

Abstract
Background
DNA tracts composed of only two bases are possible in six combinations: A+G (purines, R), C+T (pyrimidines, Y), G+T (Keto, K), A+C (Imino, M), A+T (Weak, W) and G+C (Strong, S). It is long known that all-pyrimidine tracts, complemented by all-purines tracts ("R.Y tracts"), are excessively present in analyzed DNA. We have previously shown that R.Y tracts are in vast excess in yeast promoters, and brought evidence for their role in gene regulation. Here we report the systematic mapping of all six binary combinations on the level of complete sequenced chromosomes, as well as in their different subregions.
Results
DNA tracts composed of the above binary base combinations have been mapped in seven sequenced chromosomes: Human chromosomes 21 and 22 (the major contigs); Drosophila melanogaster chr. 2R; Caenorhabditis elegans chr. I; Arabidopsis thaliana chr. II; Saccharomyces cerevisiae chr. IV and M. jannaschii. A huge over-representation, reaching million-folds, has been found for very long tracts of all binary motifs except S, in each of the seven organisms. Long R.Y tracts are the most excessive, except in D. melanogaster, where the K.M motif predominates. S (G, C rich) tracts are in excess mainly in CpG islands; the W motif predominates in bacteria. Many excessively long W tracts are nevertheless found also in the archeon and in the eukaryotes. The survey of complete chromosomes enables us, for the first time, to map systematically the intergenic regions. In human and other chromosomes we find the highest over-representation of the binary DNA tracts in the intergenic regions. These over-representations are only partly explainable by the presence of interspersed elements.
Conclusions
The over-representation of long DNA tracts composed of five of the above motifs is the largest deviation from randomness so far established for DNA, and this in a wide range of eukaryotic and archeal chromosomes. A propensity for ready DNA unwinding is proposed as the functional role, explaining the evolutionary conservation of the huge excesses observed.
Keywords: Chr. 21; Chr. 22; Drosophila chr. 2R; elegans Chr. I; Arabidopsis chr. II; Yeast Chr. IV; M. jannaschii; DNA unwinding, purine, pyrimidine
Background
In 1952, Erwin Chargaff published a paper in which he brought evidence that runs of pyrimidines are highly over-represented in eukaryotic DNA [1]. DNA was "depurinated" in formic acid and the remaining pyrimidines were subsequently size separated by the then novel technique of paper chromatography [2], see also [3]. An unexpectedly high number of pyrimidine and purine tracts ("isostichs"), 9 bases and higher, was found in human, calf, salmon and rye DNA [4,5]. These findings were subsequently corroborated by a number of techniques, incl. molecular hybridization rates [6-8]. The over-representation of long purine and pyrimidine runs could be exactly analyzed when sequences of many genes became available [9,10]. The phenomenon discovered by Chargaff turned out to be a very significant one – over-representation of the longer tracts reaches values of many ten-folds, as will be demonstrated on a genome-wide basis in this paper. Homopurine (R) and homopyrimidine (Y) tracts will be referred to jointly as "R.Y tracts", because whenever a run of pyrimidines is present on one strand, it is complemented by a run of purines on the opposite strand (the dot separates complementary strands, in accordance with IUBMB rules). It should be stressed that alternating A and G (poly A-G) are only one component of R tracts, and any combination of A's and G's an make an R tract – see Additional file: 7.
Examining increasing number of genes revealed that R.Y tracts are not the only over-represented binary DNA motif. Three additional combinations of two bases are possible [11], namely: A, T only ("W tracts"); G, C only ("S tracts"), and tracts which are G, T on one strand complemented by A, C on the opposite strand (jointly: "K.M tracts"). The S tracts, found in high concentrations in certain regions, are well studied as CpG islands [12]. The abundance of these combinations was previously established in an assortment of mammalian genes [13] and in a yeast chromosome [14]. In bacteria, the W motif, rather than the R.Y motif, was found to be the predominating binary motif [15,16].
In this paper, we shall map the occurrence of binary tracts in seven recently sequenced chromosomes, representing the major currently studied eukaryotic and archeal phyla (previous studies encompassed mainly incidentally selected gene regions). These chromosomes, especially the human and plant ones, also represent a large selection of intergenic regions not previously mapped. It will be shown that the huge over-representation is prevalent in all the selected chromosomes, in particular in their intronic and intergenic subregions. A functional significance of this remarkable departure of real DNA from random DNA has yet to be established. We have previously suggested, based on our experimental findings [17], that a DNA unwinding role, necessary for initiation of transcription, replication and other DNA directed processes, could be involved, as will be detailed in the Disscusion.
Results
R.Y tracts in chromosome 22
The chromosomes selected and their basic data are given in Table 1. Program TRACTS was applied to map the occurrence of binary DNA tracts in these chromosomes (See methods). The occurrence of R.Y tracts of different lengths in "contig 23", the main contig of human chromosome 22 (66.6% of the chromosome) is shown in Table 2. In columns 2 and 3 of the table, the number of R and Y tracts of each length found in the GenBank-listed strand is listed. Opposite each Y tract there is of course an R tract, and vice versa. The number of R tracts of each length can be seen to be roughly equal to the number Y tracts. This justifies the joint consideration of the R and Y tracts as a pair (R.Y) at this stage.
Table 1.
| Chromosome | Date | Access. No. | Length | No. of genes b | %Exons + Introns | Reference | |
| H. sapiens | 21, contig "28" | 17/4/01 | NT_011512.3 | 28,515,322 | 91 | 16 | [18] |
| H. sapiens | 22, contig "23" | 17/4/01 | NT_011620.5 | 22,998,450 | 226 | 36 | [19] |
| D. melanogaster | 2R (Right arm) | 7/11/02 | NT_033778.1 | 20,302,755 | 2687c | 57 | [20] |
| C. elegans | I | 23/4/99 | "chr_I" | 16,183,833a | 2516 | 42.6 | [21] |
| A. thaliana | II | 21/12/99 | AE002093 | 19,647,091 | 4116 | 41.7 | [22] |
| S. cerevisiae | IV | 16/6/02 | NC_001136.2 | 1,531,929 | 856 | 73.8 | [23] |
| M. jannaschii | Main | 30/1/98 | L7717 | 1,664,970 | 1715 | 87.1 | [24] |
a 1,441,828 N bases excluded. bAs found by "ANEX" in the annotation file used. c Many alternatively spliced genes.
Table 2.
R.Y Tracts in Contig "23" of Chromosome 22 (22,998,450 nt)
| No. of Tracts | No of Bases | Bases GE | |||||||
| Length | R | Y | Found(f) | Expected(e) | Difference | Found(f) | Expected(e) | f/e ratio | Ratio |
| 1 | 2,275,282 | 2,278,327 | 4,553,609 | 5,749,613 | -1,196,004 | 4,553,609 | 5,749,613 | 0.79 | 1.00 |
| 2 | 1,126,166 | 1,125,235 | 2,251,401 | 2,874,806 | -623,405 | 4,502,802 | 5,749,612 | 0.78 | 1.07 |
| 3 | 641,092 | 640,661 | 1,281,753 | 1,437,403 | -155,650 | 3,845,259 | 4,312,209 | 0.89 | 1.21 |
| 4 | 413,142 | 411,867 | 825,009 | 718,702 | 106,308 | 3,300,036 | 2,874,806 | 1.15 | 1.40 |
| 5 | 214,646 | 214,404 | 429,050 | 359,351 | 69,699 | 2,145,250 | 1,796,754 | 1.19 | 1.58 |
| 6 | 122,734 | 122,815 | 245,549 | 179,675 | 65,874 | 1,473,294 | 1,078,052 | 1.37 | 1.85 |
| 7 | 68,181 | 68,317 | 136,498 | 89,838 | 46,660 | 955,486 | 628,864 | 1.52 | 2.21 |
| 8 | 35,646 | 35,203 | 70,849 | 44,919 | 25,930 | 566,792 | 359,351 | 1.58 | 2.75 |
| 9 | 21,592 | 21,485 | 43,077 | 22,459 | 20,618 | 387,693 | 202,135 | 1.92 | 3.69 |
| 10 | 14,127 | 14,002 | 28,129 | 11,230 | 16,899 | 281,290 | 112,297 | 2.50 | 5.13 |
| 11 | 9,039 | 9,184 | 18,223 | 5614.9 | 12,608 | 200,453 | 61,763 | 3.25 | 7.32 |
| 12 | 6,173 | 6,081 | 12,254 | 2807.4 | 9,447 | 147,048 | 33,689 | 4.36 | 10.77 |
| 13 | 4,139 | 4,118 | 8,257 | 1403.7 | 6,853 | 107,341 | 18,248 | 5.88 | 16.27 |
| 14 | 2,834 | 2,811 | 5,645 | 701.9 | 4,943 | 79,030 | 9,826.0 | 8.04 | 25.27 |
| 15 | 1,982 | 2,082 | 4,064 | 350.9 | 3,713 | 60,960 | 5,263.9 | 11.58 | 40.35 |
| 16 | 1,394 | 1,539 | 2,933 | 175.5 | 2,758 | 46,928 | 2,807.4 | 16.72 | 65.73 |
| 17 | 1,131 | 1,093 | 2,224 | 87.7 | 2,136 | 37,808 | 1,491.5 | 25.35 | 109.3 |
| 18 | 913 | 932 | 1,845 | 43.9 | 1,801 | 33,210 | 789.6 | 42.06 | 184.4 |
| 19 | 710 | 695 | 1,405 | 21.9 | 1,383 | 26,695 | 416.7 | 64.06 | 312.5 |
| 20 | 568 | 568 | 1,136 | 11.0 | 1,125 | 22,720 | 219.3 | 103.6 | 537.3 |
| 21 | 480 | 478 | 958 | 5.5 | 953 | 20,118 | 115.2 | 174.7 | 931.5 |
| 22 | 405 | 369 | 774 | 2.7 | 771 | 17,028 | 60.3 | 282.3 | 1,622 |
| 23 | 305 | 339 | 644 | 1.4 | 643 | 14,812 | 31.5 | 469.8 | 2,851 |
| 24 | 302 | 292 | 594 | 0.7 | 593 | 14,256 | 16.5 | 866.6 | 5,041 |
| 25 | 277 | 251 | 528 | 0.343 | 528 | 13,200 | 8.6 | 1,540 | 8,896 |
| 26 | 220 | 222 | 442 | 0.171 | 442 | 11,492 | 4.5 | 2,579 | 15,706 |
| 27 | 194 | 202 | 396 | 8.57E-02 | 396 | 10,692 | 2.3 | 4,622 | 27,896 |
| 28 | 156 | 173 | 329 | 4.28E-02 | 329 | 9,212 | 1.2 | 7,680 | 49,564 |
| 29 | 121 | 162 | 283 | 2.14E-02 | 283 | 8,207 | 0.6 | 13,213 | 88,656 |
| 30 | 121 | 141 | 262 | 1.07E-02 | 262 | 7,860 | 3.21E-01 | 24,464 | 159,233 |
| 31 | 89 | 117 | 206 | 5.35E-03 | 206 | 6,386 | 1.66E-01 | 38,470 | 285,578 |
| 32 | 92 | 78 | 170 | 2.68E-03 | 170 | 5,440 | 8.57E-02 | 63,495 | 517,709 |
| 33 | 83 | 80 | 163 | 1.34E-03 | 163 | 5,379 | 4.42E-02 | 121,761 | 945,205 |
| 34 | 61 | 57 | 118 | 6.69E-04 | 118 | 4,012 | 2.28E-02 | 176,291 | 1,721,595 |
| 35 | 60 | 57 | 117 | 3.35E-04 | 117 | 4,095 | 1.17E-02 | 349,595 | 3,181,047 |
| 36 | 38 | 47 | 85 | 1.67E-04 | 85 | 3,060 | 6.02E-03 | 507,958 | 5,859,445 |
| 37 | 48 | 44 | 92 | 8.37E-05 | 92 | 3,404 | 3.10E-03 | 1.10E+06 | 1.09E+07 |
| 38 | 35 | 38 | 73 | 4.18E-05 | 73 | 2,774 | 1.59E-03 | 1.74E+06 | 2.03E+07 |
| 39 | 43 | 47 | 90 | 2.09E-05 | 90 | 3,510 | 8.16E-04 | 4.30E+06 | 3.78E+07 |
| 40 | 30 | 27 | 57 | 1.05E-05 | 57 | 2,280 | 4.18E-04 | 5.45E+06 | 6.97E+07 |
| 31 – 40 | 579 | 592 | 1,171a | 1.07E-02a | 1,171 | 40,340a | 3.42E-01b | 5.45E+06 | 6.97E+07b |
| 41 – 50 | 161 | 168 | 329 | 1.04E-05 | 329 | 14,669 | 4.39E-04 | 2.25E+09 | 4.22E+10 |
| 51 – 60 | 74 | 66 | 140 | 1.02E-08 | 140 | 7,643 | 5.30E-07 | 6.02E+11 | 2.92E+13 |
| 61 – 70 | 43 | 28 | 71 | 9.97E-12 | 71 | 4,647 | 6.18E-10 | 6.16E+14 | 2.24E+16 |
| 71 – 80 | 24 | 22 | 46 | 9.72E-15 | 46 | 3,445 | 6.99E-13 | 4.21E+17 | 1.78E+19 |
| 81 – 90 | 21 | 21 | 42 | 9.51E-18 | 42 | 3,603 | 7.79E-16 | 4.31E+20 | 1.41E+22 |
| 92 – 100 | 14 | 14 | 28 | 4.63E-21 | 28 | 2,675 | 4.31E-19 | 2.20E+23 | 2.26E+25 |
| 101 – 110 | 13 | 18 | 31 | 9.06E-24 | 31 | 3,280 | 0.00c | 0.00c | 0.00c |
| 111 – 120 | 3 | 7 | 10 | 8.24E-27 | 10 | 1,142 | 0 | 0.00 | 0.00 |
| 121 – 130 | 7 | 5 | 12 | 8.56E-30 | 12 | 1,509 | 0 | 0.00 | 0.00 |
| 131 – 140 | 3 | 10 | 13 | 5.92E-33 | 13 | 1,746 | 0 | 0.00 | 0.00 |
| 141 – 150 | 6 | 10 | 16 | 0.00c | 16 | 2,319 | 0 | 0.00 | 0.00 |
| 151 – 158 | 3 | 2 | 5 | 0.00 | 5 | 774 | 0 | 0.00 | 0.00 |
| 161 – 170 | 0 | 6 | 6 | 0.00 | 6 | 999 | 0 | 0.00 | 0.00 |
| 171 – 178 | 5 | 3 | 8 | 0.00 | 8 | 1,398 | 0 | 0.00 | 0.00 |
| 181 – 200 | 3 | 6 | 9 | 0.00 | 9 | 1,711 | 0 | 0.00 | 0.00 |
| 202 – 218 | 6 | 2 | 8 | 0.00 | 8 | 1,666 | 0 | 0.00 | 0.00 |
| 224 | 0 | 2 | 2 | 0.00 | 2 | 448 | 0 | 0.00 | 0.00 |
| 226 | 0 | 1 | 1 | 0.00 | 1 | 226 | 0 | 0.00 | 0.00 |
| 229 | 1 | 0 | 1 | 0.00 | 1 | 229 | 0 | 0.00 | 0.00 |
| 230 | 0 | 1 | 1 | 0.00 | 1 | 230 | 0 | 0.00 | 0.00 |
| 237 | 1 | 0 | 1 | 0.00 | 1 | 237 | 0 | 0.00 | 0.00 |
| 241 | 0 | 1 | 1 | 0.00 | 1 | 241 | 0 | 0.00 | 0.00 |
| 250 | 0 | 1 | 1 | 0.00 | 1 | 250 | 0 | 0.00 | 0.00 |
| 270 | 0 | 1 | 1 | 0.00 | 1 | 270 | 0 | 0.00 | 0.00 |
| 308 | 0 | 2 | 2 | 0.00 | 2 | 616 | 0 | 0.00 | 0.00 |
| 318 | 0 | 1 | 1 | 0.00 | 1 | 318 | 0 | 0.00 | 0.00 |
| 325 | 0 | 1 | 1 | 0.00 | 1 | 325 | 0 | 0.00 | 0.00 |
| 367 | 1 | 0 | 1 | 0.00 | 1 | 367 | 0 | 0.00 | 0.00 |
a. Found and Expected for the range of lengths b. From here, the ratios are for the last in the range, e.g. for l = 40 in the 31–40 range. c. From here, values are not computable with single precision in our setup.
Every tract length up to 78 nt is represented, and many longer tracts are present. The longest tract found is a 367 nt long, an R tract (second column). In column 5, the number of R.Y tracts that are expected in random DNA of the same length and base composition as the analyzed contig is shown (see methods). It is seen that the number of tracts expected decreases much more rapidly than the number of tracts observed (column 4). In fact, for all tracts longer than 23 nt not even a single tract is expected in randomized DNA (see column 5), while 644 such tracts are found at that length alone! (column 4). This enormous over-representation certainly calls for a biological explanation. The extent of over-representation is listed in column 9, which gives the ratio between the number of tracts (or bases) observed, to the number of tracts (or bases) expected in random DNA. This ratio is below unity for the first three rows, namely for single pyrimidines (purines) flanked by two purines (pyrimidines), their doublets and triplets. The low ratio for the short tracts compensates for the over-representation of the longer tracts, which increases steadily up to enormous figures for the higher lengths (column 9). The increase in ratios is relatively smooth, as can also be seen in Fig. 1a, which indicates that a property special to a particular length or length group is not responsible for the high excesses found. We shall use the found/expected ratio values ("f/e ratios") as the main measure for the extent of binary tract over-representation in the coming Tables. In the last column, the f/e ratio is listed for all tracts longer or equal (also "Greater or Equal", or "GE") than the length given in the first column (calculated as GE bases found divided by GE bases expected, eq. (4)). The GE value is more meaningful for the longer tract lengths, when only few tracts are encountered, so that individual f/e ratios lose their significance.
Figure 1.
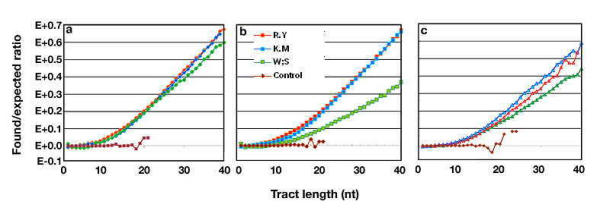
Binary tract over-representation in three chromosomes: The log ratios of found to expected number of binary tracts (f/e ratios) are plotted against tract length. Control runs are average values of five randomized DNA tracts of 1 Mb each, see Table 2, Additional Table 4 and text. a) Contig 23 of human chromosome 22, see Table 1. b) Contig 28 of human chromosome 21. c) Chromosome 2 of D. melanogaster, right arm. Tracts were plotted up to just 40 nt, to enhance resolution and to make visible the under-representation of very short tracts (f/e ratio below unity).
R.Y tracts in seven chromosomes
Tables similar to Table 2 have been constructed for a series of other genomes as well as for the other binary DNA motifs. The Data for human chr. 21 and the Drosophila chromosome are shown in Figs. 1b and 1c as well as in the Additional files: 1 and 2, also at the authors web site http://www.weizmann.ac.il/~lcyagil. The found/expected ratio values (f/e ratios) will be shown in most following tables, as the criterion for over-representation.
Table 3 gives the f/e ratios for R.Y tracts in seven chromosomes selected from sequenced genomes across the eukaryotic and archeal kingdoms. The major characteristics of the selected chromosomes have been listed in Table 1. In the last column of Table 3 a control run is shown – five random 1 Mb DNA sequences were generated and run by TRACTS, as an additional verification of the analytical formula used to calculate the expected values and f/e ratios (see methods). It can be seen that all the f/e ratios, except the longest, are close to unity. No R.Y tract longer than 21 nt was found, as it should be. (a 24 nt K.M tract was found, see below). The standard deviations are less than 5% up to 13 nt (Additional file: 4), when found tracts begin to be few. A larger SD is indeed expected for the longest tracts, because for example, for a 19 nt tract, only 19 or 38 bases, or occasionally 57 bases, are possible for that length. The detailed data can be found in Additional file: 4. The control runs thus confirm that tracts much longer than 21 nt cannot be expected in randomly composed DNA.
Table 3.
R.Y tracts in selected chromosomes. The numbers give for each length the number of found tracts divided by the number expected tracts, eq (1).
| H. sap. 22, contig 23 | H. sap. 21, contig 28 | D. mel. IIR | C. eleg. I | A. thal. II | S. cer. IV | M. jan. | Control | |
| Bases: | 22,998,450 | 28,515,322 | 20,302,755 | 14,752,005 | 19,647.091 | 1,531,929 | 1,664,970 | 5 × 1 Mb |
| %A,G: | 50.0 | 50.1 | 50.0 | 50.0 | 49.9 | 50.1 | 50.3 | 50.0 |
| Length(nt) | ||||||||
| 1 | 0.79 | 0.86 | 0.96 | 0.78 | 0.86 | 0.89 | 0.78 | 1.00 |
| 2 | 0.78 | 0.79 | 0.97 | 0.83 | 0.88 | 0.93 | 0.87 | 1.00 |
| 3 | 0.89 | 0.88 | 0.98 | 0.88 | 0.91 | 0.87 | 0.79 | 1.00 |
| 4 | 1.15 | 1.07 | 0.95 | 1.04 | 1.00 | 0.94 | 1.02 | 1.00 |
| 5 | 1.19 | 1.20 | 1.06 | 1.31 | 1.12 | 1.13 | 1.27 | 1.00 |
| 6 | 1.37 | 1.32 | 1.07 | 1.50 | 1.17 | 1.23 | 1.35 | 1.00 |
| 7 | 1.52 | 1.46 | 1.07 | 1.56 | 1.41 | 1.36 | 1.87 | 1.00 |
| 8 | 1.58 | 1.63 | 1.18 | 1.77 | 1.66 | 1.66 | 2.13 | 1.00 |
| 9 | 1.92 | 2.04 | 1.27 | 2.16 | 1.92 | 1.86 | 2.04 | 1.01 |
| 10 | 2.50 | 2.55 | 1.54 | 2.61 | 2.40 | 2.31 | 3.32 | 1.00 |
| 11 | 3.25 | 3.21 | 1.84 | 3.03 | 3.12 | 2.91 | 3.94 | 1.04 |
| 12 | 4.36 | 4.29 | 2.20 | 3.49 | 4.05 | 3.52 | 3.96 | 1.09 |
| 13 | 5.88 | 5.54 | 2.89 | 4.20 | 5.34 | 4.81 | 6.66 | 1.09 |
| 14 | 8.04 | 7.48 | 3.58 | 5.95 | 7.53 | 5.49 | 7.40 | 1.14 |
| 15 | 11.58 | 10.16 | 4.83 | 7.73 | 10.36 | 8.89 | 7.30 | 1.05 |
| 16 | 16.72 | 14.53 | 6.88 | 11.61 | 14.94 | 9.41 | 14.66 | 1.13 |
| 17 | 25.35 | 21.48 | 9.93 | 16.54 | 23.05 | 14.02 | 17.71 | 0.89 |
| 18 | 42.06 | 33.01 | 14.38 | 25.45 | 28.84 | 18.81 | 22.87 | 2.31 |
| 19 | 64.06 | 48.22 | 18.28 | 33.12 | 47.22 | 35.56 | 30.68 | 0.84 |
| 20 | 103.6 | 72.85 | 27.99 | 52.46 | 80.36 | 41.02 | 52.55 | 1.68 |
| 21 | 174.7 | 125.1 | 42.56 | 78.76 | 102.2 | 57.42 | 65.01 | 8.39 |
| 22 | 282.3 | 199.9 | 71.06 | 98.38 | 175.9 | 98.42 | 94.94 | |
| 23 | 469.8 | 335.7 | 110.7 | 217.2 | 251.0 | 54.67 | 69.90 | |
| 24 | 866.6 | 575.0 | 150.4 | 270.7 | 384.1 | 131.2 | 159.6 | |
| 25 | 1,541 | 945.3 | 231.4 | 436.7 | 662.4 | 306.1 | 159.5 | |
| 26 | 2,579 | 1,815 | 350.4 | 545.9 | 1,086 | 1,049 | 398.3 | |
| 27 | 4,622 | 2,774 | 502.4 | 1,092 | 1,680 | 874.1 | 318.3 | |
| 28 | 7,680 | 4,946 | 1,005 | 1,383 | 2,622 | 1,049 | 635.9 | |
| 29 | 13,213 | 8,650 | 1,322 | 1,747 | 4,971 | 2,796 | 3,811 | |
| 30 | 24,464 | 14,667 | 1,798 | 4,658 | 6,664 | 5,591 | 2,538 | |
| 31 | 38,470 | 24,969 | 3,808 | 6,696 | 11,143 | 2,795 | 2,535 | |
| 32 | 63,495 | 36,998 | 5,077 | 16,304 | 16,167 | 11,177 | - | |
| 33 | 121,761 | 66,771 | 8,461 | 11,646 | 23,594 | 22,348 | - | |
| 34 | 176,291 | 145,557 | 10,153 | 18,633 | 38,448 | - | - | |
| 35 | 349,595 | 204,479 | 30,460 | 32,608 | 108,348 | 178,697 | - | |
| 36 | 507,958 | 380,048 | 81,226 | 37,266 | 209,697 | 89,326 | 80,562 | |
| 37 | 1.10E+06 | 711,906 | 54,150 | 186,332 | 209,687 | - | - | |
| 38 | 1.74E+06 | 1.50E+06 | 54,150 | 372,665 | 391,396 | - | - | |
| 39 | 4.30E+06 | 2.12E+06 | 216,599 | 372,665 | 615,020 | - | - | |
| 40 | 5.45E+06 | 4.85E+06 | - | 447,198 | 1.12E+06 | - | - | |
| 41 | 1.11E+07 | 7.38E+06 | 433,193 | 1.79E+06 | 1.12E+06 | 2.85E+06 | - | |
| 42 | 1.87E+07 | 1.42E+07 | 433,190 | 1.79E+06 | 2.24E+06 | - | - | |
| 43 | 3.44E+07 | 2.15E+07 | 2.60E+06 | 2.39E+06 | 2.68E+06 | - | - | |
| 44 | 4.59E+07 | 4.80E+07 | 1.73E+06 | 4.77E+06 | 1.07E+07 | - | - | |
| 45 | 7.96E+07 | 8.12E+07 | - | 1.43E+07 | 1.79E+07 | 4.56E+07 | - | |
| 46 | 1.29E+08 | 1.38E+08 | 6.93E+06 | 9.54E+06 | 4.29E+07 | - | - | |
| 47 | 3.30E+08 | 2.75E+08 | - | - | 7.15E+07 | 1.82E+08 | 1.62E+08 | |
| 48 | 6.61E+08 | 4.52E+08 | - | 3.82E+07 | 2.86E+07 | - | - | |
| 49 | 1.13E+09 | 7.08E+08 | - | 2.29E+08 | 1.14E+08 | - | - | |
| 50 | 2.25E+09 | 1.42E+09 | 1.11E+08 | 1.53E+08 | 2.29E+08 | - | 1.29E+09 | |
| 51 | 3.13E+09 | 2.52E+09 | - | 6.11E+08 | - | 2.91E+09 | - | |
| 52 | 7.44E+09 | 6.61E+09 | - | - | 4.58E+08 | - | - | |
| 53 | 1.64E+10 | 1.07E+10 | - | - | 3.66E+09 | - | - | |
| 54 | 3.13E+10 | 1.51E+10 | - | 4.88E+09 | - | - | - | |
| 55 | 4.39E+10 | 2.01E+10 | - | 4.88E+09 | 3.66E+09 | 4.65E+10 | - | |
| 56 | 1.07E+11 | 6.04E+10 | - | 1.95E+10 | 7.32E+09 | |||
| 57 | 1.38E+11 | 1.21E+11 | - | 5.86E+10 | - | |||
| 58 | 1.75E+11 | 1.81E+11 | - | - | 5.86E+10 | |||
| 59 | 4.51E+11 | 6.84E+11 | - | - | 5.86E+10 | |||
| 60 | 6.02E+11 | 8.85E+11 | - | - | - | |||
| 61 | 1.20E+12 | 1.77E+12 | - | 3.13E+11 | 2.34E+11 | |||
| 62 | 4.01E+12 | 3.22E+12 | - | 6.25E+11 | 4.68E+11 | |||
| 63 | 3.21E+12 | 7.72E+12 | - | - | 9.36E+11 | |||
| 64 | 1.12E+13 | 1.03E+13 | - | - | 1.87E+12 | |||
| 65 | 2.89E+13 | 1.80E+13 | - | - | - | |||
| 66 | 3.21E+13 | 3.08E+13 | - | - | - | |||
| 67 | 1.41E+14 | 1.34E+14 | - | - | - | |||
| 68 | 2.05E+14 | 2.26E+14 | - | - | - | |||
| 69 | 2.57E+14 | 2.88E+14 | - | - | 5.99E+13 | |||
| 70 | 6.16E+14 | 6.57E+14 | 4.65E+14 | - | 2.40E+14 | |||
| 71 | 1.23E+15 | 1.48E+15 | 6.40E+14 | - | ||||
| 72 | 2.46E+15 | 3.61E+15 | 6.40E+14 | - | ||||
| 73 | 4.93E+15 | 5.91E+15 | - | - | ||||
| 74 | 4.93E+15 | 5.25E+15 | - | 1.92E+15 | ||||
| 75 | 1.97E+16 | 7.88E+15 | - | - | ||||
| 76 | 2.63E+16 | 2.63E+16 | 1.02E+16 | - | ||||
| 77 | 6.57E+16 | 5.25E+16 | 2.05E+16 | - | ||||
| 78 | 1.58E+17 | 6.30E+16 | - | - | ||||
| 79 | - | 2.52E+17 | - | - | ||||
| 80 | 4.21E+17 | 4.20E+17 | - | 2.45E+17 | ||||
| 81 | 6.31E+17 | 8.39E+17 | - | 2.45E+17 | ||||
| 82 | 2.52E+18 | 1.68E+18 | - | - | ||||
| 83 | 8.41E+17 | 3.35E+18 | - | - | ||||
| 84 | 6.73E+18 | 2.68E+18 | - | 1.96E+18 | ||||
| 85 | 1.35E+19 | 5.36E+18 | - | 7.84E+18 | ||||
| 86 | 3.36E+19 | 1.07E+19 | 1.05E+19 | - | ||||
| 87 | 8.07E+19 | 5.36E+19 | - | - | ||||
| 88 | 1.08E+20 | 1.29E+20 | 4.20E+19 | - | ||||
| 89 | 2.69E+20 | 1.71E+20 | - | - | ||||
| 90 | 4.31E+20 | 4.29E+20 | - | - | ||||
| 91 | 1.29E+21 | 8.57E+20 | - | - | ||||
| 92 | 1.72E+21 | 6.85E+20 | 0.00E+01 | - | ||||
| 93 | 4.31E+21 | 2.06E+21 | 0.00E+01 | - | ||||
| 94 | 6.89E+21 | 4.11E+21 | - | - | ||||
| 95 | 6.89E+21 | 2.74E+21 | 0.00E+01 | - | ||||
| 96 | 1.38E+22 | 5.48E+21 | 0.00E+01 | 3.21E+22 | ||||
| 97 | 4.13E+22 | 5.47E+22 | 0.00E+01 | 6.41E+22 | ||||
| 98 | 2.76E+22 | 1.09E+23 | ||||||
| 99 | 2.76E+23 | - | ||||||
| 100 | 2.20E+23 | 3.50E+23 | ||||||
| 101 | 0.00E+01 | 0.00E+01 | ||||||
| Beyond This point ratios are not calculable at our precision. From here, number of tracts, or lengths, are shown | ||||||||
| 102 | 1 (1Y) | - | Also: | Also: | ||||
| 103 | 4 (1Y+3R) | - | 114 nt | 108; 110 nt | ||||
| 104 | 2 (1Y+1R) | 5 (3Y+2R) | 117 nt | 111; 121 nt | ||||
| 105 | 3 (1Y+2Y) | 2 (2Y) | 120 nt | 124; 139 nt | ||||
| 106 | 5 (2Y+3R) | 2 (1Y+1R) | 134 nt | 145; 169 nt | ||||
| 107 | 8 (6Y+2R) | 3 (2Y+1R) | 140 nt | 174; 180 nt | ||||
| 108 | 1 (1R) | 8 (4Y +4R) | 161 nt | 182; 189 nt | ||||
| 109 | 3 (2Y+1R) | 1 (1R) | 194 nt | |||||
| 110 | 2 (1Y+1R) | 6 (4Y+2R) | ||||||
| Longer tracts and Summary | ||||||||
| H. sap. 22 contig 23 | H. sap. 21, contig 28 | D. mel. IIR | C. eleg. I | A. thal. II | S. cer. IV | M. jan. | ||
| All found, up to (nt): | 78 | 98 | 39 | 46 | 50 | 33 | 31 | |
| Next missing (nt) | 114 | 102 | 45 | 52 | 54 | 37 | 37 | |
| 100 to 200 nt (tracts): | 113 | 142 | - | 6 | 13 | - | - | |
| Longer than 200 nt(tracts) | 22 | 24 | - | - | - | - | - | |
| Longest (nt): | 367 | 568 | 70 | 161 | 189 | 55 | 50 | |
A – (hyphen) means that no tract of that length is present
The major conclusion from Table 3 is that the longer R.Y tracts are highly over-represented, up to extreme values, in all the seven genomes examined. In contrast, tracts of lengths up to three nt are under-represented in all the phyla studied, as already described for chromosome 22. The longest tracts found in each species are roughly related to the length of the input DNA: From 50 nt for the 1.65 Mb M. jannaschii, to 55 nt for yeast chromosome IV (the longest yeast chromosome), to 161 for the elegans chromosome (14.7 Mb); 194 nt for the 20 Mb of Arabidopsis, and up to 367; 568 nt for the two human contigs. The one exception is the Drosophila half chromosome (20 Mb) where the longest tract is just 70 nt. It will be seen that the Drosophila chromosome is exceptional in other respects as well. A correlation between the size of the longest tract up to which every length is present, with the length of the input DNA sequence is also observed (Table 3): 31 nt for jannaschii, 33 for yeast, 46 nt for elegans, 50 for Arabidopsis, 78 and 98 nt for the human contigs. Again, 39 nt for Drosophila is an outlier. The lesser over-representation in Drosophila is also evident when individual numbers are compared to the other organisms; the highest excesses of long tracts are clearly in the two human contigs. The overall result is that the two human chromosomes exhibit the highest over-representation, with most other chromosomes not far behind; the short M. jannaschii leads occasionally for 7–11 NT tracts. These conclusions can also be seen in Fig. 2a. Between the two human chromosomes, the gene poor chromosome 21 takes the lead.
Figure 2.
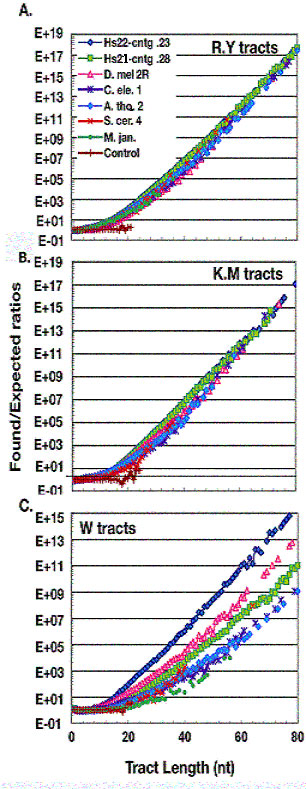
The over-representation of binary tracts in chromosomes of six organisms. The log ratios of found to expected number of binary tracts are plotted against tract length. Control runs are the same as in Fig. 1. Tracts up to 80 nt are plotted. The symbols for the seven chromosomes are given on the figure. A R.Y tracts. B K.M tracts. C W tracts
K.M tracts in seven chromosomes
It was noted earlier that not only R.Y tracts are over-represented, but, at first a bit counter-intuitively, the other three binary DNA combinations as well. Thus, K.M tracts were found in large excess in the human β globin complex and in organelle DNA [13], as well as in yeast chromosome 3 [14]. The data in Tables 4 and 5 show that these findings can be extended to the wider range of phyla studied here. In Table 4, the f/e ratios for K.M tracts are shown. As for the R.Y pair, the detailed outputs for each chromosome (see Additional files: 1, 2 and 3) show that roughly equal numbers of K tracts and M tracts are present in the analyzed strand, and justifies their joint consideration. Overall, it is clear that K.M tracts are also highly over-represented, in all seven chromosomes, even if to a lesser extent than the R.Y tracts. In humans, contig 23 of chromosome 22 shows the highest over-representations but beyond 67 nt many lengths are missing, the longest tract being just 91 nt long. In contig 28 many K.M lengths beyond 62 nt are missing; there are only two K.M tracts longer than 100 nt (101 nt, 268 nt). The f/e ratios for K.M tracts are sometimes even higher than for R.Y tracts (in chr. 21 there are 9 cases between 32 and 51 nt and 5 cases in chr. 22). Beyond 52 nt, f/e ratios are always higher for R.Y than for K.M tracts.
Table 4.
K.M Tracts in selected chromosomes. Numbers are f/e ratios, i.e. number of tracts found at each length, divided by the number expected, eq.(1)
| H. sap 22 contig 23 | H. sap 21 contig 28 | D. mel IIR | C. ele I | A. tha II | S. cer. IV | M. jan. | Control | |
| 22,998,450 | 28,515,322 | 20,302,755 | 14,752,005 | 19,647,091 | 1,531,929 | 1,664,970 | 5 × 1 Mb | |
| %AC: | 0.501 | 0.502 | 0.500 | 0.500 | 0.499 | 0.500 | 0.500 | 0.500 |
| Length | ||||||||
| 1 | 0.82 | 0.84 | 0.88 | 0.83 | 0.93 | 0.90 | .95 | 1.00 |
| 2 | 0.90 | 0.89 | 0.91 | 0.84 | 0.88 | 0.93 | .85 | 1.00 |
| 3 | 0.97 | 0.98 | 0.97 | 0.90 | 0.88 | 0.96 | .99 | 1.00 |
| 4 | 1.04 | 1.05 | 1.04 | 1.08 | 1.00 | 1.04 | 1.00 | 1.00 |
| 5 | 1.12 | 1.14 | 1.11 | 1.24 | 1.11 | 1.12 | 1.11 | 1.01 |
| 6 | 1.28 | 1.24 | 1.13 | 1.41 | 1.18 | 1.16 | 1.26 | 1.01 |
| 7 | 1.30 | 1.31 | 1.21 | 1.52 | 1.36 | 1.28 | 1.39 | 1.01 |
| 8 | 1.45 | 1.43 | 1.40 | 1.65 | 1.59 | 1.38 | 1.39 | 0.98 |
| 9 | 1.54 | 1.57 | 1.55 | 1.88 | 1.86 | 1.43 | 1.39 | 1.00 |
| 10 | 1.86 | 1.88 | 1.91 | 2.31 | 2.25 | 1.58 | 1.60 | 0.98 |
| 11 | 2.29 | 2.22 | 2.33 | 2.70 | 2.81 | 1.78 | 1.79 | 1.00 |
| 12 | 2.96 | 2.78 | 2.82 | 3.24 | 3.40 | 1.97 | 1.98 | 1.07 |
| 13 | 3.67 | 3.15 | 3.73 | 3.76 | 4.34 | 2.12 | 2.14 | 1.12 |
| 14 | 5.27 | 4.38 | 5.07 | 4.50 | 5.78 | 3.19 | 2.81 | 0.93 |
| 15 | 7.23 | 5.97 | 6.80 | 5.66 | 7.80 | 3.04 | 3.15 | 1.03 |
| 16 | 11.66 | 8.91 | 9.59 | 8.10 | 10.78 | 3.76 | 3.23 | 0.99 |
| 17 | 18.29 | 14.78 | 14.68 | 10.00 | 14.80 | 6.67 | 3.15 | 0.72 |
| 18 | 30.09 | 21.06 | 22.03 | 15.57 | 20.70 | 6.84 | 1.89 | 0.41 |
| 19 | 46.13 | 33.74 | 32.59 | 19.05 | 26.41 | 7.53 | 2.52 | 1.29 |
| 20 | 76.86 | 52.93 | 56.09 | 26.58 | 44.50 | 12.3 | 8.82 | 1.23 |
| 21 | 130.6 | 91.90 | 79.53 | 36.96 | 60.19 | 13.7 | 2.52 | 4.50 |
| 22 | 219.5 | 152.0 | 110.7 | 47.20 | 81.53 | 27.4 | 10.08 | - |
| 23 | 395.3 | 267.6 | 214.0 | 87.57 | 142 | 11.0 | 10.08 | 6.50 |
| 24 | 732.2 | 455.3 | 294.2 | 152.4 | 178 | 43.8 | - | 6.49 |
| 25 | 1,237 | 802.4 | 585.0 | 168.3 | 290 | 87.6 | ||
| 26 | 2,019 | 1,342 | 1,025 | 191.1 | 382 | 350.0 | ||
| 27 | 3,232 | 2,532 | 1,467 | 345.7 | 669 | 350.0 | ||
| 28 | 6,021 | 4,069 | 2,353 | 509.5 | 874 | 701.3 | ||
| 29 | 10,034 | 7,274 | 3,596 | 655.1 | 993 | - | ||
| 30 | 16,988 | 12,743 | 4,653 | 1,601 | 1,967 | - | ||
| 31 | 26,694 | 22,181 | 9,730 | 1,456 | 2,622 | - | ||
| 32 | 50,773 | 37,754 | 11,845 | 1,747 | 3,933 | - | ||
| 33 | 100,049 | 62,900 | 21,151 | 2,329 | 6,991 | - | ||
| 34 | 150,816 | 105,392 | 43,994 | 6,987 | 13,982 | 2.24 E+04 | ||
| 35 | 316,553 | 227,460 | 60,914 | 27,950 | 13,982 | 4.49 E+04 | ||
| 36 | 513,634 | 402,082 | 108,290 | 18,633 | 34,953 | 8.97 E+04 | ||
| 37 | 848,064 | 775,120 | 162,433 | 18,633 | 41,942 | - | ||
| 38 | 1.86E+06 | 1.26E+06 | 270,718 | 111,800 | 167,761 | - | ||
| 39 | 2.87E+06 | 2.52E+06 | 216,572 | 74,533 | 55,918 | - | ||
| 40 | 5.64E+06 | 3.82E+06 | 757,990 | - | 111,831 | - | ||
| 41 | 9.17E+06 | 8.86E+06 | 1.08E+06 | - | 223,651 | 2.87 E+06 | ||
| 42 | 1.76E+07 | 1.59E+07 | 1.30E+06 | - | 894,562 | - | ||
| 43 | 3.59E+07 | 2.50E+07 | 2.60E+06 | - | 3.58E+06 | - | ||
| 44 | 3.97E+07 | 4.76E+07 | 8.66E+06 | - | 3.58E+06 | - | ||
| 45 | 7.03E+07 | 1.00E+08 | 1.04E+07 | 9.54E+06 | 3.58E+06 | Also: | ||
| 46 | 2.14E+08 | 1.07E+08 | - | - | - | 97nt | ||
| 47 | 3.67E+08 | 3.02E+08 | 1.39E+07 | - | 1.43E+07 | 2.07 E+23 | ||
| 48 | 6.85E+08 | 4.09E+08 | 5.54E+07 | - | 5.72E+07 | |||
| 49 | 1.27E+09 | 8.18E+08 | 5.54E+07 | - | - | 155 nt | ||
| 50 | 9.78E+08 | 7.79E+08 | 2.22E+08 | 1.53E+08 | 1.14E+08 | (telomere) | ||
| 51 | 3.52E+09 | 3.11E+09 | - | - | - | |||
| 52 | 5.87E+09 | 3.42E+09 | 4.43E+08 | 1.22E+09 | - | |||
| 53 | 7.82E+09 | 4.35E+09 | - | - | - | |||
| 54 | 2.19E+10 | 1.37E+10 | 3.55E+09 | - | - | |||
| 55 | 1.25E+10 | 1.74E+10 | 3.55E+09 | - | - | |||
| 56 | 7.51E+10 | 4.47E+10 | - | 9.77E+09 | - | |||
| 57 | 7.51E+10 | 2.98E+10 | - | - | - | |||
| 58 | 1.50E+11 | 1.79E+11 | - | - | 5.86E+10 | |||
| 59 | 2.50E+11 | 2.38E+11 | 5.68E+10 | - | ||||
| 60 | 7.01E+11 | 4.75E+11 | - | 1.56E+11 | ||||
| 61 | 6.00E+11 | 3.17E+11 | 2.27E+11 | 3.13E+11 | ||||
| 62 | 1.60E+12 | 9.50E+11 | - | - | ||||
| 63 | 1.60E+12 | - | - | - | ||||
| 64 | 3.20E+12 | 2.53E+12 | - | - | ||||
| 65 | 9.61E+12 | 5.05E+12 | - | - | ||||
| 66 | 6.40E+12 | 5.05E+12 | - | - | ||||
| 67 | 1.28E+13 | - | - | - | ||||
| 68 | - | - | - | - | ||||
| 69 | 5.12E+13 | 4.03E+13 | - | 1.60E+14 | ||||
| 70 | - | 8.05E+13 | - | - | ||||
| 71 | - | 4.83E+14 | - | - | ||||
| 72 | 2.05E+14 | - | - | - | ||||
| 73 | 1.89E+14 | - | - | - | ||||
| 74 | 1.64E+15 | - | 1.86E+15 | - | ||||
| 75 | 3.28E+15 | - | ||||||
| 76 | 6.55E+15 | - | ||||||
| 80 | 1.05E+17 | - | ||||||
| 91 | 2.14E+20 | - | ||||||
| 93 | - | 6.61E+20 | ||||||
| Also Found (nt): | 101, 268 | |||||||
| Summary | ||||||||
| H. sap. 22, contig 23 | H. sap. 21, contig 28 | D. mel. IIR | C. eleg. I | A. thal. II | S. cer. IV | M. jan. | ||
| All found, up to (nt): | 67 | 62 | 45 | 39 | 45 | 28 | 23 | |
| Next missing (nt) | 70 | 67 | 51 | 41 | 49 | 30 | - | |
| Longest (nt): | 91 | 268 | 74 | 69 | 58 | 155 | 23 | |
Table 5.
W Tracts in Selected Chromosomes. The numbers give for each length the number of found tracts divided by the number expected tracts, eq (1).
| H. sap. 22 contig 23 | H. sap. 21, contig 28 | D. mel. IIR | C. eleg. I | A. thal. II | S. cer. IV | M. jan. | Control | |
| Bases | 22,998,450 | 28,515,322 | 20,302,755 | 14,752,005 | 19,647,091 | 1,531,929 | 1,664,970 | 5 × 1 Mb |
| %AT: | 52.6 | 60.9 | 56.0 | 64.0 | 64.1 | 62.1 | 68.6 | |
| 1 | 1.27 | 1.24 | 1.00 | 1.04 | 1.06 | 0.98 | 0.99 | 1.00 |
| 2 | 0.89 | 0.95 | 0.87 | 0.97 | 1.13 | 1.11 | 0.98 | 1.00 |
| 3 | 0.78 | 0.87 | 0.85 | 0.80 | 1.03 | 0.98 | 0.92 | 1.00 |
| 4 | 0.76 | 0.84 | 0.93 | 0.86 | 0.98 | 1.00 | 0.88 | 1.00 |
| 5 | 0.75 | 0.84 | 0.97 | 0.95 | 0.91 | 1.01 | 1.06 | 1.00 |
| 6 | 0.82 | 0.85 | 0.95 | 1.00 | 0.82 | 0.91 | 0.92 | 1.00 |
| 7 | 0.95 | 0.90 | 1.04 | 1.07 | 0.81 | 0.92 | 1.02 | 0.99 |
| 8 | 1.49 | 1.05 | 1.25 | 1.06 | 0.82 | 0.97 | 1.14 | 0.98 |
| 9 | 1.50 | 1.10 | 1.46 | 1.13 | 0.85 | 0.93 | 0.99 | 0.99 |
| 10 | 1.75 | 1.19 | 1.78 | 1.19 | 0.90 | 0.95 | 1.03 | 0.98 |
| 11 | 2.38 | 1.41 | 2.10 | 1.21 | 0.95 | 0.94 | 1.08 | 1.02 |
| 12 | 2.89 | 1.55 | 2.47 | 1.26 | 1.04 | 0.89 | 0.96 | 1.06 |
| 13 | 4.04 | 1.72 | 3.04 | 1.38 | 1.12 | 0.91 | 1.15 | 1.24 |
| 14 | 5.73 | 2.08 | 3.78 | 1.63 | 1.28 | 0.93 | 1.28 | 0.94 |
| 15 | 8.33 | 2.53 | 5.00 | 1.88 | 1.45 | 1.13 | 1.19 | 0.99 |
| 16 | 13.59 | 3.12 | 6.73 | 2.42 | 1.67 | 1.14 | 1.32 | 1.01 |
| 17 | 23.49 | 3.94 | 8.68 | 2.94 | 2.04 | 1.21 | 1.33 | 1.12 |
| 18 | 32.84 | 4.78 | 11.33 | 3.40 | 2.42 | 1.38 | 1.41 | 0.66 |
| 19 | 50.82 | 6.11 | 15.47 | 4.19 | 2.96 | 1.75 | 1.59 | 1.36 |
| 20 | 79.24 | 7.96 | 22.23 | 4.96 | 3.73 | 2.63 | 1.36 | 2.82 |
| 21 | 132.7 | 9.92 | 28.73 | 5.25 | 4.50 | 2.72 | 1.49 | 2.86 |
| 22 | 214.1 | 13.57 | 38.57 | 6.83 | 4.91 | 2.27 | 1.81 | 1.00 |
| 23 | 362.3 | 18.37 | 46.58 | 6.92 | 6.69 | 2.35 | 2.60 | |
| 24 | 542.3 | 24.43 | 73.95 | 8.99 | 8.74 | 6.31 | 2.19 | |
| 25 | 882.6 | 33.06 | 103.4 | 12.26 | 10.39 | 6.78 | 3.41 | |
| 26 | 1,355 | 42.51 | 150.1 | 12.39 | 12.68 | 9.83 | 3.87 | |
| 27 | 2,061 | 50.14 | 205.0 | 14.55 | 18.18 | 12.31 | 4.03 | |
| 28 | 3,323 | 74.65 | 293.6 | 16.50 | 18.70 | 8.49 | 7.29 | |
| 29 | 5,538 | 94.83 | 416.8 | 24.25 | 26.51 | 4.56 | 5.15 | |
| 30 | 6,465 | 145.7 | 618.8 | 28.41 | 36.50 | 29.37 | 6.00 | |
| 31 | 12,621 | 171.0 | 860.5 | 41.73 | 46.36 | 23.65 | 5.84 | |
| 32 | 18,990 | 277.2 | 1,217 | 31.35 | 64.04 | 19.05 | 7.45 | |
| 33 | 29,963 | 342.3 | 1,845 | 55.41 | 90.69 | 92.02 | 6.21 | |
| 34 | 49,956 | 459.7 | 2,786 | 68.44 | 104.3 | 98.80 | - | |
| 35 | 101,531 | 723.0 | 4,446 | 88.03 | 118.0 | 79.56 | 29.70 | |
| 36 | 138,383 | 805.0 | 6,586 | 103.1 | 173.6 | 256.3 | 19.25 | |
| 37 | 270,878 | 1,452 | 8,640 | 161.1 | 200.2 | 206.3 | 21.05 | |
| 38 | 522,205 | 2,172 | 9,963 | 287.6 | 253.1 | 996.9 | 30.70 | |
| 39 | 661,419 | 2,574 | 11,744 | 224.6 | 539.3 | 535.2 | - | |
| 40 | 956,122 | 4,805 | 24,918 | 292.3 | 533.2 | 1,724 | 21.76 | |
| 41 | 2.23E+06 | 7,104 | 44,059 | 502.2 | 1,087 | - | - | |
| 42 | 2.27E+06 | 11,670 | 84,394 | 499.2 | 797.6 | - | 138.84 | |
| 43 | 5.62E+06 | 12,780 | 149,221 | 334.2 | 1,554 | - | 67.49 | |
| 44 | 9.25E+06 | 20,295 | 81,183 | 869.9 | 1,454 | - | - | |
| 45 | 2.64E+07 | 40,237 | 466,518 | 1,359 | 3,778 | - | 143.52 | |
| 46 | 2.44E+07 | 49,102 | 253,807 | 1,698 | 2,945 | - | 209.29 | |
| 47 | 7.32E+07 | 80,662 | 336,576 | 663 | 5,510 | - | 305.21 | |
| 48 | 7.88E+07 | 101,928 | 991,860 | 2,071 | 10,022 | - | 445.08 | |
| 49 | 1.41E+08 | 167,440 | 1.40E+06 | 6,470 | 6,696 | - | - | |
| 50 | 2.51E+08 | 247,554 | 6.20E+05 | 5,053 | 10,440 | - | - | |
| 51 | 4.13E+08 | 338,889 | 5.48E+06 | 7,893 | 13,563 | - | - | |
| 52 | 6.04E+08 | 371,137 | 9.69E+06 | 6,164 | 29,605 | - | - | |
| 53 | 1.03E+09 | 609,680 | 1.37E+07 | - | 32,968 | - | - | |
| 54 | 3.49E+09 | 901,390 | 1.82E+07 | 15,038 | 41,119 | - | 8560.91 | |
| 55 | 3.73E+09 | 987,165 | 1.07E+07 | 23,489 | 32,053 | - | - | |
| 56 | 5.51E+09 | 2.43E+06 | 1.90E+07 | 146,758 | 99,945 | - | 9102.78 | |
| 57 | 1.05E+10 | 4.44E+06 | 6.70E+07 | 114,615 | 155,820 | - | ||
| 58 | 1.99E+10 | 8.02E+06 | 5.92E+07 | 268,534 | 60,733 | - | ||
| 59 | 3.78E+10 | 9.59E+06 | 1.05E+08 | 139,813 | 189,371 | - | ||
| 60 | 9.23E+10 | 1.38E+07 | 3.70E+08 | 436,763 | - | - | ||
| 61 | 1.36E+11 | 2.91E+07 | 3.28E+08 | 1.02E+06 | 230,146 | - | ||
| 62 | 7.40E+10 | 2.66E+07 | 1.16E+09 | - | 358,810 | - | ||
| 63 | 3.52E+11 | 6.11E+07 | - | - | 559,403 | - | ||
| 64 | 1.34E+11 | 1.00E+08 | - | - | - | 7.99E+07 | ||
| 65 | 1.52E+12 | 9.42E+07 | - | 4.06E+06 | - | - | ||
| 66 | 1.45E+12 | 1.16E+08 | - | - | 2.12E+06 | - | ||
| 67 | 9.16E+11 | 2.54E+08 | - | - | - | - | ||
| 68 | - | 3.13E+08 | 1.77E+10 | - | - | - | ||
| 69 | 6.62E+12 | 1.71E+08 | 3.13E+10 | 1.21E+07 | 8.03E+06 | - | ||
| 70 | 3.14E+13 | 2.82E+08 | - | - | - | - | ||
| 71 | 2.39E+13 | 9.25E+08 | 9.78E+10 | - | - | - | ||
| 72 | 4.54E+13 | 7.60E+08 | - | 4.61E+07 | - | - | ||
| 73 | - | 2.50E+09 | 3.06E+11 | - | 4.75E+07 | - | ||
| 74 | 8.19E+13 | 6.15E+09 | - | 2.25E+08 | - | - | ||
| 75 | 1.56E+14 | 1.01E+10 | - | - | 1.15E+08 | - | ||
| 76 | 2.96E+14 | 5.64E+09 | - | - | - | - | ||
| 77 | 5.61E+14 | 1.90E+10 | 2.99E+12 | - | - | - | ||
| 78 | - | 6.42E+10 | 5.28E+12 | - | - | - | ||
| 79 | - | 5.41E+10 | - | 1.04E+09 | - | - | ||
| 80 | - | 9.13E+10 | - | - | 1.06E+09 | - | ||
| 81 | - | 7.70E+10 | - | - | - | - | ||
| 82 | 1.39E+16 | - | - | - | - | - | ||
| 83 | 2.64E+16 | 4.39E+11 | - | - | - | - | ||
| 84 | - | 7.40E+11 | 1.61E+14 | - | - | - | ||
| 85 | - | 6.25E+11 | - | - | - | 1.77E+12 | ||
| 86 | 5.43E+17 | 1.05E+12 | 2.55E+19 | - | - | |||
| 87 | 3.44E+17 | 1.78E+12 | - | - | - | |||
| 88 | - | - | - | - | - | |||
| 89 | 1.24E+18 | - | - | 1.81E+11 | - | |||
| 90 | - | - | - | - | - | |||
| 91 | 4.48E+18 | - | - | 96: 2.05E+12 | ||||
| 92 | - | 2.44E+13 | - | 131 nt: 1.09E+17 | ||||
| 93 | 1.62E+19 | 8.26E+13 | 105: 1.61E+14 | |||||
| 94 | - | - | 112: 1.55E+21 | |||||
| 95 | - | 2.36E+14 | 126 5.92E+24 | |||||
| 96 | 1.17 E+20 | - | 168: 3.85E+30 | |||||
| 97 | 4.56 E+20 | - | ||||||
| 98 | 4.42 E+20 | - | 22/23, continued: | 21/28, continued: | ||||
| 99 | 1.72 E+21 | 1.93E+15 | ||||||
| 100 | - | - | 126 | 1.70E+21 | ||||
| 101 | - | - | 135 | 2.50E+23 | ||||
| 102 | - | 4.67E+15 | 137 | 7.86E+23 | ||||
| 103 | 1.22 E+22 | 138 | 1.40E+24 | |||||
| 104 | - | 1.34E+16 | 121 | 1.9E+27 | 139 | 2.52E+24 | ||
| 105 | - | 2.27E+16 | 181 | 0.00E+0 | 140 | 4.57E+24 | ||
| 106 | - | 7.71E+16 | 210 | 0.00E+0 | 146 | 2.15E+26 | ||
| 107 | 1.73 E+23 | - | 218 | 0.00E+0 | 152 | 1.47E+28 | ||
| 108 | - | - | 265 | 0.00E+0 | 155 | 2.10E+28 | ||
| 109 | - | 1.88E+17 | 238 | 0.00E+01 | ||||
| 110 | - | - | 242 | 0.00E+01 | ||||
| 111 | - | - | 332 | 0.00E+01 | ||||
| 112 | - | 9.25E+17 | 349 | 0.00E+01 | ||||
| 117 | - | 1.33E+19 | 473 | 0.00E+01 | ||||
| 120 | - | 6.63E+19 | ||||||
| Summary | ||||||||
| H. sap. 22, contig 23 | H. sap. 21, contig 28 | D. mel. IIR | C. eleg. I | A. thal. II | S. cer. IV | M. jan. | ||
| All up to: | 67 | 81 | 62 | 52 | 59 | 40 | 33 | |
| Next missing | 73 | 88 | 64 | 62 | 64 | 42 | 39 | |
| Longest: | 265 | 473 | 168 | 96 | 131 | 85 | 56 | |
The interesting genome is again Drosophila: Here the over-representation of K.M tracts is eventually 2–3 times higher than for the R.Y tracts (Fig. 1c) and is sometimes higher than in the human chromosomes (between 10–20 nt it is as high as in chr. 21 and not much lower at other lengths, Table 4). Whatever the function of the binary tracts may be, in Drosophila that function seems to be taken over, at least partly, by the K.M tracts. All K.M tract lengths are represented up to 45 nt, the longest K.M tract being just 74 nt. In Arabidopsis the K.M tracts are again in high excess, but to a lesser extent than the R.Y motif – there are only two tracts longer than 48 nt (50 and 58 nt). The excess of K.M tracts in elegans and in yeast is less by an order of magnitude compared to humans (Fig. 2b; except for the yeast telomere), and is marginal but still significant in the archeon. Control runs with the same 5 × 1 Mb random sequences, but for the K.M motif, remain close to unity as expected (Table 4); the longest tract in this case is 24 nt long, present in a single random 1 Mb sequence.
W and S tracts in the seven chromosomes
W and S tracts are autocomplementary each, rather than complementing one another. W and S tracts are therefore separately compared. The f/e ratios for W tracts are shown in Table 5 and Fig. 2c. It is seen immediately that W tracts are also over-represented, but to a more variable degree than R.Y or K.M tracts. A difference of more than 100 fold is evident between the two human chromosomes for W tracts longer than 32 nt: At that length, f/e = 18,990 in contig 23, vs. f/e = 227 in contig 28. This large difference is partly due to the sensitivity of the calculated value to the percentage of AT, which is 60.9% in contig 28 vs. 52.6% in contig 23 (%AC and %AG are always close to 50%, "the second Chargaff parity rule", see end of discussion). A far higher number of W tracts are thus expected in chr. 21 by eq. (1), simply due to different p and q values. In addition, the 60.9% AT of contig 28 is an average between a very gene poor half with a high %AT (~64% between coordinates 0–7 Mb, see Additional file: 8) and a gene richer half with 56% AT (towards the telomere of the chromosome). The actual f/e ratio in the gene rich domain is much closer to that of contig 23. In yeast, Arabidopsis and jannaschii (68.5 %AT!), W tracts are under-represented up to 15 nt, but then are increasingly over-represented, reaching an excess of hundred-folds for 30–40 nt tracts. The C. elegans chromosome contains few very long W tracts, up to 96 nt. Again – the relatively low excess of W is partly due to the high percent AT. The actual number of tracts, not f/e ratios, is closer to that of the R.Y or K.M motifs (Additional files: 1, 2 and 3). It should be added that the high % AT can be explained only very partly by the mere presence of many long W tracts, because more than 89% of the A's and T's reside in the majority of short, under-represented tracts, up to 10 nt; a certain compensation may be in place for strict quantitative comparison. Still, it can be concluded that the W motif in eukaryotes is also an extensively over-represented binary motif, in similarity to the situation in bacteria [15].
Finally, S tracts. There are many fewer long S tracts in all the chromosomes studied (data in Additional file: 5). S tracts are often concentrated near transcription start site, as part of the well studied CpG Islands [12,25]. Thus, in contig 23 (47.4% G,C) only five S tracts longer than 37 nt are found (56 the longest). Nevertheless, in the 12 – 37 nt range, over-representations increase from 1.12 up to 480,000 fold. In Arabidopsis (only 35.9% G,C) the longest S tract is 20 nt, but over-representation still increases steadily up 200 fold, at length 20. S tracts can thus be considered as another member of the over-represented class. Program TRACTS can be a convenient tool for detecting the CpG islands, espscially in its web version [26].
Distribution in genic subregions
In which genic subregions do the excessive tracts reside? Subprogram ANEX distributes the tracts between exon, intron and intercoding or intergenic classes. The term intergenic is appropriate when mRNA entries are parsed; in that case, UTR regions are evaluated as exons. The distribution between exons, introns and intergenic of all tracts 15 nt and longer (GE 15) is shown in Table 6. W and S tracts are presented here as a pair, but since S tracts are minority for tracts GE 15, the f/e ratios represent practically the W tracts alone. Very high over-representations are again evident: Over-representations is highest for R.Y tracts in all genomes surveyed, except for Drosophila, where K.M tracts are in the largest excess.
Table 6.
Binary tracts longer or equal to 15 nt, in 7 genomes. Ratio of found to expected tracts.
| R.Y | K.M | W;S | |
| H. sap. Chr. 22 Contig 23 | |||
| %A,Xb: | 50.0 | 49.9 | 52.6 |
| All Regions | 40.35 | 27.75 | 27.25 |
| Exonsa | 18.20 | 7.84 | 12.04 |
| Introns | 38.81 | 29.00 | 26.43 |
| Intergenic | 41.46 | 27.87 | 27.93 |
| H. sap. Chr21, Contig 28 | |||
| %A,X: | 50.1 | 50.2 | 0.96 |
| All Regions | 31.14 | 20.59 | 5.29 |
| Exonsa | 16.48 | 7.94 | 2.14 |
| Introns | 36.31 | 24.14 | 5.24 |
| Intergenic | 30.33 | 20.06 | 5.33 |
| D. mel. Chr. II-Right arm | |||
| %A,X: | 50.0 | 50.0 | 56.6 |
| All Regions | 10.32 | 16.84 | 10.52 |
| Exonsa | 4.32 | 7.37 | 3.40 |
| Introns | 12.89 | 21.95 | 12.08 |
| Intergenic | 12.14 | 18.96 | 13.78 |
| C. elegans Chr. I | |||
| %A,X: | 50.0 | 50.0 | 64.0 |
| All Regions | 15.70 | 9.20 | 3.05 |
| Exons | 16.41 | 8.91 | 3.18 |
| Introns | 16.89 | 8.93 | 3.11 |
| Intercoding | 15.09 | 9.38 | 2.99 |
| A. thal. Chr. II | |||
| %A,X: | 49.9 | 50.1 | 64.1 |
| All Regions | 23.82 | 3.39 | 2.65 |
| Exonsa | 18.57 | 2.10 | 0.12 |
| Introns | 20.76 | 4.25 | 1.81 |
| Intergenic | 27.03 | 2.04 | 4.05 |
| S. cer. Chr. IV | |||
| %A,X: | 50.1 | 50.1 | 62.1 |
| All Regions | 15.32 | 5.52 | 1.60 |
| Exons | 9.16 | 3.64 | 0.42 |
| Introns | 7.76 | 0.00 | 2.33 |
| Intercoding | 33.25 | 11.07 | 4.96 |
| M. jan. Chromosome | |||
| %A,X: | 50.3 | 50.0 | 68.6 |
| All Regions | 15.73 | 3.21 | 1.53 |
| Exons | 17.39 | 3.09 | 0.93 |
| Introns | 0.00 | 0.00 | 0.00 |
| Intercoding | 4.7 | 4.04 | 5.60 |
a – Including identified 3' and 5' UTR regions. b – A, X is: A,G for R.Y; A,C for K.M and A,T for W;S.
The f/e ratios are lowest in coding regions (exons), with the exception of R.Y in M. jannaschii, (which is 87% coding, see column 7 of Table 1), and of W tracts in elegans. The lower excess in exons can be expected, since, for instance, an oligopurine on the coding strand imposes on the coded protein mostly polar amino acids (all-purine codons code for lys, arg, gln, also for gly).
Introns are the subregion in which K.M tracts are the most excessive, except for elegans, and jannaschii. In the fly introns have more excessive K.M tracts than R.Y tracts. The introns are the subregion richest in R.Y tracts in the fly, elegans and chr. 21 by the criterion used (≤ 15 nt). The well-known oligopyrimidine close to the 3' splice site contributes to the excess of Y tracts in introns. We also observe, in the full sequence outputs, many long binary tracts in the UTR regions, particularly in the 3' UTR. An example can be seen in reference [26]: The three R.Y tracts above position 19,000 of p53 listed there are in the 3' UTR of the gene. A suggested RNA stability signal of 9 W bases [27] may explain some of the W tracts, but many other long tracts, of all motifs, are found in the 3' UTR region, appearing often in blocks, and call for an explanation.
In the intergenic regions, R.Y tracts are the highest over-represented subregion in human chr. 22, in Arabidopsis and in yeast, while in chr. 21, in fly and in worm, introns are the even somewhat richer in R.Y tracts. In the smaller, but gene rich 3.45 Mb contig of chr. 21 (data not shown) R.Y tracts are highest in the intergenic regions, as in chr. 22. The excess of K.M over R.Y tracts in intergenic regions of Drosophila is to be particularly noted, while in the Arabidopsis chromosome their contribution is not very high.
A reviewer inquired how over-representation varies along a chromosome. The data in Additional file: 8 shows that for contig 28, f/e ratios for R.Y tracts decrease somewhat from the A,T rich, gene poor "desert" in the first half, to the gene rich second half. The f/e ratio of the W tracts increase even stronger in the same direction, but that may be due to the fact, that expected values increase strongly with % A,T while actually found tracts increase much less if at all.
Interspersed elements
A major finding of the human sequencing project was that a very high portion of the human chromosomes consists of various interspersed elements introduced into the genome. To what extent can these elements be responsible for the over-represented binary tracts? For instance, most alu elements contain, at their end, 20–30 consecutive A's partly incorporated into the genome. To answer this question, several genes and chromosomal contigs were run by TRACTS after having been "masked" (interspersed elements taken out). This was done with program RepeatMasker, with parameter – nolow; this means that "simple" repeats and certain other low complexity tracts are not taken out; only LTR, MER, LINE and SINE elements were masked out (mainly alu runs, Additional file: 6). The longest sequence we could run was contig "3.45" of Chr. 21, which is the q most contig of the chromosome, a relatively gene rich contig with 51.5% GC. After masking, 2,125,818 bases out of the original 3,450,347 bases remained (61%). The masked sequence was subjected to TRACTS. The results (Table 7) show that over-representation of all three binary pairs is reduced, but only to a limited extent – over-representation remains high for all three binary compositions. The most reduced motif is the W motif – possibly because of the last bases of the alu element. This means that a certain share of the long tracts does indeed reside in the inserted elements, but that many long tracts do reside in the non-masked fraction. This was true even when masking out also the "simple" and the "low complexity" elements. It is clear that over-representation cannot be explained as stemming mainly from so far identified inserted elements.
Table 7.
Masked and non-masked frequencies of long R.Y tracts. In contig 3.45 of human chromosome 21
| Masked sequence (2,125,818 nt) | Full sequence (3,450,347 nt) | |||||
| f/e ratio GE 15 | Up to (nt) | Longest (nt) | f/e ratio GE 15 | Up to (nt) | Longest (nt) | |
| R.Y | 22.6 | 44 | 171 | 28.0 | 44 | 175 |
| K.M | 18.8 | 46 | 121 | 23.7 | 50 | 121 |
| W;S | 6.03 | 36 | 134 | 21.4 | 42 | 134 |
Discussion
The main finding reported here is that DNA tracts consisting of only two of the bases are in vast excess all over the animal and plant kingdoms, reaching mega-fold values. The highest excesses are found for R.Y tracts in humans and in other mammals, as observed originally in the pioneering work of Erwin Chargaff and coworkers [29]. In certain organisms – like in Drosophila – K.M tracts prevail. In bacteria, W tracts are the most over-represented binary motif [15], a finding also anticipated by Chargaff and coworkers [29]. One caveat – only one chromosome or contig, from a single species in a particular phylum, is discussed here, except for the two human contigs. Two yeast chromosomes and one Drosophila segment were previously reported, and all show similar abundances [14,30].
Gentles and Karlin [31] report a distinct dinucleotide signature for each of the genomes studied here. The four dinuleotides present in homopurine tracts are AA, GG, GA, and AG. These four dinucleotides are indeed over-represented in the genomes surveyed by Gentles and Karlin (except in Drosophila!). The rarity of CpG dinucleotides most probably contributes to the low number of S tracts in humans. On the other hand, only a minor percentage of all bases (and dinucleotides) resides in long tracts: For instance, only 5.5% of all bases in contig 23 of chr. 22 are in binary tracts longer than 10 nt (Table 2). Long tracts are thus not necessarily the major factor determining the dinucleotide signature. It is worth to note that the D. melanogaster chromosome, besides the high K.M ratios, manifests also the highest excess of long W tracts (Table 5), along with E. coli and H. influenzae; a closer relationship between these organisms has also been noted when dinucleotide signatures of E. coli and Drosophila were compared [31].
A comment on the equations used to calculate expected values (eqs. 1–4): It was assumed tacitly that compositional frequencies are neighbor independent (zero Markov order). Lower tract abundances would have been obtained, if higher order dependencies were introduced. For instance, we have seen that purines avoid being flanked by pyrimidines, and prefer to be flanked by purines. Specifically, a single A, or a single G prefer an A or a G base next to them. This effect is formally a first order Markov effect, but we prefer the biological viewpoint that a particular function with selective advantage, rather than an inherent neighbor effect, drives the bases together to form binary tracts. A neutral, nonfunctional driving force towards excess of purine.pyrimidine caused by different substitution mutation rates has indeed been noted [32]. The substitution rates in the direction of all-purine or all-pyrimidine tracts were however the lowest [32] and are therefore unlikely to explain the massive excesses of R.Y tracts observed.
The vast excess of long binary tracts raises two questions: Is an essential structural and/or functional role responsible for the high numbers of binary tracts in the range of species studied? And if so, has that property been conserved throughout evolution, or have convergent processes been responsible for their wide spread presence? As to the second question, the reappearance of massive W tracts in Drosophila can be quoted in favor of independent (convergent) evolution, while if conservation would be the answer, an early progenitor with only a binary code could be suspected. A previous suggestion of an early RNY or YRN progenote is not in line with an all purine or all pyrimidine progenote [33]. More comparative binary DNA mapping will be required to answer this question.
This leaves the question of what can the essential function be. We, and others, have proposed that a special propensity of the binary tracts to unwind and be strand separated may be responsible. Ready unwinding is certainly expected for W tracts, based on their established melting properties. As to R.Y tracts, Weintraub and Larsen showed, in their seminal work [34], that certain purine/pyrimidine rich sequences in the 5' promoter region of the chicken beta globin gene complex are sensitive to single-strand DNA specific nucleases. Sensitivity to single-stranded specific nucleases means that these binary DNA regions have to be strand separated, at least temporarily. Since 1982, R.Y tracts in promoters of many genes (reviewed in [35]) have been found to be attacked by single-strand specific nucleases and hence are likely to undergo a transition into a strand separated state, at least temporarily. The list of these promoters includes a number of yeast and bacterial sequences characterized as AT rich [36,37]. The single-strand nuclease sensitive regions have been called by Umek and Kowalski DNA Unwinding Elements, or DUE's [38]. Evidence from modification by chemical reagents attacking only unpaired bases, like permanganate [39,40], chloroacetaldehyde [41] and osmium tetroxide [42] support at least intermittent conversion of the attacked strands into an unwound state [43,44]. We have previously found that in yeast chromosomes III and XI [14] the highest binary tract concentrations are in the 5' promoter regions. This intriguing observation deserves a separate analysis of the promoter regions, which is in progress.
In our experimental work [17] we studied two yeast promoters that contain long oligopyrimidine tracts, namely the promoter regions of gene cyc1, which has an oligopyrimidine tracts of 40 nt, and of gene ded1, with a 32 nt pyrimidine tract (interrupted by a TATA box). These oligo Y regions, and their complementary R tracts, were found to be sensitive to the single-strand specific nuclease P1 when under normal cellular superhelical stress. Topological analysis was consistent with the opening of six turns of the primary helix. These findings strongly support the idea that binary tracts in critical regions can readily unwind and thus facilitate the transcription initiation process, possibly helped by single strand specific proteins. The notion that binary DNA tracts can lead to transitional strand opening can apply also to other DNA directed processes, including recombination, replication and segregation. We found evidence that a long W tract in the centromere yeast chromosome IV (78 nt) has a strong propensity to form an unwound structure [40]. A role in these processes can provide an explanation for the massive presence of the binary tracts in intergenic regions, far from transcriptional start sites.
As said, the early melting of W tracts is a well-established fact, while for S tracts the propensity to be methylated may be involved. It is somewhat harder to understand why R.Y or K.M tracts should readily unwind and form paranemic, unwound DNA structures [35] (also known as a local supercoil-stabilized structures [43]), especially when G or C rich. It is possible that the contribution of the different dinucleotides to stability [45] changes under superhelical stress and at ambient temperatures. Experiments to clarify this possibility have yet to be carried out. It should be noted that the bulk of the binary tracts observed here do not have the internal symmetries associated with paranemic structures such as DNA triplexes, cruciforms or even B-Z junctions. The DUE's are more likely to separate into single-strands and be stabilized by cellular proteins.
Are the observed binary tracts "simple" sequences in the usual sense, i.e. are the observed tracts composed of one or few nucleotides repeated many times, like oligo (C-T)? A detailed inspection of the tracts listed by the program demonstrates that for most tracts this is not the case: To get an idea, the last 15 longer tracts of contig 23, located beyond the last gene of the contig, are shown in Additional file: 7. The list contains a few simple sequences, for instance a 17 nt tract with GA repeated 8 times, ((GA)8G). Some longer tracts may also show simple repeats within their sequence. For example, the long 367 nt R tract has a number of GGGAGGAGAGA repeats in it (see Additional file: 7). This repeat covers however only part of the tract and the other parts are much more random. A slippage mechanism [46] would need too many "slippages" to explain this tract, or many other tracts in the lists, as generated by TRACTS. Oligo A or Oligo T tracts are partial components of quite a number of R tracts (as well as of W and M tracts) and for these an additional mechanism may be operative. Nevertheless, the bulk of the binary tracts are just as random a mixture of two nucleotide bases as can be, and cannot be regarded as simple or even cryptic elements [46]. A full quantitative analysis has yet to be undertaken.
Finally, Table 6 documents another intriguing finding connected with the name of Erwin Chargaff, namely that in single strands the percentage of purines is equal to the percentage of pyrimidines. The same equality was found for A+C bases being equal to G+T bases, again in single strands [47,48]. This phenomenon has been termed "the second parity rule of Chargaff". The percentages of A+G and A+C shown in Tables 3 and 6 demonstrate that their closeness to 50% is quite convincing. I have not encountered serious departures from that rule down to the length of individual genes, in all phyla studied, including bacteria. Two explanations have been raised: One explanation is that random inversion of homopurines during evolution caused this equality [49-51]. An alternative possibility is that there is a lot of potential or actual secondary structure in genomic DNA [52]. A definite explanation has yet to be provided and is beyond the scope of this paper. In conclusion, it thus seems that the analytical findings of the Late E. Chargaff will keep us busy for a while to come.
Conclusions
This paper documents one of the more significant departures of DNA from randomicity, namely that genomes exhibit an enormous excess of DNA tracts composed of only two bases. This phenomenon is conserved throughout evolution, and is therefore likely to reflect a specific DNA function. A most likely function is a propensity of these binary tracts (and possibly additional base combinations) to adopt under suitable condition an alternative, paranemic conformation. This notion is supported by a range of experimental evidence, detailed in the discussion part. We are presently examining whether a particularly high excess of the binary tracts is present in human promoters, as already found in yeast (R.Y tracts) and E. coli (W tracts), supporting a role for the binary DNA tracts in the regulation of transcription and other DNA directed processes.
Methods
Program TRACTS identifies all binary tracts in a given DNA sequence. The program was run in its original FORTRAN version [9,15] on an UNIX platform. A cgi web server version, in perl, is now available [26] at url: http://www.weizmann.ac.il/~lcyagil/binaries_refs.html. The program calculates overall binary tract frequencies (see Table 2) as well as distributions in genic sub regions – exons, introns and intergenic regions. A further output of TRACTS shows the sequence entered, with each exon and intron indicated and each binary tract beyond a given length shown in color on or below the line. For more details, see [26]. A preprogram, ANEX, parses GenBank/DDJB/EMBL annotation files (flat format) and produces a file with a one line entry for each gene which includes a short comment on the product/function of the gene. When only a single or a few genes is examined, a list of all exons and introns is produced.
In GenBank files that contain both mRNA and CDS entries, the mRNA entries were parsed. Consequently, UTR regions are mostly part of the exonic sub regions. In yeast, C. elegans and M. jannaschii, where no mRNA data are yet available, the UTR regions are necessarily counted as intergenic (intercoding, to be strict). The accession and version numbers of the genomes analyzed are shown in Table 1. In humans, two large contigs, making up most of chromosomes 21 and 22, were analyzed: The "28" contig of chr. 21 which goes from the centromere through most of the q arm (28,515,322 nt) and makes up 85% of the sequenced chromosome; and the "23" contig of chr. 22 (22,998,450 nt), which makes up 66.6% of the sequenced chromosome.
Expected binary tract frequencies
Frequencies of binary tracts expected in random DNA are calculated as following: N(l) gives the number of tracts of length l expected in random DNA of length L and of fractional base composition p by:
N(l) = L(plxq2 + qlxp2), (1)
where p, q are the fractions of the participating base pairs, p+q = 1 (p is e.g. the fraction of A+G). To calculate expected values for only one member of a pair, only one member of the above sum is to be used. The number of bases expected in tracts of length n(l) is simply:
n(l) = l xN(l). (2)
The expected number of tracts equal or greater (GE) than a given length l, N(≥l), can be shown to be [9]:
N(≥l) = L (p x ql + q x pl). (3)
The expected number of bases in these tracts, n(≥l), is:
n(≥l) = L {(p + ql) pl + (q + pl) ql}. (4)
The validity of these expressions was tested by generating random DNA sequences and running them by TRACTS. For this paper, five 1 Mb random sequences with exactly 25% of each nucleotide base were generated and run for each binary composition, so that standard deviations could be calculated and are listed in Additional file: 4. The percentage of W and S bases in the analyzed chromosomes is not 50%, but a control run with 62.5% AT was previously run for H. influenzae, giving the same picture [15].
Abbreviations
chr. – chromosome
GE – Greater or Equal (longer or equal)
Supplementary Material
Table 1. Binary tract frequencies in contig 28 of human chromosome 22
Table 2. Binary tract frequencies in Drosophila chromosome R2
Table 3. Binary tract frequencies of yeast chromosome IV
Table 4. Five random sequences, 1 Mb each.
Table 5. Frequencies of S tracts in seven sequenced chromosomes.
Table 6. Masked regions in contig 3.45 of human chromosome 21.
Table 7. All R.Y tracts longer than 10 nt, beyond position 22,944,530 of contig 23.
Table 8. Contig 28 in windows of 2 Mb – f/e ratio GE 13.
Acknowledgments
Acknowledgements
Dedicated to Erwin Chargaff (1905–2002), a pioneer. I am indebted to Dr. Jaime Prilusky for many helpful advices and to Dr. Shifra Ben-Dor for thoughtful comments to the manuscript. The help of many other members of the Biological Computing Unit of this Institute is gratefully acknowledged.
References
- Tamm C, Shapiro HS, Lipshitz R, Chargaff E. Distribution density of nucleotides within a deoxyribonucleic acid chain. J Biol Chem. 1953;203:673–688. [PubMed] [Google Scholar]
- Spencer JH, Chargaff E. Studies on nucleotide arrangement in deoxyribonucleic acids, VI. Pyrimidine clusters: Frequency and distribution in several species of the AT type. Bioch Bioph Acta. 1963;68:9–27. doi: 10.1016/0006-3002(63)90105-7. [DOI] [PubMed] [Google Scholar]
- Petersen GB. The distribution of nucleotides in deoxyribonucleic acid. Biochem J. 1963;87:495–500. doi: 10.1042/bj0870495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spencer JH, Chargaff E. Pyrimidine nucleotide sequences in deoxyribonucleic acids. Bioch Bioph Acta. 1961;51:209–211. doi: 10.1016/0006-3002(61)91045-9. [DOI] [Google Scholar]
- Shapiro HS, Chargaff E. Studies on nucleotide arrangement in deoxyribonucleic acids VI. Direct estimation of pyrimidine nucleotide runs. Bioch Bioph Acta. 1963;76:1–8. doi: 10.1016/0006-3002(63)90002-7. [DOI] [PubMed] [Google Scholar]
- Szybalski W, Kubinski H, Sheldrick P. Pyrimidine clusters on the transcribing strands of DNA and their possible role in the initiation of RNA synthesis. Cold Spring Harbor Symp Quant Biol. 1966;31:123–127. doi: 10.1101/sqb.1966.031.01.019. [DOI] [PubMed] [Google Scholar]
- Birnboim HC, Sederoff RR, Paterson MC. Distribution of R.Y segments in DNA from various organisms. Europ J Biochem. 1979;98:301–307. doi: 10.1111/j.1432-1033.1979.tb13189.x. [DOI] [PubMed] [Google Scholar]
- Case ST, Baker R. Detection of long eukaryote-specific pyrimidine runs in repetitive DNA sequences and their relation to single-stranded regions in DNA isolated from sea urchin embryos. J Mol Biol. 1975;98:69–92. doi: 10.1016/s0022-2836(75)80102-1. [DOI] [PubMed] [Google Scholar]
- Bucher P, Yagil G. Occurrence of oligopurine.oligopyrimidine tracts in eukaryotic and prokaryotic genes. DNA Sequence. 1991;1:27–43. doi: 10.3109/10425179109020767. [DOI] [PubMed] [Google Scholar]
- Behe MJ. An overabundance of long oligopurine tracts occurs in the genome of simple and complex eukaryotes. Nucleic Acids Res. 1995;23:689–695. doi: 10.1093/nar/23.4.689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karlin S, Ghandour G. The use of multiple alphabets in kappa-gene immunoglobulin DNA sequence comparisons. EMBO J. 1985;4:1217–1223. doi: 10.1002/j.1460-2075.1985.tb03763.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antequera F, Bird A. CpG islands as genomic footprints of promoters that are associated with replication origins. Current Biol. 1999;9:R661–R667. doi: 10.1016/S0960-9822(99)80418-7. [DOI] [PubMed] [Google Scholar]
- Yagil G. The frequency of two-base tracts in eukaryotic genomes. J Mol Evol. 1993;37:123–130. doi: 10.1007/BF02407347. [DOI] [PubMed] [Google Scholar]
- Yagil G. The frequency of oligopurine.oligopyrimidine and of other two-base tracts in yeast chromosome III. Yeast. 1994;10:603–611. doi: 10.1002/yea.320100505. [DOI] [PubMed] [Google Scholar]
- Shomer B, Yagil G. Long W tracts are over-represented in the E. coli and H. Influenzae genomes. Nucl Acids Res. 1999;27:4491–4480. doi: 10.1093/nar/27.22.4491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raghavan S, Haniharan R, Brahmachari SK. Polypurine.polypyrimidine sequences in complete bacterial genomes: Preference for polypurines in protein-coding regions. Gene. 2000;242:275–283. doi: 10.1016/S0378-1119(99)00505-3. [DOI] [PubMed] [Google Scholar]
- Yagil G, Shimron F, Tal M. DNA unwinding in the CYC1 and DED1 yeast promoters. Gene. 1998;225:152–163. doi: 10.1016/S0378-1119(98)00525-3. [DOI] [PubMed] [Google Scholar]
- Hattori M, Fujiyama A, Taylor TD, Watanabe H, Yada T, Park HS, Toyoda A, Ishii K, Totoki Y, Choi DK, et al. The DNA sequence of human chromosome 21. Nature. 2000;405:311–319. doi: 10.1038/35012518. [DOI] [PubMed] [Google Scholar]
- Dunham I, Hunt AR, Collins JE, Bruskiewich R, Beare DM, Clamp M, Smink LJ, Ainscough R, Almeida JP, Babbage A, et al. The DNA sequence of human chromosome 22. Nature. 1999;402:489–495. doi: 10.1038/990031. [DOI] [PubMed] [Google Scholar]
- Adams MD, Celniker SE, Holt RA, Evans CA, Gocayne JD, Amanatides PG, Scherer SE, Li PW, Hoskins RA, Galle RF, et al. The genome sequence of Drosophila melanogaster. Science. 2000;287:2185–2195. doi: 10.1126/science.287.5461.2185. [DOI] [PubMed] [Google Scholar]
- C. elegans Sequencing Consortium Genome sequence of the nematode C. elegans: A platform for investigating biology. Science. 1998;282:2012–2018. doi: 10.1126/science.282.5396.2012. [DOI] [PubMed] [Google Scholar]
- Lin XY, Kaul S, Rounsley S, Shea TP, Benito MI, Town CD, Fuji CY, Mason T, Bowman CL, Barnstead M, et al. Sequence and analysis of chromosome 2 of the plant Arabidopsis thaliana. Nature. 1999;402:761–768. doi: 10.1038/45471. [DOI] [PubMed] [Google Scholar]
- Jacq C, and 138 colleagues The nucleotide sequence of S. cerevisiae chromosome IV. Nature. 1997:75–78. [PubMed] [Google Scholar]
- Bult CJ, White O, Olsen GJ, Zhou L, Fleischmann RD, Sutton GG, Blake JA, FitzGerald LM, Clayton RA, Gocayne JD, et al. Complete genome sequence of the methanogenic archeon, Methanococcus jannaschii. Science. 1996;273:1058–1073. doi: 10.1126/science.273.5278.1058. [DOI] [PubMed] [Google Scholar]
- Davuluuri RV, Grosse IV, Zhang MQ. Computational identification of promoters and first exons in the human genome. Nature Genetics. 2001;29:412–417. doi: 10.1038/ng780. [DOI] [PubMed] [Google Scholar]
- Gal M, Katz T, Ovadia A, Yagil G. TRACTS: A program to map oligopurine.oligopyrimidine and other binary DNA tracts. Nucl Acids Res. 2003;31:3679–3681. doi: 10.1093/nar/gkg625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zubiaga AM, Belasco JG, Greenberger ME. The nonamer UUAUUUAUU is the key AU-rich sequence motif that mediates mRNA degradation. Mol Cell Biol. 1995;15:2219–2230. doi: 10.1128/mcb.15.4.2219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chargaff E. Essays in Nucleic Acids Amsterdam Elsevier. 1963. pp. 126ff–146ff.
- Shapiro HS, Rudner R, Miura K-I, Chargaff E. Inferences from the distribution of pyrimidine isostichs in deoxynucleic acids. Nature. 1965;205:1068–70. doi: 10.1038/2051068a0. [DOI] [PubMed] [Google Scholar]
- Yagil G. Binary DNA tracts can serve as DNA unwinding centers. J Biomolecular Struct Dyn. 2001;18:911–911. [Google Scholar]
- Gentles AJ, Karlin S. Genome-scale compositional comparisons in eukaryotes. Genome Res. 2001;11:540–546. doi: 10.1101/gr.163101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hess ST, Blake JD, Blake RD. Wide variations in neighbor-dependent substitution rates. J Mol Biol. 1994;236:1022–1033. doi: 10.1016/0022-2836(94)90009-4. [DOI] [PubMed] [Google Scholar]
- Eigen M, Osswatich-Winkler R. Transfer-RNA, an early gene? Naturwissenschaften. 1981;68:282–292. doi: 10.1007/BF01047470. [DOI] [PubMed] [Google Scholar]
- Larsen A, Weintraub H. An altered DNA conformation detected by S1 nuclease occurs at specific regions in active chick globin chromatin. Cell. 1982;29:609–616. doi: 10.1016/0092-8674(82)90177-5. [DOI] [PubMed] [Google Scholar]
- Yagil G. Paranemic structures of DNA and their role in DNA unwinding. Crit Revs Biochem Mol Biol. 1991;26:475–559. doi: 10.3109/10409239109086791. [DOI] [PubMed] [Google Scholar]
- Kowalski D, Eddy MJ. The DNA unwinding element: A novel, cis acting component that facilitates the opening of the E. Coli replication origin. EMBO J. 1989;8:4335–4339. doi: 10.1002/j.1460-2075.1989.tb08620.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bramhill D, Kornberg A. A model for initiation at origins of DNA replication. Cell. 1988;54:915–917. doi: 10.1016/0092-8674(88)90102-X. [DOI] [PubMed] [Google Scholar]
- Umek RM, Eddy MJ, Kowalski D. DNA sequences required for unwinding prokaryotic and eukaryotic replication origins. Cancer Cells. 1988;6:473–478. [Google Scholar]
- Borowiec JA, Hurwitz J. Localized melting and structural changes in the SV40 origin of replication induced by T-antigen. EMBO J. 1988;7:3149–3158. doi: 10.1002/j.1460-2075.1988.tb03182.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tal M, Shimron F, Yagil G. Unwound regions in yeast centromere DNA. J Mol Biol. 1994;243:179–189. doi: 10.1006/jmbi.1994.1645. [DOI] [PubMed] [Google Scholar]
- Kohwi Y, Kohwi-Shigematsu T. Structural polymorphism of homopurine-homopyrimidine sequences at neutral pH. J Mol Biol. 1993;231:1090–1101. doi: 10.1006/jmbi.1993.1354. [DOI] [PubMed] [Google Scholar]
- Palecek E, Robert-Nicoud M, Jovin T. Local opening of the DNA double helix in eukaryotic cells, detected by osmium probe and adduct-specific immunofluorescence. J Cell Sci. 1993;104:653–661. doi: 10.1242/jcs.104.3.653. [DOI] [PubMed] [Google Scholar]
- Palecek E. Local supercoil-stabilized DNA structures. Crit Rev Biochem Mol Biol. 1991;26:151–226. doi: 10.3109/10409239109081126. [DOI] [PubMed] [Google Scholar]
- Benham C, Kohwi-Shigematsu T, Bode J. Stress-induced duplex DNA destabilization in scaffold/matrix attachment regions. J Mol Biol. 1997;274:181–196. doi: 10.1006/jmbi.1997.1385. [DOI] [PubMed] [Google Scholar]
- SantaLucia J. A unified view of polymer, dumbbell, and oligonucleotides DNA nearest-neighbor thermodynamics. Proc Natl Acad Soc (USA) 1998;95:1460–1465. doi: 10.1073/pnas.95.4.1460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tautz D, Trick M, Dover GA. Cryptic simplicity in DNA is a major source of genetic variation. Nature. 1986;322:652–656. doi: 10.1038/322652a0. [DOI] [PubMed] [Google Scholar]
- Elson D, Chargaff E. Evidence for common regularities in the comparison of pentose nucleic acids. Bioch Bioph Acta. 1955;17:362–376. doi: 10.1016/0006-3002(55)90384-X. [DOI] [PubMed] [Google Scholar]
- Rudner R, Karkas JD, Chargaff E. Separation of B. subtilis DNA into complementary strands III. Direct analysis. Proc Natl Acad Soc (USA) 1968;63:921–922. doi: 10.1073/pnas.60.3.921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baisnee P-F, Hampson S, Baldi P. Why are complementary DNA strands symmetric? Bioinformatics. 2002;18:1021–1033. doi: 10.1093/bioinformatics/18.8.1021. [DOI] [PubMed] [Google Scholar]
- Lobry JR. Properties of a general model of DNA evolution under no-strand-bias conditions. J Mol Evol. 1995;40:326–330. doi: 10.1007/BF00163237. [DOI] [PubMed] [Google Scholar]
- Sueoka N. Intrastrand parity rules of DNA base composition and usage biases of of synonymous codons. J Mol Evol. 1995;40:318–325. doi: 10.1007/BF00163236. [DOI] [PubMed] [Google Scholar]
- Bell SJ, Forsdyke DR. Deviations from Chargaff's second parity rule correlate with direction of transcription. J Theor Biol. 1999;197:63–76. doi: 10.1006/jtbi.1998.0858. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table 1. Binary tract frequencies in contig 28 of human chromosome 22
Table 2. Binary tract frequencies in Drosophila chromosome R2
Table 3. Binary tract frequencies of yeast chromosome IV
Table 4. Five random sequences, 1 Mb each.
Table 5. Frequencies of S tracts in seven sequenced chromosomes.
Table 6. Masked regions in contig 3.45 of human chromosome 21.
Table 7. All R.Y tracts longer than 10 nt, beyond position 22,944,530 of contig 23.
Table 8. Contig 28 in windows of 2 Mb – f/e ratio GE 13.