

Abstract
Background
The ZNF217 gene, encoding a C2H2 zinc finger protein, is located at 20q13 and found amplified and overexpressed in greater than 20% of breast tumors. Current studies indicate ZNF217 drives tumorigenesis, yet the regulatory mechanisms of ZNF217 are largely unknown. Because ZNF217 associates with chromatin modifying enzymes, we postulate that ZNF217 functions to regulate specific gene signaling networks. Here, we present a large-scale functional genomic analysis of ZNF217, which provides insights into the regulatory role of ZNF217 in MCF7 breast cancer cells.
Results
ChIP-seq analysis reveals that the majority of ZNF217 binding sites are located at distal regulatory regions associated with the chromatin marks H3K27ac and H3K4me1. Analysis of ChIP-seq transcription factor binding sites shows clustering of ZNF217 with FOXA1, GATA3 and ERalpha binding sites, supported by the enrichment of corresponding motifs for the ERalpha-associated cis-regulatory sequences. ERalpha expression highly correlates with ZNF217 in lysates from breast tumors (n = 15), and ERalpha co-precipitates ZNF217 and its binding partner CtBP2 from nuclear extracts. Transcriptome profiling following ZNF217 depletion identifies differentially expressed genes co-bound by ZNF217 and ERalpha; gene ontology suggests a role for ZNF217-ERalpha in expression programs associated with ER+ breast cancer studies found in the Molecular Signature Database. Data-mining of expression data from breast cancer patients correlates ZNF217 with reduced overall survival.
Conclusions
Our genome-wide ZNF217 data suggests a functional role for ZNF217 at ERalpha target genes. Future studies will investigate whether ZNF217 expression contributes to aberrant ERalpha regulatory events in ER+ breast cancer and hormone resistance.
Electronic supplementary material
The online version of this article (doi:10.1186/1471-2164-15-520) contains supplementary material, which is available to authorized users.
Keywords: Breast cancer, ZNF217, ERalpha, GATA3, FOXA1, ChIP-seq, RNA-seq, Endocrine resistance
Background
Recurrent and common genomic amplifications have led to the discovery of important oncogenes and cancer therapeutic targets. The 20q13 amplification including the ZNF217 gene coding for a transcription factor is found in ~ 20-30% of breast cancers and is associated with aggressive tumor behavior, shorter disease-free survival, chemoresistance, and poor prognosis [1, 2]. A recent report shows that ZNF217 overexpression accelerates aberrant cell differentiation through signaling events leading to increased self-renewal capacity, a mesenchymal phenotype, motility, chemoresistance and metastasis in mammary mouse models [3]. Earlier work using ChIP-chip tiling arrays for the 5 kb DNA region surrounding the transcriptional start site (TSS) identified ZNF217 regulatory gene targets in the embryonal carcinoma cell line, Ntera2, and the breast cancer cell line MCF7 [4]. This work supported a developmental role for ZNF217 as a regulatory factor at differentiation-specific genes. Findings from this work led to the discovery that ZNF217 directly activates ERBB3 and downstream signaling events through PI3K and MAPK pathways [3, 5, 6]. Despite increasing knowledge of ZNF217-induced phenotypes and regulation of pathways promoting tumorigenesis, there is a lack of understanding of the downstream ZNF217-induced effectors driving these cell pathways.
ZNF217 encodes a transcription factor with eight C2H2 zinc finger motifs and a proline-rich transactivation domain at the C-terminus [7]. ZNF217 has been reported to physically interact with CtBP1/2 [7], an adaptor protein found in multiple regulatory complexes at both activated and repressed gene targets [5, 7, 8]. ChIP-chip studies indicate CtBP and ZNF217 are co-bound at the majority of ZNF217 DNA binding sites [4]. ZNF217 biochemically purifies with histone deacetylases HDAC1/2 [9, 10], histone demethylases LSD1 [10, 11] and Jarid1b/Plu-1 [11], and histone methyltranferases G9a and EZH2 [11], suggesting a range of regulatory functions in histone modifying complexes. Based on our current knowledge of ZNF217 and its association with DNA regulatory proteins, it has been hypothesized that ZNF217 functions as an organizer of histone chromatin modifiers [11]. The cooperating transcriptional mechanisms used by ZNF217, and its association with specific regulatory elements remains unexplored.
An important step in understanding the molecular role of ZNF217 in breast cancer is to gain a more complete understanding of the mechanisms of genome-wide gene regulation by ZNF217 in breast cancer cells, including the involvement of cooperating transcriptional partners. In the current study we employed an integrative genomics approach to uncover the mechanisms of ZNF217 target gene regulation in breast cancer cells. Using a combination of RNA- and ChIP-seq techniques in MCF7 breast cancer cells, we focused on identifying genes that were regulated by ZNF217 DNA binding. Our findings suggest a functional association for ZNF217 with estrogen receptor alpha (ERα) at co-bound ERα gene targets. This work supports further exploration into the connection between ZNF217 expression levels in breast tumors with clinical outcome and, importantly, whether ZNF217 plays a transcriptional role in aberrant ERα signaling, contributing to breast cancer and therapy resistance.
Results
ZNF217 genome-wide binding patterns
To elucidate the regulatory function of ZNF217 in breast cancer, we performed genome-wide chromatin mapping of ZNF217 by ChIP-seq using the ER+ HER2- MCF7 cell line. Accordingly, we sequenced two independent ChIP-seq replicates and determined a set of enriched ZNF217 binding sites, using the Sole-Search peak calling tool [12], by taking the shared ZNF217 binding sites (False Discovery Rate (FDR) < 0.0001) from both replicates (18, 965 binding sites; Additional file 1). These datasets agree with the ENCODE guidelines for transcription ChIP-seq overlap rules [13]. To determine the Refseq genes nearest to each ZNF217 binding site, we used the Sole-Search location analysis tool. This analysis identified ZNF217 ChIP-seq targets at a total of 6,965 target genes with multiple binding events occurring at 3,619 genes (see Additional file 2). Our earlier work from MCF7 ChIP-chip promoter arrays identified ZNF217 binding at the proximal promoter regions of 5,061 genes [4]; 45% of these genes are present in the current ZNF217 ChIP-seq dataset. Analysis of distances between ZNF217-associated regions and the nearest annotated TSSs revealed that ZNF217 binds predominantly distal to promoter regions; less than 10% of the sites are located within 1 kb of annotated TSSs. In contrast, 15%, 52%, and 26% of the sites are located within 1–10 kb, 10–100 kb, and > 100 kb from a TSS, respectively (Figure 1A). Interestingly, location analysis shows 6,348 binding sites (33%) are located within genes (introns and exons), with the majority (92%) within introns. A panel of ZNF217-bound sites located at distal regions, including intronic sites, was confirmed by ChIP qPCR (see Additional file 3). ZNF217 binding at the proximal region of TFF1 and the previously identified ZNF217 regulatory target, ERBB3 [5], are also shown.
Figure 1.
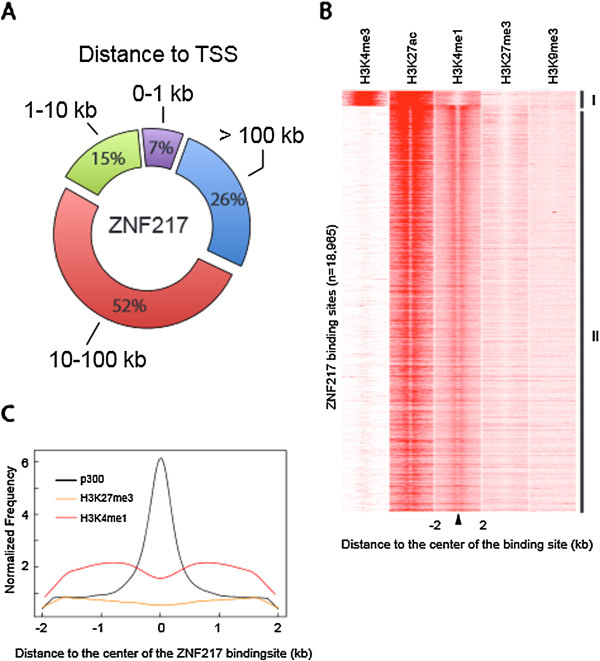
Genome-wide analysis of ZNF217chromatin occupancy. (A) Distribution of ZNF217 binding sites relative to the nearest annotated TSS. (B) Heatmaps depict H3K4me3, H3K27ac, H3K27me3, and H3K9me3 ChIP-Seq signals at regions spanning ZNF217 binding sites (±2 kb) in MCF7 cells organized according to their chromatin status: cluster I and cluster II. Active K4me3-only (cluster I) sites are predominately promoters, while K4me1 and K27ac sites (cluster II) are primarily enriched for distal regions. (C) Average p300 (black), H3K4me1 (red), and H3K27me3 (orange) ChIP-seq enrichment profiles around central position of ZNF217 binding sites.
To further explore ZNF217 binding with respect to specific regulatory elements (such as enhancers or silencers), we analyzed the ChIP-seq read density relative to the total number of ZNF217 binding sites in MCF7 using ChIP-seq datasets for a number of activating histone marks including mono- and trimethylation of histone H3 Lys4, acetyl histone H3 Lys27 (H3K4me1, H3K4me3, H3K27ac, respectively), as well as the repressive histone marks trimethylation of histone H3 Lys9 and trimethylation of histone H3 at Lys27 (H3K9me3 and H3K27me3, respectively). The heatmap in Figure 1B shows a strong enrichment for H3K27ac and H3K4me1 relative to the center of the majority of ZNF217 sites (cluster II). A small proportion of sites are also enriched for H3K4me3 and H3K4me1 (cluster I). Location analysis on these ZNF217 binding sites indicate that the cluster II binding sites are largely distal regions, whereas the H3K4me3 enriched cluster I sites are largely promoter proximal (see Additional file 4). These results show that the majority of ZNF217 binding sites are marked by active chromatin marks and that the predominant class of binding events is located at distal regulatory elements associated with H3K4me1 and H3K27ac. Recent reports demonstrated that active enhancer elements are marked by the histone acetyltransferase p300 and by enrichment of H3K4me1 at flanking nucleosomes [14, 15]. Indeed, using p300 ChIP-seq data we observe p300 enrichment at ZNF217 peaks and H3K4me1 flanked ZNF217-bound regions, whereas H3K27me3 showed no substantial enrichment (Figure 1C).
To glean insight into the function of ZNF217 binding, we performed functional annotation of the top 5,000 ZNF217-bound regions with GREAT, a software tool designed to assign biological function to noncoding genomic regions [16]. This analysis showed an association with ontological categories including mesoderm development, mammary gland development and gastrulation. The Pathway Commons category terms aligned with ERα signaling and FOXA1 transcription networks. ZNF217 binding sites associated with multiple genes in TGFβ, WNT, and ER signaling pathways (gene associations are shown in Additional file 5).
ZNF217 binding sites are occupied by multiple transcription factors
Another property of enhancer regions is their relative abundance of binding sites for multiple transcription factors [15, 17]. To gain insight into the function of ZNF217 at distal regulatory regions in relation to other transcriptional partners, we identified the predominant motifs for known transcription factors near ZNF217 binding sites. We retrieved the 200 bp DNA sequence for the ZNF217-bound regions and identified the most significant motif logos using HOMER [17] (Figure 2A). Our search identified enriched cis-regulatory sequences for FOXA1 in 31%, GATA3 in 34%, and EREs at 20% of the ZNF217 binding sites (Figure 2A) of which the density of each motif is enriched ± 200 bp relative to the center of the ZNF217 binding peak (Figure 2B). Next, we searched for de novo motifs over-represented within ZNF217-bound regions. We did not find enrichment of the ZNF217 consensus sequences reported in our earlier work [4], or the work of others [8]. We did however identify a unique, non-repetitive sequence at ZNF217 sites: 5’-TGA(G/C)TCA(T/C)-3’ (or its reverse complement 5’-(A/G)TGA(C/G)TCA-3’). This de novo ZNF217 consensus site indicates enrichment of a sequence similar to the AP-1 motif (see Additional file 6). AP-1 motifs are very often found near binding sites for many types of TFs, including ERα, and AP-1 has been considered by some as a “general” enhancer binding factor [18]. AP-1 is a transcription factor that interacts with ERα and plays a role in the recruitment of ERα upon ligand binding to specific regulatory elements within the genome [18–21]. To further investigate the transcriptional partners of ZNF217, we performed unsupervised clustering analysis of all ENCODE ChIP-seq transcription factor binding sites found in MCF7 cells. We found a group of five transcription factors with binding sites that cluster with ZNF217 binding sites, including TCF7L2, NR2F2, ERα, GATA3, and FOXA1 (Figure 2C). This occupancy and motif analysis highlights a significant interaction between ZNF217 and alternative breast cancer transcription regulators.
Figure 2.
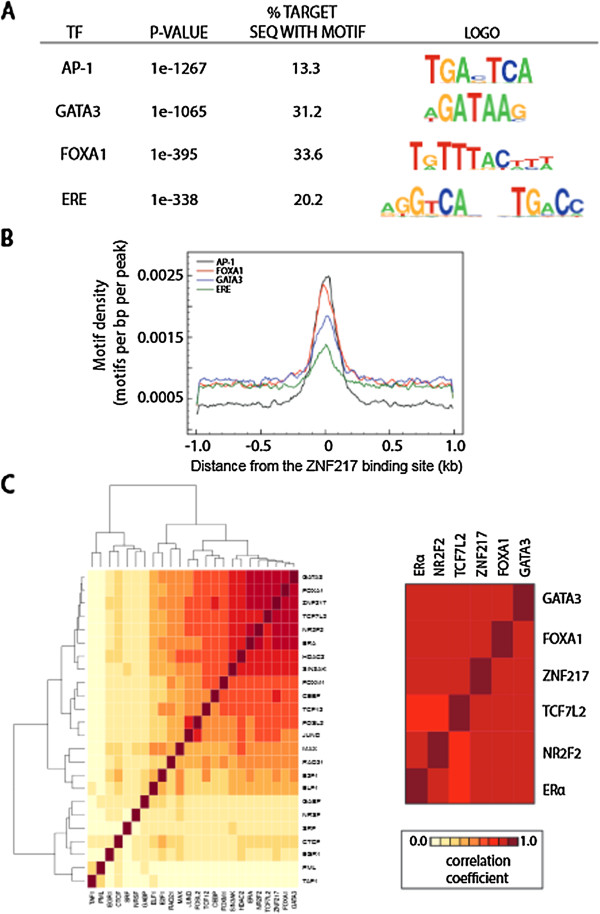
ZNF217 binding sites cluster with transcription factor binding sites in MCF7 cells. (A) Top TF motifs enriched in ZNF217 binding sites. (B) Distribution of identified motifs relative to the center of ZNF217 binding sites. Shown below are the motif logos for each TF. (C) ZNF217 shows co-binding with GATA3, FOXA1, TCF7L2, NR2F2 and ERα. Colors in the heat map reflect the colocalization frequency of each transcription factor (the descending frequency of localization ranges from red to yellow to white).
Several proteins serve as pioneer factors contributing to ERα action in breast cancer, including FOXA1 and GATA3 (reviewed in [22]). We therefore assessed the four-way overlap between these factors. Binding site overlap analysis indicate a large fraction of the total ZNF217 binding sites are shared by ERα, GATA3, and FOXA1 in asynchronous MCF7 cells (Figure 3A and C). In addition, this analysis reveals distinct combinations of binding sites shared between these factors. We found that while 41% of ZNF217 sites overlap with ERα (see Additional file 7), there are other unique associations of ZNF217 with GATA3 and FOXA1 independent of ERα in MCF7 cells (Figure 3A). The individual sets of genes bound by ZNF217 and each factor or groups of factors are listed in Additional file 8. Heatmap clustering of the read density from independent ERα, FOXA1, and GATA3 ChIP-seq experiments supports the association of these transcriptional regulators, where an enriched signal is prevalent surrounding the center of ZNF217 binding sites (Figure 3B). One example of overlap between ZNF217 with ERα, FOXA1, and GATA3 co-bound regions is shown at the TFF1 gene, a well-known ER gene target [23] (Figure 3C).
Figure 3.
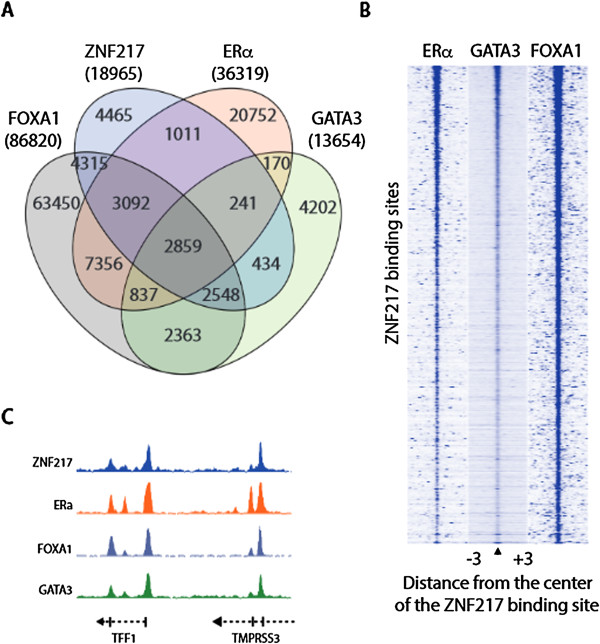
ZNF217 binding correlates with ERα, GATA3 and FOXA1. (A) A four-way comparison of binding sites shared with ZNF217 in MCF7 cells. Listed below the factor is the total number of binding sites for that dataset. (B) Heatmap depicting ERα, GATA3 and FOXA1 ChIP-seq signal density spanning ZNF217 binding sites (±2 kb) in MCF7 cells. (C) IGB snapshot of ZNF217, ERα, GATA3 and FOXA1 occupancy at the TFF1 gene.
Depletion of ZNF217 identifies a gene expression signature associated with ERα gene targets
To gain further insight into the regulation of gene expression by ZNF217, we employed an RNA-seq approach following ZNF217 silencing to characterize the direct and indirect targets of ZNF217 in MCF7 breast cancer cells. We treated cells with ZNF217-targeting short interfering RNA (siRNA) leading to ZNF217 mRNA and protein depletion (see Additional file 9) and performed gene expression profiling by RNA-seq. We identified 3,402 differentially expressed genes; 1,615 genes were up-regulated and 1,787 were down-regulated genes in the ZNF217 knockdown sample, as compared to the control (siScramble) siRNA-treated sample (at 1.25 -fold change cutoff and criteria representing statistical significance, false discovery rate (FDR) < 0.0001; genes listed in Additional file 10; Figure 4A). RNA-seq results were confirmed by RT-qPCR on a small panel of genes (Figure 4B). To determine if the genes that changed with ZNF217 silencing are also bound by ZNF217, we integrated the ChIP-seq and RNA-seq datasets and compared the expression of genes with a ZNF217 binding site. Among the 3,402 differentially expressed genes, following ZNF217 silencing, 1,230 (36%) of the ZNF217-bound genes change with ZNF217 knockdown. In addition, we find that 62% and 38% of the 1,230 differentially expressed genes with a ZNF217 binding site show increased and decreased expression, respectively (Figure 4C). We also found that among a total 4,442 genes co-bound by ZNF217 and ERα, 854 genes change in expression with ZNF217 silencing of which 60% (512 genes) show increased expression levels (Figure 4C). Independent RT-qPCR experiments validate the increased levels of the known estrogen responsive ERα-target genes LRIG1 [24] and SLC22A5 [25] and the decreased levels of AXIN1 and IGFBP4 following ZNF217 depletion (Figure 4B and Additional file 11).
Figure 4.
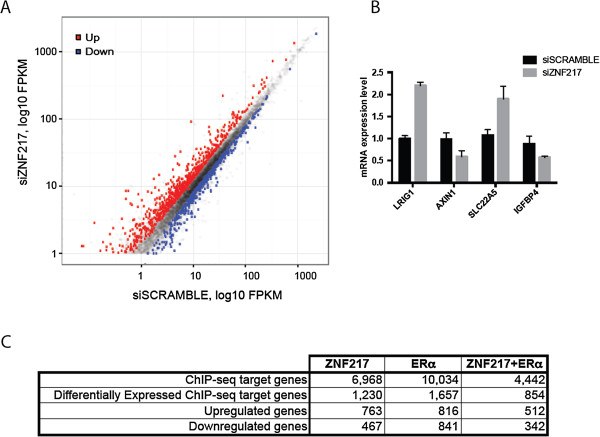
Analysis of gene expression changes upon ZNF217 knockdown in MCF7 cells. (A) A scatterplot of expression data from RNA-seq experiments. Each point corresponds to one NCBI Reference Sequence (RefSeq) transcript with fragments per kilobase of exon model per million mapped reads (FPKM) values for control and ZNF217 knockdown samples shown on an exponential scale. Significantly affected transcripts (FDR < 0.05 with a 1.25-fold change cuttoff) are depicted (red upregulated and blue downregulated upon ZNF217 knockdown). The dashed line represents no change in gene expression between the two samples. (B) The triplicate biological (sequenced) RNA samples from siScrambled- or siZNF217- treated MCF7 cells were analyzed by quantitative RT-PCR. ABI expression assay Taqman probes to measure transcript levels of six genes are indicated. Each sample was assayed in triplicate and the cT values were normalized to GAPDH. Average relative transcript level was graphed using BioRad CFX software. Columns: transcript levels, error bars: standard error of the mean. (C) Summary of differentially expressed genes, downregulated or upregulated by depletion of ZNF217 and bound by ZNF217, ERα, or both ZNF217 and ERα.
The ZNF217 gene signature identifies gene set enrichments from ER+ breast cancer studies
To understand the genes regulated by ZNF217 and the pathways associated with ERα function, we performed gene ontology analysis on the lists of differentially regulated genes following ZNF217 silencing with the GREAT classification gene ontology tool [16]. Interestingly, both the list of 1,230 ZNF217-bound differentially regulated genes and the list of 854 ZNF217-ERα co-bound differentially regulated genes show strong associations with genes associated with multiple cancer studies present in the Molecular Signatures Database (MSigDB) [26]. Genes in the ZNF217-ERα co-bound list correlate with genes identified from key cancer studies including: (a) Genes found up-regulated or down-regulated in breast cancer tumors (formed by MCF-7 xenografts) resistant to tamoxifen (Massarweh [27]), (b) Genes up-regulated in luminal-like breast cancer cell lines compared to basal or mesenchymal-like ones (Charafe [28]), (c) Genes associated with acquired endocrine therapy resistance in breast tumors expressing ESR1 but not ERBB2 (Creighton [29]), (d) Down-regulated genes in the cancer progenitor (stem) cells corresponding to side population (SP) MCF7 cells positive for MUC1 (Engelmann [30]), (e) Genes up-regulated in MCF-7 cells under hypoxia and (Elvidge [31]), (f) Genes that change according to the ESR1 status: ER positive vs ER negative tumor (Doane [32]), and (g) Genes up-regulated in MCF7 cells (breast cancer) after stimulation with NRG1 (Nagashima [33]). The enrichments of the ZNF217-ERα co-bound MSigDB categories are shown in Figure 5. A comparison of the complete list of cancer studies associated with ZNF217-bound and ZNF217-ERα co-bound genes are in Additional file 12.
Figure 5.
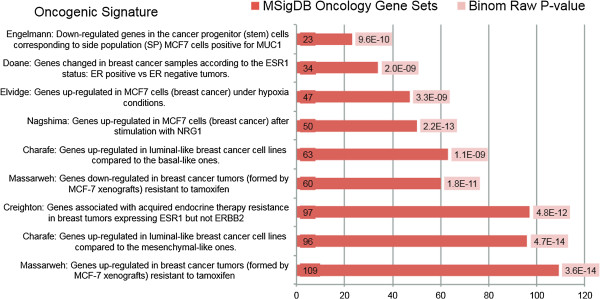
Analysis of gene expression changes upon ZNF217 knockdown in MCF7 reveals deregulation of breast cancer gene signatures. GREAT gene ontology on the 854 ZNF217-ERα co-bound gene list identifies overlap with several MSigDB Cancer Studies (p < 10−8, exact binomial test).
We were particularly interested in the combined up- and down-regulated ZNF217 differentially expressed genes (169) associated with the Massarweh expression study [27]. Gene ontology for the 169 genes showed functional classification associating with developmental pathways; many of these genes are known estrogen-responsive ERα target genes (data not shown). The gene expression changes found in tamoxifen-resistant tumors (derived from MCF7 xenografts) included several genes either up or down-regulated by ZNF217 and co-bound by ZNF217- ERα including CAV2, ELF5, ID2, TGFB2, LRIG1, FOXO3, CXCL12, and CD44.
ZNF217 and ERα precipitate in MCF7 nuclear extracts
Our genome-wide binding analysis indicates ZNF217 is associated with ERα and other regulatory factors at “active” DNA regulatory elements. It is well-known that nuclear receptors do not function in isolation but require co-factors to assist with DNA interaction; intensive work in the field of estrogen receptor biology has sought to identify critical factors that affect ERα-mediated transcription [34]. To identify potential unknown regulators of ERα, Jason Carroll’s group performed a novel approach termed RIME (Rapid Immunoprecipitation with ERα followed by Mass spectrometry of Endogenous proteins). This work identified an association between ERα and 108 proteins, including the association of ZNF217 and the binding partners, CtBP1/2, in three out of three replicate experiments [35]. To confirm this finding, we tested whether or not ZNF217 associates with ERα in MCF7 nuclear extracts by co-immunoprecipitation assays. Immunoblotting of lysates showed ERα precipitated both ZNF217 and the ZNF217-binding partner CtBP2 (see Additional file 13).
ZNF217 is enriched in ER+ breast cancer
If ZNF217 interacts with ERα and potentially contributes to the chromatin landscape associated with ERα binding, then we reasoned that ZNF217 expression might be higher in estrogen receptor positive (ER+) tumors. Using microarray expression studies of breast tumors across multiple patient cohorts, we found that ZNF217 expression levels are higher in ER+ tumors than in ER- tumors (data-mining from the Chin 2006 [36] and the Hess 2006 [37] studies are shown in Figure 6A and B). We next compared ZNF217 expression levels across patient subtypes: Luminal A, Luminal B, ERBB2+, basal, and normal. Patient data from the Chin and Hess studies indicate ZNF217 expression is consistently highest in Luminal and lowest in basal subtype tumors (Figure 6C and D).
Figure 6.
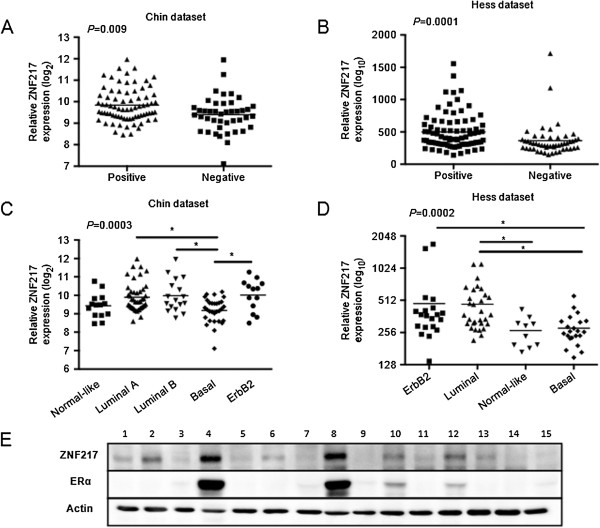
High ZNF217 expression is enriched in ER + breast cancer. (A) Estrogen receptor status from Chin et al. 2006 study [36]. ZNF217 expression levels in primary tumors that were ER positive (n = 75) or ER negative (n = 43) compared by Mann–Whitney t-test (p = 0.009). (B) Estrogen receptor status from Hess et al. 2006 study [37]. ZNF217 expression levels in primary tumors that were ER-positive (n = 82) or negative (n = 51) and compared by Mann–Whitney t-test (p ≤ 0.001). (C) ZNF217 expression levels in patients separated by gene expression molecular subtypes (p = 0.0003; ANOVA,Kruskal-Wallis) from Chin et al. 2006 [36] study. (D) ZNF217 expression levels in patients separated by gene expression molecular subtypes (p = 0.0002; ANOVA,Kruskal-Wallis) from Hess et al. 2006 [37] study. For both analyses, individual cohort combinations were compared by Dunn’s multiple comparison test, and those marked with * had p < 0.05. Each line marks the mean for the subtype. (E) Immunoblotting of 15 breast tumor lysates with antibodies to ZNF217, ER, or actin as a loading control. ZNF217 and ER protein densitometry was quantitated (r2 = 0.93).
We previously found that high ZNF217 expression is a prognostic indicator in general as well as in ER+HER2+LN− patients with breast cancer correlating with shorter overall, disease-specific, and relapse-free survival [3]. We next wanted to determine if ZNF217 overexpression had prognostic value exclusive of estrogen receptor status. We found that high ZNF217 expression consistently predicted poor prognosis across multiple tumor subtypes: Luminal, ER−, HER2+/ERBB2+, and basal cohorts ([3] and Additional file 14). Together, our data show that ZNF217 is prognostic of reduced survival in ER+, ER−, HER2+/ERBB2+, Luminal, and basal subtype patients by univariate analysis. Moreover, ZNF217 was a better predictor of survival than ERα status but not tumor size by multivariate analysis [3].
We also looked for a correlation between ZNF217 and ERα protein expression in breast tumor samples. We screened a small sample size of 15 tumor lysates and immunoblotted for ZNF217 and ERα. Densitometry indicated a high correlation for ZNF217 and ERα protein expression (r2 = 0.93) in the tumor samples (Figure 6E).
ZNF217 expression is prognostic of reduced survival in patients with ER+ and Luminal A breast tumors
To further explore the effect of ZNF217 expression on patient survival we interrogated the Kaplan Meier-plotter, an online survival analysis tool, for overall survival in breast cancer patients to rapidly assess the effect of 22,277 genes on breast cancer prognosis using microarray data of 1,809 patients [38]. We compared all ER+ patients (all intrinsic subtypes with any therapy) and found that high ZNF217 expression was associated with lower overall survival (p = 0.03, Figure 7A). To determine whether ZNF217 was simply identifying the poorer prognosis Luminal B tumors from within the Luminal cohort, we asked whether ZNF217 expression had prognostic value in the ER+ Luminal A tumors or the ER+ Luminal B tumors (with any chemo or anti-hormone therapy). As shown in Figure 7B, high ZNF217 expression is associated with lower overall survival in the Luminal A tumors (p = 0.035). Interestingly, the data for Luminal B tumors expressing high ZNF217 did not reach statistical significance (Figure 7C). This finding suggests that ZNF217 provides additional prognostic information, beyond ERα status and Luminal subtype.
Figure 7.
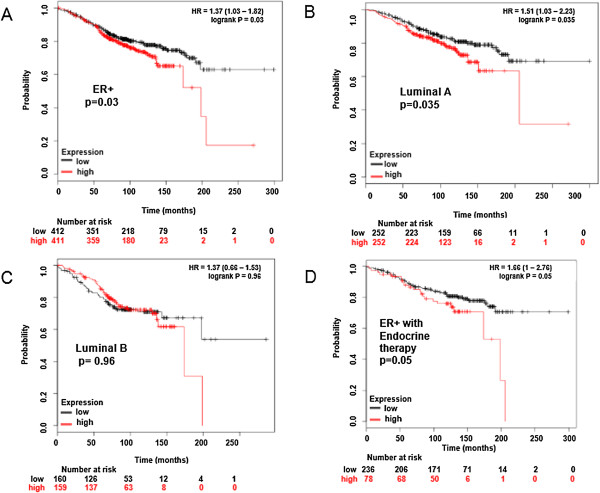
ZNF217 expression is prognostic of reduced survival in patients with ER + and Luminal A breast tumors. ZNF217 gene expression was analyzed using the Kaplan Meier-Plotter, an online survival analysis tool to analyze gene expression with breast cancer prognosis using microarray data from 1,809 patients [38]. High ZNF217 expression affects Overall Survival in Luminal A but not Luminal B ER+ breast cancer patients (any systemic anti-hormonal therapy treatment and/or chemotherapy). The prognostic value of high ZNF217 expression (median cutoff) was compared to the patient cohort with low ZNF217 expression in (A) All ER+ patients (n = 823), p = 0.03. (B) Luminal A (n = 504), p = 0.035 (C) Luminal B (n = 319), p = 0.96. (D) High ZNF217 correlates with worse survival in ER+ patients treated with any endocrine therapy (but not chemotherapy). The prognostic value of high ZNF217 (n = 78) expression (upper quartile) was compared to the patient cohort with low ZNF217 expression (n = 236), p = 0.01.
ZNF217 expression is prognostic of reduced survival after hormone treatment of ER+ breast cancer patients
The high overlap between differentially regulated genes co-bound by ZNF217 and ERα and the poor prognosis of patients with breast tumors that overexpress ZNF217 suggested that ZNF217 may have a regulatory role in expression changes that occur in tamoxifen-resistant breast tumors. To determine if high ZNF217 expression correlates with reduced overall survival in the cohort of ER+ patients that received anti-hormone therapy only (without the confounding effects of chemotherapy), we explored n = 78 patients with high versus n = 236 low ZNF217 expression in the Kaplan Meier-plotter (Figure 7D). Patients with high ZNF217 expression (upper quartile) demonstrated worse outcome p = 0.05.
Discussion
In this work we have mapped the chromatin landscape surrounding ZNF217 DNA binding sites and found significant overlap between ZNF217 binding and transcription factors associated with the ERα regulatory network. ZNF217 predominantly occupies distal regulatory regions marked by the active histone marks H3K27ac and H3K4me1 and, depending on the chromosomal context, may participate either in the repression or the activation of gene expression. A similar mode of action for ER-mediated gene regulation has been proposed where ERα associates with auxiliary TFs and co-regulators in a specific chromosomal context to control gene expression, although it is not well understood what specifies the context-dependent activity of this regulation. In support of a transcriptional role for ZNF217 in aberrant ERα signaling, we identified a ZNF217-ERα gene signature with ontological classification that aligns with multiple breast cancer studies (Figure 5). Data-mining of expression data from breast cancer patients also associates high ZNF217 expression with poor prognosis across multiple intrinsic subtypes and reduced response to hormone therapy (Figure 7).
The function of ER binding at chromatin regions and transcriptional regulation is strongly influenced by GATA3 and FOXA1 [39–42]. GATA3 appears to function as a critical regulator of transcription factor binding, chromatin structure, and long-range genomic communication [43]. FOXA1 is known as a pioneer factor, a special class of transcription factors that bind to condensed chromatin and facilitate the binding of additional transcription factors [22]. An elegant study by Ross-Innes et al., mapped the genome-wide ER-binding events by ChIP-seq in primary breast cancers from patients with different clinical outcomes [44]. They found distinct ER binding co-factor combinations linked with the different clinical outcomes, and that FOXA1 appears to mediate the differential ER-binding profiles found in clinical subtypes. However it is still unknown what dictates the differential FOXA1 and subsequent ERα binding. At the genome level approximately a quarter of all ERα -binding events are co-occupied by GATA3 and FOXA1 [43]. Similarly, we found an enrichment of GATA3 and FOXA1 factors at ZNF217-bound regions (Figure 3A). Our results suggest ZNF217-bound regions may be co-occupied with ERα and FOXA1 at multiple ERα gene targets, including TFF1, ERBB3, SLC22A5, and LRIG1 (Additional file 3: Figure S1B and Additional file 11: Figure S6). Knockdown of ZNF217 followed by expression profiling identified 3,402 differentially affected genes suggesting a functional regulatory role linked to multiple signaling pathways associated with breast cancer (Figure 5 and Additional file 12: Table S6). Future work will determine whether ZNF217 binding to ERα cis-regulatory sequences contributes an additional level of regulation to ERα-binding potential and ERα-signaling. Multiple regulatory scenarios are possible and include ZNF217 promotion of chromatin accessibility through recruitment of chromatin remodeling factors or regulation of three-dimensional chromatin to spatially connect distal enhancer regions with proximal promoters. It will be interesting to unravel the potential regulatory role for ZNF217 signaling and whether ZNF217 co-operates or competes with ERα regulatory factors such as GATA3 and FOXA1.
Interestingly, ZNF217 and GATA3-FOXA1 are associated with opposite tumor phenotypes. Well-differentiated tumors are generally less advanced and support a better prognosis than poorly differentiated tumors that are more aggressive and correlate with poorer prognosis. The regulation of cell differentiation during normal development involves transcription factors that are increasingly associated with tumorigenesis upon their aberrant re-activation and expression. Recently, GATA3 has been shown to play an important role at multiple stages of mammary gland development, including the formation of terminal end buds in luminal cell differentiation [45]. GATA3 is a strong predictor of tumor grade, ER status, well-differentiated tumors, and a marker of good prognosis. Loss of GATA3 coincides with loss of differentiation and tumor metastasis [45]. Similar to GATA3, FOXA1 plays a role in postnatal development of the mammary gland, perhaps due to its requirement for ERα chromatin binding and activity [46, 47]. Both GATA3 and FOXA1 expression are part of the Luminal A molecular subtype gene signature [48], associated with well-differentiated tumors and better prognosis. Tumors negative for GATA3 and FOXA1 expression associate with poorly differentiated tumors and malignant progression [45, 46].
In contrast to GATA3 and FOXA1, the ZNF217 gene is amplified at 20q13 in ~20% of breast tumors and is associated with aggressive breast disease [1, 3, 49]. Our recent studies show the overexpression of ZNF217 promotes cell plasticity particularly in the maintenance of the de-differentiated stem-like cell state, and accelerates tumor progression and metastasis [3]. Our findings that ZNF217 interacts physically with ERα and that ZNF217 occupies chromatin regions also occupied by ERα, GATA3, and FOXA1 will require further investigation to understand whether ZNF217 is one of the multiple factors that contribute to ERα chromatin binding and ERα gene regulation.
Data-mining of microarray expression data from primary breast tumors and the corresponding clinical data showed high ZNF217 expression correlates with shorter overall survival and relapse-free survival [3]. Our current studies demonstrate a high correlation between ZNF217 and ERα expression in breast cancer cell lines and breast tumor tissue, yet the precise role of ZNF217 in ER+ breast cancer is not fully understood. Studies conducted over 10 years ago using microarray technology revealed tumor heterogeneity at the gene expression level and led to the identification of breast cancer subtypes [48, 50, 51]. The ER+ subtypes, referred to as Luminal A and Luminal B, have different gene-expression profiles, prognosis and treatment responses [48, 50–52]. When compared to the Luminal A subtype, Luminal B tumors often have lower expression levels of ERα or ER-regulated genes, higher tumor grade, activation of growth factor receptor signaling pathways and reduced sensitivity to chemotherapy than Luminal A tumors [53]. Despite the divisions between Luminal A and B subtypes, the heterogeneity found in ER+/−and luminal tumors suggests further stratification is necessary to enable more accurate prognosis and treatment plan for individual patients [53]. Our findings suggest that ZNF217 overexpression in ER+, Luminal A, or other subtypes of breast cancer will identify a subset of patients with worse prognosis who will benefit from more aggressive or alternative therapy. The identification of the ZNF217-ER gene signature aligning with multiple breast cancer studies further supports a transcriptional role for ZNF217 in aberrant ERα signaling (Figure 5). Future investigations will explore the connection between ZNF217 expression levels in breast tumors with clinical outcome and, importantly, whether ZNF217 plays a transcriptional role in aberrant ERα signaling, contributing to breast cancer and therapy resistance.
Conclusions
In this genome-wide study we show the distribution of ZNF217 binding events coincide with GATA3, FOXA1, and ERα binding. Motif analysis and co-immunoprecipitation assays implicate ZNF217 in a co-regulatory role with ERα at co-bound regions. The understanding of the chromatin landscape at ZNF217-bound regions is beginning to provide insight into a relationship between ZNF217 and ERα to modulate enhancer accessibility, ERα-binding potential, and gene regulation. Additional studies are underway to explore the requirement of ZNF217 in ERα gene regulation at co-bound sites prior to estrogen stimulation. Our findings contribute to the genome-wide potential of ZNF217 in the hierarchy of ERα-mediated regulation by multiple transcription factors and provide a possible mechanism by which ZNF217 contributes to poor prognosis in breast cancer patients.
Methods
Cell culture
The MCF7 breast cancer cell line was obtained from American Type Culture Collection and cultured in Dulbecco’s Modified Eagle’s Medium supplemented with 10% fetal bovine serum, 2 mM L-glutamine and 1% penicillin/streptomycin.
Primary human tissue specimens
Frozen human breast tumor specimens were provided by the UCD Cancer Center Specimen Repository and the NCI Cooperative Human Tissue Network. All samples were de-identified, and the study was approved by the Institutional Review Board of the UCD School of Medicine. Frozen tissues were homogenized in ice-cold T-PER Tissue Protein Extraction Reagent (Pierce) with protease and phosphatase inhibitors, and then centrifuged to remove insoluble debris.
siRNA-mediated silencing
MCF7 cells were reverse-transfected at a density of 5 × 105 per well in 6-well culture plates. Transfection was performed using 100 nM siRNA targeting ZNF217 (OnTarget plus SMARTpool, Dharmacon, cat # L-004987-01) or non-targeting pool (OnTarget plus non-targeting pool, Dharmacon, cat # D-001810-10) using RNAiMax (Invitrogen) per the manufacturer’s instructions. RNA was collected at 48 h post-transfection.
RNA isolation and real-time qPCR
RNA samples for RNA-seq or cDNA synthesis were purified using a commercial kit (Qiagen; RNAeasy Kit). RNA (2.5ug) was converted to cDNA using the High Capacity cDNA Reverse Transcription Synthesis Kit (Applied Biosystems). Analyses were performed using TaqMan Gene Expression Assays (Applied Biosystems; listed in Additional file 15: Supplemental Methods) on the BioRad CFX 9600 thermocycler.
Co-immunoprecipitation assays
A total of 1x107 MCF7 cells were grown in complete medium and harvested after washing with PBS. The cellular nuclear extracts were isolated with a Nuclear Extraction Kit and provided protocol (Affymetrix cat #AY2002). Immunoprecipitation was performed with 5 ug of antibody or negative IgG control from rabbit on pre-cleared nuclear extracts. After overnight incubation, protein G agarose beads were added for 2 h. The beads were washed in co-IP buffer (20 mM Tris ph 7.5,150 mM NaCL, 1% NP40,10% glycerol,1 mM MgCl2), boiled 5 min at 95°C and run on an 8% acrylamide gel. Antibodies: ZNF217 column-purified rabbit polyclonal [4], ERα (Santa Cruz Biotechnology; cat #scbt-543), CtBP2 (BD Transduction cat #612044), and IgG from rabbit serum (Sigma cat #15006).
ChIP-seq assays
Chromatin immunoprecipitation (ChIP) was performed following the method previously described [54]. Briefly, crosslinked chromatin was collected from MCF7 cells asynchronously grown in full media at 80% confluence. ZNF217 antibody (previously described [4]), H3K4me3, H3K9me3, or H3K27me3 antibodies were incubated with sonicated chromatin overnight. Libraries were constructed and analyzed using an Illumina Hiseq2000. ChIP-seq experiments were performed in duplicate using MCF7 cells grown on different days using independently performed ChIP assays. Replicate input chromatin samples were also included as a control. Additional ChIP assays were performed for ERα (Santa Cruz Biotechnology; cat #scbt8002) and FOXA1 (Abcam #23738). ChIP-qPCR was performed using iQ SYBR Green Supermix (BioRad). Primers used to validate TF binding are listed in Additional file 15: Supplementary Methods.
ChIP-seq data processing
All ChIP-seq datasets were processed by first aligning sequence reads to the hg19 genome using Bowtie2 [55]. We obtained the raw read data files for the ChIP-seq experiments corresponding to p300, ERα, FOXA1 and GATA3 from asynchronously grown MCF7 cells from previously published studies [41, 56, 57]. H3K27ac and H3K4me1 ChIP-seq data generated in asynchronously grown MCF7 cells was obtained from [57]. All transcription factor and histone modification peaks were called using Sole-Search software using sequenced MCF7 input as a control [12, 58]. For ZNF217, we used the ENCODE overlap rules to evaluate the reproducibility of the two biological replicates for each factor or histone modification and cell-type combination [13]. For this, we first truncated the peak lists of the two replicates for a given factor/cell-type combination so that both the A and B replicate peak list were the same length. Then, we overlapped the top 40% of the replicate A peak list with the entire replicate B peak list (and vice versa). ENCODE standards state that approximately 80% of the top 40% set should be contained in the larger set. After determining that replicate datasets met this standard, we merged the two replicates and called peaks on the merged dataset.
TF Co-association
Co-occurance analysis to study the overlap of ZNF217 with other transcription factor binding sites was performed using the Aggregation and Correlation Tool (ACT Tool) [59]. The signal files corresponding all MCF7 transcription factor binding regions were obtained from the ENCODE project (https://genome.ucsc.edu/ENCODE/downloads.html). Using these signal tracks and peak lists for transcription factor we used the ACT tool to perform the signal aggregation over each peak list. The resulting matrix was clustered using Cluster3.0 [60] using a Pearson Correlation distance metric and was visualized in R.
RNA-seq
RNA samples from triplicate biological replicates of MCF7 cells treated with scrambled siRNA or siZNF217 were extracted using the RNAeasy Kit (Qiagen). 2 ug of RNA samples were processed with the TruSeq RNA Sample Prep Kit (Illumina) to make RNA libraries following the Low-Throughput protocol. Libraries were prepared and sequenced using the HiSeq2500 platform at the UC Davis DNA Technologies Core Facility (http://dnatech.genomecenter.ucdavis.edu).
RNA-seq data processing
RNA-seq data was processed using TopHat and Cufflinks [61, 62] as described [63].
Additional supporting data
All data are publicly available via the UCSC Genome ENCODE Browser and/or has been submitted to the Gene Expression Omnibus. The NCBI accession number for the RNAseq data is GSE58326. http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE58326. The NCBI accession number for the ZNF217 ChIPseq data is GSM935563.
Electronic supplementary material
Additional file 1: Table S1: ZNF217 binding sites from merged ChIP-seq datasets. ZNF217 peaks were called using Sole-Search peak calling software [12, 58] on replicate datasets (see Methods for details). A total of 18,965 ZNF217 binding events are listed. (XLS 2 MB)
Additional file 2: Table S2: ZNF217-associated genes (6,965) from ChIP-seq binding sites. (XLS 432 KB)
Additional file 3: Figure S1: ChIP-qPCR confirming TF binding sites identified by ChIP-seq. (A) Panel of ZNF217-bound distal sites confirmed by ChIP-qPCR. (B) Relative TF ChIP enrichment at the ERα target genes ERBB3, TFF1, and LRIG1. ChIP assays were performed on two independent biological replicates using the following antibodies ZNF217, ERα and FOXA1. IgG was used as a control. Relative DNA enrichment was calculated relative to DNA input and using the non-target ZNF10 locus as a negative control. (PDF 337 KB)
Additional file 4: Figure S2: Location Analysis of ZNF217 epigenomic clusters. Location analysis of the cluster I region and cluster II region from Figure 1B. The fraction of the ZNF217 binding sites found in cluster I (top chart) or cluster II (below chart) relative to Refseq genes is shown. (PDF 365 KB)
Additional file 5: Table S3: GREAT annotation of top 5 k ZNF217-associated genes. Table showing GO Biological Process category from gene analysis using the GREAT web tool. (XLSX 22 KB)
Additional file 6: Figure S3: de novo ZNF217 motif. Identification of de novo motif for ZNF217 binding sites in MCF7 cells. HOMER de novo motif analysis using the central most 100 base pairs of all 18,965 ChIP-seq binding regions reveal the indicated motif (shown in both forward and reverse complement). (PDF 389 KB)
Additional file 7: Figure S4: Venn diagram for ZNF217 and ER binding overlap. Overlap analysis of ZNF217 and ERα ChIP-seq binding sites in MCF7 cells. The Venn diagram illustrates the total number of genomic regions shared between these two factors (overlapping by at least 1 base pair). The total number of identified sites is indicated below each factor’s name. (PDF 355 KB)
Additional file 8: Table S4: 4-way Venn Diagram Gene Overlap for multiple TF binding. sites. Excel file contains multiple worksheets listing the ZNF217, ERα, FOXA1 and GATA3-associated genes and co-bound genes for TF combinations. (XLSX 13 MB)
Additional file 9: Figure S5: Validation of ZNF217 knockdown for RNA-sequencing. MCF7 cells were reverse transfected with scrambled or ZNF217 siRNA for 48 h. Cells were collected from each well and split into two samples for RNA isolation and protein lysate. (A) Protein lysates from triplicate samples were immunoblotted for ZNF217 or actin as a loading control. Chemoluminescence was analyzed on an Alpha-Innotech Imaging documentation system. (B) RNA samples were converted to cDNA and quantitative RT-PCR performed using ABI expression assay Taqman probes for ZNF217 and GAPDH. Each sample was assayed in triplicate and the cT values were normalized to GAPDH. Average relative transcript level was graphed using BioRad CFX software. Columns: transcript levels, error bars: standard error of the mean. (PDF 651 KB)
Additional file 10: Table S5: Differentially expressed genes from RNA-seq. List of the 3,402 differentially expressed genes; 1,615 genes were up-regulated and 1,787 were down-regulated genes in the ZNF217 knockdown sample, as compared to the control siRNA-treated sample (at 1.25 -fold change cutoff and criteria representing statistical significance, false discovery rate (FDR) < 0.0001. (XLS 992 KB)
Additional file 11: Figure S6: Integrated Genome Browser snapshots of 4 genes bound by ZNF217, ERα, FOXA1 and GATA3. Co-bound regions are shown for LRIG1, AXIN1, SLC22A5, and IGFBP4. (PDF 489 KB)
Additional file 12: Table S6: MSigDB cancer studies for 1,230 and 854 gene lists. Worksheets with complete list of all MSigDB cancer studies resulting from GREAT analysis for ZNF217-bound genes versus ZNF217-ER co-bound genes. (XLSX 17 KB)
Additional file 13: Figure S7: ERα co-immunoprecipitation with ZNF217 and CtBP2. Co-immunoprecipitation of endogenous ZNF217, ERα and CtBP2 from MCF7 nuclear extracts. Immunoprecipitation experiments with ERα antibody to co-IP ZNF217 were blotted with corresponding antibodies. CtBP2 is a ZNF217-binding partner. Input from nuclear extracts is shown in the two left lanes. (PDF 99 KB)
Additional file 14: Figure S8: Overall Survival in ER- and Basal breast cancer patient cohorts. A) Overall survival of basal subtype patients (n = 30) based on ZNF217 expression from primary tumors for low (n = 19) versus high (n = 11) ZNF217 expression (P = 0.01; Logrank). B). Overall survival in ER- patients based on ZNF217 expression for low ZNF217 (n = 24) versus high ZNF217 (n = 17) (P = 0.005; Logrank). Data was sorted first by ZNF217 expression using the median cutoff. Then the data was sorted to identify the ZNF217 high patients and the ZNF217 low patients who were ER- or basal patients. Expression Data from Chin et al., [36]. (PDF 27 KB)
Additional file 15: Supplementary Methods: Primers used to validate TF binding sites and Taqman Assays for RNA-seq confirmations. (XLSX 13 KB)
Acknowledgements
This work was supported by the Department of Defense Breast Cancer Research Program BC101740 to S. Krig; S. Frietze was supported by 1U54HG004558 as a component of the ENCODE Project from the National Cancer Institute to P. Farnham; L. Littlepage was supported by R01 CA129523 and by Simon-Harper Inter-Institutional Research Team (SHIIRT) Grant; C. Simion was supported by RO1 CA118384 to C. Sweeney.
Abbreviations
- ZNF217
zinc finger protein #217
- ER/ERα
Estrogen receptor alpha
- GATA3
GATA binding protein 3
- FOXA1
forkhead box A1
- H3K27ac
Histone H3 acetylated on Lysine 27
- H3K4me1
Histone H3 monomethylated on Lysine 4
- LSD1
Histone demethylase
- HDAC
Histone deacetylase
- Jarid1b/Plu-1
Lysine (K)-specific demethylase
- G9a
Euchromatic histone-lysine N-methyltransferase
- EZH2
Enhancer of zeste homolog 2
- siRNA
Small interfering RNA
- ChIP-qPCR
Chromatin immunoprecipiation-quantitative PCR
- MSigDB
Molecular signature database
- GREAT
Genomic Regions of Enrichment Annotation Tool
- TFs
Transcription factors.
Footnotes
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
All authors have read and approved the manuscript for publication. SF carried out genomics analyses, drafted the manuscript, and participated in design and coordination of the study. HO participated in the design and coordination of the study and the genomics analyses. LL interrogated published studies of expression array data from primary breast tumors with matching clinical data and drafted the manuscript. CS performed molecular biology experiments. CAS participated in the analyses of translational impact; Kaplan Meier plots on breast cancer cohorts. PJF coordinated the ChIP-sequencing genome analyses of ZNF217 for ENCODE submission. SK oversaw the conception and execution of the study, carried out molecular biology experiments and drafted the manuscript.
Contributor Information
Seth Frietze, Email: seth.frietze@unco.edu.
Henriette O’Geen, Email: hhorvath@ucdavis.edu.
Laurie E Littlepage, Email: laurie.littlepage@nd.edu.
Catalina Simion, Email: csimion@ucdavis.edu.
Colleen A Sweeney, Email: casweeney@ucdavis.edu.
Peggy J Farnham, Email: peggy.farnham@med.usc.edu.
Sheryl R Krig, Email: srkrig@ucdavis.edu.
References
- 1.Tanner MM, Tirkkonen M, Kallioniemi A, Holli K, Collins C, Kowbel D, Gray JW, Kallioniemi OP, Isola J. Amplification of chromosomal region 20q13 in invasive breast cancer: prognostic implications. Clin Cancer Res. 1995;1:1455–1461. [PubMed] [Google Scholar]
- 2.Werner M, Mattis A, Aubele M, Cummings M, Zitzelsberger H, Hutzler P, Hofler H. 20q13.2 amplification in intraductal hyperplasia adjacent to in situ and invasive ductal carcinoma of the breast. Virchows Arch. 1999;435:469–472. doi: 10.1007/s004280050429. [DOI] [PubMed] [Google Scholar]
- 3.Littlepage LE, Adler AS, Kouros-Mehr H, Huang G, Chou J, Krig SR, Griffith OL, Korkola JE, Qu K, Lawson DA, Xue Q, Sternlicht MD, Dijkgraaf GJ, Yaswen P, Rugo HS, Sweeney CA, Collins CC, Gray JW, Chang HY, Werb Z. The transcription factor ZNF217 is a prognostic biomarker and therapeutic target during breast cancer progression. Cancer Discov. 2012;2:638–651. doi: 10.1158/2159-8290.CD-12-0093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Krig SR, Jin VX, Bieda MC, O'Geen H, Yaswen P, Green R, Farnham PJ. Identification of genes directly regulated by the oncogene ZNF217 using chromatin immunoprecipitation (ChIP)-chip assays. J Biol Chem. 2007;282:9703–9712. doi: 10.1074/jbc.M611752200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Krig SR, Miller JK, Frietze S, Beckett LA, Neve RM, Farnham PJ, Yaswen PI, Sweeney CA. ZNF217, a candidate breast cancer oncogene amplified at 20q13, regulates expression of the ErbB3 receptor tyrosine kinase in breast cancer cells. Oncogene. 2010;29:5500–5510. doi: 10.1038/onc.2010.289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Vendrell JA, Thollet A, Nguyen NT, Ghayad SE, Vinot S, Bieche I, Grisard E, Josserand V, Coll JL, Roux P, Corbo L, Treilleux I, Rimokh R, Cohen PA. ZNF217 is a marker of poor prognosis in breast cancer that drives epithelial-mesenchymal transition and invasion. Cancer Res. 2012;72:3593–3606. doi: 10.1158/0008-5472.CAN-11-3095. [DOI] [PubMed] [Google Scholar]
- 7.Quinlan KG, Nardini M, Verger A, Francescato P, Yaswen P, Corda D, Bolognesi M, Crossley M. Specific recognition of ZNF217 and other zinc finger proteins at a surface groove of C-terminal binding proteins. Mol Cell Biol. 2006;26:8159–8172. doi: 10.1128/MCB.00680-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Thillainadesan G, Isovic M, Loney E, Andrews J, Tini M, Torchia J. Genome analysis identifies the p15ink4b tumor suppressor as a direct target of the ZNF217/CoREST complex. Mol Cell Biol. 2008;28:6066–6077. doi: 10.1128/MCB.00246-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.You A, Tong JK, Grozinger CM, Schreiber SL. CoREST is an integral component of the CoREST- human histone deacetylase complex. Proc Natl Acad Sci U S A. 2001;98:1454–1458. doi: 10.1073/pnas.98.4.1454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shi Y, Sawada J, Sui G, Affar el B, Whetstine JR, Lan F, Ogawa H, Luke MP, Nakatani Y. Coordinated histone modifications mediated by a CtBP co-repressor complex. Nature. 2003;422:735–738. doi: 10.1038/nature01550. [DOI] [PubMed] [Google Scholar]
- 11.Banck MS, Li S, Nishio H, Wang C, Beutler AS, Walsh MJ. The ZNF217 oncogene is a candidate organizer of repressive histone modifiers. Epigenetics. 2009;4:100–106. doi: 10.4161/epi.4.2.7953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Blahnik KR, Dou L, O'Geen H, McPhillips T, Xu X, Cao AR, Iyengar S, Nicolet CM, Ludascher B, Korf I, Farnham PJ. Sole-Search: an integrated analysis program for peak detection and functional annotation using ChIP-seq data. Nucleic Acids Res. 2010;38:e13. doi: 10.1093/nar/gkp1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Landt SG, Marinov GK, Kundaje A, Kheradpour P, Pauli F, Batzoglou S, Bernstein BE, Bickel P, Brown JB, Cayting P, Chen Y, DeSalvo G, Epstein C, Fisher-Aylor KI, Euskirchen G, Gerstein M, Gertz J, Hartemink AJ, Hoffman MM, Iyer VR, Jung YL, Karmakar S, Kellis M, Kharchenko PV, Li Q, Liu T, Liu XS, Ma L, Milosavljevic A, Myers RM, et al. ChIP-seq guidelines and practices of the ENCODE and modENCODE consortia. Genome Res. 2012;22:1813–1831. doi: 10.1101/gr.136184.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Heintzman ND, Hon GC, Hawkins RD, Kheradpour P, Stark A, Harp LF, Ye Z, Lee LK, Stuart RK, Ching CW, Ching KA, Antosiewicz-Bourget JE, Liu H, Zhang X, Green RD, Lobanenkov VV, Stewart R, Thomson JA, Crawford GE, Kellis M, Ren B. Histone modifications at human enhancers reflect global cell-type-specific gene expression. Nature. 2009;459:108–112. doi: 10.1038/nature07829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Heintzman ND, Stuart RK, Hon G, Fu Y, Ching CW, Hawkins RD, Barrera LO, Van Calcar S, Qu C, Ching KA, Wang W, Weng Z, Green RD, Crawford GE, Ren B. Distinct and predictive chromatin signatures of transcriptional promoters and enhancers in the human genome. Nat Genet. 2007;39:311–318. doi: 10.1038/ng1966. [DOI] [PubMed] [Google Scholar]
- 16.McLean CY, Bristor D, Hiller M, Clarke SL, Schaar BT, Lowe CB, Wenger AM, Bejerano G. GREAT improves functional interpretation of cis-regulatory regions. Nat Biotechnol. 2010;28:495–501. doi: 10.1038/nbt.1630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, Cheng JX, Murre C, Singh H, Glass CK. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol Cell. 2010;38:576–589. doi: 10.1016/j.molcel.2010.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Carroll JS, Meyer CA, Song J, Li W, Geistlinger TR, Eeckhoute J, Brodsky AS, Keeton EK, Fertuck KC, Hall GF, Wang Q, Bekiranov S, Sementchenko V, Fox EA, Silver PA, Gingeras TR, Liu XS, Brown M. Genome-wide analysis of estrogen receptor binding sites. Nat Genet. 2006;38:1289–1297. doi: 10.1038/ng1901. [DOI] [PubMed] [Google Scholar]
- 19.Webb P, Lopez GN, Uht RM, Kushner PJ. Tamoxifen activation of the estrogen receptor/AP-1 pathway: potential origin for the cell-specific estrogen-like effects of antiestrogens. Mol Endocrinol. 1995;9:443–456. doi: 10.1210/mend.9.4.7659088. [DOI] [PubMed] [Google Scholar]
- 20.Uht RM, Anderson CM, Webb P, Kushner PJ. Transcriptional activities of estrogen and glucocorticoid receptors are functionally integrated at the AP-1 response element. Endocrinology. 1997;138:2900–2908. doi: 10.1210/endo.138.7.5244. [DOI] [PubMed] [Google Scholar]
- 21.Tang Z, Treilleux I, Brown M. A transcriptional enhancer required for the differential expression of the human estrogen receptor in breast cancers. Mol Cell Biol. 1997;17:1274–1280. doi: 10.1128/mcb.17.3.1274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zaret KS, Carroll JS. Pioneer transcription factors: establishing competence for gene expression. Genes Dev. 2011;25:2227–2241. doi: 10.1101/gad.176826.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Beck S, Sommer P, dos Santos Silva E, Blin N, Gott P. Hepatocyte nuclear factor 3 (winged helix domain) activates trefoil factor gene TFF1 through a binding motif adjacent to the TATAA box. DNA Cell Biol. 1999;18:157–164. doi: 10.1089/104454999315547. [DOI] [PubMed] [Google Scholar]
- 24.Krig SR, Frietze S, Simion C, Miller JK, Fry WH, Rafidi H, Kotelawala L, Qi L, Griffith OL, Gray JW, Carraway KL, III, Sweeney C. Lrig1 is an estrogen-regulated growth suppressor and correlates with longer relapse-free survival in ERalpha-positive breast cancer. Mol Cancer Res. 2011;9:1406–1417. doi: 10.1158/1541-7786.MCR-11-0227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang C, Uray IP, Mazumdar A, Mayer JA, Brown PH. SLC22A5/OCTN2 expression in breast cancer is induced by estrogen via a novel intronic estrogen-response element (ERE) Breast Cancer Res Treat. 2012;134:101–115. doi: 10.1007/s10549-011-1925-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Liberzon A, Subramanian A, Pinchback R, Thorvaldsdottir H, Tamayo P, Mesirov JP. Molecular signatures database (MSigDB) 3.0. Bioinformatics. 2011;27:1739–1740. doi: 10.1093/bioinformatics/btr260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Massarweh S, Osborne CK, Creighton CJ, Qin L, Tsimelzon A, Huang S, Weiss H, Rimawi M, Schiff R. Tamoxifen resistance in breast tumors is driven by growth factor receptor signaling with repression of classic estrogen receptor genomic function. Cancer Res. 2008;68:826–833. doi: 10.1158/0008-5472.CAN-07-2707. [DOI] [PubMed] [Google Scholar]
- 28.Charafe-Jauffret E, Ginestier C, Monville F, Finetti P, Adelaide J, Cervera N, Fekairi S, Xerri L, Jacquemier J, Birnbaum D, Bertucci F. Gene expression profiling of breast cell lines identifies potential new basal markers. Oncogene. 2006;25:2273–2284. doi: 10.1038/sj.onc.1209254. [DOI] [PubMed] [Google Scholar]
- 29.Creighton CJ, Massarweh S, Huang S, Tsimelzon A, Hilsenbeck SG, Osborne CK, Shou J, Malorni L, Schiff R. Development of resistance to targeted therapies transforms the clinically associated molecular profile subtype of breast tumor xenografts. Cancer Res. 2008;68:7493–7501. doi: 10.1158/0008-5472.CAN-08-1404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Engelmann K, Shen H, Finn OJ. MCF7 side population cells with characteristics of cancer stem/progenitor cells express the tumor antigen MUC1. Cancer Res. 2008;68:2419–2426. doi: 10.1158/0008-5472.CAN-07-2249. [DOI] [PubMed] [Google Scholar]
- 31.Elvidge GP, Glenny L, Appelhoff RJ, Ratcliffe PJ, Ragoussis J, Gleadle JM. Concordant regulation of gene expression by hypoxia and 2-oxoglutarate-dependent dioxygenase inhibition: the role of HIF-1alpha, HIF-2alpha, and other pathways. J Biol Chem. 2006;281:15215–15226. doi: 10.1074/jbc.M511408200. [DOI] [PubMed] [Google Scholar]
- 32.Doane AS, Danso M, Lal P, Donaton M, Zhang L, Hudis C, Gerald WL. An estrogen receptor-negative breast cancer subset characterized by a hormonally regulated transcriptional program and response to androgen. Oncogene. 2006;25:3994–4008. doi: 10.1038/sj.onc.1209415. [DOI] [PubMed] [Google Scholar]
- 33.Nagashima T, Shimodaira H, Ide K, Nakakuki T, Tani Y, Takahashi K, Yumoto N, Hatakeyama M. Quantitative transcriptional control of ErbB receptor signaling undergoes graded to biphasic response for cell differentiation. J Biol Chem. 2007;282:4045–4056. doi: 10.1074/jbc.M608653200. [DOI] [PubMed] [Google Scholar]
- 34.Jozwik KM, Carroll JS. Pioneer factors in hormone-dependent cancers. Nat Rev Cancer. 2012;12:381–385. doi: 10.1038/nrc3263. [DOI] [PubMed] [Google Scholar]
- 35.Mohammed H, D'Santos C, Serandour AA, Ali HR, Brown GD, Atkins A, Rueda OM, Holmes KA, Theodorou V, Robinson JL, Zwart W, Saadi A, Ross-Innes CS, Chin SF, Menon S, Stingl J, Palmieri C, Caldas C, Carroll JS. Endogenous purification reveals GREB1 as a key estrogen receptor regulatory factor. Cell Rep. 2013;3:342–349. doi: 10.1016/j.celrep.2013.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chin K, DeVries S, Fridlyand J, Spellman PT, Roydasgupta R, Kuo WL, Lapuk A, Neve RM, Qian Z, Ryder T, Chen F, Feiler H, Tokuyasu T, Kingsley C, Dairkee S, Meng Z, Chew K, Pinkel D, Jain A, Ljung BM, Esserman L, Albertson DG, Waldman FM, Gray JW. Genomic and transcriptional aberrations linked to breast cancer pathophysiologies. Cancer Cell. 2006;10:529–541. doi: 10.1016/j.ccr.2006.10.009. [DOI] [PubMed] [Google Scholar]
- 37.Hess KR, Anderson K, Symmans WF, Valero V, Ibrahim N, Mejia JA, Booser D, Theriault RL, Buzdar AU, Dempsey PJ, Rouzier R, Sneige N, Ross JS, Vidaurre T, Gómez HL, Hortobagyi GN, Pusztai L. Pharmacogenomic predictor of sensitivity to preoperative chemotherapy with paclitaxel and fluorouracil, doxorubicin, and cyclophosphamide in breast cancer. J Clin Oncol. 2006;24:4236–4244. doi: 10.1200/JCO.2006.05.6861. [DOI] [PubMed] [Google Scholar]
- 38.Gyorffy B, Lanczky A, Eklund AC, Denkert C, Budczies J, Li Q, Szallasi Z. An online survival analysis tool to rapidly assess the effect of 22,277 genes on breast cancer prognosis using microarray data of 1,809 patients. Breast Cancer Res Treat. 2010;123:725–731. doi: 10.1007/s10549-009-0674-9. [DOI] [PubMed] [Google Scholar]
- 39.Eeckhoute J, Carroll JS, Geistlinger TR, Torres-Arzayus MI, Brown M. A cell-type-specific transcriptional network required for estrogen regulation of cyclin D1 and cell cycle progression in breast cancer. Genes Dev. 2006;20:2513–2526. doi: 10.1101/gad.1446006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Carroll JS, Liu XS, Brodsky AS, Li W, Meyer CA, Szary AJ, Eeckhoute J, Shao W, Hestermann EV, Geistlinger TR, Fox EA, Silver PA, Brown M. Chromosome-wide mapping of estrogen receptor binding reveals long-range regulation requiring the forkhead protein FoxA1. Cell. 2005;122:33–43. doi: 10.1016/j.cell.2005.05.008. [DOI] [PubMed] [Google Scholar]
- 41.Hurtado A, Holmes KA, Ross-Innes CS, Schmidt D, Carroll JS. FOXA1 is a key determinant of estrogen receptor function and endocrine response. Nat Genet. 2011;43:27–33. doi: 10.1038/ng.730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Theodorou V, Stark R, Menon S, Carroll JS. GATA3 acts upstream of FOXA1 in mediating ESR1 binding by shaping enhancer accessibility. Genome Res. 2013;23:12–22. doi: 10.1101/gr.139469.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Eeckhoute J, Keeton EK, Lupien M, Krum SA, Carroll JS, Brown M. Positive cross-regulatory loop ties GATA-3 to estrogen receptor alpha expression in breast cancer. Cancer Res. 2007;67:6477–6483. doi: 10.1158/0008-5472.CAN-07-0746. [DOI] [PubMed] [Google Scholar]
- 44.Ross-Innes CS, Stark R, Teschendorff AE, Holmes KA, Ali HR, Dunning MJ, Brown GD, Gojis O, Ellis IO, Green AR, Ali S, Chin SF, Palmieri C, Caldas C, Carroll JS. Differential oestrogen receptor binding is associated with clinical outcome in breast cancer. Nature. 2012;481(7381):389–393. doi: 10.1038/nature10730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kouros-Mehr H, Kim JW, Bechis SK, Werb Z. GATA-3 and the regulation of the mammary luminal cell fate. Curr Opin Cell Biol. 2008;20:164–170. doi: 10.1016/j.ceb.2008.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bernardo GM, Keri RA. FOXA1: a transcription factor with parallel functions in development and cancer. Biosci Rep. 2012;32:113–130. doi: 10.1042/BSR20110046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bernardo GM, Lozada KL, Miedler JD, Harburg G, Hewitt SC, Mosley JD, Godwin AK, Korach KS, Visvader JE, Kaestner KH, Abdul-Karim FW, Montano MM, Keri RA. FOXA1 is an essential determinant of ERalpha expression and mammary ductal morphogenesis. Development. 2010;137:2045–2054. doi: 10.1242/dev.043299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Perou CM, Sorlie T, Eisen MB, van de Rijn M, Jeffrey SS, Rees CA, Pollack JR, Ross DT, Johnsen H, Akslen LA, Fluge O, Pergamenschikov A, Williams C, Zhu SX, Lønning PE, Børresen-Dale AL, Brown PO, Botstein D. Molecular portraits of human breast tumours. Nature. 2000;406:747–752. doi: 10.1038/35021093. [DOI] [PubMed] [Google Scholar]
- 49.Collins C, Rommens JM, Kowbel D, Godfrey T, Tanner M, Hwang SI, Polikoff D, Nonet G, Cochran J, Myambo K, Jay KE, Froula J, Cloutier T, Kuo WL, Yaswen P, Dairkee S, Giovanola J, Hutchinson GB, Isola J, Kallioniemi OP, Palazzolo M, Martin C, Ericsson C, Pinkel D, Albertson D, Li WB, Gray JW. Positional cloning of ZNF217 and NABC1: genes amplified at 20q13.2 and overexpressed in breast carcinoma. Proc Natl Acad Sci U S A. 1998;95:8703–8708. doi: 10.1073/pnas.95.15.8703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sorlie T, Perou CM, Tibshirani R, Aas T, Geisler S, Johnsen H, Hastie T, Eisen MB, van de Rijn M, Jeffrey SS, Thorsen T, Quist H, Matese JC, Brown PO, Botstein D, Lønning PE, Børresen-Dale AL. Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proc Natl Acad Sci U S A. 2001;98:10869–10874. doi: 10.1073/pnas.191367098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sotiriou C, Neo SY, McShane LM, Korn EL, Long PM, Jazaeri A, Martiat P, Fox SB, Harris AL, Liu ET. Breast cancer classification and prognosis based on gene expression profiles from a population-based study. Proc Natl Acad Sci U S A. 2003;100:10393–10398. doi: 10.1073/pnas.1732912100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Creighton CJ. The molecular profile of luminal B breast cancer. Biologics. 2012;6:289–297. doi: 10.2147/BTT.S29923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ignatiadis M, Sotiriou C. Luminal breast cancer: from biology to treatment. Nat Rev Clin Oncol. 2013;10:494–506. doi: 10.1038/nrclinonc.2013.124. [DOI] [PubMed] [Google Scholar]
- 54.O'Geen H, Nicolet CM, Blahnik K, Green R, Farnham PJ. Comparison of sample preparation methods for ChIP-chip assays. Biotechniques. 2006;41:577–580. doi: 10.2144/000112268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Langmead B, Trapnell C, Pop M, Salzberg SL. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10:R25. doi: 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Tropberger P, Pott S, Keller C, Kamieniarz-Gdula K, Caron M, Richter F, Li G, Mittler G, Liu ET, Buhler M, Margueron R, Schneider R. Regulation of transcription through acetylation of H3K122 on the lateral surface of the histone octamer. Cell. 2013;152:859–872. doi: 10.1016/j.cell.2013.01.032. [DOI] [PubMed] [Google Scholar]
- 57.Frietze S, Wang R, Yao L, Tak YG, Ye Z, Gaddis M, Witt H, Farnham PJ, Jin VX. Cell type-specific binding patterns reveal that TCF7L2 can be tethered to the genome by association with GATA3. Genome Biol. 2012;13:R52. doi: 10.1186/gb-2012-13-9-r52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Blahnik KR, Dou L, Echipare L, Iyengar S, O'Geen H, Sanchez E, Zhao Y, Marra MA, Hirst M, Costello JF, Korf I, Farnham PJ. Characterization of the contradictory chromatin signatures at the 3' exons of zinc finger genes. PLoS One. 2011;6:e17121. doi: 10.1371/journal.pone.0017121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Jee J, Rozowsky J, Yip KY, Lochovsky L, Bjornson R, Zhong G, Zhang Z, Fu Y, Wang J, Weng Z, Gerstein M. ACT: aggregation and correlation toolbox for analyses of genome tracks. Bioinformatics. 2011;27:1152–1154. doi: 10.1093/bioinformatics/btr092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.de Hoon MJ, Imoto S, Nolan J, Miyano S. Open source clustering software. Bioinformatics. 2004;20:1453–1454. doi: 10.1093/bioinformatics/bth078. [DOI] [PubMed] [Google Scholar]
- 61.Trapnell C, Williams BA, Pertea G, Mortazavi A, Kwan G, van Baren MJ, Salzberg SL, Wold BJ, Pachter L. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol. 2010;28:511–515. doi: 10.1038/nbt.1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Trapnell C, Pachter L, Salzberg SL. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics. 2009;25:1105–1111. doi: 10.1093/bioinformatics/btp120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Trapnell C, Roberts A, Goff L, Pertea G, Kim D, Kelley DR, Pimentel H, Salzberg SL, Rinn JL, Pachter L. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat Protoc. 2012;7:562–578. doi: 10.1038/nprot.2012.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional file 1: Table S1: ZNF217 binding sites from merged ChIP-seq datasets. ZNF217 peaks were called using Sole-Search peak calling software [12, 58] on replicate datasets (see Methods for details). A total of 18,965 ZNF217 binding events are listed. (XLS 2 MB)
Additional file 2: Table S2: ZNF217-associated genes (6,965) from ChIP-seq binding sites. (XLS 432 KB)
Additional file 3: Figure S1: ChIP-qPCR confirming TF binding sites identified by ChIP-seq. (A) Panel of ZNF217-bound distal sites confirmed by ChIP-qPCR. (B) Relative TF ChIP enrichment at the ERα target genes ERBB3, TFF1, and LRIG1. ChIP assays were performed on two independent biological replicates using the following antibodies ZNF217, ERα and FOXA1. IgG was used as a control. Relative DNA enrichment was calculated relative to DNA input and using the non-target ZNF10 locus as a negative control. (PDF 337 KB)
Additional file 4: Figure S2: Location Analysis of ZNF217 epigenomic clusters. Location analysis of the cluster I region and cluster II region from Figure 1B. The fraction of the ZNF217 binding sites found in cluster I (top chart) or cluster II (below chart) relative to Refseq genes is shown. (PDF 365 KB)
Additional file 5: Table S3: GREAT annotation of top 5 k ZNF217-associated genes. Table showing GO Biological Process category from gene analysis using the GREAT web tool. (XLSX 22 KB)
Additional file 6: Figure S3: de novo ZNF217 motif. Identification of de novo motif for ZNF217 binding sites in MCF7 cells. HOMER de novo motif analysis using the central most 100 base pairs of all 18,965 ChIP-seq binding regions reveal the indicated motif (shown in both forward and reverse complement). (PDF 389 KB)
Additional file 7: Figure S4: Venn diagram for ZNF217 and ER binding overlap. Overlap analysis of ZNF217 and ERα ChIP-seq binding sites in MCF7 cells. The Venn diagram illustrates the total number of genomic regions shared between these two factors (overlapping by at least 1 base pair). The total number of identified sites is indicated below each factor’s name. (PDF 355 KB)
Additional file 8: Table S4: 4-way Venn Diagram Gene Overlap for multiple TF binding. sites. Excel file contains multiple worksheets listing the ZNF217, ERα, FOXA1 and GATA3-associated genes and co-bound genes for TF combinations. (XLSX 13 MB)
Additional file 9: Figure S5: Validation of ZNF217 knockdown for RNA-sequencing. MCF7 cells were reverse transfected with scrambled or ZNF217 siRNA for 48 h. Cells were collected from each well and split into two samples for RNA isolation and protein lysate. (A) Protein lysates from triplicate samples were immunoblotted for ZNF217 or actin as a loading control. Chemoluminescence was analyzed on an Alpha-Innotech Imaging documentation system. (B) RNA samples were converted to cDNA and quantitative RT-PCR performed using ABI expression assay Taqman probes for ZNF217 and GAPDH. Each sample was assayed in triplicate and the cT values were normalized to GAPDH. Average relative transcript level was graphed using BioRad CFX software. Columns: transcript levels, error bars: standard error of the mean. (PDF 651 KB)
Additional file 10: Table S5: Differentially expressed genes from RNA-seq. List of the 3,402 differentially expressed genes; 1,615 genes were up-regulated and 1,787 were down-regulated genes in the ZNF217 knockdown sample, as compared to the control siRNA-treated sample (at 1.25 -fold change cutoff and criteria representing statistical significance, false discovery rate (FDR) < 0.0001. (XLS 992 KB)
Additional file 11: Figure S6: Integrated Genome Browser snapshots of 4 genes bound by ZNF217, ERα, FOXA1 and GATA3. Co-bound regions are shown for LRIG1, AXIN1, SLC22A5, and IGFBP4. (PDF 489 KB)
Additional file 12: Table S6: MSigDB cancer studies for 1,230 and 854 gene lists. Worksheets with complete list of all MSigDB cancer studies resulting from GREAT analysis for ZNF217-bound genes versus ZNF217-ER co-bound genes. (XLSX 17 KB)
Additional file 13: Figure S7: ERα co-immunoprecipitation with ZNF217 and CtBP2. Co-immunoprecipitation of endogenous ZNF217, ERα and CtBP2 from MCF7 nuclear extracts. Immunoprecipitation experiments with ERα antibody to co-IP ZNF217 were blotted with corresponding antibodies. CtBP2 is a ZNF217-binding partner. Input from nuclear extracts is shown in the two left lanes. (PDF 99 KB)
Additional file 14: Figure S8: Overall Survival in ER- and Basal breast cancer patient cohorts. A) Overall survival of basal subtype patients (n = 30) based on ZNF217 expression from primary tumors for low (n = 19) versus high (n = 11) ZNF217 expression (P = 0.01; Logrank). B). Overall survival in ER- patients based on ZNF217 expression for low ZNF217 (n = 24) versus high ZNF217 (n = 17) (P = 0.005; Logrank). Data was sorted first by ZNF217 expression using the median cutoff. Then the data was sorted to identify the ZNF217 high patients and the ZNF217 low patients who were ER- or basal patients. Expression Data from Chin et al., [36]. (PDF 27 KB)
Additional file 15: Supplementary Methods: Primers used to validate TF binding sites and Taqman Assays for RNA-seq confirmations. (XLSX 13 KB)