

1. Introduction
Protein folding is the spontaneous process of assembling a polypeptide chain into a distinct three-dimensional structure. Knowledge of the details of this reaction lies at the heart of understanding some of the basic mechanisms of life, as the final conformation is normally the unique biologically active conformation. Conversely, protein unfolding is the competing reverse process that normally leads to denaturation and loss of function. Unfolding and misfolding are also linked to a variety of human diseases, including Alzheimer’s disease, Parkinson’s disease and number of other amyloid diseases [1]. Therefore, it is of fundamental importance to study the folding/unfolding process and to determine both the atomic details and the specific pathways between the native state and denatured/unfolded states.
Many experimental techniques are routinely employed to investigate the folding/unfolding process [2]. In addition, a number of theoretical techniques [3] are employed to complement experiment to provide an overall picture of the process. The importance of performing computer simulations is twofold. In some cases, it is possible to rationalize a number of experimental observations into a single, well-defined molecular representation. In other cases, a theoretical model can precede and guide experiments, making predictions on a complex system under ideal conditions and without needing to perturb it in the observation.
Among other simulation techniques, molecular dynamics (MD) plays a distinctive role in the area of protein folding [4]. MD is based on the numerical integration of the classical Newtonian equations of motions for all the atoms in a system, their interactions being described by empirical potential functions (or force fields) parameterized to capture, as realistically as possible, atomic interactions and fluctuations [5]. Bonded interactions include bond stretching, angle bending and dihedral angle torsions, and are described by harmonic or other simple potentials. Nonbonded interactions consist of the van der Waals contribution, described by a Lennard-Jones potential, and electrostatic forces among charged atoms that are computed using Coulomb’s law.
An attractive feature of MD is that it is a simple and yet accurate technique for sampling the energy landscape of a macromolecule in an unbiased way. Details of the positions of all the atoms of a protein can be obtained under simplified but biologically relevant conditions, by including solvent, osmolytes, other proteins, substrates, etc. In addition, one has the possibility to follow the time evolution of the whole system up to the millisecond timescale [6] either directly or indirectly through use of elevated temperature, as discussed below. In contrast to other simulation techniques for generating a statistical ensemble of conformations [3], such as Monte Carlo methods or simulated annealing, MD samples atomic motion in a continuous fashion, producing realistic and continuous pathways through time, provided the appropriate simulation conditions are used.
In this review we survey applications of MD in the area of protein folding, highlighting how such simulations can be combined with experiment to elucidate details of the folding/unfolding process. The structure of this paper is as follows. First a basic introduction to folding by MD is presented, with an emphasis on the possibility to explore folding directly, which can be achieved for small structural motifs and small ultrafast-folding proteins, or indirectly by investigating the reverse process using high-temperature unfolding simulations for larger proteins. The subsequent section focuses on how to apply MD to proteins that perform common biological functions, with the aim of characterizing their states along the pathway in the free-energy landscape. Finally, simulations performed under more complex conditions are discussed, which include protein folding in more realistic environments and in the presence of other molecules and unfolding due to external factors other than temperature.
2. Protein folding/unfolding in simulation
MD simulations represent a particularly convenient choice for investigating the folding/unfolding process. The empirical force field used in MD implicitly takes into account polarization and other multi-body effects, reducing the computational cost of a simulation and extending the limit of sampling of conformational space. This is especially important for folding simulations, which require massive calculations. The impossibility of describing traditional chemical reactions –a known shortcoming of MD techniques– does not represent an inconvenience in investigating folding/unfolding, which is possibly the most complex chemical process that does not involve the formation or rupture of covalent bonds.
2.1 Simulations of protein folding
In early studies [7–9], unbiased MD simulations described protein folding events that were mostly incomplete. However, these studies provided important insights into this complex process for several small proteins. With the advent of more computing power and the discovery of ultrafast [10] folding and unfolding systems, systematic and reproducible convergence from an extended conformation to a final state close to the native one has been achieved [11], which convincingly shows that protein folding can be obtained by brute force simulation, at least in principle. In the last decade, advancement in the field has been largely due to the steady increase in the computational power available, including the use of massively parallel computers [12, 13], inexpensive but fast processing units such as GPUs [14], or distributed computing [15].
The use of specially tailored fast-folding proteins has further reduced the computational cost necessary to achieve a direct comparison between simulation and experiment [16]. Recently, Shaw and co-workers have obtained impressive results by using a dedicated supercomputer called Anton [12] to fold a number of mini-proteins. For example, an equilibrium simulation of 100 μs capturing multiple folding/unfolding events for FiP35, a fast-folding variant of the three-stranded β-sheet Pin1 WW domain, was recently reported [6]. Subsequently, folding was obtained for 12 different fast-folding protein domains, spanning different structural classes, over a timescale ranging from 100 μs to 1 ms [17]. Protein refolding has also been achieved in shorter ns simulations that begin from more relevant, experimentally validated, “unfolded” states that effectively bypass the sampling of biologically unrealistic extended states [18].
2.2 Protein unfolding in simulation
Thermal unfolding is a phenomenon of biological interest in and of itself, as temperature is the most common factor determining protein denaturation. Unfolding simulations are useful to investigate this process and, concurrently, have been long used to study the folding process “in reverse” [19, 20]. The unparalleled convenience of performing high temperature (up to 500 K) simulations is due to two distinct reasons. The first one is the uniqueness of the protein folded state, which allows the simulation to begin from a relevant conformational state. The other rationale is that the process is speeded up due to high temperature to overcome energetic barriers, allowing to reduce the timescale necessary for mapping the full process and to perform multiple simulations that can capture heterogeneity in the pathway. For these reasons, high-temperature MD simulations have been applied to several proteins.
An important issue is the reliability of comparing high temperature unfolding simulations with experiments performed at lower or even room temperature. One of the criticisms is that common force fields were not parameterized for high temperature. However, MD simulations performed with different force fields and under a variety of simulation conditions have shown a consistent mutual agreement in finding similar unfolding pathways when applied to the same proteins, as first demonstrated for CI2 [21–23]. In addition, the unfolding pathway has been shown to be essentially independent of temperature by both simulations performed at a variety of temperatures [24] and comparing simulations in water to those in urea [25, 26] and with experiment [27]. Differences in the folding pathway and in the protein states along it are generally consistent with the fact that MD samples an ensemble that fluctuates around an average representing the experimental data. While adequacy of the empirical force field may represent a concern for unfolding simulations at high temperature, this is no different for folding simulations at room temperature. As an example, the force field employed was demonstrated [6] to be responsible for the incorrect folded structure previously obtained [28] in room temperature MD simulation of the aforementioned FiP35 WW domain.
Another common criticism of unfolding studies is the possibility that folding and unfolding may not follow the same pathway in the forward and reverse directions. In fact, caution has to be exercised in invoking the principle of microscopic reversibility under non-equilibrium conditions. However, simulation studies have demonstrated microscopic reversibility for both continuous trajectories at a protein’s melting temperature where unfolding and refolding occur [29] and in thermally quenched simulations in which refolding is triggered by lowering the temperature [18]. In both cases the process of unfolding mirrored the process of refolding. Recently, it has been pointed out by Bhatt and Zuckerman [30] that an approximate symmetry can be determined, even for processes out of equilibrium and without invoking the principle of microscopic reversibility, when representative states in a dynamical direct/inverse process are well determined. This condition is automatically verified when one of these states is the native structure of a protein. When the simulated system is simple enough and the computational power is sufficient to allow for “equilibrium” MD, it is possible to show directly that the pathway of folding/unfolding is not only the same, but also independent of temperature. Results have been obtained for several proteins, demonstrating both the independence of the unfolding pathway from temperature [24] and the possibility of sampling multiple folding/unfolding events [6, 11, 29], including not just recovering the native state but also demonstrating that the same intermediate and transition states are populated during unfolding and refolding [29].
3. Exploring the free energy landscape of protein folding
Prediction and validation of the correct folding/unfolding pathways and final structures, as obtained for a number of fast-folding proteins [17], constitutes a decisive demonstration of the power of MD simulations. On the other hand, fast folders are small, simple polypeptides that are far from being representative of the vast majority of proteins of biological interest. Therefore, MD simulations are often used to complement experiment by reproducing the behavior of larger proteins, but concentrating on specific stages corresponding to a more limited portion of the folding/unfolding reaction coordinate. On proceeding towards the native conformation, the folding of a protein can be described in terms of a succession of states: from the unfolded one, possibly through one or more intermediate states, passing the transition state to reach at last the final structure.
The starting point in the folding reaction is the unfolded or denatured state. This state is normally the most difficult one to characterize, because of its poorly defined structural features. However, highly unfolded denatured states are not usually obtained except under extreme denaturing conditions. Thus, for many proteins the denatured state may be identified in MD through conformational clustering techniques and described in terms of a single loose topology. Simulations have long shown the propensity to form both dynamic native-like and nonnative structures in the unfolded state [17], which has been verified by NMR and other experimental techniques. In particular, the early studies presented predictions of residual structure in the denatured state and experimental validation came after the simulations were published [31–33]. Similar studies are also expected to shed light into the structural and dynamical features of intrinsically disordered proteins, a vast and yet poorly understood class of proteins that challenges the concept of a uniquely structured native state. The importance of determining a representative unfolded conformation of a protein relies also on its specific role as a starting structure in folding simulations, which can be critical to estimate correctly the time required for the completion of the process [34, 35], because, as mentioned above, the use of extended states is unrealistic and requires increased computational time to reach regions of conformational space sampled by experimental studies for the onset of folding.
Along the unfolding pathway, the simulation may reveal the existence of one or more intermediate states. Such partially folded states are highly dynamic and could easily escape detection in the experiment. Early on, the microscopic existence of intermediate states was postulated based on observations in simulations and then through experimental validation by stabilization of said “unobservable” intermediates [27]. Now the possibility of the presence of “hidden” or “silent” intermediates is commonly accepted even for relatively simple proteins [2]. The picture is further complicated because not only on-pathway intermediate states can exist, but also off-pathway ones. Characterization of the latter are of particular interest for reproducing in MD the conditions leading to misfolded protein conformations [36]. As in the case of unfolded states, results on intermediates can be checked against NMR [37] and several other experimental techniques [2], including time-resolved single-molecule fluorescence based on Förster resonance energy transfer (FRET) [38].
On top of the high-energy barrier separating the unfolded and folded state is the transition state for folding/unfolding. From a structural point of view, the transition state is an ensemble of partially folded conformations, often closely resembling the protein topology in the native form, but with equal probability to evolve back into the denatured one. Fast-folding proteins reveal in simulation [17] an energy barrier of the order of at most a few kBT (where kB is Boltzmann’s constant and T is the temperature), i.e. comparable with thermal fluctuations. Even such small differences determine a two-state reaction, with a transition state in between the two basins representing the unfolded and folded state. The transition state is only transiently populated because protein structures do not accumulate in a single conformation. Therefore, MD simulations are employed to provide atomistic details corresponding to this transient ensemble of conformers.
The most common experimental procedure for studying the transition state is to apply the so-called Φ-value analysis [27]. This technique consists in using protein engineering to mutate a number of residues along the polypeptide chain and measuring the corresponding effect on the energetics of the denatured, transition and native state along the folding/unfolding pathway. By using the denatured state as a reference, it is possible to measure the relative destabilization energy of the transition and native state, ΔΔGT and ΔΔGN, respectively. The ratio of the two values, Φ = ΔΔGT/ΔΔGN, gives indication on the local structure at the site of mutation in the transition state. The extreme values of Φ are 0 when the transition state at the site of mutation is identical to the denatured state and 1 when is identical to the native state; intermediate values correspond (in proportion) to a partial structure. Thus, by using standard kinetic and thermodynamic experiments, the Φ-value analysis allows to deduce structural details from the energetics of the process. MD can be used to sum up the wealth of data in a single, comprehensive picture. In addition, simulations can be used to test the assumption that the mutations act as merely probes of wild-type behavior without dramatically affecting the folding/unfolding pathway [39, 40].
At the end of the entire folding pathway lies the native state, which is of importance in folding simulations because of its intrinsic nature of “kinetic hub” [41] of the entire process. The folded state is also the elect starting point for simulating protein unfolding or misfolding by using high temperature MD simulations (as discussed in the previous section). However, in these latter cases, sampled structures become progressively more labile and less significant the more a simulation departs from the starting state, unless they are analyzed in terms of an ensemble with extensive exploration of conformational space. For this reason, many MD studies focus on the early events of the unfolding reaction, which can be due to thermal effects [42, 43] or other environmental conditions, such as pH [44] and solvent [45].
The MD technique may provide important information regarding the folding process even when simulations are performed on a native protein at room temperature. Analysis of the flexibility of the protein backbone could give hints on specific regions that are prone to unfold, because denaturation often happens by selective excitation of fluctuations along modes that are already detectable in the native state [46]. Conversely, the same regions are likely to be among the last ones forming or reaching a definitive (native) structure during the folding process. More recent calculations of protein flexibility and comparison with unfolding behavior by MD have confirmed these findings on a larger scale including 187 proteins as part of the Dynameomics project [47, 48]. Comparison of the dynamics at room temperature for two or more proteins with high similarity can give information on their thermal stability and on specific regions that are critical in determining it. As an example, differences in the simulated dynamics of corresponding mesophilic and thermophilic proteins [49] can be used to rationalize differences in their stability at increasing temperature. In contrast, similarities in dynamical features can be found across a shared fold, superfamily or family of proteins [50–53], giving useful indications to reveal conserved residues that are fundamental to preserve the folding topology.
4. Simulation of folding/unfolding under more complex conditions
4.1 Folding under more complex conditions
A number of factors can increase the complexity of protein folding in vivo, hampering an accurate reproduction of the process both in the experiment and in simulation. In the sequential biosynthesis of the polypeptide chain, the exit tunnel in the ribosome from which the nascent protein emerges may determine a bias in the folding process. Pande and coworkers have used MD to investigated how the ribosome interacts with an unfolded protein [54], by probing the exit tunnel with different amino acid side chains and determining the corresponding, sequence-specific free energy barriers. In addition, both the behavior of the solvent [55] and the accessibility of Na+ and Cl− ions [54] in the tunnel were analyzed and suggested to play a role in the folding process. Research on this aspect could help to elucidate obscure and yet important biological processes, such as modulation of the rate of translation of some proteins. MD simulations [56] have been used to explore the mechanism by which the presence of the protein SecM in the exit tunnel mediates translational stalling in the ribosome, providing a rationale for cryo-electron microscopy data [57].
Molecular crowding and confinement represent closely related and additional factors affecting protein folding. Concentration of macromolecules in cells exceeds 102 g/L [58], while experimental studies are usually performed at much lower concentration. Zhou and collaborators have developed a method of postprocessing MD trajectories [59, 60] to use the simulated conformations of a protein to calculate the change in the chemical potential after transfer to a crowded solution. How this change depends on the reaction coordinates reflects the influence of crowding on the energy landscape available for the folding polypeptide. Effects on folding/unfolding rates [61] and native state stability [60] can be modest, in agreement with the experiment [62, 63]. However, results depend significantly upon the size of the crowders and are non-additive for combinations of different crowding agents, pointing out to the necessity of using composite mixtures to mimic reality both in silico and in vitro. A recent study directly addressed the effect of neighboring molecules on native state dynamics and thermal unfolding for a well-characterized system [64]. In comparing these “test-tube” simulations (18 mM protein) to conventional single-molecule simulations it was found that neighboring molecules slowed unfolding slightly but the pathway was unaffected. Carrying out the high temperature simulations further led to aggregation through nonpolar inter-protein interactions through both folded and unfolded regions of the protein.
The effect of confinement has been directly addressed in MD simulations of villin headpiece inside an inert nanopore [65]. The unfolded state is destabilized in the nanopore, thereby promoting folding. In contrast, the confinement of water in the pore leads to a solvent-mediated effect that destabilizes the native state and produces an unfolded state more compact than the one obtained in bulk solution. The competing effects influence the kinetics and thermodynamics of the folding process. Steric confinement of a protein has a special role in the folding reaction assisted by molecular chaperones in the cell. In particular, chaperonins are a subclass of chaperones with a hollow structure that provides a cage for the correct folding of a large number of proteins. It is becoming increasingly evident [66] that chaperonins not only prevent aggregation of the polypeptide chain but can also act as “foldase/unfoldase enzymes”, speeding up the folding reaction and reversing the process for misfolded conformations. MD simulations are contributing to clarifying the mechanisms of assisted folding by giving insights on long-range structural transitions in the chaperonin GroEL [67, 68] and on the behavior of other molecular chaperones [69, 70].
Many proteins require the addition of cofactors to perform their function. The presence of even a small prosthetic group can dramatically modify the folding process of a holo protein with respect to the apo form. For example, MD simulations of the Cys2His2-type zinc-finger motif [71] showed that the metal ion binds first to the two cysteine ligands and only later to the two histidines. In addition, it participates in other earlier and significant nonnative contacts with various protein residues and water molecules, contributing actively to the folding process, not only to stabilize the final structure. The effect of a number of other common post-translational modifications of proteins, such as phosphorylation and glycosylation, are currently investigated in folding/unfolding simulations [72, 73]. In particular, glycosylation can help the folding process through the formation of specific long-range contacts involving the oligosaccharide moiety in the folding nucleus [73], contributing also to enhance the thermal stability of the protein native state [74].
4.2 Unfolding under more complex conditions
Unfolding of a protein can also be induced by high pressure. The effects of pressure on protein folding/unfolding has been extensively investigated in simulation by Garcia and coworkers [75–77], showing that the pressure-denatured state of a protein is generally more compact than the temperature-denatured one and with more retained elements of secondary structure. Pressure leads to the unfolding of proteins because the molar volume of the denatured state is smaller that that of the native state [78]. This has been tentatively attributed to a combination of destabilization of internal (micro)cavities in the protein, differences between the density of water surrounding the protein with respect to bulk water and changes in the structure of the bulk solvent. Recently, the first of the three effects has emerged as being by far the most relevant [77], as demonstrated by combining MD simulations with crystallography, fluorescence and NMR spectroscopy.
The presence in solution of additional cosolvents or osmolytes may contribute to the unfolding of a polypeptide. Simulations carried out in a mixture of water and ethanol [79], acetonitrile [80] or other organic solvents [81] can determine the degree of destabilization of a protein, leading to possible applications in non-aqueous enzymology. Urea plays a special role because of its widespread use as a denaturing agent. MD is contributing to reveal how water and urea cooperate to unfold proteins [25, 82, 83]. In the first such study it was shown that urea exerts both a direct and indirect effect on protein unfolding [25]. The indirect effect involves perturbation of water structure that both facilitates the exposure of nonpolar groups and the liberation of water molecules to “attack” the protein. Then, after unfolding begins urea moves in and makes specific interactions with the protein, thereby stabilizing the unfolded state. In recent microsecond simulations of lysozyme in concentrated urea solution [82] the specific importance of Van der Waals interactions between urea and the protein chain was highlighted. The action of guanidinium chloride, another common denaturing agent, seems to be similar to urea in that it diminishes the attraction between hydrophobic surfaces [84], possibly preventing hydrophobic collapse of expanded protein conformations, as observed previously in 60% MeOH simulations [8]. However, many other details of the microscopic mechanisms of action of urea and guanidinium appear to be different [84, 85]. In addition, osmolytes can act as chemical chaperones. For example, MD simulations have been used to determine how trimethylamine oxide counteracts the effects of urea [86].
Mechanical unfolding of a protein can be obtained by applying external forces above a few tens of piconewtons [87]. Force-probe simulations, also known as steered MD, can be used to follow the structural changes obtained by applying to selected protein atoms either a constant force or a constant pulling velocity. Steered MD provides an atomistic view for experimental results obtained with single-molecule atomic force microscopy (AFM)-based force spectroscopy [87]. One well-studied model system is the immunoglobulin domain I27 of titin, a giant protein (about 30,000 amino acid residues) responsible for the contraction and extension of muscles [88–90]. In particular, the height of the potential energy barrier [89] and the additional effect of temperature during pulling was determined [88]. Steered MD and force spectroscopy have also recently cooperated to reveal multiple pathways in the unfolding of a slipknotted protein [91]. Such studies are expected to reveal important details in the folding/unfolding pathway of proteins with complex topological fold.
5. Concluding remarks
Protein folding is an intrinsically complex process whose investigation requires a concerted effort between experiment and theory. Computer simulations have been helping to describe the atomistic details of the folding/unfolding reaction and characterize the structural states occurring along the pathway. At present, the complete folding process can be reproduced only for a restricted number of fast-folding miniproteins, but many of its fundamental aspects are being revealed in simulation for a vast number of common proteins under a variety of biologically relevant conditions. In this respect, MD constitutes an effective tool to support the experiment, providing both a framework for data interpretation and a guide for further investigations. Immediate and future fallouts of the use of simulations are expected in a range of important areas, from understanding functional mechanisms occurring in the cells to the rational improvement of protein stability for preventing or treating dysfunctions.
Fig. 1.
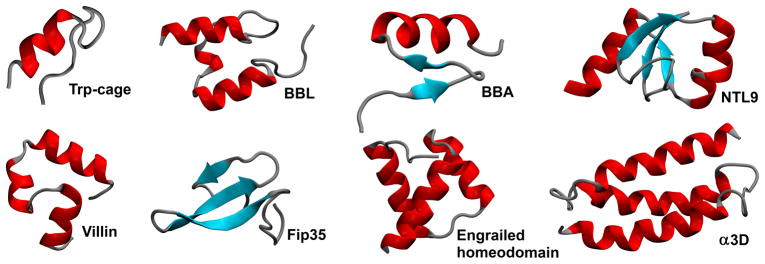
Miniproteins folded in brute-force MD simulations [6, 17, 18]. Different structural classes are present: α-helical, β-sheet and mixed α/β structure.
Fig. 2.
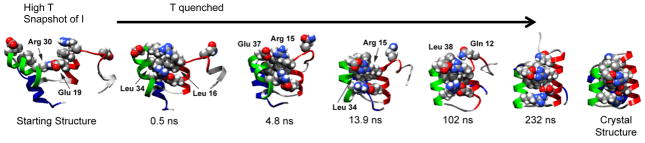
Refolding in simulation (Structures of Engrailed homeodomain showing refolding of the protein on quenching the denatured structure obtained in simulation [18]).
Fig. 3.
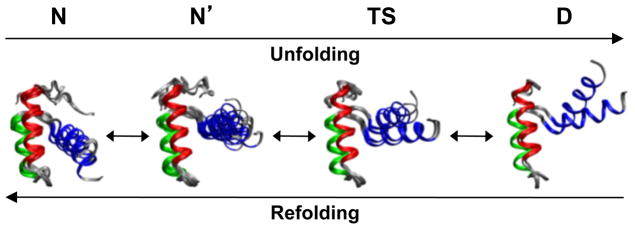
Thermal unfolding in simulation (Snapshots of protein structures at different stages along a simulated unfolding pathways [29]).
Fig. 4.

Free energy as a function of reaction coordinate for a simple, two-state folding process from denatured to native state, with no intermediate state(s).
Fig. 5.

Example of Φ-value analysis. (a) Experimental (solid line) and simulated (dotted line) Φ-values [4, 21] for selected mutants of CI2. (b) Scatter plot of the points shown in panel a. (c) Scatter plot comparing experimental Φ-values and calculated average S-values, i.e. the product of local secondary structure and the amount of native-state contacts.
Fig. 6.

Protein native state as kinetic hub of the folding process [41]. Letters indicate the unfolded (U), intermediate (I), and native state (N).
Fig. 7.

Positional fluctuations of Cα atoms as a function of residue number for amicyanin from two sources, (solid square) Paracoccus denitrificans and (open circle) Paraccocus versutus, obtained in principal component analysis: (a) first, (b) second and (c) third eigenvector [52]. Comparison of backbone flexibility gives indications on specific regions that contribute to maintain the protein folding.
Fig. 8.

Macromolecular crowding inside cells shifts the folding equilibrium toward the protein native state because of excluded volume effects [59, 60]. The stability of a protein may decrease going from (a) a crowded environment to (b) a less crowded one.
Fig. 9.

Simulated folding/unfolding of the blue copper protein azurin. The presence of a single Cu ion (in blue) influences significantly the dynamics [93, 94] and thermal stability of the protein, without affecting the folding topology.
Fig. 10.
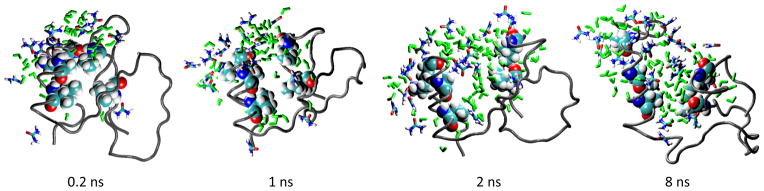
Influence of cosolvents and osmolytes on the unfolding (Effects of urea on the unfolding of CI2 in simulation [25]).
Fig. 11.

Mechanical unfolding of titin domain I27. Protein in the (a) starting structure, with native hydrogen bonds in the first β-strand, and (b) detail of the hydrogen bond breaking after applying a steering force in simulation [89, 90].
Highlights.
Molecular Dynamics (or MD) is one of the most widespread and effective simulation technique.
Protein folding can be reproduced in simulation under biologically relevant conditions.
Structural states along the folding/unfolding pathway are characterized at atomic resolution.
Simulation provides detailed structural models for interpreting experimental data.
Acknowledgments
VD is grateful for funding from the National Institutes of Health (GM 50789) that made the various simulations studies from her group cited here possible. VMD - Visual Molecular Dynamics [92] was used for protein displays.
References
- 1.Chiti F, Dobson CM. Annual Review of Biochemistry. 2006;75:333–366. doi: 10.1146/annurev.biochem.75.101304.123901. [DOI] [PubMed] [Google Scholar]
- 2.Bartlett AI, Radford SE. Nature Structural & Molecular Biology. 2009;16:582–588. doi: 10.1038/nsmb.1592. [DOI] [PubMed] [Google Scholar]
- 3.Liwo A, Czaplewski C, Oldziej S, Scheraga HA. Current Opinion in Structural Biology. 2008;18:134–139. doi: 10.1016/j.sbi.2007.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Daggett V. Accounts of Chemical Research. 2002;35:422–429. doi: 10.1021/ar0100834. [DOI] [PubMed] [Google Scholar]
- 5.Beck DAC, Daggett V. Methods. 2004;34:112–120. doi: 10.1016/j.ymeth.2004.03.008. [DOI] [PubMed] [Google Scholar]
- 6.Shaw DE, Maragakis P, Lindorff-Larsen K, Piana S, Dror RO, Eastwood MP, Bank JA, Jumper JM, Salmon JK, Shan YB, Wriggers W. Science. 2010;330:341–346. doi: 10.1126/science.1187409. [DOI] [PubMed] [Google Scholar]
- 7.Duan Y, Kollman PA. Science. 1998;282:740–744. doi: 10.1126/science.282.5389.740. [DOI] [PubMed] [Google Scholar]
- 8.Alonso DOV, Daggett V. Journal of Molecular Biology. 1995;247:501–520. doi: 10.1006/jmbi.1994.0156. [DOI] [PubMed] [Google Scholar]
- 9.Alonso DOV, Daggett V. Protein Science. 1998;7:860–874. doi: 10.1002/pro.5560070404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kubelka J, Hofrichter J, Eaton WA. Current Opinion in Structural Biology. 2004;14:76–88. doi: 10.1016/j.sbi.2004.01.013. [DOI] [PubMed] [Google Scholar]
- 11.Seibert MM, Patriksson A, Hess B, van der Spoel D. Journal of Molecular Biology. 2005;354:173–183. doi: 10.1016/j.jmb.2005.09.030. [DOI] [PubMed] [Google Scholar]
- 12.Shaw DE, Deneroff MM, Dror RO, Kuskin JS, Larson RH, Salmon JK, Young C, Batson B, Bowers KJ, Chao JC, Eastwood MP, Gagliardo J, Grossman JP, Ho CR, Ierardi DJ, Kolossvary I, Klepeis JL, Layman T, Mcleavey C, Moraes MA, Mueller R, Priest EC, Shan YB, Spengler J, Theobald M, Towles B, Wang SC. Communications of the Acm. 2008;51:91–97. [Google Scholar]
- 13.Gara A, Blumrich MA, Chen D, Chiu GLT, Coteus P, Giampapa ME, Haring RA, Heidelberger P, Hoenicke D, Kopcsay GV, Liebsch TA, Ohmacht M, Steinmacher-Burow BD, Takken T, Vranas P. Ibm Journal of Research and Development. 2005;49:195–212. [Google Scholar]
- 14.Stone JE, Hardy DJ, Ufimtsev IS, Schulten K. Journal of Molecular Graphics & Modelling. 2010;29:116–125. doi: 10.1016/j.jmgm.2010.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shirts M, Pande VS. Science. 2000;290:1903–1904. doi: 10.1126/science.290.5498.1903. [DOI] [PubMed] [Google Scholar]
- 16.Schaeffer RD, Fersht A, Daggett V. Current Opinion in Structural Biology. 2008;18:4–9. doi: 10.1016/j.sbi.2007.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lindorff-Larsen K, Piana S, Dror RO, Shaw DE. Science. 2011;334:517–520. doi: 10.1126/science.1208351. [DOI] [PubMed] [Google Scholar]
- 18.McCully ME, Beck DAC, Fersht AR, Daggett V. Biophysical Journal. 2010;99:1628–1636. doi: 10.1016/j.bpj.2010.06.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Daggett V, Levitt M. Proceedings of the National Academy of Sciences of the United States of America. 1992;89:5142–5146. doi: 10.1073/pnas.89.11.5142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Daggett V, Levitt M. Journal of Molecular Biology. 1992;223:1121–1138. doi: 10.1016/0022-2836(92)90264-k. [DOI] [PubMed] [Google Scholar]
- 21.Li AJ, Daggett V. Proceedings of the National Academy of Sciences of the United States of America. 1994;91:10430–10434. doi: 10.1073/pnas.91.22.10430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Li AJ, Daggett V. Journal of Molecular Biology. 1996;257:412–429. doi: 10.1006/jmbi.1996.0172. [DOI] [PubMed] [Google Scholar]
- 23.Lazaridis T, Karplus M. Science. 1997;278:1928–1931. doi: 10.1126/science.278.5345.1928. [DOI] [PubMed] [Google Scholar]
- 24.Day R, Bennion BJ, Ham S, Daggett V. Journal of Molecular Biology. 2002;322:189–203. doi: 10.1016/s0022-2836(02)00672-1. [DOI] [PubMed] [Google Scholar]
- 25.Bennion BJ, Daggett V. Proceedings of the National Academy of Sciences of the United States of America. 2003;100:5142–5147. doi: 10.1073/pnas.0930122100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Day R, Daggett V. Protein Science. 2005;14:1242–1252. doi: 10.1110/ps.041226005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Daggett V, Fersht A. Nature Reviews Molecular Cell Biology. 2003;4:497–502. doi: 10.1038/nrm1126. [DOI] [PubMed] [Google Scholar]
- 28.Freddolino PL, Liu F, Gruebele M, Schulten K. Biophysical Journal. 2008;94:L75–L77. doi: 10.1529/biophysj.108.131565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.McCully ME, Beck DAC, Daggett V. Biochemistry. 2008;47:7079–7089. doi: 10.1021/bi800118b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bhatt D, Zuckerman DM. Journal of Chemical Theory and Computation. 2011;7:2520–2527. doi: 10.1021/ct200086k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bond CJ, Wong KB, Clarke J, Fersht AR, Daggett V. Proceedings of the National Academy of Sciences of the United States of America. 1997;94:13409–13413. doi: 10.1073/pnas.94.25.13409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wong KB, Clarke J, Bond CJ, Neira JL, Freund SMV, Fersht AR, Daggett V. Journal of Molecular Biology. 2000;296:1257–1282. doi: 10.1006/jmbi.2000.3523. [DOI] [PubMed] [Google Scholar]
- 33.Kazmirski SL, Wong KB, Freund SMV, Tan YJ, Fersht AR, Daggett V. Proceedings of the National Academy of Sciences of the United States of America. 2001;98:4349–4354. doi: 10.1073/pnas.071054398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Paci E, Cavalli A, Vendruscolo M, Caflisch A. Proceedings of the National Academy of Sciences of the United States of America. 2003;100:8217–8222. doi: 10.1073/pnas.1331838100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Best RB, Mittal J. Proceedings of the National Academy of Sciences of the United States of America. 2011;108:11087–11092. doi: 10.1073/pnas.1016685108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.van der Kamp MW, Daggett V. Prion Proteins. 2011;305:169–197. doi: 10.1007/128_2011_158. [DOI] [PubMed] [Google Scholar]
- 37.Korzhnev DM, Kay LE. Accounts of Chemical Research. 2008;41:442–451. doi: 10.1021/ar700189y. [DOI] [PubMed] [Google Scholar]
- 38.Schuler B, Eaton WA. Current Opinion in Structural Biology. 2008;18:16–26. doi: 10.1016/j.sbi.2007.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pan YP, Daggett V. Biochemistry. 2001;40:2723–2731. doi: 10.1021/bi0022036. [DOI] [PubMed] [Google Scholar]
- 40.Daggett V, Li AJ, Fersht AR. Journal of the American Chemical Society. 1998;120:12740–12754. [Google Scholar]
- 41.Bowman GR, Pande VS. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:10890–10895. doi: 10.1073/pnas.1003962107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rizzuti B, Daggett V, Guzzi R, Sportelli L. Biochemistry. 2004;43:15604–15609. doi: 10.1021/bi048685t. [DOI] [PubMed] [Google Scholar]
- 43.Lin YW, Ni FY, Ying TL. Journal of Molecular Structure-Theochem. 2009;898:82–89. [Google Scholar]
- 44.van der Kamp MW, Daggett V. Biophysical Journal. 2010;99:2289–2298. doi: 10.1016/j.bpj.2010.07.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Borkar AN, Rout MK, Hosur RV. Plos One. 2011;6 doi: 10.1371/journal.pone.0019830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Roccatano D, Daidone I, Ceruso MA, Bossa C, Di Nola A. Biophysical Journal. 2003;84:1876–1883. doi: 10.1016/S0006-3495(03)74995-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Benson NC, Daggett V. Protein Science. 2008;17:2038–2050. doi: 10.1110/ps.037473.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.van der Kamp MW, Schaeffer RD, Jonsson AL, Scouras AD, Simms AM, Toofanny RD, Benson NC, Anderson PC, Merkley ED, Rysavy S, Bromley D, Beck DAC, Daggett V. Structure. 2010;18:423–435. doi: 10.1016/j.str.2010.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sterpone F, Melchionna S. Chemical Society Reviews. 2012;41:1665–1676. doi: 10.1039/c1cs15199a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Grottesi A, Sansom MSP. Febs Letters. 2003;535:29–33. doi: 10.1016/s0014-5793(02)03849-8. [DOI] [PubMed] [Google Scholar]
- 51.Pandini A, Bonati L. Protein Engineering Design & Selection. 2005;18:127–137. doi: 10.1093/protein/gzi017. [DOI] [PubMed] [Google Scholar]
- 52.Rizzuti B, Sportelli L, Guzzi R. Proteins-Structure Function and Bioinformatics. 2009;74:961–971. doi: 10.1002/prot.22204. [DOI] [PubMed] [Google Scholar]
- 53.Velazquez-Muriel JA, Rueda M, Cuesta I, Pascual-Montano A, Orozco M, Carazo JM. Bmc Structural Biology. 2009;9 doi: 10.1186/1472-6807-9-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Petrone PM, Snow CD, Lucent D, Pande VS. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:16549–16554. doi: 10.1073/pnas.0801795105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lucent D, Snow CD, Aitken CE, Pande VS. Plos Computational Biology. 2010;6 doi: 10.1371/journal.pcbi.1000963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Gumbart J, Schreiner E, Wilson DN, Beckmann R, Schulten K. Biophysical Journal. 2012;103:331–341. doi: 10.1016/j.bpj.2012.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bhushan S, Hoffmann T, Seidelt B, Frauenfeld J, Mielke T, Berninghausen O, Wilson DN, Beckmann R. Plos Biology. 2011;9 doi: 10.1371/journal.pbio.1000581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Luby-Phelps K. International Review of Cytology - a Survey of Cell Biology. 2000;192(192):189–221. doi: 10.1016/s0074-7696(08)60527-6. [DOI] [PubMed] [Google Scholar]
- 59.Qin SB, Minh DDL, McCammon JA, Zhou HX. Journal of Physical Chemistry Letters. 2010;1:107–110. doi: 10.1021/jz900023w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Qin SB, Zhou HX. Biophysical Journal. 2009;97:12–19. doi: 10.1016/j.bpj.2009.03.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Tjong H, Zhou HX. Biophysical Journal. 2010;98:2273–2280. doi: 10.1016/j.bpj.2010.01.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ai XJ, Zhou Z, Bai YW, Choy WY. Journal of the American Chemical Society. 2006;128:3916–3917. doi: 10.1021/ja057832n. [DOI] [PubMed] [Google Scholar]
- 63.Spencer DS, Xu K, Logan TM, Zhou HX. Journal of Molecular Biology. 2005;351:219–232. doi: 10.1016/j.jmb.2005.05.029. [DOI] [PubMed] [Google Scholar]
- 64.McCully ME, Beck DAC, Daggett V. Proceedings of the National Academy of Sciences of the United States of America. 2012 in press. [Google Scholar]
- 65.Lucent D, Vishal V, Pande VS. Proceedings of the National Academy of Sciences of the United States of America. 2007;104:10430–10434. doi: 10.1073/pnas.0608256104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Lucent D, England J, Pande V. Physical Biology. 2009;6 doi: 10.1088/1478-3975/6/1/015003. [DOI] [PubMed] [Google Scholar]
- 67.Ma JP, Karplus M. Proceedings of the National Academy of Sciences of the United States of America. 1998;95:8502–8507. doi: 10.1073/pnas.95.15.8502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Piggot TJ, Sessions RB, Burston SG. Biochemistry. 2012;51:1707–1718. doi: 10.1021/bi201237a. [DOI] [PubMed] [Google Scholar]
- 69.Colombo G, Morra G, Meli M, Verkhivker G. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:7976–7981. doi: 10.1073/pnas.0802879105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ohtaki A, Kida H, Miyata Y, Ide N, Yonezawa A, Arakawa T, Lizuka R, Noguchi K, Kita A, Odaka M, Miki K, Yohda M. Journal of Molecular Biology. 2008;376:1130–1141. doi: 10.1016/j.jmb.2007.12.010. [DOI] [PubMed] [Google Scholar]
- 71.Li WF, Zhang J, Wang J, Wang W. Journal of the American Chemical Society. 2008;130:892–900. doi: 10.1021/ja075302g. [DOI] [PubMed] [Google Scholar]
- 72.Chen HF. Plos One. 2009;4 doi: 10.1371/journal.pone.0006516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Kaushik S, Mohanty D, Surolia A. Protein Science. 2011;20:465–481. doi: 10.1002/pro.578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.DeMarco ML, Daggett V. Journal of Neurochemistry. 2009;109:60–73. doi: 10.1111/j.1471-4159.2009.05892.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Sarupria S, Ghosh T, Garcia AE, Garde S. Proteins-Structure Function and Bioinformatics. 2010;78:1641–1651. doi: 10.1002/prot.22680. [DOI] [PubMed] [Google Scholar]
- 76.Rouget JB, Aksel T, Roche J, Saldana JL, Garcia AE, Barrick D, Royer CA. Journal of the American Chemical Society. 2011;133:6020–6027. doi: 10.1021/ja200228w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Roche J, Caro JA, Norberto DR, Barthe P, Roumestand C, Schlessman JL, Garcia AE, Garcia-Moreno B, Royer CA. Proceedings of the National Academy of Sciences of the United States of America. 2012;109:6945–6950. doi: 10.1073/pnas.1200915109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Royer CA. Biochimica Et Biophysica Acta-Protein Structure and Molecular Enzymology. 2002;1595:201–209. doi: 10.1016/s0167-4838(01)00344-2. [DOI] [PubMed] [Google Scholar]
- 79.Lousa D, Baptista AM, Soares CM. Journal of Chemical Information and Modeling. 2012;52:465–473. doi: 10.1021/ci200455z. [DOI] [PubMed] [Google Scholar]
- 80.Cruz A, Ramirez E, Santana A, Barletta G, Lopez GE. Molecular Simulation. 2009;35:205–212. [Google Scholar]
- 81.Micaelo NM, Soares CM. Journal of Physical Chemistry B. 2008;112:2566–2572. doi: 10.1021/jp0766050. [DOI] [PubMed] [Google Scholar]
- 82.Hua L, Zhou RH, Thirumalai D, Berne BJ. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:16928–16933. doi: 10.1073/pnas.0808427105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Stumpe MC, Grubmuller H. Biophysical Journal. 2009;96:3744–3752. doi: 10.1016/j.bpj.2009.01.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.England JL, Pande VS, Haran G. Journal of the American Chemical Society. 2008;130:11854–11855. doi: 10.1021/ja803972g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.O’Brien EP, Dima RI, Brooks B, Thirumalai D. Journal of the American Chemical Society. 2007;129:7346–7353. doi: 10.1021/ja069232+. [DOI] [PubMed] [Google Scholar]
- 86.Bennion BJ, Daggett V. Proceedings of the National Academy of Sciences of the United States of America. 2004;101:6433–6438. doi: 10.1073/pnas.0308633101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Puchner EM, Gaub HE. Current Opinion in Structural Biology. 2009;19:605–614. doi: 10.1016/j.sbi.2009.09.005. [DOI] [PubMed] [Google Scholar]
- 88.Bung N, Priyakumar UD. Journal of Molecular Modeling. 2012;18:2823–2829. doi: 10.1007/s00894-011-1298-7. [DOI] [PubMed] [Google Scholar]
- 89.Lu H, Schulten K. Chemical Physics. 1999;247:141–153. [Google Scholar]
- 90.Lu H, Schulten K. Biophysical Journal. 2000;79:51–65. doi: 10.1016/S0006-3495(00)76273-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.He CZ, Genchev GZ, Lu H, Li HB. Journal of the American Chemical Society. 2012;134:10428–10435. doi: 10.1021/ja3003205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Humphrey W, Dalke A, Schulten K. Journal of Molecular Graphics & Modelling. 1996;14:33–38. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- 93.Rizzuti B, Swart M, Sportelli L, Guzzi R. Journal of Molecular Modeling. 2004;10:25–31. doi: 10.1007/s00894-003-0165-6. [DOI] [PubMed] [Google Scholar]
- 94.Rizzuti B, Sportelli L, Guzzi R. Biophysical Chemistry. 2007;125:532–539. doi: 10.1016/j.bpc.2006.11.003. [DOI] [PubMed] [Google Scholar]