

Abstract
Recombinases of the RecA family play vital roles in homologous recombination, a high-fidelity mechanism to repair DNA double-stranded breaks. These proteins catalyze strand invasion and exchange after forming dynamic nucleoprotein filaments on ssDNA. Increasing evidence suggests that stabilization of these dynamic filaments is a highly conserved function across diverse species. Here, we analyze the presynaptic filament formation and DNA binding characteristics of the Sulfolobus solfataricus recombinase SsoRadA in conjunction with the SsoRadA paralog SsoRal1. In addition to constraining SsoRadA ssDNA-dependent ATPase activity, the paralog also enhances SsoRadA ssDNA binding, effectively influencing activities necessary for presynaptic filament formation. These activities result in enhanced SsoRadA-mediated strand invasion in the presence of SsoRal1 and suggest a filament stabilization function for the SsoRal1 protein.
Keywords: Paralog, Presynapsis, Recombinase mutants, Nucleoprotein filament dynamics, SsoRadA, SsoRal1
1. Introduction
DNA double-strand breaks (DSBs) are one of the most deleterious forms of cellular damage and repair of these breaks is essential for genomic integrity. Homologous recombination (HR) is a complex high-fidelity mechanism that repairs DSBs through the concerted action of a number of proteins. Recombinases, including Rad51 in eukaryotes, RecA in bacteria, and RadA in archaea play a central role in DSB repair through HR [1–5]. These proteins assemble into filaments on ssDNA that are competent to search for homologous sequences in order to initiate synapsis and catalyze strand exchange [6,7]. Formation of presynaptic filaments occurs through nucleation and propagation processes characterized by dynamic protein binding, turnover, and disassembly [8]. These events are linked to the ATPase cycle and functional interactions with accessory proteins. Increasing evidence suggests that stabilization of filaments, thereby reducing their dynamic nature, is an important function necessary for efficient HR. The T4 UvsY mediator protein can stabilize the UvsX recombinase through both protein–protein and DNA-mediated interactions [9]. Eukaryotic presynaptic filament stabilization has been linked to a decrease in ATP hydrolysis by the recombinase. In vitro experiments with human Rad51 protein indicate that the addition of factors that reduce ATP turnover, including Ca+2 as well as the recombination mediator protein BRCA2, lead to stabilization of presynaptic filaments [10–12].
Archaea are chimeric microbes that comprise the third branch in the phylogenetic tree of life. While physically resembling bacteria, the mechanisms involved in genomic informational processing are more eukaryotic in nature. HR is one key process in which archaea employ proteins with strong eukaryotic homology. One of the best studied models from this domain is the hyperthermophilic crenarchaeon Sulfolobus solfataricus. The SsoRadA protein has been biochemically characterized and crystallized and, like the other RecA family recombinases, is a ssDNA-dependent ATPase that catalyzes both strand invasion and exchange [2,13]. S. solfataricus also encodes three SsoRadA recombinase paralogs, whose function in HR is not well understood [2,14–16]. In vivo, expression of the genes encoding these paralogs is induced in response to chromosomal damage resulting from exposure to actinomycin D, UV-C, and ionizing radiation, implicating involvement of the proteins in repair of DNA breaks [14,16,17]. The first of these S. solfataricus RadA paralogs to be examined in vitro is encoded by open reading frame Sso2452 in the P2 type-strain. The protein is a member of the aRadC family and has been referred to in the literature as RadB, Sso2452 (the open reading frame designation in the type-strain), and SsoRal1 [14,15,18,19]. The RadB name was widely abandoned following the description of a protein motif present only in euryarchaeal RadB sequences but absent in the crenarchaea [19]. Our laboratory has adopted the name SsoRal1 (RadA-like) to alleviate confusion when referring to this protein in S. solfataricus strains where open reading frame designations are not the same as those used for the P2 type-strain [14]. The SsoRal1 protein has been recently crystallized and shows anti-recombinase activity through inhibition of SsoRadA-mediated strand invasion [15]. To further understand the activities of this paralog, we biochemically examined the SsoRal1 protein from S. solfataricus strain P2-A [14] and its involvement in SsoRadA presynaptic filament formation.
2. Materials and methods
2.1. Expression vector construction and protein purification
The SsoRal1 gene was PCR amplified using genomic DNA from S. solfataricus strain P2 obtained from the ATCC [14] and the primers: SsoRal1F (5′-CGATATTTAAAATTATGGTAAGCC-3′) and SsoRal1R (5′-CGATATTTAAACATATGGTAAGCC-3′). The PCR product was cloned into pET21a (Novagen) at the NdeI and BamHI sites in the polylinker. SsoRadA ATPase mutant expression vectors were constructed using the QuikChange II site-directed mutagenesis kit (Stratagene) following the manufacturer’s protocol. The SsoRadA K120A vector was made using the primers RadAK2AF (5′-CTTCGGTGAGTTTGGGTCTGGTGCCACACAGCTATGTCATCAG-3′) and RadAK2AR (5′-CAGATGACATAGCTGTGTGGCACCAGACCCAAA-CTCACCGAAG-3′) with the pET3a-RadA expression vector [20] as the starting template. The SsoRadA K120R mutant was produced using the primers RadAK2RF (5′-CTTCGGTGAGTTTGGG-TCTGGTCGAACACAGCTATGTCATCAG-3′) and RadAK2RR (5′-CAGA-TGACATAGCTGTGTTCGACCAGACCCAAACTCACCGAAG-3′) using the same template. The mutations were confirmed by DNA sequencing of the expression clones at Amplicon Express, Pullman, WA.
Overexpression of the SsoRal1 protein was accomplished using the CodonPlus strain (Stratagene). Cells carrying the SsoRal1 expression construct were grown in LB medium (10 g tryptone, 5 g yeast extract, 5 g sodium chloride per liter) containing 0.1 mg/mL ampicillin and 0.03 mg/mL chloramphenicol at 37 °C until late log phase. IPTG (Sigma) was added to a final concentration of 1 mM and protein production was permitted to continue for 3 h, after which cells were collected by centrifugation and stored at −80 °C. For protein purification, all subsequent steps were performed at room temperature. Cell paste was resuspended in sonication buffer consisting of 20 mM Tris–Cl (pH 8.0), 50 mM NaCl, 10% glycerol, 0.25% N-lauroyl sarkosyl, 1 mM PMSF, and EDTA-free protease inhibitor cocktail (Roche) to a density of 6 mL/g. Cells were disrupted by sonication using a Branson Sonifier, after which the crude lysate was dialyzed for 4 h against binding buffer (20 mM Tris–Cl (pH 8.0), 50 mM NaCl, and 10% glycerol) at room temperature with two changes of buffer. Dialyzed material was heat treated by incubation at 80 °C for 20 min and clarified by centrifugation at 16,300 × g at room temperature. Clarified sonicate was batch bound overnight to ssDNA-cellulose (GE-Healthcare) in binding buffer at room temperature with gentle mixing. Bound resin was poured into a BioRad Econo-Column glass column support and was washed with 5 column volumes of the binding buffer. Protein was eluted stepwise using 200, 400, 600, and 800 mM NaCl concentrations. SsoRal1 eluted at 400 and 600 mM NaCl and these fractions were pooled and dialyzed against binding buffer at room temperature with two changes of buffer. This material was loaded onto a 1 mL HiTrapQ column (GE Healthcare) which was then washed with 10 column volumes of binding buffer. Protein was eluted stepwise using NaCl, where SsoRal1 eluted at 200 mM NaCl. The resulting protein was dialyzed against 20 mM Tris–Cl (pH 8.0), 100 mM NaCl, and 10% glycerol at room temperature with two changes of buffer before concentration using PEG 20,000 (JT Baker). The final protein stock was flash frozen and stored at −80 °C.
SsoRadA, tmLDH, and tmPK proteins were from laboratory stocks. The SsoRadA K120A and K120R proteins were purified essentially as previously described for the wild-type SsoRadA protein [20]. To ensure the K120R and K120A protein preparations were free of contaminating DNA, 2 μL of each was subjected to a 5′ end labeling reaction using 10 U T4 polynucleotide kinase (NEB) and [γ-32P] ATP in the supplier’s recommended buffer. Samples were incubated 1 h at 37 °C, followed by incubation at 75 °C for 10 min to inactivate the T4 nucleotide kinase. 4 μL of loading dye (0.025% bromophenol blue in 30% glycerol) was then added and reactions were evaluated by electrophoresis on a 130 mm long 7% acrylamide/1× TBE (9 mM Tris, 9 mM boric acid, 0.2 mM EDTA) gel. The gel was dried, exposed to a phosphor imager screen, and scanned with a Storm 850 PhosphorImager (Molecular Dynamics). Filter binding assays to determine KD values for wild-type SsoRadA, K120A and K120R proteins were performed by incubating 20 pmol of protein with ATP for 15 min at 80 °C in a buffer comprised of 40 mM HEPES (pH 7.0), 10 mM magnesium acetate, and 50 mM potassium chloride in a 10 μL volume. In cases with ssDNA present, a 63-mer oligonucleotide (5′-ACAGCACCAATGAAATCTATTAAGCTCCTCATCGTCCGCAAAAATATC-GTCACCTCAAAAGGA-3′) at a concentration of 0.68 μM was used. The reaction was then applied to a pre-moistened 13 mm MF membrane filter (Millipore) under residual vacuum. The filter was washed by vacuum filtration three times using 1 mL wash buffer (40 mM HEPES (pH 7.0), 20 mM magnesium acetate, and 50 mM potassium chloride) under vacuum. Radioactive signal retained was determined by placing the filter in 5 mL Ecoscint scintillation fluid and counting using a Packard 1900A scintillation counter. Reactions were performed in triplicate and dissociation constants were determined by saturation binding curve using Prism 4.0 (GraphPad).
2.2. ATPase assays
All assays were performed essentially as previously described [20]. Buffer was 40 mM HEPES (pH7.0), the ssDNA substrate was 3 μM ϕX174 (NEB), and the ATP concentration was 3 mM. After assay samples were equilibrated at 80 °C for 3 min, the reaction initiating component was added and incubation was continued for an additional 2 min at the same temperature. Protein concentrations for SsoRal1 were 0.03 μM or 1.0 μM (as indicated in the text) while concentrations of SsoRadA, SsoRadA-K120R, and SsoRadA-K120A were 1.0 μM. Assays were performed a minimum of 3 times and graphs were prepared using the Prism 4.0 program (GraphPad).
2.3. Electrophoretic mobility shift assays
The same 63-mer oligonucleotide that was used in the filter binding experiments was 5′ end labeled using 10 U of T4 polynucleotide kinase (NEB) and [γ-32P]ATP by incubating 1 h at 37 °C followed by enzyme inactivation by incubation at 75 °C for 10 min. The dsDNA substrate was prepared by annealing the end-labeled 63-mer oligonucleotide to its unlabeled complement mixing equimolar quantities of each and heating at 95 °C for 10 min, followed by slow cooling to room temperature over a 2 h period. Typical reactions contained 30 mM HEPES (pH 7.0), 10 mM MgOAc, 50 μg/mL bovine serum albumin, 100 mM NaCl, varying concentrations of SsoRal1, SsoRadA-K120A, SsoRadA-K120R, and/or SsoRadA, and 5 μM nucleotides radiolabeled ssDNA or 5 μM base pairs radio-labeled dsDNA substrate in a 20 μL volume. For salt midpoint experiments, concentrations of NaCl in the reactions are as noted in the text. Where indicated, ADP and ATP were added to a final concentration of 3 mM and ATPγS was added to a final concentration of 50 μM. Reactions were incubated for 30 min at 80 °C in an Eppendorf Master Cycler Gradient Thermal Cycler with a heated lid. In protein order of addition experiments, incubation with the first protein proceeded for 5 min before addition of the second protein. Cross-linking was accomplished by adding aqueous 25% glutaraldehyde (Alfa Aesar) to a final concentration of 0.75% followed by incubation for 15 min at room temperature. 4 μL of loading dye (0.025% bromophenol blue in 30% glycerol) was added and samples were loaded on a 130 mm long 7% acrylamide 1× TBE gel that had been pre-run for 15 min at 45 mA. Following electrophoresis, gels were dried and exposed to a phosphor imaging screen which was scanned with a Storm 850 PhosphorImager (Molecular Dynamics). Quantitation was performed using Image-Quant TL software. Each experimental condition was replicated a minimum of three separate times.
2.4. Western blots
Electrophoretic mobility shift samples were prepared and subjected to electrophoresis identically to those described in the previous section. Following electrophoresis, gels were exposed to X-ray film to visualize the bands. As indicated in the text, individual bands were excised from the gel using the X-ray exposure as a guide. These gel slices were inserted into the wells of a 1.5 mm 10% acrylamide Tricine gel with a 4% acrylamide stacking gel and subjected to electrophoresis. Western blots were prepared and performed essentially as previously described [20]. Primary antibodies used were: 1:1000 anti-SsoRadA polyclonal antibody (WSU antibody production facility) and 1:2500 anti-SsoRal1 peptide antibody (custom antibody, Yen Zym). Secondary antibodies were 1:1000 HRP conjugated goat anti-mouse IgG H + L (Pierce) for SsoRadA primary antibody detection and 1:400 Clean Blot IP-HRP (Pierce) for SsoRal1 primary antibody detection. The Pierce ECL Western Blotting kit was used to develop the blots which were then exposed to X-ray film.
2.5. Displacement loop assays
The pBR322 circular dsDNA substrate used for the displacement loop (D-loop) assays was prepared using a sucrose gradient without alkali lysis. DH5α E. coli cells containing the pBR322 plasmid were grown overnight in LB medium at 37 °C in a 1 L volume until they reached stationary phase. Cells were centrifuged for 15 min at 9000 rpm in an SLA3000 rotor at 4 °C, followed by washing with cold M9 buffer (22 mM KH2PO4, 42 mM Na2HPO4, 19 mM NH4Cl) and resuspension in 40 mL cold lysis buffer (50 mM Tris–Cl (pH 8.0), 10 mM EDTA, 10% sucrose). The NaCl concentration of the lysis buffer was adjusted to 1 M, and N-lauroyl sarkosyl was added to a final concentration of 0.9%. Lastly 58 μg of lysozyme (Fischer Biochemicals) was added to the lysis buffer. The lysate was centrifuged for 45 min at 34,400 rpm and 4 °C using a Ti 50.2 rotor and then subjected to phenol::chloroform extraction twice. DNA in the soluble fraction was precipitated with addition of 1/10th volume 5 M NaCl and 2 vol 100% ethanol. The mixture was centrifuged 15 min at 16,700 × g at room temperature. The resulting pellet was resuspended in 2 mL of TE (10 mM Tris–Cl (pH 8.0), 1 mM EDTA) with 100 μg of RNase A then incubated 30 min on ice. Next LiCl was added to a final concentration of 3 M and the 30 min incubation on ice was repeated. The mixture was centrifuged for 15 min at 12,000 × g and 4 °C, followed by transfer of the supernatant to a new tube. The DNA in the supernatant was ethanol precipitated and the pellet was resuspended in 1 mL TE and 0.5 mL 40% PEG 8000 with 30 mM MgCl2. The mixture was centrifuged for 30 min at 16,300 × g at room temperature. The pellet was washed twice with 70% ethanol to remove any traces of the PEG 8000 and then resuspended in 0.5 mL TE. The DNA was top-loaded onto a 5–20% sucrose gradient and centrifuged for 18 h at 25,600 rpm and 4 °C using a SW 41 Ti rotor. Fractions of approximately 0.5 mL were collected by needle puncture. 1 μL of each fraction was subjected to electrophoresis on a 1% agarose/1× TBE gel to identify fractions containing covalently closed pBR322. Those fractions were pooled, ethanol precipitated, and resuspended in TE. Purified DNA was stored at −20 °C until use. The ssDNA 101-mer substrate (5′-TGGCCTGCAACGCGGGCATCCCGATGCCGCCGGAAGCGA-GAAGAATCATAATGGGGAAGGCCATCCAGCCTCGCGTCGCGAACGC-CAGCAAGACGTAGCCC-3′) was synthesized by Sigma–Aldrich and purified by HPLC. The oligonucleotide was 5′ end labeled with [γ-32P] ATP using 10 units of T4 polynucleotide kinase (NEB) and the manufacturer recommended buffer. Reactions were incubated 1 h at 37 °C, followed incubation at 75 °C for 10 min to inactivate the enzyme. Typical D-loop reactions contained 25 mM Tris–Cl pH7.5, 10 mM MgOAc, 50 μg/μL BSA, 0.9 μM (nts) oligonucleotide, 2 mM ATP, and 0.6 μM SsoRadA, SsoRadA K120R, or SsoRadA K120A, and 0.009 μM SsoRal1 as indicated in the text. Reactions were incubated for 5 min at 65 °C, followed by addition of pBR322 to a final concentration of 30 μM (base pairs). Reactions were then incubated for the indicated times and stopped with addition of 0.5 μL 0.5 M EDTA, 0.5 μL 20 mg/mL Proteinase K (NEB), and 1.5 μL 10% SDS followed by a 30 min incubation at 65 °C for deproteinization. 4 μL of loading buffer (0.025% bromophenol blue in 30% glycerol) was added to each reaction prior to loading on a vertical 1.5% agarose/1× TAE (0.04 M TrisOAc and 0.002 M EDTA) gel which was electrophoresed at 75 V for 150 min. Gels were then dried and exposed to a Molecular Dynamics phosphor imaging screen. Images were captured using a Storm 850 phosphor imager and quantitation was performed using the ImageQuant TL program.
The maximal invasion product produced by wild-type SsoRadA (at the 1 min time point) was arbitrarily set at a value of 1. Product formation for all other conditions was compared to this value and expressed as fold change.
3. Results and discussion
3.1. SsoRal1 alters SsoRadA ssDNA-dependent ATP hydrolysis
Both SsoRal1 and SsoRadA have been shown to be ssDNA-dependent ATPases [15,20], but the influence of either protein upon the other during nucleoprotein filament formation has not been determined. DNA-dependent ATPase assays are commonly used as an indirect measure of presynaptic filament formation for RecA family proteins [20–26]. To better understand the potential involvement of SsoRal1 in filament formation, we investigated the effect of addition of SsoRal1 protein to SsoRadA ATPase activity assays (Fig. 1 and Table 1). Activity at 80 °C was measured using a real-time assay that couples ATP hydrolysis to NADH oxidation [20,27]. Our source of SsoRal1 was S. solfataricus strain P2-A, which is 95% identical at the protein sequence level to the Sso2452 protein that has been crystallized and characterized as an anti-recombinase [15,17]. Using this protein and the coupled real-time assay, SsoRadA was found to be a more active ATPase than SsoRal1, results that are similar to those obtained for the Sso2452 protein [15]. We further extended the analysis of these two proteins by performing order of addition experiments. In Fig. 1A, both proteins were present at saturating concentrations, where either protein alone would be in sufficient quantity to completely occupy all available binding sites on the ssDNA substrate as determined by published site-size information [2,15]. When SsoRadA is present first, secondary addition of SsoRal1 immediately reduces the reaction velocity, suggesting prevention of normal SsoRadA ATP hydrolysis (Fig. 1A, black circles). Addition of SsoRadA to SsoRal1 reactions has little effect, and the velocity is very close to those obtained for SsoRal1 alone (Fig. 1A, comparing blue inverted triangles to purple triangles). When both proteins are added together, the reaction velocity again resembles that of SsoRal1 alone (Fig. 1A, comparing red diamonds to purple triangles). Taken together, these results suggest that there is considerable competition for available binding sites and that at high concentration, SsoRal1 simply binds ssDNA more efficiently than SsoRadA. Indeed, this seemed likely since Sso2452 has been shown to bind ssDNA more tightly than SsoRadA [15].
Fig. 1. SsoRal1 alters SsoRadA ssDNA-dependent ATP hydrolysis.
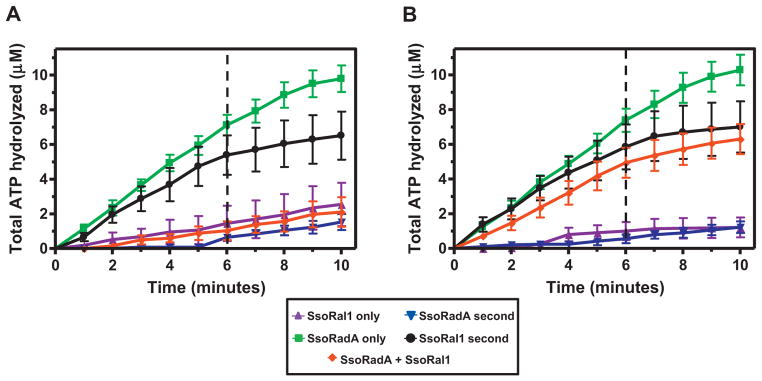
Assays were performed using the coupled ATPase assay as previously described [20] at 80 °C using 3 μM ssϕX174 as the substrate. The first protein was added prior to time 0 and, where applicable, the second protein was added after 6 min (as indicated by dashed vertical lines). Symbols are: SsoRal1, purple triangles; SsoRadA, green squares; SsoRadA second, blue inverted triangles; SsoRal1 second, black circles; SsoRadA and SsoRal1 added together at time 0, red diamonds. (A) SsoRal1 and SsoRadA used at saturating concentrations (1 μM). (B) SsoRadA at a saturating concentration (1 μM) and SsoRal1 at a subsaturating concentration (0.03 μM). (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
Table 1.
Effect of saturating or subsaturating concentrations of SsoRal1 on wild-type Sso-RadA, SsoRadA K120R, and SsoRadA K120A ATP hydrolysis at 80 °C.
| Protein concentration | Assay initiating component | Velocity (μmol ATP hydrolyzed/min) |
|---|---|---|
| Sat. SsoRadA | SsoRadA | 1.42 ± 0.35 |
| Sat. SsoRal1 | SsoRal1 | 0.41 ± 0.06 |
| Subsat. SsoRal1 | SsoRal1 | 0.37 ± 0.14 |
| Sat. SsoRal1 Sat. SsoRadA |
SsoRadA | 0.17 ± 0.16 |
| SsoRal1 | 0.27 ± 0.10 | |
| ϕX174 ssDNA | 0.23 ± 0.19 | |
| Subsat. SsoRal1 Sat. SsoRadA |
SsoRadA | 0.12 ± 0.04 |
| SsoRal1 | 0.15 ± 0.05 | |
| ϕX174 ssDNA | 0.36 ± 0.11 | |
| Sat. K120R | K120R | 0.70 ± 0.32 |
| Sat. SsoRal1 Sat. K120R |
K120R | 0.66 ± 0.11 |
| SsoRal1 | 0.05 ± 0.08 | |
| ϕX174 ssDNA | 0.31 ± 0.08 | |
| Subsat. SsoRal1 Sat. K120R |
K120R | 0.33 ± 0.03 |
| SsoRal1 | 0.42 ± 0.15 | |
| ϕX174 ssDNA | 0.49 ± 0.04 | |
| Sat. K120A | K120A | 0.15 ± 0.18 |
| Sat. SsoRal1 Sat. K120A |
K120A | 0.36 ± 0.11 |
| SsoRal1 | 0.09 ± 0.04 | |
| ϕX174 ssDNA | 0.18 ± 0.10 | |
| Subsat. SsoRal1 Sat. K120A |
K120A | 0.3 ± 0.10 |
| SsoRal1 | 0.52 ± 0.06 | |
| ϕX174 ssDNA | 0.35 ± 0.05 |
In reactions containing two proteins, velocity was calculated after addition of the second protein. In reactions containing a single protein, velocity was calculated from the initial linear portion of the curve.
To test the importance of protein level in what appeared to be competitive binding, we performed the same order of addition experiments but reduced the concentration of SsoRal1 to a subsaturating level (Fig. 1B). We again observed the rapid reduction in reaction velocity when SsoRal1 was added second (Fig. 1B, black circles). This result indicated that the change in velocity was likely not due to simple binding site competition with SsoRadA, as the concentration of SsoRadA present in the reaction was sufficient for maximal activity in the absence of SsoRal1 (Fig. 1B, green squares). When SsoRadA was added second, we saw results similar to those obtained when both proteins were present at saturating concentration (Fig. 1A and B, blue inverted triangles), suggesting that while SsoRadA will bind ssDNA and hydrolyze ATP over time, the lower SsoRal1 activity observed at early stages of the reaction is not rapidly overcome by SsoRadA. Addition of both proteins together results in a velocity above that produced with both proteins present at saturating concentrations (Fig. 1A and B, red diamonds). Thus, while the paralog protein binds ssDNA more efficiently than SsoRadA [15] and has considerably lower ATPase activity, at a subsaturating concentration where it cannot be simply outcompeting SsoRadA for binding sites it has a measurable effect on the nucleoprotein filament. SsoRal1 could influence the nucleoprotein filament in a number of ways, including disassociation of SsoRadA from ssDNA, prevention of SsoRadA from reassociation with ssDNA after ATP hydrolysis, or stabilization of the filament through a reduction in SsoRadA protein dynamics.
3.2. SsoRal1 ssDNA binding
To further address the involvement of SsoRal1 in the formation of the SsoRadA nucleoprotein filament, we more directly examined the DNA binding characteristics of both proteins using electrophoretic mobility shift assays (EMSAs) as shown in Fig. 2. Neither SsoRadA nor SsoRal1 bound dsDNA (data not shown), but both proteins could bind ssDNA (Fig. 2). SsoRadA shifted species were observed in the presence (Fig. 2A) of ATP, but chemical cross-linking was required. These shifted products were very salt sensitive and, along with the need for crosslinking, likely reflect dynamic binding like that which has been described for other RecA family recombinase proteins [11,21,28–32]. SsoRal1 protein did not require crosslinking to produce gel shift products, but binds ssDNA robustly with a salt midpoint of approximately 1.2 M and in an ATP-dependent manner (Fig. 2B and C). The gel shift pattern of SsoRal1 showed two distinct bands, and the relative abundance of each shifted as protein concentration was increased. The lower band was most prevalent at the lowest SsoRal1 concentration, while the higher band was predominant at the highest concentration. The presence of these concentration-dependent shifted products suggests that there could be two distinct modes of interaction between SsoRal1 and ssDNA. While the higher concentration of SsoRal1 generates a more slowly migrating gel shift product, this does not correlate to correspondingly higher levels of ATPase activity, as would be expected if the paralog were forming an active nucleoprotein filament like that made by SsoRadA (Fig. 1). Consequently, the higher SsoRal1 gel shift product could reflect a non-physiological state or a structural function for this protein.
Fig. 2. SsoRal1 and wild-type SsoRadA ssDNA binding characteristics.

Representative mobility shift assays are shown. (A) Salt midpoint titration of 1.7 μM SsoRadA, where ssDNA–protein complexes were formed in the presence of salt and then either cross-linked with 0.75% (final concentration) glutaraldehyde (left side of panel) or were not subjected to cross-linking (right side of panel). (B) Salt titration of 1.7 μM SsoRal1, where ssDNA–protein complexes were formed in the presence of salt but did not require cross-linking for visualization. (C) Reactions containing 100 mM NaCl and increasing concentrations of SsoRal1 were incubated with ssDNA in the presence or absence of ATP and without cross-linking. All reactions contained 5 μM nucleotides ssDNA. In all panels the position of the wells and unbound oligonucleotide are indicated.
3.3. SsoRal1 enhances SsoRadA ssDNA binding
The effect of SsoRal1 on SsoRadA ATPase activity taken together with strong ssDNA binding suggested that this protein could inhibit the SsoRadA recombinase–ssDNA interaction through binding-site competition or disruption of the nucleoprotein filament. We used EMSAs to evaluate the ssDNA binding capabilities of the proteins when both were added simultaneously and relative protein concentrations were similar to those used in the ATPase experiments in Fig. 1B, where a subsaturating concentration of SsoRal1 dramatically altered SsoRadA reaction velocity. As shown in Fig. 3, lanes 1 and 2, saturating and subsaturating concentrations of SsoRal1 yielded both the faster and slower migrating bands we observed in Fig. 2B and C. Neither saturating nor subsaturating concentrations of SsoRadA produced a gel shift product in the absence of crosslinking (Fig. 2A, right side of panel; Fig. 3A, lanes 3 and 4). When both proteins were present at saturating concentrations, a more slowly migrating species distinct from the largest Sso-Ral1 associated band was observed (Fig. 3A, lane 5). This band migrates to similar position in the gel as the cross-linked SsoRadA species shown in Supplemental Fig. 1. Accordingly, this more slowly migrating band appears to represent primarily SsoRadA protein bound to ssDNA. When the concentration of SsoRal1 is reduced to subsaturating, we detected the lower SsoRal1 band along with the SsoRadA-specific band (Fig. 3A, lane 6), indicating that even small amounts of SsoRal1 protein can influence SsoRadA ssDNA binding. Lastly, we examined binding of both proteins under subsaturating concentrations (Fig. 3A, lane 7). In this case we detected only SsoRal1 specific shift products, suggesting that SsoRadA ssDNA binding is enhanced by SsoRal1 only when SsoRadA is at a saturating concentration. We repeated all of these conditions with order of addition experiments, adding either the SsoRadA or SsoRal1 protein to the reaction second, but did not observe results that were significantly different from those produced when both proteins were added simultaneously (data not shown).
Fig. 3. SsoRal1 enhances SsoRadA–ssDNA binding.

(A) A representative mobility shift assay is shown for SsoRal1 in combination with wild-type SsoRadA. All reactions contained 5 μM ssDNA as the substrate, 100 mM NaCl, and 3 mM ATP. Where used together, SsoRadA and SsoRal1 proteins were added simultaneously. The position of shifted species and unbound oligonucleotide are indicated. (B) Western blot detection of SsoRadA and SsoRal1 in stabilized nucleoprotein filaments. Using a gel identical to that shown in (A), shifted products located in the SsoRadA position (indicated by arrow) in lanes 5 and 6 (the most slowly migrating bands) were directly excised from the gel and subjected to electrophoresis followed by Western blotting. Purified SsoRadA and SsoRal1 proteins were used as controls and protein concentrations are indicated above the lanes.
To better understand the composition of the shifted species in the EMSA, we turned to direct detection of the proteins by Western blot. We repeated the EMSA experiments shown in Fig. 3A, lanes 5 and 6 and exposed the resulting gel to X-ray film to determine the position of the shifted species. We then excised the most slowly migrating band for each reaction (designated SsoRadA to the right of the gel in Fig. 3A) and inserted these gel slices into the wells of a second denaturing acrylamide gel where they were again subjected to electrophoresis prior to Western blotting. The resulting Western blots are shown in Fig. 3B. We were able to detect the presence of both SsoRadA and SsoRal1 in the slowly migrating band in Fig. 3A, lane 5, where both proteins were present at saturating concentrations. Only the SsoRadA protein was apparent in the equivalent band (Fig. 3A, lane 6) where SsoRadA was saturating and SsoRal1 was subsaturating; our failure to detect a signal for SsoRal1 at this low molar concentration could be due to differences between the primary antibodies we used. The antibody for SsoRadA is polyclonal, but the SsoRal1 antibody is a peptide antibody and may not provide enough sensitivity to detect very low levels of the protein in this gel shift product. The presence of SsoRal1 with SsoRadA in the filament suggests that upon stabilization, some percentage of SsoRal1 remains associated. Overall these results indicate that SsoRal1 does not remove SsoRadA from ssDNA or prevent reassociation after hydrolysis, but rather enhances SsoRadA binding. As no direct interaction between purified SsoRal1 and SsoRadA has been detected ([15]; W. Graham and C. Haseltine, unpublished results), this stabilization effect is likely to be DNA-mediated.
To further understand the nature of the shifted species in Fig. 3, we examined the nucleotide cofactor requirements for SsoRal1 and SsoRadA ssDNA binding (Fig. 4). In the presence of ADP, saturating SsoRal1 produced a shift pattern that was very similar to that observed with ATP, but with less overall less shifted material (Fig. 4A, lanes 1 and 3). At a subsaturating SsoRal1 concentration no shifted product was apparent with ADP (Fig. 4A, compare lanes 2 and 4). Using the slowly hydrolyzed ATP analog ATPγS, we found that saturating SsoRal1 produced only the more slowly migrating band, although both bands were present for the subsaturating SsoRal1 condition (Fig. 4A, lanes 5 and 6). This suggested that when ATP is bound but not rapidly hydrolyzed, the larger SsoRal1 gel shift product is favored. Conversely, SsoRadA gel shift products were optimally obtained using ADP instead of ATP (Fig. 4B, lanes 7 and 9). Subsaturating SsoRadA yielded no shifted product with either ATP or ATPγS, although some signal was detected when SsoRadA was saturating (Fig. 4B, lanes 7, 8, 11, and 12). Our results suggest that cofactor requirements for ssDNA binding by SsoRadA are very different from those of SsoRal1 and that ATP hydrolysis has differential effects on SsoRal1 and SsoRadA ssDNA binding. ADP enhances SsoRadA–ssDNA binding, suggesting a reduction in filament dynamics when the recombinase is not bound to ATP or ATPγS. Conversely, SsoRal1–ssDNA binding is reduced by ADP and enhanced by ATP or ATPγS, indicating that the observed low ATPase activity of the paralog is likely due to slower turnover and more resident time bound to ssDNA.
Fig. 4. Binding of SsoRal1 and SsoRadA to ssDNA is influenced by nucleotide cofactors.
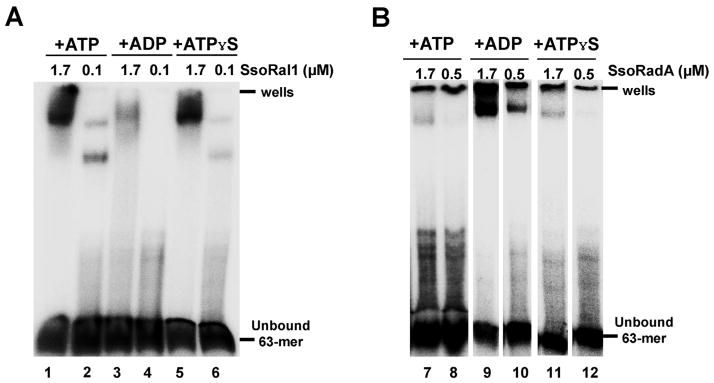
EMSAs are shown for SsoRal1 in (A) and wild-type SsoRadA (B) where binding reactions were performed using ATP, ADP, or ATPγS. All reactions included 5 μM ssDNA and 100 mM NaCl. ATP and ADP concentrations were 3 mM, while ATPγS was present at a concentration of 50 μM. All wild-type SsoRadA reactions were cross-linked by the addition of glutaraldehyde to a final concentration of 0.75%. The position of the wells and unbound oligonucleotide are indicated.
3.4. SsoRal1 affects SsoRadA Walker A mutant ATPase activity and ssDNA binding
The role of ATP binding and hydrolysis has been previously examined for RecA and Rad51 proteins through substitution of the invariant lysine residue within the Walker A motif to either alanine or arginine, but there are significant differences in the outcome of these mutations. For example, the Escherichia coli RecA K72A protein has reduced NTP hydrolysis but can still bind ssDNA [33]. A similar change in yeast Rad51 (K191A) results in a protein that does not hydrolyze ATP or bind ssDNA, while human Rad51 K133A can bind DNA but forms inactive filaments [34–37]. Both yeast Rad51 K191R and human Rad51 K133R mutant proteins can bind DNA, however [36–38]. To better understand the role of ATP in DNA binding of SsoRadA, we altered the lysine residue in the Walker A motif of SsoRadA to either alanine or arginine to generate SsoRadA K120A and SsoRadA K120R mutant proteins. We first determined the ability of each of the proteins to bind ATP by filter binding. Wild-type SsoRadA had a KD of 2.4 ± 0.6 μM, but we were unable to detect significant ATP binding for either of the Walker A motif mutants, suggesting that these mutants do not bind ATP well in the absence of DNA (data not shown). In the presence of ssDNA, the wild-type SsoRadA protein KD for ATP binding was nearly the same as in the absence of ssDNA (2.5 ± 0.7 μM). While the presence of ssDNA did not result in detectable levels of ATP binding by the K120A mutant, the K120R protein had a KD of 59.4 ± 9.4 μM. ATPase activity assays using ssDNA and the mutant proteins were performed both with and without the addition of SsoRal1 and are reported in Table 1. Similar to the results obtained with wild-type SsoRadA (Fig. 1 and Table 1), addition of SsoRal1 to SsoRadA K120R reactions resulted in a measurable reduction in reaction velocity. This effect was most pronounced, however, when SsoRal1 was added at a saturating concentration, in contrast to our observation with wild-type SsoRadA where only a subsaturating concentration of SsoRal1 was necessary for reaction velocity reduction. In assays containing both K120A and saturating SsoRal1, the observed activity was not significantly different than that observed when the protein is added first (as determined by the Student’s T-test). Secondary addition of K120A to SsoRal1 results in a significantly different velocity when compared to that observed when K120A is alone (95% confidence, Student’s T-test). When SsoRal1 was used at a subsaturating concentration, addition of this protein second to the K120A mutant results in what might initially appear to be stimulatory behavior (0.52 μmol ATP hydrolyzed/min). This is not likely the case, however, as the overall velocity of these reactions is not significantly above that which would be additive (0.37 μmol ATP hydrolyzed/min for unsaturated SsoRal1 alone +0.15 μmol ATP hydrolyzed/min for K120A alone).
Like SsoRadA, neither SsoRadA K120A nor K120R protein produced a gel shift product with either ss- or dsDNA in the absence of crosslinking (data not shown). Salt titration experiments that included crosslinking showed that SsoRadA K120R is extremely sensitive to salt and binds ssDNA poorly (Fig. 5A). SsoRadA K120A, however, binds ssDNA more strongly than K120R and wild-type SsoRadA, with a salt midpoint close to 50 mM (Fig. 5A). Alteration of the nucleotide cofactor did not improve SsoRadA K120R ssDNA binding, suggesting that the mutation may result in overall poor ssDNA binding ability (Fig. 5B, lanes 3, 4, 7, 8, 11, and 12). SsoRadA K120A was able to bind ssDNA with ATP, ADP, or ATPγS under saturating protein conditions, but failed to bind well at subsaturating concentrations with ATP or ATPγS (Fig. 5B, compare lanes 1 to 2, 5 to 6, and 9 to 10). Substitution of wild-type SsoRadA with either the K120A or K120R mutant protein in gel shift experiments with Sso-Ral1 resulted in a marked reduction in the influence of SsoRal1 on the slowest migrating species (Fig. 6). In contrast to the wild-type SsoRadA experiments in Fig. 3, we found that enhancement of the SsoRadA-specific gel shift product with a subsaturating concentration of SsoRal1 did not occur with the mutant proteins. Instead, the effect is only apparent when a saturating concentration of SsoRal1 is present (Fig. 6, lanes 7 and 10). This result is consistent with our ATPase data for the K120A and K120R proteins under conditions where SsoRal1 is saturating; the nearly immediate cessation of activity suggests a strong reduction in filament dynamics through failure of SsoRadA release from ssDNA. The reduced ATPase activity of the K120A and K120R mutant proteins likely reflects a more incomplete filament, where significantly higher concentrations of SsoRal1 are required to achieve the observed full shift indicative of recombinase binding. Differential ssDNA binding characteristics of the mutant proteins also explains the need for saturating concentrations of SsoRal1 to achieve stabilization of the nucleoprotein filament. Since SsoRadA K120R binds ssDNA poorly, more SsoRal1 is required to stabilize this weak interaction. Strong binding of K120A to ssDNA likely inhibits SsoRal1–ssDNA binding through prolonged occupation of available binding sites.
Fig. 5. ssDNA binding characteristics of SsoRadA ATPase motif mutants.
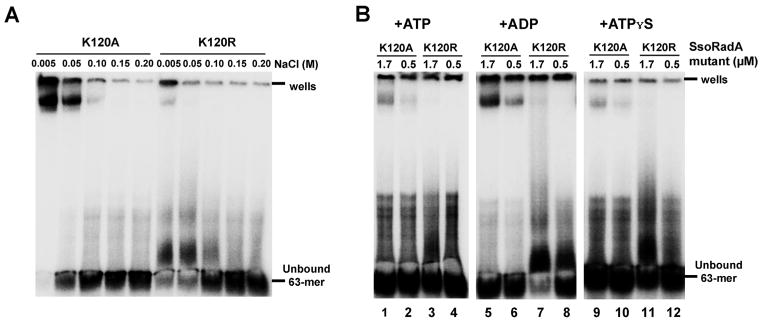
Representative mobility shift assays are shown for the SsoRadA K120A and K120R mutant proteins. (A) SsoRadA K120A and K120R were incubated in increasing concentrations of NaCl with 5 μM ssDNA in the presence of 3 mM ATP. (B) Cofactors were included in the binding reactions with 100 mM NaCl. ATP and ADP concentrations were 3 mM, while ATPγS was present at a concentration of 50 μM. All reactions included 5 μM ssDNA as the substrate, and were cross-linked by addition of glutaraldehyde to a final concentration of 0.75%. The position of the wells and unbound oligonucleotide are indicated.
Fig. 6. ssDNA binding of SsoRal1 with SsoRadA ATPase motif mutants.

A representative mobility shift assay is shown for SsoRal1 in combination with SsoRadA K120A and K120R mutant proteins. All reactions contained 5 μM ssDNA as the substrate, 100 mM NaCl, and 3 mM ATP. The position of shifted species and unbound oligonucleotide are indicated.
3.5. SsoRal1 stimulates SsoRadA-mediated strand invasion
Strand invasion occurs when the presynaptic filament invades dsDNA and a joint molecule including a displacement loop (D-loop) is formed. This invasion step is catalyzed by the RecA family of recombinases including SsoRadA [2,39,40]. Since we observed SsoRadA filament stabilization by SsoRal1, it seemed likely that strand invasion could be altered in the presence of the paralog. To test the effect of SsoRal1 on SsoRadA-mediated strand invasion, we directly measured joint molecule formation using D-loop assays as shown in Fig. 7A. In these assays the formation of strand invasion products can be directly measured, as can dissociation of the joint molecule resulting from branch migration over time during the D-loop cycle [41,42]. For all assays (Fig. 7B and C), we used protein stoichiometries identical to those that resulted in decreased ATPase activity in the previous ATPase experiments (Fig. 1) where SsoRadA and Walker motif mutant proteins were saturating and SsoRal1 was subsaturating. As shown in Fig. 7, joint molecule formation by wild-type SsoRadA was highest at early time points and lower as the D-loop cycle progressed. While SsoRadA K120A displayed similar strand invasion behavior, joint molecules persisted for a longer period than those produced using SsoRadA. This persistence may be the result of the enhanced ssDNA binding activity and reduced ATP hydrolysis of K120A relative to SsoRadA (see Figs. 4B, 5B, and Table 1). While not producing high levels of invasion products, the stability of the K120A filaments could allow more persistent D-loop products. The SsoRadA K120R mutant protein also catalyzes strand invasion but does so more slowly than either the wild-type or K120A mutant proteins, which is most likely a result of the ssDNA binding defect in this mutant reducing its ability to form presynaptic filaments (see Figs. 4B and 5B). Interestingly, it appears that SsoRal1 can also catalyze strand invasion and does so at the subsaturating concentration we tested. When both SsoRadA and SsoRal1 were added simultaneously to the reaction (and SsoRadA was saturating and SsoRal1 was subsaturating) strand invasion products were increased more than 3-fold, suggesting that the filament stabilization activity of SsoRal1 translates to enhanced joint molecule formation. We also observed stimulation of strand invasion with the SsoRadA K120R mutant, but the effect was reduced relative to wild-type SsoRadA and closer to 2-fold. Here, SsoRal1 appears to assist SsoRadAK120R in overcoming its ssDNA binding defect (see Figs. 4B and 5B) and more effectively perform strand invasion, which could occur through simple stabilization of the protein–ssDNA interaction. SsoRal1 appears to have a negligible effect on strand invasion catalysis by the SsoRadA K120A mutant, presumably because the K120A presynaptic filament is intrinsically more stable than its wild-type counterpart (Figs. 2A and 5A).
Fig. 7. SsoRal1 catalyzes strand invasion and enhances SsoRadA D-loop production.

(A) Schematic of the D-loop reaction. (B) Representative autoradiographs showing D-loop formation by various proteins or combinations of proteins at 1, 5, 10, and 20 min. NP is no protein and was taken at 20 min. Concentrations of SsoRadA, SsoRadA K120R, and SsoRadA K120A were 0.6 μM, while concentration of SsoRal1 was 0.009 μM. (C) Quantitation of D-loop formation by individual proteins or protein combinations as indicated by the labels along the X-axis. Error bars represent the standard deviation of a minimum of three repetitions. The amount of D-loop product formed by SsoRadA at 1 min was arbitrarily set at 1, and values for all other bars are compared to the value for SsoRadA at 1 min. The dashed line is set at 1 for ease of comparison across the graph.
4. Conclusions
Taken together, our results suggest that SsoRal1 can directly affect nucleoprotein filament formation through alteration of Sso-RadA binding behavior and reduction of filament dynamics and have led us to propose the model in Fig. 8. Our filter binding experiments suggest that SsoRadA can bind ATP independently of ssDNA, a result that is supported by the gel shift pattern produced by Sso-RadA in the presence of ATP (Fig. 2). The ATPase activity of SsoRadA is ssDNA-dependent, however, suggesting that the recombinase first binds ATP, then binds DNA (Fig. 8A). Also consistent with initial ATP binding is the observation that the K120R mutant only binds ATP in the presence of ssDNA as measured by filter binding and does not produce a stable gel shift product. Thus the SsoRadA recombinase is unlikely to bind ssDNA prior to binding ATP. The nearly identical gel shift patterns for SsoRadA with ATP or ATPγS suggest that ATP hydrolysis has no effect on ssDNA binding and that it is only after the protein is bound to ssDNA that it hydrolyzes ATP. Both wild-type and Walker motif mutant proteins display optimal binding with ADP, indicating that ATP is quickly hydrolyzed after ssDNA binding. Addition of SsoRal1 to the forming filament reduces the ATPase activity of SsoRadA and stabilizes recombinase binding to ssDNA (Fig. 8B and C). In the absence of SsoRal1, SsoRadA binds ssDNA more efficiently with ADP, but addition of the paralog results in stronger recombinase binding with ATP for both the wild-type and mutant proteins. In conjunction with the observed reduction in SsoRadA ATP hydrolysis in the presence of SsoRal1, this implies that the paralog drives the recombinase to bind ssDNA in an ATP-bound state instead of a predominantly ADP-bound state. The SsoRal1 protein does not appear to exert this effect and depart the filament, however, since direct examination of stabilized SsoRadA–ssDNA species from gel shift experiments by Western blot showed the presence of both SsoRadA and SsoRal1 (Fig. 3B). Instead the SsoRal1 and SsoRadA are more likely forming a mixed filament since we have not been able to detect a direct interaction between these proteins. The presence of SsoRal1 within the SsoRadA filament is also stimulatory to strand invasion (Fig. 8D). We observed increases in total D-loop products and persistence of products with the addition of SsoRal1, suggesting that enhanced filament stability results in more efficient strand invasion and greater joint molecule stability.
Fig. 8. Model of SsoRal1 stabilization of the SsoRadA nucleoprotein filament.

(A) The SsoRadA presynaptic filament is dynamic and slow to form. SsoRadA binds ssDNA and dissociates rapidly. (B) and (C) Binding of SsoRal1 to ssDNA reduces dissociation of SsoRadA and stabilizes the filament. (D) Strand invasion is enhanced through reduction of SsoRadA presynaptic filament dynamics due to SsoRal1 binding.
SsoRal1 is one of a number of proteins that have been examined for their mediator roles in HR. Included among these is the E. coli RecFOR protein complex which assists in RecA nucleation at ds/ssDNA junctions and stimulates RecA ssDNA-dependent ATPase activity [43]. It seems unlikely that the SsoRal1 protein performs the same nucleation function as RecFOR, as the protein does not stimulate SsoRadA ATPase activity but rather decreases it (Fig. 1). The Rad52 protein of S. cerevisiae destabilizes RPA–ssDNA interactions and assists in Rad51 loading onto ssDNA via direct interaction [44–48]. SsoRal1 does not appear to be equivalent to Rad52 either, as we were unable to detect a direct interaction between Sso-Ral1 and SsoRadA. UvsY protein from the bacteriophage T4 directly stabilizes UvsX recombinase presynaptic filaments [9], and this stabilization is predominantly the result of UvsY interaction with ssDNA [49]. Additionally, UvsY has been proposed to function as a nucleotide exchange factor for UvsX, allowing the recombinase to remain ssDNA-bound for longer periods of time [50]. SsoRal1 does show similarities to the UvsY mediator since SsoRal1 enhances SsoRadA filament stability by suppressing the hydrolysis of ATP to ADP. The S. cerevisiae Rad55/57 protein can stabilize the Rad51 presynaptic filament against various destabilizing challenges, form a co-complex with Rad51 on ssDNA, and stimulate the formation of Rad51-mediated strand invasion products [8,51,52]. SsoRal1 protein shares many of these characteristics, including stabilization of the SsoRadA–nucleoprotein filament as demonstrated by a reduction in ATPase activity, forming a co-complex with SsoRadA on ssDNA, and stimulation of SsoRadA-mediated strand invasion. The functional similarities of SsoRal1 to both UvsY and the Rad55/57 heterodimer suggest that presynaptic filament stabilization is universally conserved mechanism in the early steps of HR. ATP hydrolysis by SsoRadA may not be entirely sufficient for filament turnover as is the case with UvsX and E. coli RecA proteins [53,54]. Instead, like eukaryotic Rad51, the involvement of additional proteins may be required [55,56]. Further studies of the SsoRal1 protein with other S. solfataricus paralogs and the SSB protein may further elucidate additional mediator functions of SsoRal1 and yield insight into the evolution of HR mediator proteins.
Supplementary Material
Acknowledgments
We thank the anonymous reviewers for their insight and constructive comments. This work was supported by NSF grant MCB-0951125 to CAH and NIH training grant T32GM083864 to WJG.
Appendix A. Supplementary data
Supplementary data associated with this article can be found, in the online version, at http://dx.doi.org/10.1016/j.dnarep.2013.03.003.
Footnotes
Conflict of interest statement
The authors declare that there are no conflicts of interest.
References
- 1.Benson FE, Stasiak A, West SC. Purification and characterization of the human Rad51 protein, an analogue of E. coli RecA. EMBO J. 1994;13:5764–5771. doi: 10.1002/j.1460-2075.1994.tb06914.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Seitz EM, Brockman JP, Sandler SJ, Clark AJ, Kowalczykowski SC. RadA protein is an archaeal RecA protein homolog that catalyzes DNA strand exchange. Genes Dev. 1998;12:1248–1253. doi: 10.1101/gad.12.9.1248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Shibata T, Cunningham RP, DasGupta C, Radding CM. Homologous pairing in genetic recombination: complexes of recA protein and DNA. Proc Natl Acad Sci U S A. 1979;76:5100–5104. doi: 10.1073/pnas.76.10.5100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Baumann P, Benson FE, West SC. Human Rad51 protein promotes ATP-dependent homologous pairing and strand transfer reactions in vitro. Cell. 1996;87:757–766. doi: 10.1016/s0092-8674(00)81394-x. [DOI] [PubMed] [Google Scholar]
- 5.Sung P. Catalysis of ATP-dependent homologous DNA pairing and strand exchange by yeast RAD51 protein. Science. 1994;265:1241–1243. doi: 10.1126/science.8066464. [DOI] [PubMed] [Google Scholar]
- 6.Kowalczykowski SC. In vitro reconstitution of homologous recombination reactions. Experientia. 1994;50:204–215. doi: 10.1007/BF01924003. [DOI] [PubMed] [Google Scholar]
- 7.Sung P, Robberson DL. DNA strand exchange mediated by a RAD51–ssDNA nucleoprotein filament with polarity opposite to that of RecA. Cell. 1995;82:453–461. doi: 10.1016/0092-8674(95)90434-4. [DOI] [PubMed] [Google Scholar]
- 8.Liu J, Ehmsen KT, Heyer WD, Morrical SW. Presynaptic filament dynamics in homologous recombination and DNA repair. Crit Rev Biochem Mol Biol. 2011;46:240–270. doi: 10.3109/10409238.2011.576007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Liu J, Bond JP, Morrical SW. Mechanism of presynaptic filament stabilization by the bacteriophage T4 UvsY recombination mediator protein. Biochemistry. 2006;45:5493–5502. doi: 10.1021/bi0525167. [DOI] [PubMed] [Google Scholar]
- 10.Bugreev DV, Mazin AV. Ca2+ activates human homologous recombination protein Rad51 by modulating its ATPase activity. Proc Natl Acad Sci U S A. 2004;101:9988–9993. doi: 10.1073/pnas.0402105101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Carreira A, Hilario J, Amitani I, Baskin RJ, Shivji MK, Venkitaraman AR, Kowalczykowski SC. The BRC repeats of BRCA2 modulate the DNA-binding selectivity of RAD51. Cell. 2009;136:1032–1043. doi: 10.1016/j.cell.2009.02.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Carreira A, Kowalczykowski SC. Two classes of BRC repeats in BRCA2 promote RAD51 nucleoprotein filament function by distinct mechanisms. Proc Natl Acad Sci U S A. 2011;108:10448–10453. doi: 10.1073/pnas.1106971108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chen LT, Ko TP, Chang YC, Lin KA, Chang CS, Wang AH, Wang TF. Crystal structure of the left-handed archaeal RadA helical filament: identification of a functional motif for controlling quaternary structures and enzymatic functions of RecA family proteins. Nucleic Acids Res. 2007;35:1787–1801. doi: 10.1093/nar/gkl1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rolfsmeier ML, Laughery MF, Haseltine CA. Repair of DNA double-strand breaks following UV damage in three Sulfolobus solfataricus strains. J Bacteriol. 2010;192:4954–4962. doi: 10.1128/JB.00667-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.McRobbie AM, Carter LG, Kerou M, Liu H, McMahon SA, Johnson KA, Oke M, Naismith JH, White MF. Structural and functional characterisation of a conserved archaeal RadA paralog with antirecombinase activity. J Mol Biol. 2009:661–673. doi: 10.1016/j.jmb.2009.04.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Abella M, Rodriguez S, Paytubi S, Campoy S, White MF, Barbe J. The Sulfolobus solfataricus radA paralogue sso0777 is DNA damage inducible and positively regulated by the Sta1 protein. Nucleic Acids Res. 2007;35:6788–6797. doi: 10.1093/nar/gkm782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rolfsmeier ML, Laughery MF, Haseltine CA. Repair of DNA double-strand breaks induced by ionizing radiation damage correlates with upregulation of homologous recombination genes in Sulfolobus solfataricus. J Mol Biol. 2011;414:485–498. doi: 10.1016/j.jmb.2011.10.020. [DOI] [PubMed] [Google Scholar]
- 18.Seitz EM, Haseltine CA, Kowalczykowski SC. DNA recombination and repair in the archaea. Adv Appl Microbiol. 2001;50:101–169. doi: 10.1016/s0065-2164(01)50005-2. [DOI] [PubMed] [Google Scholar]
- 19.Haldenby S, White MF, Allers T. RecA family proteins in archaea: RadA and its cousins. Biochem Soc Trans. 2009;37:102–107. doi: 10.1042/BST0370102. [DOI] [PubMed] [Google Scholar]
- 20.Rolfsmeier ML, Haseltine CA. The single-stranded DNA binding protein of Sulfolobus solfataricus acts in the presynaptic step of homologous recombination. J Mol Biol. 2010;397:31–45. doi: 10.1016/j.jmb.2010.01.004. [DOI] [PubMed] [Google Scholar]
- 21.Lindsley JE, Cox MM. Assembly and disassembly of RecA protein filaments occur at opposite filament ends. Relationship to DNA strand exchange. J Biol Chem. 1990;265:9043–9054. [PubMed] [Google Scholar]
- 22.Menetski JP, Varghese A, Kowalczykowski SC. Properties of the high-affinity single-stranded DNA binding state of the Escherichia coli recA protein. Biochemistry. 1988;27:1205–1212. doi: 10.1021/bi00404a021. [DOI] [PubMed] [Google Scholar]
- 23.Pugh BF, Cox MM. High salt activation of recA protein ATPase in the absence of DNA. J Biol Chem. 1988;263:76–83. [PubMed] [Google Scholar]
- 24.Shan Q, Cox MM. RecA protein dynamics in the interior of RecA nucleoprotein filaments. J Mol Biol. 1996;257:756–774. doi: 10.1006/jmbi.1996.0200. [DOI] [PubMed] [Google Scholar]
- 25.Sugiyama T, Zaitseva EM, Kowalczykowski SC. A single-stranded DNA-binding protein is needed for efficient presynaptic complex formation by the Saccharomyces cerevisiae Rad51 protein. J Biol Chem. 1997;272:7940–7945. doi: 10.1074/jbc.272.12.7940. [DOI] [PubMed] [Google Scholar]
- 26.Zhang XP, Galkin VE, Yu X, Egelman EH, Heyer WD. Loop 2 in Saccharomyces cerevisiae Rad51 protein regulates filament formation and ATPase activity. Nucleic Acids Res. 2009;37:158–171. doi: 10.1093/nar/gkn914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Seybert A, Scott DJ, Scaife S, Singleton MR, Wigley DB. Biochemical characterisation of the clamp/clamp loader proteins from the euryarchaeon Archaeoglobus fulgidus. Nucleic Acids Res. 2002;30:4329–4338. doi: 10.1093/nar/gkf584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shivashankar GV, Feingold M, Krichevsky O, Libchaber A. RecA polymerization on double-stranded DNA by using single-molecule manipulation: the role of ATP hydrolysis. Proc Natl Acad Sci U S A. 1999;96:7916–7921. doi: 10.1073/pnas.96.14.7916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Neuendorf SK, Cox MM. Exchange of recA protein between adjacent recA protein-single-stranded DNA complexes. J Biol Chem. 1986;261:8276–8282. [PubMed] [Google Scholar]
- 30.Liu J, Renault L, Veaute X, Fabre F, Stahlberg H, Heyer WD. Rad51 paralogues Rad55–Rad57 balance the antirecombinase Srs2 in Rad51 filament formation. Nature. 2011;479:245–248. doi: 10.1038/nature10522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Li X, Zhang XP, Solinger JA, Kiianitsa K, Yu X, Egelman EH, Heyer WD. Rad51 and Rad54 ATPase activities are both required to modulate Rad51-dsDNA filament dynamics. Nucleic Acids Res. 2007;35:4124–4140. doi: 10.1093/nar/gkm412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mazin AV, Zaitseva E, Sung P, Kowalczykowski SC. Tailed duplex DNA is the preferred substrate for Rad51 protein-mediated homologous pairing. EMBO J. 2000;19:1148–1156. doi: 10.1093/emboj/19.5.1148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rehrauer WM, Kowalczykowski SC. Alteration of the nucleoside triphosphate (NTP) catalytic domain within Escherichia coli RecA protein attenuates NTP hydrolysis but not joint molecule formation. J Biol Chem. 1993;268:1292–1297. [PubMed] [Google Scholar]
- 34.Donovan JW, Milne GT, Weaver DT. Homotypic and heterotypic protein associations control Rad51 function in double-strand break repair. Genes Dev. 1994;8:2552–2562. doi: 10.1101/gad.8.21.2552. [DOI] [PubMed] [Google Scholar]
- 35.Morgan EA, Shah N, Symington LS. The requirement for ATP hydrolysis by Saccharomyces cerevisiae Rad51 is bypassed by mating-type heterozygosity or RAD54 in high copy. Mol Cell Biol. 2002;22:6336–6343. doi: 10.1128/MCB.22.18.6336-6343.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sung P, Stratton SA. Yeast Rad51 recombinase mediates polar DNA strand exchange in the absence of ATP hydrolysis. J Biol Chem. 1996;271:27983–27986. doi: 10.1074/jbc.271.45.27983. [DOI] [PubMed] [Google Scholar]
- 37.Chi P, Van Komen S, Sehorn MG, Sigurdsson S, Sung P. Roles of ATP binding and ATP hydrolysis in human Rad51 recombinase function. DNA Repair (Amst) 2006;5:381–391. doi: 10.1016/j.dnarep.2005.11.005. [DOI] [PubMed] [Google Scholar]
- 38.Morrison C, Shinohara A, Sonoda E, Yamaguchi-Iwai Y, Takata M, Weichselbaum RR, Takeda S. The essential functions of human Rad51 are independent of ATP hydrolysis. Mol Cell Biol. 1999;19:6891–6897. doi: 10.1128/mcb.19.10.6891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.McIlwraith MJ, Van Dyck E, Masson JY, Stasiak AZ, Stasiak A, West SC. Reconstitution of the strand invasion step of double-strand break repair using human rad51 rad52 and RPA proteins. J Mol Biol. 2000;304:151–164. doi: 10.1006/jmbi.2000.4180. [DOI] [PubMed] [Google Scholar]
- 40.Shibata T, DasGupta C, Cunningham RP, Radding CM. Purified Escherichia coli recA protein catalyzes homologous pairing of superhelical DNA and single-stranded fragments. Proc Natl Acad Sci U S A. 1979;76:1638–1642. doi: 10.1073/pnas.76.4.1638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shibata T, Ohtani T, Iwabuchi M, Ando T. D-loop cycle. A circular reaction sequence which comprises formation and dissociation of D-loops and inactivation and reactivation of superhelical closed circular DNA promoted by recA protein of Escherichia coli. J Biol Chem. 1982;257:13981–13986. [PubMed] [Google Scholar]
- 42.Shibata T, Nishinaka T, Mikawa T, Aihara H, Kurumizaka H, Yokoyama S, Ito Y. Homologous genetic recombination as an intrinsic dynamic property of a DNA structure induced by RecA/Rad51-family proteins: a possible advantage of DNA over RNA as genomic material. Proc Natl Acad Sci U S A. 2001;98:8425–8432. doi: 10.1073/pnas.111005198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Morimatsu K, Kowalczykowski SC. RecFOR proteins load RecA protein onto gapped DNA to accelerate DNA strand exchange: a universal step of recombinational repair. Mol Cell. 2003;11:1337–1347. doi: 10.1016/s1097-2765(03)00188-6. [DOI] [PubMed] [Google Scholar]
- 44.Sugiyama T, Kowalczykowski SC. Rad52 protein associates with replication protein A (RPA)-single-stranded DNA to accelerate Rad51-mediated displacement of RPA and presynaptic complex formation. J Biol Chem. 2002;277:31663–31672. doi: 10.1074/jbc.M203494200. [DOI] [PubMed] [Google Scholar]
- 45.Shinohara A, Ogawa H, Ogawa T. Rad51 protein involved in repair and recombination in S. cerevisiae is a RecA-like protein. Cell. 1992;69:457–470. doi: 10.1016/0092-8674(92)90447-k. [DOI] [PubMed] [Google Scholar]
- 46.Sung P. Function of yeast Rad52 protein as a mediator between replication protein A and the Rad51 recombinase. J Biol Chem. 1997;272:28194–28197. doi: 10.1074/jbc.272.45.28194. [DOI] [PubMed] [Google Scholar]
- 47.Milne GT, Weaver DT. Dominant negative alleles of RAD52 reveal a DNA repair/recombination complex including Rad51 and Rad52. Genes Dev. 1993;7:1755–1765. doi: 10.1101/gad.7.9.1755. [DOI] [PubMed] [Google Scholar]
- 48.Park MS, Ludwig DL, Stigger E, Lee SH. Physical interaction between human RAD52 and RPA is required for homologous recombination in mammalian cells. J Biol Chem. 1996;271:18996–19000. doi: 10.1074/jbc.271.31.18996. [DOI] [PubMed] [Google Scholar]
- 49.Bleuit JS, Ma Y, Munro J, Morrical SW. Mutations in a conserved motif inhibit single-stranded DNA binding and recombination mediator activities of bacteriophage T4 UvsY protein. J Biol Chem. 2004;279:6077–6086. doi: 10.1074/jbc.M311557200. [DOI] [PubMed] [Google Scholar]
- 50.Farb JN, Morrical SW. Functional complementation of UvsX and UvsY mutations in the mediation of T4 homologous recombination. Nucleic Acids Res. 2009;37:2336–2345. doi: 10.1093/nar/gkp096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Fortin GS, Symington LS. Mutations in yeast Rad51 that partially bypass the requirement for Rad55 and Rad57 in DNA repair by increasing the stability of Rad51-DNA complexes. EMBO J. 2002;21:3160–3170. doi: 10.1093/emboj/cdf293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sung P. Yeast Rad55 and Rad57 proteins form a heterodimer that functions with replication protein A to promote DNA strand exchange by Rad51 recombinase. Genes Dev. 1997;11:1111–1121. doi: 10.1101/gad.11.9.1111. [DOI] [PubMed] [Google Scholar]
- 53.Liu J, Qian N, Morrical SW. Dynamics of bacteriophage T4 presynaptic filament assembly from extrinsic fluorescence measurements of Gp32-single-stranded DNA interactions. J Biol Chem. 2006;281:26308–26319. doi: 10.1074/jbc.M604349200. [DOI] [PubMed] [Google Scholar]
- 54.Menetski JP, Kowalczykowski SC. Interaction of recA protein with single-stranded DNA. Quantitative aspects of binding affinity modulation by nucleotide cofactors. J Mol Biol. 1985;181:281–295. doi: 10.1016/0022-2836(85)90092-0. [DOI] [PubMed] [Google Scholar]
- 55.Krejci L, Van Komen S, Li Y, Villemain J, Reddy MS, Klein H, Ellenberger T, Sung P. DNA helicase Srs2 disrupts the Rad51 presynaptic filament. Nature. 2003;423:305–309. doi: 10.1038/nature01577. [DOI] [PubMed] [Google Scholar]
- 56.Veaute X, Jeusset J, Soustelle C, Kowalczykowski SC, Le Cam E, Fabre F. The Srs2 helicase prevents recombination by disrupting Rad51 nucleoprotein filaments. Nature. 2003;423:309–312. doi: 10.1038/nature01585. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.