

Summary
Fluorescent proteins are commonly used to label cells across organisms, but the unmodified forms cannot control biological activities. Using GFP-binding proteins derived from Camelid antibodies, we co-opted GFP as a scaffold for inducing formation of biologically active complexes, developing a library of hybrid transcription factors that control gene expression only in the presence of GFP or its derivatives. The modular design allows for variation in key properties such as DNA specificity, transcriptional potency and drug dependency. Production of GFP controlled cell-specific gene expression and facilitate functional perturbations in the mouse retina and brain. Further, retrofitting existing transgenic GFP mouse and zebrafish lines for GFP-dependent transcription enabled applications such as optogenetic probing of neural circuits. This work establishes GFP as a multi-functional scaffold and opens the door to selective manipulation of diverse GFP-labeled cells across transgenic lines. This approach may also be extended to exploit other intracellular products as cell-specific scaffolds in multi-cellular organisms.
Introduction
Studies of multi-cellular organisms would be greatly facilitated by the ability to manipulate the activities of genes within any tissue or cell type. This is challenging to achieve in tissues with diverse cell types, such as the nervous system (Masland, 2004). To label and provide genetic access to diverse cell types, much effort has been devoted to generating transgenic organisms in which transgenes are placed under the control of large genomic fragments or endogenous gene loci. Transgenic lines expressing driver genes such as transcription factors or site-specific recombinases can then be used to control the expression of genes in responder cassettes. However, the utility of individual lines is limited by a transgene's functional abilities; reporter lines expressing fluorescent proteins and histochemical enzymes are useful for labeling cells, but cannot currently be used to control biological activities. To replace transgenes driven by the same cis-regulatory elements requires generation of additional transgenic lines. Such a procedure can be costly and lengthy for organisms such as the mouse. Thus, a key to conducting efficient and wide-ranging studies on existing and future model organisms is to increase the versatility of transgenic resources.
Owing to their ease of detection, green fluorescent protein (GFP) and its derivatives (Tsien, 1998) have become common markers of gene expression (Chalfie et al., 1994) across model organisms. Notably, thousands of transgenic GFP lines have been generated for the mouse (Gong et al., 2003). This growing and important resource reveals the expression pattern of many genes and provides strains in which GFP selectively labels many cell types of interest (www.gensat.org) (Siegert et al., 2009). Transgenic GFP lines have enabled applications such as cell type-specific transcriptome profiling as well as targeted anatomical and physiological analysis (Huang et al., 2003; Siegert et al., 2012). However, functional manipulation of GFP-labeled cell types often requires the use of driver lines such as those that express Cre, which currently exist in limited numbers.
A system converting GFP expression into desired molecular outputs would enable existing and future transgenic GFP lines to be used directly for gene manipulation of specific cell types. Synthetic RNA devices have been engineered to convert the presence of an intracellular protein into gene expression output (Culler et al., 2010). While promising, these devices have yet to be applied in animals (Chang et al., 2012). Meanwhile, artificially-derived binding proteins, herein including antibodies and unrelated proteins with ideal structures for evolving target recognition (Wurch et al., 2012), are being used intracellularly to target proteins in cells and organisms. Thus far, these reagents are only used for target-centric purposes such as protein interference (Jobling et al., 2003), degradation (Caussinus et al., 2012) and modulation (Kirchhofer et al., 2010). Artificially-derived binding proteins could possibly be a powerful platform to co-opt intracellular proteins as cell-specific signals that control synthetic circuits, without modifications to the target protein or reliance on the target protein's natural interactions or functions.
We explored whether artificial-derived binding proteins can confer GFP with the ability to regulate genes. GFP seems relatively inert in many heterologous systems; it is freely diffusible in the cytoplasm, can enter the nucleus, confers low cytotoxicity and has few interactions with host proteins (Trinkle-Mulcahy et al., 2008). The development of GFP binding proteins (GBPs) from Camelid antibodies (Kirchhofer et al., 2010) has made possible the construction of GBP-fusion proteins non-covalently linking GFP to a variety of proteins in living cells (Caussinus et al., 2012). These reagents, termed nanobodies, are single-chain antigen binding domains that are relatively small in size (~300-400bp) and can be easily expressed in living cells (Rothbauer et al., 2006). Given the availability of multiple GBPs, we reasoned that GFP might be used as a scaffold to organize the formation of biologically active complexes. In one scheme, GFP would act like a small molecule “dimerizer”, bridging the association of distinct modular domains or protein fragments to reconstitute useful activities such as transcription and recombination (Jullien et al., 2003; Pollock and Clackson, 2002).
Here, we identified pairs of GBPs that can recruit tethered proteins onto the GFP scaffold, providing the means by which GFP-inducible systems can be built. We developed a GFP-dependent transcription system with these reagents, enabling control of any target gene for functional studies across tissues and organisms. The modular design of the transcription system allowed for straightforward and predictable changes to critical features such as DNA binding specificity, transcriptional potency and drug dependency. Our work extends the functionality of GFP into the regulatory realm, thus opening the door to selective manipulation of GFP-labeled cells across transgenic GFP lines and establishing novel components for the design of synthetic circuits.
Results
Design and isolation of GFP-dependent transcription factors
In order to use GFP as a dimerizer, one has to identify GBP pairs that can bind to GFP at the same time. Suitable GBP pairs could then bring together fusion protein partners on the GFP scaffold. We obtained six GBPs for this purpose (Kirchhofer et al., 2010). Several GBPs were reported to bind additively to a pre-formed GFP-GBP1 complex when tested as purified proteins in vitro (Kirchhofer et al., 2010). However, it was unclear whether any of the identified pairs could co-occupy GFP, tolerate the addition of fusion partners and induce the formation of biologically active complexes in cells. Furthermore, many possible GBP pair-wise combinations had not been tested for their ability to co-occupy GFP. To address these issues, we performed an in vitro reporter screen for GBP pairs that could induce the formation of an active transcription factor (Figure 1 and Extended Experimental Procedures). The Gal4 DNA binding domain (DBD) and VP16 activation domain (AD) (Sadowski et al., 1988) were separately fused to GBPs in various configurations and placed under control of the ubiquitous CAG promoter (Niwa et al., 1991) (Figure 1B). DBD-GBP (DBDG) and AD-GBP (ADG) fusion constructs were screened in pair-wise combinations for GFP-dependent activation of a UAS luciferase (luc2) reporter in 293T cells. Functional DBDG/ADG pairs will be referred to as Transcription Devices Dependent on GFP (T-DDOG). T-DDOGs employing GBP1+6 or GBP2+7 consistently gave the strongest reporter induction (Figure 1C, 2 and S1), and became the focus of this study. To specify DBDG+ADG combinations, the DBD-GBPX fusion is listed in regular font, along with the AD-GBPX fusion in superscript, giving DBD-GBPXAD-GBPY. Specific T-DDOG configurations are tabulated in Table S1.
Figure 1. In vitro screen used to identify functional GBP pairs for the GFP-dependent transcription system.

(A) Schematic of GFP-dependent transcription system. DBD, DNA-binding domain; AD, activation domain; UAS, upstream activating sequence. (B) Strategy for making DBDGBP (DBDG) or AD-GBP (ADG) fusion constructs used in the screen for T-DDOGs. All genes were controlled by the CAG promoter in pCAG vector. (C) Schematic of typical in vitro luciferase screen for functional GBP-fusion combinations capable of inducing GFP-dependent transcription. See Table S1
Figure 2. Characterization of the GFP-dependent transcription system.

(A) Schematic of Gal4-based T-DDOGs. (B) GFP-dependent activation of UAS-luc2 by Gal4-GBP6VP16-GBP1 and Gal4-GBP2VP16-GBP7. n=9. (C) Gal4-GBP6VP16-GBP1 strongly activated UAS-tdT in the presence of GFP. Mutation of GBP1-binding residues in GFP (GFPmG1) abolished tdT activity. Scale bar, 10μm. (D) Specificity of T-DDOGs for different fluorescent proteins. n=9. *, p<0.001. (E) Activity of Gal4-GBP6VP16-GBP1 in response to varying amount of transfected GFP plasmids. The transfected DNA amount was kept constant among conditions, with CAG-mCherry (bottom panel) acting as a filler plasmid to compensate for reduction in GFP (top panel) plasmids. Panels show representative GFP and mCherry fluorescence in single cells for each corresponding data point below. n=6. Plots are mean +/− SD. See also Figure S1, S2 and Table S2.
Characterization of the GFP-dependent transcription system in vitro
The induced transcription output in 293T cells was found to be dependent on all components of the system, as removal of GFP, DBDG or ADG from the transfection mixture resulted in loss of reporter activity (Figures 2B and S1). Reporter induction was further dependent on the ability of GBP to bind to GFP. Based on the GBP1 + GFP crystal structure (Kirchhofer et al., 2010), we mutated GFP residues expected to directly interact with GBP1. One such variant, GFPmG1, carries the mutations E143K and N147Q. Like GFP, GFPmG1 was localized to the nucleus by the VP16AD-GBP7 fusion protein (Figures S2). However, unlike GFP, GFPmG1 was not localized to the nucleus by the VP16AD-GBP1 fusion protein (Figure S2). In agreement with this, GFPmG1 induced strong UAS-reporter in the presence of Gal4-GBP2VP16-GBP7, but not Gal4-GBP6VP16-GBP1 (Figure 2B, 2C, and S1 and Table S2). These data confirm a requirement for GFP-GBP interactions, as well as suggest that GBP2 and GBP7 do not depend critically on residue 143 or 147 for binding to GFP.
We also tested whether T-DDOG activity can be controlled by the GFP derivatives cyano and yellow fluorescent proteins (CFP and YFP), and the Discosoma-derived red fluorescent proteins dsRed, mCherry and tdTomato (tdT) (Shaner et al., 2005). CFP and YFP induced Gal4-GBP2VP16-GBP7 activity to a similar extent as GFP (Figure 2D). However, CFP had reduced ability to activate Gal4-GBP6VP16-GBP1. This was expected because CFP differs from GFP at the GBP1 interacting residue 147 (Rothbauer et al., 2008). Also as expected, none of the red fluorescent proteins could induce T-DDOG activity. In support of this, red fluorescent proteins were diffusely distributed in the cell even when T-DDOG components were clearly localizing GFP to the nucleus (Figures 2C, 2E).
To evaluate the effect of GFP level on T-DDOG activity, we varied the amount of GFP plasmid delivered to 293T cells and examined UAS-luc2 expression in the presence of Gal4-GBP6VP16-GBP1. The observations were consistent with those reported for small molecule dimerizers (Ho et al., 1996). T-DDOG activity increased linearly with the amount of transfected GFP until a certain point, beyond which further increases in GFP led to reduction of activity. The reduced activity is likely due to titration of T-DDOG components by GFP. Interestingly, GFP was highly enriched in the nucleus at levels correlating with the rising phase of the dosage curve, but spread into the cytoplasm at levels correlated with the declining phase of the curve (Figure 2E).
Modularity of GFP-dependent transcription system permits various adjustments and fine-tuning
Transcription factors are highly modular (Luan et al., 2006; Sadowski et al., 1988). To exploit this feature for creating a diversity of T-DDOGs with varying properties, we substituted the transcription domains in our original GBP fusion library with other commonly used ones and conducted additional in vitro screens. Indeed, we were able expand and diversify the functional repertoire of T-DDOGs. T-DDOGs using the rTetR and LexA DBDs activated reporters bearing their respective binding sequences, tetO (included in tetracycline response element, or TRE) and lexAop, only when GFP was present (Figure 3A-3E) (Butala et al., 2009; Schonig et al., 2010). The activities of rTetR-based T-DDOGs were further found to depend on doxycycline levels (Figure 3D). This drug dependency provides temporal control for the system.
Figure 3. T-DDOGs are highly adjustable.

(A-E) T-DDOGs based on LexA (A) and rTetR (C) DBDs. Doxycycline is “D” in (C). Tetracycline response element (TRE) includes seven tetO sequences (C). (B) LexA-GBP1VP16-GBP6 activated a lexAop-luc2 reporter only in the presence of GFP. n=9. (D) rTetR-GBP1VP16-GBP6 activated TRE-luc2 in a GFP and doxycycline-dependent manner. n=6-9. (E) Similar results were seen with TRE-tdT. Doxycycline was used at 1μg/ml. Images were taken 16 hours post-transfection. (F and G) Tuning T-DDOGs with adjustable DBDs and ADs. (F) Increasing the number of GBP1 on Gal4DBD (n=6-9) enhanced the transcriptional potency for each ADG (n=9). (G) Potency of p65AD compared to VP16AD. T-DDOGs used are Gal4-GBP1p65-GBP6 and Gal4-GBP1-BVP16-GBP6. n=9. Scale bar, 100μm. Plots are mean +/− SD.
T-DDOGs can also be adjusted to alter their transcriptional potency. The critical region for VP16AD function lies within a 12 amino acid peptide (VPmin) (Baron et al., 1997). We could predictably adjust the transcription activity of Gal4-GBP1AD-GBP6 by either varying the number of VPmin repeats or the number of GBPs fused to the DBD (Figure 3F). We further isolated potent T-DDOGs bearing the p65AD (Schmitz and Baeuerle, 1991), an alternative to VP16AD in synthetic transcription systems (Rivera, 1998) (Figure 3G). Overall, we consistently isolated potent T-DDOG variants using the GBP1+6 and GBP2+7 combinations, suggesting that these pairs can effectively recruit various combinations of fusion partners onto the GFP scaffold.
The GFP-dependent transcription system can be used in the mouse for cell-specific gene regulation
To evaluate whether GFP can control the activity of T-DDOGs in vivo, we used electroporation to introduce GFP, T-DDOGs and UAS-tdT into the murine retina. In our initial tests, we found that overexpression of VP16AD caused mis-positioning of rod photoreceptors in the outer nuclear layer (ONL), likely due to squelching of transcription machinery (Figure S3) (Gill and Ptashne, 1988). To address this, we screened T-DDOGs with alternative ADs described above for their effects in the retina (Extended Experimental Procedures). We found that T-DDOGs made with VPminx2 and p65 ADs induced little to no disruption of normal rod positioning in the ONL (Figure S3). T-DDOGs bearing p65ADs were used in all subsequent experiments.
We examined how T-DDOG activity would respond to changes in GFP expression in the retina (Figures 4, S4, S5 and Table S3-S6). When GFP was expressed under the ubiquitous CAG promoter, UAS-tdT was induced in GFP-expressing cell types of both the ONL and inner nuclear layer (INL) (Figure 4C). In contrast, little to no tdT signal was detected in electroporated retinas when GFP was excluded (Figures 4C and S4B). When the GFP expression pattern was manipulated with promoters specifically active in rods (Rho-GFP) (Matsuda and Cepko, 2004) or in ON bipolar cells (mGluR6-GFP) (Kim et al., 2008), the tdT expression pattern shifted accordingly, and was highly restricted to GFP-expressing cells (Figures 4D, 4E, S4 and S5). Cells labeled by the electroporation marker, nuclear β-galactosidase (n-βgal), but not GFP, did not express tdT (Figures C-4E). The efficiency of UAS-tdT activation, adjusting for the probability of a cell receiving all 4 necessary components for tdT activation, was ~56-93%. Despite the lack of GFP signal amplification with antibodies, more than 90% to 95% of tdT positive cells were positive for GFP expression in all cases (Table S6). Unexpectedly, we detected faint mGluR6-GFP expression in the ONL, which was not seen without the introduction of T-DDOGs. This low level of GFP induced little to no tdT expression (Figures 4E and S4G). Follow-up experiments indicated that Gal4-GBP1p65-GBP6, but not Gal4-GBP2p65-GBP7, stabilized a low level of ONL GFP leaking from the mGluR6 promoter (see Supplemental Information). This suggests that GBP1+6 can reveal GFP expression that is normally below the threshold of detection, whereas the GBP2+7 pair allows for gene manipulation without revealing sub-detection levels of the native GFP expression pattern. Overall, these results showed that GFP could be used as a cell-specific regulator of T-DDOG activities in the mouse.
Figure 4. GFP controls the spatial expression of genes in vivo.

(A) Schematic of experiment. (B) (left) In electroporated retinas, CAG-GFP expresses in multiple cell types (green outline). Rho-GFP expresses in photoreceptors of the outer nuclear layer (ONL) (beige fill). mGluR6-GFP expresses in ON bipolar cells of the inner nuclear layer (INL) (orange fill). GCL, ganglion cell layer. (right) Anticipated UAS-tdT expression pattern, aligned to left diagram (C) Gal4-GBP1p65-GBP6 induces UAS-tdT only in the presence of GFP. Nuclear β-galactosidase (n-βgal) (magenta) is an electroporation marker. (D and E) (top) Rho-GFP and mGluR6-GFP induce tdT expression in rods and ON bipolar cells, respectively. (bottom) tdT activation depends on ADG. Inset of (E) shows GFP and tdT co-localization upon GFP intensity enhancement. Merge panels includes GFP, tdT and DAPI channels. Scale bar, 20μm. See also Figure S3, S4, S5 and Table S3-S6.
Utility of GFP-dependent transcription system for electrophysiological studies and gene perturbations
To evaluate whether T-DDOGs altered the properties of neurons, we electroporated GFP, T-DDOGs and UAS-tdT into the somatosensory cortex and examined various properties of cortical neurons from ~1.5 week old mouse brains. We compared pyramidal neurons expressing the full set of T-DDOGs, showing both GFP and tdT, with those that expressed GFP alone, as well as with neighboring neurons that lacked fluorescence (Figure 5A-5D). TdT signal was not observed in GFP-negative neurons in the acute slices (data not shown). We found that excitability and passive membrane properties were similar for the three groups of neurons (Figure 5C-D and Table S7) and were consistent with intrinsic cellular properties previously reported for cortical neurons of this age (Oswald and Reyes, 2008). Moreover, transducing T-DDOGs did not impact morphological features such as dendritic spine density and length (Figure S6 and Table S7). Thus, T-DDOGs are compatible with electrophysiological assays and do not induce functional and structural alterations in the developing brain, within the tested time frame.
Figure 5. T-DDOGs support electrophysiological and gene perturbation studies in the central nervous system.

(A) Electroporation setup for neuronal recordings. Micrograph shows GFP in electroporated primary somatosensory cortex (S1). (B-F) (B) Image of an acute brain slice from an electroporated mouse. Scale bar, 10μm. Three categories of pyramidal layer 2/3 S1 neurons were recorded from brain slices: non-fluorescent controls (gray), GFP+ (green), and GFP+/tdT+ (yellow). (C) Representative single current-clamp trace of action potentials in response to a 50pA, 1000ms-long step current injection. (D) Plots show action potential (AP) frequency upon current injection, as well as input resistance and membrane capacitance of recorded cell classes. p>0.5 for all comparisons, (n=8-10 neurons per condition). Plots show mean +/− SEM. (E-H) GFP-dependent excision of Otx2fl/fl in the retina. (E) P0, Otx2fl/fl mouse retina was electroporated with T-DDOG components and UAS-Cre, and either CAG-GFP or CAG-dsRed, (F) Loss of Otx2 was confirmed by Otx2 immunostaining and (G,H) ectopic Pax6+ ONL cells (n=10 stacks, 5 retinas per electroporated condition. For non-electroporated retina, n=19 stacks, 10 retinas). Boxplots show median, maximum and minimum values. Retinal stacks are 12μm thick confocal images. *, p<0.001. n-βgal marks electroporated cells in (F). Scale bar, 5μm in (F), 20μm in (G,H). See also Figure S6, S7 and Table S7.
We further evaluated the utility of T-DDOGs for deriving biological effects in developing tissues. Otx2 is a homeobox gene that is necessary for photoreceptor specification in the retina (Nishida et al., 2003). We used GFP to induce Cre-mediated excision of a floxed Otx2 allele (Otx2fl/fl) (Tian et al., 2002) in mouse retinas ex vivo, with Cre being under the regulation of the UAS promoter (Figure 5E-5H). This led to the loss of Otx2 protein and the expected ectopic gain of Pax6 in the ONL (Nishida et al., 2003) (Figure 5F and 5G). Conversely, Otx2 levels were not significantly perturbed when GFP was replaced in the same experiment with dsRed (Figure S7). A slight increase in Pax6+ ONL cells above background values was likely due to leakage of UAS-Cre under experimental conditions. Thus, T-DDOGs will be useful for converting GFP expression into desired Cre-mediated genetic changes using a variety of existing conditional alleles. Taken together, these results showed that T-DDOGs are suitable for gene perturbations in the mouse and compatible with assays of cellular function.
Retrofitting transgenic GFP lines for GFP-dependent manipulation of genes and neural circuits
We examined whether T-DDOGs can retrofit transgenic GFP lines for cell-specific gene manipulations. In the mouse retina, visual information detected by rod and cone photoreceptors is transmitted to bipolar cells and ultimately to ganglion cells. Amongst bipolar cells, the rod bipolar cell type receives input from rods, whereas many types of cone bipolar cells receive input primarily from cones. Currently, almost none of the cone bipolar types can be singly isolated for genetic manipulation, but multiple GFP lines do label subsets of bipolar types (Siegert et al., 2009) (Wassle et al., 2009). The α-gustducin-GFP transgenic line, Tg(GUS8.4-GFP) (Huang et al., 2003), expresses GFP in type 7 cone bipolar cells and in rod bipolar cells (Figure 6A). Both cell types respond to light increments and are called ON bipolar cells. Introduction of T-DDOGs and UAS-tdT into Tg(GUS8.4-GFP) retinas resulted in tdT induction selectively in these 2 cell types; identification was based on morphology and axonal stratification in the inner plexiform layer (IPL), aligned to the IPL markers Calbindin or Calretinin (Ghosh et al., 2004) (Figure 6A-6C). Importantly, 98.9% of tdT+ cells were positive for GFP expression (n=91 cells, sampled from 3 retinas).
Figure 6. Retrofitting a transgenic GFP mouse line for GFP-dependent manipulation of gene expression and neural circuit activities.

(A) Tg(GUS8.4GFP) expresses GFP in type 7 cone bipolar and rod bipolar cell types (green fill) of the retina. Adopted schematic (Ghosh et al., 2004). (B) Cryosection of electroporated, Tg(GUS8.4GFP) retina expressing Gal4-GBP2p65-GBP7 and UAS-tdT. Scale bar, 20μm. (C) Type 7 (left) and rod bipolar (right) cell types labeled by UAS-tdT. Anti-Calretinin (left) or anti-Calbindin (right) staining identify specific layers of the IPL. Scale bar, 10μm. GFP was immunostained in (B, C). (D) Schematic of ChR2 experiment. Electroporated Tg(GUS8.4-GFP) retinas expressing 10xUAS-ChR2/H134R-mCherry and 5xUAS-tdT were analyzed for ChR2-mediated responses in random GCL cells. (E) Cumulative plot of ON responses in GCl cells. Number of spikes counted during the first 300ms after stimulus onset, normalized to control (minus APB). APB blocks ON responses originating from photoreceptors. Plots are mean +/− SEM (n=4 per condition) (F) Spiking response of a GCL cell. Gray bar, duration of light stimulus. Response to normal light stimuli under control condition (top), or in the presence of APB (middle). Light stimuli focused on INL activate ChR2/H134R in the presence of APB (lower). (G and H) Top and side views of a neurobiotin-filled (green) ganglion cell identified by light-stimulation of ChR2. Magenta lines indicate level of anti-Chat bands (not shown). Scale bar, 20μm.
One exciting use of T-DDOGs would be to express light-sensing ion channels in cell types labeled by transgenic GFP for refined, optogenetic probing of neural circuits (Yizhar et al., 2011). We explored this possibility by expressing a UAS-regulated channelrhodopsin-2 (ChR2) variant, H134R (Nagel et al., 2005) in Tg(GUS8.4-GFP)-labeled cells. We asked whether light-driven ChR2 activation in GFP-labeled bipolar cells could trigger downstream spiking responses in cells of the GCL (Figure 6D). Electroporated retinas were presented with two different light stimuli and recordings were performed on GCL cells. The first stimulus had low light intensity and could evoke photoreceptor-mediated responses in GCL cells, but was not bright enough to activate ChR2. We used this stimulus to select GCL cells that responded to both light increments and decrements (ON/OFF cells) (Figure 6E-6F). We next blocked synaptic communication between photoreceptors and ON bipolar cells with 2-amino-4-phosphonobutyrate (APB) (Slaughter and Miller, 1981), and presented the retina with a brighter light stimulus that could activate ChR2. Since ON/OFF GCL cells receive excitatory input from ON bipolar cells, some of these cells should be connected via excitatory synapses (directly or indirectly) to ChR2-expressing ON bipolar cells. Indeed, the brighter stimulus elicited ON responses in some recorded GCL cells in the presence of APB (Figure 6G-6H). In contrast, recordings made from ON and ON/OFF GCL cells in non-electroporated regions of multiple retinas did not reveal any response after the onset of the brighter stimulus, in the presence of APB (data not shown). Thus, ChR2 activation in rod bipolar or ON cone bipolar cells was robust enough to evoke neurotransmitter release from bipolar cells. Further, the resulting current in GCL cells was large enough to reach spike threshold and evoke spiking responses. These results showed that T-DDOGs could turn on optogenetic tools in transgenic GFP cells, permitting functional interrogation of neural circuits.
Utility of T-DDOGs in zebrafish
In order to determine if T-DDOGs can direct GFP-dependent activities in other organisms, zebrafish were tested. Here, T-DDOG components were translated from one bicistronic transcript by linking DBDG and ADG components with an IRES element. We microinjected RNAs with this structure into ubiquitin-GFP transgenic (Tg(ubi-GFP) (Mosimann et al., 2011) zebrafish embryos in a transient reporter assay. Indeed, mosaic UAS-tdT expression was clearly induced in 78 of 90 injected GFP+ embryos but not in the 136 injected GFP- embryos (Figure 7). This demonstrates the utility of this system across species.
Figure 7. GFP-dependent transcription in transgenic zebrafish.
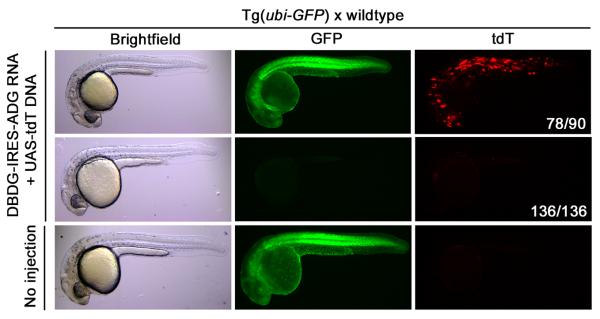
Embryos from Tg(ubi-GFP) × wildtype outcrosses were microinjected with DBDG-IRES-ADG (Gal4-GBP1p65-GBP6) RNA and UAS-tdT DNA at the 1-2 cell stage and examined 1-2 days post-fertilization. Images represent X number of embryos out of Y number of injected embryos (X/Y), shown in white font in tdT panel.
Discussion
Fluorescent proteins are useful for illuminating cells and cellular processes. Moreover, their apparent lack of connection to many host protein networks make them ideal scaffolds upon which one can build synthetic complexes with desirable biological activities. We demonstrated this principle here, using GFP to induce formation of a hybrid transcription factor for gene regulation purposes. The ability to use GFP for gene regulation now enables one to experiment with many GFP-labeled cell types without the need to create new cell-specific driver lines or to discover new cell-specific promoters. This system can be used for gene overexpression and gene deletion (Figure 4-7) and should be able to perform RNAi knockdown (Dickins et al., 2007; Dietzl et al., 2007). Activities of the system can be controlled by GFP and its derivatives but not by red fluorescent proteins, thereby allowing the two types of fluorescent proteins to be used independently in the same experiment. Red fluorescent proteins can likely be used as scaffolds as well. In particular, monomeric variants such as mCherry would be straightforward to use, as they do not undergo obligate dimerization or tetramerization (Campbell et al., 2002).
Selective control of GFP-labeled cells in transgenic GFP organisms
The development and functions of complex multi-cellular organisms depend upon the activities of a large number of distinct cell types. To investigate these activities in the nervous system, for example, many molecular tools have been developed for anatomical circuit tracing as well as physiological control (Wickersham and Feinberg, 2012; Yizhar et al., 2011). However, the full potential of these tools can only be realized when one can selectively control them in any cell type in the nervous system. We demonstrated that the diverse transgenic GFP lines available in the mouse and other organisms would be useful for cell-specific manipulation of genes and neural circuits. In the mouse, such manipulations are performed primarily with recombinases such as Cre. While we anticipate the number of cell-specific Cre mouse lines will continually increase along with that of GFP lines, each collection of lines will be independently useful for some applications and will be complementary for other applications. As discussed below, the use of GFP is not necessarily limited to transcriptional control. Nevertheless, gene regulatory systems based on transcription factors differ fundamentally from those based on recombination strategies. As recombination induces alterations to DNA sequences, this typically results in permanent changes in gene expression. In contrast, transcription systems are reversible. Recombination systems are especially useful for targeting cells with a common gene expression history and for long-term transgene expression independent of initial induction signals. However, the irreversibility of recombination events can result in the manipulation of undesired cell types. This should be less of a problem for a transcription system, as continual expression of GFP, in our case, is required to maintain the transcription of target genes. Although GFP may persist for a prolonged period of time after its own transcription has been shut off, this effect may be advantageous in certain applications, such as when it is desirable to achieve a moderately prolonged, but reversible gene expression effect. When temporal control of gene expression is desired regardless of GFP expression, rTetR-based T-DDOGs should be useful as they are additionally controlled by drug treatment (Figure 3C-E), and could take advantage of the various TRE reagents available (Schonig et al., 2010).
There are additional reasons to use GFP lines for cell-specific targeting. First, not all definable cell types can be specifically targeted by a single driver line (Dymecki et al., 2010). Restriction of target gene expression may be accomplished by intersecting GFP expression with expression of T-DDOG components, other transcription systems, and/or recombination systems. Second, position effects can sometimes unexpectedly activate GFP in unique cell types. For example, this is thought to be the case for Tg(GUS8.4-GFP) line and the Tg(Thy1-XFP) collections (Huang et al., 2003) (Feng et al., 2000). Replacement of transgenes typically requires the generation of new transgenic lines and can result in changes to cell-specificity of transgene expression. Emerging site-specific, genome-editing strategies hold promise for enabling efficient swapping of transgenes while minimizing changes to cell-specificity (Cong et al., 2013; Gohl et al., 2011; Mali et al., 2013). However, it still takes a relatively long time, as well as significant expense, to generate, characterize and maintain modified transgenic mouse lines.
The GFP-dependent transcription system should find applications beyond mice and zebrafish. As T-DDOGs are built from protein parts commonly used in other model organisms such as Drosophila, other communities can easily adopt T-DDOG components for use in concert with existing GFP driver and responder lines, as well as transient gene delivery vectors. In addition, the modularity of this system allows for a seemingly unlimited number of T-DDOGs to be created according to user demands. Notably, T-DDOGs with customizable DNA binding specificity (Hsu and Zhang, 2012) would allow for targeted control of endogenous loci without the need for responder cassettes.
Practical considerations for T-DDOG use
Although T-DDOG activities are highly dependent on GFP expression, whether one succeeds in converting an observed GFP expression pattern into corresponding gene output pattern depends on several factors. First, cells expressing GFP at low levels, or transiently, may evade detection in the initial stages of characterization. Such “background” GFP expression may be detected by certain T-DDOG configurations. Differing affinities between GBPs for GFP probably contribute to differences in T-DDOG sensitivity. Specifically, we found that GBP1+6 pair promoted detection of normally undetectable GFP expression from mGluR-GFP in photoreceptors. However, little to no T-DDOG-mediated expression occurred in these cells. As an mGluR6-driven Cre construct was found to induce recombination in photoreceptors, in addition to bipolar cells (data not shown), we interpret the photoreceptor GFP signal as reflecting leakage from the mGluR6 promoter as well as stabilization of the leaky GFP by the GBP1+6 pair. Since the GBP2+7 pair did not reveal the leaky mGluR6-GFP expression, and its corresponding T-DDOG did not induce reporter output in photoreceptors, the GBP2+7 pair may be used for cases when it is not desirable to reveal GFP expression normally below detection threshold. A second issue regards T-DDOG detection of transient GFP expression during early development. This could be addressed by using rTetR-based T-DDOGs for temporal control, or by restricting T-DDOGs expression to late progenitors or post-mitotic cells, as is possible with electroporation, viral vectors and/or late-expressing promoters.
Very high levels of GFP expression also require consideration, as too much GFP may saturate GBP binding sites, thereby preventing assembly of T-DDOGs (Figure 2E). Nonetheless, we could induce strong T-DOGG read-out in Tg(CRX-GFP), a very strong GFP-expressing mouse line (Samson et al., 2009) (data not shown). When necessary, there are approaches to overcome the issue of excessive GFP expression. First, one can capture T-DDOG activity using a recombinase as the T-DDOG read-out. We demonstrated that T-DOGGs could drive expression of Cre to induce irreversible gene expression changes regardless of changes in GFP level. Second, we showed that one could increase the number of GBPs on the DBDG component; this is expected to enhance the GFP binding capacity of the system. Lastly, higher levels of expression of T-DOGGs should be able to balance high GFP levels. Transgenic lines expressing T-DDOGs at high, medium and low levels should be sufficient for a research community to manipulate a broad range of transgenic GFP lines.
Here, we demonstrated that electroporation or microinjection could immediately be used to deliver T-DDOGs to receptive tissues and organisms for manipulation of GFP-labeled cell types. Additionally, it should require little effort to extend the delivery route to viral vectors. The two components of T-DDOGs can be linked by IRES elements or 2a peptides for expression from a single promoter (Figure 7). Each component is relatively small, ranging from ~500bp to ~1.2kbp in length, allowing both T-DDOG components to fit into popular viral vectors such as AAVs (Yizhar et al., 2011). Responder cassettes can also be delivered virally or by electroporation (Figure 4-6). Transient delivery methods usually do not provide access to all possible cell types within a tissue or organism; this can be taken into account during experimental design. For example, any retinal cell class is accessible given the right choice of AAV serotypes or electroporation method (Matsuda and Cepko, 2004, 2007; Watanabe et al., 2013). Furthermore, the inherent cell type-specificity of certain gene delivery methods can be exploited to subtract undesired GFP-labeled cell types from being manipulated. For some applications, it will be desirable to deliver T-DDOGs to all cells in a tissue, or to entire organisms. Transgenic lines expressing T-DDOGs under the control of broadly active promoters would meet these needs, and these tools are under development.
Whenever exogenous components are delivered to cells, one should be cautious of unintended effects. Potential side effects from AD overexpression have long been recognized (Gill and Ptashne, 1988), but applications with transcription factors continue to grow (Schonig et al., 2010; Venken et al., 2011) and drive meaningful discoveries (Kinoshita et al., 2012; Miyamichi et al., 2011). As we found here, problems with an AD may be overcome by testing ADs differing in origin and transcriptional potency. Alternatively, one can reduce expression of a given T-DOGG. IRES-linked cassettes typically give lower expression of genes in the second position relative to those in the first position (Mizuguchi et al., 2000). This could be used to express the DBDG component at high levels while keeping the ADG levels relatively low.
Fluorescent proteins as multi-functional switches for heterologous systems
As GFP do not have any committed regulatory function in host cells, it may be co-opted for other regulatory purposes. To realize GFP's full potential, additional GFP-binding reagents and engineering efforts will be needed to expand its functionality and to improve the performance of GFP-dependent devices. Beyond transcription, the GFP scaffold should be able to regulate other activities such as recombination (Jullien et al., 2003) and proteolysis (Wehr et al., 2006). Although the GBP1 binding epitope on GFP has been revealed by x-ray crystallography (Kirchhofer et al., 2010), it is unclear how GBP2, 6 or 7 bind to GFP. Structural understanding of how GBP pairs co-occupy GFP would facilitate the design of other GFP-inducible complexes, such as when protein fragments have to be positioned in strict orientations.
Eventually, fluorescent proteins may become preferred transgenes for organisms with long generation times, such as the mouse and primates. Since the expression pattern of fluorescent proteins can be characterized from the first set of transgene carriers, any experimental manipulation of labeled cells could be conducted within the same generation by transient device delivery, or in the next generation by mating with transgenic device carriers. Also, as one can retroactively build systems to exploit fluorescent proteins for different purposes, it may become unnecessary to generate redundant lines driving different transgenes selectively in the same cell populations. Lastly, engineering of small-molecule ligands that regulate fluorescence would even enable one to use fluorescent proteins exclusively for gene control without interfering with the imaging of other spectrally overlapping probes (Kumagai et al., 2013).
Perspective on targeting intracellular products for cell-specific control
Many intracellular products, such as RNA and proteins, are expressed in a cell-specific manner, and could potentially be exploited as spatial signals to control synthetic circuits in multi-cellular organisms. Here, we demonstrated that artificially-derived binding proteins are useful for co-opting an intracellular protein, GFP, for this purpose. Since this approach does not require any modification of the target molecule or rely on the molecule's natural interactions or functions, it may be generalizable to any intracellular product for which artificially-derived binding proteins can be selected. Certainly, GFP seems to be an ideal target because it is an exogenous molecule that shows little connection to host protein networks. However, other exogenous molecules, such as β-galactosidase or Cre recombinase, should also be useful as scaffold proteins. Furthermore, endogenous molecules probably exhibit a spectrum of connectivity within the host interactome, and a subset might be appropriate for conferring cell-specific manipulations in multi-cellular organisms. The ability to use intracellular products simply as cell-specific scaffolds would provide new ways to target and control cells in non-model organisms where transgenic lines are not available.
Experimental procedures
Detailed methods, data analysis and reagents used can be found in Extended Experimental Procedures.
Animals
All animal experiments performed were approved by the Institutional Animal Care and Use Committee at Harvard University. Animal information is in Extended Experimental Procedures.
Molecular biology and screens
Using standard techniques, coding sequences of GBPs were fused to those of DBDs or ADs in many configurations, and the products were inserted into pCAG (Niwa et al., 1991). Pairwise combination of DBDG and ADG constructs were then introduced into 293T cells or the mouse retina for screens. See Extended Experimental Procedures for details.
In vitro luciferase assays
Plasmids encoding CAG-driven GFP, DBDG and ADG were transfected via polyethyleneimine (PEI) into 293T cells along with plasmids encoding UAS-luc2 and Renilla luciferase or UAS-tdT. Cells were harvested 24 hours later for Dual Luciferase Assay (Promega), or imaged 16 hours later. All transfections, except for the dosage curve, were done at a 1:1:1 (GFP:DBDG:ADG) plasmid molar ratio.
In vivo electroporations
P0 CD1 retinas were microinjected with plasmids into the subretinal space and subjected to electroporation (Matsuda and Cepko, 2004). Plasmids encoded DBDG, ADG, CAG-nlacZ (expresses the electroporation marker n-βgal), UAS-tdT and different promoter-GFP constructs. Electroporated retinas were harvested at P14, immunostained for n-βgal and imaged by confocal microscopy. At P3-5, Tg(GUS8.4GFP) retinas were electroporated with plasmids encoding T-DDOG components, UAS-driven constructs and CAG-nlacZ; retinas were harvested between 3-4 weeks of age for UAS-tdT detection and between 8-10 weeks of age for 10×UAS-ChR2/134R stimulation. Wherever applicable, retinas were immunostained with anti-GFP or anti-dsRed to visualize processes. Anti-Calbindin or anti-Calretinin label layers in the IPL.
Neuronal recordings
C57BL/6 embryos were electroporated with CAG-driven GFP, DBDG, ADG and UAS-tdT into the lateral ventricle at embryonic day 15.5. Acute brain slices were prepared from electroporated 1-2 week old mice using standard procedures. Whole-cell current clamp recordings were performed on GFP-, GFP+ and GFP+/tdT+ cortical layer 2/3 pyramidal neurons in regions of dense electroporation. For ChR2/H134R experiment, electroporated retinas from 8-10 weeks old Tg(GUS8.4GFP) mice were flat-mounted, and loose cell-attached patch clamp was performed on GCL cells that had mCherry/tdT+ bipolar cells in their dendritic fields. Photoreceptors were stimulated by light focused on the outer segments of photoreceptors, at a light intensity of 1.3 × 103 R*/s. 20μM APB was used whenever applicable. ChR2 was stimulated by light focused on the bipolar cell layer at ~108 R*/s for 2s. Epifluorescence or 2-photon microscopes were used to identify fluorescent cells.
Otx2 removal experiment
P0 Otx2fl/fl (Tian et al., 2002) retinas were electroporated ex vivo (Emerson and Cepko, 2011) with plasmids bearing Gal4-GBP1p65-GBP6, UAS-Cre, CAG-nlacZ along with either GFP or dsRed-expressing plasmids, Retinas were cultured ex vivo, harvested at P8, immunostained for Otx2, Pax6 and/or n-βgal expression, and subjected to confocal imaging.
Zebrafish microinjections
DBDG and ADG coding sequences of Gal4-GBP1p65-GBP6 were linked by an IRES element, subcloned into pCS2+, and transcribed in vitro from the SP6 promoter (mMessage mMachine SP6 RNA Kit, Ambion). RNAs were subjected to LiCl precipitation. 1-2 cell embryos were injected with 40ng/μl RNA encoding IRES-linked T-DDOG and 25ng/μl of NotI-linearized UAS-tdT DNA. GFP+ and GFP- embryos, obtained from outcrosses of heterozygote Tg(ubi-GFP) (Mosimann et al., 2011) males to wildtype Tubingen females, were blindly injected with the same RNA/DNA mixture in the same experiment such that the injection success rate for both genotypes should be similar. Injected embryos were incubated at 28 degrees Celsius and survivors were analyzed for GFP and tdT expression 1-2 days post-fertilization.
Statistical analysis
2-tailed Student t-test assuming unequal variance was used for all comparisons except cortical recording analysis, for which one-way ANOVA was used. p>0.05 is judged as statistical significant.
Supplementary Material
Research Highlights.
- A synthetic strategy using GFP as a scaffold for control of protein association
- GFP-dependent transcription factors are modular and highly adjustable
- GFP is useful for cell-specific manipulation and functional perturbation in animals
- Retrofitting transgenic GFP lines enables functional interrogation of neural circuits
Acknowledgements
We thank Matt Harris and Katrin Henke for assistance with zebrafish injections. Mark Emerson for discussions for Otx2 experiment. Quentin Gilly of Perrimon lab for access to microplate reader. Kailun Jiang, Jonathan Coleman, Susan Dymecki, Jacob Hodgson, Douglas Allan, Andreas Mayer, Jesse Gray, Ben Huang and members of the Cepko/Tabin/Dymecki lab for comments. We are grateful to Heinrich Leonhardt and Ulrich Rothbauer for advice and GBP reagents. We thank Liqun Luo, David Bartel, Gerald Rubin, Benjamin White, Dominic Esposito, Warner Greene and Matija Peterlin for plasmids from Addgene, and Robert Margolskee for Tg(GUS8.4GFP) line. Y.K. was supported by the Leonard and Isabelle Goldenson Research Fellowship. B.R. was supported by a European Research Council grant, a Swiss-Hungarian grant, TREATRUSH, SEEBETTER and OPTONEURO grants from the European Union. C.L.C. and B.L.S. were supported by the Howard Hughes Medical Institute.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
See the Supplemental Information for detailed methods and reagents used.
Reference
- Baron U, Gossen M, Bujard H. Tetracycline-controlled transcription in eukaryotes: novel transactivators with graded transactivation potential. Nucleic Acids Res. 1997;25:2723–2729. doi: 10.1093/nar/25.14.2723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butala M, Zgur-Bertok D, Busby SJ. The bacterial LexA transcriptional repressor. Cell Mol Life Sci. 2009;66:82–93. doi: 10.1007/s00018-008-8378-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell RE, Tour O, Palmer AE, Steinbach PA, Baird GS, Zacharias DA, Tsien RY. A monomeric red fluorescent protein. Proc Natl Acad Sci U S A. 2002;99:7877–7882. doi: 10.1073/pnas.082243699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caussinus E, Kanca O, Affolter M. Fluorescent fusion protein knockout mediated by anti-GFP nanobody. Nat Struct Mol Biol. 2012;19:117–121. doi: 10.1038/nsmb.2180. [DOI] [PubMed] [Google Scholar]
- Chalfie M, Tu Y, Euskirchen G, Ward WW, Prasher DC. Green fluorescent protein as a marker for gene expression. Science. 1994;263:802–805. doi: 10.1126/science.8303295. [DOI] [PubMed] [Google Scholar]
- Chang AL, Wolf JJ, Smolke CD. Synthetic RNA switches as a tool for temporal and spatial control over gene expression. Curr Opin Biotechnol. 2012 doi: 10.1016/j.copbio.2012.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cong L, Ran FA, Cox D, Lin S, Barretto R, Habib N, Hsu PD, Wu X, Jiang W, Marraffini LA, et al. Multiplex genome engineering using CRISPR/Cas systems. Science. 2013;339:819–823. doi: 10.1126/science.1231143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Culler SJ, Hoff KG, Smolke CD. Reprogramming cellular behavior with RNA controllers responsive to endogenous proteins. Science. 2010;330:1251–1255. doi: 10.1126/science.1192128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickins RA, McJunkin K, Hernando E, Premsrirut PK, Krizhanovsky V, Burgess DJ, Kim SY, Cordon-Cardo C, Zender L, Hannon GJ, et al. Tissue-specific and reversible RNA interference in transgenic mice. Nat Genet. 2007;39:914–921. doi: 10.1038/ng2045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dietzl G, Chen D, Schnorrer F, Su KC, Barinova Y, Fellner M, Gasser B, Kinsey K, Oppel S, Scheiblauer S, et al. A genome-wide transgenic RNAi library for conditional gene inactivation in Drosophila. Nature. 2007;448:151–156. doi: 10.1038/nature05954. [DOI] [PubMed] [Google Scholar]
- Dymecki SM, Ray RS, Kim JC. Mapping cell fate and function using recombinase-based intersectional strategies. Methods Enzymol. 2010;477:183–213. doi: 10.1016/S0076-6879(10)77011-7. [DOI] [PubMed] [Google Scholar]
- Emerson MM, Cepko CL. Identification of a retina-specific Otx2 enhancer element active in immature developing photoreceptors. Dev Biol. 2011;360:241–255. doi: 10.1016/j.ydbio.2011.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng G, Mellor RH, Bernstein M, Keller-Peck C, Nguyen QT, Wallace M, Nerbonne JM, Lichtman JW, Sanes JR. Imaging neuronal subsets in transgenic mice expressing multiple spectral variants of GFP. Neuron. 2000;28:41–51. doi: 10.1016/s0896-6273(00)00084-2. [DOI] [PubMed] [Google Scholar]
- Ghosh KK, Bujan S, Haverkamp S, Feigenspan A, Wassle H. Types of bipolar cells in the mouse retina. J Comp Neurol. 2004;469:70–82. doi: 10.1002/cne.10985. [DOI] [PubMed] [Google Scholar]
- Gill G, Ptashne M. Negative effect of the transcriptional activator GAL4. Nature. 1988;334:721–724. doi: 10.1038/334721a0. [DOI] [PubMed] [Google Scholar]
- Gohl DM, Silies MA, Gao XJ, Bhalerao S, Luongo FJ, Lin CC, Potter CJ, Clandinin TR. A versatile in vivo system for directed dissection of gene expression patterns. Nat Methods. 2011;8:231–237. doi: 10.1038/nmeth.1561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong S, Zheng C, Doughty ML, Losos K, Didkovsky N, Schambra UB, Nowak NJ, Joyner A, Leblanc G, Hatten ME, et al. A gene expression atlas of the central nervous system based on bacterial artificial chromosomes. Nature. 2003;425:917–925. doi: 10.1038/nature02033. [DOI] [PubMed] [Google Scholar]
- Ho SN, Biggar SR, Spencer DM, Schreiber SL, Crabtree GR. Dimeric ligands define a role for transcriptional activation domains in reinitiation. Nature. 1996;382:822–826. doi: 10.1038/382822a0. [DOI] [PubMed] [Google Scholar]
- Hsu PD, Zhang F. Dissecting neural function using targeted genome engineering technologies. ACS Chem Neurosci. 2012;3:603–610. doi: 10.1021/cn300089k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang L, Max M, Margolskee RF, Su H, Masland RH, Euler T. G protein subunit G gamma 13 is coexpressed with G alpha o, G beta 3, and G beta 4 in retinal ON bipolar cells. J Comp Neurol. 2003;455:1–10. doi: 10.1002/cne.10396. [DOI] [PubMed] [Google Scholar]
- Jobling SA, Jarman C, Teh MM, Holmberg N, Blake C, Verhoeyen ME. Immunomodulation of enzyme function in plants by single-domain antibody fragments. Nat Biotechnol. 2003;21:77–80. doi: 10.1038/nbt772. [DOI] [PubMed] [Google Scholar]
- Jullien N, Sampieri F, Enjalbert A, Herman JP. Regulation of Cre recombinase by ligand-induced complementation of inactive fragments. Nucleic Acids Res. 2003;31:e131. doi: 10.1093/nar/gng131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim DS, Matsuda T, Cepko CL. A core paired-type and POU homeodomain-containing transcription factor program drives retinal bipolar cell gene expression. J Neurosci. 2008;28:7748–7764. doi: 10.1523/JNEUROSCI.0397-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinoshita M, Matsui R, Kato S, Hasegawa T, Kasahara H, Isa K, Watakabe A, Yamamori T, Nishimura Y, Alstermark B, et al. Genetic dissection of the circuit for hand dexterity in primates. Nature. 2012;487:235–238. doi: 10.1038/nature11206. [DOI] [PubMed] [Google Scholar]
- Kirchhofer A, Helma J, Schmidthals K, Frauer C, Cui S, Karcher A, Pellis M, Muyldermans S, Casas-Delucchi CS, Cardoso MC, et al. Modulation of protein properties in living cells using nanobodies. Nat Struct Mol Biol. 2010;17:133–138. doi: 10.1038/nsmb.1727. [DOI] [PubMed] [Google Scholar]
- Kumagai A, Ando R, Miyatake H, Greimel P, Kobayashi T, Hirabayashi Y, Shimogori T, Miyawaki A. A Bilirubin-Inducible Fluorescent Protein from Eel Muscle. Cell. 2013 doi: 10.1016/j.cell.2013.05.038. [DOI] [PubMed] [Google Scholar]
- Luan H, Peabody NC, Vinson CR, White BH. Refined spatial manipulation of neuronal function by combinatorial restriction of transgene expression. Neuron. 2006;52:425–436. doi: 10.1016/j.neuron.2006.08.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mali P, Yang L, Esvelt KM, Aach J, Guell M, DiCarlo JE, Norville JE, Church GM. RNA-guided human genome engineering via Cas9. Science. 2013;339:823–826. doi: 10.1126/science.1232033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masland RH. Neuronal cell types. Curr Biol. 2004;14:R497–500. doi: 10.1016/j.cub.2004.06.035. [DOI] [PubMed] [Google Scholar]
- Matsuda T, Cepko CL. Electroporation and RNA interference in the rodent retina in vivo and in vitro. Proc Natl Acad Sci U S A. 2004;101:16–22. doi: 10.1073/pnas.2235688100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuda T, Cepko CL. Controlled expression of transgenes introduced by in vivo electroporation. Proc Natl Acad Sci U S A. 2007;104:1027–1032. doi: 10.1073/pnas.0610155104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyamichi K, Amat F, Moussavi F, Wang C, Wickersham I, Wall NR, Taniguchi H, Tasic B, Huang ZJ, He Z, et al. Cortical representations of olfactory input by trans-synaptic tracing. Nature. 2011;472:191–196. doi: 10.1038/nature09714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizuguchi H, Xu Z, Ishii-Watabe A, Uchida E, Hayakawa T. IRES-dependent second gene expression is significantly lower than cap-dependent first gene expression in a bicistronic vector. Mol Ther. 2000;1:376–382. doi: 10.1006/mthe.2000.0050. [DOI] [PubMed] [Google Scholar]
- Mosimann C, Kaufman CK, Li P, Pugach EK, Tamplin OJ, Zon LI. Ubiquitous transgene expression and Cre-based recombination driven by the ubiquitin promoter in zebrafish. Development. 2011;138:169–177. doi: 10.1242/dev.059345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagel G, Brauner M, Liewald JF, Adeishvili N, Bamberg E, Gottschalk A. Light activation of channelrhodopsin-2 in excitable cells of Caenorhabditis elegans triggers rapid behavioral responses. Curr Biol. 2005;15:2279–2284. doi: 10.1016/j.cub.2005.11.032. [DOI] [PubMed] [Google Scholar]
- Nishida A, Furukawa A, Koike C, Tano Y, Aizawa S, Matsuo I, Furukawa T. Otx2 homeobox gene controls retinal photoreceptor cell fate and pineal gland development. Nat Neurosci. 2003;6:1255–1263. doi: 10.1038/nn1155. [DOI] [PubMed] [Google Scholar]
- Niwa H, Yamamura K, Miyazaki J. Efficient selection for high-expression transfectants with a novel eukaryotic vector. Gene. 1991;108:193–199. doi: 10.1016/0378-1119(91)90434-d. [DOI] [PubMed] [Google Scholar]
- Pollock R, Clackson T. Dimerizer-regulated gene expression. Curr Opin Biotechnol. 2002;13:459–467. doi: 10.1016/s0958-1669(02)00373-7. [DOI] [PubMed] [Google Scholar]
- Rivera VM. Controlling gene expression using synthetic ligands. Methods. 1998;14:421–429. doi: 10.1006/meth.1998.0596. [DOI] [PubMed] [Google Scholar]
- Rothbauer U, Zolghadr K, Muyldermans S, Schepers A, Cardoso MC, Leonhardt H. A versatile nanotrap for biochemical and functional studies with fluorescent fusion proteins. Mol Cell Proteomics. 2008;7:282–289. doi: 10.1074/mcp.M700342-MCP200. [DOI] [PubMed] [Google Scholar]
- Rothbauer U, Zolghadr K, Tillib S, Nowak D, Schermelleh L, Gahl A, Backmann N, Conrath K, Muyldermans S, Cardoso MC, et al. Targeting and tracing antigens in live cells with fluorescent nanobodies. Nat Methods. 2006;3:887–889. doi: 10.1038/nmeth953. [DOI] [PubMed] [Google Scholar]
- Sadowski I, Ma J, Triezenberg S, Ptashne M. GAL4-VP16 is an unusually potent transcriptional activator. Nature. 1988;335:563–564. doi: 10.1038/335563a0. [DOI] [PubMed] [Google Scholar]
- Samson M, Emerson MM, Cepko CL. Robust marking of photoreceptor cells and pinealocytes with several reporters under control of the Crx gene. Dev Dyn. 2009;238:3218–3225. doi: 10.1002/dvdy.22138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitz ML, Baeuerle PA. The p65 subunit is responsible for the strong transcription activating potential of NF-kappa B. EMBO J. 1991;10:3805–3817. doi: 10.1002/j.1460-2075.1991.tb04950.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schonig K, Bujard H, Gossen M. The power of reversibility regulating gene activities via tetracycline-controlled transcription. Methods Enzymol. 2010;477:429–453. doi: 10.1016/S0076-6879(10)77022-1. [DOI] [PubMed] [Google Scholar]
- Shaner NC, Steinbach PA, Tsien RY. A guide to choosing fluorescent proteins. Nat Methods. 2005;2:905–909. doi: 10.1038/nmeth819. [DOI] [PubMed] [Google Scholar]
- Siegert S, Cabuy E, Scherf BG, Kohler H, Panda S, Le YZ, Fehling HJ, Gaidatzis D, Stadler MB, Roska B. Transcriptional code and disease map for adult retinal cell types. Nat Neurosci. 2012;15:487–495. S481–482. doi: 10.1038/nn.3032. [DOI] [PubMed] [Google Scholar]
- Siegert S, Scherf BG, Del Punta K, Didkovsky N, Heintz N, Roska B. Genetic address book for retinal cell types. Nat Neurosci. 2009;12:1197–1204. doi: 10.1038/nn.2370. [DOI] [PubMed] [Google Scholar]
- Tian E, Kimura C, Takeda N, Aizawa S, Matsuo I. Otx2 is required to respond to signals from anterior neural ridge for forebrain specification. Dev Biol. 2002;242:204–223. doi: 10.1006/dbio.2001.0531. [DOI] [PubMed] [Google Scholar]
- Trinkle-Mulcahy L, Boulon S, Lam YW, Urcia R, Boisvert FM, Vandermoere F, Morrice NA, Swift S, Rothbauer U, Leonhardt H, et al. Identifying specific protein interaction partners using quantitative mass spectrometry and bead proteomes. J Cell Biol. 2008;183:223–239. doi: 10.1083/jcb.200805092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsien RY. The green fluorescent protein. Annu Rev Biochem. 1998;67:509–544. doi: 10.1146/annurev.biochem.67.1.509. [DOI] [PubMed] [Google Scholar]
- Venken KJ, Simpson JH, Bellen HJ. Genetic manipulation of genes and cells in the nervous system of the fruit fly. Neuron. 2011;72:202–230. doi: 10.1016/j.neuron.2011.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wassle H, Puller C, Muller F, Haverkamp S. Cone contacts, mosaics, and territories of bipolar cells in the mouse retina. J Neurosci. 2009;29:106–117. doi: 10.1523/JNEUROSCI.4442-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe S, Sanuki R, Ueno S, Koyasu T, Hasegawa T, Furukawa T. Tropisms of AAV for subretinal delivery to the neonatal mouse retina and its application for in vivo rescue of developmental photoreceptor disorders. PLoS One. 2013;8:e54146. doi: 10.1371/journal.pone.0054146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wehr MC, Laage R, Bolz U, Fischer TM, Grunewald S, Scheek S, Bach A, Nave KA, Rossner MJ. Monitoring regulated protein-protein interactions using split TEV. Nat Methods. 2006;3:985–993. doi: 10.1038/nmeth967. [DOI] [PubMed] [Google Scholar]
- Wickersham IR, Feinberg EH. New technologies for imaging synaptic partners. Curr Opin Neurobiol. 2012;22:121–127. doi: 10.1016/j.conb.2011.12.001. [DOI] [PubMed] [Google Scholar]
- Wurch T, Pierre A, Depil S. Novel protein scaffolds as emerging therapeutic proteins: from discovery to clinical proof-of-concept. Trends Biotechnol. 2012 doi: 10.1016/j.tibtech.2012.07.006. [DOI] [PubMed] [Google Scholar]
- Yizhar O, Fenno LE, Davidson TJ, Mogri M, Deisseroth K. Optogenetics in neural systems. Neuron. 2011;71:9–34. doi: 10.1016/j.neuron.2011.06.004. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.