

Abstract
Human alkyladenine-DNA glycosylase (AAG) initiates base excision repair (BER) of alkylated and deaminated bases in DNA. Here, we assessed the mutability of the AAG substrate binding pocket, and the essentiality of individual binding pocket amino acids for survival of methylation damage. We used oligonucleotide-directed mutagenesis to randomize 19 amino acids, 8 of which interact with substrate bases, and created more than 4.5 million variants. We expressed the mutant AAG's in repair-deficient E. coli and selected for protection against the cytotoxicity of either methylmethane sulfonate (MMS) or methyl-lexitropsin (Me-lex), an agent that produces 3-methyladenine as the predominant base lesion. Sequence analysis of 116 methylation-resistant mutants revealed no substitutions for highly conserved Tyr127and His136. In contrast, one mutation, L180F, was greatly enriched in both the MMS- and Me-lex-resistant libraries. Expression of the L180F single mutant conferred 4.4-fold enhanced survival at the high dose of MMS used for selection. The homogeneous L180F mutant enzyme exhibited 2.2-fold reduced excision of 3-methyladenine and 7.3-fold reduced excision of 7-methylguanine from methylated calf thymus DNA. Decreased excision of methylated bases by the mutant glycosylase could promote survival at high MMS concentrations, where the capacity of downstream enzymes to process toxic BER intermediates may be saturated. The mutant also displayed 6.6-, and 3.0-fold reduced excision of 1,N6-ethenoadenine and hypoxanthine from oligonucleotide substrates, respectively, and a 1.7-fold increase in binding to abasic site-containing DNA. Our work provides in vivo evidence for the substrate binding mechanism deduced from crystal structures, illuminates the function of Leu180 in wild-type human AAG, and is consistent with a role for balanced expression of BER enzymes in damage survival.
Keywords: Alkylating agents, base excision repair, 3-methyladenine, 7-methylguanine, methyl-lexitropsin, random mutagenesis
1. Introduction
Alkylating agents are widely dispersed in the environment, occur endogenously, and are frequently used in the treatment of cancer [1,2]. If not repaired, alkylated bases in DNA can be mutagenic and cytotoxic [3]. For example, modification at the N3 position of adenine can disrupt interaction of DNA polymerases with the template DNA strand [4,5], apparently leading to arrest of the replication machinery and cell death [6-8]. Base excision repair (BER) is a primary mechanism by which cells remove damaged bases from the genome, including alkylated bases, and has been conserved in evolution. BER pathways are initiated by a DNA glycosylase that recognizes the adducted base and hydrolyzes the N-glycosylic bond linking the damaged base to the sugar-phosphate backbone [9]. The resulting abasic site is then processed by AP endonuclease or Ap lyase, DNA polymerase and DNA ligase activities [10]. Importantly, abasic sites and the downstream interrupted-strand intermediates can lead to double-strand breaks and are therefore potentially cytotoxic. DNA glycosylase action thus commits cells to a pathway in which each step produces a different and possibly lethal form of damaged DNA, until repair is complete. As a consequence, the balanced expression [11,12] and coordinated action [10] of enzymes in BER pathways are important for survival of DNA damage.
Alkyladenine DNA glycosylases are found in organisms ranging from bacteria and plants to humans [2,13]. The alkyladenine-DNA glycosylase AAG, [also known as N-methylpurine DNA glycosylase (MPG), or ANPG] is the only known human DNA glycosylase that removes alkylated bases [1,9]. Human AAG is active on a broad range of chemically and structurally diverse substrates. In addition to 3-methyladenine (3-meA), AAG also excises the alkylated purines 7-methylguanine (7-meG) and 3-methylguanine (3-meG) [13]; the oxidative purine deamination products hypoxanthine, xanthine, and oxanine [14,15]; the lipid peroxidation-derived 1,N6-ethenoadenine (εA) and 1,N2-ethenoguanine (εG); the oxidative lesion 8-hydroxyguanine [16]; and, very inefficiently, normal guanine [13,17-19]. AAG has been reported to excise hypoxanthine most efficiently, followed by εA and 3-meA, leading to the suggestion that AAG may have primarily evolved as a hypoxanthine-DNA glycosylase [20,21].
Crystallographic studies and molecular modeling [22,23], together with site-specific mutagenesis [24], have provided substantial information concerning the mechanism of action of human AAG and the structural basis for binding adducted purines. Co-crystal structures of AAG complexed with lesion-containing DNA reveal that AAG, like other DNA glycosylases, uses a nucleotide flipping mechanism to recognize target bases in an extrahelical conformation [22,23]. AAG binds in the minor groove of DNA, and a single tyrosine (Tyr165) inserts into the minor groove, intercalating between the bases adjacent to, and occupying the space left by, the flipped-out nucleotide. The flipped-out nucleotide is captured in a substrate binding pocket, and, in the case of εA, stacks between the side-chains of Tyr127 on one side and His136 and Tyr159 on the other [23]. Cleavage of the glycosylic bond is catalyzed by a nucleophilic water molecule that is activated by the essential Glu125 [20,23]. Many evolutionarily conserved amino acids form the substrate binding pocket [22,24], and site-directed mutagenesis has begun to illuminate their functions. For example, Asn169 has been proposed to play an important role in substrate specificity. When a normal guanine is modeled into the substrate-binding pocket, the exocyclic amino group clashes with the Asn169 side chain [23]; in accord, replacements for Asn169 can enhance excision of guanine from mispairs [25]. Other substrate binding pocket residues include Leu180, which lies adjacent to the N3 position of εA, and is expected to pack closely with the methyl adduct of 3-meA [22].
Our present understanding of the roles of individual substrate binding pocket residues in substrate discrimination by AAG, and their importance for damage survival, is incomplete. Here, we used oligonucleotide-directed random mutagenesis (e.g., 26) and selection for methylating agent survival to examine the replaceability of amino acids that line the AAG substrate binding pocket. Analysis of methylation-resistant mutants identified mutable and immutable amino acids, yielding in vivo evidence for an essential role of Tyr127 and His136 in survival of methylation damage. Our results provide functional validation of crystallographically observed interactions, suggest functions of Leu180 in substrate discrimination by wild-type human AAG, and provide a presumptive example of the importance for damage survival of coordinated processing of intermediates in BER pathways.
2. Materials and methods
2.1. Strains and materials
The alkylation repair-deficient strain E. coli MV1932 (alkA1 tag) is a derivative of strain AB1157 [see [12]]. E. coli BL21(DE3) was purchased from Agilent technologies (Santa Clara, CA). Enzymes were from NEB (Beverly, MA), and oligonucleotides were from IDT (Coralville, IA). E. coli endonuclease IV was kindly provided by Dr. S. Bennett (Oregon State University). Anti-AAG (MPG) antibody was obtained from Imgenex (San Diego, CA).
2.2. Creation of pUC-AAG-SK and KX mutant libraries
The starting vector encoded a full length human AAG cDNA in a pUC19-derived plasmid. A schematic diagram of the construction of the pUC-AAG-SK and pUC-AAG-KX random mutant libraries is shown in Supplementary Fig. S1. The library designations SK and KX refer to the SacII and KpnI, and KpnI and XbaI silent restriction sites, respectively, that were introduced in the AAG coding sequence to allow substitution with oligonucleotide cassettes containing randomized codons. The SK library encodes 9 randomized amino acids (Ala126, Tyr127, Leu128, Gly129; Glu133, Ala134, Ala135, His136, and Ser137) and the KX library 10 randomized amino acids (Val158, Tyr159, Ile160; Cys167, Met168, Asn169, Ile170; Val179, Leu180, and Leu181). To permit library construction, the pre-existing SacII and XbaI sites in the pUC19 multiple cloning region were eliminated via restriction digestion, followed by mung bean exonuclease digestion to remove non-complementary termini, filling with Klenow fragment, and religation. To generate silent restriction sites in the AAG reading frame, site-directed mutagenesis using a QuickChange kit (Stratagene) was performed, introducing the silent mutations A354C to create a SacII site; G462T to create a KpnI site; and A549T, G552A to create an XbaI site (numbers refer to the wild type AAG cDNA sequence.)
A hexahistidine tag and the factor Xa cleavage sequence were inserted at the N-terminus of AAG by PCR amplification with the forward primer 5′-GCA CGC GGA TCC ATG GGT GGT TCT CAT CAC CAT CAC CAT CAC GGT GGG ATC GAA GGT CGT GCT GTC ACC CCC GCT TTG CAG ATG AAG AAA CCA AAG CAG-3′ and the reverse primer 5′-GAT GCG GCC GCG GAG TTC TGT GCC ATT AGG AAG TCG CCG-3′. The amplified product was gel purified, cut with BamHI and SacII and religated into the correspondingly cut vector to create pUC-AAG (Fig. S1A).
To avoid contamination of the libraries with re-circularized vector containing wild-type AAG, inactive constructs (“dummy vectors”) with internal deletions in the AAG cDNA were made prior to library creation (Fig. S1A). To generate the internal deletion inserts for inactive SK and KX “dummy” constructs, the sense oligonucleotide 5′-CCG CGG CCT GCA CGT GGT CGG TAC CTG TGC TAG CAA GTA TCT AGA-3′ was annealed to the antisense oligonucleotide 5′-CTT AGA TAC TTG CTA GCA CAG GTA CCG ACC ACG TGC AGG CCG C-3′ to generate a DNA fragment with SacII and XbaI sticky ends and internal PmlI, KpnI, and NheI restriction sites. The fragment was digested with KpnI. To generate the pUC-AAG-SK and pUC-AAG-SK dummy vectors, the digestion products were ligated (T4 DNA ligase, Invitrogen) into pUC-AAG that was pre-cut with SacII and KpnI, or with KpnI and XbaI, respectively. The constructs were confirmed by sequencing. The SK random oligonucleotide cassette (Fig. S1B) was constructed by annealing the single-stranded oligomers SK sense: 5′-ACA GAA CTC CGC GGC CGC ATC GTG GAG ACC GAG GCA TAC CTG GGT CCA GAG GAT GAA GCC GCC CAC TCA CGC GGT GGC CGG CAG ACG-3′ and SK antisense: 5′-CAC GTA CAG GGT ACC CGG CTT CAT GAA CAT GCC TCG GTT GCG AGG CGT CTG CCG GCC ACC GCG-3′. The KX random oligonucleotide cassette (Fig. S1B) was constructed by annealing KX sense: 5′-G AAG CCG GGT ACC CTG TAC GTG TAC ATC ATT TAC GGC ATG TAC TTC TGC ATG AAC ATC TCC AGC CAG GGC GAC GGG-3′ with KX antisense: 5′-CAG CGG CTC TAG AGC TCG CAG CAA GAC GCA AGC CCC GTC GCC CTG GCT GGA-3′. Oligomers were synthesized to contain 88% wild-type nucleotide and 4% of each of the other three nucleotides at each underlined position; the 18 bp regions for hybridization are italicized. Annealed oligomers were extended with Klenow fragment, and the fully double-stranded SK random oligonucleotides were incubated with SacII and KpnI, and the KX random oligonucleotides with KpnI and XbaI. The resulting SK and KX random oligonucleotide cassettes were ligated into the correspondingly pre-cut pUC-AAG-SK and pUC-AAG-KX dummy constructs. Ligation products were cut with restriction enzymes (PmlI for the SK dummy vector and NheI for the KX dummy vector) to linearize recircularized dummy constructs lacking the randomized oligonucleotide inserts, but not mutant AAG constructs with the inserted cassettes; subsequent sequencing of unselected and selected mutants revealed no carry-over dummy constructs. To estimate library size, i.e., the number of variants subsequently screened for MMS survival, aliquots of plasmid DNA harboring the randomized oligonucleotide inserts were electroporated into E. coli XL1-Blue cells and the number of transformants was determined by plating on LB-carbenicillin agar.
2.3. Construction of the YH library
Tyr127 and His136 were targeted for further mutagenesis by generation of the YH library containing fully randomized nucleotides at the codons for these two amino acids. Procedures followed those for construction of the SK and KX libraries. The YH sense oligonucleotide [5′-ACA GAA CTC CGC GGC CGC ATC GTG GAG ACC GAG GCA NNN CTG GGT CCA GAG GAT GAA GCC GCC NNN TCA CGC GGT GGC CGG CAG ACG-3′] contained 25% of each nucleotide at the positions underlined. The YH anti-sense oligonucleotide was 5′-CAC GTA CAG GGT ACG CGG CTT CAT GAA CAT GCC TCG GTT GCG AGG CGT CTG CCG GCC ACC GCG-3′. The annealed oligomers were cut with SacII and KpnI and ligated into the correspondingly cut SK dummy vector.
2.4. Selection and characterization of active AAG mutant libraries
MV1932 cells were transformed with the pUC-AAG-SK, pUC-AAG-KX or pUC-AAG-YH mutant libraries or dummy vectors, and grown to mid-log phase in LB-carbenicillin medium. Aliquots of cultures were plated on LB-carbenicillin agar, and clones were subsequently isolated in order to characterize the unselected libraries. Mid-log phase cells were incubated with 0.2% (w/v) MMS for l hr at 37 °C, serially diluted into M9 minimal medium and spread on LB-carbenicillin plates. Survival was estimated by counting surviving colonies after incubation at 37 °C for 24 hr. Plasmids from a total of 72 unselected and 118 selected clones from the SK and KX libraries, and 15 unselected and 27 selected clones from the YH libraries were isolated. The 198 bp SacII-XbaI region of AAG was sequenced using the primer 5′-ACTGGG GTTGGAGTTCTTCG-3′ and BigDye 3.0 (ABI). Mutant sequences were aligned and identified with Sequencher 3.0 (Gene Codes Corporation, Ann Arbor, MI).
Mutants resistant to Me-lex were isolated by growing cells to mid-logarithmic phase in LB-carbenicillin medium. Prior to Me-lex selection, library aliquots were plated on LB-carbenicillin, and unselected clones were isolated and sequenced in order to characterize the composition of the unselected SK and KX libraries. MV2157 cells (1 mL) expressing wild-type pUC-AAG, the SK or KX library, or SK dummy were washed with phosphate-buffered saline, resuspended in citrate buffer, and treated with 200 μM Me-lex for 1 hr. Cells were then diluted into M9 minimal media, spread on LB-carbenicillin plates and incubated for 24 hr at 37 °C. Plasmids from surviving colonies were isolated and sequenced.
2.5. Purification of wild-type and L180F AAG
Full length wild-type AAG and the mutant L180F were cloned into the expression vector pET101/D-TOPO via directional TOPO cloning (Invitrogen). The resulting AAGs are fused to a C-terminal hexahistidine tag via a V5 epitope site and expressed under the control of the T7 promoter. The pET-AAGs were transformed into E. coli BL21(DE3) and grown at 30 °C in 6 L of 2×YT medium containing 0.5 % glucose and 50 μg/ml carbenicillin; IPTG (1 mM final concentration) was added at OD600 = ∼0.6. After further incubation for 16 h at 30 °C, cells were harvested by centrifugation and resuspended in 120 ml of buffer SB [50 mM Tris-HCl (pH 8.0), 10 mM NaCl, 1 mM PMSF, 1 mM DTT and 10 % (w/v) glycerol], and adjusted to 500 μg/ml lysozyme prior to storage at -80 °C. The cell suspension was thawed, immediately supplemented with 1× protease inhibitor cocktail (EMD Biosciences) and sonicated on ice. The following procedures were carried out at 4°C. After centrifugation at 20,000 ×g for 30 min, the supernatant fraction was reserved, and the cell pellet resuspended in 30 ml of buffer SB; the sonification and centrifugation steps were repeated and the supernatant fractions were combined (Fraction I). Fraction I was loaded onto a DEAE cellulose (Sigma) column (4.9 cm2 × 4 cm) equilibrated in buffer A [30 mM Tris-HCl (pH 8.0), 10 mM NaCl, 1 mM DTT, 1 mM EDTA, 2 mM benzamidine (EMD Biosciences) and 10 % (w/v) glycerol] and the column was washed with 60 ml of equilibration buffer. Fractions containing AAG activity were pooled and dialyzed against buffer A (Fraction II). Fraction II was applied to a SP-Sepharose column (1.8 cm2 × 4.2 cm) equilibrated in buffer A. The column was washed with 40 ml of buffer A, followed by a 45 ml linear gradient of 10-500 mM NaCl in buffer A. The fractions were analyzed in 12.5% SDS-polyacrylamide gels, and fractions containing AAG were pooled and dialyzed against buffer B [50 mM sodium phosphate (pH 8.0), 300 mM NaCl, 10 mM imidazole, 1 mM β-mercatoethanol, 2 mM benzamidine and 5 % (w/v) glycerol] (Fraction III). Fraction III was loaded onto a Qiagen nickel-nitrilotriacetic acid (Ni-NTA) affinity column (1.77 cm2 × 3 cm) equilibrated in buffer B. The column was washed with 20 ml of buffer B and then with a 30 ml linear gradient of 10-300 mM imidazole in buffer SB. Fractions containing AAG, identified by electrophoresis in 12.5 % SDS-polyacrylamide gels, were pooled (fraction IV). After dialysis against storage buffer [30 mM Tris-HCl (pH 7.4), 1 mM DTT, 1 mM EDTA, 2 mM benzamidine and 10 % (w/v) glycerol], the preparation was concentrated using an Amicon filter unit (MW cut-off 30,000), aliquoted and stored at -80 °C. Protein concentration was determined by the Bradford reaction [26] using the Bio-Rad Protein Assay with bovine serum albumin as a standard. Equivalent amounts of wild-type and mutant enzymes, as assessed in the Bradford assay, exhibited equivalent absorbance at A280.
2.6. DNA glycosylase assays
DNA glycosylase activity was measured by quantifying the release of [3H]-labeled N-methylpurines from calf thymus DNA methylated with [3H]-methylnitrosourea (1 mCi, GE Healthcare) as previously described [27]. Reaction mixtures (100 μl) contained [3H]-calf thymus DNA (20,000 cpm), 50 nM or 500 nM AAG or L180F mutant enzyme as specified, 20 mM Tris-HCl (pH 7.6), 100 mM KCl, 5 mM EDTA, 2 mM EGTA, 5 mM β-mercaptoethanol; protein concentrations were equalized by addition of bovine serum albumin. Following incubation at 37 °C for 0, 1, 2, 4, 8, or 16 min, reactions were stopped by addition of 0.1 volumes of 3 M sodium acetate (pH 5.2) and 3 volumes of ethanol followed by incubation at −20 °C for 1 hr to precipitate DNA. Non-radioactive 3-meA and 7-meG standards were added to the supernatant containing released 3H-methylated bases, and the solution was taken to dryness by vacuum desiccation. Samples were dissolved in 15 ml 0.1 M HCl and spotted on Whatman 3MM paper. Descending paper chromatography was performed for 18 hr in 70% isopropanol, 20% water and 10% concentrated ammonium hydroxide to separate 3-meA and 7-meG. Standards were visualized under UV light, and the paper was cut into 1 cm strips. Radioactivity was measured by scintillation counting.
DNA glycosylase activity was also measured by using a double-stranded 5′-32P end-labeled 36-mer DNA substrate containing either a εA•T or Hx•T base pair [15]. The lesion-containing strand was 5′ TTGGCAGCAGAATATTGCTXGCGGG-AATTCGGCGCG-3′ where X= εA or Hx. Reaction mixtures (10 μl) contained 1 pmol 32P-labeled DNA substrate, 62.5 fmol to 1.6 pmol of AAG or L180F mutant enzyme in 30 mM Tris-Hcl (pH 7.4), 1 mM EDTA, 1 mM DTT, 100 mM KCl. Following incubation at 37 °C for 30 min, reactions were stopped by heating at 70 °C for 5 min, brought to 50 mM KCl by addition of 1.2 μl 0.5 M KCl, and incubated with E. coli endonuclease IV (1 unit) at 37 °C for 30 min. Reactions were terminated by heating at 70 °C for 5 min and placed on ice for 3 min. Samples were then combined with an equal volume of 2X denaturing buffer containing 95% de-ionized formamide, 1 mM EDTA, and 0.05% bromophenol blue, heated at 97 °C for 3 min, and resolved by electrophoresis in 12% polyacrylamide, 8.3 M urea gels. Gels were dried and visualized by phosphor imaging, and the reaction products were analyzed by ImageQuant software (GE Healthcare).
2.7. DNA binding assay
The relative binding affinity for lesion-containing oligonucleotides was evaluated by using an electrophoretic mobility shift assay as previously described [25]. The 5′-32P end-labeled 36-mer double-stranded DNA ligand contained the lesion opposite T. The lesion-containing strand was 5′TTGGCAGCAGAATA-TTGCTXGCGGGAATTCGGCGCG-3′ where X was either εA, Hx or a tetrahydrofuran abasic site analog. Briefly, reaction mixtures (10 μl) contained 1 pmol of DNA ligand and 1-15 pmol AAG in 50 mM Tris-HCl (pH 7.4), 1 mM EDTA, 1 mM DTT, and 100 μg/ml acetylated bovine serum albumin. Following incubation on ice for 30 min, samples were analyzed by electrophoresis in 7.5% polyacrylamide gels. Gels were dried, visualized by phosphor imaging, and the reaction products were analyzed by ImageQuant software (GE Healthcare).
2.8. Fluorescence polarization binding assay
Steady-state fluorescence polarization assays were performed to compare the dissociation constants for binding of wild-type AAG vs. the L180F mutant enzyme to double-stranded DNA containing a single abasic (AP) site. The AP site-containing strand was the same as in the DNA binding assay, except that the DNA was 5′-end labeled with 6-carboxyfluorescein (FAM). The fluorescence polarization of FAM was measured at 25 °C with a SpectraMax M5 Microplate Reader (Molecular Devices Co., Sunnyvale, CA) by exciting at 495 nm and monitoring emission at 518 nm. Binding reactions (150 μl) were carried out in binding buffer (30 mM Tris-HCl [pH 7.4], 1 mM EDTA, 1 mM DTT, 25 mg/ml acetylated bovine serum albumin) and contained 100 nM FAM-labeled DNA and increasing concentrations of enzyme. Reaction mixtures were equilibrated at 25 °C for 15 min prior to fluorescence measurements. Assays were performed in triplicate. After subtracting the background DNA fluorescence polarization, measurements were fit to the following equation to determine the apparent dissociation constant Kd (41).
| Eq. (1) |
In Eq. (1), P represents the intensity of fluorescence polarization enhancement, c is the maximum value of the fluorescence polarization, Lo and Ro the total concentrations of DNA substrate and wild-type AAG or L180F mutant protein, respectively, Kd the overall dissociation constant for binding of wild-type or mutant protein to the FAM-AP·T-36-mer.
3. Results
3.1. Creation of AAG substrate binding pocket mutant libraries
Crystal structures and molecular modeling of AAG complexed with lesion-containing oligonucleotides have revealed an active site pocket that binds substrate bases while discriminating against normal purines. A model of AAG bound to a DNA oligomer containing εA is shown in Fig. 1A, and a more detailed view of the substrate binding pocket is illustrated in Fig. 1B. The proposed pocket is formed mainly by aromatic and hydrophobic residues, and is believed to bind substrate bases in part via stacking interactions involving the conjugated bases Tyr127, His136, and Tyr159 [22,23]. In order to provide evidence for the in vivo function of binding pocket residues, and to assess their essentiality for methylating agent resistance, we created libraries of mutants containing random substitutions for 19 wild-type amino acids. We mutated eight residues that contact the εA substrate in a co-crystal complex [Tyr127, Ala134, Ala135, His136, Tyr159, Cys167, Asn169, Leu180 [23]] (Fig. 1B), together with flanking residues in the primary sequence. We used random oligonucleotide replacement mutagenesis to create two libraries, designated SK and KX. The SK library targets nine residues in the top half of the model in Fig. 1B, including Tyr127, Ala134, Ala135, and His136; the KX library targets ten residues in the bottom half including Tyr159, Cys167, Asn169, and Leu180. Construction of the mutant libraries is described in Experimental Procedures and is represented schematically in Supplementary Fig. S1. Oligonucleotide mixtures that contained 12% random nucleotides at each position encoding a targeted amino acid replaced the wild-type codon in the libraries; i.e., each targeted position contained 88% wild-type nucleotide and 3% each of the other three nucleotides. The unselected SK and KX libraries contained 2.8 × 106 and 1.7 × 106 mutant AAGs, respectively.
Figure 1. Modelled AAG bound to eA-containing DNA and close-up view of the substrate binding pocket and randomized amino acids.
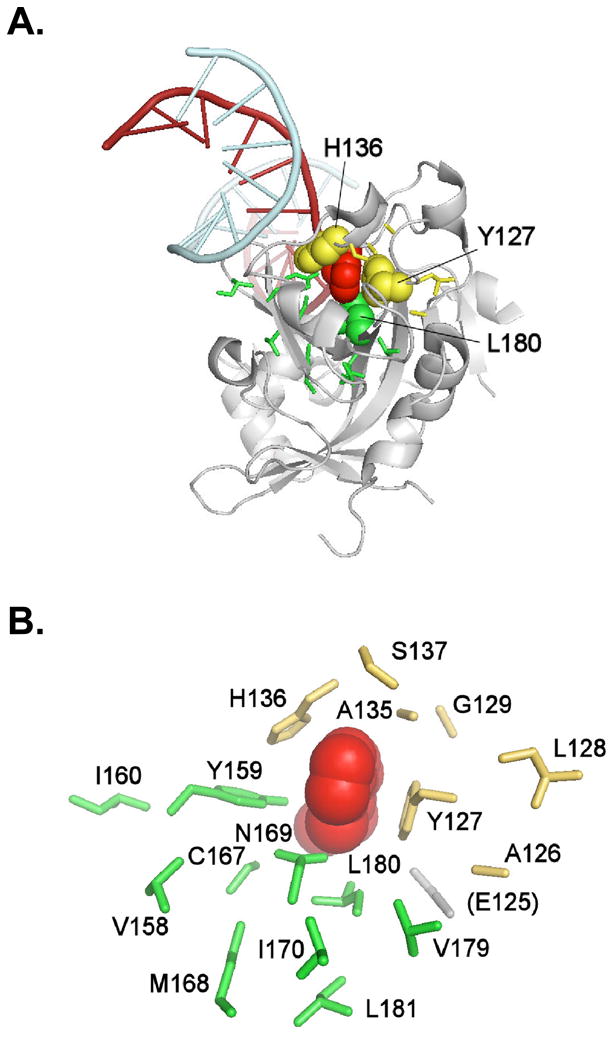
A, ribbon drawing of AAG illustrating the context of the residues that are mutated in this work and that surround the flipped-out base [coordinate set 1F4R (Lau et al (23) in the Protein Data Bank (40)]. The flipped-out base (εA, in 1F4R) is depicted as space-filling (colored red), as are His136 and Tyr127 (colored yellow), and Leu180 (colored green). The flipped-out base fits closely in the binding pocket and is stacked between Tyr127 on one side, and His136 and Tyr159 on the other. B, the substrate binding pocket and randomized residues. We randomized residues contacting the εA substrate (Tyr127, Ala134, Ala135, His136, Tyr159, Cys167, Asn169, Leu180), together with flanking amino acids in the primary sequence, by using oligonucleotide-directed mutagenesis. Residues randomized in the SK library are shown in yellow, residues randomized in the KX library in green. Ala134 is not shown. The figure was drawn using PyMOL (DeLano, W.L. The PyMOL Molecular Graphics System (2002) DeLano Scientific, Palo Alto, CA, USA. http://www.pymol.org).
We sequenced the randomized regions of 37 unselected clones from the SK library and 35 from the KX library. The majority of clones contained multiple nucleotide substitutions, yielding an average of 2.5 amino acid changes per SK mutant cDNA and 2.3 amino acid changes per KX mutant cDNA (Fig. 2A,B). Multiple nucleotide substitutions were observed at each targeted codon in both libraries (Fig. 3B). Approximately 15% and 4% of the unselected SK and KX libraries harbored frameshift mutations, respectively, presumably derived from incomplete extension during random oligonucleotide synthesis. No mutant cDNA was observed more than once.
Figure 2. Characterization of unselected and selected AAG mutant libraries.
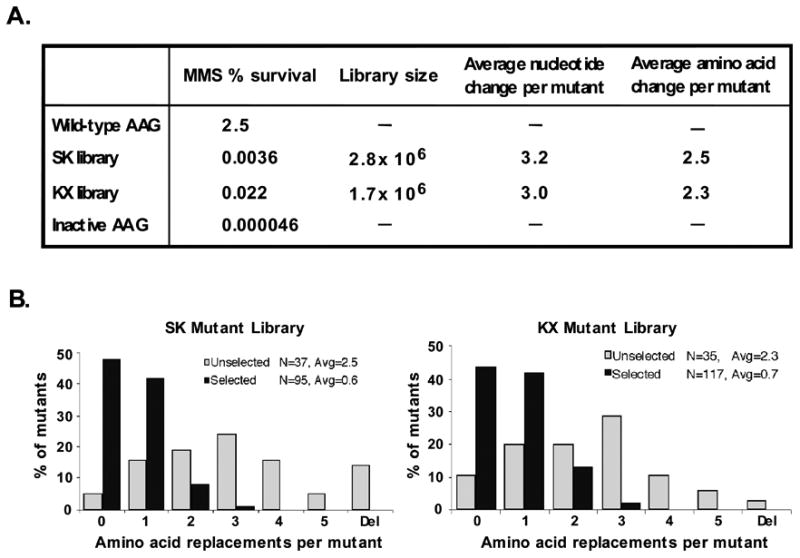
A, unselected SK and KX libraries. Library size was measured by plating on LB-carbenicillin medium and counting colonies; nucleotide and amino acid changes were determined by sequencing 72 individual mutants. For assessment of MMS resistance, cultures of log phase MV1932 cells expressing either wild type AAG, the SK or KX library, or inactive AAG (SK dummy vector) were treated with 0.2% MMS for 1 hr at 37 °C, and surviving colonies were counted. B, The distribution of amino acid changes in the SK and KX libraries was assessed from the sequences of the 73 unselected mutants and 118 selected mutants that survived the MMS treatment described above. The distribution shows post-selection enrichment for the wild-type AAG sequence and a shift toward fewer substitutions per clone.
Figure 3. Amino acid substitutions in unselected and selected AAG mutants.
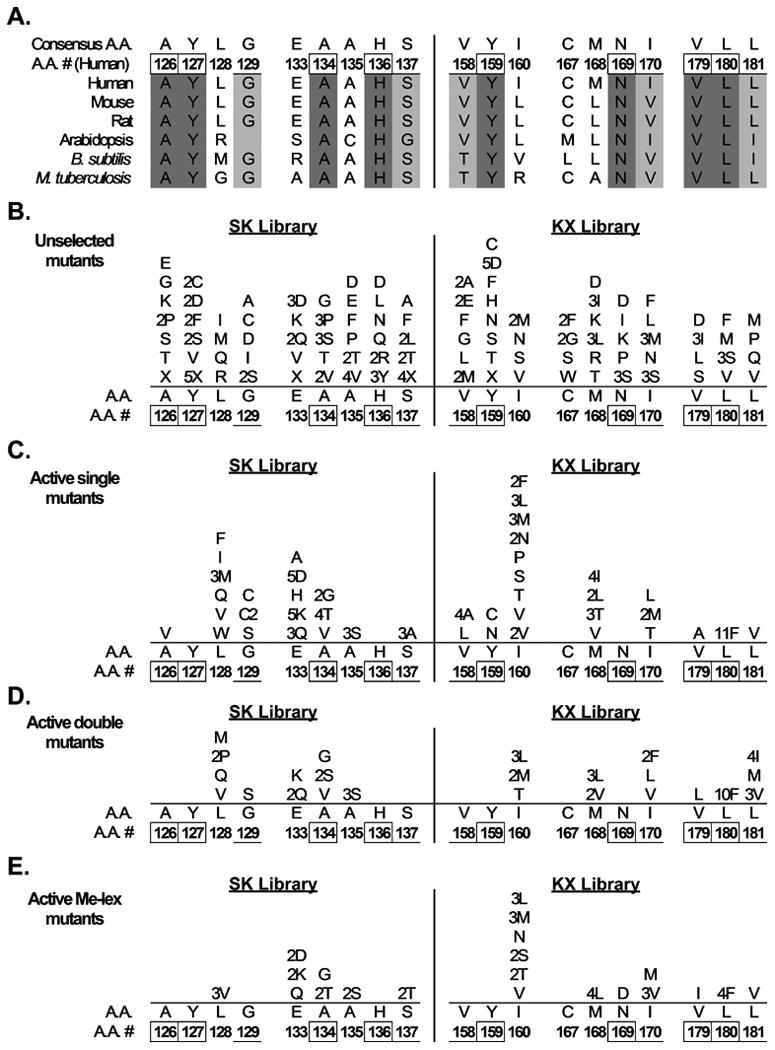
A, Conservation of AAG primary sequence in natural evolution. Aligned sequences of human AAG and homologs are shown; numbering refers to the human sequence. Absolutely conserved residues are indicated in dark gray and the numbers are boxed. Residues showing only conservative substitutions are indicated in light gray and the numbers are underlined. B-E, Plasmid libraries harboring mutant AAGs created by random mutagenesis were expressed in glycosylase-deficient E. coli MV1932. Mutants that complemented the methylating agent sensitivity of MV1932 cells were selected and the plasmid sequences were determined. Amino acid substitutions shown were observed in 72 mutants from the unselected AAG SK and KX libraries (B), 89 single mutants selected for MMS resistance (C), 25 double mutants selected for MMS resistance (D), and 42 mutants selected for Me-lex resistance (E). Mutations in 4 MMS-resistant triple mutants are not shown.
3.2. Genetic complementation yields mutant libraries that confer MMS resistance
The well-characterized DNA repair-deficient E. coli strain MV1932 lacks the 3-meA DNA glycosylase AlkA and the O6-methylguanine-DNA methyltransferase Ada [12], and is highly sensitive to the methylating agent MMS. Expression of human AAG rescues MV1932 cells from MMS cytoxicity [6], and is the basis of our functional complementation system for isolation of active AAG mutants. AAG excises both 3-meA and 7-meG from double-stranded DNA [13]. 3-meA comprises about 10% of MMS-induced base lesions, is highly cytotoxic, and is efficiently cleaved; 7-meG comprises about 70% of base lesions, appears to be relatively innocuous, and is less efficiently cleaved. We found that incubation of mid-logarithmic phase MV1932 cells with 0.2% MMS for 1 hr at 37° C yielded 2.5 % survival of cells expressing wild-type AAG, and 4.6 × 10-5 % survival of cells expressing an inactive construct with an internal deletion in the AAG cDNA, a 54,000-fold difference in resistance (Fig. 2A). To select for active AAG mutants, MV1932 cells were transformed with the SK and KX libraries, and then incubated with 0.2 % MMS for 1 hr and plated on medium lacking MMS. The surviving fraction was 3.6 × 10-3 % and 2.2 × 10-2 % of the SK and KX libraries, respectively, (Fig. 2A), indicating that only a small proportion of mutants was able to confer MMS resistance. Surviving colonies were picked, and plasmids harboring mutant AAG genes were sequenced. The two independent libraries were comparable in several respects (Fig. 2A,B). Selected mutants contained 3- to 4-fold fewer mutations than unselected mutants. The selected SK and KX libraries averaged 0.6 and 0.7 amino acid substitutions per mutant, respectively, compared with 2.5 and 2.3 for the unselected libraries. No frameshifts, nonsense mutations or deletions were observed among the survivors. AAG with zero amino acid changes was enriched in both libraries, from an initial 5% and 11% in the unselected SK and KX libraries, respectively, to 49% and 44% in the selected libraries. The large majority of mutants that were wild-type at the amino acid level harbored different silent mutations at the nucleotide level. The post-selection shift toward lower mutational load, noted in previous studies with other enzymes, e.g., [28], the observed absence of frameshifts, nonsense mutations and deletions, and enrichment for the wild-type amino acid sequence are all indicative of strong selective pressure for active AAG.
3.3. Mutability of AAG substrate binding pocket residues
Selection for MMS resistance in our cross-species functional complementation system revealed the mutability of the substrate binding pocket and showed striking parallels with natural evolution (Fig. 3A), as well as differences. DNA sequencing of 116 mutants from the active SK and KX libraries yielded the spectrum of amino acid substitutions shown in Fig. 3C,D. On average, strictly conserved residues showed fewer replacements than relatively conserved residues, and, in turn, relatively conserved residues showed fewer replacements than non-conserved residues. This finding holds for the number of different replacements, and for total replacements, at a single position. Most notably, no replacements were found for the three strictly conserved residues Tyr127, His136, and Asn169. The absence of substitutions in both single mutants and in double mutants (which have the potential for compensatory substitutions) emphasizes the functional importance of the respective side chains implicit in both evolutionary conservation (Fig. 3A) and X-ray structures [[22,23] and Fig. 1]. These results, together with the proposed key roles of Tyr127 and His136 in substrate binding [23], led us to examine these residues in greater depth. The partially random oligonucleotide mutagenesis described above represents an incomplete search for amino acid substitutions. In order to test all possible substitutions for Tyr127 and His136, we constructed the YH library, in which each wild-type nucleotide encoding these residues was synthesized with a mixture of 25% each A, T, G, and C. Over 16,000 mutants from this totally random library were transformed into MV1932 cells and subjected to MMS selection. Sequencing of twenty surviving colonies revealed that all were wild-type at the amino acid level, and that most harbored silent mutations (data not shown). These results further document the critical importance of Tyr127 and His136 for MMS resistance.
We noted interesting and potentially informative exceptions to the general evolutionary parallels noted above. The most striking is that of Leu180, a strictly conserved residue whose side chain lies adjacent to the N3 position of the εA substrate [23] and is expected to pack closely with the methyl adduct in 3-meA [22]. Leu180 to Phe substitutions were found in fully 33% of sequenced mutants from the selected KX library, representing a 6-fold enrichment from 3% of sequenced clones in the unselected library to 19% in the selected library. In comparison, enrichment for the wild-type AAG sequence was 4-fold, from 11% of sequenced clones in the unselected library to 44% in the selected library (Fig. 2B). The observed selection for L180F AAG suggests that the Leu to Phe replacement confers as great or greater MMS resistance than that of wild-type in our complementation system. Another, contrasting, exception to the evolutionary conservation parallel is found in the results for Cys167. We observed no replacements for this amino acid in single or double mutants, although it is not conserved in natural evolution (Fig. 3A).
3.4. Me-lex and MMS survivors share a similar spectrum of substitutions
We also subjected the SK and KX libraries to selection for resistance to the site-specific methylating agent Me-lex. Me-lex selectively binds to AT-rich sequences and preferentially generates 3-meA lesions (93% of base adducts) [29-31]. Selection for Me-lex survival enriched for the wild-type AAG amino acid sequence, which comprised 63% and 54% of the selected SK and KX libraries, respectively. Fig. 3E shows the sequence of 15 SK and 27 KX library mutants. The overall pattern is comparable to that found after MMS selection (Fig. 3C,D). Again, no substitutions were found for Tyr127 and His136, nor for Cys167. One substitution was observed for Asn169, as part of the I160S, N169D, V179I triple mutant. Interestingly, Leu180 to Phe substitutions were once again highly enriched, appearing in 4 of the 19 mutants in the selected KX library. The similarity in spectrum of substitutions produced by MMS and Me-lex selection suggests that protection against the lethality of 3-meA may be a key factor governing MMS resistance in our complementation system.
3.5. Effect of the L180F substitution on MMS survival
In accord with the notable enrichment for the L180F mutant in the MMS- and Me-lex-selected libraries (Fig. 3B-E), we found that expression of the L180F mutant conferred improved MMS protection relative to that of wild-type AAG (Fig. 4A). Although the LD50 was essentially unchanged, we observed a statistically significant 4.4-fold increase in survival (P < 0.005) at the high concentration of MMS used for selection of active mutants (0.2% for 1 hr at 37 °C); overall survival was reduced more than three orders of magnitude under these stringent conditions. The difference between mutant and wild-type viability was greatest at high MMS concentrations, presumably because compensatory survival mechanisms, such as translesion DNA synthesis and recombination, were saturated or approaching saturation. Immunoblotting showed that expression of the L180F mutant protein was comparable to that of the wild-type at the start of MMS exposure, and up to 4 hr thereafter (Fig. 4B,C).
Figure 4. MMS resistance conferred by wild-type and L180F mutant AAG.
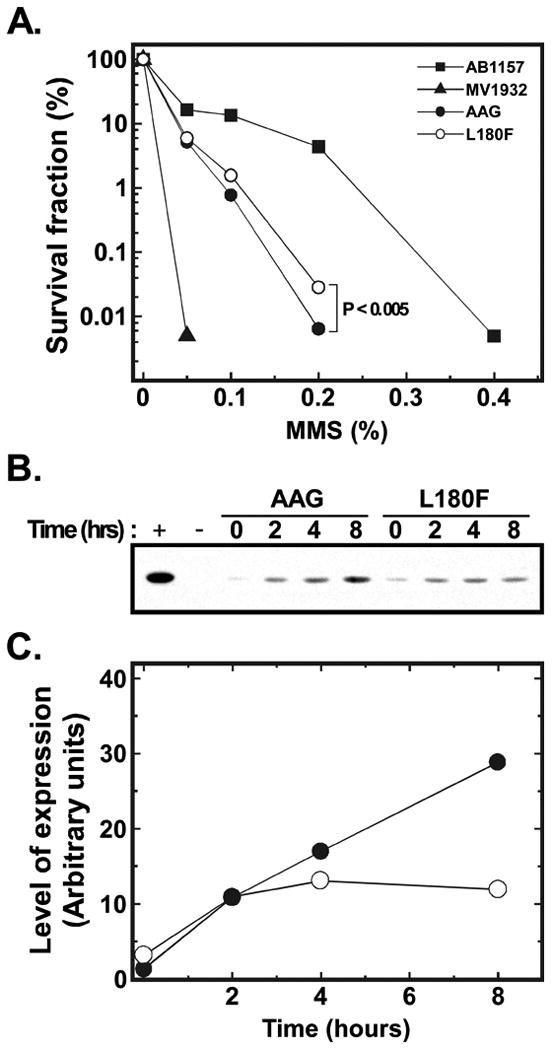
A, MMS survival of repair-proficienct E. coli AB1157, glycosylase-deficient MV1932, and MV1932 expressing either wild-type or L180F mutant AAG. The AAG proteins were expressed from the vector pTrcHis2 (Invitrogen) under control of the T7 promoter. Cells were grown to OD600 0.6-0.85, adjusted to ∼108 cells/ml in LB-carbenicillin medium and treated with various concentrations of MMS for 1 hr at 37 °C. Cells were diluted and plated in triplicate, and colonies were counted after 24 hr. The results are the average of two different experiments, each carried out with a different clone of wild-type and L180F AAG, that yielded very similar results. Error bars are not shown because the standard deviations are too small to be indicated on the y-axis that ranges over 5 orders of magnitude. At 0.2% MMS, the concentration used for selection, the difference in surviving fraction of the wild-type strain (0.0064% ± 0.0003%; mean and standard deviation) and the mutant strain (0.0283% ± 0.0011%) is highly significant (P < 0.005, two sample t-test, as calculated using OriginPro software, OriginLab Corporation, Northampton, MA). B, Western blot. Aliquots of cells were taken at various times after MMS treatment and subjected to immunoblotting using an anti-AAG polyclonal antibody as previously described [39]. C, Expression of wild-type AAG (closed circles) and the L180F mutant protein (open circles) was quantified by phosphor imaging using ImageQuant software (GE Healthcare).
3.6. Effect of the L180F substitution on excision of methyl adducts
To examine the effect of the L180F substitution on removal of methylated bases, we purified the wild-type and L180F AAG proteins. The final preparations were essentially homogeneous (>98% pure) in SDS-polyacrylamide gels and exhibited mobility consistent with the predicted molecular weight (Fig. 5A). Both the wild-type and mutant preparations were free of contaminating nuclease activity, measured in assays utilizing 5′-end 32P-labeled single- and double-stranded oligonucleotides (data not shown). DNA glycosylase activity was assayed by quantifying release of chromatographically separated radiolabelled N-methylpurines from calf thymus DNA methylated with [3H]-methylnitrosourea [27]. Previous measurements have shown that the potentially lethal lesion 3-meA is a preferred substrate of wild-type human AAG, while the presumptively benign lesion 7-meG is removed at a slower rate [32]. As illustrated in Fig. 5B,C, the L180F mutant enzyme displayed reduced activity, the rate of release of [3H]-labeled 3-meA and 7-meG being lower than that observed for wild type AAG. The rate of excision of 3-meA by the mutant enzyme was reduced 2.2-fold relative to that of wild-type AAG (0.82 vs. 0.38 fmol excised/min/pmol enzyme, respectively), whereas the rate of excision of 7-meG was reduced 7.3-fold (0.049 vs. 0.0068 fmol excised/min/pmol enzyme, respectively). Thus, the mutant enzyme exhibited modestly increased discrimination against 7-meG. Decreased generation of cytotoxic abasic sites resulting from reduced excision of 3-meA and 7-meG may be a factor promoting survival at high MMS concentrations (Fig. 4A), where downstream enzymes in BER pathways may be saturated; this factor could be particularly relevant in the case of 7-meG, which is believed to be much less toxic than the abasic site reaction product. Possible structural determinants of the altered substrate preference are presented in the Discussion.
Figure 5. Excision of 3-meA and 7-meG from DNA by homogeneous wild-type and L180F AAG.
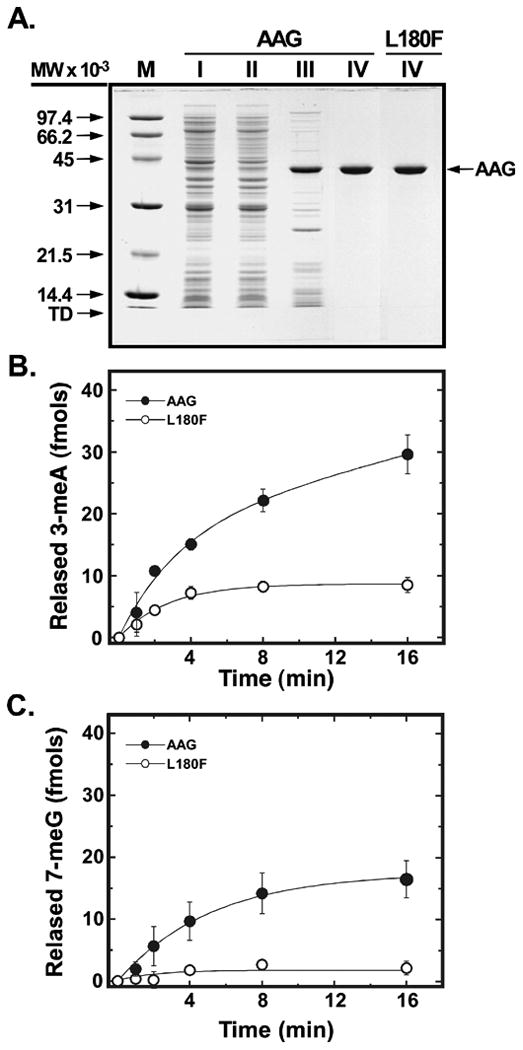
A, wild-type AAG and the L180F mutant protein were purified as described in Experimental Procedures. Samples (4 μg) of wild-type fractions I-IV and L180F fraction IV were subjected to electrophoresis in a 12.5% SDS-polyacrylamide gel and protein was visualized by staining with Coomassie Brilliant Blue G-250. The mobility of molecular weight standards and tracking dye is indicated by arrows. B,C, DNA glycosylase activity of wild-type (closed circles) and L180F (open circles). AAG was assayed as a function of time by measuring the release of [3H]-labelled 3-meA and 7-meG from methylated calf thymus DNA, as described in Experimental Procedures. Excision of 3-meA and 7-meG was measured in reactions containing 50 and 500 nM of enzyme, respectively.
3.7. Effect of the L180F substitution on excision and binding of neutral adducted bases
The methylated bases derived from MMS and Me-lex are electron deficient, and carry a delocalized positive charge that is presumably important for binding and excision by AAG [23]. However adducted bases that have no net charge are also efficiently cleaved. To examine the effect of the L180F substitution on activity toward neutral adducted bases, we measured excision of εA from an oligonucleotide substrate in a coupled assay in which excision of the damaged base is followed by the endonuclease-mediated cleavage of the abasic site product (Fig. 6A). Quantification of the resulting, cleaved oligonucleotide in a denaturing polyacrylamide gel (Fig. 6B) revealed a 6.6-fold decrease in activity of the L180F mutant enzyme, calculated by using regression analysis of 5 points on the initial, linear portion of curves. The specific activity of wild-type AAG was 68 fmol εA excised/min/fmol enzyme and that of the L180F mutant 10 fmol εA excised/min/fmol enzyme. Similarly, we observed a 3.0-fold decrease in excision of Hx by the mutant enzyme (Fig. 6C,D); the oligomer was the same as in the εA assays, except for the lesion. The specific activity of wild-type AAG was 111 fmol Hx excised/min/fmol enzyme and that of the L180F mutant 37 fmol Hx excised/min/fmol enzyme. We also examined binding of the wild-type and L180F mutant enzymes to lesion-containing oligonucleotides in electrophoretic mobility shift assays (Fig. S2). In the case of εA (Fig. S2A), quantification of bound enzyme (Fig. S2B) revealed a 1.2-fold increase in relative binding affinity for the mutant enzyme. Relative binding affinity to a Hx-containing oligonucleotide (Figs. S2C,D) was the same for both enzymes.
Figure 6. Excision of εA and Hx by homogeneous wild-type and L180F AAG.
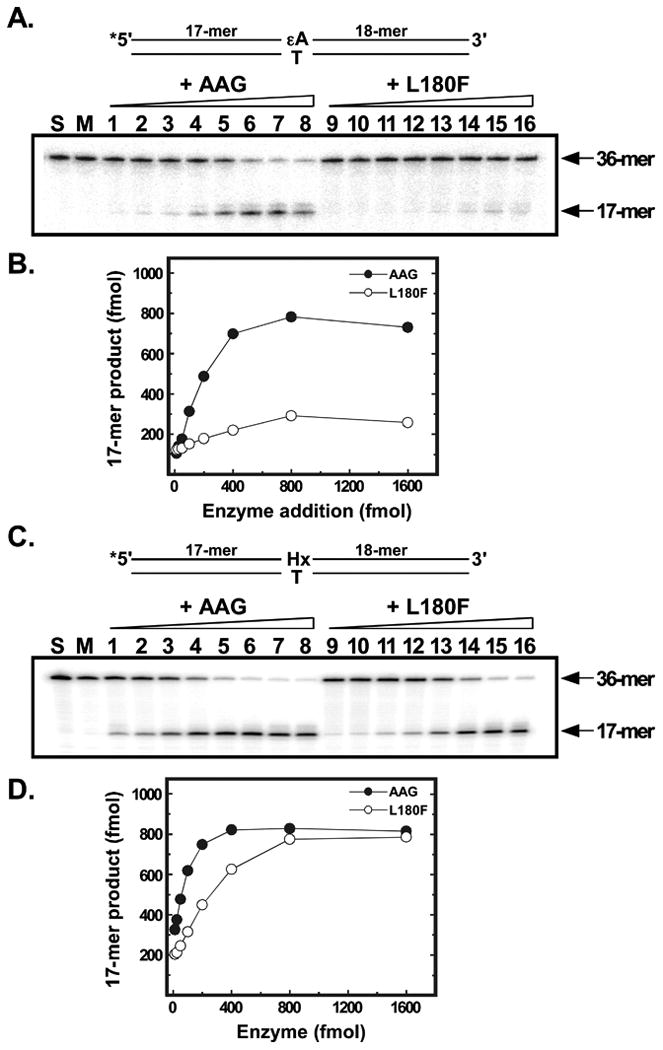
Excision of εA (A,B) or Hx (C,D) from 5′-32P end-labeled 36-mer oligonucleotides was measured in a coupled assay where excision is followed by cleavage of the abasic site product by E. coli endonuclease IV (see Experimental Procedures for details). A, Reaction mixtures contained 1 pmol substrate plus either 1, 2, 3, 4, 5, 6, 8, or 10 pmol wild-type AAG (lanes 1-8) or L180F mutant protein (lanes 9-16). Following incubation and treatment with endonuclease IV, samples were analyzed by electrophoresis in 12% denaturing polyacrylamide gels. Gels were dried and the products visualized by phosphor imaging. The positions of the 36-mer substrate and 17-mer excision/cleavage product are indicated by arrows. Lane S contained 1 pmol of substrate alone. Lane M contained a mock reaction carried through the assay with all components except glycosylase. B, activity of wild-type (closed circles) and L180F (open circles) AAG was quantified by phosphor imaging using ImageQuant software (GE Healthcare). C, as in A above. D, as in B above.
3.8. Effect of the L180F substitution on binding to abasic site-containing DNA
The abasic site product of AAG cleavage is highly cytotoxic. Therefore the enhanced survival of methylation damage mediated by the L180F mutant could be due to tighter binding and retarded release of this potentially lethal intermediate. To compare the relative binding affinity of the wild-type and mutant proteins to AP-DNA, we performed electrophoretic mobility shift assays using a double-stranded 5′-32P end-labeled 36 base-pair oligonucleotide containing an abasic site analog opposite thymine; the oligomer was the same as in the εA and Hx assays, except for the lesion. We observed that affinity of the L180F mutant for AP-DNA was slightly (∼1.5 fold) higher than that of wild-type AAG (data not shown). In order to further compare the difference between the wild-type and mutant proteins, we performed sensitive fluorescence polarization assays to determine the apparent steady-state equilibrilium dissociation constant for binding to a duplex 36-mer containing an abasic site opposite T and 5′-end labeled with the fluorescent dye FAM. When a protein binds to a fluorescently-labeled DNA, the rotational freedom of the DNA is usually decreased, leading to increased polarization. As shown in Fig. 7A, when the duplex, FAM-labeled AP·T-36-mer was titrated with increasing amounts of wild-type AAG, a concentration-dependent increase of fluorescence polarization was observed that saturated at approximately 1 μM AAG. The net polarization enhancement at each concentration was fit to Eq. (1), yielding an apparent Kd of 824 ± 104 nM for binding under our assay conditions. The magnitude of polarization enhancement at saturation observed for the F180L mutant enzyme (Fig. 7B), was comparable to that for wild-type AAG, yielding an apparent Kd of 479± 38 nM. Hence, the binding affinity of L180F for the duplex AP-DNA, as measured by 1/Kd, is ∼ 1.7 fold greater than that of wild-type AAG.
Figure 7. Binding of wild-type and L180F AAG to abasic site-containing DNA.
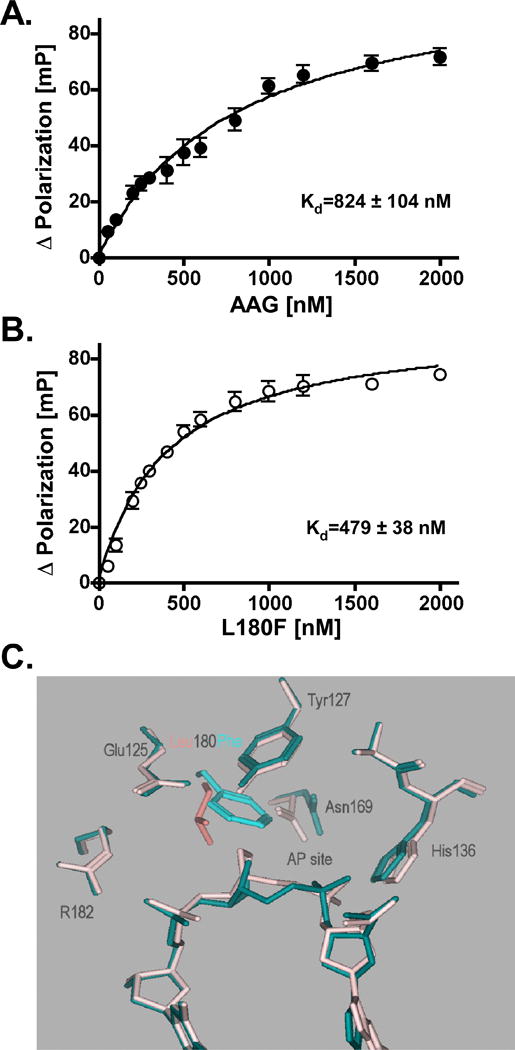
The apparent binding affinity for abasic site-containing DNA was evaluated by using a fluorescence polarization assay as described in Experimental Procedures. The 36-mer double-stranded DNA ligand was the same as in Fig. S2, but was 5′-end labeled with the fluorescent dye FAM. A, fluoresence polarization (mP) of FAM-labeled DNA titrated with increasing concentrations of wild-type AAG. Final reaction mixtures contained binding buffer, 100 nM FAM-labeled DNA, and 50, 100, 200, 250, 300, 400, 500, 600, 800, 1200, 1600, or 2000 nM enzyme. Reaction mixtures were equilibrated at 25 °C for 15 min prior to fluorescence polarization measurements. Assays were performed in triplicate; the mean and standard deviation are indicated. B, fluoresence polarization of FAM-labeled DNA titrated with increasing concentrations of L180F mutant protein. Procedures were as described in A. C, modeled complexes of wild-type (colored pink) and F180L (colored blue) AAG with abasic DNA. A model of the L180F mutant AAG complexed with a pyrrolidone abasic site-containing oligonucleotide was derived from coordinate set 1F6O (22) in the Protein Data Bank (40), by replacing Leu180 with phenylalanine and energy minimizing, as described in Section 3.8. This model, in which the Phe side chain projects into the abasic site, is superposed with the un-mutated coordinate set. The image is drawn using the Molecular Operating Environment package.
To explore the possible structural basis for enhanced abasic site binding, we modeled the Leu180 to Phe substitution in the co-crystal structure of wild-type human AAG complexed to a pyrrolidine abasic nucleotide-containing DNA [22]. The model was derived by replacing Leu180 with phenylalanine and energy-minimizing, using Amber parameters with no chiral restraints in the Molecular Operating Environment package of computer programs [Chemical Computing Group, Montreal (www.chemcorp.com)]. A conformation could be adopted in which the Phe side chain simply replaced the Leu side chain; the total potential energy calculated for the conformer was -7197 kcal mol-1. A n energetically equivalent conformation (-7287 kcal mol-1) could be adopted by choosing an alternate rotamer, which resulted in the Phe side chain projecting into the abasic site. This model, superposed with that of the un-mutated protein, is illustrated in Fig. 7C. As shown, the aromatic side chain of Phe180 rotates toward the abasic residue from the position of Leu180 in wild-type AAG, and partially fills the space occupied by a substrate base. Greater proximity of the Phe180 side chain to the baseless site may provide more favorable geometry for interaction, e.g., van der Waals contact of the epsilon and zeta carbons of Phe with deoxyribose, and may thereby increase binding affinity of the L180F protein for AP-DNA. Modeling also shows that the abasic residue is displaced outward in the mutant, causing loss of a possible, weak H bond between O2P of the 5′ phosphate and the hydroxyl of Tyr159 (not shown).
4. Discussion
Human AAG has broad substrate specificity, acting on variously modified purines arising from methylation, oxidation and lipid peroxidation. Substrate discrimination by the wild-type enzyme likely represents a compromise that promotes protection against differing sources of DNA damage, and is governed, in part, by the detailed architecture of the substrate binding pocket. Here, we used oligonucleotide-directed mutagenesis to simultaneously randomize eight residues that contribute to the substrate binding pocket and have been shown in crystal structures to contact substrate bases. Our mutagenesis strategy affords an experimental opportunity to remodel the active site, from one optimized during natural evolution for defense against diverse damaging agents to one optimized for defense against one specific agent. We used a bacterial complementation system to select for variant AAGs that protect against killing by the methylating agent MMS. Our selection for methylation-resistant mutants recapitulated the conservation of certain key amino acids in natural evolution, providing in vivo evidence in support of the active site model deduced from crystal structures [22,23]. We also observed departures from natural evolution that may reflect optimization for methylation survival in our complementation system.
Crystallographic data have indicated a requirement for the strictly conserved aromatic residues Tyr127, His136, and Tyr159 which appear to provide crucial stacking interactions of AAG with an εA substrate [23]. Consistent with these data, we found no replacements for Tyr127 among single or double MMS- and Me-lex-resistant mutants, suggesting that Tyr127 has essential interactions with methylated bases as well. We also found no replacements for His136, although it has been suggested that His136 may be dispensable for excision of 3-meA, based in part on a model in which the methylated base does not reach far enough into the substrate binding cavity to stack against His136 [22]. Our data indicate that His136 is required for survival of methylation damage in our complementation system, and modeling suggests it functions to impede accommodation of 7-meG in the binding pocket, thus limiting conversion of a relatively innocuous base lesion to a potentially lethal abasic site. Superimposing 7-meG on the crystal structures of AAG complexed with an abasic site- or εA-containing oligonucleotide [22,23] reveals steric interference of the His136 imidazole side chain with the exocyclic N7 methyl group (Fig. 8C). The position of His136 is fixed by numerous H bonds of main chain and side chain atoms (e.g., with the OH group of the Tyr157 side chain and the phosphate upstream of the baseless deoxyribose), thus limiting the size of the binding pocket and disfavoring accommodation of 7-meG. Interestingly, an H136Q mutant was found to allow survival, albeit diminished relative to wild-type AAG, in a yeast complementation system [42]; the difference between this result and ours likely reflects differences in the species employed and in the expression levels of AAG relative to downstream BER enzymes. We observed two non-conservative (non-planar) substitutions for Tyr159, namely Asn and Cys, among MMS-resistant single mutants, indicating that the function(s) of Tyr159 is not essential under our selection conditions.
Figure 8. Comparison of the active sites of wild-type and mutant AAG complexed with DNA containing 3-meA- or 7-meG.
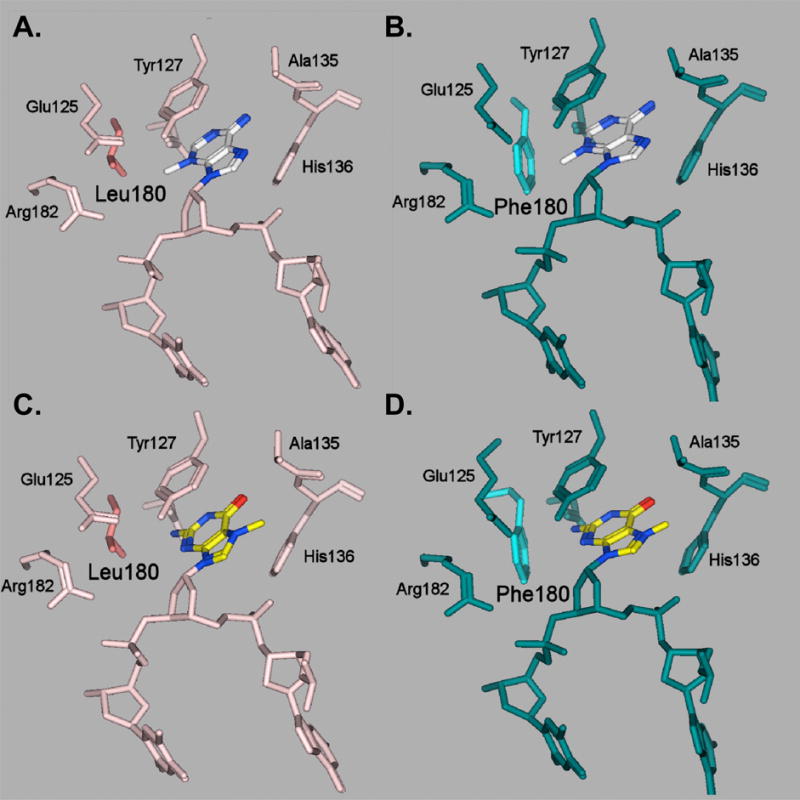
Modeled active sites of wild-type (A,C) and mutant L180F proteins (B,D) with 3-meA (A,B) and 7-meG (C,D) were derived from coordinate set 1F4R (23) in the Protein Data Bank (40). The mutant protein L180F was modeled by replacing Leu180 with phenylalanine, while the substrates were modeled by suitable addition and subtraction of functional groups on the εA moiety in the coordinate set 1F4R for 3meA, and for 7meG, by superposing a 7-meG on to the εA site. All four resultant models were independently energy minimized using the Molecular Operating Environment package of programs. Re-orientation of the Glu125 side chain, postulated to mediate catalysis, may reduce the activity of the L180F mutant enzyme on both 3-meA- and 7-meG-containing substrates. Additional factors, such as increased steric hindrance enforced by the planar Phe180 ring, may further reduce activity of the mutant enzyme on 7-meG-containing substrates.
The only substitution we found for highly conserved Asn169 among single or double mutants was Asp, a replacement we observed once among Me-lex survivors. Preservation of Asn is concordant with the proposed role of the side chain in excluding normal guanine from the active site via a steric clash with the exocyclic N2 amino group [23]. Interestingly, we observed no substitutions for Cys167 among single or double methylation-resistant mutants, indicating that it may be required for excision of methylated bases in our complementation system.
An unexpected finding of this work is the remarkable selection for Phe in place of strictly conserved Leu180. The L180F mutant was enriched from a random library to an ∼50% greater extent than wild-type AAG, dominating the spectrum of MMS-resistant mutants. Expression of the L180F mutant yielded survival equivalent to that of wild-type AAG at low doses of MMS, and up to 4.4-fold better survival at high doses where viability was severely compromised. Purification of the L180F mutant protein and examination of its catalytic properties support the conclusion that the increased survival is mediated by a reduction in excision activity, a change in substrate discrimination, and a slight increase in binding affinity for abasic sites. These alterations are consistent with the idea that, relative to human cells, bacterial host cells have reduced capacity to repair potentially lethal BER intermediates arising from glycosylase action, and are better protected from methylators by a variant AAG that generates abasic sites at a slower-than-wild-type rate. We observed that the overall rate of removal of adducted bases from methylated DNA by the homogenous L180F protein was reduced relative to that of wild type AAG. The rate of excision of 3-meA from calf thymus DNA was decreased 2.2-fold, while the rate for 7-meG was decreased 7.3-fold. Because 7-meG represents ∼70% - 80% of the base lesions generated by MMS, increased discrimination against 7-meG could contribute to enhanced survival at high levels of methylation where abasic sites and other potentially cytotoxic BER intermediates might saturate downstream enzymes, leading to lethal double strand breaks and cell death [33]. Also of possible importance for survival, we found that the apparent binding affinity of the L180F mutant for abasic-site-containing DNA was increased about 70%, as assessed by fluorescence polarization; a reduced rate of release might relieve toxicity associated with unrepaired abasic sites.
Several previous studies have suggested that imbalanced BER caused by excess DNA glycosylase activity may increase sensitivity to alkylating agents. A genetic study in yeast [11] has demonstrated that the spontaneous mutation rate in apurinic endonuclease-deficient apn1 cells can be increased 600-fold by a simple imbalance between the 3-methyladenine DNA glycosylase Mag1 and apurinic endonuclease. The increased spontaneous mutation is due to the generation of abasic sites resulting mainly from Mag1 glycosylase activity, and exogenous Apn1 expression dramatically suppresses the elevation of mutation. Another report [34] has suggested that repair of some damaged lesions can be regulated by DNA glycosylase activity if the glycosylase reaction is the rate-limiting step in BER. Importantly, recent work suggests that, in human cells, coordination of the activities of AAG and APE1 depends, at least in part, on slow excision by AAG and rapid incision of AP sites by the much more abundant APE1 (34). This balance of BER activities, reached in natural evolution, is lost in cross-species complementation systems such as ours, presumably leading to selection of variant AAGs that are better matched with the capacity of host cells to repair potentially lethal BER intermediates arising from glycosylase action.
In addition to alteration in the rates of hydrolysis of methylated bases by the L180F mutant, we observed a 3- to 7-fold decrease in the rate of excision of the neutral adducted bases ethenoA, which arises from lipid peroxidation, and hypoxanthine, which arises by oxidative deamination of adenine. Overall, our data indicate that the L180F substitution alters discrimination of methylated bases to better protect against MMS in our complementation system, with concomitant reduction of activity on damage from other sources.
How does the L180F substitution mediate the altered substrate specificity we observed? Introduction of the bulky aromatic Phe side chain decreases the space available in the substrate binding pocket, and can thereby limit accommodation of modified bases. Fig. 8 shows a model of the wild-type and mutant substrate binding pocket, occupied by either 3-meA or 7-meG. In the case of 3-meA, the wild-type Leu180 side chain makes hydrophobic contact with the methyl adduct (Fig. 8A, top left panel). In the L180F mutant (Fig. 8B, top right panel), the modeled Phe180 benzyl ring is located 4.2-4.6 Å from the methyl adduct, within favorable range for π-cation stacking interaction [35,36]. In addition, the orientation of the Glu125 side chain is almost perpendicular to that in the wild-type enzyme. Glu125 may function to deprotonate a water molecule (not shown) for nucleophilic attack on the glycosydic bond [22], and re-orientation of this residue suggests a possible mechanism for the slight reduction in the rate of excision of 3-meA. In the important case of 7-meG, the mutant Phe180 may decrease excision by several mechanisms. The first is re-orientation of the Glu125 side chain, as described above (Fig. 8B, compare panels A and B). Second, Phe180 may decrease accommodation, because the adducted substrate is tightly boxed into the mutant binding pocket, between His136 imidazole on one side and the planar benzyl ring on the other. Yet more pronounced steric effects have been described for excluding 7-meG from the substrate binding pocket of the highly specific 3-methyladenine DNA glycosylase from H. pylori, a glycosylase that belongs to a different superfamily and has a different overall architecture [37]. Third, substitution of Phe for Leu180 also increases the surface area for van der Waal's interactions, apparently resulting in increased contact with deoxyribose that may mediate tighter binding and slower release of the abasic site product.
By illustrating the consequences of replacement with Phe, our work illuminates the roles of Leu180 in wild-type human AAG. As might be expected for a residue that lines the substrate binding pocket and contacts substrate bases, Leu contributes, albeit modestly, to wild-type excision activity, to the pattern of substrate discrimination, and to the overall compromise for protection against diverse sources of DNA damage. Of particular interest, Leu180 appears to modulate excision of 7-meG, hence contributing to the balance within BER pathways that remove methylation damage [33,38], and raising the question of whether the wild-type rate of excision of 7-meG, a relatively benign lesion, is a necessary structural consequence of preserving activity on more deleterious adducts.
Supplementary Material
{kind=link}
{kind=link}
Acknowledgments
We thank Dr. Elinor Adman for molecular modeling and valuable discussions and Yunny Yunita, K. Marinov Georgi and Dr. Eric Althoff for technical assistance. This work was supported by NIH grants CA78885, CA77852, and ES07033 to L.A.L. and CA55042 to L.D.S. H.H.G was supported by the Medical Scientist Training Program. L.D.S. is an American Cancer Society Research Professor.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Sedgwick B. Repairing DNA-methylation damage. Nat Rev Mol Cell Biol. 2004;5:148–157. doi: 10.1038/nrm1312. [DOI] [PubMed] [Google Scholar]
- 2.Wyatt MD, Pittman DL. Methylating agents and DNA repair responses: Methylated bases and sources of strand breaks. Chem Res Toxicol. 2006;19:1580–1594. doi: 10.1021/tx060164e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Friedberg EC, Walker GC, Siede W, Wood RD, Schultz RA, Ellenberger T. DNA Repair and Mutagenesis. American Society for Microbiology; Washington, D.C.: 2005. [Google Scholar]
- 4.Doublie S, Tabor S, Long AM, Richardson CC, Ellenberger T. Crystal structure of a bacteriophage T7 DNA replication complex at 2.2 A resolution. Nature. 1998;391:251–258. doi: 10.1038/34593. [DOI] [PubMed] [Google Scholar]
- 5.Beard WA, Osheroff WP, Prasad R, Sawaya MR, Jaju M, Wood TG, Kraut J, Kunkel TA, Wilson SH. Enzyme-DNA interactions required for efficient nucleotide incorporation and discrimination in human DNA polymerase beta. J Biol Chem. 1996;271:12141–12144. doi: 10.1074/jbc.271.21.12141. [DOI] [PubMed] [Google Scholar]
- 6.Samson LD, Derfler B, Boosalis M, Call K. Cloning and characterization of a 3-methyladenine DNA glycosylase cDNA from human cells whose gene maps to chromosome 16. Proc Natl Acad Sci U S A. 1991;88:9127–9131. doi: 10.1073/pnas.88.20.9127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Engelward BP, Dreslin A, Christensen J, Huszar D, Kurahara C, Samson LD. Repair-deficient 3-methyladenine DNA glycosylase homozygous mutant mouse cells have increased sensitivity to alkylation-induced chromosome damage and cell killing. EMBO J. 1996;15:945–952. [PMC free article] [PubMed] [Google Scholar]
- 8.Engelward BP, Allan JM, Dreslin AJ, Kelly JD, Wu MM, Gold B, Samson LD. A chemical and genetic approach together define the biological consequences of 3-methyladenine lesions in the mammalian genome. J Biol Chem. 1998;273:5412–5418. doi: 10.1074/jbc.273.9.5412. [DOI] [PubMed] [Google Scholar]
- 9.Scharer OD, Jiricny J. Recent progress in the biology, chemistry and structural biology of DNA glycosylases. Bioessays. 2001;23:270–281. doi: 10.1002/1521-1878(200103)23:3<270::AID-BIES1037>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- 10.Hosfield DJ, Daniels DS, Mol CD, Putnam CD, Parikh SS, Tainer JA. DNA damage recognition and repair pathway coordination revealed by the structural biochemistry of DNA repair enzymes. Prog Nucleic Acid Res Mol Biol. 2001;68:315–347. doi: 10.1016/s0079-6603(01)68110-8. [DOI] [PubMed] [Google Scholar]
- 11.Glassner BJ, Rasmussen LJ, Najarian MT, Posnick LM, Samson LD. Generation of a strong mutator phenotype in yeast by imbalanced base excision repair. Proc Natl Acad Sci U S A. 1998;95:9997–10002. doi: 10.1073/pnas.95.17.9997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Posnick LM, Samson LD. Imbalanced base excision repair increases spontaneous mutation and alkylation sensitivity in Escherichia coli. J Bacteriol. 1999;181:6763–6771. doi: 10.1128/jb.181.21.6763-6771.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wyatt MD, Allan JM, Lau AY, Ellenberger TE, Samson LD. 3-methyladenine DNA glycosylases: structure, function, and biological importance. Bioessays. 1999;21:668–676. doi: 10.1002/(SICI)1521-1878(199908)21:8<668::AID-BIES6>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- 14.Wuenschell GE, O'Connor TR, Termini J. Stability, miscoding potential, and repair of 2′-deoxyxanthosine in DNA: implications for nitric oxide-induced mutagenesis. Biochemistry. 2003;42:3608–3616. doi: 10.1021/bi0205597. [DOI] [PubMed] [Google Scholar]
- 15.Hitchcock TM, Dong L, Connor EE, Meira LB, Samson LD, Wyatt MD, Cao W. Oxanine DNA glycosylase activity from Mammalian alkyladenine glycosylase. J Biol Chem. 2004;279:38177–38183. doi: 10.1074/jbc.M405882200. [DOI] [PubMed] [Google Scholar]
- 16.Bessho T, Roy R, Yamamoto K, Kasai H, Nishimura S, Tano K, Mitra S. Repair of 8-hydroxyguanine in DNA by mammalian N-methylpurine-DNA glycosylase. Proc Natl Acad Sci U S A. 1993;90:8901–8904. doi: 10.1073/pnas.90.19.8901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dosanjh MK, Roy R, Mitra S, Singer B. 1,N6-ethenoadenine is preferred over 3-methyladenine as substrate by a cloned human N-methylpurine-DNA glycosylase (3-methyladenine-DNA glycosylase) Biochemistry. 1994;33:1624–1628. doi: 10.1021/bi00173a002. [DOI] [PubMed] [Google Scholar]
- 18.Saparbaev M, Langouet S, Privezentzev CV, Guengerich FP, Cai H, Elder RH, Laval J. 1,N(2)-ethenoguanine, a mutagenic DNA adduct, is a primary substrate of Escherichia coli mismatch-specific uracil-DNA glycosylase and human alkylpurine-DNA-N-glycosylase. J Biol Chem. 2002;277:26987–26993. doi: 10.1074/jbc.M111100200. [DOI] [PubMed] [Google Scholar]
- 19.Berdal KG, Johansen RF, Seeberg E. Release of normal bases from intact DNA by a native DNA repair enzyme. EMBO J. 1998;17:363–367. doi: 10.1093/emboj/17.2.363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.O'Brien PJ, Ellenberger T. Dissecting the broad substrate specificity of human 3-methyladenine-DNA glycosylase. J Biol Chem. 2004;279:9750–9757. doi: 10.1074/jbc.M312232200. [DOI] [PubMed] [Google Scholar]
- 21.Aamodt RM, Falnes PO, Johansen RF, Seeberg E, Bjoras M. The Bacillus subtilis counterpart of the mammalian 3-methyladenine DNA glycosylase has hypoxanthine and 1,N6-ethenoadenine as preferred substrates. J Biol Chem. 2004;279:13601–13606. doi: 10.1074/jbc.M314277200. [DOI] [PubMed] [Google Scholar]
- 22.Lau AY, Scharer OD, Samson L, Verdine GL, Ellenberger T. Crystal structure of a human alkylbase-DNA repair enzyme complexed to DNA: mechanisms for nucleotide flipping and base excision. Cell. 1998;95:249–258. doi: 10.1016/s0092-8674(00)81755-9. [DOI] [PubMed] [Google Scholar]
- 23.Lau AY, Wyatt MD, Glassner BJ, Samson LD, Ellenberger T. Molecular basis for discriminating between normal and damaged bases by the human alkyladenine glycosylase, AAG. Proc Natl Acad Sci U S A. 2000;97:13573–13578. doi: 10.1073/pnas.97.25.13573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Guo HH, Choe J, Loeb LA. Protein tolerance to random amino acid change. Proc Natl Acad Sci U S A. 2004;101:9205–9210. doi: 10.1073/pnas.0403255101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Connor EE, Wyatt MD. Active-site clashes prevent the human 3-methyladenine DNA glycosylase from improperly removing bases. Chem Biol. 2002;9:1033–1041. doi: 10.1016/s1074-5521(02)00215-6. [DOI] [PubMed] [Google Scholar]
- 26.Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 27.Samson LD, Linn S. DNA alkylation repair and the induction of cell death and sister chromatid exchange in human cells. Carcinogenesis. 1987;8:227–230. doi: 10.1093/carcin/8.2.227. [DOI] [PubMed] [Google Scholar]
- 28.Encell LP, Landis DM, Loeb LA. Improving enzymes for cancer gene therapy. Nat Biotechnol. 1999;17:143–147. doi: 10.1038/6142. [DOI] [PubMed] [Google Scholar]
- 29.Fronza G, Gold B. The biological effects of N3-methyladenine. J Cell Biochem. 2004;91:250–257. doi: 10.1002/jcb.10698. [DOI] [PubMed] [Google Scholar]
- 30.Encell L, Shuker DE, Foiles PG, Gold B. The in vitro methylation of DNA by a minor groove binding methyl sulfonate ester. Chem Res Toxicol. 1996;9:563–567. doi: 10.1021/tx9501849. [DOI] [PubMed] [Google Scholar]
- 31.Kelly JD, Inga A, Chen FX, Dande P, Shah D, Monti P, Aprile A, Burns PA, Scott G, Abbondandolo A, Gold B, Fronza G. Relationship between DNA methylation and mutational patterns induced by a sequence selective minor groove methylating agent. J Biol Chem. 1999;274:18327–18334. doi: 10.1074/jbc.274.26.18327. [DOI] [PubMed] [Google Scholar]
- 32.O'Connor TR. Purification and characterization of human 3-methyladenine-DNA glycosylase. Nucleic Acids Res. 1993;21:5561–5569. doi: 10.1093/nar/21.24.5561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rinne ML, He Y, Pachkowski BF, Nakamura J, Kelley MR. N-methylpurine DNA glycosylase overexpression increases alkylation sensitivity by rapidly removing non-toxic 7-methylguanine adducts. Nucleic Acids Res. 2005;33:2859–2867. doi: 10.1093/nar/gki601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Allinson SL, Sleeth KM, Matthewman GE, Dianov GL. Orchestration of base excision repair by controlling the rates of enzymatic activities. DNA Repair (Amst) 2004;3:23–31. doi: 10.1016/j.dnarep.2003.09.002. [DOI] [PubMed] [Google Scholar]
- 35.Gallivan JP, Dougherty DA. Cation-pi interactions in structural biology. Proc Natl Acad Sci U S A. 1999;96:9459–9464. doi: 10.1073/pnas.96.17.9459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hu G, Gershon PD, Hodel AE, Quiocho FA. mRNA cap recognition: dominant role of enhanced stacking interactions between methylated bases and protein aromatic side chains. Proc Natl Acad Sci U S A. 1999;96:7149–7154. doi: 10.1073/pnas.96.13.7149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Eichman BF, O'Rourke EJ, Radicella JP, Ellenberger T. Crystal structures of 3-methyladenine DNA glycosylase MagIII and the recognition of alkylated bases. EMBO J. 2003;22:4898–4909. doi: 10.1093/emboj/cdg505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Maher RL, Vallur AC, Feller JA, Bloom LB. Slow base excision by human alkyladenine DNA glycosylase limits the rate of formation of AP sites and AP endonuclease 1 does not stimulate base excision. DNA Repair (Amst) 2007;6:71–81. doi: 10.1016/j.dnarep.2006.09.001. [DOI] [PubMed] [Google Scholar]
- 39.Fishel ML, Seo YR, Smith ML, Kelley MR. Imbalancing the DNA base excision repair pathway in the mitochondria; targeting and overexpressing N-methylpurine DNA glycosylase in mitochondria leads to enhanced cell killing. Cancer Res. 2003;63:608–615. [PubMed] [Google Scholar]
- 40.Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE. The Protein Data Bank. Nucleic Acids Res. 2000;28:235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yang Y, Sass LE, Du C, Hsieh P, Erie DA. Determination of protein-DNA binding constants and specificities from statistical analyses of single molecules: MutS-DNA interactions. Nucleic Acids Res. 2005;33:4322–4334. doi: 10.1093/nar/gki708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.O'Brien PJ, Ellenberger T. Human alkyladenine DNA glycosylase uses acid-base catalysis for selective excision of damaged purines. Biochemistry. 2003;42:12418–12429. doi: 10.1021/bi035177v. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.