

Abstract
The ExPortal protein secretion organelle in Streptococcus pyogenes is an anionic phospholipid-containing membrane microdomain enriched in Sec translocons and postsecretion protein biogenesis factors. Polymyxin B binds to and disrupts ExPortal integrity, resulting in defective secretion of several toxins. To gain insight into factors that influence ExPortal organization, a genetic screen was conducted to select for spontaneous polymyxin B-resistant mutants displaying enhanced ExPortal integrity. Whole-genome resequencing of 25 resistant mutants revealed from one to four mutations per mutant genome clustered primarily within a core set of 10 gene groups. Construction of mutants with individual deletions or insertions demonstrated that 7 core genes confer resistance and enhanced ExPortal integrity through loss of function, while 3 were likely due to gain of function and/or combinatorial effects. Core resistance genes include a transcriptional regulator of lipid biosynthesis, several genes involved in nutrient acquisition, and a variety of genes involved in stress responses. Two members of the latter class also function as novel regulators of the secreted SpeB cysteine protease. Analysis of the most frequently isolated mutation, a single nucleotide deletion in a track of 9 consecutive adenine residues in pstS, encoding a component of a high-affinity Pi transporter, suggests that this sequence functions as a molecular switch to facilitate stress adaptation. Together, these data suggest the existence of a membrane stress response that promotes enhanced ExPortal integrity and resistance to cationic antimicrobial peptides.
INTRODUCTION
Numerous host-pathogen interactions depend upon the timely coordination of secretion and processing of multiple proteins, including adhesion proteins and toxins. For Gram-positive pathogens, this represents a significant problem because these bacteria lack a periplasmic space with which to spatially coordinate the interaction of nascently secreted polypeptides with the accessory factors required for their folding and processing. One solution found among certain species of streptococci and enterococci is to cluster secretion and accessory factors at a defined membrane microdomain known as the ExPortal (1–5). However, the factors that promote the integrity and organization of the ExPortal microdomain are not well understood.
The ExPortal has been most extensively analyzed in Streptococcus pyogenes, an important human pathogen and the causative agent of numerous diseases ranging from superficial infections of soft tissue (e.g., pharyngitis) to toxigenic diseases (e.g., scarlet fever) to those destructive of tissue (e.g., necrotizing fasciitis). Infection can also trigger several postinfection sequelae that may arise from an inappropriate immune response (e.g., rheumatic fever) (6). To cause this array of diseases, S. pyogenes secretes a multitude of extracellular toxins and cell wall-associated proteins (7). Many of these require accessory factors to facilitate their biogenesis into an active form or for their incorporation into the cell wall and presentation at the cell surface (8). Of the few secretion systems identified in Gram-positive bacteria, including twin-arginine translocation (TAT), ESX/type VII secretion, and the accessory SecA2 system, S. pyogenes encodes only the ubiquitous and highly conserved canonical Sec (SecA1) pathway (9). Components of the Sec translocon are enriched at the ExPortal microdomain (1, 2) along with accessory factors required for toxin biogenesis, including the HtrA protease/chaperone (1). Mutations that mislocalize HtrA away from the ExPortal result in significant defects in the postsecretion processing of certain secreted toxins, including the SpeB cysteine protease (1). These data suggest that ExPortal organization functions to facilitate the postsecretory interaction of substrate and accessory biogenesis factors. Similar studies showing that mislocalization of sortase away from the ExPortal in Enterococcus faecalis impairs the efficiency of pilus biogenesis (5) also support a critical role for the ExPortal in the spatial coordination of virulence factor biogenesis.
While the mechanism that underlies the structural organization of the ExPortal is not understood, some insight has come from the observation that its lipid composition is asymmetric with respect to the peripheral membrane and is enriched in its content of anionic phospholipids (10). Microdomains enriched for anionic phospholipids have been observed in numerous bacterial species (11–13) and have been implicated in multiple cellular functions, including cell division (11), and in maintenance of Sec translocons along a longitudinal spiral pattern in certain rod-shaped bacteria (14). In the case of the E. faecalis pilus sortase (SrtC), mutations that reduce the density of positively charged residues of the cytoplasmic tail of its single transmembrane domain result in redistribution of SrtC from the ExPortal to the peripheral membrane (5). Together, these observations implicate protein-lipid charge interactions as an organizing principle responsible for retention of at least some proteins at the anionic ExPortal microdomain.
In a similar fashion, redistribution of ExPortal anionic lipids also results in redistribution of ExPortal-retained proteins. The anionic character of the microdomain renders it a target of cationic antimicrobial peptides (CAPs), and at low sublethal concentrations, certain CAPs, including polymyxin B (PB) and human neutrophil peptide 1 (HNP-1), preferentially target the ExPortal over the peripheral membrane (15). At higher but still sublethal concentrations, these CAPs disrupt the ExPortal, resulting in the redistribution of both anionic lipids and retained proteins from a focal location at the ExPortal to a more uniform circumferential distribution around the peripheral membrane (15). As a consequence, this redistribution is associated with the inhibition of secretion of several toxins important for virulence (15). These data further support a role for protein-lipid interaction in ExPortal organization. However, how the anionic phospholipids themselves are organized and maintained as part of a discrete microdomain is not understood.
In the present study, we have exploited the ability of CAPs to disrupt the ExPortal in order to gain insight into factors that participate in ExPortal assembly and maintenance. Our approach was to first select for spontaneous mutants of S. pyogenes resistant to the lethal effects of PB and then to identify those that bound PB in a manner indistinguishable from the wild type (WT). This pool was then analyzed to identify mutants that displayed enhanced ExPortal integrity in the face of PB challenge. This strategy generated a variety of mutant classes, including those with altered lipid metabolism, nutrient acquisition, and stress responses. Together, these data demonstrate that numerous cellular processes are involved in promoting the integrity of the ExPortal.
MATERIALS AND METHODS
Escherichia coli strains, media, and growth conditions.
Routine molecular cloning and plasmid propagation utilized E. coli DH5α, which was cultured in Luria-Bertani medium at 37°C. When appropriate, antibiotics were added to media at the following concentrations: 500 μg ml−1 erythromycin, 100 μg ml−1 spectinomycin, and 15 μg ml−1 chloramphenicol.
S. pyogenes strains, media, and growth conditions.
Experiments utilized Streptococcus pyogenes HSC5 (16, 17) and mutant derivatives of this strain. The spontaneous PB-resistant (PBr) mutants derived in this study are listed in Table 1, and the full genome sequence data are listed in Table S6 in the supplemental material. Other mutants used, including the various insertion and deletion mutants in various core PBr genes, are listed in Tables S1 and S2. Gene deletion mutants were generated using standard techniques (18) except in the case of the ΔPtsI::cat mutant, whereby a promoterless chloramphenicol cassette (cat) was used to select for ptsI gene replacement upon plasmid integration at the nonpermissive temperature. Routine culture employed Todd-Hewitt medium (Difco) supplemented with 0.2% yeast extract (Difco) (THY medium). When indicated, strains were cultured in C medium (0.5% protease peptone no. 3 [Difco], 1.5% yeast extract [Difco], 10 mM K2HPO4, 0.4 mM MgSO4, 17 mM NaCl). Salt-reduced C medium was identical, except that it lacked all added salts. For experiments involving carbohydrate supplementation, filter-sterilized 20% (wt/vol) stock solutions were used to add each of the indicated carbohydrates (Sigma) to a final concentration of 0.2% (wt/vol) in medium that had been sterilized in an autoclave, with the exception of raffinose, malate, and maltodextrin, which were prepared in 10%, 5%, and 0.4% (wt/vol) stock solutions, respectively, and added to sterilized 2× C medium which was further diluted with sterile water to reach finally 1× C medium. Unless otherwise indicated, all growth experiments utilized sealed culture tubes at 37°C under static conditions. Solid medium was prepared by the addition of 1.4% Bacto agar (Difco) and was cultured anaerobically in sealed jars using a commercial gas generator (GasPak; BBL) as described previously (19). When appropriate, antibiotics were added at the following concentrations: 1 μg ml−1 erythromycin, 100 μg ml−1 spectinomycin, and 3 μg ml−1 chloramphenicol.
TABLE 1.
Spontaneous PBr mutants
| Straina | PB cultureb |
PB bindingc | Locusd | Predicted proteind | Annotationd | Mutatione |
||
|---|---|---|---|---|---|---|---|---|
| Growth | SpeB | Type | Change | |||||
| WT | − | − | + | NA | NA | NA | NA | NA |
| PBr 1.1 | + | + | + | 04725 | PstS | Phosphate ABC transporter | InDel | fs-aa6 |
| PBr 2.1 | + | + | + | 04725 | PstS | InDel | fs-aa6 | |
| PBr 3.1 | + | + | + | 03260 | EbsA | Pore-forming protein | SNP | I95N |
| 04415 | TopA | DNA topoisomerase I | InDel | fs-aa661 | ||||
| 04725 | PstS | InDel | fs-aa6 | |||||
| PBr 3.2 | + | + | + | 00065 | FtsH | Cell division protein | InDel | Δ293 |
| 04720 | PstC | Phosphate ABC transporter permease | SNP | Q126* | ||||
| PBr 3.3 | + | + | + | 04725 | PstS | InDel | fs-aa6 | |
| 09125 | GdpP | GGDEF domain phosphodiesterase | InDel | fs-aa3 | ||||
| PBr 3.5 | + | + | + | 07215 | FabT | Transcription repressor, fatty acid biosynthesis | SNP | S84L |
| 09125 | GdpP | InDel | fs-aa218 | |||||
| 09180 | YfmH | M16 family zinc protease | InDel | fs-aa140 | ||||
| PBr 3.6 | + | + | + | 02925 | AgaS | Galactosamine-6P isomerase | SNP | G72D |
| 07145 | ManN | Mannose-specific PTS IID protein | SNP | I75F | ||||
| 07215 | FabT | SNP | *145L | |||||
| 09125 | GdpP | InDel | fs-aa76 | |||||
| PBr 3.7 | + | + | + | 00065 | FtsH | SNP | V291I | |
| 04725 | PstS | InDel | fs-aa6 | |||||
| PBr 4.1 | + | − | + | 03620 | ClpX | Clp protease ATP-binding subunit | SNP | Q311* |
| PBr 4.4 | + | + | + | 04725 | PstS | InDel | fs-aa6 | |
| 09125 | GdpP | InDel | Δ642–645 | |||||
| PBr 4.5 | + | + | + | 03645 | DeoB | Phosphopentomutase | SNP | S87Y |
| 04725 | PstS | InDel | fs-aa6 | |||||
| PBr 6.3 | + | + | + | 07145 | ManN | SNP | G166D | |
| PBr 6.7 | + | + | + | 00060 | Hpt | Hypoxanthine-guanine phosphoribosyltransferase | InDel | fs-aa20 |
| 09125 | GdpP | InDel | fs-aa563 | |||||
| PBr 6.10 | + | + | + | 00055 | TilS | tRNAIle-lysidine synthase | InDel | Δ428 |
| 00060 | Hpt | InDel | ΔRBS-53 | |||||
| 07135 | ManL | Mannose-specific PTS IIAB protein | SNP | N284K | ||||
| PBr 9.21 | + | − | + | 03620 | ClpX | InDel | Δ352–356 | |
| 07215 | FabT | SNP | H39N | |||||
| PBr 10.6 | + | + | + | 07215 | FabT | SNP | S84L | |
| 07750 | NA | Phage repressor protein | InDel | (−24) | ||||
| 09125 | GdpP | SNP | Q547* | |||||
| PBr 10.9 | + | + | + | 04415 | TopA | InDel | fs-aa27 | |
| 04715 | PstA | Phosphate ABC transporter permease | InDel | fs-aa35 | ||||
| 06715 | Gmk | Guanylate kinase | SNP | L131I | ||||
| PBr 10.11 | + | − | + | 01235 | NanH | N-Acetylneuraminate lyase | SNP | [V120V] |
| 03620 | ClpX | InDel | fs-aa178 | |||||
| 07215 | FabT | SNP | A56V | |||||
| PBr 10.14 | + | + | + | 04545 | GuaA | GMP synthase | SNP | A248D |
| 04725 | PstS | InDel | fs-aa6 | |||||
| PBr 10.15 | + | + | + | 00055 | TilS | SNP | G428E | |
| 00060 | Hpt | SNP | (−9) RBS | |||||
| PBr 11.1 | + | − | + | 03260 | EbsA | InDel | Δ21–25 | |
| 03620 | ClpX | SNP | E263* | |||||
| 04650 | NupP | Guanosine ABC transporter permease | SNP | W209* | ||||
| 07215 | FabT | SNP | (−9) RBS | |||||
| PBr 11.2 | + | + | + | 00055 | TilS | SNP | *429R | |
| 00060 | Hpt | SNP | (−7) RBS | |||||
| 04415 | TopA | SNP | R646* | |||||
| PBr 11.3 | + | + | + | 05585 | PtsI | PEPf-protein phosphotransferase | SNP | A510V |
| 08845 | LuxR | Transcriptional regulator, LuxR family | SNP | D693Y | ||||
| 09125 | GdpP | SNP | A574D | |||||
| PBr 11.5 | + | + | + | 07135 | ManL | SNP | N284K | |
| 08035 | Fba | Fructose-bisphosphate aldolase | SNP | A6V | ||||
| PBr 11.6 | + | + | + | 04415 | TopA | SNP | V29D | |
| 05585 | PtsI | SNP | D164Y | |||||
WT is HSC5. Mutants whose designations begin with different numbers were isolated from independent experiments.
Ability (+) or inability (−) to form colonies (growth) and express SpeB protease activity (SpeB) when grown on protease indicator plates supplemented with 400 μg/ml PB.
Focal (+) or undetectable (−) membrane binding when stained with 5 μM dansyl-PB.
Genetic loci, the predicted proteins that an ORF encodes, and annotations are based on the genome of HSC5 (16). Loci follow the format L897_xxxxx, where xxxxx are numbered. NA, not applicable. Annotations are listed only at first instance. In subsequent examples, the field is left blank.
InDel, insertion/deletion; SNP, single nucleotide polymorphism; “fs-aa” followed by numeral, frameshift at the indicated codon; *, stop codon; number in parentheses, distance upstream of start codon; RBS, ribosome binding site; data in brackets, synonymous mutation; Δ, deletion. Note that tilS and hpt mutations result from a single alteration since the 3′ end of tilS overlaps the RBS of hpt.
PEP, phosphoenolpyruvate.
Selection of polymyxin B-resistant mutants.
Wild-type HSC5 was cultured overnight in 50 ml THY medium. The following day, cultures were centrifuged at 6,000 × g for 5 min and the bacterial pellet was resuspended in 5 ml fresh THY medium. One-hundred-microliter aliquots (yielding approximately 5 × 107 CFU when plated under nonselective conditions) were plated onto protease indicator plates (solidified C medium supplemented with 2% milk) containing 400 μg ml−1 (289 μM) polymyxin B (Sigma) and incubated at 37°C for up to 48 h under anaerobic conditions. Colonies that emerged were retested for growth on fresh polymyxin B-containing plates. Colonies verified as resistant were cultured overnight in THY medium and stored at −80°C with 20% glycerol for further analysis.
Resequencing analyses.
Genomic DNA was extracted using the FastDNA Spin kit (MP Biomedical; catalog no. 116540-600) and subjected to DNA sequence analysis using the Illumina HiSeq 2000 platform (conducted by GTAC, Washington University, St. Louis, MO) by standard methods as described previously (16). Sequence reads were aligned to the reference HSC5 scaffold sequence using DNAstar SeqMan NGen 4.0.0 (DNAstar) using the default stringency parameters. Single nucleotide polymorphisms (SNPs) and single nucleotide insertions and deletions (InDels) were identified by setting the SNP filter to a minimum of 30% coverage and manually verified by inspection of the Illumina sequence data. On average, SNPs had ∼80-fold coverage, but those with <15-fold coverage were manually verified by PCR and Sanger sequencing. Large deletions (deletions of >1 nucleotide) were either verified by realigning Illumina reads to a scaffold sequence altered to contain the deletion or manually verified by PCR and Sanger sequencing. Genomic loci follow the format L897_xxxxx, where xxxx are numbered. Gene names and annotations either are based on a consensus of all annotated S. pyogenes genomes available on www.BioCyc.org (20), or in the absence of consensus, are based on homology to other genomes—Streptococcus pneumoniae (fabT) and Bacillus subtilis 168 (gdpP [yybT]; yfmH; nupP [yufP])—or are based on annotated function (luxR family transcriptional activator).
Analysis of polymyxin B binding and disruption of SpeB secretion.
Various mutant and wild-type S. pyogenes strains were cultured in C medium and challenged with a sublethal concentration of polymyxin B as described in detail elsewhere (15). Briefly, 30 μM polymyxin B was added to late-logarithmic-phase cells and allowed to grow for an additional 2 h before supernatants were collected and monitored for SpeB secretion by Western blotting using polyclonal anti-SpeB rabbit serum (Abcam; ab53403). For analysis of polymyxin B binding, late-logarithmic-phase cells were stained with dansyl-polymyxin B (catalog no. P13238; Invitrogen) at low (5 μM) or high (30 μM) concentrations and examined by fluorescence microscopy as described previously (15). Focal localization in images of cells was scored as staining at a unique focus, staining at multiple foci, or circumferential staining (staining was of homogeneous intensity around the cellular circumference) (5). A strain was considered to have an intact ExPortal if >80% of cells examined demonstrated focal binding. Each mutant was examined in a minimum of three independent experiments with examination of 1,000 total stained cells.
DNA techniques.
Plasmid DNA was isolated via standard techniques and used to transform S. pyogenes or E. coli as described previously (21). In-frame deletion mutations in genes were generated using the vector pJRS233 (22) or pGCP213 (23), and insertion mutations were generated using the vector pSPC18 (24), to construct the mutagenic plasmids listed in Table S3 in the supplemental material. Each deletion or insertion allele was generated by PCR through a process of overlap extension PCR (25), and modified alleles were inserted directly into plasmids at the M13F and M13R universal primer binding sites utilizing the overlap extension PCR cloning method described in detail elsewhere (26) using primers listed in Table S4. The fidelity of all molecular constructs and mutated chromosomal loci was confirmed by PCR and determination of DNA sequences (Genewiz) using oligonucleotide primers (IDT) of the appropriate sequences. Putative −35 and −10 promoter elements were identified using the online software BProm (Softberry, Inc., Mount Kisco, NY) (27), while ribosome binding sites (RBS) were manually inspected. The consensus binding site for the FabT transcriptional regulator was calculated using the online software RegPrecise (http://regprecise.lbl.gov) (28) from a consensus of 56 putative binding sites from a variety of streptococcal and lactococcal species. The fab operon from the S. pneumoniae strain D39 was used for comparison (29).
Analysis of transcript.
Transcript abundances of select genes were analyzed by real-time reverse transcription-PCR (RT-PCR) as described previously (30). Briefly, for speB gene expression analysis, overnight cultured cells were diluted 1:25 or 1:10 into fresh C medium and harvested at mid-logarithmic phase (50% of final optical density at 600 nm [OD600]), late logarithmic phase (75% of final OD600), or stationary phase (100% of final OD600) or 2 h after onset of stationary phase for mRNA extraction. For fatty acid and anionic lipid biosynthetic gene expression analysis, overnight-cultured cells were diluted 1:25 into fresh THY medium and harvested at early logarithmic phase (20% of final OD600) for mRNA extraction. Select genes were analyzed by real-time RT-PCR normalized to recA using the primers listed in Table S5 in the supplemental material. speB transcript is displayed as threshold cycle (−ΔCT), normalized to recA for each strain and time point, while fatty acid and anionic lipid biosynthetic genes are displayed as ΔΔCT, normalized to the wild type. RNA data shown are the means and the standard errors of the means derived from triplicate determinations of samples from at least three independent experiments.
Analysis of SpeB protease activity.
The ability of various strains to express active SpeB protease was assessed on protease indicator plates (solidified C medium supplemented with 2% milk) (19) as an activity titer, determined as follows: cells cultured overnight in C medium were centrifuged and concentrated 20× to 200× into fresh C medium. A series of seven 10-fold dilutions were prepared, and 5-μl aliquots were spotted onto protease indicator plates and incubated at 37°C for 40 h under anaerobic conditions. CFU were quantified at dilutions which resulted in 3 to 30 CFU, and CFU were estimated to be 10-fold greater at each preceding dilution. Regions surrounding bacterial growth were monitored every 4 h for zones of clearance, which is indicative of SpeB proteolysis activity. Activity was quantified as the lowest dilution at which clearing occurred upon each observation, or if no clearing was observed, the CFU value was given as 10-fold greater than the highest dilution plated, and data were plotted as 7-log10 (CFUclear). Activity data shown are the means and the standard errors of the means derived from triplicate determinations of samples and are representative of at least three independent experiments. As opposed to the fluorescein isothiocyanate (FITC)-casein cleavage assay, protease indicator plates were utilized to measure SpeB activity over time, since this assay is technically simple to perform and is easily scalable to allow for testing of multiple strains simultaneously in order to monitor expression and activation kinetics.
Growth rate and yield analyses.
Indicated bacterial strains were back-diluted 1:50 into 1 ml of fresh C medium (unmodified or altered as indicated in the text). Cultures were mixed briefly by vortexing, 200 μl was placed in triplicate into a 96-well plate (catalog no. 655-180; Greiner), and plates were sealed using transparent adhesive film (catalog no. 60941-078; VWR). Monitoring of growth at 37°C was performed in a Tecan Infinite M200 Pro plate reader. During growth, the plate was shaken every 5 min for 30 s, followed by a 5-s wait period and measurement of the OD600. Data were normalized by subtracting the OD600 value of a blank well (200 μl uninoculated C medium or C medium supplemented with ascorbic acid, which is oxygen sensitive and darkens the medium over time), and the first derivative of the log10 [(1,000) OD600] of each time point was input into Prism (GraphPad). The 13 time points (60 min) corresponding to the maximum slope were determined to define the time of peak growth. This peak growth time (approximately 15 to 30% of maximum OD600) was used to calculate growth rate (t1/2) as follows: normalized data were input into Prism. Next, the defined 60-min time period of exponential growth was fit to a growth equation (Y = Y0ekt; Y = OD600, Y0 = OD600 at time zero, k = exponential rate constant, and t = time) using nonlinear regression curve analysis to determine the exponential rate constant (k), which was then used to calculate the doubling time. Percent growth yield in sugar-supplemented versus unmodified medium was determined using normalized OD600 values from stationary-phase cultures, calculated as OD600+sugar/OD600:unmod and displayed as a percentage. The average doubling time and percent growth yield were calculated from each replicate from at least three independent experiments.
Polymyxin B challenge and heat challenge on solid medium.
Strains cultured overnight in C medium were used to prepare a series of six 10-fold dilutions for challenge studies. For heat challenge, 5 μl of each dilution was spotted onto solidified THY medium in duplicate and incubated at either 37°C or 43°C for 48 to 72 h under anaerobic conditions. CFU were quantified, and the efficiency of plating (EOP) was calculated as (log10 CFU43°C) − (log10 CFU37°C). For polymyxin B challenge, 5 μl of each dilution was spotted onto solidified C medium unmodified or supplemented with 40 μg ml−1 (29 μM) polymyxin B. CFU were quantified, and EOP was calculated as (log10 CFU+PB) − (log10 CFU−PB). Data shown are the means and the standard errors of the means derived from at least four independent experiments. Determination of EOP proved to be a more reproducible and sensitive metric of resistance than a standard culture-based MIC assay, as continued exposure of cultures to higher concentrations of polymyxin B tended to select for variants with higher levels of resistance. It is important to note that due to the higher lipid content from the added milk, the concentration of PB required to inhibit the growth of wild-type S. pyogenes is ∼10-fold greater on protease indicator plates than on C medium plates.
Statistical analyses.
Differences between wild-type and mutant strains were tested for significance using the Dunnett test available in InStat (GraphPad) software where an asterisk indicates P < 0.01. For all tests, the null hypothesis was rejected for P > 0.05.
RESULTS
Selection of PB-resistant mutants.
Resistance to CAPs in most species of Gram-positive bacteria is associated with neutralization of the cell's highly negative surface charge, reducing its affinity for binding positively charged CAPs. Common mechanisms include lysinylation of anionic phospholipids by MprF (31) and d-alanine esterification of lipoteichoic acid by DltA (32). However, S. pyogenes does not encode mprF, making it highly susceptible to inhibition by most CAPs, including PB (15). This property was exploited to select for mutants capable of growth at a normally inhibitory concentration of PB. We also made use of the fact that disruption of ExPortal integrity by PB is associated with inhibition of secretion of the SpeB cysteine protease, whose expression can be conveniently assessed on indicator plates. To select for spontaneous PB-resistant mutants, cells from overnight cultures of the M14 serotype S. pyogenes HSC5 were plated onto protease indicator plates containing a concentration of PB (400 μg ml−1) that prevents formation of colonies. Approximately 2 × 1010 CFU (as determined on unsupplemented medium) representing 11 independent pools was plated onto PB-protease indicator plates. Candidate mutants capable of forming discrete colonies were subjected to several rounds of restreaking to identify those with a reproducible and stable resistance phenotype. A total of 25 distinct PB-resistant mutants (designated PBr mutants) were isolated (spontaneous mutation rate of ∼10−9), of which 21 also displayed SpeB activity (Table 1).
Mutations in a core set of 10 gene groups confer resistance to PB.
In order to identify the genetic alterations that gave rise to PB resistance, each of the PBr mutants was subjected to whole-genome resequencing and compared to the recently completed genome of the wild-type strain HSC5 (16). Sequence alignments of the 25 mutants revealed a total of 31 single nucleotide polymorphisms (SNPs) and 24 insertion/deletions (InDels) that were identified in the open reading frames (ORFs) or putative RBS of a predicted gene. Individual PBr mutants contained between 1 and 4 mutations (average 2.2 mutations/isolate [Table 1; see also Tables S6 and S11 in the supplemental material]). SNPs displayed a bias toward transitions over transversions (17 and 14, respectively), similar to what has been reported in other organisms (33). However, 30/31 SNPs resulted in mutation to either A or T, reflecting the low GC content of S. pyogenes (38.5%) (see Table S7). The majority of InDels consisted of single nucleotide deletions in predicted ORFs resulting in frameshifts, although a few contained larger deletions of up to 167 bp (see Table S8). Insertions were rare and were limited solely to 1-bp insertions (see Table S8). The majority of InDels occurred from loss of repetitive elements, particularly in homopolymeric stretches of adenines, although loss of larger and more complex repetitive elements also occurred (see Table S9). SNPs caused a variety of genetic lesions, including nonsynonymous mutations and premature stop codons, as well as the loss of stop codons that generated larger fusion proteins. In 3 independent cases, a single mutation affected the same 2 overlapping genes, tilS and hpt, as the final 3 codons in the 3′ end of tilS overlap the putative RBS of hpt (PBr 6.10, 10.15, and 11.2 [Table 1]). Of the 55 total mutations identified, only one mutation occurred outside a predicted ORF or RBS (PBr 10.6, which contains 2 other SNPs) and only one PBr mutant contained a synonymous mutation (PBr 10.11, which also has an SNP and an InDel) (Table 1), suggesting that mutations without functional consequences were rarely associated with mutations that promoted resistance. Surprisingly, the majority of mutations were localized within a set of only 10 gene groups. These “core PBr genes” (defined as individual genes or sets of genes with closely related annotations that were mutated in >1 PBr mutant) accounted for 49 of the 55 total SNPs and InDels identified (Fig. 1A). Each PBr mutant contained between 1 and 4 core PBr mutations (Fig. 1B). Furthermore, a single mutation in any one of a set of 4 core PBr genes conferred resistance in the absence of any other mutation, including pstS (PBr 1.1 and 2.1), clpX (PBr 4.1), manN (PBr 6.3), and tilS/hpt (PBr 10.15) (Table 1).
FIG 1.
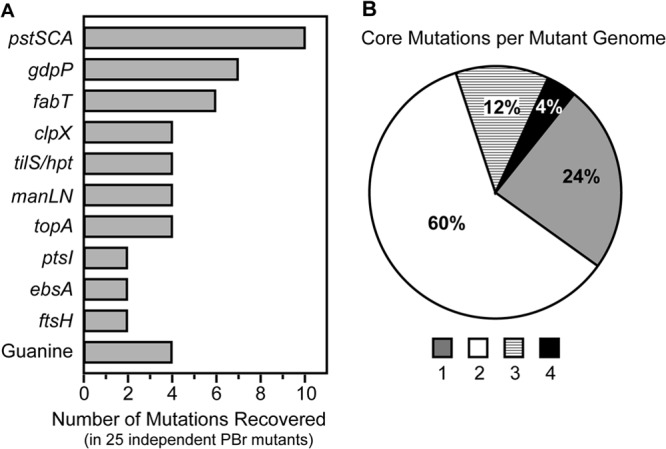
Core PBr genes cluster within 10 gene groups. (A) Independently isolated PBr mutants contain mutations within a set of 10 core gene groups. Genes involved in guanine metabolism other than hpt (including guaA, deoB, gmk, and nupP) were represented by single PBr mutants and are displayed separately from hpt for clarity. (B) Each PBr mutant contains between 1 and 4 mutated core genes.
Construction of mutations in individual PBr genes.
Given the rich diversity of mutations, resistance may have developed from loss- or gain-of-function mutations or from the combinatorial effect of multiple mutations. To gain further insight into the role of each core PBr gene, an attempt was made to construct individual loss-of-function mutations in the wild-type background. An initial attempt was made to construct in-frame deletions within each gene group, and this strategy yielded mutations in 5 PBr gene groups (gdpP, pstS, manLMN, ebsA, and clpX [Table 2]). Since recovery of a mutant using this method requires that it compete successfully with the wild type (18), an alternative replacement strategy to place a promoterless chloramphenicol resistance gene under the control of the native gene's promoter was employed. If the promoter is of sufficient strength, chloramphenicol selection will promote recovery of the null mutant. This approach was successful for an additional gene (ptsI [Table 2]). Mutations in hpt and ftsH were successfully constructed by a one-step duplication-inactivation method (18) that allows for direct selection for insertion of a spectinomycin resistance determinant (Table 2). For fabT, it was noted that all spontaneous PBr fabT mutations were also associated with mutations of either gdpP or clpX (PBr 3.5, 3.6, 9.21, 10.6, 10.11, and 11.1 [Table 1]), suggesting that the latter mutations may compensate for a deleterious effect of loss of fabT on growth rate or viability. This idea was then supported by our ability to generate an in-frame deletion in fabT in the context of deletion of either gdpP or clpX (Table 2) but not in wild-type cells. However, a similar strategy to generate a topA mutation in the context of deletion of pstS was not successful. Finally, deletion of tilS was not attempted since the mutations in the original mutants likely confer PB resistance by alteration of the hpt RBS (see below). Analysis of growth rates and yields of the resulting mutants was mostly consistent with the method required to generate each mutant. In-frame deletion mutants had either a growth rate or yield (or both) near that of the wild type, while those requiring selection (ΔPtsI::cat, ΩHpt, and ΩFtsH) had both a longer growth time and a lower growth yield (Table 2).
TABLE 2.
Characterization of mutants with deletion or disruption of PBr core genes
| Mutant | Straina | Growth rate (% yield)b | PB bindingc | ExPortald | SpeB secretione |
|
|---|---|---|---|---|---|---|
| −PB | +PB | |||||
| WT | HSC5 | 50.1 (100) | + | Disrupted | + | − |
| ΔClpX | GCP688 | 70.1 (97) | + | Intact | − | − |
| ΔGdpP | GCP751 | 51.9 (119) | + | Intact | +/− | +/− |
| ΔPstS | GCP754 | 58.0 (99) | + | Intact | + | + |
| ΔGdpP-ΔFabT | GCP766 | 66.0 (76) | + | Intact | +/− | +/− |
| ΔClpX-ΔFabT | GCP767 | 70.9 (87) | + | Intact | − | − |
| ΔManLMN | GCP771 | 51.7 (109) | + | Heterogeneous | + | − |
| ΔEbsA | GCP784 | 53.4 (99) | + | Heterogeneous | + | − |
| ΔPtsI::cat | GCP953 | 68.5 (80) | + | Intact | + | + |
| ΩHpt | GCP859 | 84.8 (68) | + | Intact | + | + |
| ΩFtsH | GCP862 | 88.5 (67) | + | Intact | +/− | − |
| ΩCon | GCP292 | 59.2 (96) | + | Disrupted | + | − |
Name of strain with the indicated mutant phenotype. See Table S1 in the supplemental material.
Doubling time (t1/2) in minutes in C medium calculated from peak growth at mid-exponential phase between 15 and 30% of maximum growth yield. Yield is percentage of WT value based on final OD600.
Absence (−) or focal pattern (+) of binding when stained with 5 μM dansyl-PB.
Staining with 30 μM dansyl-PB results in a focal (intact) or dispersed (disrupted) pattern of binding or a mixture of cells with an intact or dispersed (heterogeneous) pattern of binding.
Level of secreted SpeB cysteine protease in culture supernatant detected by Western blotting following challenge during the late logarithmic phase of growth with (+PB) or without (−PB) 30 μM PB and harvested 2 h later after the onset of stationary phase: +, ≥80%; +/−, 50 to 80%; −, ≤10%, all compared to WT.
PB resistance primarily arises through loss of function of core PBr genes.
To gain insight into the contribution of individual core genes to the PB resistance phenotype, the panel of null mutants was tested for sensitivity to PB challenge. Using a quantitative efficiency-of-plating assay, both the wild-type (WT) strain and a previously described control strain with an intergenic insertion downstream of a gene not associated with PB resistance (ΩCon [24]) demonstrated over 5 logs of sensitivity to PB (Fig. 2A). In contrast, the various core PBr mutants were highly resistant and typically had plating efficiencies on the order of 4 to 5 logs higher than that of the wild type (Fig. 2A). The exceptions were ΔManLMN and ΩFtsH, which were as sensitive as the WT and ΩCon strains (Fig. 2A). To test the contribution of FabT toward PB resistance, ΔGdpP-ΔFabT and ΔClpX-ΔFabT double mutants were complemented with plasmids expressing either GdpP or ClpX. Although empty vector (pABG5) had no effect on PB resistance in any strain, pGdpP and pClpX were able to restore PB sensitivity to wild-type levels in their respective single deletion mutant strains (Fig. 2B). In contrast, these plasmids had no effect on PB resistance for either ΔGdpP-ΔFabT or ΔClpX-ΔFabT (Fig. 2B), demonstrating that loss of FabT alone is sufficient to confer PB resistance. PB binding and enhanced ExPortal integrity were then assessed by challenge of mid-logarithmic-growth-phase cultures with fluorescently labeled PB at a low concentration that results in a focal pattern of binding and at a high but still sublethal concentration that results in a dispersed pattern of binding in the WT strain. At low concentrations, dansyl-PB bound in a focal pattern to all mutants (Table 2). However, when examined at a concentration that disrupts the ExPortal in the WT strain, all mutants demonstrated enhanced ExPortal integrity. Of the mutants, 5 gained the ability to secrete the SpeB protease at concentrations that inhibit its expression in the wild type or ΩCon (Table 2), although those with mutations in gdpP or ftsH had an overall reduction in SpeB expression under all conditions. In contrast, those mutants lacking ClpX function (ΔClpX and ΔClpX-ΔFabT) failed to express SpeB even in the absence of PB (Table 2). Thus, while most spontaneous mutants have mutations in multiple genes, PB resistance and enhanced ExPortal integrity can be recapitulated by the loss of function in almost all core PBr genes. Exceptions are a lack of resistance for ftsH and manLMN null mutants. However, spontaneous ftsH mutations were always found in the context of pstS or pstC mutations (Table 1) and spontaneous manLMN missense mutations were found in isolation (e.g., PBr 6.3 [Table 1]). Taken together, these data indicate that the PBr phenotype for the latter mutations likely represents a gain of function, while the former may contribute to a combinatorial effect.
FIG 2.
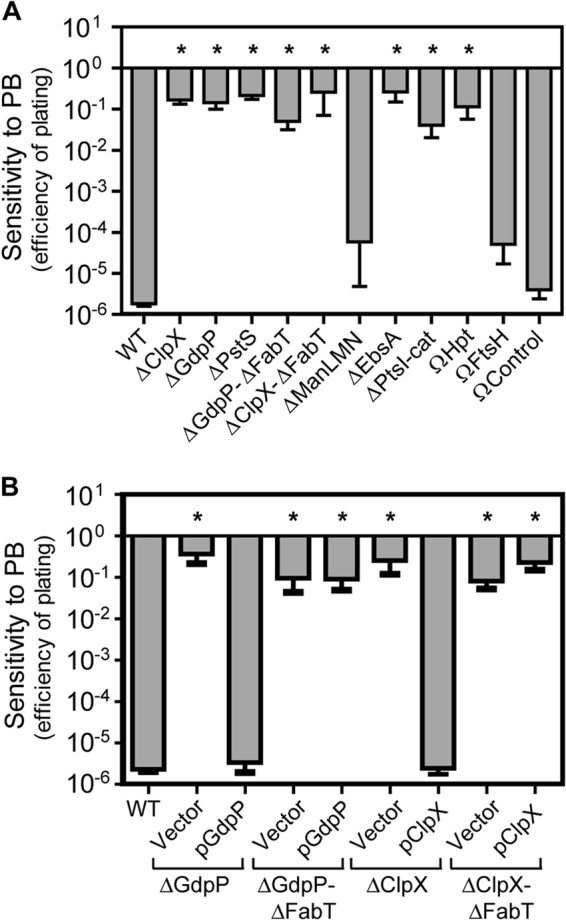
Most core PBr genes confer resistance through loss of function. Polymyxin B resistance of deletion or insertional disruption mutants (A) and selected mutants transformed with plasmids expressing GdpP (pGdpP), ClpX (pClpX), or empty pABG5 vector (Vector) (B) was determined by efficiency of plating on solid medium. Stationary-phase cultures of indicated strains were serially diluted and plated onto solidified C medium either unmodified or containing 40 μg ml−1 polymyxin B. Efficiency of plating was calculated as log10 CFU+PB/log10 CFUunmod. Data shown are the means and the standard errors of the means derived from at least four independent experiments. Differences between wild-type and mutant strains were tested for significance using the Dunnett test (*, P < 0.01).
Altered expression of the Fab operon in PBr fabT mutants.
In streptococci, the expression of the genes encoding enzymes involved in fatty acid biosynthesis is under the control of the transcriptional regulator FabT (34). Since the ExPortal has an asymmetric content of anionic phospholipids (10), any PBr genes affecting lipid biosynthesis are of particular interest. In addition, the fact that fabT mutations were common among PBr mutants (Table 1) but that it was possible to construct null fabT mutations only in the context of other mutations complicates understanding how mutation of fabT may contribute to PB resistance. Similar to Streptococcus pneumoniae (35), the genes for fatty acid biosynthesis are clustered together in S. pyogenes (Fab operon). The two operons are nearly identical in sequence, although several of the FabT binding sites differ in location relative to their promoters and the S. pyogenes sites lack the sequence diversity of S. pneumoniae (Fig. 3A). Consistent with its function as a repressor of transcription (34), deletion of fabT resulted in a 2- to 8-fold increase in transcription of the genes adjacent to each of the 4 FabT sites when monitored by real-time RT-PCR (Fig. 3B). This increase was strictly due to the loss of fabT, as mutation of the companion genes alone (ΔClpX and ΔGdpP) did not result in a significant increase in expression of any Fab operon gene (ΔClpX and ΔGdpP [Fig. 3A]). Fatty acid biosynthesis also produces precursors required for synthesis of anionic phospholipids. However, the loss of fabT did not impact expression of genes required for phosphatidylglycerol (pgsA) or cardiolipin (cls) synthesis (Fig. 3B). Interestingly, although fabT mutations were present in approximately one-fourth of the PBr isolates, they consisted entirely of missense mutations or alterations to the RBS; there were no frameshift, nonsense, or InDel mutations. This limited distribution of mutation type was in contrast to the collection of mutations obtained in other core genes like clpX and pstS (see Table S10 in the supplemental material). While this suggests that PB resistance required a gain of function in FabT, this was not supported by analysis of gene expression, which indicated that all fabT PBr mutations derepressed expression of Fab genes to levels nearly approaching those observed for fabT deletion mutants (Fig. 3C). The contribution of fabT alteration to the PBr phenotype is discussed in more detail below.
FIG 3.

Mutation of the FabT transcriptional repressor results in upregulation of fatty acid biosynthetic genes but not anionic lipid genes. (A) The fatty acid biosynthesis (fab) region carried within S. pyogenes contains 3 predicted operons (thin black arrows) and 4 putative FabT binding sites upstream (black boxes) or within fab genes (hollow arrows). Genes interrogated for mRNA expression levels are shaded in gray. Predicted FabT binding sites are shown in comparison with S. pneumoniae, and nucleotide distances in relation to translational start codons (ATG or TTG) are indicated in parentheses. Distances between FabT binding sites and predicted −35 and −10 promoter elements and the ribosome binding site (RBS) are indicated in the diagram. The consensus 13-nucleotide palindromic FabT binding site is shown (Con), and deviations are indicated in lowercase. (B and C) The relative level of message of select genes in deletion mutants (B) and PBr mutants containing FabT SNPs (C) was determined using real-time RT-PCR and normalized to the wild-type strain. Data shown are the means and the standard errors of the means derived from triplicate determinations of samples from at least three independent experiments. The dashed lines indicate >2-fold or <2-fold changes. An asterisk indicates an undetectable level due to the absence of the appropriate gene in the deletion mutant.
Several core PBr mutants exhibit enhanced multistress resistance.
In other bacterial species, mutations in several of the core PBr genes have been implicated in resistance to multiple stresses, including heat (36) and acid (37). To investigate whether the S. pyogenes core PBr mutations can confer resistance to additional stresses, the panel of PBr mutants was tested for resistance to heat stress. The WT strain used here (HSC5) is typical of most S. pyogenes strains and cannot grow at temperatures over 39°C. When examined quantitatively, the WT strain was unable to grow at an elevated temperature and had an efficiency of plating below the limit of detection for the assay (over 5 logs of sensitivity) when tested at 43°C versus 37°C (Fig. 4). In contrast, several of the core PBr mutants, including ΔClpX, ΔClpX/ΔFabT, and ΩHpt, gained the ability to grow at this high temperature and had plating efficiencies that were reduced less than 1 log (Fig. 4). Other core mutants remained heat sensitive (Fig. 4). Examination of several spontaneous PBr isolates with mutations in clpX (PBr 4.1, 9.21, 10.11, and 11.1) or hpt (PBr 6.7, 6.10, 10.15, and 11.2) revealed that they were similarly heat resistant (data not shown). Since hpt functions in guanine metabolism, other PBr isolates with mutations in genes involved in guanine metabolism were tested, including deoB (PBr 4.5) and guaA (PBr 10.14), and these were also heat resistant, as was PBr 11.5 with mutations in fba and manL (data not shown). Thus, some mutations that promote PB resistance, most notably in clpX and in guanine metabolism genes, also can confer resistance to other types of stress.
FIG 4.
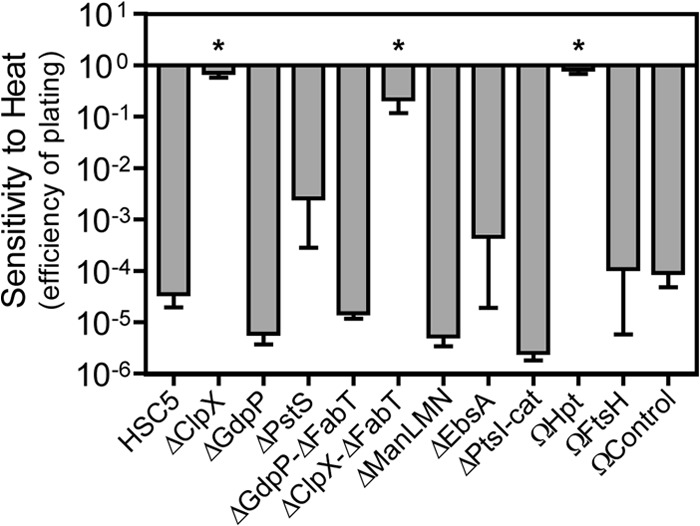
Loss of ClpX or Hpt confers heat resistance. Stationary-phase cultures were serially diluted, plated onto solid medium, and incubated at permissive (37°C) or restrictive (43°C) temperature. After 48 h, CFU were quantified and efficiency of plating was calculated as log10 CFU43°C/log10 CFU37°C. Data shown are the means and the standard errors of the means derived from at least four independent experiments. Differences between wild-type and mutant strains were tested for significance using the Dunnett test (*, P < 0.01).
A subset of PBr mutants display altered carbohydrate metabolism.
Many core PBr genes encode putative transporters of nutrients, suggesting that mutation of these genes may confer a starvation state that is associated with PB resistance (Table 1). To test this, the WT strain and selected mutants were cultured in a carbohydrate-poor medium (C medium) that was supplemented with various sugars to determine if PB resistance is associated with defects in carbohydrate utilization. The sugars tested were selected on the basis of the predicted repertory of S. pyogenes HSC5 carbohydrate transporters (see Table S12 in the supplemental material), and it was considered that a sugar could be utilized if supplementation resulted in at least a 20% increase in growth yield. A selection of sugars not predicted to be utilized by S. pyogenes was included for comparison. Generally consistent with predictions, the addition of 14 different sugars increased growth yields (Fig. 5A). Similarly to a recently described ptsI deletion mutant of S. pyogenes (38), ΔPtsI lost the ability to utilize 9 of these 14 sugars (Fig. 5A). Its growth was enhanced by glucose, the amino sugar N-acetylglucosamine (GlcNAc), and multimeric forms of glucose linked by α(1→4) glycosidic bonds (maltose, maltotriose, and maltodextrin) but not α,α-(1→1) and β(1→4) glycosidic bonds (disaccharides trehalose and cellobiose, respectively) (Fig. 5A). Since this mutant lacks a key component of the sugar phosphotransferase system (PTS), these data define the classes of PTS-dependent and -independent sugars for S. pyogenes. However, growth profiles differed markedly for spontaneous ptsI PBr mutants. These mutants had no obvious defect in utilization of representative PTS (mannose and fructose) and non-PTS (glucose) sugars (PBr 11.3 and 11.6 [Fig. 5B]). Since both of these have missense mutations (Table 1), their contribution to PB resistance must not arise from a loss of function.
FIG 5.
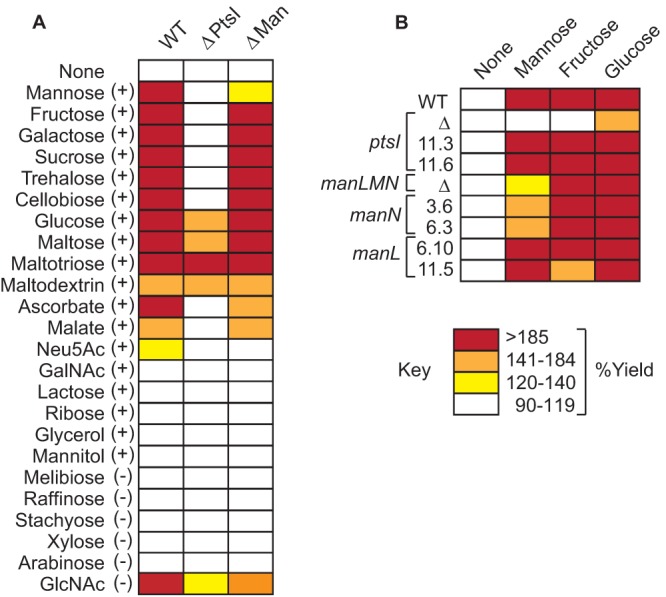
Some PBr mutants show altered sugar utilization. (A) Wild-type or deletion mutant strains were tested for sugar utilization by supplementing C medium with various sugars that HSC5 was either predicted (+) or not (−) to utilize based on genome annotation. (B) PBr mutants containing SNPs in ptsI or manL/N were tested for sugar utilization with select sugars in comparison to deletion mutants. Percent yield was calculated as OD600+sugar/OD600:unmod. Data shown are from triplicate determinations of samples from at least three independent experiments.
Another series of carbohydrate-related core PBr genes are those of the manLMN operon putatively associated with transport of mannose (Table 1). However, the PBr phenotype was not recapitulated in the loss-of-function ΔManLMN mutant (see above). Thus, it was of interest to compare profiles of carbohydrate metabolism in this group of mutants. The ΔManLMN mutant was able to utilize most of the same sugars as those utilized by the WT strain with the primary exception being that it was much less efficient in its ability to utilize mannose, suggesting that while ManLMN is not the only mannose transporter, it makes a major contribution to mannose metabolism in S. pyogenes (Fig. 5A). This phenotype differed for all spontaneous PBr Man operon mutants. The manN mutants had only a slight defect in mannose utilization, while the manL mutants had no defect and achieved enhanced growth yields equivalent to those of the WT (Fig. 5B). Since there was no direct correlation with a loss of mannose metabolism, this indicates that the PBr phenotype is a gain of function associated with the mannose membrane transporter complex.
A subset of PBr mutants display altered salt metabolism.
Another nutrient acquisition pathway associated with PB resistance involves the uptake of inorganic phosphate (Pi). Mutations in a putative high-affinity Pi ABC-type transporter encoded by the Pst operon were the most commonly isolated mutations, appearing in 40% of the total number of PBr mutants isolated (Fig. 1A). The most common lesion was a frameshift in pstS (Table 1), which encodes the extracellular substrate-binding lipoprotein component of the transporter. Independent mutants were also isolated with mutations in pstA (frameshift) and pstC (nonsense) (Table 1), which encode membrane permease components. To test if these mutations resulted in alterations of salt metabolism that may correlate with PB resistance, the growth characteristics of WT and the various mutants were compared in media with an altered content of salts. In unaltered C medium, the ΔPstS mutant had a growth rate and yield equivalent to those of the WT (Table 2). In C medium prepared without its normal complement of salts (10 mM K2HPO4, 0.4 mM MgSO4, 17 mM NaCl), the growth rate of the WT strain was unaltered; however, ΔPstS failed to grow (none detected [ND] [Table 3]). Although MgSO4 had no effect on growth, addition of NaCl alone was sufficient to restore ΔPstS growth at a slightly reduced rate, while the addition of K2HPO4 by itself also restored growth, although at a substantially reduced rate and growth yield (Table 3). Furthermore, the addition of both K2HPO4 and NaCl restored growth of ΔPstS to levels equivalent to all 3 salts (Table 3). Finally, swapping cations by adding KCl did not restore growth; however, the addition of Na2HPO4 restored growth to a rate and yield equivalent to those for complete medium (Table 3). Thus, full growth of ΔPstS was restored only either by the presence of K2HPO4 plus NaCl or by Na2HPO4, indicating a requirement for both Na+ and Pi. This same requirement for salts was observed for all spontaneous PBr PstS/A/C-containing mutants (Table 4), although pstS frameshift mutants did show a low level of growth following an extended lag period of several hours (data not shown). Taken together, this analysis indicates that an altered salt metabolism may lead to the acquisition of PB resistance.
TABLE 3.
Growth rates in presence and absence of salts
| Salt(s)b | Growth rate (% yield)a |
|
|---|---|---|
| WT | ΔPstSc | |
| None | 42.0 | ND |
| K2HPO4, MgSO4, NaCl | 53.8 | 57.2 |
| K2HPO4 | 58.3 | 99.5 (81) |
| MgSO4 | 49.8 | ND |
| NaCl | 47.2 | 68.6 |
| K2HPO4, MgSO4 | 56.8 | 85.4 (81) |
| K2HPO4, NaCl | 54.0 | 55.4 |
| MgSO4, NaCl | 46.4 | 75.8 |
| Na2HPO4 | 57.5 | 59.2 |
| KCl | 41.1 | ND |
| MgSO4, KCl | 45.7 | ND |
Growth rate (t1/2) was measured in salt-reduced C medium supplemented with indicated salts.
Salts were added to salt-reduced C medium as described in Materials and Methods.
Final yields for ΔPstS reached approximately 100% of the WT value except for those indicated in parentheses or labeled ND (none detected), which were <5% of the WT value.
TABLE 4.
Growth rates for salt-dependent PstS, PstC, and PstA PBr mutants
| Salts | Growth ratea |
|||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| WT | PstS mutantb: |
PstC mutant 3.2b | PstA mutant 10.9b | |||||||||
| Δ | 1.1 | 2.1 | 3.1 | 3.3 | 3.7 | 4.4 | 4.5 | 10.14 | ||||
| Nonec | 46.1 | ND | 99.4 | 127.0 | 173.1 | 133.0 | 158.8 | 104.0 | 186.3 | 177.6 | 200.3 | ND |
| K2HPO4, MgSO4, NaCl | 51.9 | 58.7 | 54.0 | 55.6 | 58.2 | 55.5 | 52.9 | 52.5 | 65.3 | 56.8 | 59.7 | 59.0 |
Growth rate (t1/2) was measured in salt-reduced C medium supplemented with indicated salts.
PBr mutants containing mutations within pstS, pstC, and pstA are described in Table 1 and in Table S6 in the supplemental material. ND, none detected.
Final yields for PBr mutants containing SNPs in pstS or pstC reached approximately 60% of the WT value after 24 h of culture in the absence of salts and 100% in the presence of salts.
Growth in low-salt medium selects for pstS revertants.
Overnight cultures of the various PBr mutants containing the pstS frameshift mutation would occasionally demonstrate reduced but detectable growth in C medium lacking salts. To determine if this was due to reversion, 10 independent cultures from each of the 8 pstS-containing mutants were prepared in reduced-salt C medium and incubated for an extended time period. When examined after 48 h, a wild-type growth yield was obtained for every culture of each mutant (Table 5). When growth from these cultures was tested again in reduced-salt C medium, they now demonstrated a growth rate indistinguishable from that of the wild type (Table 5). DNA sequence analyses of each of these cultures revealed reversion to the wild-type pstS sequence (see Table S2 in the supplemental material). Furthermore, reversion was specific to pstS, as each individual revertant retained all other associated mutations corresponding to their parental PBr strain. In contrast, salt-revertant mutants were rarely obtained in strains with mutations in pstC or pstA and were never recovered from ΔPstS (Table 5). DNA sequence analysis revealed that the one revertant derived from PBr 10.9 gained a single nucleotide insertion at the site of the original 19-bp deletion within pstA, thus resulting in an 18-bp in-frame deletion and generating a PstA open reading frame lacking 6 amino acids, while the one revertant of PBr 3.2 contained an SNP within pstC reverting the nonsense mutation (premature stop codon) to a missense mutation (altered amino acid), thus restoring the PstC open reading frame (see Table S2). The pstS frameshift mutation (fs-aa6 [Table 1]) occurs at the terminal position of a track of 9 consecutive adenine residues. As will be discussed below, the frequency with which this mutation arises and reverts and its robust PB-resistant phenotype suggest that this homopolymeric track functions as a molecular switch to facilitate adaptation to stressful environments.
TABLE 5.
Auxotrophic Pi revertants recovered following 48 h of culture in salt-reduced C medium
| Mutanta | Growthb | Ratec |
|---|---|---|
| PstS | ||
| Δ | 0/10 | ND |
| 1.1 | 10/10 | 46.3 |
| 2.1 | 10/10 | 49.0 |
| 3.1 | 10/10 | 45.3 |
| 3.3 | 10/10 | 48.0 |
| 3.7 | 10/10 | 45.7 |
| 4.4 | 10/10 | 53.8 |
| 4.5 | 10/10 | 48.7 |
| 10.14 | 10/10 | 48.6 |
| PstC mutant 3.2 | 1/10 | 46.3 |
| PstA mutant 10.9 | 1/10 | 50.2 |
PBr mutants containing mutations within pstS, pstC, and pstA are described in Table 1 and in Table S6 in the supplemental material. Pst revertant mutants are described in detail in Table S2.
PBr mutants were subcultured 1:1,000 from C medium into salt-reduced C medium. Ability to grow in salt-reduced C medium was scored in each of 10 replicate culture tubes after 24 h by measurement of OD600.
Growth rate (t1/2) of revertant mutants was measured in salt-reduced C medium. ND, none detected.
Core PBr genes include novel regulators of SpeB expression.
As discussed above, the processing of the secreted SpeB cysteine protease zymogen to its intact form depends upon an intact ExPortal. However, several of the mutants with enhanced ExPortal integrity had a defect in SpeB expression when examined on protease indicator plates (Table 1). Furthermore, several of the core PBr deletion mutants had decreased levels of the SpeB polypeptide in culture supernatant when examined at a time point when expression in the WT strain is maximal (Table 2). Expression of SpeB is highly regulated by factors that act at multiple levels, including transcription, secretion, and postsecretory folding and maturation (for a review, see reference 39). For example, we have previously reported that a mutant with an insertional disruption of the core PBr gene gdpP has a kinetic defect in the processing of 40-kDa SpeB zymogen to its 27-kDa active form, even though it expresses normal levels of the speB transcript and is not defective for secretion (30). To gain insight into how other core PBr genes affect SpeB expression, the panel of SpeB-defective mutants was compared to the gdpP deletion mutant (ΔGdpP) mutant constructed for this study. Examination of ΩFtsH revealed that it had an even greater kinetic defect than ΔGdpP when both are compared to WT and to a mutant with an accelerated pattern of expression (ΔVfr) that results from uncoupling speB transcription from its normal mode of growth phase control (Fig. 6A; see also Fig. S1 in the supplemental material). In contrast, ΔClpX was completely deficient in expression of SpeB and more closely resembled a strain that expresses a catalytically inactive SpeB protein (SpeBC192S) or one that lacks RopB (ΔRopB), the transcriptional activator required for SpeB expression (Fig. 6A; see also Fig. S1). Examination of speB transcript levels by real-time RT-PCR revealed a significant defect at the level of speB transcription for the ΔClpX mutant, and to a lesser extent, the ΩFtsH mutant (Fig. 6B). In contrast, transcript levels were unaltered in ΔGdpP, indicating a posttranscriptional defect (Fig. 6B). Taken together, these data indicate that several core PBr mutants (ΩFtsH, ΔClpX, and ΔGdpP) have SpeB defects at the transcriptional and posttranscriptional level that are independent of ExPortal integrity.
FIG 6.
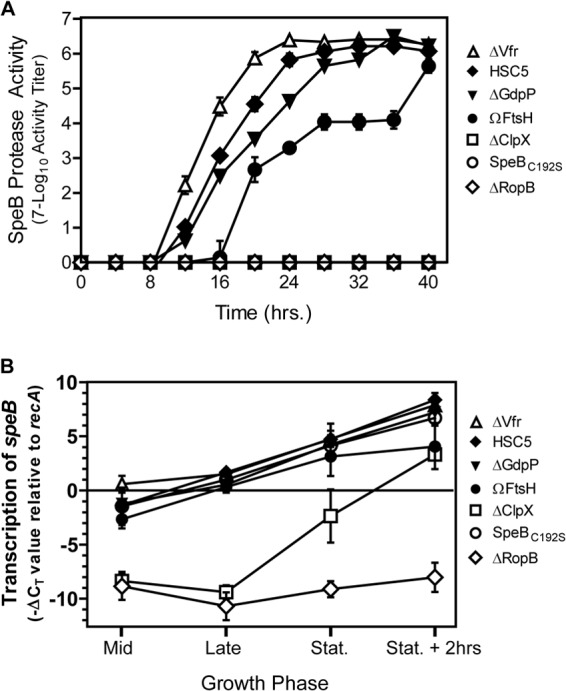
Several core PBr genes regulate SpeB expression. (A) SpeB protease activity was determined for various strains by plating serial dilutions onto protease indicator plates and monitoring for protease activity over time. Activity data shown are the means and the standard errors of the means derived from triplicate determinations of samples and are representative of at least three independent experiments. (B) The relative level of speB transcript over the course of growth was determined using real-time RT-PCR. RNA data shown are the means and the standard errors of the means derived from triplicate determinations of samples from at least three independent experiments.
DISCUSSION
It has only recently been appreciated that CAPs can have significant biological effects at sublethal concentrations, including an ability to alter patterns of protein secretion by targeting and disrupting the anionic phospholipid-enriched microdomain of the ExPortal (15). In the present study, we have used this property of CAPs to design a genetic screen to gain insight into the mechanism(s) by which S. pyogenes organizes and maintains the ExPortal. Analysis of the resulting collection of mutants implicated a role for an assortment of genes whose loss or gain of function influenced ExPortal organization, including those involved in nutrient acquisition, stress responses, and virulence gene regulation.
Since S. pyogenes exclusively relies upon the Sec system for secretion of virulence factors, understanding how the ExPortal is maintained and organized is of central importance for understanding how the Sec pathway has been adapted to support the secretion and processing of proteins which have numerous roles in pathogenesis. The observation that disruption of the anionic phospholipid-enriched microdomain leads to the redistribution of ExPortal components (15, 40) indicates that a key question involves the mechanism that clusters these lipids into a focal microdomain. At the concentrations found in physiological membranes, various lipid species only transiently associate into microdomains. The stabilization of these microdomains then becomes dependent on a number of different organizing factors. In eukaryotic cells, numerous proteins have been described that function to extend the half-lives of lipid microdomains in order to promote specific functionality, which typically involves endocytosis or cell signaling (41). In rod-shaped bacteria, the stable segregation of anionic phospholipids into functional helix-like domains is organized via interaction with the cytoskeletal protein MreB and its various homologs (11, 42–44). However, S. pyogenes, like all other streptococcal species, lacks any MreB-like protein (9). Similarly, how CAPs can disrupt these normally stable microdomains in the absence of significant membrane damage is also not understood. In model membranes, rather than dispersing anionic phospholipids, challenge with CAPs tends to promote the transition of anionic lipids from a liquid-disordered state to a more clustered liquid-ordered configuration (45). Thus, further analysis of how the various mutations identified in the current study can promote enhanced ExPortal integrity will provide fundamental insights into the mechanisms that support the segregation of lipids into functional domains.
Analysis of this collection of mutants should also contribute to our understanding of mechanisms of resistance to CAPs. Many bacterial species possess factors that confer CAP resistance that most commonly act by modification of negative surface charges to abrogate CAP binding. These include MprF, which lysinylates anionic phospholipids (31), and DltA, which acts via d-alanine esterification of teichoic acids. However, S. pyogenes does not encode MprF, and loss of DltA results in increased susceptibility to CAPs (32). Alteration of surface charge also did not appear to be a common mechanism among the PB-resistant mutants isolated in the current study, as their PB-binding properties were unaltered. Another common mechanism of resistance to antibiotics that target anionic phospholipids involves alterations in the biosynthesis pathways for these lipids. For example, resistance to daptomycin can result from mutations that alter residues in the enzyme Cls or PgsA or from mutations that affect their transcriptional regulators (46–51). In the current study, no mutations were identified in the anionic phospholipid biosynthetic genes or their presumed regulators. However, multiple PBr isolates were found to have a mutation in fabT, which encodes the master regulator of fatty acid biosynthesis in streptococci (52). The frequency and diversity of fabT mutations suggest that alterations to membrane composition may contribute to resistance.
An understanding of the role of fabT mutations in the PB resistance phenotype was complicated by the fact that null mutations could be recreated only in the context of mutations in either clpX or gdpP. However, both comutations could be restored through expression off a plasmid in the fabT deletion strains, demonstrating that loss of fabT was sufficient for resistance to PB. Although it is unclear why these particular comutations facilitate the genetic manipulation of fabT, it has been noted that the generation of fabT deletions in Streptococcus pneumoniae is notoriously difficult (34), suggesting that the comutations may restore some measure of fatty acid homeostasis. However, since the comutations by themselves did not alter transcription of the fatty acid biosynthesis operon, some other compensatory mechanism must be involved. The S. pneumoniae fabT mutants display a 2- to 4-fold increase in transcription of the fatty acid biosynthesis operon and an increase in the ratio of saturated to unsaturated fatty acids, as well as a predominance of longer fatty acid chains (34). Alteration to pools of anionic phospholipids also may result since these fatty acids are precursors in their biosynthesis. This modified membrane composition may both contribute to enhanced ExPortal integrity and trigger a membrane stress response to a deleterious level. A damping of this may explain the compensatory function of the comutations, as ClpX and GdpP have both been associated with regulation of stress responses (37, 53–55). Interestingly, two independent PBr mutations altered the same serine residue at position 84 of FabT (S84L), a potential target of the Stk/Stp serine/threonine kinase/phosphatase of S. pyogenes (56, 57). Stk regulation of FabT alters membrane fatty acid composition (56) and contributes to resistance to several types of stress (57), further implicating a link between FabT, stress response, and ExPortal integrity.
An involvement in stress response may be a property shared by many of the other genes identified as associated with PB resistance. For example, transposon mutagenesis screens designed to isolate Lactococcus lactis mutants resistant to heat (36) or acid stress (37) yielded resistant mutants containing transposon insertions in many of the same genes that were identified here, including core PBr genes pstS, hpt, and gdpP. Also identified among the genes that were represented only once among the current collection of mutants were those involved in guanine metabolism, including deoB and guaA. A general characteristic of mutants with these insertionally inactivated genes is that they are resistant to a variety of different stresses, suggesting that their loss of function results in the activation of a general stress response pathway(s). Several global regulatory pathways that link metabolic stress to guanine metabolism have been identified, including (p)ppGpp and the stringent response (37) and the transcription regulator CodY (58). Other mutants represent pathways that link other aspects of nucleotide metabolism to general stress responses, including the second messenger cyclic-di-AMP phosphodiesterase GdpP (55, 59, 60), which has recently been linked to potassium import under osmotic stress (61–63). If triggering a stress response is a characteristic shared by other mutations in this collection, then it is possible that those involving the loss of function of a single subunit of a multicomponent membrane transporter may cause a membrane perturbation that induces a stress response. However, the collection also includes examples of membrane transporter mutations that were a gain of function, such as the manL/N mutants that conferred PB resistance while retaining their ability to transport mannose. In this case, it is possible that these mutations had a more peripheral effect that induced a stress response, including altering how the transporter interacts with other membrane proteins or causing it to mislocalize or cluster at an inappropriate location in the membrane.
Similar to the mannose transporter, mutations conferring PB resistance that were apparently the result of a gain of function occurred in FtsH, a membrane-bound protease that degrades SecY when it becomes uncoupled from other Sec translocon components (64), or upon protein secretion stress which can occur when the Sec translocon becomes jammed (65). As PB causes the redistribution of the Sec translocons and may also impart significant secretion stress, these ftsH mutations may result in the stabilization of SecY. Interestingly, both spontaneous ftsH mutations localized to a specific region of the gene, immediately downstream of the region encoding its Walker B motif. Combined with the observation that the FtsH null mutant was not PB resistant, the localization of these mutations to a specific motif may suggest that one of FtsH's other known regulatory functions (66) contributes to PB resistance.
An unusual core PB-resistant gene identified was the EbsA gene. Although deletion of EbsA was sufficient to confer PB resistance, the ΔEbsA mutant displayed disrupted ExPortal staining and was unable to secrete SpeB following PB challenge. Spontaneous EbsA mutants were identified twice in the screen (PBr 3.1 and 11.1), both of which maintained nearly full-length ORFs (I95N and Δ21–25, respectively), and were both isolated in the context of several additional comutations. Therefore, either the spontaneous EbsA mutants may impart a gain of function to EbsA or the comutations may impart a synergistic effect with respect to ExPortal integrity, as PBr 3.1 maintained SpeB expression upon PB challenge. EbsA was originally identified in Enterococcus faecalis as being required for expression of enterococcal binding substance and necessary pheromone-inducible plasmid conjugation (67). Although the mechanism is still unclear, EbsA has homology to pore-forming proteins associated with phage endolysins and is therefore predicted to affect cell wall metabolism. The recent association of peptidoglycan metabolism and ExPortal organization supports this model (40).
From the class of null mutations in membrane transporters, perhaps the most interesting case involves the Pst Pi uptake transporter. Although the genes encoding three of its five components were represented in the collection, pstS was the most commonly mutated gene and was found altered in 8 of the 25 PBr mutants. Significantly, each isolate possessed the exact same mutation even when isolated from multiple independent experiments, a frameshift in the codon for the lysine residue at position number 6. This repetitively occurring mutation was most likely due to the fact that pstS carries a homopolymeric stretch of 9 consecutive adenines encoding 3 lysine residues which may be subject to slipped-strand mispairing during DNA replication (68). The AT-rich S. pyogenes HSC5 genome (38.5% GC content) contains 25 homopolymer A or T tracks of ≥9 bp. However, the frequency of mutation at this homopolymer track far surpassed that of any other locus in the PBr isolates (see Table S9 in the supplemental material), demonstrating a significant association between slippage at this locus and PB resistance. As expected for a slippage mechanism, wild-type pstS revertant mutants were readily isolated at high frequency under selective pressure. Thus, strand slipping at this locus may represent a form of phase variation whereby S. pyogenes can transiently enter a Pi-starved state to induce a PB resistance response and then revert to wild type when the selective pressure is removed. Additional evidence to support this hypothesis comes from the observation that despite the availability of two codons encoding lysine (AAA and AAG), the homopolymeric 9-adenine track is conserved in 14 out of 17 of the available sequenced S. pyogenes genomes, while the remaining three contain an 8-adenine track (AAA-AAA-AAG), in which the guanine residue is replaced at the terminal adenine position. In contrast, S. mutans also contains a polylysine track in pstS but circumvents slippage with the sequence AAA-AAG-AAA-AAA. The ability of Na+ to restore growth of the ΔPstS mutant suggests that the NptA Na+/Pi cotransporter (L897_06955) may sustain viability in the absence of PstS, except under conditions of extreme Pi limitation. Since the pst mutations are all loss-of-function mutations, they could confer PB resistance either through the inappropriate assembly of the transporter's remaining components or through an effect on phosphate metabolism. For lactic acid bacteria, alterations of phosphate metabolism have been implicated in regulation of central metabolism and stress resistance (69, 70).
In addition to potential roles in regulation of stress responses, several of the core PBr genes were found to have a role in regulation of the SpeB cysteine protease. This protease can account for >95% of the total protein found in the culture supernatant (71). As a relatively nonspecific protease, it was anticipated that SpeB would be required for PB resistance, as has been reported for other CAPs (72). However, several of the PB-resistant mutants had a defect in SpeB expression associated with mutations in gdpP and clpX. It has previously been reported that inactivation of gdpP is associated with decreased SpeB activity associated with a kinetic delay in processing the SpeB zymogen to its active form (30, 59). The recently described second messenger molecule cyclic-di-AMP is the substrate of the GdpP phosphodiesterase and has been linked to regulation of cell wall and cell envelope homeostasis (55, 59, 73–75), suggesting an overlap of these activities with regulation of SpeB expression. In addition, DarR, a recently identified cyclic-di-AMP-responsive transcription regulator, controls expression of fatty acid biosynthesis genes in Mycobacterium smegmatis (76), consistent with other data implicating regulation of membrane lipid composition, stress response, PB resistance, and ExPortal integrity. In contrast to gdpP, previous transposon mutagenesis screens for SpeB regulators have failed to identify clpX (30), which is likely due to a polar effect on the gene immediately downstream, engB (L897_03625), encoding YsxC, a GTPase essential for ribosome assembly (77, 78). In addition to the regulation of toxin expression in other Gram-positive pathogens (79, 80), regulation of several stress responses has also been recognized as one of the multiple functions of ClpX, a component of the ClpXP AAA+ protease (54). Together, these data are consistent with the idea that many core PBr genes confer enhanced ExPortal integrity via activation of stress responses.
Intriguingly, like ExPortal proteins, several of the proteins encoded by core PBr genes have been found to localize to specific subcellular regions in other bacteria. These include FtsH, which localizes to the septum in B. subtilis (81); PtsI, which clusters at the poles in E. coli and B. subtilis (82–84); and ClpX, which clusters in cytoplasmic foci in multiple bacteria (85–88) and whose localization is crucial for its function (89). Further work will address whether there is similar compartmentalization of proteins encoded by core PBr genes, along with the contribution of fatty acid metabolism, nutrient acquisition, and stress responses in how S. pyogenes maintains its ability to secrete toxins in response to challenge with CAPs.
Supplementary Material
ACKNOWLEDGMENTS
We thank Zachary Cusumano for providing plasmid pClpX.
This work was supported by Public Health Service grant AI046433 from the NIH (to M.G.C.), 1F31AI081504 (to L.A.V.), and a W. M. Keck Fellowship (to G.C.P.).
Footnotes
Published ahead of print 2 May 2014
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JB.01596-14.
REFERENCES
- 1.Rosch JW, Caparon MG. 2005. The ExPortal: an organelle dedicated to the biogenesis of secreted proteins in Streptococcus pyogenes. Mol. Microbiol. 58:959–968. 10.1111/j.1365-2958.2005.04887.x [DOI] [PubMed] [Google Scholar]
- 2.Rosch J, Caparon M. 2004. A microdomain for protein secretion in Gram-positive bacteria. Science 304:1513–1515. 10.1126/science.1097404 [DOI] [PubMed] [Google Scholar]
- 3.Huang M, Meng L, Fan M, Hu P, Bian Z. 2008. Effect of biofilm formation on virulence factor secretion via the general secretory pathway in Streptococcus mutans. Arch. Oral Biol. 53:1179–1185. 10.1016/j.archoralbio.2008.07.007 [DOI] [PubMed] [Google Scholar]
- 4.Hu P, Bian Z, Fan M, Huang M, Zhang P. 2008. Sec translocase and sortase A are colocalised in a locus in the cytoplasmic membrane of Streptococcus mutans. Arch. Oral Biol. 53:150–154. 10.1016/j.archoralbio.2007.08.008 [DOI] [PubMed] [Google Scholar]
- 5.Kline KA, Kau AL, Chen SL, Lim A, Pinkner JS, Rosch J, Nallapareddy SR, Murray BE, Henriques-Normark B, Beatty W, Caparon MG, Hultgren SJ. 2009. Mechanism for sortase localization and role in efficient pilus assembly in Enterococcus faecalis. J. Bacteriol. 191:3237–3247. 10.1128/JB.01837-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cunningham MW. 2000. Pathogenesis of group A streptococcal infections. Clin. Microbiol. Rev. 13:470–511. 10.1128/CMR.13.3.470-511.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mitchell TJ. 2003. The pathogenesis of streptococcal infections: from tooth decay to meningitis. Nat. Rev. Microbiol. 1:219–230. 10.1038/nrmicro771 [DOI] [PubMed] [Google Scholar]
- 8.Scott JR, Barnett TC. 2006. Surface proteins of gram-positive bacteria and how they get there. Annu. Rev. Microbiol. 60:397–423. 10.1146/annurev.micro.60.080805.142256 [DOI] [PubMed] [Google Scholar]
- 9.Ferretti JJ, McShan WM, Ajdic D, Savic DJ, Savic G, Lyon K, Primeaux C, Sezate S, Suvorov AN, Kenton S, Lai HS, Lin SP, Qian Y, Jia HG, Najar FZ, Ren Q, Zhu H, Song L, White J, Yuan X, Clifton SW, Roe BA, McLaughlin R. 2001. Complete genome sequence of an M1 strain of Streptococcus pyogenes. Proc. Natl. Acad. Sci. U. S. A. 98:4658–4663. 10.1073/pnas.071559398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rosch JW, Hsu FF, Caparon MG. 2007. Anionic lipids enriched at the ExPortal of Streptococcus pyogenes. J. Bacteriol. 189:801–806. 10.1128/JB.01549-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Barak I, Muchova K, Wilkinson AJ, O'Toole PJ, Pavlendova N. 2008. Lipid spirals in Bacillus subtilis and their role in cell division. Mol. Microbiol. 68:1315–1327. 10.1111/j.1365-2958.2008.06236.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kawai F, Shoda M, Harashima R, Sadaie Y, Hara H, Matsumoto K. 2004. Cardiolipin domains in Bacillus subtilis Marburg membranes. J. Bacteriol. 186:1475–1483. 10.1128/JB.186.5.1475-1483.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mileykovskaya E, Dowhan W. 2000. Visualization of phospholipid domains in Escherichia coli by using the cardiolipin-specific fluorescent dye 10-N-nonyl acridine orange. J. Bacteriol. 182:1172–1175. 10.1128/JB.182.4.1172-1175.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Campo N, Tjalsma H, Buist G, Stepniak D, Meijer M, Veenhuis M, Westermann M, Muller JP, Bron S, Kok J, Kuipers OP, Jongbloed JD. 2004. Subcellular sites for bacterial protein export. Mol. Microbiol. 53:1583–1599. 10.1111/j.1365-2958.2004.04278.x [DOI] [PubMed] [Google Scholar]
- 15.Vega LA, Caparon MG. 2012. Cationic antimicrobial peptides disrupt the Streptococcus pyogenes ExPortal. Mol. Microbiol. 85:1119–1132. 10.1111/j.1365-2958.2012.08163.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Port GC, Paluscio E, Caparon MG. 2013. Complete genome sequence of emm type 14 Streptococcus pyogenes strain HSC5. Genome Announc. 1(4):e00612–13. 10.1128/genomeA.00612-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hanski E, Horwitz PA, Caparon MG. 1992. Expression of protein F, the fibronectin-binding protein of Streptococcus pyogenes JRS4, in heterologous streptococcal and enterococcal strains promotes their adherence to respiratory epithelial cells. Infect. Immun. 60:5119–5125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cho KH, Caparon M. 2006. Genetics of group A streptococci, p 59–73 In Fischetti VA, Novick RP, Ferretti JJ, Portnoy DA, Rood JI. (ed), Gram-positive pathogens, 2nd ed. ASM Press, Washington, DC [Google Scholar]
- 19.Lyon WR, Gibson CM, Caparon MG. 1998. A role for trigger factor and an rgg-like regulator in the transcription, secretion and processing of the cysteine proteinase of Streptococcus pyogenes. EMBO J. 17:6263–6275. 10.1093/emboj/17.21.6263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Caspi R, Altman T, Dale JM, Dreher K, Fulcher CA, Gilham F, Kaipa P, Karthikeyan AS, Kothari A, Krummenacker M, Latendresse M, Mueller LA, Paley S, Popescu L, Pujar A, Shearer AG, Zhang P, Karp PD. 2010. The MetaCyc database of metabolic pathways and enzymes and the BioCyc collection of pathway/genome databases. Nucleic Acids Res. 38:D473–D479. 10.1093/nar/gkp875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Caparon MG, Stephens DS, Olsen A, Scott JR. 1991. Role of M protein in adherence of group A streptococci. Infect. Immun. 59:1811–1817 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Perez-Casal J, Price JA, Maguin E, Scott JR. 1993. An M protein with a single C repeat prevents phagocytosis of Streptococcus pyogenes: use of a temperature-sensitive shuttle vector to deliver homologous sequences to the chromosome of S. pyogenes. Mol. Microbiol. 8:809–819. 10.1111/j.1365-2958.1993.tb01628.x [DOI] [PubMed] [Google Scholar]
- 23.Nielsen HV, Guiton PS, Kline KA, Port GC, Pinkner JS, Neiers F, Normark S, Henriques-Normark B, Caparon MG, Hultgren SJ. 2012. The metal ion-dependent adhesion site motif of the Enterococcus faecalis EbpA pilin mediates pilus function in catheter-associated urinary tract infection. mBio 3(4):e00177–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cho KH, Caparon MG. 2005. Patterns of virulence gene expression differ between biofilm and tissue communities of Streptococcus pyogenes. Mol. Microbiol. 57:1545–1556. 10.1111/j.1365-2958.2005.04786.x [DOI] [PubMed] [Google Scholar]
- 25.Horton RM, Cai ZL, Ho SN, Pease LR. 1990. Gene splicing by overlap extension: tailor-made genes using the polymerase chain reaction. Biotechniques 8:528–535 [PubMed] [Google Scholar]
- 26.Bryksin AV, Matsumura I. 2010. Overlap extension PCR cloning: a simple and reliable way to create recombinant plasmids. Biotechniques 48:463–465. 10.2144/000113418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Solovyev V, Salamov A. 2011. Automatic annotation of microbial genomes and metagenomic sequences, p 61–78 In Li RW. (ed), Metagenomics and its applications in agriculture, biomedicine and environmental studies. Nova Science Publishers, Hauppauge, NY [Google Scholar]
- 28.Novichkov PS, Laikova ON, Novichkova ES, Gelfand MS, Arkin AP, Dubchak I, Rodionov DA. 2010. RegPrecise: a database of curated genomic inferences of transcriptional regulatory interactions in prokaryotes. Nucleic Acids Res. 38:D111–D118. 10.1093/nar/gkp894 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lanie JA, Ng WL, Kazmierczak KM, Andrzejewski TM, Davidsen TM, Wayne KJ, Tettelin H, Glass JI, Winkler ME. 2007. Genome sequence of Avery's virulent serotype 2 strain D39 of Streptococcus pneumoniae and comparison with that of unencapsulated laboratory strain R6. J. Bacteriol. 189:38–51. 10.1128/JB.01148-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cho KH, Caparon MG. 2008. tRNA modification by GidA/MnmE is necessary for Streptococcus pyogenes virulence: a new strategy to make live attenuated strains. Infect. Immun. 76:3176–3186. 10.1128/IAI.01721-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Peschel A, Jack RW, Otto M, Collins LV, Staubitz P, Nicholson G, Kalbacher H, Nieuwenhuizen WF, Jung G, Tarkowski A, van Kessel KP, van Strijp JA. 2001. Staphylococcus aureus resistance to human defensins and evasion of neutrophil killing via the novel virulence factor MprF is based on modification of membrane lipids with l-lysine. J. Exp. Med. 193:1067–1076. 10.1084/jem.193.9.1067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kristian SA, Datta V, Weidenmaier C, Kansal R, Fedtke I, Peschel A, Gallo RL, Nizet V. 2005. D-alanylation of teichoic acids promotes group A streptococcus antimicrobial peptide resistance, neutrophil survival, and epithelial cell invasion. J. Bacteriol. 187:6719–6725. 10.1128/JB.187.19.6719-6725.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ochman H. 2003. Neutral mutations and neutral substitutions in bacterial genomes. Mol. Biol. Evol. 20:2091–2096. 10.1093/molbev/msg229 [DOI] [PubMed] [Google Scholar]
- 34.Lu YJ, Rock CO. 2006. Transcriptional regulation of fatty acid biosynthesis in Streptococcus pneumoniae. Mol. Microbiol. 59:551–566. 10.1111/j.1365-2958.2005.04951.x [DOI] [PubMed] [Google Scholar]
- 35.Jerga A, Rock CO. 2009. Acyl-acyl carrier protein regulates transcription of fatty acid biosynthetic genes via the FabT repressor in Streptococcus pneumoniae. J. Biol. Chem. 284:15364–15368. 10.1074/jbc.C109.002410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Duwat P, Ehrlich SD, Gruss A. 1999. Effects of metabolic flux on stress response pathways in Lactococcus lactis. Mol. Microbiol. 31:845–858. 10.1046/j.1365-2958.1999.01222.x [DOI] [PubMed] [Google Scholar]
- 37.Rallu F, Gruss A, Ehrlich SD, Maguin E. 2000. Acid- and multistress-resistant mutants of Lactococcus lactis: identification of intracellular stress signals. Mol. Microbiol. 35:517–528. 10.1046/j.1365-2958.2000.01711.x [DOI] [PubMed] [Google Scholar]
- 38.Hondorp ER, Hou SC, Hause LL, Gera K, Lee CE, McIver KS. 2013. PTS phosphorylation of Mga modulates regulon expression and virulence in the group A streptococcus. Mol. Microbiol. 88:1176–1193. 10.1111/mmi.12250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nelson DC, Garbe J, Collin M. 2011. Cysteine proteinase SpeB from Streptococcus pyogenes—a potent modifier of immunologically important host and bacterial proteins. Biol. Chem. 392:1077–1088. 10.1515/BC.2011.208 [DOI] [PubMed] [Google Scholar]
- 40.Vega LA, Port GC, Caparon MG. 2013. An association between peptidoglycan synthesis and organization of the Streptococcus pyogenes ExPortal. mBio 4(5):e00485–13. 10.1128/mBio.00485-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Boscher C, Nabi IR. 2012. Caveolin-1: role in cell signaling. Adv. Exp. Med. Biol. 729:29–50. 10.1007/978-1-4614-1222-9_3 [DOI] [PubMed] [Google Scholar]
- 42.Garner EC, Bernard R, Wang W, Zhuang X, Rudner DZ, Mitchison T. 2011. Coupled, circumferential motions of the cell wall synthesis machinery and MreB filaments in B. subtilis. Science 333:222–225. 10.1126/science.1203285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Dominguez-Escobar J, Chastanet A, Crevenna AH, Fromion V, Wedlich-Soldner R, Carballido-Lopez R. 2011. Processive movement of MreB-associated cell wall biosynthetic complexes in bacteria. Science 333:225–228. 10.1126/science.1203466 [DOI] [PubMed] [Google Scholar]
- 44.Divakaruni AV, Baida C, White CL, Gober JW. 2007. The cell shape proteins MreB and MreC control cell morphogenesis by positioning cell wall synthetic complexes. Mol. Microbiol. 66:174–188. 10.1111/j.1365-2958.2007.05910.x [DOI] [PubMed] [Google Scholar]
- 45.Epand RM, Epand RF. 2011. Bacterial membrane lipids in the action of antimicrobial agents. J. Pept. Sci. 17:298–305. 10.1002/psc.1319 [DOI] [PubMed] [Google Scholar]
- 46.Hachmann AB, Sevim E, Gaballa A, Popham DL, Antelmann H, Helmann JD. 2011. Reduction in membrane phosphatidylglycerol content leads to daptomycin resistance in Bacillus subtilis. Antimicrob. Agents Chemother. 55:4326–4337. 10.1128/AAC.01819-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Peleg AY, Miyakis S, Ward DV, Earl AM, Rubio A, Cameron DR, Pillai S, Moellering RC, Jr, Eliopoulos GM. 2012. Whole genome characterization of the mechanisms of daptomycin resistance in clinical and laboratory derived isolates of Staphylococcus aureus. PLoS One 7:e28316. 10.1371/journal.pone.0028316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Palmer KL, Daniel A, Hardy C, Silverman J, Gilmore MS. 2011. Genetic basis for daptomycin resistance in enterococci. Antimicrob. Agents Chemother. 55:3345–3356. 10.1128/AAC.00207-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Arias CA, Panesso D, McGrath DM, Qin X, Mojica MF, Miller C, Diaz L, Tran TT, Rincon S, Barbu EM, Reyes J, Roh JH, Lobos E, Sodergren E, Pasqualini R, Arap W, Quinn JP, Shamoo Y, Murray BE, Weinstock GM. 2011. Genetic basis for in vivo daptomycin resistance in enterococci. N. Engl. J. Med. 365:892–900. 10.1056/NEJMoa1011138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mehta S, Cuirolo AX, Plata KB, Riosa S, Silverman JA, Rubio A, Rosato RR, Rosato AE. 2012. VraSR two-component regulatory system contributes to mprF-mediated decreased susceptibility to daptomycin in in vivo-selected clinical strains of methicillin-resistant Staphylococcus aureus. Antimicrob. Agents Chemother. 56:92–102. 10.1128/AAC.00432-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hachmann AB, Angert ER, Helmann JD. 2009. Genetic analysis of factors affecting susceptibility of Bacillus subtilis to daptomycin. Antimicrob. Agents Chemother. 53:1598–1609. 10.1128/AAC.01329-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Fujita Y, Matsuoka H, Hirooka K. 2007. Regulation of fatty acid metabolism in bacteria. Mol. Microbiol. 66:829–839. 10.1111/j.1365-2958.2007.05947.x [DOI] [PubMed] [Google Scholar]
- 53.Smith WM, Pham TH, Lei L, Dou J, Soomro AH, Beatson SA, Dykes GA, Turner MS. 2012. Heat resistance and salt hypersensitivity in Lactococcus lactis due to spontaneous mutation of llmg_1816 (gdpP) induced by high-temperature growth. Appl. Environ. Microbiol. 78:7753–7759. 10.1128/AEM.02316-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Baker TA, Sauer RT. 2012. ClpXP, an ATP-powered unfolding and protein-degradation machine. Biochim. Biophys. Acta 1823:15–28. 10.1016/j.bbamcr.2011.06.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Corrigan RM, Abbott JC, Burhenne H, Kaever V, Grundling A. 2011. c-di-AMP is a new second messenger in Staphylococcus aureus with a role in controlling cell size and envelope stress. PLoS Pathog. 7:e1002217. 10.1371/journal.ppat.1002217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Agarwal S, Pancholi P, Pancholi V. 2011. Role of serine/threonine phosphatase (SP-STP) in Streptococcus pyogenes physiology and virulence. J. Biol. Chem. 286:41368–41380. 10.1074/jbc.M111.286690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bugrysheva J, Froehlich BJ, Freiberg JA, Scott JR. 2011. Serine/threonine protein kinase Stk is required for virulence, stress response, and penicillin tolerance in Streptococcus pyogenes. Infect. Immun. 79:4201–4209. 10.1128/IAI.05360-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Brinsmade SR, Sonenshein AL. 2011. Dissecting complex metabolic integration provides direct genetic evidence for CodY activation by guanine nucleotides. J. Bacteriol. 193:5637–5648. 10.1128/JB.05510-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Cho KH, Kang SO. 2013. Streptococcus pyogenes c-di-AMP phosphodiesterase, GdpP, influences SpeB processing and virulence. PLoS One 8:e69425. 10.1371/journal.pone.0069425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Bai Y, Yang J, Eisele LE, Underwood AJ, Koestler BJ, Waters CM, Metzger DW, Bai G. 2013. Two DHH subfamily 1 proteins in Streptococcus pneumoniae possess cyclic di-AMP phosphodiesterase activity and affect bacterial growth and virulence. J. Bacteriol. 195:5123–5132. 10.1128/JB.00769-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Bai Y, Yang J, Zarrella TM, Zhang Y, Metzger DW, Bai G. 2014. Cyclic ci-AMP impairs potassium uptake mediated by a cyclic di-AMP binding protein in Streptococcus pneumoniae. J. Bacteriol. 196:614–623. 10.1128/JB.01041-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Corrigan RM, Grundling A. 2013. Cyclic di-AMP: another second messenger enters the fray. Nat. Rev. Microbiol. 11:513–524. 10.1038/nrmicro3069 [DOI] [PubMed] [Google Scholar]
- 63.Corrigan RM, Campeotto I, Jeganathan T, Roelofs KG, Lee VT, Grundling A. 2013. Systematic identification of conserved bacterial c-di-AMP receptor proteins. Proc. Natl. Acad. Sci. U. S. A. 110:9084–9089. 10.1073/pnas.1300595110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kihara A, Akiyama Y, Ito K. 1995. FtsH is required for proteolytic elimination of uncomplexed forms of SecY, an essential protein translocase subunit. Proc. Natl. Acad. Sci. U. S. A. 92:4532–4536. 10.1073/pnas.92.10.4532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.van Stelten J, Silva F, Belin D, Silhavy TJ. 2009. Effects of antibiotics and a proto-oncogene homolog on destruction of protein translocator SecY. Science 325:753–756. 10.1126/science.1172221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Narberhaus F, Obrist M, Fuhrer F, Langklotz S. 2009. Degradation of cytoplasmic substrates by FtsH, a membrane-anchored protease with many talents. Res. Microbiol. 160:652–659. 10.1016/j.resmic.2009.08.011 [DOI] [PubMed] [Google Scholar]
- 67.Bensing BA, Dunny GM. 1993. Cloning and molecular analysis of genes affecting expression of binding substance, the recipient-encoded receptor(s) mediating mating aggregate formation in Enterococcus faecalis. J. Bacteriol. 175:7421–7429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.McDonald MJ, Wang WC, Huang HD, Leu JY. 2011. Clusters of nucleotide substitutions and insertion/deletion mutations are associated with repeat sequences. PLoS Biol. 9:e1000622. 10.1371/journal.pbio.1000622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Levering J, Musters MW, Bekker M, Bellomo D, Fiedler T, de Vos WM, Hugenholtz J, Kreikemeyer B, Kummer U, Teusink B. 2012. Role of phosphate in the central metabolism of two lactic acid bacteria—a comparative systems biology approach. FEBS J. 279:1274–1290. 10.1111/j.1742-4658.2012.08523.x [DOI] [PubMed] [Google Scholar]
- 70.Cesselin B, Ali D, Gratadoux JJ, Gaudu P, Duwat P, Gruss A, El Karoui M. 2009. Inactivation of the Lactococcus lactis high-affinity phosphate transporter confers oxygen and thiol resistance and alters metal homeostasis. Microbiology 155:2274–2281. 10.1099/mic.0.027797-0 [DOI] [PubMed] [Google Scholar]
- 71.Chaussee MS, Cole RL, van Putten JP. 2000. Streptococcal erythrogenic toxin B abrogates fibronectin-dependent internalization of Streptococcus pyogenes by cultured mammalian cells. Infect. Immun. 68:3226–3232. 10.1128/IAI.68.6.3226-3232.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Schmidtchen A, Frick IM, Andersson E, Tapper H, Bjorck L. 2002. Proteinases of common pathogenic bacteria degrade and inactivate the antibacterial peptide LL-37. Mol. Microbiol. 46:157–168. 10.1046/j.1365-2958.2002.03146.x [DOI] [PubMed] [Google Scholar]
- 73.Witte CE, Whiteley AT, Burke TP, Sauer JD, Portnoy DA, Woodward JJ. 2013. Cyclic di-AMP is critical for Listeria monocytogenes growth, cell wall homeostasis, and establishment of infection. mBio 4(3):e00282–13. 10.1128/mBio.00282-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Luo Y, Helmann JD. 2012. Analysis of the role of Bacillus subtilis sigma(M) in beta-lactam resistance reveals an essential role for c-di-AMP in peptidoglycan homeostasis. Mol. Microbiol. 83:623–639. 10.1111/j.1365-2958.2011.07953.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Oppenheimer-Shaanan Y, Wexselblatt E, Katzhendler J, Yavin E, Ben-Yehuda S. 2011. c-di-AMP reports DNA integrity during sporulation in Bacillus subtilis. EMBO Rep. 12:594–601. 10.1038/embor.2011.77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Zhang L, Li W, He ZG. 2013. DarR, a TetR-like transcriptional factor, is a cyclic-di-AMP responsive repressor in Mycobacterium smegmatis. J. Biol. Chem. 288:3085–3096. 10.1074/jbc.M112.428110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Cooper EL, Garcia-Lara J, Foster SJ. 2009. YsxC, an essential protein in Staphylococcus aureus crucial for ribosome assembly/stability. BMC Microbiol. 9:266. 10.1186/1471-2180-9-266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Wicker-Planquart C, Foucher AE, Louwagie M, Britton RA, Jault JM. 2008. Interactions of an essential Bacillus subtilis GTPase, YsxC, with ribosomes. J. Bacteriol. 190:681–690. 10.1128/JB.01193-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Zemansky J, Kline BC, Woodward JJ, Leber JH, Marquis H, Portnoy DA. 2009. Development of a mariner-based transposon and identification of Listeria monocytogenes determinants, including the peptidyl-prolyl isomerase PrsA2, that contribute to its hemolytic phenotype. J. Bacteriol. 191:3950–3964. 10.1128/JB.00016-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.McGillivray SM, Ebrahimi CM, Fisher N, Sabet M, Zhang DX, Chen Y, Haste NM, Aroian RV, Gallo RL, Guiney DG, Friedlander AM, Koehler TM, Nizet V. 2009. ClpX contributes to innate defense peptide resistance and virulence phenotypes of Bacillus anthracis. J. Innate Immun. 1:494–506. 10.1159/000225955 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Wehrl W, Niederweis M, Schumann W. 2000. The FtsH protein accumulates at the septum of Bacillus subtilis during cell division and sporulation. J. Bacteriol. 182:3870–3873. 10.1128/JB.182.13.3870-3873.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Govindarajan S, Elisha Y, Nevo-Dinur K, Amster-Choder O. 2013. The general phosphotransferase system proteins localize to sites of strong negative curvature in bacterial cells. mBio 4(5):e00443–13. 10.1128/mBio.00443-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Lopian L, Elisha Y, Nussbaum-Shochat A, Amster-Choder O. 2010. Spatial and temporal organization of the E. coli PTS components. EMBO J. 29:3630–3645. 10.1038/emboj.2010.240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Patel HV, Vyas KA, Li X, Savtchenko R, Roseman S. 2004. Subcellular distribution of enzyme I of the Escherichia coli phosphoenolpyruvate:glycose phosphotransferase system depends on growth conditions. Proc. Natl. Acad. Sci. U. S. A. 101:17486–17491. 10.1073/pnas.0407865101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Dziedzic R, Kiran M, Plocinski P, Ziolkiewicz M, Brzostek A, Moomey M, Vadrevu IS, Dziadek J, Madiraju M, Rajagopalan M. 2010. Mycobacterium tuberculosis ClpX interacts with FtsZ and interferes with FtsZ assembly. PLoS One 5:e11058. 10.1371/journal.pone.0011058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Kirstein J, Strahl H, Moliere N, Hamoen LW, Turgay K. 2008. Localization of general and regulatory proteolysis in Bacillus subtilis cells. Mol. Microbiol. 70:682–694. 10.1111/j.1365-2958.2008.06438.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Kain J, He GG, Losick R. 2008. Polar localization and compartmentalization of ClpP proteases during growth and sporulation in Bacillus subtilis. J. Bacteriol. 190:6749–6757. 10.1128/JB.00589-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.McGrath PT, Iniesta AA, Ryan KR, Shapiro L, McAdams HH. 2006. A dynamically localized protease complex and a polar specificity factor control a cell cycle master regulator. Cell 124:535–547. 10.1016/j.cell.2005.12.033 [DOI] [PubMed] [Google Scholar]
- 89.Iniesta AA, McGrath PT, Reisenauer A, McAdams HH, Shapiro L. 2006. A phospho-signaling pathway controls the localization and activity of a protease complex critical for bacterial cell cycle progression. Proc. Natl. Acad. Sci. U. S. A. 103:10935–10940. 10.1073/pnas.0604554103 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.