

Abstract
Multiple factors are involved in the etiology of cardiovascular disease (CVD). Pathological changes occur in a variety of cell types long before symptoms become apparent and diagnosis is made. Dysregulation of physiological functions are associated with the activation of immune cells, leading to local and finally systemic inflammation that is characterized by production of high levels of reactive oxygen species (ROS). Patients suffering from inflammatory diseases often present with diminished levels of antioxidants either due to insufficient dietary intake or, and even more likely, due to increased demand in situations of overwhelming ROS production by activated immune effector cells like macrophages. Antioxidants are suggested to beneficially interfere with diseases-related oxidative stress, however the interplay of endogenous and exogenous antioxidants with the overall redox system is complex. Moreover, molecular mechanisms underlying oxidative stress in CVD are not fully elucidated. Metabolic dybalances are suggested to play a major role in disease onset and progression. Several central signaling pathways involved in the regulation of immunological, metabolic and endothelial function are regulated in a redox-sensitive manner. During cellular immune response, interferon γ-dependent pathways are activated such as tryptophan breakdown by the enzyme indoleamine 2,3-dioxygenase (IDO) in monocyte-derived macrophages, fibroblasts, endothelial and epithelial cells. Neopterin, a marker of oxidative stress and immune activation is produced by GTP-cyclohydrolase I in macrophages and dendritic cells. Nitric oxide synthase (NOS) is induced in several cell types to generate nitric oxide (NO). NO, despite its low reactivity, is a potent antioxidant involved in the regulation of the vasomotor tone and of immunomodulatory signaling pathways. NO inhibits the expression and function of IDO. Function of NOS requires the cofactor tetrahydrobiopterin (BH4), which is produced in humans primarily by fibroblasts and endothelial cells. Highly toxic peroxynitrite (ONOO-) is formed solely in the presence of superoxide anion (O2-). Neopterin and kynurenine to tryptophan ratio (Kyn/Trp), as an estimate of IDO enzyme activity, are robust markers of immune activation in vitro and in vivo. Both these diagnostic parameters are able to predict cardiovascular and overall mortality in patients at risk. Likewise, a significant association exists between increase of neopterin concentrations and Kyn/Trp ratio values and the lowering of plasma levels of vitamin-C, -E and -B. Vitamin-B deficiency is usually accompanied by increased plasma homoycsteine. Additional determination of NO metabolites, BH4 and plasma antioxidants in patients with CVD and related clinical settings can be helpful to improve the understanding of redox-regulation in health and disease and might provide a rationale for potential antioxidant therapies in CVD.
Keywords: Atherogenesis, Cardiovascular disease, Neopterin, Nitric oxide, Tetrahydrobiopterin, Tryptophan, Oxidative stress, Homocysteine, Vitamins, Antioxidative therapy
Core tip: Crosstalk between a number of pathways involved in the regulation of immune and endothelial homeostasis is strongly coordinated by redox processes. Underlying molecular mechanisms of atherogenesis include metabolic imbalances that are linked to the onset and progression of endothelial dysfunction and inflammation, finally leading to a status of heightened oxidative stress. Decrease of plasma antioxidants may develop secondarily due to an increased demand for oxidation-sensitive vitamins during inflammation. Antioxidant and vitamin supplementation therapy is controversially discussed and success might depend of an individual patient’s demand.
INTRODUCTION
Despite the availability of successful treatment strategies for dyslipidemia and hypertension, cardiovascular diseases (CVD) account for one third of all deaths worldwide, and prevalence still increases[1,2].
CVD comprise a class of diseases that involve heart and systemic blood vessels[3]. In coronary heart disease, cerebrovascular disease or peripheral arterial disease, impaired blood vessel function leads to an inadequate blood supply of organs. Deep vein thrombosis and pulmonary embolism are usually caused by blood clots in the leg veins.
Avoiding risk factors such as smoking, obesogenic lifestyle, e.g., unhealthy diet, physical inactivity, high blood pressure, diabetes and dyslipidemia, is strongly recommended for disease prevention. Nevertheless, beside lifestyle, genetic, epigenetic and environmental factors may essentially influence the risk of CVD.
The multifactorial background makes it difficult to unravel initial pathological events, which are suggested to occur in a very early phase of disease, where symptoms are subclinical. Inflammation is considered to play a key role in both disease initiation and progression[4]. Chronic inflammatory conditions attenuate endogenous antioxidant capacities due to continuous production of high levels of reactive oxygen species (ROS). Patients often represent with low blood levels of antioxidants[5] and enhanced oxidative stress markers[6]. This is usually due to increased demand in situations of overwhelming ROS production by activated immune effector cells like macrophages. Also insufficient nutritional intake may play a role. Uptake of exogenous antioxidants is suggested to beneficially interfere with diseases-related oxidative stress, however the interplay of endogenous and exogenous antioxidants with the overall redox system is complex.
The object of this review is to give an overview on immunobiochemical pathways activated in atherogenesis, which lead to oxidative stress-related pathological consequences. Understanding of the mechanisms will be helpful in the establishment of new preventive and therapeutic strategies.
MAIN FEATURES OF ATHEROGENESIS
Atherosclerosis is the most common pathological process that leads to CVD including myocardial infarction (MI), heart failure, stroke and claudication. A central event is the development of atherosclerotic plaques in the inner lining of arteries. Irritative inflammatory stimuli, hypertension, hyperglycemia and dyslipidaemia cause endothelial stress leading to expression of adhesion molecules and recruitment of leukocytes[7].
Atherosclerotic plaques are characterized by necrotic cores, calcification, accumulation of modified lipids and foam cells, but also other cell types such as smooth muscle cells, vascular dendritic cells, T cells and endothelial cells are involved in lesion formation[8]. The “oxidative modification hypothesis” of atherogenesis implies that low-density lipoprotein (LDL) oxidation is an early event in atherosclerosis[9]. Cholesterol-containing LDL particles are retained in the artery wall and biochemically modified components of these particles in turn induce leukocyte adhesion but also intracellular cholesterol accumulation in invaded macrophages[10]. Chronic inflammatory conditions are maintained due to the production of pro-inflammatory mediators through immune competent cells in the lesions[11]. Activation of macrophages is a key factor in atherosclerotic plaque formation, fibrous cap disruption and thrombus formation.
While in the past atherosclerosis was viewed primarily as passive process of cholesterol accumulation, recent evidence indicates that it is a highly active process involving components of the vascular, immune, metabolic and endocrine system[12]. Initial pathological changes occur in a variety of cell types long before symptoms become apparent and diagnosis is made[13,14]. Of note, also in a large sample of cardiovascular disease-free adults, Chrysohoou et al[15] revealed an association of pre-hypertension with reduced serum antioxidant capacity and increased oxidized LDL probably indicating initial pathological changes.
Atherosclerotic plaque composition, endothelial erosion, intraplaque hemorrhage, adventitial and intraplaque neovascularization, rather than the percentage of stenosis, turned out to be critical predictors for both risk of plaque rupture and subsequent thrombogenicity[2,16,17]. Disruption of a vulnerable or unstable plaque may lead to a complete occlusion, to plaque progression or result in an acute coronary syndrome, i.e., acute MI (AMI), unstable angina and sudden cardiac death or stroke in case of carotid plaque destabilization.
OXIDATIVE STRESS AND IMMUNE ACTIVATION
Although substantial efforts have been made to dissect molecular details of atherogenesis, a full understanding of the underlying mechanisms is still missing. However, activation of immune competent cells, leading to local and finally systemic inflammatory phenomena and the associated status of heightened oxidative stress are central events[4].
Monocyte/macrophage accumulation at the lesion is a key factor in the pathology and involves several steps, such as monocyte recruitment by expression of adhesion molecules and chemotactic factors, induction of activation and differentiation processes as well as proliferation, and immobilization of macrophages in the inflamed plaque[18]. Current data indicate that due to the presence of variable differentiation stimuli, different macrophage populations reside in the atherosclerotic plaque[19]. Both M1 and M2 macrophages are present in atherosclerotic regions. Macrophage colony-stimulating factor (M-CSF), which is continuously present in circulation, induces predominantly M2-type macrophages with increased phagocytic activity, characterized by expression of interleukin (IL)-10, scavenger receptor A and mannose receptor[18,19]. Granulocyte-macrophage CSF (GM-CSF) induces M1-polarized cells with antigen presentation capacities, which express tumor necrosis factor alpha (TNFα) and pro-inflammatory cytokines such as IL-1β, IL-6, IL-8 and IL-12 upon stimulation with interferon gamma (IFN-γ) or lipopolysaccharides (LPS)[18,20]. While M1 macrophages were found to predominate in rupture-prone plaque regions, M2-type are located in the vascular adventitial tissue[21]. However, also other macrophage phenotypes are suggested to contribute to the inflammatory process in atherosclerosis, such as the recently described platelet chemokine CXCL4-induced M4 cells[22].
Immune reactions in atherosclerotic lesions are mainly T helper (Th1) type responses, as indicated by the dominance of pro-inflammatory and macrophage-stimulating cytokines found in advanced plaques[11,23,24]. During Th1-type response, IFN-γ is probably the most important trigger for high ROS production in macrophages[25] by phagocytic NADPH oxidase (NOX)[26]. Main reactive species are hydrogen peroxide (H2O2), superoxide anion (O2-), but also reactive nitrogen species such as peroxynitrite (ONOO-), nitrogen dioxide and trioxide[27]. IFN-γ signaling initiates a variety of cellular defense mechanisms such as pro-inflammatory cytokine production via nuclear factor kappa B (NF-κB) signaling, enhancement of antigen presentation[28] and other important pathways, e.g., neopterin formation via guanosine triphosphate (GTP)-cyclohydrolase I (GTP-CH-I) and indoleamine 2,3-dioxygenase (IDO)-mediated tryptophan breakdown[29] (Figure 1).
Figure 1.
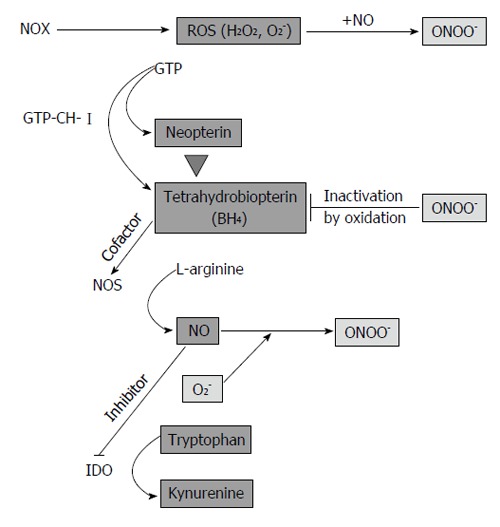
Regulatory circuits in inflammation and endothelial dysfunction. During inflammation, NADPH oxidase (NOX) produces high levels of reactive oxygen species (ROS). T cells and natural killer cells produce interferon-γ, which activates enzyme GTP-cyclohydrolase I (GTP-CH-I), indoleamine 2,3-dioxygenase (IDO) and inducible nitric oxide synthase (iNOS) in monocyte-derived macrophages (M) and dendritic cells (DC). In endothelial cells, endothelial NOS (eNOS) is constitutively expressed and GTP-CH-I produces tetrahydrobioterin (BH4), which is a NOS cofactor. BH4 deficiency leads to NOS uncoupling and superoxide anion (O2-) formation, which reacts with NO to form peroxynitrite (ONOO-). In a vicious cycle, ONOO- oxidizes BH4. In M/DC, GTP-CH-I synthesizes neopterin at expense of BH4, which contributes to the low activity of iNOS in human M/DC. Furthermore, NO is a reversible inhibitor of the immunoregulatory enzyme IDO. IDO degrades the essential amino acid tryptophan to kynurenine.
Under normal conditions, low levels of ROS are mainly byproducts from electron transport chain reactions in the mitochondria[30]. They are important regulators of several redox-sensitive pathways involved in the maintenance of cellular homeostasis[31], and act by modifying molecules, enzymes and transcription factors as well by interfering with the endogenous antioxidant pool[27,31,32]. Depletion of endogenous redox buffer systems in conditions with overwhelming oxidative stress is critical, not only due to triggering of immune responses but also through leading to endothelial and smooth muscle dysfunction, and thus to the progression of atherosclerosis[33,34].
ROLE OF LIPOPROTEINS IN ATHEROSCLEROSIS
Proatherogenic oxidized LDL (oxLDL) accumulates in the vascular wall and contributes to the pathogenesis of vascular dysfunction early in the development of atherosclerosis. After incorporation via scavenger receptors of macrophages, oxLDL leads to their transformation into foam cells[35]. Foam cells accumulate a variety of lipids in droplets in the cytoplasm and secrete extracellular matrix proteins that further support the retention of lipoproteins and attraction of immune cells, thus leading to an enlargement of the lesion[10].
Oxidation of LDL is considered to occur primarily in the vascular wall[36], but also circulating oxLDL was detected in CVD patients[37]. Although the amount of circulating oxLDL is small compared to oxLDL present in vessels[38], enhanced serum levels of oxLDL are predictive for endothelial dysfunction and coronary heart disease[36-39]. The role of oxLDL as a relevant pro-atherogenic marker is further supported by the study of Meisinger et al[40], who found elevated oxLDL to be predictive for future coronary heart disease events in apparently healthy men. Oxidation of LDL contributes to the prooxidant environment in atherosclerotic lesions. OxLDL is a potent stimulus of vascular ROS formation, mainly through activation of NOX and uncoupling of endothelial nitric oxide (NO)-synthase (NOS) that promotes endothelial dysfunction[36].
High-density lipoprotein (HDL) is another potential biomarker with anti-atherogenic properties due to its function in the reverse cholesterol transport and in decreasing lipoprotein oxidation[41]. HDL is involved in several biological processes that counteract inflammation and oxidative stress, by beneficially influencing, e.g., pancreatic beta-cell function, endothelial vasoreactivity, endothelial apoptosis, restorative processes and monocyte activation as well as adhesion molecules expression, thus being highly vasculoprotective[42]. Paraoxonase-1, a calcium dependent enzyme, is located at the surface of HDL particles and contributes to the antioxidant and anti-inflammatory role of HDL[43]. In particular, HDL-associated paraoxonase was shown to inhibit the formation of “minimally oxidized” LDL[44]. Nevertheless, also other mechanisms are suggested to be involved in HDL-associated inhibition of LDL oxidation[45].
Plasma HDL cholesterol (HDL-C) levels are inversely associated with CVD risk in preclinical and large epidemiologic studies. Low HDL-C level was identified as a robust predictor of lipid peroxidation irrespective of gender, age, obesity and inflammatory or metabolic biomarkers in the Styrian Juvenile Obesity/ Early DEteCTion of Atherosclerosis study employing 797 participants aged from 5 to 50 years[46]. However, HDL is highly heterogeneous and the atheroprotective functions of the different HDL subpopulations are not completely understood. Furthermore, current data indicate that therapeutically increased HDL-C levels per se do not always correlate with enhanced HDL functions in vivo[47,48].
Of note, accumulation of free, unesterified cholesterol can lead to crystal formation both in vitro and in vivo[49]. Crystalized cholesterol in atherosclerotic plaques was shown to activate the NLR family, pyrin domain containing 3 (NLRP3) inflammasome complex by employing the complement system, thereby leading to the release of proinflammatory cytokine IL-1β[50,51]. Cholesterol crystals were mainly found in advanced plaques, however the inflammatory responses caused by NLRP3 inflammasome activation might represent an important trigger in disease progression and could thus represent an important pharmaceutical target[52].
NEOPTERIN FORMATION
Neopterin, a marker of immune system activation, is produced by GTP-CH-I in macrophages and dendritic cells (DC)[53,54] and has emerged as an important independent and predictive marker in cardiovascular risk assessment[6]. IFN-γ is the major stimulus for neopterin formation. Other cytokines have only limited stimulatory potential in vitro but some, e.g., TNFα, can indirectly enhance IFN-γ induced neopterin formation[55]. Of note, also pro-inflammatory compounds like LPS can elevate neopterin levels[55]. The amount of neopterin secreted by human macrophages correlates with their ROS-generation capacity in vitro[56] and neopterin concentration in body fluids is considered as an indicator for immune activation-associated oxidative stress[57].
GTP-CH-1 catalyzes the conversion of guanosine triphosphate (GTP) to 7,8-dihydroneopterin triphosphate finally leading to the formation of neopterin, 7,8-dihydroneopterin and 5,6,7,8-tetrahydrobiopterin (BH4)[57]. Human monocyte-derived macrophages and DCs are the most important source of neopterin and its partially reduced derivative 7,8-dihydroneopterin, both present in relative constant ratio in human serum[57], but not of BH4, due to the relative deficiency of pyruvoyl-tetrahydropterin synthase in this cell types[58] (Figure 1). In contrast, cells from other animal species and other human cell types such as endothelial cells or fibroblasts preferentially produce BH4, which is needed as a cofactor by several monooxygenases including NOS, phenylalanine hydroxylase or tyrosine hydroxylase[59].
Elevated neopterin concentrations were reported to be associated with chronic immune activation in several diseases such as viral, bacterial and parasite infections, autoimmune or malignant tumor diseases and during rejection episodes in allograft recipients[60-63]. Also patients with CVD present with increased neopterin concentrations, supporting the crucial involvement of chronic immune activation, in particular of macrophages, in atherogenesis. Several studies (Table 1) strengthened the impact of neopterin as an independent marker for CVD, with predictive value for coronary artery disease (CAD) progression[6].
Table 1.
Selected studies investigating neopterin concentrations in cardiovascular disease patients
| Ref. | Condition | n | Result |
| Melichar et al[64], 1994 | AMI | 13 | Increased urinary neopterin |
| Anwaar et al[65], 1999 | Acute cerebral ischemia or transient ischemic attack, 1-yr follow-up | 59 (57) | Increase of plasma neopterin after acute cerebral ischemia |
| Tatzber et al[66], 1991 | Different clinical stages of atherosclerosis | 61 | Elevated plasma neopterin in about 50% of hospitalized patients undergoing conservative or surgical therapy, higher neopterin levels were overrepresented in patients with higher Frederickson type |
| Weiss et al[67], 1994 | Cross-sectional community-based screening study (Ischemic Heart Disease and Stroke Prevention Study, Bruneck, Italy) | 561 (total) | Serum neopterin correlated with the extent of carotid atherosclerosis |
| Schumacher et al[68], 1997 | AMI Stable CAD | 21 62 | Neopterin levels were highest in AMI patients but also elevated in those with CAD |
| Gurfinkel et al[69], 1999 | Unstable angina pectoris (non-Q-wave AMI) | 52 (26) | Serum neopterin correlated with score of atherosclerotic extension (angiography) |
| Zouridakis et al[70], 2004 | Chronic stable angina pectoris | 124 | CAD progression correlated with increased neopterin and high-sensitivity C-reactive protein as well endothelial activation markers |
| Avanzas et al[71], 2005 | Patients with chronic stable chest pain undergoing diagnostic coronary angiography, 1-yr follow-up | 297 | Elevated serum neopterin correlated with adverse coronary events during follow-up (17.2%) |
| Kaski et al[72], 2008 | NSTE ACS (unstable angina and NSTE MI), 6-mo follow-up | 397 (147,250) | Baseline neopterin in unstable angina and NSTE MI comparable, increased neopterin was associated with adverse cardiac events |
| Johnston et al[73], 2006 | ACS (treatments: medication, uncoated or rapamycin-eluting coronary stents) and stable CAD | 70 (35, 25, 10) 36 | Serum neopterin correlated with thrombolysis in myocardial infarction; mean changes in serum neopterin higher in uncoated stent group |
| Barani et al[74], 2006 | Critical limb ischemia, 1-yr follow-up | 232 | Neopterin was elevated in patients with atrial fibrillation or flutter and with ischemic electrocardiogram changes which were at risk for adverse cardiac events |
| Ray et al[75], 2007 | ACS (PROVE IT–TIMI 22) 2-yr follow-up | 3946 | Increased neopterin was associated with increased risk of death and of acute coronary events after ACS |
NSTE: Non-ST-segment elevation; PROVE IT-TIMI: PRavastatin or atorVastatin Evaluation Infection Therapy-Thrombolysis In Myocardial Infarction; AMI: Acute myocardial infarction; MI: Myocardial infarciationnon; CAD: Coronary artery disease; ACS: Acute coronary syndrome. Table adapted and extended from Fuchs et al[6].
Of note, neopterin-positive macrophages were found in coronary atherectomy specimens from patients with stable angina pectoris and to a lesser extent in those with unstable angina pectoris, and the number of these macrophages correlated with T cell and neutrophil count in the lesions[76]. Furthermore, neopterin was shown to induce an atherothrombotic phenotype in human coronary endothelial cells in vitro by promoting cellular adhesion molecules (intercellular adhesion molecule 1 and vascular cell adhesion molecule 1) and tissue factor (TF) expression mediated by activation of NF-κB[77]. These data suggest that neopterin is not only associated with the systemic inflammation process in atherosclerosis, but might also be of importance for the inflammatory process within the plaque and thus for plaque destabilisation[6,76].
Neopterin concentrations correlate with IFN-γ-induced ROS production[56]. In addition, neopterin has pro-oxidant properties itself by intensifying the effects of ROS as well as of reactive chlorine and nitrogen species[78]. Of note, Herpfer et al[79] showed that neopterin is able to enhance ONOO- as well as Cu(II)-mediated LDL oxidation, whereas 7,8-dihydroneopterin may protect LDL from oxidation under certain conditions[79,80]. Neopterin may also enhance the effects of ONOO- in the processes of protein nitration[81]. This pro-oxidant property of neopterin indicates a potential involvement in the antimicrobial and antitumoral action of macrophages[82]. The property of neopterin to interfere with and enhance the effects of various ROS might be of central relevance also in atherogenesis.
Inflammation-associated oxidative stress may lead to a rapid consumption of circulating antioxidants. In patients with CAD, higher neopterin concentrations were associated with a decline in levels of several antioxidant compounds and vitamins such as ascorbic acid, α-tocopherol, lycopene, lutein and zeaxanthin[5].
TRYPTOPHAN BREAKDOWN
In parallel to neopterin formation, other IFN-γ-dependent pathways are activated during cellular immune response such as tryptophan breakdown by IDO. IDO catalyzes the rate-limiting step in the conversion of tryptophan (Trp) and other indole derivates to kynurenine (Kyn)[83] and is induced in monocyte-derived macrophages but also in fibroblast, endothelial and epithelial cells[84,85] (Figure 1). Both expression and activity of the haeme-containing enzyme IDO is sensible to redox-regulation and IDO enzyme itself can exert antioxidant activity by scavenging of O2-[86,87]. The estimation of Kyn to Trp ratio (Kyn/Trp), expressed as μmol kynurenine per mmol tryptophan, can be used as measure of IDO enzyme activity both in vitro and in vivo[60,88]. Simultaneous measurement of immune activation markers such as neopterin, IFN-γ or soluble interleukin receptors, allow to relate circulating Trp levels with inflammation-induced IDO activity, as also hepatic tryptophan 2,3-dioxygenase (TDO) could degrade Trp. TDO, however, is regulated via tryptophan content and steroid hormones such as glucocorticoids[89,90], while IDO is strongly induced in response to several pro-inflammatory stimuli such as IFN-γ, TNFα or LPS[55,85].
Depletion of the essential amino acid Trp contributes to the development of an antiproliferative environment and represents an effective antimicrobial and antitumoral strategy[91]. Also T cell proliferation depends on Trp availability, thus IDO activation is a metabolic checkpoint of immunoregulation[92]. IDO activity is crucially involved in the control of T cell proliferation and in the generation of regulatory T cells, and thus in the suppression of autoimmune responses and promotion of tolerance[92,93].
Metabolic control by reduction of Trp levels may slow down hematopoiesis in addition to other proinflammatory stimuli by affecting the growth and differentiation of erythroid progenitor cells. In line with this, in patients with inflammation-induced anemia, Kyn/Trp was found to inversely correlate with haemoglobin levels[94,95].
Accelerated Trp breakdown was reported in patients with coronary heart disease[96] and IDO activity correlated significantly with several risk factors for atherosclerosis in the Cardiovascular Risk in Young Finns Study[97]. Niinsalo et al[98] reported that IDO activity positively correlated with carotid artery intima/media thickness, an early marker of atherosclerosis, although this association did not remain significant after adjustment with classical risk factors in this patient group.
In inflammatory diseases including CVD, a concurrent increase of neopterin production and tryptophan degradation is usually observed. The prognostic ability of neopterin is likely to relate to the association with IFN-γ in the atherogenic process[6]. IDO-mediated tryptophan breakdown is suggested to be responsible for several additional aspects observed during disease progression[29], e.g., the development of depression. Because tryptophan is a precursor for the biosynthesis of serotonin, the lowered tryptophan availability under inflammatory conditions may limit serotonin formation and thus enhance the susceptibility for lowered mood and depression[99]. Of note, development of depressive symptoms have been associated with increased CAD risk and poor prognosis[100]. Estimation of Trp breakdown rate could contribute to a better understanding of the interplay between inflammation, metabolic syndrome, mood disturbance, and anemia, all previously described as significant predictors of an unfavorable outcome in patients with CVD[101].
NITRIC OXIDE
NOS converts L-arginine into citrulline, thereby synthesizing NO. Free NO can migrate through cell membranes by diffusion, and although it relatively low reactivity, NO is a potent antioxidant molecule that can protect from ROS damage[102]. However, NO is a free radical, and can undergo oxidation to nitrite and nitrate, react with O2- to form ONOO-, or bind to transition metals[103]. NO signaling is strongly concentration dependent and although endogenous NO is essentially involved in many physiological processes and beneficial in a variety of circumstances, its reaction products may mediate nitrosative and oxidative stress. However, NO products can have also protective effects. In plasma, NO circulates primarily complexed in S-nitrosothiol species[104] that are suggested to be a transport and buffer system that controls intercellular NO exchange. S-nitrosylation of the proteome is a unique form of posttranslational modification that can have significant consequences for protein function and cell phenotype. In particular in the cardiovascular system, S-nitrosothiols were shown to exert many actions, including promoting vasodilation, inhibiting platelet aggregation, and regulating Ca(2+) channel function of myocytes[105]. The impact of S-nitroso but also N-nitroso protein formation on the reduction of free NO under inflammatory conditions in vivo has still to be investigated[106,107].
Endothelial and neuronal NOS are constitutively expressed and produce NO at low concentrations, while inducible NOS is activated, e.g., in macrophages of several species in response to pro-inflammatory stimuli giving rise to higher NO output[108]. Endothelial dysfunction, e.g., vasodilation and/or platetelet inhibition, a key feature of early atherosclerosis, is associated with the reduced availability of endothelium-derived NO[109]. Defects in NO production, metabolism and response have been described to be responsible mechanisms.
In the presence of O2-, ONOO- formation may be a factor that limits NO bioavailability. Beside being strongly vasoconstrictory, ONOO- has been shown to oxidize the NOS cofactor BH4, thereby leading to eNOS uncoupling and O2- production[110], thus starting a vicious cycle (Figure 1). Reduced vascular BH4 levels were found in rat and mice models of atherosclerosis and diabetes[111].
High NO output and generation of reactive nitrogen species via iNOS contribute to cytocidal defense strategies in inflammation. However, although this has been reported for several species, including mice, until now, large output of NO by iNOS could not be equally demonstrated in human macrophages[112,113]. Human macrophages produce neopterin at the expense of BH4, and low BH4 leads to NOS enzyme uncoulpling. Furthermore, the pro-oxidant properties of neopterin may compensate for deficient NO and ONOO- production[114].
Of note, NO inhibits IDO expression and function by reversibly binding to the active site heme[115]. Induction of IDO and NOS in IFN-γ-mediated inflammatory response is suggested to be functionally cross-regulated[116]. The absence of NO-mediated immunoregulation may support enhanced IDO activity at the site of inflammation.
POTENTIAL ROLE OF TH2 RESPONSES IN CVD
Th1 responses are in general proinflammatory and known to be proatherogenic, while Th2 cells are usually involved in helmintic and allergic responses. The role of Th2 cells in atherosclerosis seems to be very complex and even contradictory. A potential protective role of Th2 response is discussed in few studies[117,118], while Ait-Oufella et al[24] assume a potential proatherogenic function of Th2 cells within the complex interaction theater of CD4+ T cell subsets in atherosclerosis. Thus, the exact role of the Th2 response remains to be elucidated based on an improved understanding of the complex interplay between Th1, Th2, and other T cell populations such as Th17 and Tregs within the atherosclerotic scenario[18,24]. Overall, Th cell subset polarization in atherosclerosis is less distinct in humans compared to mice[119].
High cholesterol diet of ApoE(-/-) mice with different T cell subset polarization resulted in increased development of atherosclerosis in the aortic root and abdominal aorta in mice with predominantly Th1-like immune responses [ApoE(-/-) BL/6 mice] in comparison to animals with Th2 predominance [ApoE(-/-) BALB/c][120]. A potential of IL-4 to limit Th1 cell responses and reducing lesion size was observed in several murine atherosclerotic models[121,122].
Only recently, Engelbertsen et al[123] reported an association between Th2 immunity and reduced risk of MI, as high numbers of Th2 cells were associated with decreased mean common carotid intima-media thickness, reduced risk of AMI in women and IL-4 was independently associated with reduced risk of CVD. Although some limitations, as, e.g., differences in lymphocyte number between healthy man and women or the use of long-term cryo-conserved cells, this study provides first hints for the clinical importance of an improved understanding of Th2-type responses in CVD. However, again in contrast to these positive, protective attributes, Shimizu et al[124] suggested a role for Th2 cells and cytokines in the promotion of arterial aneurysm formation.
ANTIOXIDANTS IN CVD THERAPY
Oxidative stress triggers inflammation and endothelial disruption in atherogenesis. A number of studies showed that exogenous antioxidants can modulate endothelium-dependent vasodilation responses, endothelium-leukocyte interactions as well as balance between pro- and anti-thrombotic properties[125]. Accordingly, antioxidant therapy was suggested to beneficially interfere with development and progression of atherosclerosis.
Th1/Th2 balance is crucially dependent on redox-events; while Th1 responses prevail at oxidative conditions, Th2 responses were shown to be supported by “antioxidative stress”[126]. Thus, disequilibrium of Th1/Th2 cytokines may be involved in CVD as a mechanism of immunotoxicity. As Th1 and Th2 reactions crossregulate each other to balance immune responses[127], suppression of Th1-type response by antioxidants would favour Th2-type reactions. Of note, several types of antioxidant were shown to reduce IFN-γ-stimulated tryptophan degradation and neopterin in peripheral blood mononuclear cells in vitro[87,128].
A number of studies reported an inverse relationship between plasma antioxidants, or total antioxidant capacity and cardiovascular diseases[5,15]. Low intake of antioxidants, in particular of vitamins, was suggested to be associated with an increased risk of CVD[129,130]. Thus, the finding of an inverse correlation between concentrations of antioxidant compounds and vitamins and disease risk could relate to an increased requirement for antioxidant molecules during inflammatory diseases and insufficient supply with these compounds may further accelerate disease process. However, this assumption has not been conclusively proven in clinical trials and is still controversially discussed in the literature[131-134]. Likewise, equivocal effects of antioxidant supplementation with vitamin E, beta-carotene, alpha lipoic acid, coenzyme Q10, alone or in combination, on cardiovascular health were reported[135].
Major effects were expected from vitamin E therapy. Due to its fat-solubility, vitamin E is part of cell membranes and lipoprotein particles, where it counteracts oxidation events. Vitamin E-mediated protection from oxidative stress and atherosclerotic plaque formation has been shown both in vitro and in mouse models. However, while in several clinical trials vitamin E supplementation lead to a reduction of risk of fatal and nonfatal AMI, others reported even a slight increase of mortality upon high dose vitamin E treatment[136]. Thus, no final suggestion can be made about the impact of vitamin E supplementation and even recent metanalysis including a large trial number lead to inconsistent results[137].
So far, although a general association of low vitamin levels and oxidative stress related conditions is established, no clear evidence for a beneficial effect of vitamin supplementation exists. The association between vitamin deficiency in patients and disease symptoms is suggested to result mainly from the inflammation-associated consumption of oxidation-sensitive vitamins[29,132,138], which may lead to a variety of secondary effects.
Apart from being part of the antioxidant defense system, some vitamins act as enzyme cofactors. Low B vitamin availability (B6, B12 and folic acid) leads to impaired remethylation of homocysteine to methionine and thus to homocysteine accumulation, as they are essential cofactors in homocysteine-methionine metabolism. Increased homocysteine levels were found to be associated with arteriosclerotic outcomes and risk of stroke in elderly individuals[139], and are considered as an independent risk marker for CVD[140]. However, lowering homocysteine levels by B-vitamin supplementation failed to demonstrate beneficial effects in CVD[141]. Also, in open-label study with demented patients on B vitamins, a decline of homocysteine has been observed, while neopterin levels were not affected[142]. Recent data indicate that homocysteine accumulates secondarily due to heightened oxidative stress associated with immune activation[143-145]. Thus, also the impact of the selected marker has to be critically evaluated when assessing the effect of vitamin supplementation.
A broader understanding of antioxidant action is clearly warranted. Beside their direct effects in the prevention of biomolecule oxidation by being oxidized themselves, several antioxidants mediate a variety of effects that are of longer duration, as they may induce signaling changes in the biological system[146]. However, a variety of drugs may act also as antioxidants, thus antioxidant vitamins could interfere with pharmaco-relevant signaling pathways. This is of particular relevance for multi-target drugs such commonly used statins.
A major aim in the treatment of atherosclerosis is the prevention of LDL oxidation. Lipid-lowering compounds such as statins and niacin (vitamin B3, nicotinic acid) are in use for a long time, alone or together, for cardiovascular protection in patients with coronary disease and low plasma levels of HDL[147]. However, combination therapies with other antioxidant vitamins seemed even to counteract the beneficial effect of statin/niacin therapy[147,148].
Statins are inhibitors of 3-hydroxy-3methylglutaryl-co-enzyme A (HMG-CoA) reductase, and their lipid-lowering effects are suggested to reduce the risk of coronary heart disease[149], although therapeutic efficacy is controversially discussed[150].
The primary mechanism of statin action is suggested to be the reduction of LDL cholesterol, but several clinical trials indicate that statins exert pleiotropic effect that contribute to therapeutic efficacy. Statins act as effective antioxidants by inhibiting generation of ROS, but also by interfering with NOX and NOS, antioxidant enzymes, lipid peroxidation and LDL cholesterol oxidation[151]. In in vitro studies with vascular smooth muscle and mononuclear cells, treatment with atorvastatin could reduce NF-κB activation and expression of pro-inflammatory cytokines and chemokines[152]. In human peripheral blood mononuclear cells and in monocytic cell lines, atorvastatin was shown to suppresses stimulation-induced neopterin formation and tryptophan degradation, suggesting that both immunoreactivity of T cells and of monocyte-derived macrophages are down-regulated by this statin[153]. Treatment with several statins could promote Th2 polarization of CD4+ T cells primed in vitro with anti-CD3 antibody and splenic antigen-presenting cells[154]. These findings strongly suggest that statins contribute to the regulation of Th1/Th2 cell balance also in vivo. In endothelial cells, statins were shown to be involved in restorative processes by increasing NO-bioavailability and promoting re-endothelialization[155]. Of note, lovastatin was able to prevent neopterin-induced activation of human coronary artery endothelial cells in vitro by interfering with NF-κB activation and decreasing expression of cellular adhesion molecules and TF[77]. Furthermore, lovastatin reduced C-reactive protein -induced NF-κB activation in human umbilical vein endothelial cells[156]. Beside NF-κB, activation of inflammatory transcription factors activator protein 1 and hypoxia-inducible factor 1 alpha were shown to be downregulated in human endothelial and vascular smooth muscle cells upon treatment with HMG-CoA reductase inhibitors[157]. In line with the reported antioxidant and anti-inflammatory properties, statin use has been associated with lower neopterin levels in patients[158,159].
The influence on redox-balance and Th1-type signalling pathways such as neopterin formation and tryptophan breakdown has been described for a variety of (potentially) cardioprotective antioxidant drugs and vitamins, e.g., aspirin[160], atorvastatin[153], vitamins C and E[161] and seems to be a common mechanism at least in vitro. Furthermore, circulating vitamin E was shown to increase upon statin therapy[162,163]. Thus, due to inferences with common pathways, therapeutic efficacy might change when combining several antioxidant drugs and supplements.
Furthermore, antioxidant composition may differ between patients, and estimation of antioxidant profiles before therapy could be useful to select candidate patients that would profit from antioxidant therapies[164,165] and to avoid overdosage. Excessive antioxidant consumption may lead to adverse reactions ranging from favoring of Th2-type responses such as allergy and asthma to an increase of mortality[166-168]. So far, for patients who respond well to statin/niacin therapy, additional supplementation might only be advantageous when nutritional deficiencies are still detectable, however this hypothesis has to be investigated in more detail.
Another question is, if moderate vitamin deficiencies cannot be better (and safer) regulated by changes of lifestyle factors, e.g., by increasing the consumption of antioxidant-rich food.
NUTRITION, ANTIOXIDANTS AND CVD
The strong relationship between redox-status, immune response and metabolism is supported by the close association of metabolic diseases such as diabetes, obesity and metabolic syndrome with CVD[169]. Tissues that are important in metabolism are suggested to have an evolutionary potential to mediate inflammatory responses[170]. Metabolic and immune response pathways are closely cross-regulated to respond to the energetic demands necessary during immune activation. Several metabolic and immune cells show similarities on genetic and functional level, e.g., pre-adipocytes can transdifferentiate into macrophages[171] and activate similar transcriptional responses[172].
In contrast to classical activation of the immune system, e.g., by infection or tissue injury, inflammation may also be induced by metabolic triggers. So called metaflammation or para-inflammation is crucially involved in the development of chronic diseases such as diabetes, fatty liver disease and CVD[172,173].
A variety of dietary factors are able to produce cardiometabolic imprints that predispose to disease development. E.g., increased consumption of trans fatty acids (TFA) is supposed to activate pathways that are linked to insulin resistance syndrome. High TFA intake was found to be associated with harmful changes in serum lipids, systemic inflammation, endothelial function, and prospective observational studies demonstrated strong positive associations with the risk of MI, coronary heart disease death, and sudden death[174]. Changes of traditional nutrition patterns, as it is the case, e.g., in India, where “Westernization” led to an increase in uptake of sugar, salt, high fat diary products, and TFA-rich food, are suggested to be at least partially responsible for an about 3-fold increase in the prevalence of CVD and diabetes in the latter part of the 20th century[175].
But also excessive intake of antioxidants is a burden of modern life due to the omnipresence of preservatives, food colorants and vitamin supplements in the “Western diet”. Although still nutritional deficiencies may exist for some specific vitamins or other antioxidants, overall antioxidant stress may favour a Th2 environment by suppressing Th1 responses (Figure 2). In combination with high caloric diet and low physical activity, this may contribute to the development of obesity[133]. Food additives such as sodium benzoate, propionic acid, sodium sulfite, sorbic acid and curcumin were shown to suppress Th1-type immune response in vitro[176]. Antioxidant food additives also interfere with satiety saturation circuits, as they have shown to inhibit leptin release in cultured lipopolysaccharide-stimulated murine adipocytes in a dose- and time dependent manner[177]. Lowering the amount of circulating leptin is suggested to contribute to a obesogenic environment, as the reduced satiety effect in turn could lead to compensatory antioxidant craving and thus even more food intake[133]. Leptin is considered as a proinflammatory cytokine with proatherogenic features, as it increases monocyte chemoattractant protein-1 and endothelin-1 secretion by endothelial cells, enhances oxidative stress, promotes migration and proliferation of smooth muscle cells and increases platelet aggregation, thus facilitating thrombosis[178]. In the initial phase of obesity-related inflammation, leptin is predictively associated with interleukin 6 plasma levels in juveniles[179]. However, leptin resistance, which later develops during obesity, does also favor atherogenesis.
Figure 2.
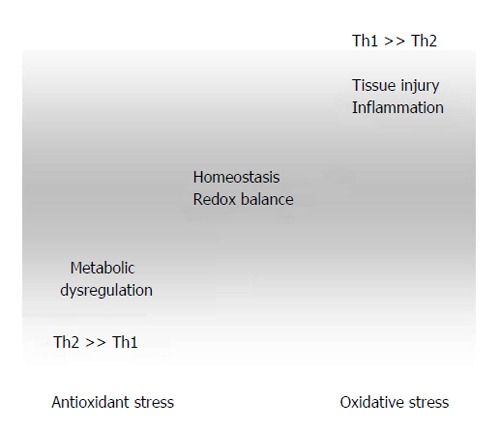
Dysregulation of redox- and Th1/Th2-balance in the course of atherogenesis. Excessive antioxidant intake in combination with other risk factors such as high caloric diet and low physical exercise lead to suppression of Th1-type immunity, thereby favoring Th2-associated development of allergies and asthma and promoting juvenile obesity. Factors such as high blood pressure and hyperlipidemia lead to shear stress and tissue injury. Inflammatory reactions are associated with high reactive oxygen species generation, which results in immunotoxicity due to oxidation of biomolecules (lipids, proteins, etc.).
Obesity-related immune mediated systemic inflammation was found to be associated with the development of the metabolic syndrome and altered Trp metabolism. However, across lifespan from juvenility to adulthood, differences in the Trp breakdown rate were observed. While juvenile overweight/obese individuals showed a decreased to unaltered Kyn/Trp ratio in comparison to normal weight controls, obese adults had significantly elevated Kyn serum levels and an increased Kyn/Trp ratio[180]. Thus, while in younger patients Th2-type responses might be favored, potentially due to the high antioxidant intake, overwhelming inflammation with Th1-type cytokines may predispose for the development of atherosclerosis in adult age.
Epidemiological observations suggest that consumption of certain foods rich in bioactive compounds, e.g., vitamins E and C, polyphenols and carotenoids such as lycopene and beta-carotene, and coenzyme Q10, is associated with decrease of atherosclerotic risk and such antioxidant-rich diet is supposed to be particularly effective in the early stages of atherosclerosis by preventing LDL oxidation and the oxidative lesion of endothelium[181,182]. However, a balanced died cannot always be translated into clinical benefit, despite its beneficial impact on human health.
There is accumulating evidence about the importance of maternal diet and early nutrition on different epigenetic mechanisms that promote the susceptibility to the development of metabolic diseases in adulthood, such as metabolic syndrome, insulin resistance, type 2 diabetes, obesity, dyslipidaemia, hypertension, and also CVD. Of note, both under-and overnutrition have been associated with adverse responses[183,184]. Several studies indicate that impaired foetal growth, and/or in utero exposure to risk factors, especially maternal hypercholesterolaemia, may be relevant for the early onset of cardiovascular damage. Translational studies support this hypothesis; however, a direct causality in humans has not been ascertained[185].
The influence of epigenetic mechanisms on the developmental induction of chronic diseases raises the possibility that nutritional or pharmaceutical interventions may be used to modify long-term cardio-metabolic disease risk and combat this rapid rise in chronic non-communicable diseases[186].
CONCLUSION
Adaptive and innate immune responses are centrally involved in the chronic inflammatory process, which leads to destabilization of atherosclerotic lesions, these processes are tightly connected to metabolic factors, which are essentially influenced by life style and also the genetic/epigenetic frame. Inflammation-induced oxidative modifications contribute to all important clinical manifestations of CVD such as endothelial dysfunction and plaque disruption. However, due the poor performance of antioxidant strategies in limiting atherosclerosis and cardiovascular events, it remains to be answered if oxidative modification is causal for the initiation or is an injurious response to atherogenesis[96]. Disease underlying interactions are too complex and the understanding is too fragmentary that clear, reliable therapeutic recommendations can be given[101].
The strong interconnection of metabolic and inflammatory pathways suggests that metabolically induced inflammatory processes should be considered as early, or even primary events[171]. Many data support that there is a large time span between initial pathological changes and the onset of clinical manifestations. This time frame could be used for preventive strategies, however a better understanding of disease development and more sensitive detection methods would be a prerequisite.
A detailed knowledge on inflammatory and redox-regulated processes would also allow a better adaption of treatment regimes. Stable biochemical markers are necessary to control disease courses and treatment efficacy. In this context, e.g., neopterin is a useful indicator of the immune activation status and oxidative stress[6] and Kyn/Trp ratio accounts for aspects of immunoregulation via IDO and represents an important metabolic checkpoint. Normalization of tryptophan metabolism represents an important goal to improve the outcome of patients suffering from CVD, whereby treatments with IDO inhibitors such as 1-methyl tryptophan could be considered[101]. However, IDO is well known for its immunosuppressive properties, and its inhibition by medications may also lead to adverse effects.
Also several antioxidant drugs, botanical extracts, phytocompounds and vitamins but also food-contained preservatives and colorants have been shown to negatively interfere with IDO[87,166]. Both inhibition of enzymatic activity as well as downregulation of activatory signals may lead to a normalization of tryptophan breakdown ratio. Thus, nutrition might be considered as a major factor that influences tryptophan metabolism and underlying inflammation in a more gentle and balanced manner than medication.
Measurement of tryptophan and kynurenine concentrations, and calculation of the Kyn/Trp ratio are important predictors of an unfavourable outcome in patients with CVD. It will be important to investigate if these parameters can provide a basis for more successful and precise biologically grounded therapeutic protocols to further reduce cardiovascular morbidity and mortality[101]. Combined measurements of multiple markers, such as additional determination of lipoproteins, NO metabolites, BH4 and plasma antioxidants, will also be helpful to understand redox-regulation in health and disease and may allow to discriminate best between different clinical diagnostic categories and to evaluate treatment strategies.
In summary, a general evaluation of the effect of an “antioxidant therapy” is not possible at the moment. While vitamin supplementation might be beneficial under certain circumstances, a variety of studies indicate no or even adverse effects when administered alone and even more when used in combination with lipid-lowering agents. However, also for statin and niacin treatment a panel of adverse effects has been described[187,188]. Although antioxidant supplementation may have some benefit to counteract secondary symptoms, their role in CAD seems to be of moderate importance[145]. Surveillance of the antioxidant status before and during therapy would allow seek out patients that could benefit from vitamin supplementation[164,165]. Impact of lifestyle factors such as nutrition and physical exercise, however, has turned out as a major factor in CVD prevention and also in influencing treatment efficacy.
Footnotes
P- Reviewers: Kindy MS, Omboni S S- Editor: Wen LL L- Editor: A E- Editor: Liu SQ
References
- 1.World Health Organization. Global status report on noncommunicable disaeses 2010. Geneva: 2010. [Google Scholar]
- 2.Hansson GK. Inflammation, atherosclerosis, and coronary artery disease. N Engl J Med. 2005;352:1685–1695. doi: 10.1056/NEJMra043430. [DOI] [PubMed] [Google Scholar]
- 3.World Health Organization. Cardiovascular diseases (CVDs) 2013. Available from: http: //www.who.int/mediacentre/factsheets/fs317/en/index.html.
- 4.Libby P, Ridker PM, Maseri A. Inflammation and atherosclerosis. Circulation. 2002;105:1135–1143. doi: 10.1161/hc0902.104353. [DOI] [PubMed] [Google Scholar]
- 5.Murr C, Winklhofer-Roob BM, Schroecksnadel K, Maritschnegg M, Mangge H, Böhm BO, Winkelmann BR, März W, Fuchs D. Inverse association between serum concentrations of neopterin and antioxidants in patients with and without angiographic coronary artery disease. Atherosclerosis. 2009;202:543–549. doi: 10.1016/j.atherosclerosis.2008.04.047. [DOI] [PubMed] [Google Scholar]
- 6.Fuchs D, Avanzas P, Arroyo-Espliguero R, Jenny M, Consuegra-Sanchez L, Kaski JC. The role of neopterin in atherogenesis and cardiovascular risk assessment. Curr Med Chem. 2009;16:4644–4653. doi: 10.2174/092986709789878247. [DOI] [PubMed] [Google Scholar]
- 7.Libby P, Ridker PM, Hansson GK. Progress and challenges in translating the biology of atherosclerosis. Nature. 2011;473:317–325. doi: 10.1038/nature10146. [DOI] [PubMed] [Google Scholar]
- 8.Galkina E, Ley K. Immune and inflammatory mechanisms of atherosclerosis (*) Annu Rev Immunol. 2009;27:165–197. doi: 10.1146/annurev.immunol.021908.132620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chisolm GM, Steinberg D. The oxidative modification hypothesis of atherogenesis: an overview. Free Radic Biol Med. 2000;28:1815–1826. doi: 10.1016/s0891-5849(00)00344-0. [DOI] [PubMed] [Google Scholar]
- 10.Tabas I. Macrophage death and defective inflammation resolution in atherosclerosis. Nat Rev Immunol. 2010;10:36–46. doi: 10.1038/nri2675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Frostegård J. Immunity, atherosclerosis and cardiovascular disease. BMC Med. 2013;11:117. doi: 10.1186/1741-7015-11-117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ghazalpour A, Doss S, Yang X, Aten J, Toomey EM, Van Nas A, Wang S, Drake TA, Lusis AJ. Thematic review series: The pathogenesis of atherosclerosis. Toward a biological network for atherosclerosis. J Lipid Res. 2004;45:1793–1805. doi: 10.1194/jlr.R400006-JLR200. [DOI] [PubMed] [Google Scholar]
- 13.Ramsey SA, Gold ES, Aderem A. A systems biology approach to understanding atherosclerosis. EMBO Mol Med. 2010;2:79–89. doi: 10.1002/emmm.201000063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Libby P. Inflammation in atherosclerosis. Arterioscler Thromb Vasc Biol. 2012;32:2045–2051. doi: 10.1161/ATVBAHA.108.179705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chrysohoou C, Panagiotakos DB, Pitsavos C, Skoumas J, Economou M, Papadimitriou L, Stefanadis C. The association between pre-hypertension status and oxidative stress markers related to atherosclerotic disease: the ATTICA study. Atherosclerosis. 2007;192:169–176. doi: 10.1016/j.atherosclerosis.2006.04.030. [DOI] [PubMed] [Google Scholar]
- 16.Corti R, Hutter R, Badimon JJ, Fuster V. Evolving concepts in the triad of atherosclerosis, inflammation and thrombosis. J Thromb Thrombolysis. 2004;17:35–44. doi: 10.1023/B:THRO.0000036027.39353.70. [DOI] [PubMed] [Google Scholar]
- 17.Tanaka K, Nagata D, Hirata Y, Tabata Y, Nagai R, Sata M. Augmented angiogenesis in adventitia promotes growth of atherosclerotic plaque in apolipoprotein E-deficient mice. Atherosclerosis. 2011;215:366–373. doi: 10.1016/j.atherosclerosis.2011.01.016. [DOI] [PubMed] [Google Scholar]
- 18.Fenyo IM, Gafencu AV. The involvement of the monocytes/macrophages in chronic inflammation associated with atherosclerosis. Immunobiology. 2013;218:1376–1384. doi: 10.1016/j.imbio.2013.06.005. [DOI] [PubMed] [Google Scholar]
- 19.Wolfs IM, Donners MM, de Winther MP. Differentiation factors and cytokines in the atherosclerotic plaque micro-environment as a trigger for macrophage polarisation. Thromb Haemost. 2011;106:763–771. doi: 10.1160/TH11-05-0320. [DOI] [PubMed] [Google Scholar]
- 20.Fleetwood AJ, Lawrence T, Hamilton JA, Cook AD. Granulocyte-macrophage colony-stimulating factor (CSF) and macrophage CSF-dependent macrophage phenotypes display differences in cytokine profiles and transcription factor activities: implications for CSF blockade in inflammation. J Immunol. 2007;178:5245–5252. doi: 10.4049/jimmunol.178.8.5245. [DOI] [PubMed] [Google Scholar]
- 21.Stöger JL, Gijbels MJ, van der Velden S, Manca M, van der Loos CM, Biessen EA, Daemen MJ, Lutgens E, de Winther MP. Distribution of macrophage polarization markers in human atherosclerosis. Atherosclerosis. 2012;225:461–468. doi: 10.1016/j.atherosclerosis.2012.09.013. [DOI] [PubMed] [Google Scholar]
- 22.Gleissner CA. Macrophage Phenotype Modulation by CXCL4 in Atherosclerosis. Front Physiol. 2012;3:1. doi: 10.3389/fphys.2012.00001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Uyemura K, Demer LL, Castle SC, Jullien D, Berliner JA, Gately MK, Warrier RR, Pham N, Fogelman AM, Modlin RL. Cross-regulatory roles of interleukin (IL)-12 and IL-10 in atherosclerosis. J Clin Invest. 1996;97:2130–2138. doi: 10.1172/JCI118650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ait-Oufella H, Taleb S, Mallat Z, Tedgui A. Cytokine network and T cell immunity in atherosclerosis. Semin Immunopathol. 2009;31:23–33. doi: 10.1007/s00281-009-0143-x. [DOI] [PubMed] [Google Scholar]
- 25.Nathan CF, Murray HW, Wiebe ME, Rubin BY. Identification of interferon-gamma as the lymphokine that activates human macrophage oxidative metabolism and antimicrobial activity. J Exp Med. 1983;158:670–689. doi: 10.1084/jem.158.3.670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rossi F, Zatti M. Biochemical aspects of phagocytosis in polymorphonuclear leucocytes. NADH and NADPH oxidation by the granules of resting and phagocytizing cells. Experientia. 1964;20:21–23. doi: 10.1007/BF02146019. [DOI] [PubMed] [Google Scholar]
- 27.Wink DA, Hines HB, Cheng RY, Switzer CH, Flores-Santana W, Vitek MP, Ridnour LA, Colton CA. Nitric oxide and redox mechanisms in the immune response. J Leukoc Biol. 2011;89:873–891. doi: 10.1189/jlb.1010550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Szabo SJ, Sullivan BM, Peng SL, Glimcher LH. Molecular mechanisms regulating Th1 immune responses. Annu Rev Immunol. 2003;21:713–758. doi: 10.1146/annurev.immunol.21.120601.140942. [DOI] [PubMed] [Google Scholar]
- 29.Schroecksnadel K, Frick B, Winkler C, Fuchs D. Crucial role of interferon-gamma and stimulated macrophages in cardiovascular disease. Curr Vasc Pharmacol. 2006;4:205–213. doi: 10.2174/157016106777698379. [DOI] [PubMed] [Google Scholar]
- 30.Le Bras M, Clément MV, Pervaiz S, Brenner C. Reactive oxygen species and the mitochondrial signaling pathway of cell death. Histol Histopathol. 2005;20:205–219. doi: 10.14670/HH-20.205. [DOI] [PubMed] [Google Scholar]
- 31.Valko M, Leibfritz D, Moncol J, Cronin MT, Mazur M, Telser J. Free radicals and antioxidants in normal physiological functions and human disease. Int J Biochem Cell Biol. 2007;39:44–84. doi: 10.1016/j.biocel.2006.07.001. [DOI] [PubMed] [Google Scholar]
- 32.Gostner JM, Becker K, Fuchs D, Sucher R. Redox regulation of the immune response. Redox Rep. 2013;18:88–94. doi: 10.1179/1351000213Y.0000000044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Griendling KK, FitzGerald GA. Oxidative stress and cardiovascular injury: Part II: animal and human studies. Circulation. 2003;108:2034–2040. doi: 10.1161/01.CIR.0000093661.90582.c4. [DOI] [PubMed] [Google Scholar]
- 34.Griendling KK, FitzGerald GA. Oxidative stress and cardiovascular injury: Part I: basic mechanisms and in vivo monitoring of ROS. Circulation. 2003;108:1912–1916. doi: 10.1161/01.CIR.0000093660.86242.BB. [DOI] [PubMed] [Google Scholar]
- 35.Peiser L, Mukhopadhyay S, Gordon S. Scavenger receptors in innate immunity. Curr Opin Immunol. 2002;14:123–128. doi: 10.1016/s0952-7915(01)00307-7. [DOI] [PubMed] [Google Scholar]
- 36.Galle J, Hansen-Hagge T, Wanner C, Seibold S. Impact of oxidized low density lipoprotein on vascular cells. Atherosclerosis. 2006;185:219–226. doi: 10.1016/j.atherosclerosis.2005.10.005. [DOI] [PubMed] [Google Scholar]
- 37.Holvoet P, Mertens A, Verhamme P, Bogaerts K, Beyens G, Verhaeghe R, Collen D, Muls E, Van de Werf F. Circulating oxidized LDL is a useful marker for identifying patients with coronary artery disease. Arterioscler Thromb Vasc Biol. 2001;21:844–848. doi: 10.1161/01.atv.21.5.844. [DOI] [PubMed] [Google Scholar]
- 38.Fraley AE, Tsimikas S. Clinical applications of circulating oxidized low-density lipoprotein biomarkers in cardiovascular disease. Curr Opin Lipidol. 2006;17:502–509. doi: 10.1097/01.mol.0000245255.40634.b5. [DOI] [PubMed] [Google Scholar]
- 39.Holvoet P, Vanhaecke J, Janssens S, Van de Werf F, Collen D. Oxidized LDL and malondialdehyde-modified LDL in patients with acute coronary syndromes and stable coronary artery disease. Circulation. 1998;98:1487–1494. doi: 10.1161/01.cir.98.15.1487. [DOI] [PubMed] [Google Scholar]
- 40.Meisinger C, Baumert J, Khuseyinova N, Loewel H, Koenig W. Plasma oxidized low-density lipoprotein, a strong predictor for acute coronary heart disease events in apparently healthy, middle-aged men from the general population. Circulation. 2005;112:651–657. doi: 10.1161/CIRCULATIONAHA.104.529297. [DOI] [PubMed] [Google Scholar]
- 41.Khera AV, Cuchel M, de la Llera-Moya M, Rodrigues A, Burke MF, Jafri K, French BC, Phillips JA, Mucksavage ML, Wilensky RL, et al. Cholesterol efflux capacity, high-density lipoprotein function, and atherosclerosis. N Engl J Med. 2011;364:127–135. doi: 10.1056/NEJMoa1001689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chapman MJ, Ginsberg HN, Amarenco P, Andreotti F, Borén J, Catapano AL, Descamps OS, Fisher E, Kovanen PT, Kuivenhoven JA, et al. Triglyceride-rich lipoproteins and high-density lipoprotein cholesterol in patients at high risk of cardiovascular disease: evidence and guidance for management. Eur Heart J. 2011;32:1345–1361. doi: 10.1093/eurheartj/ehr112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ferretti G, Bacchetti T, Masciangelo S, Bicchiega V. HDL-paraoxonase and membrane lipid peroxidation: a comparison between healthy and obese subjects. Obesity (Silver Spring) 2010;18:1079–1084. doi: 10.1038/oby.2009.338. [DOI] [PubMed] [Google Scholar]
- 44.Watson AD, Berliner JA, Hama SY, La Du BN, Faull KF, Fogelman AM, Navab M. Protective effect of high density lipoprotein associated paraoxonase. Inhibition of the biological activity of minimally oxidized low density lipoprotein. J Clin Invest. 1995;96:2882–2891. doi: 10.1172/JCI118359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Graham A, Hassall DG, Rafique S, Owen JS. Evidence for a paraoxonase-independent inhibition of low-density lipoprotein oxidation by high-density lipoprotein. Atherosclerosis. 1997;135:193–204. doi: 10.1016/s0021-9150(97)00162-7. [DOI] [PubMed] [Google Scholar]
- 46.Zelzer S, Fuchs N, Almer G, Raggam RB, Prüller F, Truschnig-Wilders M, Schnedl W, Horejsi R, Möller R, Weghuber D, et al. High density lipoprotein cholesterol level is a robust predictor of lipid peroxidation irrespective of gender, age, obesity, and inflammatory or metabolic biomarkers. Clin Chim Acta. 2011;412:1345–1349. doi: 10.1016/j.cca.2011.03.031. [DOI] [PubMed] [Google Scholar]
- 47.Escolà-Gil JC, Cedó L, Blanco-Vaca F. High-density lipoprotein cholesterol targeting for novel drug discovery: where have we gone wrong? Expert Opin Drug Discov. 2014;9:119–124. doi: 10.1517/17460441.2014.871257. [DOI] [PubMed] [Google Scholar]
- 48.Singh IM, Shishehbor MH, Ansell BJ. High-density lipoprotein as a therapeutic target: a systematic review. JAMA. 2007;298:786–798. doi: 10.1001/jama.298.7.786. [DOI] [PubMed] [Google Scholar]
- 49.Small DM. George Lyman Duff memorial lecture. Progression and regression of atherosclerotic lesions. Insights from lipid physical biochemistry. Arteriosclerosis. 1988;8:103–129. doi: 10.1161/01.atv.8.2.103. [DOI] [PubMed] [Google Scholar]
- 50.Duewell P, Kono H, Rayner KJ, Sirois CM, Vladimer G, Bauernfeind FG, Abela GS, Franchi L, Nuñez G, Schnurr M, et al. NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature. 2010;464:1357–1361. doi: 10.1038/nature08938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Samstad EO, Niyonzima N, Nymo S, Aune MH, Ryan L, Bakke SS, Lappegård KT, Brekke OL, Lambris JD, Damås JK, et al. Cholesterol crystals induce complement-dependent inflammasome activation and cytokine release. J Immunol. 2014:192; 2837–2845. doi: 10.4049/jimmunol.1302484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Grebe A, Latz E. Cholesterol crystals and inflammation. Curr Rheumatol Rep. 2013;15:313. doi: 10.1007/s11926-012-0313-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Huber C, Batchelor JR, Fuchs D, Hausen A, Lang A, Niederwieser D, Reibnegger G, Swetly P, Troppmair J, Wachter H. Immune response-associated production of neopterin. Release from macrophages primarily under control of interferon-gamma. J Exp Med. 1984;160:310–316. doi: 10.1084/jem.160.1.310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wirleitner B, Reider D, Ebner S, Böck G, Widner B, Jaeger M, Schennach H, Romani N, Fuchs D. Monocyte-derived dendritic cells release neopterin. J Leukoc Biol. 2002;72:1148–1153. [PubMed] [Google Scholar]
- 55.Werner-Felmayer G, Werner ER, Fuchs D, Hausen A, Reibnegger G, Wachter H. Tumour necrosis factor-alpha and lipopolysaccharide enhance interferon-induced tryptophan degradation and pteridine synthesis in human cells. Biol Chem Hoppe Seyler. 1989;370:1063–1069. doi: 10.1515/bchm3.1989.370.2.1063. [DOI] [PubMed] [Google Scholar]
- 56.Nathan CF. Peroxide and pteridine: a hypothesis on the regulation of macrophage antimicrobial activity by interferon gamma. Interferon. 1986;7:125–143. [PubMed] [Google Scholar]
- 57.Murr C, Widner B, Wirleitner B, Fuchs D. Neopterin as a marker for immune system activation. Curr Drug Metab. 2002;3:175–187. doi: 10.2174/1389200024605082. [DOI] [PubMed] [Google Scholar]
- 58.Werner ER, Werner-Felmayer G, Fuchs D, Hausen A, Reibnegger G, Yim JJ, Pfleiderer W, Wachter H. Tetrahydrobiopterin biosynthetic activities in human macrophages, fibroblasts, THP-1, and T 24 cells. GTP-cyclohydrolase I is stimulated by interferon-gamma, and 6-pyruvoyl tetrahydropterin synthase and sepiapterin reductase are constitutively present. J Biol Chem. 1990;265:3189–3192. [PubMed] [Google Scholar]
- 59.Werner-Felmayer G, Golderer G, Werner ER. Tetrahydrobiopterin biosynthesis, utilization and pharmacological effects. Curr Drug Metab. 2002;3:159–173. doi: 10.2174/1389200024605073. [DOI] [PubMed] [Google Scholar]
- 60.Fuchs D, Forsman A, Hagberg L, Larsson M, Norkrans G, Reibnegger G, Werner ER, Wachter H. Immune activation and decreased tryptophan in patients with HIV-1 infection. J Interferon Res. 1990;10:599–603. doi: 10.1089/jir.1990.10.599. [DOI] [PubMed] [Google Scholar]
- 61.Murr C, Fuith LC, Widner B, Wirleitner B, Baier-Bitterlich G, Fuchs D. Increased neopterin concentrations in patients with cancer: indicator of oxidative stress? Anticancer Res. 1999;19:1721–1728. [PubMed] [Google Scholar]
- 62.Sucher R, Schroecksnadel K, Weiss G, Margreiter R, Fuchs D, Brandacher G. Neopterin, a prognostic marker in human malignancies. Cancer Lett. 2010;287:13–22. doi: 10.1016/j.canlet.2009.05.008. [DOI] [PubMed] [Google Scholar]
- 63.Reibnegger G, Aichberger C, Fuchs D, Hausen A, Spielberger M, Werner ER, Margreiter R, Wachtehr H. Posttransplant neopterin excretion in renal allograft recipients--a reliable diagnostic aid for acute rejection and a predictive marker of long-term graft survival. Transplantation. 1991;52:58–63. doi: 10.1097/00007890-199107000-00012. [DOI] [PubMed] [Google Scholar]
- 64.Melichar B, Gregor J, Solichová D, Lukes J, Tichý M, Pidrman V. Increased urinary neopterin in acute myocardial infarction. Clin Chem. 1994;40:338–339. [PubMed] [Google Scholar]
- 65.Anwaar I, Gottsäter A, Lindgärde F, Mattiasson I. Increasing plasma neopterin and persistent plasma endothelin during follow-up after acute cerebral ischemia. Angiology. 1999;50:1–8. doi: 10.1177/000331979905000101. [DOI] [PubMed] [Google Scholar]
- 66.Tatzber F, Rabl H, Koriska K, Erhart U, Puhl H, Waeg G, Krebs A, Esterbauer H. Elevated serum neopterin levels in atherosclerosis. Atherosclerosis. 1991;89:203–208. doi: 10.1016/0021-9150(91)90061-7. [DOI] [PubMed] [Google Scholar]
- 67.Weiss G, Willeit J, Kiechl S, Fuchs D, Jarosch E, Oberhollenzer F, Reibnegger G, Tilz GP, Gerstenbrand F, Wachter H. Increased concentrations of neopterin in carotid atherosclerosis. Atherosclerosis. 1994;106:263–271. doi: 10.1016/0021-9150(94)90131-7. [DOI] [PubMed] [Google Scholar]
- 68.Schumacher M, Halwachs G, Tatzber F, Fruhwald FM, Zweiker R, Watzinger N, Eber B, Wilders-Truschnig M, Esterbauer H, Klein W. Increased neopterin in patients with chronic and acute coronary syndromes. J Am Coll Cardiol. 1997;30:703–707. doi: 10.1016/s0735-1097(97)00172-1. [DOI] [PubMed] [Google Scholar]
- 69.Gurfinkel EP, Scirica BM, Bozovich G, Macchia A, Manos E, Mautner B. Serum neopterin levels and the angiographic extent of coronary arterial narrowing in unstable angina pectoris and in non-Q-wave acute myocardial infarction. Am J Cardiol. 1999;83:515–518. doi: 10.1016/s0002-9149(98)00905-9. [DOI] [PubMed] [Google Scholar]
- 70.Zouridakis E, Avanzas P, Arroyo-Espliguero R, Fredericks S, Kaski JC. Markers of inflammation and rapid coronary artery disease progression in patients with stable angina pectoris. Circulation. 2004;110:1747–1753. doi: 10.1161/01.CIR.0000142664.18739.92. [DOI] [PubMed] [Google Scholar]
- 71.Avanzas P, Arroyo-Espliguero R, Quiles J, Roy D, Kaski JC. Elevated serum neopterin predicts future adverse cardiac events in patients with chronic stable angina pectoris. Eur Heart J. 2005;26:457–463. doi: 10.1093/eurheartj/ehi111. [DOI] [PubMed] [Google Scholar]
- 72.Kaski JC, Consuegra-Sanchez L, Fernandez-Berges DJ, Cruz-Fernandez JM, Garcia-Moll X, Marrugat J, Mostaza J, Toro-Cebada R, González-Juanatey JR, Guzmán-Martínez G. Elevated serum neopterin levels and adverse cardiac events at 6 months follow-up in Mediterranean patients with non-ST-segment elevation acute coronary syndrome. Atherosclerosis. 2008;201:176–183. doi: 10.1016/j.atherosclerosis.2008.01.009. [DOI] [PubMed] [Google Scholar]
- 73.Johnston DT, Gagos M, Raio N, Ragolia L, Shenouda D, Davis-Lorton MA, De Leon JR. Alterations in serum neopterin correlate with thrombolysis in myocardial infarction risk scores in acute coronary syndromes. Coron Artery Dis. 2006;17:511–516. doi: 10.1097/00019501-200609000-00003. [DOI] [PubMed] [Google Scholar]
- 74.Barani J, Mattiasson I, Lindblad B, Gottsäter A. Cardiac function, inflammatory mediators and mortality in critical limb ischemia. Angiology. 2006;57:437–444. doi: 10.1177/0003319706290743. [DOI] [PubMed] [Google Scholar]
- 75.Ray KK, Morrow DA, Sabatine MS, Shui A, Rifai N, Cannon CP, Braunwald E. Long-term prognostic value of neopterin: a novel marker of monocyte activation in patients with acute coronary syndrome. Circulation. 2007;115:3071–3078. doi: 10.1161/CIRCULATIONAHA.106.666511. [DOI] [PubMed] [Google Scholar]
- 76.Adachi T, Naruko T, Itoh A, Komatsu R, Abe Y, Shirai N, Yamashita H, Ehara S, Nakagawa M, Kitabayashi C, et al. Neopterin is associated with plaque inflammation and destabilisation in human coronary atherosclerotic lesions. Heart. 2007;93:1537–1541. doi: 10.1136/hrt.2006.109736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Cirillo P, Pacileo M, DE Rosa S, Calabrò P, Gargiulo A, Angri V, Granato-Corigliano F, Fiorentino I, Prevete N, DE Palma R, et al. Neopterin induces pro-atherothrombotic phenotype in human coronary endothelial cells. J Thromb Haemost. 2006;4:2248–2255. doi: 10.1111/j.1538-7836.2006.02125.x. [DOI] [PubMed] [Google Scholar]
- 78.Weiss G, Fuchs D, Hausen A, Reibnegger G, Werner ER, Werner-Felmayer G, Semenitz E, Dierich MP, Wachter H. Neopterin modulates toxicity mediated by reactive oxygen and chloride species. FEBS Lett. 1993;321:89–92. doi: 10.1016/0014-5793(93)80627-7. [DOI] [PubMed] [Google Scholar]
- 79.Herpfer I, Greilberger J, Ledinski G, Widner B, Fuchs D, Jürgens G. Neopterin and 7,8-dihydroneopterin interfere with low density lipoprotein oxidation mediated by peroxynitrite and/or copper. Free Radic Res. 2002;36:509–520. doi: 10.1080/10715760290025898. [DOI] [PubMed] [Google Scholar]
- 80.Greilberger J, Oettl K, Cvirn G, Reibnegger G, Jürgens G. Modulation of LDL oxidation by 7,8-dihydroneopterin. Free Radic Res. 2004;38:9–17. doi: 10.1080/10715760310001623322. [DOI] [PubMed] [Google Scholar]
- 81.Widner B, Baier-Bitterlich G, Wede I, Wirleitner B, Fuchs D. Neopterin derivatives modulate the nitration of tyrosine by peroxynitrite. Biochem Biophys Res Commun. 1998;248:341–346. doi: 10.1006/bbrc.1998.8856. [DOI] [PubMed] [Google Scholar]
- 82.Hoffmann G, Wirleitner B, Fuchs D. Potential role of immune system activation-associated production of neopterin derivatives in humans. Inflamm Res. 2003;52:313–321. doi: 10.1007/s00011-003-1181-9. [DOI] [PubMed] [Google Scholar]
- 83.Taylor MW, Feng GS. Relationship between interferon-gamma, indoleamine 2,3-dioxygenase, and tryptophan catabolism. FASEB J. 1991;5:2516–2522. [PubMed] [Google Scholar]
- 84.Byrne GI, Lehmann LK, Kirschbaum JG, Borden EC, Lee CM, Brown RR. Induction of tryptophan degradation in vitro and in vivo: a gamma-interferon-stimulated activity. J Interferon Res. 1986;6:389–396. doi: 10.1089/jir.1986.6.389. [DOI] [PubMed] [Google Scholar]
- 85.Werner ER, Bitterlich G, Fuchs D, Hausen A, Reibnegger G, Szabo G, Dierich MP, Wachter H. Human macrophages degrade tryptophan upon induction by interferon-gamma. Life Sci. 1987;41:273–280. doi: 10.1016/0024-3205(87)90149-4. [DOI] [PubMed] [Google Scholar]
- 86.Thomas SR, Stocker R. Redox reactions related to indoleamine 2,3-dioxygenase and tryptophan metabolism along the kynurenine pathway. Redox Rep. 1999;4:199–220. doi: 10.1179/135100099101534927. [DOI] [PubMed] [Google Scholar]
- 87.Schroecksnadel K, Fischer B, Schennach H, Weiss G, Fuchs D. Antioxidants suppress Th1-type immune response in vitro. Drug Metab Lett. 2007;1:166–171. doi: 10.2174/187231207781369816. [DOI] [PubMed] [Google Scholar]
- 88.Widner B, Werner ER, Schennach H, Wachter H, Fuchs D. Simultaneous measurement of serum tryptophan and kynurenine by HPLC. Clin Chem. 1997;43:2424–2426. [PubMed] [Google Scholar]
- 89.Knox WE, Piras MM, Tokuyama K. Induction of tryptophan pyrrolase in rat liver by physiological amounts of hydrocortisone and secreted glucocorticoids. Enzymol Biol Clin (Basel) 1966;7:1–10. doi: 10.1159/000457200. [DOI] [PubMed] [Google Scholar]
- 90.Knox WE. The regulation of tryptophan pyrrolase activity by tryptophan. Adv Enzyme Regul. 1966;4:287–297. doi: 10.1016/0065-2571(66)90023-9. [DOI] [PubMed] [Google Scholar]
- 91.Brandacher G, Winkler C, Schroecksnadel K, Margreiter R, Fuchs D. Antitumoral activity of interferon-gamma involved in impaired immune function in cancer patients. Curr Drug Metab. 2006;7:599–612. doi: 10.2174/138920006778017768. [DOI] [PubMed] [Google Scholar]
- 92.Munn DH, Mellor AL. Indoleamine 2,3 dioxygenase and metabolic control of immune responses. Trends Immunol. 2013;34:137–143. doi: 10.1016/j.it.2012.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Sucher R, Fischler K, Oberhuber R, Kronberger I, Margreiter C, Ollinger R, Schneeberger S, Fuchs D, Werner ER, Watschinger K, et al. IDO and regulatory T cell support are critical for cytotoxic T lymphocyte-associated Ag-4 Ig-mediated long-term solid organ allograft survival. J Immunol. 2012;188:37–46. doi: 10.4049/jimmunol.1002777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Fuchs D, Hausen A, Reibnegger G, Werner ER, Werner-Felmayer G, Dierich MP, Wachter H. Immune activation and the anaemia associated with chronic inflammatory disorders. Eur J Haematol. 1991;46:65–70. doi: 10.1111/j.1600-0609.1991.tb00524.x. [DOI] [PubMed] [Google Scholar]
- 95.Weiss G, Schroecksnadel K, Mattle V, Winkler C, Konwalinka G, Fuchs D. Possible role of cytokine-induced tryptophan degradation in anaemia of inflammation. Eur J Haematol. 2004;72:130–134. doi: 10.1046/j.0902-4441.2003.00197.x. [DOI] [PubMed] [Google Scholar]
- 96.Wirleitner B, Rudzite V, Neurauter G, Murr C, Kalnins U, Erglis A, Trusinskis K, Fuchs D. Immune activation and degradation of tryptophan in coronary heart disease. Eur J Clin Invest. 2003;33:550–554. doi: 10.1046/j.1365-2362.2003.01186.x. [DOI] [PubMed] [Google Scholar]
- 97.Pertovaara M, Raitala A, Juonala M, Lehtimäki T, Huhtala H, Oja SS, Jokinen E, Viikari JS, Raitakari OT, Hurme M. Indoleamine 2,3-dioxygenase enzyme activity correlates with risk factors for atherosclerosis: the Cardiovascular Risk in Young Finns Study. Clin Exp Immunol. 2007;148:106–111. doi: 10.1111/j.1365-2249.2007.03325.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Niinisalo P, Raitala A, Pertovaara M, Oja SS, Lehtimäki T, Kähönen M, Reunanen A, Jula A, Moilanen L, Kesäniemi YA, et al. Indoleamine 2,3-dioxygenase activity associates with cardiovascular risk factors: the Health 2000 study. Scand J Clin Lab Invest. 2008;68:767–770. doi: 10.1080/00365510802245685. [DOI] [PubMed] [Google Scholar]
- 99.Widner B, Laich A, Sperner-Unterweger B, Ledochowski M, Fuchs D. Neopterin production, tryptophan degradation, and mental depression--what is the link? Brain Behav Immun. 2002;16:590–595. doi: 10.1016/s0889-1591(02)00006-5. [DOI] [PubMed] [Google Scholar]
- 100.Wellenius GA, Mukamal KJ, Kulshreshtha A, Asonganyi S, Mittleman MA. Depressive symptoms and the risk of atherosclerotic progression among patients with coronary artery bypass grafts. Circulation. 2008;117:2313–2319. doi: 10.1161/CIRCULATIONAHA.107.741058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Mangge H, Stelzer I, Reininghaus EZ, Weghuber D, Postolache TT, Fuchs D. Disturbed tryptophan metabolism in cardiovascular disease. Curr Med Chem. 2014;21:1931–1937. doi: 10.2174/0929867321666140304105526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Wink DA, Vodovotz Y, Grisham MB, DeGraff W, Cook JC, Pacelli R, Krishna M, Mitchell JB. Antioxidant effects of nitric oxide. Methods Enzymol. 1999;301:413–424. doi: 10.1016/s0076-6879(99)01105-2. [DOI] [PubMed] [Google Scholar]
- 103.Stamler JS, Singel DJ, Loscalzo J. Biochemistry of nitric oxide and its redox-activated forms. Science. 1992;258:1898–1902. doi: 10.1126/science.1281928. [DOI] [PubMed] [Google Scholar]
- 104.Stamler JS, Jaraki O, Osborne J, Simon DI, Keaney J, Vita J, Singel D, Valeri CR, Loscalzo J. Nitric oxide circulates in mammalian plasma primarily as an S-nitroso adduct of serum albumin. Proc Natl Acad Sci USA. 1992;89:7674–7677. doi: 10.1073/pnas.89.16.7674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Maron BA, Tang SS, Loscalzo J. S-nitrosothiols and the S-nitrosoproteome of the cardiovascular system. Antioxid Redox Signal. 2013;18:270–287. doi: 10.1089/ars.2012.4744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Rassaf T, Bryan NS, Kelm M, Feelisch M. Concomitant presence of N-nitroso and S-nitroso proteins in human plasma. Free Radic Biol Med. 2002;33:1590–1596. doi: 10.1016/s0891-5849(02)01183-8. [DOI] [PubMed] [Google Scholar]
- 107.Feelisch M, Rassaf T, Mnaimneh S, Singh N, Bryan NS, Jourd’Heuil D, Kelm M. Concomitant S-, N-, and heme-nitros(yl)ation in biological tissues and fluids: implications for the fate of NO in vivo. FASEB J. 2002;16:1775–1785. doi: 10.1096/fj.02-0363com. [DOI] [PubMed] [Google Scholar]
- 108.Thomas DD, Ridnour LA, Isenberg JS, Flores-Santana W, Switzer CH, Donzelli S, Hussain P, Vecoli C, Paolocci N, Ambs S, et al. The chemical biology of nitric oxide: implications in cellular signaling. Free Radic Biol Med. 2008;45:18–31. doi: 10.1016/j.freeradbiomed.2008.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Stocker R, Keaney JF. Role of oxidative modifications in atherosclerosis. Physiol Rev. 2004;84:1381–1478. doi: 10.1152/physrev.00047.2003. [DOI] [PubMed] [Google Scholar]
- 110.Laursen JB, Somers M, Kurz S, McCann L, Warnholtz A, Freeman BA, Tarpey M, Fukai T, Harrison DG. Endothelial regulation of vasomotion in apoE-deficient mice: implications for interactions between peroxynitrite and tetrahydrobiopterin. Circulation. 2001;103:1282–1288. doi: 10.1161/01.cir.103.9.1282. [DOI] [PubMed] [Google Scholar]
- 111.Bendall JK, Douglas G, McNeill E, Channon KM, Crabtree MJ. Tetrahydrobiopterin in cardiovascular health and disease. Antioxid Redox Signal. 2014;20:3040–3077. doi: 10.1089/ars.2013.5566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Fang FC, Nathan CF. Man is not a mouse: reply. J Leukoc Biol. 2007;81:580. doi: 10.1189/jlb.1206715. [DOI] [PubMed] [Google Scholar]
- 113.Schneemann M, Schoeden G. Macrophage biology and immunology: man is not a mouse. J Leukoc Biol. 2007;81:579; discussion 580. doi: 10.1189/jlb.1106702. [DOI] [PubMed] [Google Scholar]
- 114.Fuchs D, Murr C, Reibnegger G, Weiss G, Werner ER, Werner-Felmayer G, Wachter H. Nitric oxide synthase and antimicrobial armature of human macrophages. J Infect Dis. 1994;169:224–225. doi: 10.1093/infdis/169.1.224. [DOI] [PubMed] [Google Scholar]
- 115.Thomas SR, Terentis AC, Cai H, Takikawa O, Levina A, Lay PA, Freewan M, Stocker R. Post-translational regulation of human indoleamine 2,3-dioxygenase activity by nitric oxide. J Biol Chem. 2007;282:23778–23787. doi: 10.1074/jbc.M700669200. [DOI] [PubMed] [Google Scholar]
- 116.Alberati-Giani D, Malherbe P, Ricciardi-Castagnoli P, Köhler C, Denis-Donini S, Cesura AM. Differential regulation of indoleamine 2,3-dioxygenase expression by nitric oxide and inflammatory mediators in IFN-gamma-activated murine macrophages and microglial cells. J Immunol. 1997;159:419–426. [PubMed] [Google Scholar]
- 117.Olson NC, Sallam R, Doyle MF, Tracy RP, Huber SA. T helper cell polarization in healthy people: implications for cardiovascular disease. J Cardiovasc Transl Res. 2013;6:772–786. doi: 10.1007/s12265-013-9496-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Magen E, Borkow G, Bentwich Z, Mishal J, Scharf S. Can worms defend our hearts? Chronic helminthic infections may attenuate the development of cardiovascular diseases. Med Hypotheses. 2005;64:904–909. doi: 10.1016/j.mehy.2004.09.028. [DOI] [PubMed] [Google Scholar]
- 119.Libby P, Lichtman AH, Hansson GK. Immune effector mechanisms implicated in atherosclerosis: from mice to humans. Immunity. 2013;38:1092–1104. doi: 10.1016/j.immuni.2013.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Schulte S, Sukhova GK, Libby P. Genetically programmed biases in Th1 and Th2 immune responses modulate atherogenesis. Am J Pathol. 2008;172:1500–1508. doi: 10.2353/ajpath.2008.070776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.George J, Shoenfeld Y, Gilburd B, Afek A, Shaish A, Harats D. Requisite role for interleukin-4 in the acceleration of fatty streaks induced by heat shock protein 65 or Mycobacterium tuberculosis. Circ Res. 2000;86:1203–1210. doi: 10.1161/01.res.86.12.1203. [DOI] [PubMed] [Google Scholar]
- 122.Huber SA, Sakkinen P, David C, Newell MK, Tracy RP. T helper-cell phenotype regulates atherosclerosis in mice under conditions of mild hypercholesterolemia. Circulation. 2001;103:2610–2616. doi: 10.1161/01.cir.103.21.2610. [DOI] [PubMed] [Google Scholar]
- 123.Engelbertsen D, Andersson L, Ljungcrantz I, Wigren M, Hedblad B, Nilsson J, Björkbacka H. T-helper 2 immunity is associated with reduced risk of myocardial infarction and stroke. Arterioscler Thromb Vasc Biol. 2013;33:637–644. doi: 10.1161/ATVBAHA.112.300871. [DOI] [PubMed] [Google Scholar]
- 124.Shimizu K, Shichiri M, Libby P, Lee RT, Mitchell RN. Th2-predominant inflammation and blockade of IFN-gamma signaling induce aneurysms in allografted aortas. J Clin Invest. 2004;114:300–308. doi: 10.1172/JCI19855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Praticò D. Antioxidants and endothelium protection. Atherosclerosis. 2005;181:215–224. doi: 10.1016/j.atherosclerosis.2005.03.005. [DOI] [PubMed] [Google Scholar]
- 126.Poljsak B, Milisav I. The neglected significance of “antioxidative stress”. Oxid Med Cell Longev. 2012;2012:480895. doi: 10.1155/2012/480895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Romagnani S. Type 1 T helper and type 2 T helper cells: functions, regulation and role in protection and disease. Int J Clin Lab Res. 1991;21:152–158. doi: 10.1007/BF02591635. [DOI] [PubMed] [Google Scholar]
- 128.Jenny M, Klieber M, Zaknun D, Schroecksnadel S, Kurz K, Ledochowski M, Schennach H, Fuchs D. In vitro testing for anti-inflammatory properties of compounds employing peripheral blood mononuclear cells freshly isolated from healthy donors. Inflamm Res. 2011;60:127–135. doi: 10.1007/s00011-010-0244-y. [DOI] [PubMed] [Google Scholar]
- 129.Rimm EB, Stampfer MJ, Ascherio A, Giovannucci E, Colditz GA, Willett WC. Vitamin E consumption and the risk of coronary heart disease in men. N Engl J Med. 1993;328:1450–1456. doi: 10.1056/NEJM199305203282004. [DOI] [PubMed] [Google Scholar]
- 130.Stampfer MJ, Hennekens CH, Manson JE, Colditz GA, Rosner B, Willett WC. Vitamin E consumption and the risk of coronary disease in women. N Engl J Med. 1993;328:1444–1449. doi: 10.1056/NEJM199305203282003. [DOI] [PubMed] [Google Scholar]
- 131.Katsiki N, Manes C. Is there a role for supplemented antioxidants in the prevention of atherosclerosis? Clin Nutr. 2009;28:3–9. doi: 10.1016/j.clnu.2008.10.011. [DOI] [PubMed] [Google Scholar]
- 132.Fuchs D, Sperner-Unterweger B. Can intake of extra antioxidants delay the development and progression of atherosclerosis? Atherosclerosis. 2013;226:43–44. doi: 10.1016/j.atherosclerosis.2012.08.019. [DOI] [PubMed] [Google Scholar]
- 133.Mangge H, Summers K, Almer G, Prassl R, Weghuber D, Schnedl W, Fuchs D. Antioxidant food supplements and obesity-related inflammation. Curr Med Chem. 2013;20:2330–2337. doi: 10.2174/0929867311320180004. [DOI] [PubMed] [Google Scholar]
- 134.Riccioni G, Bucciarelli T, Mancini B, Corradi F, Di Ilio C, Mattei PA, D’Orazio N. Antioxidant vitamin supplementation in cardiovascular diseases. Ann Clin Lab Sci. 2007;37:89–95. [PubMed] [Google Scholar]
- 135.Sachidanandam K, Fagan SC, Ergul A. Oxidative stress and cardiovascular disease: antioxidants and unresolved issues. Cardiovasc Drug Rev. 2005;23:115–132. doi: 10.1111/j.1527-3466.2005.tb00160.x. [DOI] [PubMed] [Google Scholar]
- 136.Saremi A, Arora R. Vitamin E and cardiovascular disease. Am J Ther. 2010;17:e56–e65. doi: 10.1097/MJT.0b013e31819cdc9a. [DOI] [PubMed] [Google Scholar]
- 137.Gerss J, Köpcke W. The questionable association of vitamin E supplementation and mortality--inconsistent results of different meta-analytic approaches. Cell Mol Biol (Noisy-le-grand) 2009;55 Suppl:OL1111–OL1120. [PubMed] [Google Scholar]
- 138.Mangge H, Weghuber D, Prassl R, Haara A, Schnedl W, Postolache TT, Fuchs D. The Role of Vitamin D in Atherosclerosis Inflammation Revisited: More a Bystander than a Player? Curr Vasc Pharmacol. 2013:Epub ahead of print. doi: 10.2174/1570161111666131209125454. [DOI] [PubMed] [Google Scholar]
- 139.Bostom AG, Rosenberg IH, Silbershatz H, Jacques PF, Selhub J, D’Agostino RB, Wilson PW, Wolf PA. Nonfasting plasma total homocysteine levels and stroke incidence in elderly persons: the Framingham Study. Ann Intern Med. 1999;131:352–355. doi: 10.7326/0003-4819-131-5-199909070-00006. [DOI] [PubMed] [Google Scholar]
- 140.McCully KS. Homocysteine and vascular disease. Nat Med. 1996;2:386–389. doi: 10.1038/nm0496-386. [DOI] [PubMed] [Google Scholar]
- 141.Debreceni B, Debreceni L. Why do homocysteine-lowering B vitamin and antioxidant E vitamin supplementations appear to be ineffective in the prevention of cardiovascular diseases? Cardiovasc Ther. 2012;30:227–233. doi: 10.1111/j.1755-5922.2011.00266.x. [DOI] [PubMed] [Google Scholar]
- 142.Frick B, Gruber B, Schroecksnadel K, Leblhuber F, Fuchs D. Homocysteine but not neopterin declines in demented patients on B vitamins. J Neural Transm. 2006;113:1815–1819. doi: 10.1007/s00702-006-0539-x. [DOI] [PubMed] [Google Scholar]
- 143.Schroecksnadel K, Grammer TB, Boehm BO, März W, Fuchs D. Total homocysteine in patients with angiographic coronary artery disease correlates with inflammation markers. Thromb Haemost. 2010;103:926–935. doi: 10.1160/TH09-07-0422. [DOI] [PubMed] [Google Scholar]
- 144.Schroecksnadel K, Walter RB, Weiss G, Mark M, Reinhart WH, Fuchs D. Association between plasma thiols and immune activation marker neopterin in stable coronary heart disease. Clin Chem Lab Med. 2008;46:648–654. doi: 10.1515/cclm.2008.121. [DOI] [PubMed] [Google Scholar]
- 145.Fuchs D, Jaeger M, Widner B, Wirleitner B, Artner-Dworzak E, Leblhuber F. Is hyperhomocysteinemia due to the oxidative depletion of folate rather than to insufficient dietary intake? Clin Chem Lab Med. 2001;39:691–694. doi: 10.1515/CCLM.2001.113. [DOI] [PubMed] [Google Scholar]
- 146.Virmani A, Pinto L, Binienda Z, Ali S. Food, nutrigenomics, and neurodegeneration--neuroprotection by what you eat! Mol Neurobiol. 2013;48:353–362. doi: 10.1007/s12035-013-8498-3. [DOI] [PubMed] [Google Scholar]
- 147.Brown BG, Zhao XQ, Chait A, Fisher LD, Cheung MC, Morse JS, Dowdy AA, Marino EK, Bolson EL, Alaupovic P, et al. Simvastatin and niacin, antioxidant vitamins, or the combination for the prevention of coronary disease. N Engl J Med. 2001;345:1583–1592. doi: 10.1056/NEJMoa011090. [DOI] [PubMed] [Google Scholar]
- 148.Cheung MC, Zhao XQ, Chait A, Albers JJ, Brown BG. Antioxidant supplements block the response of HDL to simvastatin-niacin therapy in patients with coronary artery disease and low HDL. Arterioscler Thromb Vasc Biol. 2001;21:1320–1326. doi: 10.1161/hq0801.095151. [DOI] [PubMed] [Google Scholar]
- 149.Hausenloy DJ, Yellon DM. Targeting residual cardiovascular risk: raising high-density lipoprotein cholesterol levels. Heart. 2008;94:706–714. doi: 10.1136/hrt.2007.125401. [DOI] [PubMed] [Google Scholar]
- 150.Baginsky P. Should we treat all patients with coronary heart disease or the equivalent with statins? Curr Atheroscler Rep. 2009;11:28–35. doi: 10.1007/s11883-009-0005-y. [DOI] [PubMed] [Google Scholar]
- 151.Stoll LL, McCormick ML, Denning GM, Weintraub NL. Antioxidant effects of statins. Drugs Today (Barc) 2004;40:975–990. doi: 10.1358/dot.2004.40.12.872573. [DOI] [PubMed] [Google Scholar]
- 152.Ortego M, Bustos C, Hernández-Presa MA, Tuñón J, Díaz C, Hernández G, Egido J. Atorvastatin reduces NF-kappaB activation and chemokine expression in vascular smooth muscle cells and mononuclear cells. Atherosclerosis. 1999;147:253–261. doi: 10.1016/s0021-9150(99)00193-8. [DOI] [PubMed] [Google Scholar]
- 153.Neurauter G, Wirleitner B, Laich A, Schennach H, Weiss G, Fuchs D. Atorvastatin suppresses interferon-gamma -induced neopterin formation and tryptophan degradation in human peripheral blood mononuclear cells and in monocytic cell lines. Clin Exp Immunol. 2003;131:264–267. doi: 10.1046/j.1365-2249.2003.02021.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Hakamada-Taguchi R, Uehara Y, Kuribayashi K, Numabe A, Saito K, Negoro H, Fujita T, Toyo-oka T, Kato T. Inhibition of hydroxymethylglutaryl-coenzyme a reductase reduces Th1 development and promotes Th2 development. Circ Res. 2003;93:948–956. doi: 10.1161/01.RES.0000101298.76864.14. [DOI] [PubMed] [Google Scholar]
- 155.Wolfrum S, Jensen KS, Liao JK. Endothelium-dependent effects of statins. Arterioscler Thromb Vasc Biol. 2003;23:729–736. doi: 10.1161/01.ATV.0000063385.12476.A7. [DOI] [PubMed] [Google Scholar]
- 156.Lin R, Liu J, Peng N, Yang G, Gan W, Wang W. Lovastatin reduces nuclear factor kappaB activation induced by C-reactive protein in human vascular endothelial cells. Biol Pharm Bull. 2005;28:1630–1634. doi: 10.1248/bpb.28.1630. [DOI] [PubMed] [Google Scholar]
- 157.Dichtl W, Dulak J, Frick M, Alber HF, Schwarzacher SP, Ares MP, Nilsson J, Pachinger O, Weidinger F. HMG-CoA reductase inhibitors regulate inflammatory transcription factors in human endothelial and vascular smooth muscle cells. Arterioscler Thromb Vasc Biol. 2003;23:58–63. doi: 10.1161/01.atv.0000043456.48735.20. [DOI] [PubMed] [Google Scholar]
- 158.van Haelst PL, Liem A, van Boven AJ, Veeger NJ, van Veldhuisen DJ, Tervaert JW, Gans RO, Zijlstra F. Usefulness of elevated neopterin and C-reactive protein levels in predicting cardiovascular events in patients with non-Q-wave myocardial infarction. Am J Cardiol. 2003;92:1201–1203. doi: 10.1016/j.amjcard.2003.07.031. [DOI] [PubMed] [Google Scholar]
- 159.Walter RB, Fuchs D, Weiss G, Walter TR, Reinhart WH. HMG-CoA reductase inhibitors are associated with decreased serum neopterin levels in stable coronary artery disease. Clin Chem Lab Med. 2003;41:1314–1319. doi: 10.1515/CCLM.2003.200. [DOI] [PubMed] [Google Scholar]
- 160.Schroecksnadel K, Winkler C, Wirleitner B, Schennach H, Fuchs D. Aspirin down-regulates tryptophan degradation in stimulated human peripheral blood mononuclear cells in vitro. Clin Exp Immunol. 2005;140:41–45. doi: 10.1111/j.1365-2249.2005.02746.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Winkler C, Schroecksnadel K, Schennach H, Fuchs D. Vitamin C and E suppress mitogen-stimulated peripheral blood mononuclear cells in vitro. Int Arch Allergy Immunol. 2007;142:127–132. doi: 10.1159/000096438. [DOI] [PubMed] [Google Scholar]
- 162.Cangemi R, Loffredo L, Carnevale R, Pignatelli P, Violi F. Statins enhance circulating vitamin E. Int J Cardiol. 2008;123:172–174. doi: 10.1016/j.ijcard.2006.11.115. [DOI] [PubMed] [Google Scholar]
- 163.Cangemi R, Loffredo L, Carnevale R, Perri L, Patrizi MP, Sanguigni V, Pignatelli P, Violi F. Early decrease of oxidative stress by atorvastatin in hypercholesterolaemic patients: effect on circulating vitamin E. Eur Heart J. 2008;29:54–62. doi: 10.1093/eurheartj/ehm565. [DOI] [PubMed] [Google Scholar]
- 164.Violi F, Loffredo L, Musella L, Marcoccia A. Should antioxidant status be considered in interventional trials with antioxidants? Heart. 2004;90:598–602. doi: 10.1136/hrt.2003.026930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Vardi M, Levy NS, Levy AP. Vitamin E in the prevention of cardiovascular disease: the importance of proper patient selection. J Lipid Res. 2013;54:2307–2314. doi: 10.1194/jlr.R026641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Gostner J, Ciardi C, Becker K, Fuchs D, Sucher R. Immunoregulatory impact of food antioxidants. Curr Pharm Des. 2014;20:840–849. doi: 10.2174/13816128113199990047. [DOI] [PubMed] [Google Scholar]
- 167.Zaknun D, Schroecksnadel S, Kurz K, Fuchs D. Potential role of antioxidant food supplements, preservatives and colorants in the pathogenesis of allergy and asthma. Int Arch Allergy Immunol. 2012;157:113–124. doi: 10.1159/000329137. [DOI] [PubMed] [Google Scholar]
- 168.Bjelakovic G, Nikolova D, Gluud C. Antioxidant supplements and mortality. Curr Opin Clin Nutr Metab Care. 2014;17:40–44. doi: 10.1097/MCO.0000000000000009. [DOI] [PubMed] [Google Scholar]
- 169.Katagiri H, Yamada T, Oka Y. Adiposity and cardiovascular disorders: disturbance of the regulatory system consisting of humoral and neuronal signals. Circ Res. 2007;101:27–39. doi: 10.1161/CIRCRESAHA.107.151621. [DOI] [PubMed] [Google Scholar]
- 170.Hotamisligil GS. Inflammation and metabolic disorders. Nature. 2006;444:860–867. doi: 10.1038/nature05485. [DOI] [PubMed] [Google Scholar]
- 171.Charrière G, Cousin B, Arnaud E, André M, Bacou F, Penicaud L, Casteilla L. Preadipocyte conversion to macrophage. Evidence of plasticity. J Biol Chem. 2003;278:9850–9855. doi: 10.1074/jbc.M210811200. [DOI] [PubMed] [Google Scholar]
- 172.Hotamisligil GS, Erbay E. Nutrient sensing and inflammation in metabolic diseases. Nat Rev Immunol. 2008;8:923–934. doi: 10.1038/nri2449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Medzhitov R. Origin and physiological roles of inflammation. Nature. 2008;454:428–435. doi: 10.1038/nature07201. [DOI] [PubMed] [Google Scholar]
- 174.Mozaffarian D, Willett WC. Trans fatty acids and cardiovascular risk: a unique cardiometabolic imprint? Curr Atheroscler Rep. 2007;9:486–493. doi: 10.1007/s11883-007-0065-9. [DOI] [PubMed] [Google Scholar]
- 175.Sivasankaran S. The cardio-protective diet. Indian J Med Res. 2010;132:608–616. [PMC free article] [PubMed] [Google Scholar]
- 176.Maier E, Kurz K, Jenny M, Schennach H, Ueberall F, Fuchs D. Food preservatives sodium benzoate and propionic acid and colorant curcumin suppress Th1-type immune response in vitro. Food Chem Toxicol. 2010;48:1950–1956. doi: 10.1016/j.fct.2010.04.042. [DOI] [PubMed] [Google Scholar]
- 177.Ciardi C, Jenny M, Tschoner A, Ueberall F, Patsch J, Pedrini M, Ebenbichler C, Fuchs D. Food additives such as sodium sulphite, sodium benzoate and curcumin inhibit leptin release in lipopolysaccharide-treated murine adipocytes in vitro. Br J Nutr. 2012;107:826–833. doi: 10.1017/S0007114511003680. [DOI] [PubMed] [Google Scholar]
- 178.Yang R, Barouch LA. Leptin signaling and obesity: cardiovascular consequences. Circ Res. 2007;101:545–559. doi: 10.1161/CIRCRESAHA.107.156596. [DOI] [PubMed] [Google Scholar]
- 179.Stelzer I, Zelzer S, Raggam RB, Prüller F, Truschnig-Wilders M, Meinitzer A, Schnedl WJ, Horejsi R, Möller R, Weghuber D, et al. Link between leptin and interleukin-6 levels in the initial phase of obesity related inflammation. Transl Res. 2012;159:118–124. doi: 10.1016/j.trsl.2011.10.001. [DOI] [PubMed] [Google Scholar]
- 180.Mangge H, Summers KL, Meinitzer A, Zelzer S, Almer G, Prassl R, Schnedl WJ, Reininghaus E, Paulmichl K, Weghuber D, et al. Obesity-related dysregulation of the tryptophan-kynurenine metabolism: role of age and parameters of the metabolic syndrome. Obesity (Silver Spring) 2014;22:195–201. doi: 10.1002/oby.20491. [DOI] [PubMed] [Google Scholar]
- 181.Kaliora AC, Dedoussis GV, Schmidt H. Dietary antioxidants in preventing atherogenesis. Atherosclerosis. 2006;187:1–17. doi: 10.1016/j.atherosclerosis.2005.11.001. [DOI] [PubMed] [Google Scholar]
- 182.Kaliora AC, Dedoussis GV. Natural antioxidant compounds in risk factors for CVD. Pharmacol Res. 2007;56:99–109. doi: 10.1016/j.phrs.2007.04.018. [DOI] [PubMed] [Google Scholar]
- 183.Godfrey KM, Gluckman PD, Hanson MA. Developmental origins of metabolic disease: life course and intergenerational perspectives. Trends Endocrinol Metab. 2010;21:199–205. doi: 10.1016/j.tem.2009.12.008. [DOI] [PubMed] [Google Scholar]
- 184.Lillycrop KA, Burdge GC. The effect of nutrition during early life on the epigenetic regulation of transcription and implications for human diseases. J Nutrigenet Nutrigenomics. 2011;4:248–260. doi: 10.1159/000334857. [DOI] [PubMed] [Google Scholar]
- 185.Napoli C. Developmental mechanisms involved in the primary prevention of atherosclerosis and cardiovascular disease. Curr Atheroscler Rep. 2011;13:170–175. doi: 10.1007/s11883-010-0156-x. [DOI] [PubMed] [Google Scholar]
- 186.Lillycrop KA, Burdge GC. Epigenetic changes in early life and future risk of obesity. Int J Obes (Lond) 2011;35:72–83. doi: 10.1038/ijo.2010.122. [DOI] [PubMed] [Google Scholar]
- 187.Sakamoto K, Kimura J. Mechanism of statin-induced rhabdomyolysis. J Pharmacol Sci. 2013;123:289–294. doi: 10.1254/jphs.13r06cp. [DOI] [PubMed] [Google Scholar]
- 188.Rhodes T, Norquist JM, Sisk CM, McQuarrie K, Trovato A, Liao J, Miller T, Maccubbin D, Watson DJ. The association of flushing bother, impact, treatment satisfaction and discontinuation of niacin therapy. Int J Clin Pract. 2013;67:1238–1246. doi: 10.1111/ijcp.12213. [DOI] [PubMed] [Google Scholar]