

Abstract
We present a gene expression atlas of early mouse craniofacial development. Laser capture microdissection (LCM) was used to isolate cells from the principal critical micro-regions, whose development, differentiation and signaling interactions are responsible for the construction of the mammalian face. At E8.5, as migrating neural crest cells begin to exit the neural fold/epidermal ectoderm boundary, we examined the cranial mesenchyme, composed of mixed neural crest and paraxial mesoderm cells, as well as cells from adjacent neuroepithelium. At E9.5 cells from the cranial mesenchyme, overlying olfactory placode/epidermal ectoderm, and underlying neuroepithelium, as well as the emerging mandibular and maxillary arches were sampled. At E10.5, as the facial prominences form, cells from the medial and lateral prominences, the olfactory pit, multiple discrete regions of underlying neuroepithelium, the mandibular and maxillary arches, including both their mesenchymal and ectodermal components, as well as Rathke’s pouch, were similarly sampled and profiled using both microarray and RNA-seq technologies. Further, we performed single cell studies to better define the gene expression states of the early E8.5 pioneer neural crest cells and paraxial mesoderm. Taken together, and analyzable by a variety of biological network approaches, these data provide a complementing and cross-validating resource capable of fueling discovery of novel compartment specific markers and signatures whose combinatorial interactions of transcription factors and growth factors/receptors are responsible for providing the master genetic blueprint for craniofacial development.
Keywords: mammalian craniofacial development, gene expression atlas, RNA-seq, microarrays
Introduction
An atlas of gene expression profiles can provide a valuable resource for the research community. A prime example is the Allen Brain Atlas, which was initiated in 2003 to create a comprehensive expression dataset to advance fundamental discovery into brain function (Lein et al., 2007). Hundreds of thousands of in situ hybridizations were carried out to define gene expression patterns in the developing and adult mouse brain, human brain, and mouse spinal cord.
The GenitoUrinary Molecular Anatomy Project (GUDMAP.ORG) provides another example of a gene expression atlas (Harding et al., 2011; McMahon et al., 2008). A few thousand in situ hybridizations were carried out. In addition, however, the diverse compartments of the kidney were gene expression profiled using a combination of laser capture microdissection (LCM) and microarrays (Brunskill et al., 2008) as well as RNA-seq (Brunskill et al., 2011a; Brunskill et al., 2011b; Brunskill and Potter, 2010; Brunskill et al., 2011c). The results define the changing waves of gene expression as the kidney progenitor cells progress through the different stages of nephrogenesis.
The FACEBASE consortium was established by NIH to provide a resource for the craniofacial research community (Hochheiser et al., 2011). One purpose of this consortium is to generate a gene expression atlas of mouse craniofacial development. In this report we describe the results of an extensive LCM/microarray/RNA-seq analysis of the gene expression patterns of early mouse craniofacial development, during E8.5, E9.5 and E10.5. At each developmental stage the multiple craniofacial compartments were isolated by LCM and gene expression patterns characterized by microarray and RNA-seq. The results define the gene expression blueprint of early craniofacial development. All growth factor, receptor and transcription factor domains of expression are defined. Novel compartment specific molecular markers are identified. In addition the RNA-seq data defines RNA splice patterns and provides a comprehensive catalog of noncoding transcripts, including those derived from enhancers. In summary, this is an extensive gene expression compendium meant to augment craniofacial research.
Materials and Methods
LCM protocols
In brief, embryos were rapidly collected from CD1 outbred mice (Charles River) with the day of vaginal plug designated E0.5. Embryos were flash frozen in O.C.T (Sakura Finetek) with liquid nitrogen cooled isopentan. Cryostat sections were made and processed, and LCM was carried out with an Arcturus Veritas machine, with membrane slides and using a combination of UV cutting and IR capture lasers as previously described (Potter and Brunskill, 2012). For a typical sample approximately 10–30 LCM collected tissue sections were pooled for anlaysis. Detailed protocols, with representative LCM images, are available at https://www.facebase.org/node/154.
RNA Purifications and Amplifications
RNA was purified using the ZR RNA MicroPrep kit (Zymo). Nugen RiboSpia Ovation Pico WTA System V2 was used for target amplification for RNA-seq. For microarrays we used the SCAMP method previously described (Brunskill et al., 2011c). RNA-seq was carried out using 50b single end reads on the Illumina Hi-Seq 2500 machine according to Illumina protocols, with read depths > 40 million. For microarrays we examined a minimum of three biological replicates, and commonly four to six. Over one hundred microarrays in total were used. Exact replicate numbers are shown in heatmaps and at FaceBase.Org. For RNA-seq single samples were analyzed.
Single Cell
Single, neural-crest cells were isolated from the cranial mesenchyme using WNT1CRE-Rosa26GFP reporter mice. The cranial mesenchyme and overlying epidermal ectoderm were separated from the neural fold, transferred to an eppendorf tube, digested with 0.05% trypsin for 5 min and the digestion stopped with ice-cold 1%FBS/PBS. Under a fluorescent microscope GFP-positive cells neural crest cells, representing neural crest, and GFP-negative paraxial mesoderm and epidermal ectoderm cells, were isolated using pulled glass pipettes. Each cell was serially transferred through several petri dishes to confirm that only a single-cell was isolated. The cell was then transferred to an eppendorf tube containing lysis buffer and quick-frozen on dry-ice for later SCAMP amplification and microarray analysis.
Data Analysis
Microarray and RNA-seq data were analyzed by a combination of Bowtie, Tophat, and GeneSpring versions 7.3.1 and 12.6 software. A standard workflow for RNA-seq analysis included removal of probesets that did not map uniquely to the mm9 ENSEMBL genome or ENSEMBL-annotated genes. For the microarray samples, probesets were selected whose RMA- estimated expression level was at least 6.2 in at least one sample. Differentially expressed genes were identified using both unpaired t-tests for pairwise comparisons and one way ANOVA for multiple compartment comparisons (FDR < 0.05), with subsequent fold-change of 2–5 fold depending on the comparisons so as to optimize the identification of expression signatures for each compartment. RNA-seq BAM files generated using the Bowtie-Tophat2 pipeline were analyzed for the expression of known and unknown genes/transcripts using both Cufflinks2 and GeneSpring 12.6. Workflows included filtering to remove duplicate reads, and those with post-aligned read metrics mapping quality below 40, as well as the exclusion of samples that exhibited strong outlier 3′/5′ read distribution ratios. Transcript/isoform and gene summarized expression tables were filtered to identify entities whose expression was at least 3 FPKM (Cufflinks) or 3 nRPKM (GeneSpring) in at least one sample. Differentially expressed gene signatures were identified using Audic Claverie tests (P < 0.05) and Welch t-tests (FDR<0.05) followed by a two to five fold change requirements. Gene Ontology and other enrichment and biological network analysis was carried out with GeneSpring, ToppGene (http://toppgene.cchmc.org), and ToppCluster (http://toppcluster.cchmc.org/) yielding both similar and complementary results.
Biological replicate microarray Pearson correlation coefficients were generally in the range of 0.93 to 0.99. For example, at E10.5, we observed for the lateral nasal prominence (0.944, 0.937, 0.962), medial nasal prominence (0.950, 0.950, 0.943), olfactory pit (0.966, 0.956, 0.955), mandibular arch (0.943, 0.947, 0.978), and maxillary arch (0.976, 0.992, 0.972).
Data is available on the FaceBase.Org website and in GEO under superset GSE55967, which includes individual data series GSE55964, GSE55965, and GSE55966 for ST1.0 single cell samples, LCM microarray, and LCM RNA-seq samples, respectively.
Generating a Global Facebase Datamine
In order to establish integrated, comprehensive, mineable, and community-useful tables and maps of gene expression patterns across the series of cell types, regions and developmental stages that were profiled in this project, we followed a strategy that allowed us to generate two large normalized data matrix files (one being RNA-seq, the other Affymetrix GeneChip ST1.0) of all the measured values in each sample in the study. Prior to “baselining”, these normalized “raw” values correspond to the estimated expression level of each gene, transcript, or probeset as measured using either technology. The RNA-seq values are FPKM values, and the microarray are based on the RMA normalization approach. These absolute expression value tables were then baselined to a global median expression value.
The RMA microarray data algorithm provides a log2 based relative intensity value for each probeset for each microarray. Our baseline referencing approach for these RMA values was to convert these into relative ratios per sample using the transform of Intensity = 2^^RMA, and then to define the ratio of each gene/probeset’s expression relative to its median expression value across all samples in the dataset. Thus, the relative expression profile for a given gene is relative to this baseline. The data submitted to GEO described above includes the global normalized value data matrices that we used in these analyses. For the FPKM-based RNA-seq data, we carried out a similar strategy, but used a denominator for relative expression that was the ratio of FPKM per sample versus the global median of (FPKM+1) so as to define the lowest level of log2FPKM = 0. Thus, the final normalized and baselined expression values represent a ratio of expression in each sample relative to the median of that gene or transcript across the sample series.
To define compartment-specific gene lists, we subjected the two expression tables to a “shredding protocol” in which a series of expression signatures per sample type were generated using relative expression rank cutoffs of 100, 250, 500 and 1000 transcripts per compartment. Additional sub-signatures from these parent lists were then generated by subjecting each gene list to K-means clustering using 5 Pearson-correlation-based K-groups, thus providing gene sets that are both highly ranked for a given compartment, and similarly expressed across the other samples in the datasets when they belong to the same K-means cluster (note that different K-means clusters have different numbers of genes per cluster). For each list of mouse genes, Toppgene only lists those that have an NCBI Homologene-mapped human gene ortholog. This generates a reference signature database composed of the series of genelists that were then placed into the ToppGene (http://toppgene.cchmc.org) and ToppCluster (http://toppcluster.cchmc.org) web site data analysis resources. Each of the genelists from this “shredding procedure” are named according to a scheme that generates descriptors such as “Facebase RNA-seq E10.5 Mandibular Arch 500”, or “Facebase ST1 E10.5 ColumnEpith Mandib 500 #4”, where the first number refers to the number of genes in the relative expression-ranked list, and the second number, if present, would refer to the K-means cluster to which it belonged. In this way, any single gene of interest can be queried through Toppgene to see if appears in one of these top-ranked gene lists. After a user inspects the names of the genelists that contain it, one or more of those lists can be downloaded or directly re-analyzed for its functional associations and enrichments with respect to all Gene Ontologies, pathways, protein-protein interactions, and known mouse and human gene knockout phenotypes as per normal analysis protocols of ToppGene and ToppCluster3 (Chen et al., 2009; Kaimal et al., 2010). By our registering the data from the present studies into Toppgene data tables, we thereby enable the analysis of a list of genes submitted by user as a query to be compared to the Facebase datasets themselves. The goal of our enabling this approach is to provide a flexible and focused analysis resource for the craniofacial research community to detect compartments and study a group of genesets that have a high likelihood of being coexpressed in specific developing craniofacial compartments. And then by using connections provided by all of the other Toppgene data features (such as mouse knockout phenotypes, gene ontology terms, protein-protein interactions, etc.) Facebase gene expression signatures can be analyzed for their relationships to known or putative functional, mechanistic, and interactions-based features, allowing for new hypotheses to be formulated.
Results and Discussion
E8.5 gene expression profiles
The neural crest cells are a multipotent and migratory cell population that will give rise to several lineages of the developing face, including melanocytes, cartilage, bone, smooth muscle and neurons (Bush and Jiang, 2012; Chai and Maxson, 2006; Kulesa et al., 2010; Szabo-Rogers et al., 2010). At around E8.5 the first neural crest cells depart the neural fold/epidermal ectoderm boundary and migrate as streams between the outer epidermal ectoderm, and the neural fold. The neural crest cells then join with resident paraxial mesoderm cells. To better understand this initial stage of craniofacial development we defined gene expression profiles of several compartments, including the mesenchyme (neural crest and paraxial mesoderm), dorsal neural fold, ventral neural fold (including the floor plate region), as well as the total caudal neural fold region (Fig. 1). A total of 18 samples were analyzed using Affymetrix mouse Gene 1.0 ST arrays, providing four or five biological replicates per compartment.
Fig. 1. Compartments isolated by LCM.
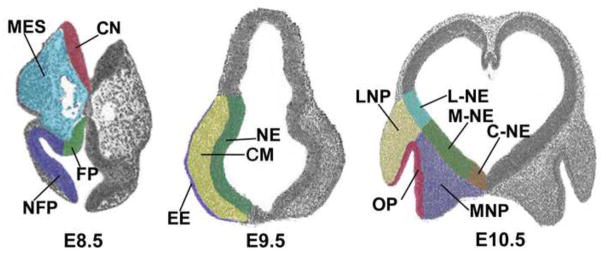
Some of the compartments isolated by LCM are illustrated. At E8.5 the cranial mesenchyme (MES), with neural crest and paraxial mesoderm, as well as the flanking neuroepithelium, divided into floorplate (FP) and more dorsal non-floor plate (NFP), as well as more caudal neuroepithelium (CN). At E9.5 LCM was used to purify the cranial mesenchyme (MES), the flanking neuroepithelium (NE), the epidermal ectoderm, including the olfactory placode (EE), as well as the otic vesicle, mandibular and maxillary arches (not shown). At E10.5 we collected the lateral nasal prominence (LNP), medial nasal prominence (MNP), flanking neuroepithelia (L-NE, M-NE, C-NE), olfactory pit (OP), and, Rathke’s pouch, both mandibular and maxillary arches, including both mesenchyme and epidermal ectoderm compartments (not shown).
The mesenchyme cells are of particular interest, as they construct the major components of the face, while the other compartments examined are primarily of interest because of their possible function in signaling to the flanking neural crest and paraxial mesoderm cells. The mesenchyme gene expression state at E8.5 was first compared to the dorsal neural fold, yielding a list of 331 upregulated genes (Table S1). These include genes up-regulated as cells from the dorsal neural fold undergo epithelial to mesenchymal transformation and generate the neural crest. Gene ontologies analysis of these 331 genes identified the top ranked molecular function as sequence specific DNA binding, with 28 transcription factor genes, including Snai1, Snai2, Foxc1, Foxc2, Foxf2, Foxd3, Alx1, Prrx1, Prrx2, Bcl2, Irx3, Irx5, Msx1, Msx2, Sp5, Etv2, Hhex, Six1, Six2, Nr2f1, Lmo2, Sox10, Nfil3, Atf3, Nkx6-1 and Pitx2. Many of these represent previously identified neural crest markers. The data suggest that these encoded transcription factors combine to determine the early mesenchyme gene expression state. Key biological processes identified included vascular development (57 associated genes), mesenchyme development (23 associated genes), and neural crest development (11 genes) (Table S2).
Of the 331 genes called by microarrays to be more abundantly expressed in mesenchyme, 312 were annotated genes with assigned gene symbols. 92% of these (all but 26) were validated as significantly (P < 0.05) changed in expression by independent RNA-seq analysis (Table S1). All but 40 of these also showed greater than two-fold change in expression with RNA-seq. These two independent technologies therefore showed quite good agreement for the genes called differently expressed.
Of interest, however, RNA-Seq called many more genes differentially expressed between mesenchyme and dorsal neural fold, with 1849 showing more than two fold change. It has been commonly observed that RNA-seq gives more genes called differently expressed than microarrays, and this is often attributed to the lower background with RNA-seq, which improves the signal to noise ratio (Wang et al., 2009). Arrays give a resulting fold change compression compared to RNA-seq. It has been observed that RNA-seq with a four fold change cutoff can identify as many differentially expressed genes as microarrays with a much less stringent two fold change (Raghavachari et al., 2012). A large scale comparison of RNA-seq and microarray technologies, however, found that in general they show excellent concordance, producing comparable results, consistent with what we observed (Guo et al., 2013).
Several of the genes showing the greatest fold change by RNA-seq, however, were not even called differently expressed by microarrays. For example the Ddr2 gene gave 2,542 raw reads for mesenchyme, and only one read for dorsal neural fold, with a fold change of 707 called by RNA-seq, while this gene was not called differently expressed by array. Similarly, Pear1 (nRPKM of 10.6 vs zero), Fzd10 (nRPKM of 13.8 versus zero) and Ebf2 (nRPKM of 22.6 vs 0.06), were not detected as differently expressed by microarray. This failure could reflect the different amplification technologies used (SCAMP for microarray versus Nugen RiboSpia for RNA-seq), and/or imperfections in microarray design.
Therefore, the independent dual technology analyses used in this study, with both microarray and RNA-seq, does provide a high throughput cross validation, with the RNA-seq independent technology typically confirming about 90% of the genes called differently expressed by arrays. The RNA-seq dataset, however, provides more accurate fold change calls, identifies a greater number of differently expressed genes, examines gene expression in an unbiased manner that does not depend on array design, and defines RNA splicing patterns (Wang et al., 2009).
The RNA-seq data showed an interesting co-expression of 5′ flanking non-spliced sequences for a number of genes, including Six1 (Fig. 2). At E8.5 these Six1 5′ flanking transcripts were restricted to the mesenchymal neural crest/paraxial mesoderm, but at both E9.5 and E10.5 the Six1 gene and its 5′ transcripts were widely expressed, in all compartments assayed. These 5′ transcripts represent candidate enhancer RNAs, since it is now established that many enhancers are transcribed, giving rise to short transcripts (Core et al., 2008; Hah et al., 2011; Kim et al., 2010; Wang et al., 2011). Further, recent results indicate that these enhancer transcripts can be functional (Lam et al., 2013; Li et al., 2013; Melo et al., 2013). Because we carried out the RNA-seq using a combined dT plus random primer reverse transcription reaction we detected nonpolyadenylated transcripts from “non-canonical” regions, which do not correspond to exons of the reference genome. It is interesting to note that Six1/Eya1 have been shown to play an important role in craniofacial development as upstream regulators of FGF8 signaling (Guo et al., 2011).
Fig. 2. RNA-seq analysis of Six1 expression.

The positions of the Six1 exons are shown at the bottom in blue. The lightest shade is intron, intermediate shade is coding, and darker blue represents 5′ and 3′ UTR. The cranial mesenchyme, top panel, shows strong expression with many RNA-seq reads, marked by small tan rectangles. The lines indicate positions of introns, where single cDNA RNA-seq reads align to the two flanking exons. Note the many reads from the 5′ region of Six1 in the top panel, coincident with high expression of the Six1 one gene itself. In the dorsal neural fold, bottom panel, there is very low expression of both the Six1 gene and the 5′ flanking region.
As mentioned, the cranial neural crest cells first migrate in close apposition to the dorsal neural fold, which may secrete important growth factor signals. Interesting signaling candidates identified included Fgf8, and Wnt8b, as well as Bmp5, which was made by both the flanking neuroepithelium and the mesenchyme cells themselves, suggesting possible paracrine and autocrine functions.
The transcription factor expression code of the dorsal neuroepithelium at E8.5 is of particular interest, as this includes the region of origin of neural crest cells. We identified 70 transcription factors with elevated expression in comparison to the ventral region of the neuroepithelium (Table S3). These included a number of genes with previously established expression in this zone, including Sp8 and Dlx5, providing positive control historic validation of the dataset.
As the neural crest cells continue to migrate they pass by the ventral region of the neuroepithelium, and again are exposed to flanking growth factors, in this case including SHH, BMP2 and BMP4, FGFs 7,11, 14, 15, and 18, as well as TGFβ1 and 2. In addition the RNA-seq data defined a transcription factor code of the ventral floor plate region, with 144 transcription factors expressed at significantly higher levels (>2FC) compared to the more dorsal neuroepithelium (Table S4).
The E8.5 mesenchymal RNA-seq gene signature was then analyzed using a screen that required a minimum 2 FC enrichment of transcripts against all other E8.5 compartments profiled, yielding a list of 704 genes (Table S5). This more stringent analysis of the RNA-seq dataset identified a total of 83 transcription factors defining the E8.5 mesenchyme (neural crest plus paraxial mesoderm) transcription factor code (Table S6), a number significantly larger than the 28 mentioned previously, defined with microarrays and using a less stringent screen. This transcription factor code for E8.5 mesenchyme included Alx1,4, Ebf1,2, the Fox genes c1, c2, d2, d3, f2, p1, p2, Gata2, Lhx6, Msx1, Myocd, Nkx3-1, Pax9, Pitx2, Six1, Snai1, Sox7,9,10,17,18, Stat3,6, Twist1,2, and many others.
E9.5 expression data
At E9.5 we used LCM to isolate six separate spatial compartments. These included the cranial mesenchyme (CM), as well as the underlying neuroepithelium (NE) and overlying epidermal ectoderm (EE) (Fig. 1). The mandibular and maxillary arches, as well as otic vesicles were also captured and gene expression profiling was carried out with Affymetrix mouse gene 1.0 ST arrays. In addition we gene expression profiled the cranial mesenchyme, overlying ectoderm, mandibular arch, and maxillary arch using RNA-seq.
We first analyzed the microarray data, removing probesets with only low expression levels, performing oneway ANOVA (P < 0.05), and requiring FC > 2 compared to other regions sampled. For example, when the cranial mesenchyme gene expression pattern was compared to mandibular arch, maxillary arch, neuroepithelium, overlying ectoderm and otic vesicle, 26 cranial mesenchyme compartment enriched genes were identified (Table S7). It is interesting to note that two of these comparison compartments, mandibular arch and maxillary arch, are also primarily made up of neural crest cells. The similarities in cell type likely partially explain the relative paucity of genes with restricted expression. This is also in part the result of microarray technology, with more differences found by RNA-seq, as described later. The list of cranial mesenchyme enriched transcription factors included Alx1, Alx3, Batf3, Six2 and Sox8 (Table S7).
The overlying ectoderm, including primarily but not exclusively the olfactory placode, showed elevated expression of 68 genes, when compared to all other E9.5 compartments, excepting otic vesicle. These cells signal to the underlying cranial mesenchyme, and subsequently invaginate to form the olfactory pit. The enriched gene transcripts encoded ALDH1a3, thought to play an important role in establishing retinoic acid gradients, as well as the growth factors BMP4, PDGFA and PDGFC. Enriched transcription factor genes included Isl1, Pax6, Pitx1, and Six3. In addition there were a number of claudins (3,4,6,7,9), keratins (8,14,18,19), and Epcam, all associated with the epithelial nature of these cells. For a complete list see (Table S8).
The mandibular arch, when compared to cranial mesenchyme, overlying ectoderm, neuroepithelium and otic vesicle, gave 232 genes with enriched transcripts. A series graph of these enriched genes shows that the expression patterns for the mandibular and maxillary arches were quite similar, with genes showing elevated expression in one generally also showing elevated expression in the other (Fig. 3). Five genes gave particularly robust elevated expression in these two compartments, Barx1, Emcn, Lhx8, Mab21l1 and Kcne3. Barx1 and Lhx8 encode homeobox transcription factors. Endomucin (Emcn) interferes with the assembly of focal adhesion complexes and inhibits interaction between cells and the extracellular matrix (Kinoshita et al., 2001). Mab21l1 is closely related to the mab21 gene of C. elegans, which has been shown to play a critical role in multiple cell fate decisions, with mutants showing posterior to anterior homeotic transformations (Chow et al., 1995). Kcne3 encodes a voltage gated ion channel. For a complete list of mandibular/maxillary arch enriched gene transcripts see (Table S9).
Fig. 3. Series graph of genes with elevated expression in the E9.5 mandibular arch.
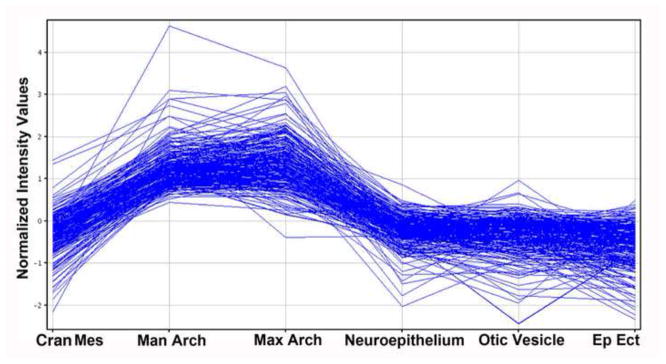
Genes with elevated expression in the mandibular arch were identified by comparing its microarray defined gene expression profile with those of all other E9.5 compartments examined, excepting the maxillary arch. As shown, genes with elevated expression in the mandibular arch are, with rare exception, also elevated in expression in the maxillary arch. It is interesting to note that the cranial mesenchyme, mandibular arch and maxillary arch are all composed primarily of neural crest cells. Ep Ect; epidermal ectoderm, including olfactory placode. Affymetrix Gene 1.0 ST microarray data.
The underlying neuroepithelium notably expressed Fgf15, Fgf17, Fgf18, and Wnt1, as well as the chemokine Ccl19, which could all provide important signals to the flanking cranial mesenchyme (Table S10).
A heatmap of the E9.5 compartment specific gene lists is shown in Fig. 4. This was generated with GeneSpring software, using Pearson’s centered distance metric and Ward’s linkage rule. The mandibular arch and maxillary arch profiles cluster together, reflecting their very similar gene expression profiles. The otic vesicle and olfactory placode/overlying ectoderm also show strongly overlapping gene expression patterns, as might be expected given their common epidermal ectodermal origin. In contrast the cranial mesenchyme and underlying neuroepithelium produced lists of genes with more distinct expression patterns. The reproducibility was generally excellent, with some expected variability due to the sampling differences resulting from random sets of LCM sections through the compartments of interest.
Fig. 4. Heatmap of genes with compartment elevated expression at E9.5.
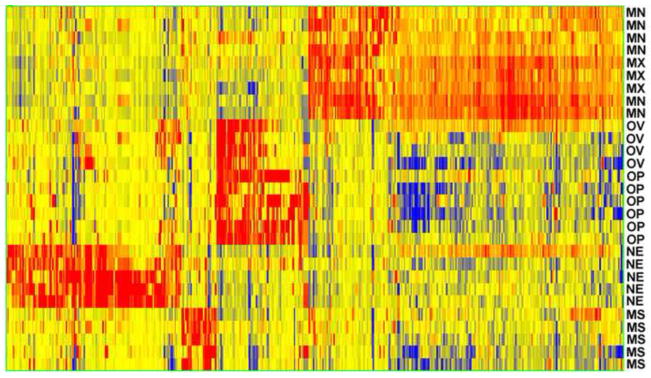
The mandibular (MN) and maxillary MX) arches cluster together, as do the otic vesicle (OV) and olfactory placode (OP), which are both derived from the epidermal ectoderm. The neuroepithelium (NE) shows a very distinct gene expression signature, and there are a limited number of genes that distinguish the cranial mesenchyme (MS). Based on microarray data. See Fig. 1 for compartment locations. Red indicates high expression and blue indicates low expression. Based on Affymetrix Gene 1.0 ST microarray data.
E9.5 RNA-seq
As noted previously, E9.5 craniofacial gene expression patterns were also examined with RNA-seq. Cranial mesenchyme, mandibular arch, maxillary arch, and overlying ectoderm, including the olfactory placode, were profiled. Although representing a more limited set than examined with microarray, these are the compartments that directly construct the face.
Once again, as for E8.5, the RNA-seq analysis provided an independent high throughput technology for validation of the microarray data. For example, in comparing the microarray and RNA-seq cranial mesenchyme data, 13/26 genes called enriched by arrays were similarly called differently expressed by RNA-Seq. Another 5/26 genes were also called differently expressed by RNA-seq, but with low expression levels (nRPKM < 3), therefore not making our normal expression threshold cutoff. Other genes showed the same directional change, but did not quite make the two fold change cutoff. In the end only three of the 26 genes showed a complete failure of validation, with either no transcripts detected by RNA-seq (one gene), or no corresponding directional change in expression observed (two genes). So, as for the E8.5 gene expression data, about 90% of genes called differently expressed by microarray were validated in significant measure by the RNA-seq data.
The RNA-seq data again produced lists with more differently expressed genes, and with generally greater fold changes than seen with microarrays. For example 1,009 genes were called cranial mesenchyme enriched (greater than two fold change compared to other compartments)(Table S11). A Gene Ontologies analysis using ToppGene gave a very strong protein synthesis signature, with many of the top molecular functions and biological processes relating to ribosomes, translation, and protein targeting to the ER (Table S12). Growth factors expressed included midkine, neurturin, and colony stimulating factor1. The list of cranial mesenchyme enriched transcription factors was expanded compared to that seen with arrays, now including Alx1, Alx3, Alx4, Btf3, Rarb, Shox2, Six2, Sox7 and Sox8. For the complete list see (Table S11).
A similar RNA-seq analysis was carried out to find genes with enriched expression in the overlying ectoderm, including the olfactory placode. A list of 1270 genes emerged (Table S13). The gene with the greatest fold transcript enrichment was Sp8, which encodes a zinc finger transcription factor, previously shown to be highly expressed in the olfactory placode (Zembrzycki et al., 2007), and previously shown to be a key regulator of craniofacial development (Kasberg et al., 2013), providing historic validation of the dataset. The epidermal ectoderm was clearly a rich source of growth factors, including BMPs 2,4,5,7,8b, FGFs 8,9,11,12,16,17,18, as well as WNTs 4 and 5b. A gene ontologies analysis confirmed this, with the top molecular function (P = 5 ×10−5) being growth factor activity. The top biological process was neuron differentiation (P = 6 × 10−10), which likely reflects the beginning stages of the generation of the olfactory neuroepithelium.
The RNA-seq data further defined distinct transcription patterns for certain genes. For example, consider the homeobox transcription factor gene Dlx6 (Fig. 5). The cranial mesenchyme showed very low expression, with almost no reads (top panel). The region of the Dlx6 gene is shown at the bottom, with introns in light blue. In contrast the epidermal ectoderm/olfactory placode showed robust expression, with standard RNA splicing as shown by the lines that span the sequences of the introns, which are deleted in the RNA-seq reads of the cDNA. Also note the abundant reads from the 5′ promoter region of Dlx6, representing candidate enhancer transcripts. These transcripts did not show evidence of RNA splicing since none of the RNA-seq reads spanned introns. Dlx6 was also strongly expressed in the maxillary arch, but in this case there were abundant spliced antisense transcripts in the region 5′ of Dlx6, with introns again represented by lines. In the mandibular arch the expression pattern was yet more complex, with the presence of two species of spliced antisense transcripts, with one spanning the entire Dlx6 gene region, representing the 3′ Opposite Strand (3′ OS) transcript shown at the bottom of Fig. 5. Both the 3′ and 5′ antisense transcripts spliced to an exon located further 5′ and not included in Fig. 5. In summary, each of the four compartments examined showed a distinct transcription/RNA processing pattern in the region of Dlx6.
Fig. 5. Compartment specific expression patterns of the Dlx6 gene at E9.5.

The position of the Dlx6 gene is shaded, with exons shown at the bottom of the figure in darker blue and introns in light blue. The E9.5 cranial mesenchyme (top) shows very low expression, with few RNA-seq reads (small rectangles). The olfactory placode, bottom, shows strong Dlx6 expression, with normal splicing. The lines show RNA-seq reads that spanned introns. In addition there are multiple RNA-seq reads from the 5′ flanking region of the gene (to the left), that did not showing splicing (no lines). The maxillary arch showed strong expression of Dlx6, as well as strong expression of a spliced antisense noncoding transcript in the 5′ promoter region, indicated by the lines marking the opposite strand (5′ OS) intron. In the mandibular arch there is strong expression of the Dlx6 gene, the 5′ antisense spliced transcript, and also an antisense transcript that initiates 3′ of the Dlx6 gene (3′ OS), with both antisense transcripts splicing to exons further 5′ and not shown in this figure.
The RNA-seq data also revealed novel transcripts and unusual RNA splicing patterns. One example is a novel transcript flanking the Rps29 gene. This transcript has two exons separated by a very large single intron, of approximately 196 Kb. More than six genes reside within this intron, including Klhdc1, Klhdc2, Nemf, Pde2, Mgat and Lrr1. The two exons of this novel transcript showed multiple splice site alternatives, including one noncanonical AT-AC junction sequence (data not shown). This RNA was robustly expressed, with raw reads of over one thousand in all compartments examined. Of interest, the splicing pattern observed in the cranial mesenchyme for this transcript was unique to that compartment. In summary, the RNA-seq dataset provides a useful resource for the analysis of the RNA-splicing patterns for all genes expressed in these compartments at E9.5.
E10.5 Microarray analysis
At E10.5 craniofacial development has progressed to form the lateral nasal process, medial nasal process, and olfactory pit. (Fig. 1). We used LCM to capture samples from these compartments for microarray analysis. We also isolated the maxillary arch, mandibular arch, and the neuroepithelium underlying the Lateral and Medial Nasal Processes, as well as the most medial neuroepithelium, which is a known growth factor signaling center. In addition we LCM purified Rathke’s pouch, the epidermal ectodermal layers of the maxillary and mandibular arches, as well as neuroepithelium that did not underly the facial processes as a control. A minimum of three biological replicates were examined for each compartment, with a total of 47 microarrays used to examine gene expression in the E10.5 developing face.
This dataset was first examined by performing ANOVA (P < 0.05), minimum fold change of at least five for any pairwise comparison, giving 1516 differentially expressed probesets (Table S14). Cluster analysis showed excellent reproducibility, with some variation expected as a result of the LCM sampling of distinct subregions of compartments (Fig. 6). Although all of the compartments were predominantly ectodermal in origin there were similarities in the heatmap that reflected their distinct lineages. Each of the neurepithelial (NE) compartments, for example, shared a large number of active genes distinct from the other regions. The epidermal ectodermal cells of the mandibular and maxillary arches also showed similar gene expression patterns, which partially overlapped the nasal pit and Rathke’s pouch, which are derived from the invagination of epidermal ectoderm. Moving to the far right of the heatmap of Fig. 6 are genes expressed predominantly by neural crest rich compartments, including the lateral nasal prominence, medial nasal prominence, mandibular and maxillary arches.
Fig. 6. Heatmap of genes with compartment elevated expression at E10.5.
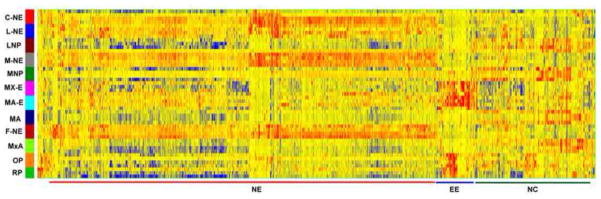
Compartments with neuroepithelial cells show closely related gene expression signatures, as marked at the bottom with NE (red line). Similarly, the compartments made of, or derived from, epidermal ectoderm show very closely related gene expression patterns, as marked with EE at the bottom (blue line). The NC denotes genes with elevated expression in a compartment made up primarily of neural crest cells (green line). See Fig. 1 for positions of most compartments. C-NE, central neuroepithelia; L-NE, neuroepithelia flanking the lateral nasal prominence; M-NE, neuroepithelia flanking the medial nasal prominence; MNP, medial nasal prominence; LNP, lateral nasal prominence; MxA, maxiallary arch; MA Mandibular arch; OP, olfactory pit; RP, Rathke’s pouch; MX-E, epidermal ectoderm of maxillary arch; MA-E, epidermal ectoderm of mandibular arch; F-NE, control neuroepithelium from dorsal region of brain. Microarray data. Red indicates high expression and blue indicates low expression.
Relatively few genes showed compartment specific expression, as might be expected given their strongly overlapping cellular compositions. It is not surprising therefore that transcripts for only 24 probesets, including 17 annotated genes, were identified, for example, as lateral nasal prominence enriched (Table S15). This screen required two fold enrichment compared to all other compartments, excepting the medial nasal prominence, which showed strongly overlapping gene expression with the lateral prominence. The lateral nasal prominence enriched list of genes encoded the transcription factors GSC and ALX1, as well as the vacuolar sorting protein VPS13d, the chemokine PF4. Enriched genes also included Bicc1, which encodes an RNA that regulates protein translation during development. The further requirement for two fold enrichment in the lateral nasal prominence versus the medial nasal prominence reduced the list to ten annotated genes (Bicc1, Gsc, Nfe2, Snhg1, S100a13, Wdr65, Vps13d, Inmt, Tyrobp and Hpgd).
A similar analysis of the olfactory pit identified the growth factor FGF17, the transcription factor neurogenin1, likely involved in differentiation of the olfactory system, Aldh1a3, again involved in establishing retinoic acid gradients, and the ephrin gene efna3.
The neuroepithelium underlying the lateral nasal prominence showed elevated expression for several transcription factors, but little evidence of FGF, BMP or SHH signaling. In contrast the more medial neuroepithelium showed expression of the Noggin, Fgf8, Fgf15, and Fgf17 genes, which likely play important roles in the development of the nearby neural crest cells of the medial nasal prominence.
The epithelia surrounding the mandibular arches expressed Igf2, Pdgfa (very high expression), Pdgfc, Wnt4 and Wnt6, which likely signal the underlying neural crest/paraxial mesoderm.
The gene expression patterns of the E10.5 craniofacial compartments were also examined by RNA-seq. Once again, this provides an independent high throughput global strategy for validation of the microarray data, as well as lending the many advantages of RNA-seq.
RNA-seq analysis of the olfactory pit confirmed and extended the microarray results. The data once again showed highly elevated levels of the Efna3, Aldh1a3, and Fgf17, as seen with microarrays. The RNA-seq further defined the signaling center function of the nasal pit, showing for example higher expression of multiple FGFs, including FGF 8, 9,12, 15,16, 17 and 18, compared to the other compartments. The olfactory pit is clearly a major FGF signaling source, with seven FGF genes showing striking elevated expression, and with FGFs 8, 9, 17 and 18 showing robust nRPKM expression levels above nine. In addition the olfactory pit expressed WNT4 (nRPKM 7), and connective tissue growth factor CTGF (nRPKM 15), which plays a role in driving chondrocyte proliferation and differentiation. Other growth factor genes expressed included inhibin alpha (Inha: nRPKM 6), Pdgfa (a very high nRPKM of 144) and Pdgfc (nRPKM 50), Tgfα (nRPKM 14), Nrg1 (nRPKM 15), and Notch1 (nRPKM 24).
The RNA-seq dataset can be screened in a rich variety of manners to yield different insights. Compartment specific growth factors, transcription factors and receptors can be defined. For example, an analysis of the mandibular arch elevated genes, compared to all other compartments excepting the maxillary arch, which shows a strongly overlapping gene expression pattern, identified the strongly expressed transcription factor genes Hand1, Hand2, Osr1, Twist2, Lhx6, Lhx8, Barx1, and Dlx1. Because of their restricted and robust expression these genes are therefore strong candidates for representing the transcription factor code that defines the mandibular arch. Of interest, all of these gene have been previously implicated in craniofacial development (Barbosa et al., 2007; Denaxa et al., 2009; Jeong et al., 2008; Liu et al., 2013; Nichols et al., 2013; Tukel et al., 2010; Zhao et al., 1999). It is also interesting to note that one of the genes with the highest expression level in the mandibular arch encodes the growth factor IGF2. In the different compartments Igf2 showed nRPKM values of: olfactory pit, 108; medial eminence 137; lateral eminence 94; maxillary arch 139, and mandibular arch 310. These high Igf2 expression levels likely help drive the rapid growth of these compartments. Of interest, altered expression of Igf2 has been associated with Russell-Silver syndrome, which includes a distinctive craniofacial phenotype (Chopra et al., 2010).
In situ hybridizations
The use of two independent high throughput gene expression profiling technologies, microarrays and RNA-seq, provided the primary method of data validation for this study. In addition, however, we carried out in situ hybridizations for a group of genes predicted to have elevated expression levels in restricted sets of compartments. The results were quite consistent with predictions, as illustrated in Fig. 7. At E8.5 Pou3f2 and Hes3 were expressed in the caudal brain neuroepithelium. At E9.5 Epcam, Aldh1a3 and Pitx2 were expressed in the epidermal ectoderm, Alx1, Alx3, Lum and Six2 were expressed in the cranial mesenchyme, and Rap2c and Pth1r were expressed in the mandibular arch. At E10.5 Barx1 was expressed in the mandibular arch, and also at a somewhat lower level in the maxillary arch. Cited 1 showed elevated expression in the mandibular arch, while Epcam, Fezf1 and Cldn3 showed elevated expression in the nasal pits. For Barx1, Rap2c, Lum, Aldh1a3, Alx1 and Alx3 we generated section in situ hybridization data to better localize expression (Fig. S2). Together these results provide significant data validation, although for a more restricted gene set than possible with the RNA-seq/microarray comparison.
Fig. 7. In situ hybridizations showing elevated gene expression in select craniofacial compartments.

Whole mount in situ hybridizations at E8.5, E9.5 and E10.5. At E8.5 Pou3f2 and Hes3 show elevated expression in the caudal brain neuroepithelium. At E9.5 Epcam, Aldh1a3 and Pitx1 show elevated expression in the olfactory placode/epidermal ectoderm, Alx1, Alx3, Lum, Six2 and Sox8 show elevated expression in the cranial mesenchyme, while Rap2c and Pth1r show elevated expression in the mandibular arch. At E10.5 Barx1 and Cited1 show elevated expression in the mandibular arch, while Epcam, Fezf1 and Cldn3 show elevated expression in the olfactory pits.
Single Cell Analyses
To better define cell-type specific gene expression profiles that correlate with individual cell types within the early tissue layers responsible for craniofacial development, we compared a series of E8.5 cells sampled from three compartments critical for craniofacial development; epidermal ectoderm, paraxial mesoderm and neural crest. Although as previously described we had used LCM to isolate and profile populations of cells that are present within each of these compartments at this stage, these profiles are complicated by the admixture of different cell types within these compartments, for example the presence of both neural crest and paraxial mesoderm cells in cranial mesenchyme. This complicates the issues of identifying cell type-specific transcriptional programs and leads to results that provide ensemble averages of the mixed populations of cells, here for example based on the ratio of neural crest and paraxial mesoderm cells that are present as well as the distribution of varying degrees of cellular maturation. The separation of these programs requires the use of single cell based profiling methodologies.
The neural crest cells were GFP labeled by crossing the Wnt1-Cre and floxed stop Rosa26-GFP reporter transgenic mice. We used manual microdissection coupled with mild trypsin treatment to isolate GFP positive single cells from the E8.5 cranial mesenchyme, flanking the neural folds, representing neural crest, as well as GFP negative cells, representing paraxial mesoderm and overlying epidermal ectoderm. In total eleven cells were purified, microscopically confirmed to be single cell, rinsed repeatedly in PBS, and then used for SCAMP amplification as previously described (Brunskill et al., 2011a) and hybridized to Affymetrix Mouse Gene 1.0 ST arrays, CEL files from which were quantitated using the RMA method.
Probesets were filtered for those that exhibited RMA –estimated expression level of at least 6.0 in at least one sample, and were then subjected to ANOVA, fold-difference, and clustering based analyses to identify probesets that exhibited differential compartment specific expression with p < 0.05, without FDR to allow identification of genes that were not necessarily expressed by all of the individual cells of the distinct sampled compartments. The resulting 377 probesets with compartment specificity of expression, were then subjected to hierarchical clustering using median baseline referenced relative expression values (Fig 8). We identified 90 candidate epidermal ectoderm genes, 217 candidate neural crest genes, and 70 candidate paraxial mesoderm genes (Table S16). The significant degree of variability among the individual cells is likely in part due to the pulsatile nature of individual cell-level gene expression (Chubb et al., 2006; Losick and Desplan, 2008; Raj et al., 2006), which can cause the gene expression profile of even a single cell to fluctuate significantly as a function of time. Regional heterogeneities of the single cells sampled will further contribute to the observed differences in gene expression. In addition the low number of transcripts per gene in a cell results in technical noise, as it is impossible to capture each transcript for analysis with 100% efficiency.
Fig. 8. Heatmap of gene expression patterns of single cells at E8.5.

We performed single cell gene expression studies to begin to define the gene expression profiles of the early neural crest (NC), paraxial mesoderm (PM), and flanking epidermal ectoderm (EE). The data defines the gene expression programs of the early neural crest cells. Red is high expression, blue is low, and black is intermediate. Affymetrix Gene 1.0 ST microarray data.
Toppgene analysis for shared properties and connections among the genes in each genelist revealed both known/expected associations as well as unknown genes and gene functions. These included known neural crest genes, Gng3, Lmo4, Myc, Snai1, Sox9, Sox10, and Twist1, as well as significant numbers of genes associated with transcriptional control, signal transduction, cell cycle regulation, and mRNA splicing. Many of the neural crest genes play key roles in mesenchymal development and the determination of multiple lineages including neuronal (Gemin2, Sox9, Sox10, Ddx20, Wrap53), and craniofacial bone development (Itgb1bp1, Lmo4, Myc1, Nkd1, Plekha1, Snai1, Sox9, Twist1). Other genes were known to be critical to prevent facial or palatal clefting, but were not previously known to be associated with neural crest-specific/enhanced expression (Arhgap29, B3gat3, G6pc3, Gsk3a, Lasp1, Orc1, Pex7, Phf6, Polr1d, Pqbp1, Ugdh). Interesting early candidate neural crest markers to emerge in the analysis included Pnp2, Lxn, Ddx47, Riok3, Tgds, Zfp105 and Zfp259. These results provide, to our knowledge, the first single cell resolution global analysis of the gene expression profiles of these early neural crest cells and provide a resource through which we can improve our understanding of the genomic basis of their function.
Data Mining the Integrated Craniofacial Gene Expression Atlas
In order to enable flexible use of the data from this project for data mining and signature comparisons across the entire series of cell types, regions, and developmental stages that we have profiled, we combined, normalized, and baseline referenced all of the data into a single data matrix per technology (RNA-seq or Affymetrix GeneChip ST1.0). To illustrate the range of patterns manifested in these data, Fig 9 illustrates gene expression clusters in the RNA-seq dataset of 511 genes that are known from mouse knockout phenotypes to result in abnormal craniofacial morphology or development. For this map we used the relatively high cutoff of 5 FPKM in order to identify the most robust clusters and patterns among these genes with known effects on craniofacial development. The heatmap is formed by hierarchical clustering by genes, and by samples, of the relative gene expression values as measured in each sample (Table S17). This map also provides a framework for considering which samples and genes are similar to each other and what types of patterns represent major and minor differences. For example, note the strong similarity of E8.5 MES, E9.5 CM, and the E9.5 Ma-AR and Mx-AR samples in the portion of the heatmap that includes Twist1, Prrx1, etc. Outside of this area, however, the similarities of these samples are less prominent. Note also that this map is only showing the patterning of known craniofacial genes, and that a much larger number of other genes that are not yet known to be essential for craniofacial development follow similar types of patterns and constitute a potential discovery resource for important genes and their interactions.
Fig. 9. Heatmap of gene expression patterns revealed by known craniofacial genes as measured in the normalized RNA-seq dataset.

Hierarchically clustered gene expression patterns in the RNA-seq dataset by genes that are already known to result in abnormal craniofacial morphology or development suggest that the shared membership of genes within a given pattern may be highly informative as to specific modules of expression associated with different cell types and stages of developing craniofacial structures. Representative genes within each cluster are shown to the right of the heatmap. Yellow indicates high expression and blue indicates low expression.
To divide the complete datasets, both ST1.0 and RNA-seq, into specific geneset modules, which allows for both analyses of these modules themselves or comparisons of a given list of genes in relation to these, we subjected the normalized data matrices to a shredding protocol as described in Methods in which sample-type-specific signatures were computationally generated and then placed into the Toppgene/Toppfun database for further use. For example, querying Toppgene for Twist1, (http://toppgene.cchmc.org/enrichment.jsp), (for single gene queries, do not use FDR statistical correction), returns a list of genelists that each contain Twist1, including a list of 226 genes titled “Facebase ST1 E10.5 MaxilArch 500”, and another a list of 207 genes titled “Facebase_ST1_E8.5_ParaxMesoderm_250”. The lists can include fewer than the stated 500/250 genes because they represent non-redundant genes that map to human orthologs and there are also sublists that were formed from these parent lists based on K-means clustering (eg cluster#1). Clicking through the listed numbers of genes in Toppgene allows for detailed view of the genes in each list and allows for immediate analysis of the enriched functions, pathways, and properties that are shared among the genes in each list. For these two particular gene lists above, only 14 genes overlap, indicating a potentially interesting shift in the genes that Twist1 interacts with in the two developmental stages and compartments. To test this hypothesis using the independent samples and platform offered by the FaceBase RNA-seq dataset, we used Pearson correlation to first identify genes most tightly correlated with Twist1’s pattern of expression and then subjected the 193 top-correlated genes to hierarchical gene clustering as shown in Fig. 10. The heatmap illustrates the difference in the E8.5 and E.10.5 patterns of expression for these Twist1 related genes. We subdivided these 193 genes into three clusters based the hierarchical tree structure (not shown), giving genes showing similar levels of expression in both stages, or predominantly only E8.5 or E10.5 (Fig 10, Table S17). Using Toppgene to carry out comparative enrichment analysis of these sets is extremely revealing about each of those two compartments, and strongly suggests context-specific roles for Twist1. In the E8.5 cranial mesenchyme (including paraxial mesoderm) gene list are 16 genes that are known to regulate mesenchyme development (P< 10−9; Bmp7, Cyp26A1, Dab2, Efna1, Eng, Foxc1, Foxc2, Foxd1, Foxf2, Frzb, Pitx2, Six1, Snai1, Snai2, Sox10, Twist1), 31+ genes associated with vascular development, 18+ genes associated with craniofacial development, 6+ genes associated with palate bone morphology (P<2E-4; 5 from above plus Pdss2) and 18+ genes with additional roles in craniofacial development (including Dkk1, Prrx1, Hesx1, Hapln1, Wls, etc). 25 of these genes in the E8.5 module are sequence specific transcription factors. Overall the group of genes that Twist1 is correlated with in the E8.5 mesenchyme is richly involved in the morphogenesis and vascularization of extracellular matrix that shapes the morphology of a broad range of craniofacial structure. In contrast, the group of genes associated with the E10.5 maxillary arch module that contains Twist1 is also rich in craniofacial phenotype determining genes, but has little enrichment in extracellular matrix biology or vascular development and rather is richly involved in bony growth and development with upwards of 15 genes associated with cleft palate in mice or humans (P<4E-6; Alx1, Barx1, Chd7, Dlx1, Fgf10, Gsc, Lhx8, Msx1, Prrx1, Tbx2, Tbx3, Tmem107, Twist1). Whereas this cluster of genes is also enriched in sequence specific transcription factors (26 genes, P < 4E-9) only 6 of these are in common with the E8.5 Paraxial Mesoderm geneset. To demonstrate how these data can be further mined and used to develop new hypotheses, Fig. 11 demonstrates the combined use of ToppCluster and Cytoscape to compare the properties and functions of the Twist1-correlated genesets shown in Fig 10 (Table S17). As shown in the Fig. 10 heatmap, these signatures overlap for one group of genes as well as have their own additional genes and thus can be used to form three genelists that are then functionally compared using ToppCluster, which similar to ToppGene carries out GO and other functional analyses. The top-ranked features from a variety of functional categories can be selected within ToppCluster and then exported as an XGMML document that can then be analyzed by Cytoscape network-based algorithms and visualization [Smoot, 2011 #44]. In contrast to the single gene list queries that are discussed above, network-based representation approaches allow functional co-relationships to be demonstrated such that the connections of one set of specific molecular or functional pathways can be linked to others by shared genes, protein-protein interactions, and other properties including specific phenotypes shared by multiple genes per cluster. These connections can thus provide highly instructive relationships and implications for the existence of larger scale gene networks that are responsible for specific aspects of craniofacial development.
Figure 10. Heatmap of the top 193 genes whose expression correlates with Twist1 in the RNA-seq dataset.

Hierarchically clustered heatmap reveals three different patterns that correspond to genes with strongest expression in relevant samples from E8.5, E10.5, or both. The E8.5 cranial mesenchyme-prominent geneset is at the top (blue), and E10.5 maxillary arch-prominent set is at the bottom (green). Twist1 itself is strong in both, red group). In the heatmap, yellow indicates high expression and blue indicates low expression.
Fig. 11. Cytoscape-ToppCluster-based network analysis of genes that differentially co-express with Twist1 expression at E8.5, E10.5 or at both stages.

Twist1-correlated genes shown in Fig. 10 were divided into three clusters corresponding to expression in E8.5 cranial mesenchyme, E10.5 maxillary arch, or both. These three patterns are represented as the clusters of genes (hexagons) that are on the left (E10.5), center (E10.5+E8.5), or the right (E8.5). These three sets of genes are further divided into those previously associated with abnormal craniofacial or vascular development, shown above, and those not yet known to be associated with those phenotypes, shown below below (mouse phenotypes are lower case, human phenotypes are capitalized). Distinct classes of gene-associated properties are shown as different colored squares around the outside of the diagram. All E8.5-specific cranio/vascular functionally known genes are selected and appear as yellow hexagons, and the functions and properties that they are connected to are indicated by the red edges. The gray edges that go into those connected concepts or the concepts that have no red edges, are thus not linked to the core set of early genes. For example the facial bony abnormalities that are in the upper left are not connected to any of the E8.5 specific genes. Similarly, only 5 of the genes associated with integrin function are known to be craniofacial phenotype associated, but 9 additional genes are also associated with integrin signaling and function. This implicates a more significant role for integrin signaling and function in craniofacial mesenchyme development than might have been anticipated. The specific modules and connectivities of genes in these networks provide powerful suggestions about the gene interactions and overarching biological processes that drive craniofacial development. The importance of these additional gene function categories is suggested by the number known craniofacial connections that they have (red edges) or the number of unknown craniofacial genes that they are connected to (gray edges).
Twist1-correlated genes shown in Fig. 10 were divided into three clusters corresponding to expression in E8.5 cranial mesenchyme, E10.5 maxillary arch, or both. These three patterns are represented as the clusters of genes (hexagons) that are on the left (E10.5), center (E10.5+E8.5), or the right (E8.5) (Fig. 11). These three sets of genes are further divided into those previously associated with abnormal craniofacial or vascular development, shown above, and those not yet known to be associated with those phenotypes, shown below below (mouse phenotypes are lower case, human phenotypes are capitalized). Distinct classes of gene-associated properties are shown as different colored squares around the outside of the diagram. All E8.5-specific cranio/vascular functionally known genes are selected and appear as yellow hexagons, and the functions and properties that they are connected to are indicated by the red edges. The gray edges that go into those connected concepts or the concepts that have no red edges, are thus not linked to the core set of early genes. For example the facial bony abnormalities that are in the upper left are not connected to any of the E8.5 specific genes. Similarly, only 5 of the genes associated with integrin function are known to be craniofacial phenotype associated, but 9 additional genes are also associated with integrin signaling and function. This implicates a more significant role for integrin signaling and function in craniofacial mesenchyme development than might have been anticipated. The specific modules and connectivities of genes in these networks provide powerful suggestions about the gene interactions and overarching biological processes that drive craniofacial development. The importance of these additional gene function categories is suggested by the number known craniofacial connections that they have (red edges) or the number of unknown craniofacial genes that they are connected to (gray edges).
In summary, in this report we describe a murine craniofacial atlas of gene expression for the E8.5, E9.5 and E10.5 developmental time points. We used LCM to capture the multiple compartments, which were then used for gene expression profiling with both microarrays and RNA-seq. The use of two independent global gene expression technologies provided a high throughput cross validation of the resulting dataset. The RNA-seq data confirmed approximately 90% of genes found differentially expressed by microarray. As might be expected, however, the RNA-seq data identified more differentially expressed genes, with greater fold changes. In addition the RNA-seq data provides a global view of gene expression that extends to non-polyadenylated RNAs, including long intergenic noncoding RNAs and enhancer transcripts, as well as defining RNA-splicing patterns.
The results create a comprehensive atlas of gene expression during the early stages of craniofacial development. The changing waves of gene expression that occur during this process are defined. The transcription patterns of genes encoding all transcription factors, growth factors and receptors are characterized in a sensitive and quantitative manner. The results identify novel compartment specific markers, guide the construction of new transgenic mouse tools and globally characterize potential inter compartmental cross talk. The gene expression blueprints of the elements of craniofacial construction are delineated. This atlas component resource of the FACEBASE Consortium is intended to facilitate further discovery by the craniofacial research community (FACEBASE.ORG).
Supplementary Material
{kind=link}
Highlights.
A gene expression atlas resource of craniofacial development is presented
Laser capture microdissection isolation of craniofacial compartments
Gene expression profiling by both microarray and RNA-seq
Defining expression patterns of all growth factors and transcription factors
Acknowledgments
This work was supported by NIH grant UO1 DE020049. We thank Paul Trainor for insightful advice.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Barbosa AC, Funato N, Chapman S, McKee MD, Richardson JA, Olson EN, Yanagisawa H. Hand transcription factors cooperatively regulate development of the distal midline mesenchyme. Developmental biology. 2007;310:154–168. doi: 10.1016/j.ydbio.2007.07.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunskill EW, Aronow BJ, Georgas K, Rumballe B, Valerius MT, Aronow J, Kaimal V, Jegga AG, Yu J, Grimmond S, McMahon AP, Patterson LT, Little MH, Potter SS. Atlas of gene expression in the developing kidney at microanatomic resolution. Developmental cell. 2008;15:781–791. doi: 10.1016/j.devcel.2008.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunskill EW, Georgas K, Rumballe B, Little MH, Potter SS. Defining the molecular character of the developing and adult kidney podocyte. PloS one. 2011a;6:e24640. doi: 10.1371/journal.pone.0024640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunskill EW, Lai HL, Jamison DC, Potter SS, Patterson LT. Microarrays and RNA-Seq identify molecular mechanisms driving the end of nephron production. BMC developmental biology. 2011b;11:15. doi: 10.1186/1471-213X-11-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunskill EW, Potter SS. Gene expression programs of mouse endothelial cells in kidney development and disease. PloS one. 2010;5:e12034. doi: 10.1371/journal.pone.0012034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunskill EW, Sequeira-Lopez ML, Pentz ES, Lin E, Yu J, Aronow BJ, Potter SS, Gomez RA. Genes that confer the identity of the renin cell. Journal of the American Society of Nephrology: JASN. 2011c;22:2213–2225. doi: 10.1681/ASN.2011040401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bush JO, Jiang R. Palatogenesis: morphogenetic and molecular mechanisms of secondary palate development. Development. 2012;139:231–243. doi: 10.1242/dev.067082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chai Y, Maxson RE., Jr Recent advances in craniofacial morphogenesis. Developmental dynamics: an official publication of the American Association of Anatomists. 2006;235:2353–2375. doi: 10.1002/dvdy.20833. [DOI] [PubMed] [Google Scholar]
- Chen J, Bardes EE, Aronow BJ, Jegga AG. ToppGene Suite for gene list enrichment analysis and candidate gene prioritization. Nucleic acids research. 2009;37:W305–311. doi: 10.1093/nar/gkp427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chopra M, Amor DJ, Sutton L, Algar E, Mowat D. Russell-Silver syndrome due to paternal H19/IGF2 hypomethylation in a patient conceived using intracytoplasmic sperm injection. Reproductive biomedicine online. 2010;20:843–847. doi: 10.1016/j.rbmo.2010.02.025. [DOI] [PubMed] [Google Scholar]
- Chow KL, Hall DH, Emmons SW. The mab-21 gene of Caenorhabditis elegans encodes a novel protein required for choice of alternate cell fates. Development. 1995;121:3615–3626. doi: 10.1242/dev.121.11.3615. [DOI] [PubMed] [Google Scholar]
- Chubb JR, Trcek T, Shenoy SM, Singer RH. Transcriptional pulsing of a developmental gene. Current biology: CB. 2006;16:1018–1025. doi: 10.1016/j.cub.2006.03.092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Core LJ, Waterfall JJ, Lis JT. Nascent RNA sequencing reveals widespread pausing and divergent initiation at human promoters. Science. 2008;322:1845–1848. doi: 10.1126/science.1162228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denaxa M, Sharpe PT, Pachnis V. The LIM homeodomain transcription factors Lhx6 and Lhx7 are key regulators of mammalian dentition. Developmental biology. 2009;333:324–336. doi: 10.1016/j.ydbio.2009.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo C, Sun Y, Zhou B, Adam RM, Li X, Pu WT, Morrow BE, Moon A, Li X. A Tbx1-Six1/Eya1-Fgf8 genetic pathway controls mammalian cardiovascular and craniofacial morphogenesis. The Journal of clinical investigation. 2011;121:1585–1595. doi: 10.1172/JCI44630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo Y, Sheng Q, Li J, Ye F, Samuels DC, Shyr Y. Large scale comparison of gene expression levels by microarrays and RNAseq using TCGA data. PloS one. 2013;8:e71462. doi: 10.1371/journal.pone.0071462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hah N, Danko CG, Core L, Waterfall JJ, Siepel A, Lis JT, Kraus WL. A rapid, extensive, and transient transcriptional response to estrogen signaling in breast cancer cells. Cell. 2011;145:622–634. doi: 10.1016/j.cell.2011.03.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harding SD, Armit C, Armstrong J, Brennan J, Cheng Y, Haggarty B, Houghton D, Lloyd-MacGilp S, Pi X, Roochun Y, Sharghi M, Tindal C, McMahon AP, Gottesman B, Little MH, Georgas K, Aronow BJ, Potter SS, Brunskill EW, Southard-Smith EM, Mendelsohn C, Baldock RA, Davies JA, Davidson D. The GUDMAP database--an online resource for genitourinary research. Development. 2011;138:2845–2853. doi: 10.1242/dev.063594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hochheiser H, Aronow BJ, Artinger K, Beaty TH, Brinkley JF, Chai Y, Clouthier D, Cunningham ML, Dixon M, Donahue LR, Fraser SE, Hallgrimsson B, Iwata J, Klein O, Marazita ML, Murray JC, Murray S, de Villena FP, Postlethwait J, Potter S, Shapiro L, Spritz R, Visel A, Weinberg SM, Trainor PA. The FaceBase Consortium: a comprehensive program to facilitate craniofacial research. Developmental biology. 2011;355:175–182. doi: 10.1016/j.ydbio.2011.02.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeong J, Li X, McEvilly RJ, Rosenfeld MG, Lufkin T, Rubenstein JL. Dlx genes pattern mammalian jaw primordium by regulating both lower jaw-specific and upper jaw-specific genetic programs. Development. 2008;135:2905–2916. doi: 10.1242/dev.019778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaimal V, Bardes EE, Tabar SC, Jegga AG, Aronow BJ. ToppCluster: a multiple gene list feature analyzer for comparative enrichment clustering and network-based dissection of biological systems. Nucleic acids research. 2010;38:W96–102. doi: 10.1093/nar/gkq418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasberg AD, Brunskill EW, Steven Potter S. SP8 regulates signaling centers during craniofacial development. Developmental biology. 2013;381:312–323. doi: 10.1016/j.ydbio.2013.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim TK, Hemberg M, Gray JM, Costa AM, Bear DM, Wu J, Harmin DA, Laptewicz M, Barbara-Haley K, Kuersten S, Markenscoff-Papadimitriou E, Kuhl D, Bito H, Worley PF, Kreiman G, Greenberg ME. Widespread transcription at neuronal activity-regulated enhancers. Nature. 2010;465:182–187. doi: 10.1038/nature09033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinoshita M, Nakamura T, Ihara M, Haraguchi T, Hiraoka Y, Tashiro K, Noda M. Identification of human endomucin-1 and -2 as membrane-bound O-sialoglycoproteins with anti-adhesive activity. FEBS letters. 2001;499:121–126. doi: 10.1016/s0014-5793(01)02520-0. [DOI] [PubMed] [Google Scholar]
- Kulesa PM, Bailey CM, Kasemeier-Kulesa JC, McLennan R. Cranial neural crest migration: new rules for an old road. Developmental biology. 2010;344:543–554. doi: 10.1016/j.ydbio.2010.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lam MT, Cho H, Lesch HP, Gosselin D, Heinz S, Tanaka-Oishi Y, Benner C, Kaikkonen MU, Kim AS, Kosaka M, Lee CY, Watt A, Grossman TR, Rosenfeld MG, Evans RM, Glass CK. Rev-Erbs repress macrophage gene expression by inhibiting enhancer-directed transcription. Nature. 2013;498:511–515. doi: 10.1038/nature12209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lein ES, Hawrylycz MJ, Ao N, Ayres M, Bensinger A, Bernard A, Boe AF, Boguski MS, Brockway KS, Byrnes EJ, Chen L, Chen L, Chen TM, Chin MC, Chong J, Crook BE, Czaplinska A, Dang CN, Datta S, Dee NR, Desaki AL, Desta T, Diep E, Dolbeare TA, Donelan MJ, Dong HW, Dougherty JG, Duncan BJ, Ebbert AJ, Eichele G, Estin LK, Faber C, Facer BA, Fields R, Fischer SR, Fliss TP, Frensley C, Gates SN, Glattfelder KJ, Halverson KR, Hart MR, Hohmann JG, Howell MP, Jeung DP, Johnson RA, Karr PT, Kawal R, Kidney JM, Knapik RH, Kuan CL, Lake JH, Laramee AR, Larsen KD, Lau C, Lemon TA, Liang AJ, Liu Y, Luong LT, Michaels J, Morgan JJ, Morgan RJ, Mortrud MT, Mosqueda NF, Ng LL, Ng R, Orta GJ, Overly CC, Pak TH, Parry SE, Pathak SD, Pearson OC, Puchalski RB, Riley ZL, Rockett HR, Rowland SA, Royall JJ, Ruiz MJ, Sarno NR, Schaffnit K, Shapovalova NV, Sivisay T, Slaughterbeck CR, Smith SC, Smith KA, Smith BI, Sodt AJ, Stewart NN, Stumpf KR, Sunkin SM, Sutram M, Tam A, Teemer CD, Thaller C, Thompson CL, Varnam LR, Visel A, Whitlock RM, Wohnoutka PE, Wolkey CK, Wong VY, Wood M, Yaylaoglu MB, Young RC, Youngstrom BL, Yuan XF, Zhang B, Zwingman TA, Jones AR. Genome-wide atlas of gene expression in the adult mouse brain. Nature. 2007;445:168–176. doi: 10.1038/nature05453. [DOI] [PubMed] [Google Scholar]
- Li W, Notani D, Ma Q, Tanasa B, Nunez E, Chen AY, Merkurjev D, Zhang J, Ohgi K, Song X, Oh S, Kim HS, Glass CK, Rosenfeld MG. Functional roles of enhancer RNAs for oestrogen-dependent transcriptional activation. Nature. 2013;498:516–520. doi: 10.1038/nature12210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu H, Lan Y, Xu J, Chang CF, Brugmann SA, Jiang R. Odd-skipped related-1 controls neural crest chondrogenesis during tongue development. Proceedings of the National Academy of Sciences of the United States of America. 2013;110:18555–18560. doi: 10.1073/pnas.1306495110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Losick R, Desplan C. Stochasticity and cell fate. Science. 2008;320:65–68. doi: 10.1126/science.1147888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMahon AP, Aronow BJ, Davidson DR, Davies JA, Gaido KW, Grimmond S, Lessard JL, Little MH, Potter SS, Wilder EL, Zhang P G project. GUDMAP: the genitourinary developmental molecular anatomy project. Journal of the American Society of Nephrology: JASN. 2008;19:667–671. doi: 10.1681/ASN.2007101078. [DOI] [PubMed] [Google Scholar]
- Melo CA, Drost J, Wijchers PJ, van de Werken H, de Wit E, Oude Vrielink JA, Elkon R, Melo SA, Leveille N, Kalluri R, de Laat W, Agami R. eRNAs are required for p53-dependent enhancer activity and gene transcription. Molecular cell. 2013;49:524–535. doi: 10.1016/j.molcel.2012.11.021. [DOI] [PubMed] [Google Scholar]
- Nichols JT, Pan L, Moens CB, Kimmel CB. barx1 represses joints and promotes cartilage in the craniofacial skeleton. Development. 2013;140:2765–2775. doi: 10.1242/dev.090639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raghavachari N, Barb J, Yang Y, Liu P, Woodhouse K, Levy D, O’Donnell CJ, Munson PJ, Kato GJ. A systematic comparison and evaluation of high density exon arrays and RNA-seq technology used to unravel the peripheral blood transcriptome of sickle cell disease. BMC medical genomics. 2012;5:28. doi: 10.1186/1755-8794-5-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raj A, Peskin CS, Tranchina D, Vargas DY, Tyagi S. Stochastic mRNA synthesis in mammalian cells. PLoS biology. 2006;4:e309. doi: 10.1371/journal.pbio.0040309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smoot ME, Ono K, Ruscheinski J, Wang PL, Ideker T. Cytoscape 2.8: new features for data integration and network visualization. Bioinformatics. 2011;27:431–432. doi: 10.1093/bioinformatics/btq675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szabo-Rogers HL, Smithers LE, Yakob W, Liu KJ. New directions in craniofacial morphogenesis. Developmental biology. 2010;341:84–94. doi: 10.1016/j.ydbio.2009.11.021. [DOI] [PubMed] [Google Scholar]
- Tukel T, Sosic D, Al-Gazali LI, Erazo M, Casasnovas J, Franco HL, Richardson JA, Olson EN, Cadilla CL, Desnick RJ. Homozygous nonsense mutations in TWIST2 cause Setleis syndrome. American journal of human genetics. 2010;87:289–296. doi: 10.1016/j.ajhg.2010.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang D, Garcia-Bassets I, Benner C, Li W, Su X, Zhou Y, Qiu J, Liu W, Kaikkonen MU, Ohgi KA, Glass CK, Rosenfeld MG, Fu XD. Reprogramming transcription by distinct classes of enhancers functionally defined by eRNA. Nature. 2011;474:390–394. doi: 10.1038/nature10006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Gerstein M, Snyder M. RNA-Seq: a revolutionary tool for transcriptomics. Nature reviews Genetics. 2009;10:57–63. doi: 10.1038/nrg2484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zembrzycki A, Griesel G, Stoykova A, Mansouri A. Genetic interplay between the transcription factors Sp8 and Emx2 in the patterning of the forebrain. Neural development. 2007;2:8. doi: 10.1186/1749-8104-2-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y, Guo YJ, Tomac AC, Taylor NR, Grinberg A, Lee EJ, Huang S, Westphal H. Isolated cleft palate in mice with a targeted mutation of the LIM homeobox gene lhx8. Proceedings of the National Academy of Sciences of the United States of America. 1999;96:15002–15006. doi: 10.1073/pnas.96.26.15002. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.