography can definitively determine RNA structure, they cannot keep pace with the rapid generation of genomic data. Thus, faster methods are required to allow accurate modeling of RNA structures. The first step in this modeling is prediction of secondary structure.
Prediction of RNA secondary structure is done mainly by two methods. One is sequence comparison (1–5). This method is based on finding structural features conserved during evolution. Most of the currently accepted RNA secondary structures were generated by this method, which is limited by sequence diversity and structural similarity within an RNA family. The second method uses thermodynamics to predict secondary structure by free energy minimization. For example, computer programs, such as MFOLD and RNAstructure (6, 7), use a nearest neighbor energetic model (7–11) to generate a lowest free energy RNA secondary structure, as well as suboptimal structures with higher free energies. When tested against a database of about 150,000 nucleotides of known structures, the predicted lowest free energy structure on average contains 73% of the known canonical base pairs (7). There are many RNA structures that are poorly predicted, however. Algorithms can include nuclease (6, 12) or chemical mapping (13) data to constrain certain nucleotides and thereby improve predictions. Presumably, additional data from other types of experiments can further improve predictions. Here, we demonstrate that oligonucleotide microarrays allow rapid probing of RNA secondary structure and thus comparisons that can improve RNA structure prediction by computational methods.
Doty, Uhlenbeck and Lewis (14–16) described an equilibrium dialysis binding assay that revealed single stranded nucleotides in tRNA and 5S rRNA by binding of trimer and tetramer oligonucleotides. Although these dialysis experiments yield valuable information, they are slow. A faster way to obtain similar data is to use microarrays of oligonucleotides, which allow many more oligonucleotides to be screened in parallel for binding to an RNA. For example, Southern and coworkers used microarrays to develop antisense candidates for mRNA (17–19). Activated glass slides are used for clever in-situ synthesis of oligonucleotides that vary in length from 1–20 nucleotides and are complementary to the RNA of interest. Thus, a microarray is custom prepared for a given mRNA.
If oligonucleotide length is restricted, then it becomes feasible to make a microarray with all possible sequences of a given length. For example, Mirzabekov and coworkers studied the stability and specificity of all hexamers immobilized on a chip made with activated polyacrylamide gel pads (20). Relative to arrays coated on a 2D surface, gel coated slides provide an advantage for detection because the 3D matrix allows more probe to be loaded.
Here we show that immobilized heptamers can hybridize to Escherichia coli 5S rRNA, and can be used to probe its secondary structure. The native secondary structure, form A, of E. coli 5S rRNA is known from sequence alignments (1, 21), and has been confirmed by NMR of fragments (22) by x-ray crystallography of a fragment (23), and by comparison to the x-ray structure of the large ribosomal subunit of Haloarcula marismortui (24). Current versions of the computer programs MFOLD (7) and RNAstructure (13), however, only predict 27% of the canonical base pairs in the accepted structure. We show that comparisons with experimental data generated with an oligonucleotide array can improve this prediction. Under non-native conditions, E. coli 5S rRNA folds into a different secondary structure, denoted as form B (25–28). Form B binds to microarrays very differently from form A.
MATERIALS AND METHODS
Materials
Standard phosphoramidites for oligonucleotide synthesis and C6-aminolinker were purchased from Glen Research. The γ 32P-ATP, T4 polynucleotide kinase, and T4 RNA ligase were purchased from Perkin Elmer, Invitrogen, and Fermentas, respectively. RNases V1 and T1 were from Ambion and nuclease S1 from Fermentas. Dimethyl sulfate (DMS) and 1-cyclohexyl-3-(2-morpholinoethyl)carbodiimide metho-p-toluenesulfonate (CMCT) were from Aldrich and kethoxal was from ICN Biomedicals Inc. N-methylisatoic anhydride (NMIA) was from Molecular Probes. Reverse transcriptase SuperScript III was from Invitrogen, dNTPs and ddNTPs from Amersham Biosciences. Agarose was a product of Invitrogen. Silanized slides and probe-clip press seal incubation chambers for hybridization experiments were purchased from Sigma. RNAsin and ribonuclease H were from Promega.
Chemical synthesis of oligonucleotides
Oligonucleotides were synthesized by the phosphoramidite approach on an ABI 392 synthesizer (29, 30). The 2′-O-methyl-oligoribonucleotides used as probes for microarrays were synthesized with a C6-aminolinker on the 5′-end. Oligonucleotides were deprotected and purified according to published procedures (9), and molecular weights were confirmed by mass spectrometry (LC MS Hewlett Packard series 1100 MSD with API-ES). Concentrations of all oligonucleotides were determined from predicted extinction coefficients for RNA (31) and measured absorbance at 260 nm at 80 °C.
Isolation of 5S rRNA
The E. coli 5S rRNA was prepared from E. coli carrying the overproducing plasmid pKK5-1 as previously reported (32) with some modification. Total RNA was fractionated by electrophoresis on an 8% polyacrylamide denaturing gel. The band corresponding to 5S rRNA was cut out and extracted with water by crush and soak procedure with stirring at 4 °C overnight. The aqueous solution was concentrated with an equal volume of 2-butanol and the RNA was precipitated with ethanol (ethanol:water 2.5:1 v/v). After centrifugation, the pellet was dissolved in water and the 5S rRNA concentration was determined by measuring the absorbance at 260 nm at room temperature using an extinction coefficient of 1.2 × 106 M−1cm−1. The sequence of the 5S rRNA was confirmed by reverse transcription dideoxy sequencing in a manner similar to that described by Merino et al. (33).
Folding of 5S rRNA
The 5S rRNA was annealed by heating for 5 min at 65 °C and then slowly cooled (0.5 °C/min) to room temperature (25). For most experiments, one of the following buffers was used for folding: (A) buffer 1Na+/4Mg2+/10T (1 M NaCl, 4 mM MgCl2, 10 mM Tris-HCl pH 7.43), (B) buffer 0.15Na+/4Mg2+/10T (150 mM NaCl, 4 mM MgCl2, 10 mM Tris-HCl pH 7.43) or (C) buffer 0.04Na+/10Mg2+/10T (40 mM NaCl, 10 mM MgCl2, 10 mM Tris-HCl pH 7.43). For some experiments, the 5S rRNA was folded into form B according to published procedure (25): 5S rRNA was incubated in 20 mM sodium borate, 7 M urea, 250 mM Tris-HCl pH 7.8 at 25 °C for 45 min, then quickly chilled at −15 °C for 3 min and precipitated with 2.5 volumes of ethanol. The 5S rRNA precipitate was diluted in buffer 1Na+/250T (1 M NaCl, 250 mM Tris-HCl pH 7.8) at 0 °C.
Native gel electrophoresis
Native gel electrophoresis was done at 4 °C on a 10% polyacrylamide nondenaturing gel containing 40 mM Tris-acetate, pH 8.3, with a running buffer of 40 mM Tris-acetate, pH 8.3. The gel was preelectrophoresed at 150 V for 2 h. Electrophoresis was at 100–200 V for 48 h.
Enzymatic mapping
RNase V1, RNase T1 and nuclease S1 were used for enzymatic mapping. The 5S rRNA was 32P radiolabeled at the 5′- or 3′-end. For enzymatic digestion, 2 pmol of 5S rRNA was used for each reaction and tRNA carrier was added to give a total RNA concentration of 8 μM. The following concentrations of enzymes were used for cleavage reactions: RNase V1 (0.005U/μL), RNase T1 (0.02U/μL) and nuclease S1 (2.5U/μL). After annealing of 5S rRNA in buffer 1Na+/4Mg2+/10T, adding ZnCl2 to a final concentration of 1 mM for S1 reaction, and adding of the appropriate enzyme, the solution was incubated at room temperature for 30 min. Reactions were stopped by ethanol precipitation on dry ice. After centrifugation, the pellet was dissolved in loading buffer and the reaction mixtures were analyzed on a 12% polyacrylamide denaturing gel. Cleavage products were analyzed by comparison to ladders obtained from formamide treatment and from partial T1 nuclease digestion in denaturing conditions.
Chemical mapping
DMS was used to modify adenosine and cytidine, kethoxal to modify guanosine and CMCT to modify uridine. Chemical mapping of 5S rRNA was performed according to published procedure (34) with several changes.
The salt dependence of reactivity of DMS on adenosine and cytosine and of CMCT on uridine was tested in buffers 1Na+/4Mg2+/10T, 0.04Na+/10Mg2+/10T, and 1Na+/250T. Uridine (409 μM) in buffer was reacted with 102 mM CMCT for 15 min at room temperature. Adenosine (657 μM) and cytosine (722 μM) were reacted with 359 mM of DMS (3.9 μL of DMS in 10 μL of ethanol added to 100 μL of nucleoside solution) for 48 h at 37 °C. Reactions were stopped by freezing at −80 °C. Results were analyzed by HPLC. The extent of reaction was typically between 30 and 50% and was independent of buffer composition.
For chemical mapping, 2 pmol of 5S rRNA was taken for each reaction and annealed in buffers 1Na+/4Mg2+/10T or 0.04Na+/10Mg2+/10T, as described above. Then tRNA carrier was added to give a total RNA concentration of 8 μM and the solution was incubated for 10 min at the temperature of chemical mapping. Form B of 5S rRNA was prepared for chemical mapping as described above and modification reactions were performed in buffer 1Na+/250T at 4 °C. To a 9 μL sample, 1 μL of dimethyl sulfate or kethoxal solution was added. Dimethyl sulfate was diluted in ethanol and used at final concentrations of 60, 30 and 15 mM. Kethoxal was diluted in ethanol: water (1:3 v/v) to give final concentrations of 320, 160 and 80 mM. After modification with kethoxal, 3 μL of 35 mM sodium borate solution was added to stabilize products of modification. For modification with CMCT, 9 μL of CMCT solution was added to the 9 μL sample of RNA. CMCT was diluted in appropriate buffer (1Na+/4Mg2+/10T, 0.04Na+/10Mg2+/10T or 1Na+/250T) to give final concentrations in the reaction mixture of: 625 (room temperature) or 500 (4 °C), 250, and 100 mM. Chemical modification reactions were performed for 20 min for chemical mapping at room temperature and 1.5 h for mapping at 4 °C. Reactions were stopped by ethanol precipitation on dry ice.
Chemical mapping was also done with N-methylisatoic anhydride (NMIA) according to published procedure (33, 35) with some changes. For each reaction, 2 pmol of 5S rRNA was annealed in buffers 1Na+/4Mg2+/10T, 0.15Na+/4Mg2+/10T, or 0.04Na+/10Mg2+/10T as described above. To a 9 μL sample, 1 μL of NMIA solution (1 or 0.5 mg NMIA/42 μL DMSO) was added. Samples were incubated for 3 h at room temperature. Reactions were stopped by ethanol precipitation on dry ice.
Primer extension reactions
DNA primers for primer extension reactions were 5′ATGCCTGGCAGTTCCCT3′ and 5′GCGCTACGGCGTTTCAC3′, which are complementary to nucleotides 104–120 and 54–70 of 5S rRNA, respectively. Primers were labeled on the 5′-end with γ 32P-ATP according to standard procedure. For each reaction, 2 pmol of primer was used. Primer extension was performed at 55 °C with reverse transcriptase SuperScript III and Invitrogen’s buffers and protocol. Reactions were stopped by adding loading buffer containing dye (Bromophenol Blue), and chilling to 0 °C. Products were separated on a 12% polyacrylamide denaturing gel. The gels were analyzed with the ImageQuant 5.2 program and products were identified by comparing to sequencing lanes for A, C, T and G and to control lanes. Modifications were initially identified by visual inspection of autoradiograms and were considered strong or medium when the band corresponding to chemical modification had at least 6 times or 2–6 times, respectively, the integrated intensity of the equivalent band in the control lane, as quantified with ImageQuant 5.2.
Preparation of microarrays
Microarrays were prepared on agarose coated slides according to the method described by Afanassiev et al. (36). Silanized slides were coated with 1% agarose activated by NaIO4. On dried microarrays, 0.5 μL of 100 μM of each probe were spotted and incubated for 4 h at 37 °C in a 100% humidity chamber. The remaining aldehyde groups on microarrays were reduced with 35 mM NaBH4 solution in PBS buffer and ethanol (3:1 v/v). Then slides were washed in water at room temperature (3 washes for 30 min each), and in 1% SDS solution at 55 °C for 1 h, and finally in water at room temperature (3 washes for 30 min each) and dried at room temperature overnight.
Hybridization conditions
E. coli 5S rRNA was radioactively labeled with [32P]Cp on the 3′-end according to standard procedure and purified on an 8% polyacrylamide denaturing gel. For hybridization, labeled 5S rRNA was used at approximately 0.01 μM. Prior to hybridization, 5S rRNA was refolded as described above. Hybridization was performed in the same buffers used for folding. For hybridization, 200 μL of hybridization buffer containing target 5S rRNA was placed in a probe-clip press seal incubation chamber and incubated for 18 h at room temperature or 4 °C. At least 4 h was required to see strong signals and 18 h was optimal. After hybridization, buffers with 5S rRNA were poured out and slides were washed in buffers with the same salt concentrations for 1 min at 0 °C. Then, slides were dried by slow centrifugation in a clinical centrifuge and covered with saran wrap. Hybridization was visualized by exposure to a phosphorimager screen, which was then scanned on a Molecular Dynamics 840 Storm Phosphorimager. Quantitative analysis was done with ImageQuant 5.2 software. Binding was considered strong when the integrated intensity was ≥ 1/3 of the strongest integrated intensity for a given condition. Medium binding had integrated intensity from 1/3 to 1/7 of the strongest.
Ribonuclease H assays
The 5S rRNA (4 pmol) was folded in buffer 0.15Na+/4Mg2+/10T and then a 10-fold excess of appropriate DNA oligomer in buffer 0.15Na+/4Mg2+/10T was added at room temperature to give a final DNA concentration of about 70 μM. The mixture was incubated for 1 h at room temperature. Then DTT to a final concentration of 1 mM and 1.25 U RNAsin and 1.3 U ribonuclease H were added. The reaction was incubated for 20 min at room temperature. The reaction mixture was extracted twice with equal volumes of phenol/chloroform/isoamyl alcohol (25:24:1) (Aldrich). The residual phenol was removed by two chloroform extractions and the RNA was precipitated with ethanol. Primer extension was used to identify sites of ribonuclease H cleavages.
Thermodynamic measurements
Oligonucleotides were melted in buffer containing 100 mM NaCl, 20 mM sodium cacodylate, 0.5 mM Na2EDTA, pH 7.0. Absorbance vs. temperature melting curves were measured at 260 nm with a heating rate of 1 °C/min from 0 to 90 °C on a Beckman DU 640 spectrometer with a water cooled Peltier thermoprogrammer. Melting curves were analyzed and thermodynamic parameters were calculated with the program MeltWin 3.5 (37). Optical melting curves for 5S rRNA were measured in the same way except that the RNA was first folded as described above.
RESULTS
Chemical mapping of 5S rRNA from E. coli
The secondary structure of 5S rRNA was chemically mapped under several conditions (Figure 1): (A) buffer 1Na+/4Mg2+/10T at room temperature; (B) buffer 0.04Na+/10Mg2+/10T at room temperature, which is known to fold 5S rRNA into native form A (25); (C) buffer 1Na+/4Mg2+/10T at 4 °C and (D) buffer 1Na+/250T at 4 °C, which is used for probing form B. Native gel electrophoresis of 5S rRNA gave a single band for folding in each buffer.
FIGURE 1.

Chemical mapping results. Conditions are: (A) 1Na+/4Mg2+/10T (1 M NaCl, 4 mM MgCl2, 10 mM Tris-HCl pH 7.43), room temperature, (B) 0.04Na+/10Mg2+/10T (40 mM NaCl, 10 mM MgCl2, 10 mM Tris-HCl pH 7.43), room temperature (C) 1Na+/4Mg2+/10T (1 M NaCl, 4 mM MgCl2,10 mM Tris-HCl pH 7.43), 4 °C, and (D) 1Na+/250T (1 M NaCl, 250 mM Tris-HCl pH 7.8), 4 °C. Also shown are results from nuclease mapping under condition (A). Nucleases V1 and S1 are specific for double and single stranded regions, respectively. T1 is specific for single stranded G. Symbols: ● - strong DMS; ○ - medium DMS; ■ - strong CMCT; □ - medium CMCT; ▲ - strong kethoxal; △ - medium kethoxal; ◆ - strong NMIA; ◇ - medium NMIA.
The chemical reagents can modify nucleotides that are not in Watson-Crick pairs flanked by Watson-Crick pairs (33, 38, 39). Thus, the modifications in buffers 1Na+/4Mg2+/10T and 0.04Na+/10Mg2+/10T are consistent with the native structure A of 5S rRNA from E. coli, but in buffer 1Na+/250T the modifications of A29 and U32 are not consistent with the native structure A (Figure 1). The modifications in buffer 0.04Na+/10Mg2+/10T are consistent with those reported previously under similar conditions (26, 40–42).
Although the modifications in buffers 1Na+/4Mg2+/10T and 0.04Na+/10Mg2+/10T are consistent with the native structure A of 5S rRNA from E. coli, the reactivities are buffer dependent. In particular, in buffer 1Na+/4Mg2+/10T at room temperature (Figure 1A) there are medium instead of strong DMS modifications of A15, A73 and A78, an additional medium kethoxal modification of G18, medium instead of strong NMIA modifications at U40 and C42, lack of modification of G41 and strong and medium NMIA modifications at A66 and A73, respectively, instead of a medium and no modification. Evidently, there are subtle changes in folding that are dependent on NaCl and MgCl2 concentrations. It is unlikely that these differences reflect any significant change in stability of secondary structure, however. Optical melting curves showed that the melting temperatures of the first transition were 54.9, 64.8 and 67.7 °C in buffer 1Na+/4Mg2+/10T, 0.15Na+/4Mg2+/10T and 0.04Na+/10Mg2+/10T, respectively. All are more than 30 °C higher than the temperature of the chemical mapping experiments, so the secondary structure is very stable in the presence of Mg2+. Thus, the differences in chemical reactivity suggest only subtle changes in folding that are dependent on NaCl and MgCl2 concentrations.
Figure 1C shows chemical mapping results in buffer 1Na+/4Mg2+/10T at 4 °C. In comparison with room temperature (Figure 1A) there are fewer reactive bases, but the general pattern of modifications is very similar. Evidently, the secondary structure of 5S rRNA in buffer 1Na+/4Mg2+/10T is close or identical to form A at room temperature and 4 °C but there are subtle differences in local folding.
The structure of 5S rRNA in buffer 1Na+/4Mg2+/10T at room temperature was also probed by enzymatic mapping. As shown in Figure 1A, the results are consistent with the secondary structure of form A. The results also agree with those in the literature under different buffer conditions giving form A (26, 27, 40, 41).
Native gel electrophoresis of E. coli 5S rRNA in buffer 1Na+/250T gave a single band that migrated faster than the band of form A. In comparison to other conditions, many more chemical modifications were observed in buffer 1Na+/250T at 4 °C (see Figure 1D). The structure in buffer 1Na+/250T is clearly different from that in the other buffers.
Probing form A of 5S rRNA from E. coli with microarrays of 2′-O-methylated heptamers
Form A of 5S rRNA from E. coli was probed under different conditions with microarrays having all possible complementary 2′-O-methylated RNA heptamers (Figures 2 and 3 and Table 1 and Supporting Information). Each 2′-O-methylated RNA heptamer is identified by a number corresponding to the number of the 5S rRNA nucleotide in the middle of the 5S rRNA sequence completely complementary to the heptamer probe.
FIGURE 2.

Hybridization results on 2′-O-methyl heptamer microarrays for 5S rRNA from E. coli in buffer 1Na+/4Mg2+/10T (1 M NaCl, 4 mM MgCl2, 10 mM Tris-HCl pH 7.43), room temperature. On microarrays shown, the 2′-O-methyl heptamers from left to right are: (A) row 1: 4→ 15 (no binding detected), row 2: 16→ 27, row 3: 28 → 39, row 4: 40 → 51, row 5: 52 → 60, (B) row 1: 61→ 72 (no binding detected), row 2: 73→ 84 (no binding detected), row 3: 85 → 96, row 4: 97 → 108 (no binding detected), row 5: 109 → 117 (no binding detected).
FIGURE 3.

Hybridization results for 5S rRNA on 2′-O-methyl-heptamer microarrays. Conditions: (A) 1Na+/4Mg2+/10T (1 M NaCl, 4 mM MgCl2, 10 mM Tris-HCl pH 7.43), room temperature; (B) 1Na+/250T (1 M NaCl, 250 mM Tris-HCl, pH 7.8), 4 °C. Note that probes 16 and 67, 38 and 93, and also 39 and 94 have identical sequences and that probe 92 likely binds to loop C. Symbols:
 - middle site of strong binding;
- middle site of strong binding;
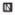 - middle site of medium binding.
- middle site of medium binding.
Table 1.
Hybridization results in 1 M NaCl, 4 mM MgCl2, 10 mM Tris-HCl pH 7.43 (1Na+/4Mg2+/10T), RNAstructure calculations, and ribonuclease H cleavages a
| center of binding site | sequence of probe (5′-3′) | 5S rRNA binding | ΔG°37 predicted (kcal/mol) | alternative binding site | ΔG°37 predicted (kcal/mol) for alternative binding site (no structure of 5S rRNA) | sites of ribonuclease H cleavage | ||
|---|---|---|---|---|---|---|---|---|
| break local structure of 5S rRNA | no structure of 5S rRNA | |||||||
| 4°C | RT | |||||||
| 27 | GUGGGAC | S | S | −9.1 | −12.2 | - | - | |
| 28 | GGUGGGA | S | S | −7.2 | −13.4 | - | - | M A29 |
| 29 | AGGUGGG | S | S | −5.4 | −12.5 | - | - | M C30 M C31 |
| 30 | CAGGUGG | M | M | −1.0 | −12.4 | 29/30 | −9.9 | |
| 35 | GGGGUCA | S | M | −6.3 | −13.8 | - | - | |
| 36 | UGGGGUC | S | S | −7.8 | −12.6 | - | - | |
| 37 | AUGGGGU | S | S | −10.0 | −11.3 | 92/93 | −9.9 | - |
| 38/93 | CAUGGGG | M | M | −11.1 | −12.0 | 93 | −11.6 | |
| 39/94 | GCAUGGG | - | - | −11.2 | −12.3 | 94 | −13.2 | M A39 M U40 M C42 |
| 40 | GGCAUGG | S | M | −11.2 | −13.2 | 94/95 | −9.9 | |
| 41 | CGGCAUG | M | M | −10.3 | −11.7 | - | - | S A39 S U40 S C42 |
| 42 | UCGGCAU | S | - | −10.6 | −10.7 | 71 | −9.2 | |
| 43 | UUCGGCA | M | - | −10.4 | −10.2 | 71 | −9.4 | S C42 S G44 S A45 |
| 44 | GUUCGGC | M | M | −10.5 | −11.7 | - | - | |
| 70 | UCGGCGC | M | - | −4.6 | −13.5 | 1) 42/43 2) 62 |
1) −9.4 2) −9.2 |
|
| 71 | AUCGGCG | M | - | −2.9 | −11.3 | 42/43 | −9.4 | |
| 90 | GGGGAGA | M | - | 0.2 | −14.1 | - | - | |
| 91 | UGGGGAG | S | - | 2.3 | −12.6 | 37 | −9.1 | - |
| 92 | AUGGGGA | S | M | 2.9 | −11.2 | 37/38 | −10.3 | - |
| 93/38 | CAUGGGG | M | M | 5.1 | −11.6 | 38 | −12.0 | |
| 94/39 | GCAUGGG | - | - | 6.5 | −13.2 | 39 | −12.3 | M A39 M U40 M C42 |
Each 2′-O-methylated RNA heptamer is identified by the number corresponding to the number of the 5S rRNA nucleotide complementary to the middle nucleotide of the heptamer probe (center of binding site). Alternative binding sites are defined by the middle nucleotide of the binding region on 5S rRNA. S - strong binding (from 1 to 1/3 integrated intensity of the strongest integrated intensity in a given condition). M - medium binding (from 1/3 to 1/7 integrated intensity of the strongest). Ribonuclease H cleavage occurs 3′ of the nucleotides listed in the right most column. “–” indicates that the probe does not bind or ribonuclease H does not cleave. The predicted values of ΔG°37 for 5S rRNA with no structure include contributions from the first 5′ and 3′ dangling ends which sometimes results in more favorable binding to alternative binding sites relative to sites fully complementary to the probe.
The best discrimination between binding and non-binding probes was achieved in buffer 1Na+/4Mg2+/10T at room temperature (Figures 2 and 3 and Table 1). There was strong binding of probes 27–29, 36 and 37, and medium binding of probes 30, 35, 38/93 (which have the same sequence), 40, 41, 44 and 92. Similar relative binding was observed in buffers 0.15Na+/4Mg2+/10T and 0.04Na+/10Mg2+/10T at room temperature but generally the binding was weaker (see Supporting Information). The strongest signal in buffers 0.15Na+/4Mg2+/10T and 0.04Na+/10Mg2+/10T was 4-fold weaker than in buffer 1Na+/4Mg2+/10T. Evidently, the secondary structure of 5S rRNA in buffers 1Na+/4Mg2+/10T, 0.15Na+/4Mg2+/10T and 0.04Na+/10Mg2+/10T is the same at room temperature (form A), but the interactions between 5S rRNA and the probes are weaker in buffers 0.15Na+/4Mg2+/10T and 0.04Na+/10Mg2+/10T than in buffer 1Na+/4Mg2+/10T.
The hybridization of 5S rRNA from E. coli to heptamer probes on microarrays was also studied in buffers 1Na+/4Mg2+/10T, 0.15Na+/4Mg2+/10T and 0.04Na+/10Mg2+/10T at 4 °C (Table 1 and Supporting Information). Hybridization patterns and amplitudes at 4 °C change relative to room temperature. Usually, stronger binding is observed at 4 °C. The binding patterns for buffers 0.15Na+/4Mg2+/10T and 0.04Na+/10Mg2+/10T at 4 °C were similar to buffer 1Na+/4Mg2+/10T at 4 °C (Supporting Information), but the strongest binding signal was 4-fold weaker.
The 4-fold difference between maximum signal intensity in buffer 1Na+/4Mg2+/10T and in buffers 0.15Na+/4Mg2+/10T and 0.04Na+/10Mg2+/10T shows that the concentration of NaCl is important, and hybridization is optimized at 1 M NaCl. The melting temperature of polymer duplexes in the presence of Mg2+ is reduced as Na+ is added (43, 44) and this was also observed for optical melting curves of E. coli 5S rRNA, as mentioned above. Perhaps oligomer-polymer duplexes behave differently and/or the reduced stability of tertiary structure of 5S rRNA in 1M Na+ facilitates binding to oligonucleotides.
Scanning for alternative binding sites with the RNAstructure program
There are only 16,384 heptamers with different sequences and heptamer duplexes can be stable even when containing a mismatch. Thus, it is possible for a given heptamer to have more than one binding site on an RNA. The RNAstructure 4.11 program (13) was used in bimolecular folding mode to predict formation of stable duplexes between unstructured E. coli 5S rRNA and probe oligonucleotides. The calculation of alternative binding sites included stability increments from mismatches and dangling ends. For E. coli 5S rRNA, probes 16 and 67, 38 and 93, and also 39 and 94 have identical sequences and thus have two completely complementary binding sites. Mismatched binding with free energy more favorable than −9 kcal/mol at 37 °C to completely unfolded 5S rRNA was also considered a possible alternative binding site. This corresponds to a dissociation constant of about 0.5 μM, which is 50-fold higher than the roughly 0.01 μM concentration of 5S rRNA on the microarray. Calculations by RNAstructure 4.11 are restricted to 37 °C and assume that both nucleic acid strands are not immobilized before binding, however, so the predictions will underestimate binding to an immobilized strand at room temperature. These calculations revealed several alternative binding sites. For example, probe 37 could bind tightly at position 92/93; probes 91 and 92 could bind at positions 37 and 37/38, respectively. The identical sequences of probes 38/93 and 39/94 along with possible mismatch binding of probes 37, 91, and 92 suggests that binding of probes 37–39 and 91–94 is to either positions 37–39 or 91–94 or both. As described below, experiments with ribonuclease H support the interpretation that binding is to positions 37–39.
Ribonuclease H cleavage assay
To test the hypothesis that some probes have multiple binding sites, a ribonuclease H assay in 0.15Na+/4Mg2+/10T buffer at room temperature was used to determine the position of binding for probes: 28, 29, 37, 39/94, 41, 43, 91, and 92. Because 2′-O-methylated oligoribonucleotides do not induce ribonuclease H cleavage of RNA (45, 46), the DNA analogues were used. The results are listed in Table 1. Oligonucleotide 28 induces medium cleavage after A29, and oligonucleotide 29 induces medium cleavage after C30 and C31. Oligonucleotide 41 induces strong cleavage after A39, U40, and C42 and oligonucleotide 43 induces strong cleavage after C42, G44 and A45. These patterns correlate well with expected binding that is fully complementary (Table 1).
Oligonucleotide 39 has the same sequence as 94, and both only bind 5S rRNA on microarrays under certain conditions (Supporting Information). The DNA analogue of 39/94 induces medium ribonuclease H cleavages only after A39, U40, and C42, corresponding to the binding site at position 39 (Table 1). Probes 38 and 93 have the same sequence and presumably also bind to loop C. Thus, it is likely that loop C is responsible for the binding observed for probes 91–94. Oligonucleotides 37, 91, and 92 did not induce ribonuclease H cleavages, however. All these oligonucleotides have G4 sequences, which probably form stable quadruplexes in solution (47–49) thus prohibiting binding to RNA. Clearly, interpretation of microarray binding results must consider potential alternative binding sites. Selected assays with ribonuclease H can sometimes identify actual binding sites.
Comparisons to predictions from the OligoWalk program
The OligoWalk program (50) was used to predict the free energies at 37 °C for binding of probes to the accepted secondary structure of form A of 5S rRNA (Table 1 and Supporting Information). The free energies for breaking target structure and binding probe to target RNA were only considered. The possibility of intermolecular association of probes was omitted because the probes are immobilized. The 2′-O-methylated heptamers were assumed to have the same energetics as RNA, which is consistent with optical melting studies on a series of 37 heteroduplexes (51).
With the exception of probe 30, oligonucleotides that bind strongly or moderately at room temperature are always predicted to have a ΔG°37 for hybridization to form A of E. coli 5S rRNA that is more favorable than −5 kcal/mol, although some of these favorable sites form mismatches and/or overhangs with the probe. For example, probe 92 has a predicted ΔG°37 that is unfavorable for binding at its fully complementary site, but a predicted favorable ΔG°37 of −10.3 kcal/mol for binding at position 38 (see Table 1). Probe 30 can bind more favorably at site 29 than at site 30 but still has a predicted ΔG°37 less favorable than −3 kcal/mol after allowing for breaking of target structure. Perhaps probe 30 binds to site 29 with coaxial stacking to flanking helixes. Coaxial stacking is not included in the OligoWalk algorithm, but is favorable for duplex formation (52).
Binding to shorter versions of probe reveals the number of base pairs required for binding at a particular site
As a test of the number of base pairs required to observe binding, the binding was measured to sequentially shortened versions of probe 35, 5′GMGMGMGMUMCMAM, which strongly and moderately binds 5S rRNA at 4 °C and room temperature, respectively. In buffer 1Na+/4Mg2+/10T at 4 °C, probes 5′GMGMGMGMUMCM, 5′GMGMGMGMUM and 5′GMGMGMGM gave signals with 0.8, 0.6 and 0.4 times the intensity of probe 35, respectively. In contrast, hexamer 5′GMGMGMUMCMAM gave only 0.1 times the intensity of probe 35, and 5′GMGMUMCMAM and 5′GMUMCMAM gave no signal. At room temperature, 5′GMGMGMGMUMCM gave 0.4 times the intensity of probe 35, but the other shortened probes gave no signal. Evidently, only the four consecutive GC pairs are required for binding of probe 35 at 4 °C, but at least six base pairs are required for binding at room temperature.
Probe 90, 5′GMGMGMGMAMGMAM, binds E. coli 5S rRNA in buffers 1Na+/4Mg2+/10T, 0.15Na+/4Mg2+/10T and 0.04Na+/10Mg2+/10T at 4 °C (Table 1 and Supporting Information). Further insight into this binding was provided by shorter probes 5′GMGMGMAMGMAM and 5′GMGMAMGMAM. Neither binds to 5S rRNA at 4 °C in any of the three buffers. These results, together with those for 5′GMGMGMGM, which binds with 0.5 times the intensity of probe 90, are consistent with probe 90 binding only to loop C. The results suggest that comparison between possible secondary structures and binding of different length oligonucleotides can help identify correct structures. A related approach was used with equilibrium dialysis data (15).
Binding of form B of E. coli 5S rRNA to microarrays
The 5S rRNA was folded into form B as described in Materials and Methods and hybridization was performed in buffer 1Na+/250T at 4 °C to avoid refolding into form A during hybridization. Many more probes bound in buffer 1Na+/250T than in buffers giving form A (see Figure 3). Evidently, oligonucleotide binding to E. coli 5S rRNA is facilitated by the absence of Mg2+.
Application of probes containing 2-thiouridine to enhance stability of A-U pairs and discrimination between A-U and G-U pairs
Two problems that arise in interpretation of oligonucleotide binding data are the sequence dependence of stabilities for perfectly matched helixes and the stabilities of helixes with non-Watson-Crick pairs. The sequence dependence of stabilities is illustrated by the predicted range of ΔG°37 from −6 to −15 kcal/mol for binding of the heptamers to their unfolded heptamer complements (see table in Supporting Information). This corresponds to over a million-fold range in binding constant. The problem of non-Watson-Crick pairs is most acute for G-U pairs because they have stabilities similar to A-U pairs (7, 9, 53). Modified nucleotides provide one way to achieve more uniform and specific binding of probes to target RNA. For example, 2-thiouridine enhances the thermodynamic stability of A-U pairs (54–57). In the RNA/RNA duplex, 5′GAGUGAG/3′CUCACUC, 2-thiouridine stabilizes the middle A-U pair by 1.1 kcal/mol and destabilizes the corresponding G-U pair by 0.3 kcal/mol at 37 °C (57). To test this in RNA/2′-O-methyl RNA hybrids, 5′-O-dimethoxytrityl-2′-O-methyl-2-thiouridine 3′-O-phosphoramidite (58) was synthesized and used for preparing model probes. Several RNA/2′-O-methyl RNA duplexes without C6-aminolinker were melted and thermodynamic parameters are collected in Table 2. The 2′-O-methylated probe 40, 5′GMGMCMAMUMGMGM, was chosen as a reference for duplex formation with natural bases. Probe 40 was selected because on microarrays at room temperature and 1 M NaCl it hybridized with medium strength to 5S rRNA, and is expected to only hybridize to position 40. For the complementary duplex 5′GMGMCMAMUMGMGM/3′CCGUACC in solution, the measured ΔG°37 is −9.69 kcal/mol and the Tm is 53.1 °C. When 2′-O-methyl uridine was replaced by 2′-O-methyl-2-thiouridine, the complementary duplex was stabilized by 1.87 kcal/mol and the Tm increased by 3.9 °C. When the RNA was changed to 3′CCGUGCC so as to form a G-U pair, the ΔG°37 was about −7.75 kcal/mol with either U or s2UM in the 2′-O-methyl strand. Thus discrimination for A-U over G-U is enhanced by about 1.9 kcal/mol in this context.
Table 2.
Thermodynamic parameters of heteroduplexes of RNA and 2′-O-methyl oligoribonucleotides a
| Duplexes | Average of curve fits
|
TM−1 vs log CT plots
|
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| RNA | 2′OMeRNA | −ΔH° (kcal/mol) |
−ΔS° (eu) | −ΔG°37 (kcal/mol) |
TMb (°C) |
−ΔH° (kcal/mol) |
−ΔS° (eu) | −ΔG°37 (kcal/mol) |
TMb (°C) |
ΔΔG°c37 (kcal/mol) |
ΔTMb,c (°C) |
|
| Probe 40 | mimics | |||||||||||
| A-U | 5′CCAUGCC | 5′GGCAUGG | 69.1±2.5 | 190.9±7.7 | 9.86±0.13 | 52.7 | 64.1±3.1 | 175.6±9.6 | 9.69±0.11 | 53.1 | ||
| A-s2U | 5′CCAUGCC | 5′GGCAs2UGG | 75.1±13.5 | 206.2±40.6 | 11.16±0.90 | 57.4 | 83.1±13.8 | 230.6±41.8 | 11.56±0.87 | 57.0 | −1.87 | 3.9 |
|
| ||||||||||||
| G-U | 5′CCGUGCC | 5′GGCAUGG | 61.5±2.2 | 173.1±7.1 | 7.78±0.05 | 43.4 | 58.4±1.6 | 163.2±4.9 | 7.76±0.01 | 43.7 | ||
| G-s2U | 5′CCGUGCC | 5′GGCAs2UGG | 61.1±3.7 | 171.9±11.9 | 7.80±0.07 | 43.6 | 56.9±4.02 | 158.6±12.7 | 7.73±0.09 | 43.7 | +0.03 | 0 |
|
| ||||||||||||
| C→U in | probe 40 | |||||||||||
| A-U | 5′CCAUACC | 5′GGUAUGG | 64.7±2.1 | 181.4±6.8 | 8.41±0.06 | 46.3 | 57.2±2.7 | 157.4±8.7 | 8.34±0.04 | 47.2 | ||
| A-s2U | 5′CCAUACC | 5′GGs2UAUGG | 65.1±3.3 | 179.9±10.3± | 9.27±0.19 | 50.6 | 58.8±0.9 | 160.5±2.9 | 9.05±0.03 | 50.9 | −0.71 | 3.7 |
|
| ||||||||||||
| G-U | 5′CCAUGCC | 5′GGUAUGG | 77.9±17.3 | 231.2±56.1 | 6.19±0.10 | 35.7 | 69.6±4.2 | 204.5±13.9 | 6.22±0.08 | 35.6 | ||
| G-s2U | 5′CCAUGCC | 5′GGs2UAUGG | 76.9±14.7 | 229.1±47.7 | 5.80±0.07 | 34.1 | 77.9±8.6 | 232.6±27.9 | 5.82±0.13 | 34.2 | +0.4 | −1.7 |
|
| ||||||||||||
| Probe 56 | mimics | |||||||||||
| A-U | 5′AGUGAAA | 5′UUUCACU | 59.7±6.7 | 177.7±22.6 | 4.60±0.35 | 27.3 | 56.4±3.5 | 166.3±11.9 | 4.79±0.16 | 27.7 | ||
| A-s2U | 5′AGUGAAA | 5′Us2UUCACs2U | 53.9±8.0 | 154.4±25.8 | 6.07±0.13 | 34.4 | 48.5±6.5 | 136.7±21.4 | 6.11±0.30 | 34.3 | −1.32 | 6.6 |
| A-s2U | 5′AGUGAAA | 5′s2UUs2UCACs2U | 59.4±3.8 | 169.8±12.2 | 6.70±0.06 | 37.9 | 54.5±1.7 | 153.7±5.6 | 6.77±0.03 | 38.4 | −1.98 | 10.7 |
| A-s2U | 5′AGUGAAA | 5′s2Us2Us2UCACs2U | 64.5±4.8 | 181.8±15.4 | 8.10±0.09 | 44.8 | 60.2±1.6 | 168.1±5.1 | 8.04±0.02 | 45.0 | −3.25 | 17.3 |
Solutions are 100 mM NaCl, 20 mM sodium cacodylate, and 0.5 mM Na2EDTA, pH 7.
Calculated for 10−4 M oligonucleotide concentration.
Differences due to substitution of U with s2U.
The oligonucleotides, 5′GMGMUMAMUMGMGM and 5′GMGMs2UMAMUMGMGM, were synthesized to test the effect of s2UM in another context. In this case, the duplex with the A-s2UM pair is 0.71 kcal/mol more stable than that with the A-U pair. The duplex with the G-s2UM pair is less stable by 0.40 kcal/mol relative to the duplex with a G-U pair (Table 2). Thus discrimination for A-U over G-U is enhanced by 1.1 kcal/mol at 37 °C in this context.
Preliminary microarray results demonstrated that 5′GMGMCMAMs2UMGMGM exhibits strong binding of E. coli 5S rRNA in 1Na+/4Mg2+/10T and 0.15Na+/4Mg2+/10T at 4 °C and medium binding at room temperature, which is the same pattern observed for the unmodified probe 40. The mismatched probe 5′GMGMs2UMAMUMGMGM does not bind under any of the conditions tested.
Various combinations of s2UM incorporated into probe 56, 5′UMUMUMCMAMCMUM, were also tested. Probe 56 is complementary to loop B, but did not bind in microarray experiments. To determine if this was due to the large AU content and if s2UM substitution could enhance binding, the following oligomers were synthesized: 5′UMs2UMUMCMAMCMs2UM, 5′s2UMUMs2UMCMAMCMs2UM, and 5′s2UMs2UMs2UMCMAMCMs2UM. The thermodynamics of these probes and probe 56 binding to their RNA complement, 5′AGUGAAA, were measured (Table 2).
Two 2′-O-methyl-2-thiouridines in probe 56 increase stability of the duplex by 1.32 kcal/mol at 37 °C and the Tm by 6.6 °C. Three modifications increase stability by 1.98 kcal/mol and Tm by 10.7 °C. Four s2UM nucleotides increase stability by 3.25 kcal/mol, to give a ΔG°37 for duplex formation of −8.04 kcal/mol and a melting temperature of 45.0 °C, which is 17.3 °C higher than the duplex without modifications. On average, each 2′-O-methyl-2-thiouridine stabilized this RNA/2′-O-methyl RNA duplex by 0.7 kcal/mol. Thus 2′-O-methyl-2-thiouridine stabilizes A-UM pairs in RNA/2′-O-methyl RNA duplexes in a context dependent manner.
The modified probe 56, 5′s2UMs2UMs2UMCMAMCMs2UM, was immobilized on microarrays. In buffer 1Na+/4Mg2+/10T, this probe hybridized to E. coli 5S rRNA with medium and weak strength at 4 °C and room temperature, respectively. In buffer 0.15Na+/4Mg2+/10T at 4 °C, it bound weakly. The unmodified probe did not bind under any conditions. Thus 2′-O-methyl-2-thiouridine substitutions can enhance binding to microarrays.
Improving secondary structure predictions by RNAstructure
One goal of this work is to improve secondary structure predictions by programs such as RNAstructure (13). Constraints from chemical mapping in vivo improve prediction for E. coli 5S rRNA secondary structure from 26% to 87% accuracy (13). Using constraints from the in vitro chemical probing of form A 5S rRNA from this work, the predicted lowest free energy structure has 100% of the known base pairs. This was found separately for constraints from buffer 1Na+/4Mg2+/10T at room temperature or at 4 °C, or from buffer 0.04Na+/10Mg2+/10T at room temperature. No constraints from NMIA reactivity were included in these calculations because the rules for NMIA reactivity have not been fully determined yet.
Microarray data may be used as constraints if the middle nucleotide of probes with clear binding sites is treated the same as a site of chemical modification. Data from hybridization of 5S rRNA at room temperature were chosen for this approach because of better discrimination against mismatches. Probes with alternative binding sites were omitted from constraints if the probe completely complementary to the alternative binding site also bound 5S rRNA at least as strongly as the probe in question. Exception was made for probe 39 on the basis of ribonuclease H cleavages indicating a single binding site for probe 39. The middle nucleotide of probes binding 5S rRNA with strong and medium intensity and which can bind tightly only by forming perfect duplexes were used as chemical modification constraints in predictions of secondary structure by RNAstructure 4.11 (see caption to Figure 4). For each buffer, the lowest free energy structure has 92% of the known canonical base pairs. As shown in Figure 4B, only part of helix III is predicted to be in an alternative fold. The crystal structure of 5S rRNA from H. marismortui has Watson-Crick base pairs with unusual geometries in this region (24), suggesting a pliable local structure.
FIGURE 4.

RNAstructure 4.11 prediction for E. coli 5S rRNA secondary structure: (A) without constraints, and (B) with constraints from hybridization results. Correct base pairs are bolded. Nucleotides constrained were: (buffer 1Na+/4Mg2+/10T) 27–29, 35, 36, 40, 41, 44; (buffer 0.15Na+/4Mg2+/10T) 27–29, 35, 36, 39, 40, 44; (buffer 0.04Na+/4Mg2+/10T) 27–29, 35 36, 39, 40, 41, 44, 90. All three sets of constraints gave the same predicted structure.
The structure of form B was also predicted with RNAstructure 4.11. Constraints from chemical mapping and from hybridization in buffer 1Na+/250T were used separately and together (Figure 5). In all cases, the predicted lowest free energy structure had a helix, which is also present in form A: U1-G10/C110-A119. Helix U1-G10/C110-A119 in both A and B forms is in agreement with proposed earlier structures (25, 28). Helix C35-C42/G79-G86 appears when only chemical mapping constraints are used (Figure 5A). Formation of helix G33-C42/G79-C88 (25) or G33-A39/U82-C88 (28) for form B was proposed previously. This helix is also consistent with V1 cuts in region G33-C42 (27). This secondary structure is not consistent, however, with the binding observed to oligonucleotides 36 and 90. When hybridization constraints alone or together with chemical mapping constraints are used, the structure shown in Figure 5B is predicted. This structure is predicted to be only 2 kcal/mol less favorable than the structure in Figure 5A. This suggests that Figure 5B is a structure of form B. It is possible that the two structures in Figure 5 and others are able to convert in the absence of Mg2+. Form B exhibits many more sites of chemical modifications (Figure 1) and heptamer binding (Figure 3) as compared to form A. Perhaps the density of oligonucleotide binding sites can provide insight into the rigidity of a structure.
FIGURE 5.

RNAstructure 4.11 predictions for form B of E. coli 5S rRNA in 1Na+/250T (1 M NaCl, 250 mM Tris-HCl, pH 7.8), 4 °C. (A) Using constraints from chemical maping data alone, ΔG°37 = −46.8 kcal/mol, and (B) using constraints shown from oligonucleotide binding data alone, ΔG°37 = −44.8 kcal/mol. The structure shown in (B) is also predicted when both chemical mapping and oligonucleotide binding data are used as constraints. Note that all binding oligonucleotides shown in Figure 3 are not used as constraints because some have potential alternative binding sites (Supporting Information Table S1). Symbols: ● - strong DMS; ○ - medium DMS; ■ - strong CMCT; □ - medium CMCT; ▲ - strong kethoxal; △ - medium kethoxal;
- middle site of strong binding;
- middle site of medium binding.
DISCUSSION
In this paper, probing of secondary structure by microarrays of oligonucleotides is explored as an approach to improve prediction of RNA secondary structure. Free energy minimization typically predicts correctly only about 70% of known canonical base pairs (7). One way to improve modeling of RNA secondary structure is to couple computations with experimental data. For example, enzymatic (34, 59) or chemical (34, 38, 39) mapping can reveal nucleotides that are not involved in canonical base pairs and this information can be used as constraints in computations (7, 12, 13). We show here that hybridization on microarrays with heptamer 2′-O-methyl oligoribonucleotides can provide additional experimental constraints that allow prediction of a nearly true secondary structure by a free energy minimization algorithm. In principle, microarrays can interrogate RNA secondary structure in a way that is faster and more easily automated than chemical probing, while also providing complementary information. The general method should also be applicable to systems that are not amenable to chemical probing, e.g. any system that relies on complementary, non-covalent interactions for folding, but does not have a backbone that allows readout by a polymerase of reactivity to chemicals.
The influence of RNA structure on hybridization to microarrays was first studied by Mir et al. (60). They used DNA oligonucleotides with lengths of 2 to 12 nucleotides immobilized on a glass surface and tRNAPhe as target. For probing of secondary structure, small probes have several advantages relative to longer probes. The first is better discrimination between Watson-Crick complementary binding and binding involving mismatches. The second is less possibility to disrupt secondary structure in the target. A third is that self-folding of the probe can be ignored. Moreover, there are only 16,384 possible heptamers, so microarrays containing all possible heptamers could be mass produced. Even a microarray containing all possible heptamers, hexamers, pentamers, and tetramers would have only 21,760 oligonucleotides. As demonstrated for probe 90, comparisons of relative binding by different length oligonucleotides can provide insight into secondary structure. The disadvantage of short oligonucleotides is less specificity, because long RNAs are likely to have multiple regions with the same sequence. Heptamers are expected to be ideal for probing the structures of RNAs shorter than roughly 1,000 nucleotides because on average a given seven nucleotide sequence will be present twice only 6% of the time in a 1,000 nucleotide sequence.
For this study, 2′-O-methyl RNA probes were selected because 2′-O-methyl RNA/RNA duplexes are thermodynamically more stable than DNA/RNA duplexes (51, 61, 62). Moreover, chemical synthesis of 2′-O-methylated oligoribonucleotides is efficient and inexpensive and the oligonucleotides are chemically stable. Such 2′-O-methyl RNA probes have been used in microarrays previously (61) although not for probing structure.
E. coli 5S rRNA was used to test oligonucleotide probing of secondary structure because only 27% of its canonical base pairs are predicted correctly by free energy minimization (7, 13). This is likely due to unusual stabilization of loop E by about −8.8 and −5.0 kcal/mol relative to 0.1 and 1 M Na+, respectively, when compared to 50 mM Mg2+ and 0.1 M Na+ (63). Thus E. coli 5S rRNA provides a good test of whether experimental data can compensate for incomplete knowledge of RNA thermodynamics.
Hybridization was tested in several buffers at room temperature and at 4 °C. Chemical mapping showed that folding E. coli 5S rRNA in buffers 1Na+/4Mg2+/10T, 0.15Na+/4Mg2+/10T or 0.04Na+/10Mg2+/10T gave native form A. The best condition for hybridization experiments was buffer 1Na+/4Mg2+/10T at room temperature. This condition gave both high signal intensity and good discrimination against mismatches.
Binding of A form E. coli 5S rRNA to immobilized oligonucleotides is consistent with the known secondary structure
E. coli 5S rRNA binds to microarrays very similarly in buffers 1Na+/4Mg2+/10T, 0.15Na+/4Mg2+/10T and 0.04Na+/10Mg2+/10T (Supporting Information). Differences in intensity are mostly observed. The most accessible region for binding of probes was loop C. In buffer 1Na+/4Mg2+/10T at room temperature, probes 35–38, 40, 41 and 44 hybridized to loop C (Figure 3A). Differences in strength of binding and in chemical modification at positions in the loop may come from tertiary interactions. For example, extension of helix III by three non-canonical base pairs into loop C has been proposed (41). A crystal structure of the equivalent loop in the large ribosomal subunit from H. marismortui shows that this loop has a Watson-Crick pair between C38 and G44 as well as tertiary interactions between A39 and A46 and between U40 and A45 (24). Probes probably break some tertiary interactions to allow hybridization but the free energy cost is small. Experiments with truncated versions of probe 35 showed that it had to invade helix III to bind at room temperature but not at 4 °C.
The 5′ side of loop B is the only other region giving strong binding to probes in buffer 1Na+/4Mg2+/10T (Figure 3A). Probes complementary to the 3′ side of loop B did not bind well, except for modified probe 56, 5′s2UMs2UMs2UMCMAMCMs2UM, at 4 °C. This is likely due to the high AU content and therefore weak base pairing of probes binding to the 3′ side. Published experiments and a molecular dynamics study suggest lability of the helix III/loop B region (25, 64, 65). DNA analogues of probes 28 and 29 induce ribonuclease H cleavage between nucleotides 29–31 indicating that the base pairs in that region are labile. Thus, the structure shown in Figure 4B may be similar in stability to the known structure shown in Figure 3A. In the crystal structure of the large ribosomal subunit from H. marismortui (24), the base pairs equivalent to helix C28-C30/G54-G56 have unusual geometries. Perhaps a dynamic structure in that region is important for function.
E. coli 5S rRNA also bound to probes 92 and 93. The sequence of 93, however, is identical to 38, which is complementary to loop C. Probe 92 can form six complementary base pairs with loop C, having only a single A dangling at the 3′ end. Thus binding to probes 92 and 93 is consistent with native structure A.
Other regions of 5S rRNA did not bind to probes. Lack of binding to loop E is consistent with chemical mapping (41, 66) and NMR (22) and crystal (23) structures of loop E which show non-canonical base pairs: G72-A104, U74-G102, G75-A101, G76-G100, U77-A99 and A78-G98. Thermodynamic measurements in the presence of magnesium revealed unusual stability for loop E (63). NMR results also show that Mg2+ is required for forming a structured loop E (22). As shown in Figure 1, nucleotides in the “loop E” region are more reactive during chemical mapping in buffer 1Na+/250T, without magnesium. Thus, the microarray data showing that probes do not bind to loop E in the presence of Mg2+ are consistent with its known properties.
Microarray probes 12 and 14 did not bind to the multibranch loop, although binding is predicted by the OligoWalk program. A12, U14 and A15 were accessible to chemicals, however (Figure 1). This suggests that the multibranch loop is also more stable than predicted. Thermodynamic measurements have revealed extra stability for a 5S rRNA multibranch loop with a different sequence (67).
Microarray data provide new insight into the structure of form B of E. coli 5S rRNA
Chemical mapping and hybridization to probes under conditions known to give form B of E. coli 5S rRNA gave results dramatically different from those under conditions giving form A. More regions are accessible for both chemical reaction and binding to microarray probes. Interestingly, structure prediction with constraints from microarray data alone or from both microarray and chemical mapping data suggests a secondary structure different from that generated when only chemical modification data are used as constraints (Figure 5). The presence of the strong helix U1-G10/C110-A119 in both structural models is consistent with previous suggestions, however (25, 28). While a single structure is implied by the observation of a single band on a nondenaturing gel, it is possible that the absence of Mg2+ allows this structure to be more dynamic. Comparison of the microarray data for forms A and B (Figure 3) suggests that binding to microarrays may allow identification of regions in an RNA that are particularly rigid or dynamic.
Substitution with 2-thiouridine can enhance binding and specificity of 2′-O-methyl oligonucleotides
Interpretation of microarray data is potentially confounded by binding involving mismatches. The different stabilities of complementary duplexes formed by probes with different AU and GC contents may also limit the information content of microarray data. In principle, modified nucleotides can overcome these limitations. To illustrate this, 2′-O-methyl-2-thiouridine was tested to improve binding and discrimination in base pairing. As shown in Table 2, 2′-O-methyl-2-thiouridine substitutions enhance the stability of an A-U pair by an average of 0.8 kcal/mol of substitution at 37 °C. For the two cases tested, s2UM substitution also enhanced discrimination of A-U over G-U pairs by 1.1 and 1.9 kcal/mol. Preliminary results with microarrays suggest that 2′-O-methyl-2-thiouridine is a promising modification for microarray technology.
Interpretation of binding to microarrays can provide insight into RNA structure and dynamics
Microarrays with all possible heptamers, hexamers, pentamers, and tetramers together with programs such as RNAstructure (13) can be a useful tool to deduce the secondary structure and dynamics of RNA. Microarrays rapidly interrogate oligonucleotide binding to an RNA. The bimolecular folding mode in RNAstructure can predict which probes that bind may also have more than one binding site including those with mismatches. These can still be used as constraints if the oligonucleotide fully complementary to the alternative binding site binds more weakly than the mismatched probe. Otherwise, probes with alternative sites can be omitted during initial modeling of structure or, if there are many such ambiguous probes, then ribonuclease H digestion can be used to identify actual binding sites. After such filtering, the middle nucleotide of hybridized, single binding site probes can be used as equivalent to a site of chemical modification to deduce a model for the secondary structure using RNAstructure. To be acceptable, the structure generated also has to have at least one reasonable binding site for probes that bind but have multiple potential binding sites. If probes of different lengths are included on the microarray, then binding as a function of length could be compared to the predicted structure as an additional test (15). Comparison of probe binding to loops found in the modeled structure may also provide insight into which loops are rigid and unusually stable as opposed to dynamic. For example, lack of binding to loop E of E. coli 5S rRNA in the presence of Mg2+ indicates that loop E is rigid, which is consistent with its known properties (22, 23, 63, 66). For E. coli 5S rRNA, this approach was able to provide a structure that contains 92% of known canonical base pairs, whereas free energy minimization alone predicted only 27% of the known base pairs (Figure 4).
Interpretation of oligonucleotide binding data will be facilitated in the future by the incorporation of modified nucleotides that equalize binding to unstructured RNA for all sequences, by improved understanding of the thermodynamics of binding, including the temperature dependence, and by the development of algorithms to take advantage of these advances. For example, programs such as OligoWalk (50) could be expanded to include contributions from coaxial stacking involving oligonucleotide, and could be used to automatically compare binding observed to a microarray to that predicted for all possible secondary structures generated by other algorithms (6, 7, 13, 68–72). New methods for detecting binding to microarrays eliminate the necessity for labeling target (73) and provide the possibility of further automating readout and interpretation of microarray results. The method should also provide insight into loops that provide good targets for antisense (74–77) and RNAi (78, 79) approaches to therapeutics. For example rigid loops such as loop E will not provide good targets for therapeutics composed of nucleic acids.
Acknowledgments
I. E. Catrina and D. H. Turner thank Drs. M. D. Disney and J. L. Childs for help in initiating this project. We also thank Professor Peter B. Moore for the gift of E. coli plasmid pKK-1.
Abbreviations
- HPLC
high performance liquid chromatography
- DMS
dimethyl sulfate
- CMCT
1-cyclohexyl-3-(2-morpholinoethyl)carbodiimide metho-p-toluenesulfonate
- NMIA
N-methylisatoic anhydride
Footnotes
This work was supported by NIH Grants GM2939 (to D.H.T.) and 1R03 TW1068 (R.K. and D.H.T.)
SUPPORTING INFORMATION AVAILABLE
Table with results for all tested probes. This material is available free of charge via the internet at http://pubs.asc.org.
References
- 1.Fox GE, Woese CR. 5S-RNA secondary structure. Nature. 1975;256:505–507. doi: 10.1038/256505a0. [DOI] [PubMed] [Google Scholar]
- 2.Pace NR, Thomas BC, Woese CR. Probing RNA structure, function, and history by comparative analysis. In: Gesteland RF, Cech TR, Atkins JF, editors. The RNA World. Cold Spring Harbor Press; New York: 1999. pp. 113–141. [Google Scholar]
- 3.Goertzen LR, Cannone JJ, Gutell RR, Jansen RK. ITS secondary structure derived from comparative analysis: implications for sequence alignment and phylogeny of the Asteraceae. Molecular Phylogenetics and Evolution. 2003;29:216–234. doi: 10.1016/s1055-7903(03)00094-0. [DOI] [PubMed] [Google Scholar]
- 4.Cannone JJ, Subramanian S, Schnare MN, Collett JR, D’Souza LM, Du YS, Feng B, Lin N, Madabusi LV, Muller KM, Pande N, Shang ZD, Yu N, Gutell RR. The Comparative RNA Web (CRW) Site: an online database of comparative sequence and structure information for ribosomal, intron, and other RNAs. Bmc Bioinformatics. 2002:3. doi: 10.1186/1471-2105-3-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Macke TJ, Ecker DJ, Gutell RR, Gautheret D, Case DA, Sampath R. RNAMotif, an RNA secondary structure definition and search algorithm. Nucleic Acids Res. 2001;29:4724–4735. doi: 10.1093/nar/29.22.4724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zuker M. On finding all suboptimal foldings of an RNA molecule. Science. 1989;244:48–52. doi: 10.1126/science.2468181. [DOI] [PubMed] [Google Scholar]
- 7.Mathews DH, Sabina J, Zuker M, Turner DH. Expanded sequence dependence of thermodynamic parameters improves prediction of RNA secondary structure. J Mol Biol. 1999;288:911–940. doi: 10.1006/jmbi.1999.2700. [DOI] [PubMed] [Google Scholar]
- 8.Tinoco I, Borer PN, Dengler B, Levine MD, Uhlenbeck OC, Crothers DM, Gralla J. Improved estimation of secondary structure in ribonucleic-acids. Nature-New Biology. 1973;246:40–41. doi: 10.1038/newbio246040a0. [DOI] [PubMed] [Google Scholar]
- 9.Xia TB, SantaLucia J, Burkard ME, Kierzek R, Schroeder SJ, Jiao XQ, Cox C, Turner DH. Thermodynamic parameters for an expanded nearest-neighbor model for formation of RNA duplexes with Watson-Crick base pairs. Biochemistry. 1998;37:14719–14735. doi: 10.1021/bi9809425. [DOI] [PubMed] [Google Scholar]
- 10.Xia TB, Mathews DH, Turner DH. Thermodynamics of RNA secondary structure formation. In: Soll DG, Nishimura, Moore PB, editors. Prebiotic Chemistry, Molecular Fossils, Nucleosides, and RNA. Elsevier; New York: 1999. pp. 21–48. [Google Scholar]
- 11.Turner DH. Conformational changes. In: Bloomfield VA, Crothers DM, Tinoco I, editors. Nucleic Acids. University Science Book; Sausalito, California: 2000. pp. 259–334. [Google Scholar]
- 12.Zuker M, Stiegler P. Optimal computer folding of large RNA sequences using thermodynamics and auxiliary information. Nucleic Acids Res. 1981;9:133–148. doi: 10.1093/nar/9.1.133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mathews DH, Disney MD, Childs JL, Schroeder SJ, Zuker M, Turner DH. Incorporating chemical modification constraints into a dynamic programming algorithm for prediction of RNA secondary structure. Proc Natl Acad Sci U S A. 2004;101:7287–7292. doi: 10.1073/pnas.0401799101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Uhlenbeck OC, Baller J, Doty P. Complementary oligonucleotide binding to the anticodon loop of fMet transfer RNA. Nature. 1970;225:508–510. doi: 10.1038/225508a0. [DOI] [PubMed] [Google Scholar]
- 15.Uhlenbeck OC. Complementary oligonucleotide binding to transfer RNA. J Mol Biol. 1972;65:25–41. doi: 10.1016/0022-2836(72)90489-5. [DOI] [PubMed] [Google Scholar]
- 16.Lewis JB, Doty P. Derivation of the secondary structure of 5S RNA from its binding of complementary oligonucleotides. Nature. 1970;225:510–512. doi: 10.1038/225510a0. [DOI] [PubMed] [Google Scholar]
- 17.Milner N, Mir KU, Southern EM. Selecting effective antisense reagents on combinatorial oligonucleotide arrays. Nat Biotechnol. 1997;15:537–541. doi: 10.1038/nbt0697-537. [DOI] [PubMed] [Google Scholar]
- 18.Sohail M, Akhtar S, Southern EM. The folding of large RNAs studied by hybridization to arrays of complementary oligonucleotides. RNA. 1999;5:646–655. doi: 10.1017/s1355838299982195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sohail M, Hochegger H, Klotzbucher A, Le Guellec R, Hunt T, Southern EM. Antisense oligonucleotides selected by hybridization to scanning arrays are effective reagents in vivo. Nucleic Acids Res. 2001;29:2041–2051. doi: 10.1093/nar/29.10.2041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chechetkin VR, Turygin AY, Proudnikov DY, Prokopenko DV, Kirillov EV, Mirzabekov AD. Sequencing by hybridization with the generic 6-mer oligonucleotide microarray: An advanced scheme for data processing. J Biomol Struct Dyn. 2000;18:83–101. doi: 10.1080/07391102.2000.10506649. [DOI] [PubMed] [Google Scholar]
- 21.Szymanski M, Specht T, Barciszewska MZ, Barciszewski J, Erdmann VA. 5S rRNA Data Bank. Nucleic Acids Res. 1998;26:156–159. doi: 10.1093/nar/26.1.156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dallas A, Moore PB. The loop E loop D region of Escherichia coli 5S rRNA: the solution structure reveals an unusual loop that may be important for binding ribosomal proteins. Structure. 1997;5:1639–1653. doi: 10.1016/s0969-2126(97)00311-0. [DOI] [PubMed] [Google Scholar]
- 23.Correll CC, Freeborn B, Moore PB, Steitz TA. Metals, motifs, and recognition in the crystal structure of a 5S rRNA domain. Cell. 1997;91:705–712. doi: 10.1016/s0092-8674(00)80457-2. [DOI] [PubMed] [Google Scholar]
- 24.Ban N, Nissen P, Hansen J, Moore PB, Steitz TA. The complete atomic structure of the large ribosomal subunit at 2.4 angstrom resolution. Science. 2000;289:905–920. doi: 10.1126/science.289.5481.905. [DOI] [PubMed] [Google Scholar]
- 25.Ciesiolka J, Lorenz S, Erdmann VA. Different conformational forms of Escherichia-Coli and Rat-Liver 5S Ribosomal-RNA revealed by Pb(II)-induced hydrolysis. Eur J Biochem. 1992;204:583–589. doi: 10.1111/j.1432-1033.1992.tb16671.x. [DOI] [PubMed] [Google Scholar]
- 26.Goringer HU, Wagner R. 5S RNA structure and function. Method Enzymol. 1988;164:721–747. doi: 10.1016/s0076-6879(88)64081-x. [DOI] [PubMed] [Google Scholar]
- 27.Christensen A, Mathiesen M, Peattie D, Garrett RA. Alternative conformers of 5S ribosomal-RNA and their biological relevance. Biochemistry. 1985;24:2284–2291. doi: 10.1021/bi00330a024. [DOI] [PubMed] [Google Scholar]
- 28.Weidner H, Yuan R, Crothers DM. Does 5S RNA function by a switch between two secondary structures? Nature. 1977;266:193–194. doi: 10.1038/266193a0. [DOI] [PubMed] [Google Scholar]
- 29.Caruthers MH, Beaton G, Wu JV, Wiesler W. Chemical synthesis of deoxyoligonucleotides and deoxyoligonucleotide analogs. Methods Enzymol. 1992;211:3–20. doi: 10.1016/0076-6879(92)11003-2. [DOI] [PubMed] [Google Scholar]
- 30.Matteucci MD, Caruthers MH. Nucleotide Chemistry. 1 Synthesis of oligodeoxypyrimidines on a polymer support. Tetrahedron Lett. 1980;21:719–722. [Google Scholar]
- 31.Puglisi JD, Tinoco I. Absorbency melting curves of RNA. Methods Enzymol. 1989;180:304–325. doi: 10.1016/0076-6879(89)80108-9. [DOI] [PubMed] [Google Scholar]
- 32.Moore PB, Abo S, Freeborn B, Gewirth DT, Leontis NB, Sun G. Preparation of 5S RNA-related materials for nuclear magnetic resonance and crystallography studies. Methods Enzymol. 1988;164:158–174. doi: 10.1016/s0076-6879(88)64041-9. [DOI] [PubMed] [Google Scholar]
- 33.Merino EJ, Wilkinson KA, Coughlan JL, Weeks KM. RNA structure analysis at single nucleotide resolution by selective 2′-hydroxyl acylation and primer extension (SHAPE) J Am Chem Soc. 2005;127:4223–4231. doi: 10.1021/ja043822v. [DOI] [PubMed] [Google Scholar]
- 34.Ziehler WA, Engelke DR. Probing RNA structure with chemical reagents and enzymes. Current Protocols in Nucleic Acid Chemistry. 2000;2:6.1.1–6.1.21. doi: 10.1002/0471142700.nc0601s00. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wilkinson KA, Merino EJ, Weeks KM. RNA SHAPE chemistry reveals nonhierarchical interactions dominate equilibrium structural transitions in tRNA(Asp) transcripts. J Am Chem Soc. 2005;127:4659–4667. doi: 10.1021/ja0436749. [DOI] [PubMed] [Google Scholar]
- 36.Afanassiev VHV, Wölfl S. Preparation of DNA and protein micro arrays on glass slides coated with an agarose film. Nucleic Acids Res. 2000;28:e66. doi: 10.1093/nar/28.12.e66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.McDowell JA, Turner DH. Investigation of the structural basis for thermodynamic stabilities of tandem GU mismatches: Solution structure of (rGAGGUCUC)2 by two-dimensional NMR and simulated annealing. Biochemistry. 1996;35:14077–14089. doi: 10.1021/bi9615710. [DOI] [PubMed] [Google Scholar]
- 38.Ehresmann C, Baudin F, Mougel M, Romby P, Ebel JP, Ehresmann B. Probing the structure of RNAs in solution. Nucleic Acids Res. 1987;15:9109–9128. doi: 10.1093/nar/15.22.9109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Moazed D, Stern S, Noller HF. Rapid chemical probing of conformation in 16-S ribosomal-RNA and 30-S ribosomal-subunits using primer extension. J Mol Biol. 1986;187:399–416. doi: 10.1016/0022-2836(86)90441-9. [DOI] [PubMed] [Google Scholar]
- 40.Douthwaite S, Garrett RA. Secondary structure of prokaryotic 5S ribosomal ribonucleic-acids - a study with ribonucleases. Biochemistry. 1981;20:7301–7307. doi: 10.1021/bi00528a039. [DOI] [PubMed] [Google Scholar]
- 41.Brunel C, Romby P, Westhof E, Ehresmann C, Ehresmann B. 3-Dimensional model of Escherichia-coli ribosomal 5-S RNA as deduced from structure probing in solution and computer modeling. J Mol Biol. 1991;221:293–308. doi: 10.1016/0022-2836(91)80220-o. [DOI] [PubMed] [Google Scholar]
- 42.Goringer HU, Wagner R. Does 5S RNA from Escherichia-coli have a pseudoknotted structure? Nucleic Acids Res. 1986;14:7473–7485. doi: 10.1093/nar/14.18.7473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Manning GS. Molecular theory of polyelectrolyte solutions with applications to electrostatic properties of polynucleotides. Q Rev Biophys. 1978;11:179–246. doi: 10.1017/s0033583500002031. [DOI] [PubMed] [Google Scholar]
- 44.Demarky N, Manning GS. Application of polyelectrolyte limiting laws to helix-coil transition of DNA. 3 Dependence of helix stability on excess univalent salt and on polynucleotide phosphate concentration for variable equivalent ratios of divalent metal-ion to phosphate. Biopolymers. 1975;14:1407–1422. doi: 10.1002/bip.1975.360140708. [DOI] [PubMed] [Google Scholar]
- 45.Crooke ST, Lemonidis KM, Neilson L, Griffey R, Lesnik EA, Monia BP. Kinetic characteristics of Escherichia-coli Rnase H1 - cleavage of various antisense oligonucleotide-RNA duplexes. Biochem J. 1995;312:599–608. doi: 10.1042/bj3120599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Monia BP, Lesnik EA, Gonzalez C, Lima WF, McGee D, Guinosso CJ, Kawasaki AM, Cook PD, Freier SM. Evaluation of 2′-modified oligonucleotides containing 2′-deoxy gaps as antisense inhibitors of gene-expression. J Biol Chem. 1993;268:14514–14522. [PubMed] [Google Scholar]
- 47.Jin RZ, Gaffney BL, Wang C, Jones RA, Breslauer KJ. Thermodynamics and structure of a DNA tetraplex - a spectroscopic and calorimetric study of the tetramolecular complexes of d(TG3T) and d(TG3T2G3T) Proc Natl Acad Sci U S A. 1992;89:8832–8836. doi: 10.1073/pnas.89.18.8832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sen D, Gilbert W. A sodium-potassium switch in the formation of 4-stranded G4-DNA. Nature. 1990;344:410–414. doi: 10.1038/344410a0. [DOI] [PubMed] [Google Scholar]
- 49.Sen D, Gilbert W. Formation of parallel 4-stranded complexes by guanine-rich motifs in DNA and its implications for meiosis. Nature. 1988;334:364–366. doi: 10.1038/334364a0. [DOI] [PubMed] [Google Scholar]
- 50.Mathews DH, Burkard ME, Freier SM, Wyatt JR, Turner DH. Predicting oligonucleotide affinity to nucleic acid targets. RNA. 1999;5:1458–1469. doi: 10.1017/s1355838299991148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kierzek E, Ciesielska A, Pasternak K, Mathews DH, Turner DH, Kierzek R. The influence of locked nucleic acid residues on the thermodynamic properties of 2′-O-methyl RNA/RNA heteroduplexes. Nucleic Acids Res. 2005;33:5082–5093. doi: 10.1093/nar/gki789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Walter AE, Turner DH. Sequence dependence of stability for coaxial stacking of RNA helixes with Watson-Crick base paired interfaces. Biochemistry. 1994;33:12715–12719. doi: 10.1021/bi00208a024. [DOI] [PubMed] [Google Scholar]
- 53.He L, Kierzek R, SantaLucia J, Walter AE, Turner DH. Nearest-neighbor parameters for GU mismatches - 5′GU3′/3′UG5′ is destabilizing in the contexts CGUG/GUGC, UGUA/AUGU, and AGUU/UUGU but stabilizing in GGUC/CUGG. Biochemistry. 1991;30:11124–11132. doi: 10.1021/bi00110a015. [DOI] [PubMed] [Google Scholar]
- 54.Agris PF, Sierzputowska-Gracz H, Smith W, Malkiewicz A, Sochacka E, Nawrot B. Thiolation of uridine carbon-2 restricts the motional dynamics of the transfer-RNA wobble position nucleoside. J Am Chem Soc. 1992;114:2652–2656. [Google Scholar]
- 55.Smith WS, Sierzputowska-Gracz H, Sochacka E, Malkiewicz A, Agris PF. Chemistry and structure of modified uridine dinucleosides are determined by thiolation. J Am Chem Soc. 1992;114:7989–7997. [Google Scholar]
- 56.Kumar RK, Davis DR. Synthesis and studies on the effect of 2-thiouridine and 4-thiouridine on sugar conformation and RNA duplex stability. Nucleic Acids Res. 1997;25:1272–1280. doi: 10.1093/nar/25.6.1272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Testa SM, Disney MD, Turner DH, Kierzek R. Thermodynamics of RNA-RNA duplexes with 2-or 4-thiouridines: Implications for antisense design and targeting a group I intron. Biochemistry. 1999;38:16655–16662. doi: 10.1021/bi991187d. [DOI] [PubMed] [Google Scholar]
- 58.Okamoto I, Shohda K, Seio K, Sekine M. A new route to 2′-O-alkyl-2-thiouridine derivatives via 4-O-protection of the uracil base and hybridization properties of oligonucleotides incorporating these modified nucleoside derivatives. J Org Chem. 2003;68:9971–9982. doi: 10.1021/jo035246b. [DOI] [PubMed] [Google Scholar]
- 59.Knapp G. Enzymatic approaches to probing of RNA secondary and tertiary structure. Methods Enzymol. 1989;180:192–212. doi: 10.1016/0076-6879(89)80102-8. [DOI] [PubMed] [Google Scholar]
- 60.Mir KU, Southern EM. Determining the influence of structure on hybridization using oligonucleotide arrays. Nat Biotechnol. 1999;17:788–792. doi: 10.1038/11732. [DOI] [PubMed] [Google Scholar]
- 61.Majlessi M, Nelson NC, Becker MM. Advantages of 2′-O-methyl oligoribonucleotide probes for detecting RNA targets. Nucleic Acids Res. 1998;26:2224–2229. doi: 10.1093/nar/26.9.2224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Sugimoto N, Nakano S, Katoh M, Matsumura A, Nakamuta H, Ohmichi T, Yoneyama M, Sasaki M. Thermodynamic parameters to predict stability of RNA/DNA hybrid duplexes. Biochemistry. 1995;34:11211–11216. doi: 10.1021/bi00035a029. [DOI] [PubMed] [Google Scholar]
- 63.Serra MJ, Baird JD, Dale T, Fey BL, Retatagos K, Westhof E. Effects of magnesium ions on the stabilization of RNA oligomers of defined structures. RNA. 2002;8:307–323. doi: 10.1017/s1355838202024226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Gabashvili IS, Whirl-Carrillo M, Bada M, Banatao DR, Altman RB. Ribosomal dynamics inferred from variations in experimental measurements. RNA. 2003;9:1301–1307. doi: 10.1261/rna.5141503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Pieler T, Digweed M, Erdmann VA. RNA structural dynamics - pre-melting and melting transitions in Escherichia-coli 5S ribosomal-RNA. J Biomol Struct Dyn. 1985;3:495–514. doi: 10.1080/07391102.1985.10508437. [DOI] [PubMed] [Google Scholar]
- 66.Leontis NB, Westhof E. The 5S rRNA loop E: Chemical probing and phylogenetic data versus crystal structure. RNA. 1998;4:1134–1153. doi: 10.1017/s1355838298980566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Diamond JM, Turner DH, Mathews DH. Thermodynamics of three-way multibranch loops in RNA. Biochemistry. 2001;40:6971–6981. doi: 10.1021/bi0029548. [DOI] [PubMed] [Google Scholar]
- 68.Wuchty S, Fontana W, Hofacker IL, Schuster P. Complete suboptimal folding of RNA and the stability of secondary structures. Biopolymers. 1999;49:145–165. doi: 10.1002/(SICI)1097-0282(199902)49:2<145::AID-BIP4>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- 69.Gultyaev AP, Vanbatenburg FHD, Pleij CWA. The computer-simulation of RNA folding pathways using a genetic algorithm. J Mol Biol. 1995;250:37–51. doi: 10.1006/jmbi.1995.0356. [DOI] [PubMed] [Google Scholar]
- 70.Rivas E, Eddy SR. A dynamic programming algorithm for RNA structure prediction including pseudoknots. J Mol Biol. 1999;285:2053–2068. doi: 10.1006/jmbi.1998.2436. [DOI] [PubMed] [Google Scholar]
- 71.Ding Y, Lawrence CE. A statistical sampling algorithm for RNA secondary structure prediction. Nucleic Acids Res. 2003;31:7280–7301. doi: 10.1093/nar/gkg938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Dirks RM, Pierce NA. A partition function algorithm for nucleic acid secondary structure including pseudoknots. J Comput Chem. 2003;24:1664–1677. doi: 10.1002/jcc.10296. [DOI] [PubMed] [Google Scholar]
- 73.Lu JH, Strohsahl CM, Miller BL, Rothberg LJ. Reflective interferometric detection of label-free oligonucleotides. Anal Chem. 2004;76:4416–4420. doi: 10.1021/ac0499165. [DOI] [PubMed] [Google Scholar]
- 74.Stephenson ML, Zamecnik PC. Inhibition of Rous-Sarcoma viral-RNA translation by a specific oligodeoxyribonucleotide. Proc Natl Acad Sci U S A. 1978;75:285–288. doi: 10.1073/pnas.75.1.285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Zamecnik PC, Stephenson ML. Inhibition of Rous-Sarcoma virus-replication and cell transformation by a specific oligodeoxynucleotide. Proc Natl Acad Sci U S A. 1978;75:280–284. doi: 10.1073/pnas.75.1.280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Agrawal S, Mayrand SH, Zamecnik PC, Pederson T. Site-specific excision from RNA by Rnase-H and mixed-phosphate-backbone oligodeoxynucleotides. Proc Natl Acad Sci U S A. 1990;87:1401–1405. doi: 10.1073/pnas.87.4.1401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Agrawal S, Jiang ZW, Zhao QY, Shaw D, Cai QY, Roskey A, Channavajjala L, Saxinger C, Zhang RW. Mixed-backbone oligonucleotides as second generation antisense oligonucleotides: In vitro and in vivo studies. Proc Natl Acad Sci U S A. 1997;94:2620–2625. doi: 10.1073/pnas.94.6.2620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Schwarz DS, Hutvagner G, Du T, Xu ZS, Aronin N, Zamore PD. Asymmetry in the assembly of the RNAi enzyme complex. Cell. 2003;115:199–208. doi: 10.1016/s0092-8674(03)00759-1. [DOI] [PubMed] [Google Scholar]
- 79.Khvorova A, Reynolds A, Jayasena SD. Functional siRNAs and rniRNAs exhibit strand bias. Cell. 2003;115:209–216. doi: 10.1016/s0092-8674(03)00801-8. [DOI] [PubMed] [Google Scholar]