

Abstract
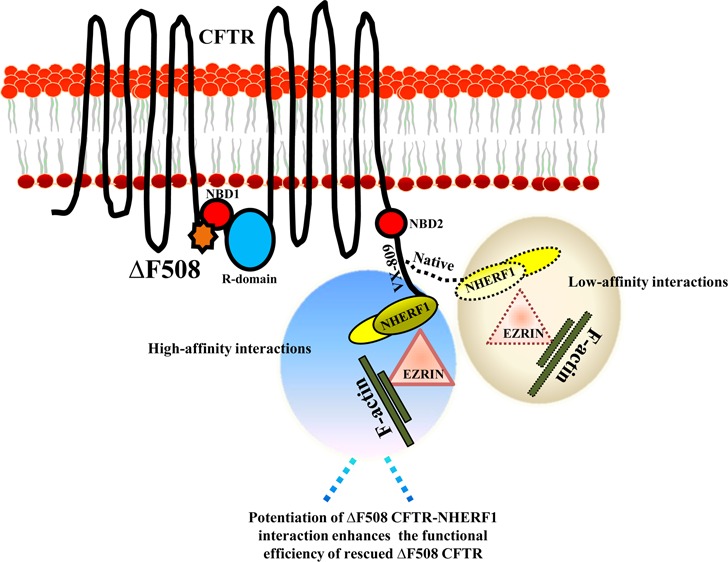
Cystic fibrosis (CF) is a recessive genetic disease caused by mutations in CFTR, a plasma-membrane-localized anion channel. The most common mutation in CFTR, deletion of phenylalanine at residue 508 (ΔF508), causes misfolding of CFTR resulting in little or no protein at the plasma membrane. The CFTR corrector VX-809 shows promise for treating CF patients homozygous for ΔF508. Here, we demonstrate the significance of protein–protein interactions in enhancing the stability of the ΔF508 CFTR mutant channel protein at the plasma membrane. We determined that VX-809 prolongs the stability of ΔF508 CFTR at the plasma membrane. Using competition-based assays, we demonstrated that ΔF508 CFTR interacts poorly with Na+/H+ exchanger regulatory factor 1 (NHERF1) compared to wild-type CFTR, and VX-809 significantly increased this binding affinity. We conclude that stabilized CFTR–NHERF1 interaction is a determinant of the functional efficiency of rescued ΔF508 CFTR. Our results demonstrate the importance of macromolecular-complex formation in stabilizing rescued mutant CFTR at the plasma membrane and suggest this to be foundational for the development of a new generation of effective CFTR-corrector-based therapeutics.
Cystic fibrosis (CF) is a recessive genetic disorder prevalent among Caucasians arising from certain mutations in the chloride channel, CF transmembrane-conductance regulator (CFTR) protein.1 CFTR regulates transepithelial chloride and bicarbonate levels across various secretory epithelia, and it is thought that CFTR function is indispensable for many aspects of fluid regulation.2 Over 1900 disease-causing CF mutations have been identified, and it has been estimated that approximately 70% of CF chromosomes worldwide contain at least one copy of the mutational deletion of phenylalanine at residue 508 (ΔF508) in the CFTR protein, which is categorized as a Class II defect3−5 (http://www.genet.sickkids.on.ca/cftr). The ΔF508 mutation results in misfolding of the protein, causing CFTR to be trapped and degraded by the endoplasmic reticulum (ER)-associated degradation (ERAD) pathway.6 A small population of ΔF508 CFTR rescued to the plasma membrane is only partially functional compared to wild-type (WT) CFTR.7 Mislocalization and insufficient function of ΔF508 CFTR protein at the plasma membrane leads to a generalized loss of hydration on extracellular surfaces and defective mucocilliary clearance on the lung surface, and the subsequent complications in the lung function can lead to mortality.1 There are many challenges to achieving highly efficient CF phenotypic correction. Even after rescue to the plasma membrane, ΔF508 CFTR has persistent gating defects manifested as reduced open probability (Po). The current understanding of how loss-of-function CFTR mutations progressively lead to severe CF pathologies, especially in the lung, is incomplete.8 Due to the heterogeneity of disease severity among individuals with CF, there is a need to develop increasingly efficient and eventually personalized CF correction. In addition to establishing improvement of defective CFTR expression and function by small-molecule CFTR modulators, it is critical to establish a clinical correlation. The efficiency of CF correction can be clinically manifested as the change in sweat chloride levels (<40 mM/L in normal individuals vs >60 mM/L in symptomatic CF patients) and significant improvement in the lung function (as measured by FEV1) of CF patients.
Based on the fact that rescued ΔF508 CFTR can function as a chloride channel once it reaches the cell surface, many pharmacological agents termed CFTR-correctors have been designed to increase cell-surface levels of ΔF508 CFTR.6,9,10 Also, CFTR potentiators ameliorate the regulation defects associated with many other CFTR mutations.11 The CFTR-corrector VX-809 can achieve 15% of WT CFTR function and protein levels in primary human bronchial epithelial (HBE) airway cells isolated from patients homozygous for the ΔF508 CFTR mutation.12 VX-809 exhibited high potency and selectivity for CFTR correction compared to the previous correctors. The CFTR-potentiator VX-770 stimulates passage of chloride ions from the G551D CFTR mutant which exhibits defective gating. VX-809 treatment alone was shown to be less effective in improving lung function in CF patients, although sweat chloride concentration was reduced (CF patients have salty sweat).13 The combination of VX-809 and VX-770 in recent Phase III clinical trials achieved improvement of 10 percentage points or more in the lung function of CF patients (http://investors.vrtx.com/releasedetail.cfm?ReleaseID=677520).
Low-temperature incubation (24–30 °C) of ΔF508 CFTR facilitates its retrieval from the ER and localization to the plasma membrane. However, the rescued ΔF508 CFTR is conformationally destabilized at physiological temperature and undergoes accelerated ubiquitination, endocytosis, and lysosomal degradation.14−16 Consequently, rescued ΔF508 CFTR has a very short life (<4 h) compared to WT CFTR (18–24 h).17 Macromolecular complex formations of CFTR regulate the function and stability of the protein at the plasma membrane.18−22 Association of CFTR with the scaffolding PSD-95/Dlg-5/ZO-1 (PDZ) protein, Na+/H+ exchanger regulatory factor 1 (NHERF1), stabilizes CFTR by coupling it to the actin cytoskeleton and regulates CFTR function by governing the phosphorylation status of the protein.20,21,23,24 Recent studies have shown that NHERF1 overexpression reduces surface ΔF508 CFTR internalization and can bring ΔF508 CFTR to the level of WT CFTR, correlating with the modulation of CFTR-NHERF1-ezrin-actin protein–protein interactions.25 Thus, parameters affecting trafficking, stability, and the regulation of surface CFTR would determine the functional output of CFTR. In the current study, we explored the contribution of macromolecular formations of CFTR in extending the surface retention of ΔF508 CFTR after conformational stabilization in the presence of VX-809. We demonstrate that the otherwise weak interaction of ΔF508 CFTR with NHERF1 is improved in the presence of VX-809, which would reduce CFTR-protein turnover and enhance the stability and functional efficiency of the protein at the plasma membrane. Macromolecular formations of CFTR could be critical to enhance the efficiency of correction of mutant CFTR and may prove beneficial in designing CFTR modulators with greater efficacy to target CF.
Materials and Methods
Reagents
VX-809 was purchased from Selleck Chemicals (Houston, TX). All VX-809 incubations were carried out at 5 μM concentration for 48 h, unless otherwise indicated.
Cell Cultures
HEK 293 and CFBEo– cells were cultured in DMEM-F12 and MEM media, respectively (Invitrogen; Carlsbad, CA), containing 10% fetal bovine serum and 1% penicillin/streptomycin. Cultures were maintained in a 5% CO2 incubator at 37 °C. Temperature rescue of ΔF508 CFTR protein was carried out at 28 °C for 24–48 h.
Mice
Cftr (+/+), Cftr (+/ΔF508), and Cftr (ΔF508/ ΔF508) C57BL/6 mice littermates were used for isolating enterospheres. All mice were maintained on colyte water and regular mouse chow.
In Vitro Organoid Culture and Fluid-Secretion Measurement in Enterospheres
The organoid-culture protocol26−28 has been standardized in our laboratory by Moon et al. (unpublished data) for monitoring CFTR-mediated fluid secretion. Briefly, the small intestines were removed from 8-week-old male mice and rinsed with cold PBS. Villi were discarded by scraping with a cover slide and the tissues were minced, followed by incubation with 2 mM EDTA in 1X PBS at 4 °C for 30 min, and subsequently passed through a 70 μm cell strainer. The crypts were collected by centrifugation at 1200 rpm for 5 min and cultured on Matrigel (BD; San Jose, CA) with organoid media (Advanced DMEM/F12 with glutamax, penicillin, streptomycin, 10 mM HEPES, B27, N2, 1 mM NAC, 100 ng/mL Noggin, 1 μg/mL R-spondin-1, and 50 ng/mL EGF) and incubated in a humidified 10% CO2 chamber at 37 °C. The Matrigel-embedded enterospheres were pretreated with DMSO or VX-809 (5 μM) at 37 °C for 48 h and placed in prewarmed HBSS in glass-bottomed dishes (MatTek; Ashland, MA). CFTR-agonists forskolin (Fsk) (10 μM) and IBMX (100 μM) were added to the enterospheres at 37 °C for 30 min, and phase-contrast images were taken using an IX-51 microscope (Olympus; Tokyo, Japan) both before and 30 min after the addition of the CFTR agonists. The radii of the whole enterospehere and luminal sphere before and after stimulation were calculated using ImageJ software (http://rsbweb.nih.gov/ij/). The volume of the luminal sphere was normalized with the volume of the entire enterosphere to calculate fluid secretion. In some experiments, the highly selective CFTR inhibitor, CFTRinh172 (50 μM), was added to the organoids to verify that the secretions were CFTR-mediated.
Proximity Ligation Assay
Matrigel-embedded mice enterospheres were used for performing the proximity ligation assay (PLA) (Olink Bioscience; Uppsala, Sweden). Briefly, enterospheres [pretreated with DMSO or VX-809 (5 μM) at 37 °C for 48 h] were incubated with rabbit 1314 CFTR polyclonal antibody (1:50) and mouse NHERF1 monoclonal antibody (1:50) (BD Biosciences) at 4 °C for overnight. The PLA was performed with anti-rabbit (plus) and anti-mouse (minus) Duolink In Situ PLA probes on the organoids following the manufacturer’s instructions. The images were taken using a Zeiss LSM-5 Pascal confocal microscope, and the PLA signals that represent the site of the protein–protein interaction appear in red.
Iodide Efflux Assay
CFTR-mediated halide efflux was measured using an iodide efflux assay. HEK 293 cells stably expressing either WT CFTR or ΔF508 CFTR were grown in 60 mm dishes and incubated with DMSO or VX-809 (5 μM) with or without temperature rescue for 48 h. Briefly, cells were loaded for 60 min at room temperature with loading buffer (136 mM NaI prepared in buffer: 137 mM NaCl, 4.5 mM KH2PO4, 1 mM CaCl2, 1 mM MgCl2, 10 mM glucose, 5 mM HEPES, pH 7.2). Extracellular NaI was washed away thoroughly (5–7 times) with efflux buffer (loading buffer with 136 mM NaNO3 replacing the NaI). Cells were equilibrated for 1 min each, and aliquots were collected four times to establish a stable baseline in the efflux buffer alone. After establishing basal equilibrium with the nitrate buffer, cells were treated with CFTR agonists (Fsk and IBMX) prepared in the nitrate buffer and aliquots were collected six times at 1 min intervals. The iodide concentration of each aliquot collected was determined using an iodide-selective electrode (Thermo Scientific, Waltham, MA) and converted to iodide efflux rate (nanomol/min) as described previously.29
Short-Circuit Current (Isc) Measurements (Ussing Chamber Experiments)
Polarized CFBEo– cell monolayers were grown on Costar Transwell permeable supports (Cambridge, MA; filter diameter 6.5 mm) until the monolayers reached confluency, and the transwells were mounted in an Ussing chamber maintained at 37 °C. Isc was measured as described previously.21 CFBEo– cells pretreated with DMSO or VX-809 (5 μM for 48 h; both apical and basolateral sides) were bathed in Ringer’s solution (mM) (Basolateral side: 140 NaCl, 5 KCl, 0.36 K2HPO4, 0.44 KH2PO4, 1.3 CaCl2, 0.5 MgCl2, 4.2 NaHCO3, 10 HEPES, 10 glucose, pH 7.2, [Cl–] = 149) and low Cl– Ringer’s solution (mM) (Apical side: 133.3 Na-gluconate, 2.5 NaCl, 0.36 K2HPO4, 0.44 KH2PO4, 5.7 CaCl2, 0.5 MgCl2, 4.2 NaHCO3, 10 HEPES, 10 mannitol, pH 7.2, [Cl–] = 14.8) with VX-809 (5 μM) in the bath solutions at 37 °C and saturated with 95% O2 and 5% CO2. A 2 mV pulse was applied every 1 min throughout the experiment to check for the integrity of the epithelia. After stabilization of basal Isc, cells were treated with Fsk and IBMX on the apical side. CFTRinh172 (20–50 μM) was added to the apical side at the end of the experiment to verify that the Isc were CFTR-dependent.
Pull-Down Assay
Parental and Flag-WT CFTR-, Flag-ΔF508 CFTR-expressing HEK 293 cells were maintained in 60 or 100 mm dishes at 37 °C until confluent.
For some experiments, Flag-WT CFTR and Flag-ΔF508 CFTR were immunopurified from HEK 293 cells that were grown in 100 mm dishes at 28 or 37 °C in the presence of DMSO or VX-809 (5 μM) for 48 h. Cells were lysed in 12 mL lysis buffer (PBS 0.2% Triton X-100 + protease inhibitors ±5 μM VX-809). Cell lysates were mixed for 30 min, followed by centrifugation at 15,000 rpm at 4 °C for 30 min. Flag-beads (400 μL of 10% slurry) were then added and mixed overnight. The mixtures were centrifuged at 3000 rpm for 2 min, the beads were washed three times with lysis buffer, and the proteins were eluted with glycine buffer containing 0.2% Triton X-100 (pH 2.2), followed by immediate neutralization with 1.5 M Tris-Cl pH 8.8.
A competition-based strategy was adopted to determine the specific effects of VX-809 on CFTR-NHERF1 binding. HEK 293 cells expressing Flag-ΔF508 CFTR were grown at 37 °C in the presence of DMSO or VX-809 (5 μM) for 24 h and lysed in PBS + 0.2% Triton X-100. The binding of ΔF508 CFTR with NHERF1 was serially disrupted in the presence of GST-NHERF1-PDZ2 (0, 2.5, 5, and 10 μg) for 2 h. CFTR protein complexes were immunoprecipitated overnight with Flag-beads (15 μL of 10% slurry) at 4 °C and then eluted with sample buffer. All of the samples were appropriately immunoblotted with mouse R1104 CFTR and NHERF1 (BD Biosciences) monoclonal antibodies. The levels of NHERF1 relative to CFTR protein were quantified using ImageJ software.
Surface-Labeling Assay
HEK 293 cells expressing Flag-WT CFTR, ΔF508 CFTR, and ΔF508 CFTR His 10 were grown on 35 mm dishes in the presence or absence of VX-809 (5 μM) at 37 °C for 48 h, fixed with 3.7% formaldehyde for 10 min, blocked with 1% BSA for 90 min, and treated with α-Flag HRP (0.2 μg/mL) for 90 min. The HRP substrate, 1-Step Ultra TMB (Pierce; Waltham, MA), was then added to the dishes and observed for color development (usually 10–15 min). The reaction was stopped by adding an equal amount of 2 N H2SO4. Absorbance was read at 450 nm.
The Hanrahan laboratory was the first to generate cells (BHK) expressing WT CFTR His10.30 We used a similar strategy to clone both WT and ΔF508 into the pLenti-Puro vector. We have used the WT CFTR His 10 in our previous studies (in collaboration with Hanrahan laboratory)20 and found that although it traffics to the plasma membrane (not as efficiently as WT), the His10 tag on the C-terminus of CFTR disrupts the binding of PDZ proteins (i.e., EBP50/NHERF1 interaction was perturbed), and therefore the macromolecular complexes between CFTR and its PDZ partners are perturbed. The first published description of His10-tagged F508del on the C-terminus (ΔF508 CFTR His10) is from Bear laboratory31 wherein they purified the protein from insect cells (Sf-9 cells).
AlphaScreen Assay for CFTR-NHERF1-PDZ2 Interactions
We used the AlphaScreen (amplified luminescent proximity homogeneous assay) GST Detection Kit (PerkinElmer; Waltham, MA) to study interactions of purified full-length Flag-WT CFTR and Flag-ΔF508 CFTR with the GST-NHERF1-PDZ2 domain. All proteins used were of high quality and purified under nondenaturing conditions (we tested protein interactions with syntaxin 1A at the N-terminus and protein binding to Azido ATP; data not shown). In brief, starting with a 100 nM concentration, CFTR protein (WT and ΔF508) was serially diluted (in 1/2 log dilution series) in the assay buffer (1x HEPES, 0.1% BSA, 0.05% Tween 20 [v/v], pH 7.2) containing GST-NHERF1-PDZ-2 (100 nM final concentration) and biotin-CFTR-C-tail peptide (100 nM final concentration). The resulting solutions were incubated at room temperature for 30 min. In triplicate, each sample solution (15 μL) was transferred to a white opaque 384-well microplate (OptiPlate-384; PerkinElmer) into which anti-GST acceptor beads (5 μL; 20 μg/mL final concentration) were added and incubated at room temperature for 30 min. Streptavidin donor beads (5 μL; 20 μg/mL final concentration) were then added and incubated at room temperature for 2 h. The plate was read on a FLUOstar-Omega plate reader (Ortenberg, Germany).
Single-Particle Tracking
Single-particle tracking (SPT) on HEK 293 cells was performed as described previously.29 HEK 293 cells stably expressing Flag-WT CFTR, Flag-ΔF508 CFTR and Flag-ΔF508 CFTR His 10 were grown on 35 mm glass-bottom dishes (MatTek). Cells were washed twice with HBSS and blocked with HBSS containing 4% BSA for 15 min. Cells were then incubated at 37 °C with biotin α-Flag antibody (1 μg/mL, Sigma) for 30 min, washed five times followed by an incubation with streptavidin-conjugated Qdot-655 (0.1 nM, Invitrogen) for 2 min, washed extensively eight times, and immediately mounted on an Olympus inverted microscope (IX51). The images were captured with a Hamamatsu EM-CCD camera at 1–3 frames per second for 1–3 min (50 ms exposure time, 100× oil-immersion objective (NA 1.40), xenon (300-W lamp) light source) controlled by SlideBook 4.2 software. SPT was performed using the particle-tracking module of SlideBook 4.2 software, which generates trajectories and calculates the mean squared displacement. Five to ten cells per dish were used for plotting histograms of the diffusion coefficients.
Statistical Analysis
Results are presented as mean ± SEM for the indicated number of experiments. Statistical analysis was performed using Student’s t test. A value of P < 0.05 was considered statistically significant.
Results and Discussion
There has been dedicated research to evolve therapies for CF, including newly generated CFTR-correctors such as VX-809. VX-809 has been demonstrated to partially overcome the processing defects of ΔF508 CFTR, enabling more CFTR molecules to reach the plasma membrane.12 VX-809 has been advanced into clinical studies, both alone and in combination with VX-770, to evaluate efficacy in patients with CF who are homozygous for the ΔF508 mutation. However, VX-809 alone has shown only modest efficacy and failed to improve lung functions in CF patients.7 Therefore, insights into the potential of VX-809 will help improve the efficacy of the drug in targeting CF.
ΔF508 CFTR is a temperature-sensitive mutant that can be rescued to the plasma membrane at low temperature (24–30 °C) and when transferred to 37 °C, in view of the high instability of the mutant protein at the cell surface, ΔF508 CFTR becomes internalized and subsequently degraded.17 It is well documented that the PDZ motif of CFTR is essential in prolonging the stability of CFTR at the plasma membrane. Haggie et al. reported that deletion of the PDZ motif results in increased lateral mobility and decreased stability of CFTR at the plasma membrane, as it disrupts the coupling of CFTR to the actin cytoskeleton via NHERF1 and ezrin.32 Also, ΔF508 CFTR molecules are less stable compared to WT CFTR at the plasma membrane.17 In view of these observations, we hypothesized that the instability of ΔF508 CFTR at the plasma membrane is due to its weak association with NHERF1. Flag-WT CFTR and Flag-ΔF508 CFTR proteins were purified, and the integrity of the purified proteins was confirmed on Coomassie-stained gels (Figure 1A). We used the AlphaScreen competition assay to determine the IC50 of Flag-ΔF508 CFTR and Flag-WT CFTR that competitively inhibit the interaction between the PDZ2 domain of NHERF1(NHERF1-PDZ2) and a biotinylated CFTR peptide (containing the last 10 amino acids at the C-terminus of CFTR). NHERF1 has two PDZ domains, and CFTR can bind to both PDZ1 and PDZ2 domains.33 The AlphaScreen data indicated a weaker-affinity binding between NHERF1-PDZ2 and ΔF508 CFTR compared to WT CFTR, with the respective IC50 of 442 and 89.3 nM, equivalent to ∼5-fold lower affinity of ΔF508 CFTR for NHERF1 (Figure 1B). To further support this observation, Flag-WT CFTR and Flag-ΔF508 CFTR proteins were immunoprecipitated from the HEK 293 cells overexpressing these proteins using Flag beads (±temperature rescue at 28 °C). The immunoprecipitated proteins were later immunoblotted and probed for interaction with NHERF1. ΔF508 CFTR purified from the cells maintained at 37 or 28 °C exhibited much weaker binding to NHERF1 (almost undetectable for 37 °C) than WT CFTR (Figure 2A and B; left, and the bindings were quantified in the right panel). These data strongly support our hypothesis that ΔF508 CFTR exhibits weaker binding affinity toward NHERF1 that may render surface CFTR unstable at physiologic temperature.
Figure 1.
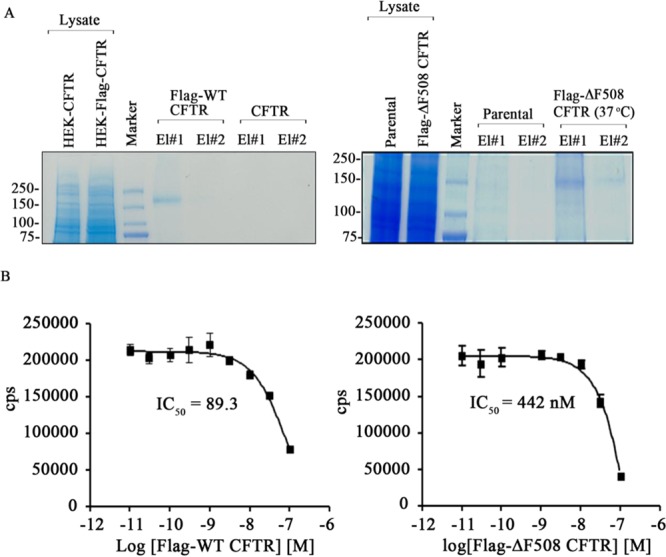
ΔF508 CFTR interacts poorly with NHERF1. (A) Coomassie-stained gel showing purified Flag-WT CFTR (left) and Flag-ΔF508 CFTR (right) at 37 °C. El#1–2 refer to the number of eluted fractions of the purified proteins. (B) Protein-binding curve derived from the Alpha Screen competition assay to measure the affinity of GST-NHERF1-PDZ2 for Flag-WT CFTR (left panel) and Flag-ΔF508 CFTR (right panel) purified from HEK 293 cells overexpressing these proteins and maintained at 37 °C.
Figure 2.

VX-809 potentiates the weaker interaction of ΔF508 CFTR with NHERF1. (A) and (B) Western blot data depicting the relative binding of Flag-WT CFTR (37 °C) and Flag-ΔF508 CFTR (37 or 28 °C) immunopurified proteins with NHERF1. The amounts of CFTR protein purified using Flag-beads are indicated. Flag-ΔF508 CFTR shows extremely weak binding with NHERF1 compared to WT-CFTR at 37 °C, as well as at 28 °C. (C) Western blots to demonstrate that VX-809 potentiates the interaction of Flag-ΔF508 CFTR with NHERF1. Bar graphs on the right side of each immunoblot represent the quantitation of NHERF1-CFTR interaction. Input refers to the cell lysates used prior to immunoprecipitation. (D) A competitive assay, in which GST-NHERF1-PDZ2 competes against endogenous NHERF1 to bind to Flag-ΔF508 CFTR in the presence or absence of VX-809, demonstrates that VX-809 improves CFTR-NHERF1 interaction affinity.
Based on these observations, we reasoned that the functional efficiency of ΔF508 CFTR can be significantly manipulated by increasing the stability of the rescued mutant protein at the plasma membrane and questioned whether ΔF508 CFTR correction by VX-809 would involve potentiating the otherwise weak interaction of ΔF508 CFTR with NHERF1, leading to prolonged retention of the mutant protein at the plasma membrane. To demonstrate this, Flag-ΔF508 CFTR was pretreated with either DMSO as a control or VX-809 at 37 °C for 48 h, and the proteins were purified and normalized. Upon immunoblotting, and probing the immunopurified proteins with NHERF1 antibody, we observed higher amounts of NHERF1 associated with Flag-ΔF508 CFTR in the presence of VX-809 compared to control (Figure 2C). Note that VX-809 always was included in the buffers during the immunoprecipitation procedures. To specifically determine that VX-809 facilitates increased binding of ΔF508 CFTR with NHERF1, we utilized a competition-based assay. The binding of Flag-ΔF508 CFTR with NHERF1 was serially disrupted in the presence of GST-NHERF1-PDZ2 (Figure 2D). Upon incubation with VX-809 for 24 h, 50% of ΔF508 CFTR-NHERF1 binding was disrupted at a lower dilution of GST-NHERF1-PDZ2 protein (∼2 μg for DMSO vs ∼4 μg for VX-809 treated) compared to DMSO alone (Figure 2D).
We next tested whether VX-809 can enhance the functional retention of temperature-rescued ΔF508 CFTR at the plasma membrane. We performed Isc measurements in CFBEo– cells stably expressing ΔF508 CFTR and ΔF508 CFTR His 10 (note: the His 10 tag on the C-terminus of CFTR disrupts binding to PDZ proteins) and temperature-rescued at 28 °C in the presence of either DMSO as a control or VX-809 (5 μM) for 48 h. The cells were mounted in the Ussing chamber and maintained at 37 °C to monitor the functional outcomes on Isc of depleting surface levels of ΔF508 CFTR and ΔF508 CFTR His 10 molecules over a period of time in the presence or absence of VX-809. CFTR function was stimulated with cAMP-elevating agents Fsk (10 μM) and the phosphodiesterase inhibitor IBMX (100 μM) added to the apical side of the cells. At the end of 4 h Isc measurement, ΔF508 CFTR retained approximately 72% of the function of that at 0 h in the presence of VX-809 compared to approximately 45% with DMSO alone in a statistically significant manner. However, the phenotype of CFTR function was not statistically significant for ΔF508 CFTR His 10 when comparing DMSO and VX-809 treatment (Supporting Information Figure S1). The data suggest that VX-809 enhances the stability and function of ΔF508 CFTR at the plasma membrane by involving certain PDZ-mediated interactions.
Several studies have demonstrated that compounds capable of modulating CFTR activity partially correct multiple structural defects in CFTR that are caused by the ΔF508 mutation.35−37 Other CF correctors (VRT-325 and Corr4a) facilitate ΔF508 CFTR maturation by restoring the otherwise poor interaction between the two halves of the molecule.36 Also, VX-770 (a CFTR potentiator) can partially restore the functional defect(s) associated with nucleotide-binding domain 1 (NBD1), which contains the mutation in ΔF508 CFTR, so that the channel can function more efficiently.37 In contrast to these findings, we demonstrated that CF correctors stabilize ΔF508 CFTR molecules at the plasma membrane via a mechanism involving protein–protein interaction. However, it cannot be ruled out that the structural defect in NBD1 of ΔF508 CFTR would confer compromised capacity of the protein to be involved in protein–protein interactions and that VX-809 acts to partially overcome this structural limitation.
We also tested the effects of VX-809 in our culture model system. HEK 293 cells stably expressing Flag-ΔF508 CFTR and Flag-ΔF508 CFTR His 10 were maintained at 28 °C for 48 h in the presence of DMSO or VX-809 (5 μM), and the surface levels of CFTR were measured using an HRP-based surface-labeling method. We found that there were fewer (approximately 2-fold less) Flag-ΔF508 CFTR His 10 molecules than Flag-ΔF508 CFTR molecules at the plasma membrane (Supporting Information Figure S2). VX-809 increased the surface levels of temperature-rescued Flag-ΔF508 CFTR and Flag-ΔF508 CFTR His 10 molecules by 1.3- and 2-fold, respectively, compared to the DMSO-treated cells (Supporting Information Figure S2). Iodide efflux measurements were performed in HEK 293 cells that overexpressed ΔF508 CFTR, and VX-809 pretreatment potentiated the response of ΔF508 CFTR and temperature-rescued ΔF508 CFTR by 3- and 2-fold, respectively, in the presence of the CFTR agonists-Fsk and IBMX (Supporting Information Figure S3A and B). VX-809 also potentiated ΔF508 CFTR function in CFBEo– cells. Polarized ΔF508 CFTR CFBEo– cells were either maintained at 37 °C or temperature-rescued in the presence or absence of VX-809, and CFTR-dependent Isc were subsequently monitored in the Ussing chamber in response to Fsk and IBMX (Supporting Information Figure S4A-D). VX-809-pretreated cells responded 10- and 3-fold higher, respectively, to the agonists compared to controls, corresponding to ΔF508 CFTR and temperature-rescued ΔF508 CFTR function (Supporting Information Figure S4B and D).
Our laboratory and others have demonstrated the feasibility of using intestinal organoids derived from crypt intestinal stem cells (also referred to as organospheres) as a model for studying CFTR-dependent fluid secretion.38,39 Intestinal organoids demonstrate luminal expansion in response to CFTR-activating agents Fsk and IBMX resulting from augmented fluid secretion, and the expansion is inhibited in the presence of a CFTR-specific inhibitor (CFTRinh172) (Supporting Information Figure S5) . Crypts were isolated from the small intestines of Cftr (+/+), Cftr (+/ΔF508), and Cftr (ΔF508/ΔF508) male mice and cultured to form intestinal organoids as described in detail in the Methods section. Cftr (ΔF508/ ΔF508) organoids were treated with VX-809 (5 μM) at 37 °C for 24 h. Fsk-stimulated luminal secretion rates were comparable between Cftr (+/+) and Cftr (+/ΔF508) intestinal organoids. VX-809 significantly improved the secretion rate in organoids from Cftr (ΔF508/ΔF508) mice by 2.5-fold compared to the DMSO-treated controls upon Fsk treatment (Figure 3A and B). The Fsk-induced fluid secretion was CFTR dependent, as Fsk failed to elicit significant secretion in the presence of CFTRinh172 (Supporting Information Figure S5). The potentiated CFTR-dependent fluid secretion in the presence of VX-809 in an in vivo stem-cell-culture model denotes possible prenatal therapeutic intervention with VX-809 that may improve the condition of CF neonates.
Figure 3.

VX-809 potentiates ΔF508 CFTR function in mice organoids in a mechanism involving potentiation of CFTR-NHERF1 interaction at the plasma membrane. (A) Representative images of intestinal organoids isolated from Cftr (+/+), Cftr (+/ΔF508), and Cftr (ΔF508/ΔF508) male mice depicting Fsk-stimulated CFTR-dependent fluid secretion. (B) Bar graph representing luminal-size change after 30 min of Fsk treatment as a measure of fluid secretion in organoids. Addition of VX-809 stimulates CFTR-dependent fluid secretion in Cftr (ΔF508/ΔF508) organoids. Basal refers to the organoids which were not treated with Fsk. Data represents mean of luminal-size measurements of 3–12 organoids of various groups (***P < 0.001). (B) Confocal images of organoids isolated from Cftr (+/+) (upper panel) and Cftr (ΔF508/ΔF508) (lower panel) mice show proximity ligation assay signal in red (marked arrows) representative of CFTR-NHERF1 interaction at the plasma membrane. VX-809 potentiates CFTR-NHERF1 interaction in Cftr (ΔF508/ΔF508) organoids (right lower panel). neg, negative control (only CFTR antibody was used) and pos, positive control (both CFTR and NHERF1 antibodies were used) in Cftr (+/+) organoids. Cftr (+/+) or simply +/+ refers to homozygous for WT CFTR. Cftr (+/ΔF508) or simply +/Δ refers to one WT and one ΔF508 CFTR allele. Cftr (ΔF508/ΔF508) or simply Δ/Δ refers to homozygous for ΔF508 CFTR.
The potentiating effect of VX-809 on ΔF508 CFTR-NHERF1 interactions was demonstrated in vivo in ΔF508 CFTR and WT CFTR organoids using the PLA. The interaction between two proteins results in PLA signals (in red), if the interacting proteins are within 40 Å of each other. We found strong PLA signals in VX-809-treated Cftr (ΔF508/ΔF508) organoids (5 μM VX-809 for 24 h) and very weak signals in the untreated organoids (Figure 3C, lower panel). WT CFTR organoids were used for positive (pos) and negative (neg) assay conditions (Figure 3C, upper panel). Therefore, the VX-809-mediated correction of ΔF508 CFTR that we observed was not merely due to localization of more ΔF508 CFTR protein to the plasma membrane, but also attributable to the stabilization of the protein at the plasma membrane via potentiation of interactions with NHERF1.
Potentiation of protein–protein interactions in the presence of an agent can be indirectly tested by monitoring the reduced mobility of a protein at the plasma membrane using SPT, which allows monitoring of the diffusive behavior of cell-surface proteins with nanometer spatial and millisecond temporal resolution.16,40 Previous studies demonstrated that deletion of the PDZ motif increased lateral mobility of WT CFTR by approximately 50% and that the increase in mobility was a consequence of compromised CFTR-ezrin interaction mediated via NHERF1.32,41 Since we had established that ΔF508 CFTR interacts poorly with NHERF1 and that VX-809 enhances the association of ΔF508 CFTR with NHERF1, we proceeded to test whether ΔF508 CFTR exhibits decreased mobility at the plasma membrane in the presence of VX-809. Diffusiveness of a protein can be measured in terms of change in the diffusion constant determined using SPT. HEK 293 cells expressing Flag-ΔF508 CFTR and Flag-ΔF508 CFTR His 10 were temperature-rescued in the presence of DMSO or VX-809 and labeled with biotin α-Flag antibody followed by streptavidin-conjugated quantum dot-655 for monitoring CFTR lateral mobility on the plasma membrane (Figure 4A). In accordance with the surface-labeling data, there were fewer ΔF508 CFTR His 10 molecules than ΔF508 CFTR molecules at the plasma membrane. HEK 293 cells expressing no CFTR (parental cells) or WT CFTR were used as negative and positive controls, respectively. Consistent with previous reports, ΔF508 CFTR exhibited higher mobility compared to WT CFTR, with mean diffusion coefficients of 0.0139 vs 0.00417 μm2/s (Figures 4B–D and 5A and B). The mean diffusion coefficient of ΔF508 CFTR molecules that were subjected to VX-809 decreased substantially, by 41% (0.00819 vs 0.0139 μm2/s), as also indicated by the more confined trajectories of ΔF508 CFTR molecules treated with VX-809 (Figures 4B, D, E and 5B). As expected, ΔF508 CFTR His 10 exhibited higher diffusiveness compared to ΔF508 CFTR (Figures 4B, F, G and 5C).40 The trajectories of ΔF508 CFTR represented in histogram form trended toward lower diffusion coefficients in VX-809-treated cells compared to DMSO-treated cells, and the effect was mitigated in ΔF508 CFTR His 10 (20%; 0.035 vs 0.028 μm2/s; Figures 4B, D–G and 5B and C). These results clearly indicate that VX-809 restricts the movement of ΔF508 CFTR at the plasma membrane involving a PDZ interaction that also would plausibly decrease protein internalization.
Figure 4.

VX-809 restricts the mobility of ΔF508 CFTR at the plasma membrane in a PDZ-interaction-based mechanism. (A) Representative bright-field and quantum dot-labeled images of HEK-293 cells expressing no CFTR (referred to as HEK-P), Flag-WT CFTR, Flag-ΔF508 CFTR, and Flag-ΔF508 CFTR His 10. Flag-ΔF508 CFTR and Flag-ΔF508 CFTR His 10 were temperature rescued in the presence or absence of VX-809 for 48 h at 28 °C. (B) Representative mean square displacement curve to show the mobility kinetics of different groups of CFTR molecules. (C–G) Histograms representing the diffusion coefficients of a population of CFTR in various groups. 1000–4000 total objects were selected for analysis pooled from two to five independent experiments.
Figure 5.

ΔF508 CFTR shows restricted trajectories in the presence of VX-809. Single-particle tracking images of HEK-293 cells (left panel) showing the overall mobility profile of (A) Flag-WT CFTR and (B) Flag-ΔF508 CFTR or (C) Flag-ΔF508 CFTR His 10 in the presence of DMSO or VX-809. Also shown are representative trajectories of individual CFTR molecules (right panel of each figure).
New aspects of ΔF508 CFTR conformational instability that have been elucidated are the impaired interactions of NBDs of CFTR with membrane-spanning domains (MSD).7 A recent study describes the action of VX-809 on the stabilization of the NBD1 (contains the ΔF508 mutation)–MSD1 interface.42 It has been proposed that VX-809 mediates this conformational stabilization by binding to CFTR inside the ER, leading to two major outcomes. First, ΔF508 CFTR subjected to VX-809 would exhibit improved folding inside the ER that impedes the degradative process and rescues more ΔF508 CFTR molecules to the cell surface. This resonates with the finding of increased resistance of ER-trapped ΔF508 CFTR to degradation in the presence of VX-809.12 Second, a conformationally better stabilized ΔF508 CFTR at the plasma membrane would exhibit improved interaction with NHERF1, less mobility, and reduced susceptibility to ubiquitination and endocytosis, and therefore, all these factors would cumulatively lead to longer retention of the protein at the membrane. Rescued non-native ΔF508 CFTR is under the surveillance of a peripheral quality control system involving chaperones, co-chaperones, and ubiquitination machinery.14,43 Therefore, it is very likely that VX-809-rescued ΔF508 CFTR proceeds better in the peripheral checkpoints compared to that with the vehicle treatment. We demonstrated that VX-809 considerably reduces the median diffusion coefficient of ΔF508 CFTR, but not significantly that of ΔF508 CFTR His 10, which suggests that VX-809-restrained CFTR binds more strongly with NHERF1 compared to the native molecule. This would further initiate the regulatory effects of NHERF1 on the peripheral profile of ΔF508 CFTR, including phosphorylation-mediated regulation of the protein. Initial studies indicated that CFTR-NHERF1 interaction is critical for polarized expression of CFTR protein.44 However, subsequent studies of C-terminal deletions in CFTR including removal of the PDZ-motif were shown not to affect the apical surface expression of the protein; nonetheless, it has been argued that these PDZ interactions are likely to affect the steady state levels of the protein.45 NHERF1 overexpression was found to decrease CFTR internalization that was accompanied by cytoskeletal changes in the cell.25 Overall, NHERF1 has been shown to mediate regulation of the phosphorylated status of the protein, surface expression, mobility, and henceforth durability of the protein.16,20,41 Insights into the transitions of VX-809-bound ΔF508 CFTR globular structure will conclusively determine the structural changes that VX-809 brings about in ΔF508 CFTR that enhance its stability at the plasma membrane.
Using VX-809 as a tool, we have evaluated an important determinant of rescued ΔF508 CFTR functional efficiency, and therefore, new schemes for the generation of drugs should emphasize mechanisms to strengthen peripheral protein–protein interactions of ΔF508 CFTR, which likely would make the drugs more effective. Development of an improved and more selective CFTR corrector that transforms into clinical studies would facilitate the development of potential new therapies to treat the severe physiological outcomes associated with the ΔF508 CFTR mutation.
Acknowledgments
The authors are grateful to J. Denise Wetzel, CCHMC Medical Writer, for critical review of the manuscript. We thank Ms. Jin Emerson-Cobb for editing the manuscript.
Glossary
Abbreviations
- CF
cystic fibrosis
- CFBEo– cells
cystic fibrosis bronchial epithelial cells
- CFTR
cystic fibrosis transmembrane-conductance regulator
- CFTRinh172
CFTR inhibitor-172
- NHERF1
Na+/H+ exchanger regulatory factor 1
- PDZ (PSD-95/Dlg-5/ZO-1)
post synaptic density protein, Drosophila disc large tumor suppressor, and zonula occludens-1 protein
- ΔF508
deletion of phenylalanine at residue 508
- ER
endoplasmic reticulum
- ERAD
endoplasmic-reticulum-associated degradation
- Fsk
forskolin
- HBE
human bronchial epithelia
- HBSS
Hank’s balanced salt solution
- IBMX
isobutyl-1-methylxanthine
- Isc
short-circuit current
- MSD
membrane-spanning domain
- NBD
nucleotide-binding domain
- PLA
proximity-ligation assay
- SPT
single-particle tracking
- WT
wild type
Supporting Information Available
Supplementary figures as described in the text. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Contributions
Kavisha Arora and Changsuk Moon contributed equally to this work.
This work was supported by the National Institutes of Health [DK080834 and DK093045] and the US Cystic Fibrosis Foundation [NAREN12PO].
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Rowe S. M.; Miller S.; Sorscher E. J. (2005) Cystic fibrosis. N. Engl. J. Med. 352, 1992–2001. [DOI] [PubMed] [Google Scholar]
- Choi J. Y.; Muallem D.; Kiselyov K.; Lee M. G.; Thomas P. J.; Muallem S. (2001) Aberrant CFTR-dependent HCO3-transport in mutations associated with cystic fibrosis. Nature 410, 94–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowntree R. K.; Harris A. (2003) The phenotypic consequences of CFTR mutations. Ann. Hum. Genet. 67, 471–485. [DOI] [PubMed] [Google Scholar]
- Kerem B.; Rommens J. M.; Buchanan J. A.; Markiewicz D.; Cox T. K.; Chakravarti A.; Buchwald M.; Tsui L. C. (1989) Identification of the cystic fibrosis gene: genetic analysis. Science 245, 1073–1080. [DOI] [PubMed] [Google Scholar]
- Bobadilla J. L.; Macek M. Jr.; Fine J. P.; Farrell P. M. (2002) Cystic fibrosis: a worldwide analysis of CFTR mutations--correlation with incidence data and application to screening. Hum. Mutat. 19, 575–606. [DOI] [PubMed] [Google Scholar]
- Gee H. Y.; Noh S. H.; Tang B. L.; Kim K. H.; Lee M. G. (2011) Rescue of DeltaF508-CFTR trafficking via a GRASP-dependent unconventional secretion pathway. Cell 146, 746–760. [DOI] [PubMed] [Google Scholar]
- Rabeh W. M.; Bossard F.; Xu H.; Okiyoneda T.; Bagdany M.; Mulvihill C. M.; Du K.; di Bernardo S.; Liu Y.; Konermann L.; Roldan A.; Lukacs G. L. (2012) Correction of both NBD1 energetics and domain interface is required to restore DeltaF508 CFTR folding and function. Cell 148, 150–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okiyoneda T.; Lukacs G. L. (2012) Fixing cystic fibrosis by correcting CFTR domain assembly. J. Cell Biol. 199, 199–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Namkung W.; Kim K. H.; Lee M. G. (2005) Base treatment corrects defects due to misfolding of mutant cystic fibrosis transmembrane conductance regulator. Gastroenterology 129, 1979–1990. [DOI] [PubMed] [Google Scholar]
- Pedemonte N.; Lukacs G. L.; Du K.; Caci E.; Zegarra-Moran O.; Galietta L. J.; Verkman A. S. (2005) Small-molecule correctors of defective DeltaF508-CFTR cellular processing identified by high-throughput screening. J. Clin. Invest. 115, 2564–2571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Goor F.; Hadida S.; Grootenhuis P. D.; Burton B.; Cao D.; Neuberger T.; Turnbull A.; Singh A.; Joubran J.; Hazlewood A.; Zhou J.; McCartney J.; Arumugam V.; Decker C.; Yang J.; Young C.; Olson E. R.; Wine J. J.; Frizzell R. A.; Ashlock M.; Negulescu P. (2009) Rescue of CF airway epithelial cell function in vitro by a CFTR potentiator, VX-770. Proc. Natl. Acad. Sci. U. S. A. 106, 18825–18830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Goor F.; Hadida S.; Grootenhuis P. D.; Burton B.; Stack J. H.; Straley K. S.; Decker C. J.; Miller M.; McCartney J.; Olson E. R.; Wine J. J.; Frizzell R. A.; Ashlock M.; Negulescu P. A. (2011) Correction of the F508del-CFTR protein processing defect in vitro by the investigational drug VX-809. Proc. Natl. Acad. Sci. U. S. A. 108, 18843–18848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clancy J. P.; Rowe S. M.; Accurso F. J.; Aitken M. L.; Amin R. S.; Ashlock M. A.; Ballmann M.; Boyle M. P.; Bronsveld I.; Campbell P. W.; De Boeck K.; Donaldson S. H.; Dorkin H. L.; Dunitz J. M.; Durie P. R.; Jain M.; Leonard A.; McCoy K. S.; Moss R. B.; Pilewski J. M.; Rosenbluth D. B.; Rubenstein R. C.; Schechter M. S.; Botfield M.; Ordonez C. L.; Spencer-Green G. T.; Vernillet L.; Wisseh S.; Yen K.; Konstan M. W. (2012) Results of a phase IIa study of VX-809, an investigational CFTR corrector compound, in subjects with cystic fibrosis homozygous for the F508del-CFTR mutation. Thorax 67, 12–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma M.; Pampinella F.; Nemes C.; Benharouga M.; So J.; Du K.; Bache K. G.; Papsin B.; Zerangue N.; Stenmark H.; Lukacs G. L. (2004) Misfolding diverts CFTR from recycling to degradation: quality control at early endosomes. J. Cell Biol. 164, 923–933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jurkuvenaite A.; Chen L.; Bartoszewski R.; Goldstein R.; Bebok Z.; Matalon S.; Collawn J. F. (2010) Functional stability of rescued delta F508 cystic fibrosis transmembrane conductance regulator in airway epithelial cells. Am. J. Respir. Cell Mol. Biol. 42, 363–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valentine C. D.; Lukacs G. L.; Verkman A. S.; Haggie P. M. (2012) Reduced PDZ interactions of rescued DeltaF508CFTR increases its cell surface mobility. J. Biol. Chem. 287, 43630–43638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukacs G. L.; Chang X. B.; Bear C.; Kartner N.; Mohamed A.; Riordan J. R.; Grinstein S. (1993) The delta F508 mutation decreases the stability of cystic fibrosis transmembrane conductance regulator in the plasma membrane. Determination of functional half-lives on transfected cells. J. Biol. Chem. 268, 21592–21598. [PubMed] [Google Scholar]
- Naren A. P.; Cormet-Boyaka E.; Fu J.; Villain M.; Blalock J. E.; Quick M. W.; Kirk K. L. (1999) CFTR chloride channel regulation by an interdomain interaction. Science 286, 544–548. [DOI] [PubMed] [Google Scholar]
- Naren A. P.; Quick M. W.; Collawn J. F.; Nelson D. J.; Kirk K. L. (1998) Syntaxin 1A inhibits CFTR chloride channels by means of domain-specific protein-protein interactions. Proc. Natl. Acad. Sci. U. S. A. 95, 10972–10977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naren A. P.; Cobb B.; Li C.; Roy K.; Nelson D.; Heda G. D.; Liao J.; Kirk K. L.; Sorscher E. J.; Hanrahan J.; Clancy J. P. (2003) A macromolecular complex of beta 2 adrenergic receptor, CFTR, and ezrin/radixin/moesin-binding phosphoprotein 50 is regulated by PKA. Proc. Natl. Acad. Sci. U. S. A. 100, 342–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li C.; Krishnamurthy P. C.; Penmatsa H.; Marrs K. L.; Wang X. Q.; Zaccolo M.; Jalink K.; Li M.; Nelson D. J.; Schuetz J. D.; Naren A. P. (2007) Spatiotemporal coupling of cAMP transporter to CFTR chloride channel function in the gut epithelia. Cell 131, 940–951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li C.; Naren A. P. (2010) CFTR chloride channel in the apical compartments: spatiotemporal coupling to its interacting partners. Integr. Biol. (Camb.) 2, 161–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guerra L.; Fanelli T.; Favia M.; Riccardi S. M.; Busco G.; Cardone R. A.; Carrabino S.; Weinman E. J.; Reshkin S. J.; Conese M.; Casavola V. (2005) Na+/H+ exchanger regulatory factor isoform 1 overexpression modulates cystic fibrosis transmembrane conductance regulator (CFTR) expression and activity in human airway 16HBE14o- cells and rescues DeltaF508 CFTR functional expression in cystic fibrosis cells. J. Biol. Chem. 280, 40925–40933. [DOI] [PubMed] [Google Scholar]
- Kwon S. H.; Pollard H.; Guggino W. B. (2007) Knockdown of NHERF1 enhances degradation of temperature rescued DeltaF508 CFTR from the cell surface of human airway cells. Cell Physiol. Biochem. 20, 763–772. [DOI] [PubMed] [Google Scholar]
- Favia M.; Guerra L.; Fanelli T.; Cardone R. A.; Monterisi S.; Di Sole F.; Castellani S.; Chen M.; Seidler U.; Reshkin S. J.; Conese M.; Casavola V. (2010) Na+/H+ exchanger regulatory factor 1 overexpression-dependent increase of cytoskeleton organization is fundamental in the rescue of F508del cystic fibrosis transmembrane conductance regulator in human airway CFBE41o-cells. Mol. Biol. Cell 21, 73–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ootani A.; Li X.; Sangiorgi E.; Ho Q. T.; Ueno H.; Toda S.; Sugihara H.; Fujimoto K.; Weissman I. L.; Capecchi M. R.; Kuo C. J. (2009) Sustained in vitro intestinal epithelial culture within a Wnt-dependent stem cell niche. Nat. Med. 15, 701–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCracken K. W.; Howell J. C.; Wells J. M.; Spence J. R. (2011) Generating human intestinal tissue from pluripotent stem cells in vitro. Nat. Protoc. 6, 1920–1928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buske P.; Przybilla J.; Loeffler M.; Sachs N.; Sato T.; Clevers H.; Galle J. (2012) On the biomechanics of stem cell niche formation in the gut--modelling growing organoids. FEBS J. 279, 3475–3487. [DOI] [PubMed] [Google Scholar]
- Penmatsa H.; Zhang W.; Yarlagadda S.; Li C.; Conoley V. G.; Yue J.; Bahouth S. W.; Buddington R. K.; Zhang G.; Nelson D. J.; Sonecha M. D.; Manganiello V.; Wine J. J.; Naren A. P. (2010) Compartmentalized cyclic adenosine 3′,5′-monophosphate at the plasma membrane clusters PDE3A and cystic fibrosis transmembrane conductance regulator into microdomains. Mol. Biol. Cell 21, 1097–1110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu T.; Dahan D.; Evagelidis A.; Zheng S.; Luo J.; Hanrahan J. W. (1999) Association of cystic fibrosis transmembrane conductance regulator and protein phosphatase 2C. J. Biol. Chem. 274, 29102–29107. [DOI] [PubMed] [Google Scholar]
- Eckford P. D.; Li C.; Ramjeesingh M.; Bear C. E. (2012) Cystic fibrosis transmembrane conductance regulator (CFTR) potentiator VX-770 (ivacaftor) opens the defective channel gate of mutant CFTR in a phosphorylation-dependent but ATP-independent manner. J. Biol. Chem. 287, 36639–36649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haggie P. M.; Stanton B. A.; Verkman A. S. (2004) Increased diffusional mobility of CFTR at the plasma membrane after deletion of its C-terminal PDZ binding motif. J. Biol. Chem. 279, 5494–5500. [DOI] [PubMed] [Google Scholar]
- Raghuram V.; Mak D. O.; Foskett J. K. (2001) Regulation of cystic fibrosis transmembrane conductance regulator single-channel gating by bivalent PDZ-domain-mediated interaction. Proc. Natl. Acad. Sci. U. S. A. 98, 1300–1305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He L.; Kota P.; Aleksandrov A. A.; Cui L.; Jensen T.; Dokholyan N. V.; Riordan J. R. (2013) Correctors of DeltaF508 CFTR restore global conformational maturation without thermally stabilizing the mutant protein. FASEB J. 27, 536–545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molinski S.; Eckford P. D.; Pasyk S.; Ahmadi S.; Chin S.; Bear C. E. (2012) Functional Rescue of F508del-CFTR Using Small Molecule Correctors. Front. Pharmacol. 3, 160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loo T. W.; Bartlett M. C.; Clarke D. M. (2009) Correctors enhance maturation of DeltaF508 CFTR by promoting interactions between the two halves of the molecule. Biochemistry 48, 9882–9890. [DOI] [PubMed] [Google Scholar]
- Yu W.; Kim Chiaw P.; Bear C. E. (2011) Probing conformational rescue induced by a chemical corrector of F508del-cystic fibrosis transmembrane conductance regulator (CFTR) mutant. J. Biol. Chem. 286, 24714–24725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dekkers J. F.; Wiegerinck C. L.; de Jonge H. R.; Bronsveld I.; Janssens H. M.; de Winter-de Groot K. M.; Brandsma A. M.; de Jong N. W.; Bijvelds M. J.; Scholte B. J.; Nieuwenhuis E. E.; van den Brink S.; Clevers H.; van der Ent C. K.; Middendorp S.; Beekman J. M. (2013) A functional CFTR assay using primary cystic fibrosis intestinal organoids. Nat. Med. 19, 939–945. [DOI] [PubMed] [Google Scholar]
- Liu J.; Walker N. M.; Cook M. T.; Ootani A.; Clarke L. L. (2012) Functional Cftr in crypt epithelium of organotypic enteroid cultures from murine small intestine. Am. J. Physiol. Cell Physiol. 302, C1492–1503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bates I. R.; Hebert B.; Luo Y.; Liao J.; Bachir A. I.; Kolin D. L.; Wiseman P. W.; Hanrahan J. W. (2006) Membrane lateral diffusion and capture of CFTR within transient confinement zones. Biophys. J. 91, 1046–1058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haggie P. M.; Kim J. K.; Lukacs G. L.; Verkman A. S. (2006) Tracking of quantum dot-labeled CFTR shows near immobilization by C-terminal PDZ interactions. Mol. Biol. Cell 17, 4937–4945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okiyoneda T.; Veit G.; Dekkers J. F.; Bagdany M.; Soya N.; Xu H.; Roldan A.; Verkman A. S.; Kurth M.; Simon A.; Hegedus T.; Beekman J. M.; Lukacs G. L. (2013) Mechanism-based corrector combination restores DeltaF508-CFTR folding and function. Nat. Chem. Biol. 9, 444–454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okiyoneda T.; Barriere H.; Bagdany M.; Rabeh W. M.; Du K.; Hohfeld J.; Young J. C.; Lukacs G. L. (2010) Peripheral protein quality control removes unfolded CFTR from the plasma membrane. Science 329, 805–810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moyer B. D.; Denton J.; Karlson K. H.; Reynolds D.; Wang S.; Mickle J. E.; Milewski M.; Cutting G. R.; Guggino W. B.; Li M.; Stanton B. A. (1999) A PDZ-interacting domain in CFTR is an apical membrane polarization signal. J. Clin. Invest. 104, 1353–1361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ostedgaard L. S.; Randak C.; Rokhlina T.; Karp P.; Vermeer D.; Ashbourne Excoffon K. J.; Welsh M. J. (2003) Effects of C-terminal deletions on cystic fibrosis transmembrane conductance regulator function in cystic fibrosis airway epithelia. Proc. Natl. Acad. Sci. U. S. A. 100, 1937–1942. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.