

Abstract
The Cpx two-component regulatory system has been shown in Escherichia coli to alleviate stress caused by misfolded cell envelope proteins. The Vibrio cholerae Cpx system was previously found to respond to cues distinct from those in the E. coli system, suggesting that this system fulfills a different physiological role in the cholera pathogen. Here, we used microarrays to identify genes that were regulated by the V. cholerae Cpx system. Our observations suggest that the activation of the V. cholerae Cpx system does not induce expression of genes involved in the mitigation of stress generated by misfolded cell envelope proteins but promotes expression of genes involved in antimicrobial resistance. In particular, activation of the Cpx system induced expression of the genes encoding the VexAB and VexGH resistance-nodulation-division (RND) efflux systems and their cognate outer membrane pore protein TolC. The promoters for these loci contained putative CpxR consensus binding sites, and ectopic cpxR expression activated transcription from the promoters for the RND efflux systems. CpxR was not required for intrinsic antimicrobial resistance, but CpxR activation enhanced resistance to antimicrobial substrates of VexAB and VexGH. Mutations that inactivated VexAB or VexGH efflux activity resulted in the activation of the Cpx response, suggesting that vexAB and vexGH and the cpxP-cpxRA system are reciprocally regulated. We speculate that the reciprocal regulation of the V. cholerae RND efflux systems and the Cpx two-component system is mediated by the intracellular accumulation of an endogenously produced metabolic by-product that is normally extruded from the cell by the RND efflux systems.
INTRODUCTION
Vibrio cholerae is a facultative human pathogen that causes cholera, a severe acute diarrheal disease that is estimated to afflict 3 to 5 million people annually (1). People acquire cholera by ingestion of V. cholerae-contaminated food or water (2). Once in the host environment, V. cholerae produces a variety of virulence factors that enable the pathogen to colonize the small intestine and to cause diarrhea. Two critical virulence factors coregulated by the virulence activator ToxR are the toxin-coregulated pilus (TCP), a type IV pilus that is essential for colonization, and cholera toxin (CT), an enterotoxin that causes the secretory diarrhea that is the hallmark of cholera (3–8). Like the expression of TCP, intestinal colonization is dependent upon V. cholerae overcoming host barriers in the human gastrointestinal tract. These barriers include antimicrobial compounds, such as bile salts, fatty acids, and components of the innate immune system. V. cholerae resistance to these factors is largely dependent upon the production of the resistance-nodulation-division (RND) family of efflux systems (9, 10).
RND efflux systems are tripartite transporters that are ubiquitous among Gram-negative bacteria. Each RND efflux system is made up of three components: an outer membrane porin homologous to Escherichia coli tolC, an integral cytoplasmic membrane pump protein belonging to the RND superfamily of transporters, and a periplasmic membrane fusion protein that links the outer membrane pore to the cytoplasmic membrane pump protein (11–15). These RND systems have garnered much attention with regard to xenobiotic resistance, as a number of RND systems have been shown to efflux numerous chemically unrelated antimicrobial compounds, including dyes, detergents, antibiotics, and antimicrobial peptides (16, 17). As such, many of these broad-spectrum RND efflux systems are intricately linked to the development of multiple-drug-resistant organisms. Although antibiotic resistance provides an easily scored phenotype for many efflux systems, phylogenetic analysis indicates that the RND efflux systems evolved independently of antibiotic selection (18, 19). Thus, the native role of the RND efflux systems in bacterial physiology remains unclear, but there is accumulating evidence to suggest that they influence bacterial physiology independently of their role in xenobiotic resistance. This is exemplified by reports implicating RND efflux systems in diverse phenotypes (reviewed in reference 20), such as biofilm formation (21, 22), iron acquisition (23), plant-bacterium interactions (24), lipid transport (25, 26), bacterial virulence (9, 10, 27), extrusion of toxic metal effectors (28), and removal of metabolic by-products from within the cell (29).
V. cholerae encodes six RND efflux systems that are required for antimicrobial resistance, virulence factor production, and intestinal colonization (9, 30, 31). Functional characterization of the RND systems revealed that four of the six systems (i.e., VexAB, VexCD, VexGH, and VexIJK) mediate resistance to antimicrobial compounds in vitro (9, 10, 31). The VexAB RND efflux system exhibits a high basal level of activity and provides V. cholerae with its intrinsic antimicrobial resistance, a function analogous to that of E. coli's AcrAB. The VexAB system is a multiple-drug efflux system that mediates resistance to bile salts, nonionic detergents, and a variety of antibiotics (e.g., ampicillin, erythromycin, novobiocin, and polymyxin B). The three other RND efflux systems are functionally redundant with VexAB. VexCD is a bile-specific efflux system; VexGH mediates resistance to bile acids, nonionic detergents, ampicillin, and novobiocin; and VexIJK effluxes bile acids, nonionic detergents, and novobiocin. The two remaining RND efflux systems, VexEF and VexLM, do not appear to influence resistance to antimicrobials. All six of the RND efflux systems contribute to virulence (9, 30), and strains lacking the RND efflux systems are severely attenuated in vivo (9, 30). Inactivation of the RND efflux systems results in downregulation of the ToxR regulon, diminished CT and TCP production, and severe attenuation of growth in suckling mice (9, 32). The mechanism(s) by which the RND efflux systems influence virulence gene expression is unknown.
The Cpx two-component system is widely distributed among Gammaproteobacteria, including the Enterobacteriaceae and Vibrionaceae. In this regulatory system, CpxA functions as a membrane-associated sensor histidine kinase. Upon stimulation, CpxA autophosphorylates itself and then transfers the phosphate to a conserved aspartate residue on the cytoplasmic CpxR response regulator (reviewed in references 33 and 34). Phosphorylated CpxR (CpxR∼P) then modulates the expression of its target genes by binding to a consensus binding sequence located in the genes' promoter regions. CpxR∼P also regulates its own expression (i.e., the cpxRA operon) and the divergently transcribed cpxP. CpxP is a periplasmic protein associated with the cpxRA operon that appears to repress CpxR activation by interacting with CpxA and inhibiting its kinase activity and may also exhibit chaperone activity (35). CpxA* mutants lead to constitutive activation of the Cpx system and have been useful in analyses of Cpx regulons. The cpxA* mutation inactivates CpxA phosphatase activity, resulting in the accumulation of activated CpxR (i.e., CpxR∼P) (36–38). The Cpx system has been most extensively studied in E. coli, where it has been shown to alleviate extracytoplasmic stress resulting from cell envelope perturbations that are generally associated with misfolded cell envelope proteins (39, 40). Consistent with this idea, the majority of stimuli that activate the Cpx system have been predicted to result in the production of misfolded or damaged cell envelope proteins (reviewed in references 33 and 34).
Although a number of Cpx-inducing stimuli have been described in E. coli, studies suggest that these stimuli are not conserved in V. cholerae (36). For example, the E. coli Cpx system is activated by increased osmolarity but not by increased salinity (36, 41). In contrast, the V. cholerae Cpx system functions in an opposite manner; it is not responsive to osmolarity but is activated by increased salinity (36). Additionally, the E. coli Cpx system is active under standard laboratory growth conditions, whereas the V. cholerae Cpx system is inactive. The differences in the physiological roles of the Cpx systems in E. coli and V. cholerae may be related to the distinct environmental niches that these organisms occupy and appear to be reflected in amino acid sequence variability in the signaling domain of CpxA (36, 42).
While the physiological roles of the V. cholerae and E. coli Cpx systems appear to differ, deletion of tolC activated the Cpx system in both organisms (36, 43). In E. coli, the activation of the Cpx system was linked to loss of TolC-dependent efflux (43). TolC functions as the outer membrane pore component of several V. cholerae transport systems, including RND family transporters (44). Thus, we speculated that the tolC-dependent activation of the V. cholerae Cpx system results from the loss of RND efflux activity. Here, we explored the linkage between RND efflux activity and the expression of the Cpx system. We show that CpxR functions as a positive regulator of the VexAB and VexGH RND efflux systems. Conversely, we found that mutation of vexRAB or vexGH resulted in the activation of the Cpx system, suggesting that the VexAB and VexGH RND efflux systems function in the regulation of the Cpx system. While the V. cholerae VexAB and VexGH RND efflux systems and the Cpx system were reciprocally regulated, the defect in virulence factor production in V. cholerae RND efflux mutants was independent of the Cpx system. Together, our findings revealed a genetic linkage between the V. cholerae Cpx system and RND-mediated efflux and suggested that the V. cholerae Cpx system is activated in response to the accumulation of RND efflux substrates.
MATERIALS AND METHODS
Bacterial strains and culture conditions.
The bacterial strains used in this study are listed in Table 1. Escherichia coli EC100Dpir+ and SM10λpir were used for cloning and plasmid mobilization, respectively. E. coli K-12 strains BW25113 and JW3883-1 (cpxR::Km) were used as hosts for the E. coli vexRAB and vexGH reporter assays (45). V. cholerae strains used in this study were derivatives of O1 El Tor strain N16961 (46). V. cholerae N16961 ΔlacZ Smr was used as the wild-type (WT) control strain for all experiments. Bacterial strains were grown at 37°C in LB broth or on LB agar. AKI broth was used for virulence-inducing conditions as described previously (10, 47). Bacterial stocks were maintained at −80°C in LB broth containing 25% glycerol. Growth medium was supplemented with carbenicillin (Cb) and streptomycin (Sm) at 100 μg/ml, kanamycin (Km) at 50 μg/ml, or chloramphenicol (Cml) at 1 μg/ml (for V. cholerae) or at 20 μg/ml (for E. coli) as required. 5-Bromo-4-chloro-3-indolyl-β-d-galactopyranoside (X-gal) was added to LB agar plates at 40 μg/ml unless otherwise indicated.
TABLE 1.
Strains, plasmids, and primers used in this study
| Strain, plasmid, or primer | Characteristic(s) or sequencea | Source |
|---|---|---|
| Strains | ||
| E. coli | ||
| EC100Dpir | supE44 ΔlacU169 (ϕ80 lacZΔM15) hsdR17 recA1 endA1 gyrA96 thi-1 relA1 (λpirR6K) | Epicenter |
| SM10λpir | thi-1 thr leu tonA lacY supE recA::RP4-2-Tc::Mu km (λpirR6K) | Lab collection |
| BW25113 | F− Δ(araD-araB)567 lacZ4787Δ::rrnB3 LAM− rph-1 Δ(rhaD-rhaB)568 hsdR514 | 45 |
| JW3883-1 | F− Δ(araD-araB)567 lacZ4787Δ::rrnB3 LAM− rph-1 Δ(rhaD-rhaB)568 hsdR514 cpxR::km | 45 |
| V. cholerae | ||
| JB58 | V. cholerae O1 El Tor strain N16961 ΔlacZ Smr | Lab collection |
| cpxA* mutant | JB58::cpxA* | 36 |
| ΔcpxR mutant | JB58 ΔcpxR | 36 |
| JB485 | JB58 ΔvexB ΔvexD ΔvexF ΔvexH ΔvexK ΔvexM | 9 |
| DT1452 | JB485 ΔcpxR | This study |
| MKW589 | ΔcpxR lacZ::cpxP-lacZEc | 36 |
| DT1458 | JB58 lacZ::cpxP-lacZEc | This study |
| DT1572 | JB58 ΔvexB lacZ::cpxP-lacZEc | This study |
| DT1574 | JB58 ΔvexD lacZ::cpxP-lacZEc | This study |
| DT1578 | JB58 ΔvexH lacZ::cpxP-lacZEc | This study |
| DT1687 | JB58 ΔvexK lacZ::cpxP-lacZEc | This study |
| DT1576 | JB58 ΔvexB ΔvexD lacZ::cpxP-lacZEc | This study |
| DT1580 | JB58 ΔvexB ΔvexH lacZ::cpxP-lacZEc | This study |
| DT1689 | JB58 ΔvexB ΔvexK lacZ::cpxP-lacZEc | This study |
| DT1582 | JB58 ΔvexD ΔvexH lacZ::cpxP-lacZEc | This study |
| DT1584 | JB58 ΔvexB ΔvexD ΔvexH lacZ::cpxP-lacZEc | This study |
| DT1691 | JB58 ΔvexB ΔvexD ΔvexK lacZ::cpxP-lacZEc | This study |
| DT1478 | JB58 ΔvexB ΔvexD ΔvexH ΔvexK lacZ::cpxP-lacZEc | This study |
| DT1480 | JB58 ΔvexB ΔvexD ΔvexF ΔvexH ΔvexK lacZ::cpxP-lacZEc | This study |
| DT1482 | JB58 ΔvexB ΔvexD ΔvexH ΔvexK ΔvexM lacZ::cpxP-lacZEc | This study |
| DT1462 | JB58 ΔvexB ΔvexF ΔvexH ΔvexK ΔvexM lacZ::cpxP-lacZEc | This study |
| DT1476 | JB58 ΔvexD ΔvexF ΔvexH ΔvexK ΔvexM lacZ::cpxP-lacZEc | This study |
| DT1586 | DT1452 lacZ::cpxP-lacZEc | This study |
| DT1460 | JB485 lacZ::cpxP-lacZEc | This study |
| Plasmids | ||
| pTL61T | lacZ transcriptional reporter plasmid, Cbr oriRK2 | 79 |
| pXB228 | pTL61T containing the vexEF promoter region | This study |
| pXB229 | pTL61T containing the vexGH promoter region | This study |
| pXB230 | pTL61T containing the vexIJK promoter region | This study |
| pXB231 | pTL61T containing the vexCD promoter region | 9 |
| pXB232 | pTL61T containing the vexLM promoter region | This study |
| pXB233 | pTL61T containing the vexRAB promoter region | 9 |
| pXB265 | pTL61T containing the breR promoter region | This study |
| pΔR | cpxR::Km allelic-exchange vector | 36 |
| pJL1P'Z | Allelic-exchange vector for placing cpxP-lacZ into the V. cholerae genome | 36 |
| pBAD33-cpxR | pBAD33 expressing cpxR | 36 |
| pBAD33 | Arabinose-regulated expression plasmid, Cmlr, p15A origin of replication | 80 |
| PCR primers | ||
| cpxR-F | GGTCAAGTGACGTATAGGGAGCG | |
| cpxR-R | GAGGTAGGGTCAATACCGCGAAC | |
| lacZ5 | CTCTAGAAGCTTCTAGCTAGAGGG | |
| lacZ6 | CCGCCACCTGACGTCTAAGAAACC | |
| P-166c-F-XhoI | TTCTCGAGGGGTCCGGAGACGTACT | |
| P-166c-R-XbaI | CGTCTAGAGGAGCTGTTTATCGCCG | |
| P-VC0628-F-XhoI | GGCTCGAGATATTTGATCGGCGGAGT | |
| P-VC0628-R-XbaI | GGCTCGAGATATTTGATCGGCGGAGT | |
| P-VC0914-F-XhoI | GCCTCGAGCACATCGCTCAAGTGCGC | |
| P-VC0914-R-XbaI | CGTCTAGATCTTTGGCCGATAGCACA | |
| P-VC1673-F-XhoI | GGCTCGAGACCGCAGCCTTGCTGGG | |
| P-VC1673-R-XbaI | AATCTAGACCCACCAGCAAAGTGGA | |
| P-VC1746-F-SmaI | AACCCGGGAATTCGGCTTTTTCTTTCCAAATCGGCAGTG | |
| P-VC1746-R-BamHI | AAGGATCCAATCAGCGCCAACCGTTTTTGCTCACTGAG | |
| P-VCA638-F-XhoI | GGCTCGAGGGGTTTGGTCGGCATCT | |
| P-VCA638-R-XbaI | CGTCTAGAGTGCGATACTCCAACTTA |
Δ, with a deletion; lacZEc, lacZ from E. coli.
Plasmid construction.
Plasmids and oligonucleotides used in this study are listed in Table 1. β-Galactosidase reporter constructs for vexEF, vexGH, vexIJK, and vexLM were constructed as follows. Briefly, the gene-specific PCR primer pairs (i.e., P-VC0628-F-XhoI/P-VC0628-R-XbaI, P-VC0914-F-XhoI/P-VC0914-R-XbaI, P-VC1673-F-XhoI/P-VC1673-R-XbaI, and P-VCA0638-F-XhoI/P-VCA0638-R-XbaI) were used to amplify the promoter region for each operon from the V. cholerae N16961 genome. The resulting PCR amplicons were then digested with XhoI and XbaI restriction endonucleases before being ligated into similarly digested pTL61T to generate pXB228 (vexEF-lacZ), pXB229 (vexGH-lacZ), pXB230 (vexIJK-lacZ), and pXB232 (vexLM-lacZ). The breR-lacZ reporter (pXB265) was similarly created using the PCR primer pair P-VC1746-F-SmaI/P-VC1746-R-BamHI and the restriction endonucleases indicated in the primer names. The DNA sequence of the reporter constructs were subsequently verified by DNA sequencing.
Mutant construction.
Deletion of V. cholerae cpxR was performed as previously described (36). Briefly, pΔR was conjugated into V. cholerae, and cointegrants were selected for Sm/Km resistance. Several Sm/Km-resistant colonies were streaked onto LB agar (without salt) containing 5% sucrose to select for resolution of the integrated plasmid. Several resistant colonies were screened for Cb sensitivity to verify the loss of the plasmid and for Km resistance to verify the presence of the Km cassette inserted into the cpxR gene. PCR using the cpxR-F and cpxR-R primers was then used to confirm the presence of the cpxR::Km insert.
Introduction of the chromosomal cpxP-lacZ reporter into V. cholerae was performed as previously described (42). Briefly, the pJL1P'Z reporter construct was conjugated into V. cholerae strains, and cointegrants were selected for Sm/Cb resistance. Several Sm/Cb-resistant colonies were then streaked onto LB agar (without salt) containing 5% sucrose to select for resolution of the integrated plasmid. Sucrose-resistant colonies were then screened for Cb sensitivity to verify plasmid loss before the cpxP-lacZ insertion was confirmed by PCR using the lacZ5 and lacZ6 primers. Construction of the V. cholerae RND efflux mutant strains was previously reported (9, 30, 31, 48).
Transcriptional reporter assays.
Strains were grown under the indicated growth conditions, and culture aliquots were taken in triplicate at various time points postinoculation to quantify β-galactosidase activity as previously described (49). All reporter experiments were performed independently at least three times. The V. cholerae lacZ::cpxP-lacZ reporter strains were cultured under AKI conditions as follows. Overnight cultures were diluted 1:10,000 into 73 ml of AKI broth in a 30-mm-diameter glass cylinder. The cultures were then incubated statically at 37°C for 4 h, after which 10-ml aliquots were transferred into 125-ml Erlenmeyer flasks and grown at 37°C with shaking. Aliquots were then collected at the times indicated below for the β-galactosidase assay. The two-plasmid reporter system utilized overnight cultures of V. cholerae containing pBAD33-cpxR or pBAD33 (negative control) and a lacZ reporter (i.e., vexRAB-lacZ or vexGH-lacZ). The cultures were diluted 1:100 in LB broth with or without arabinose to achieve the final concentrations indicated (Fig. 4), and the cultures were incubated with shaking at 37°C. Aliquots were then collected at 4 h to measure gene expression using the β-galactosidase assay.
FIG 4.
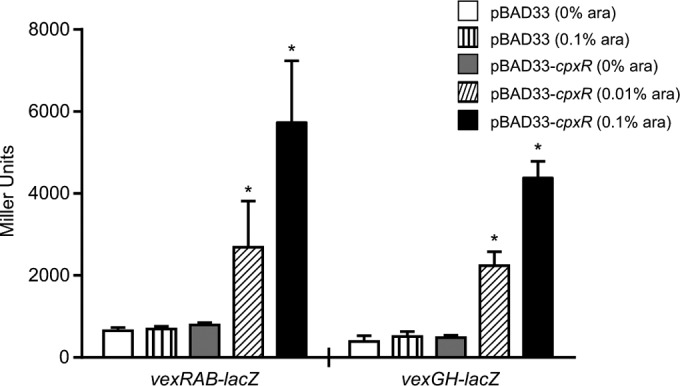
Ectopic expression of cpxR activates vexRAB and vexGH expression in V. cholerae. V. cholerae strains containing the indicated RND efflux system reporters and either pBAD33-cpxR or pBAD33 were grown in LB broth at 37°C in the presence or absence of arabinose (ara) as described in Materials and Methods. After 4 h of growth, triplicate aliquots were taken and assayed for β-galactosidase activity. The presented data are the means ± SD of results from three independent experiments. Statistical significance was determined by ANOVA with the Tukey-Kramer multiple-comparison test. *, P < 0.001.
Microarray analysis.
RNA was isolated for the cpxA* microarrays from the WT and the cpxA* mutant strain that were grown in LB broth at 37°C with shaking to an optical density at 600 nm (OD600) of ∼1.0. The subsequent steps of cDNA preparation and labeling, microarray hybridization, and data collection were carried out as previously described (50). The experiment was repeated independently three times, with two technical replicates for each individual experiment. The resulting data were processed as follows. The background subtracted spot intensities for each gene were subjected to global normalization before being averaged across all experimental samples. Genes exhibiting a change in expression of ≥2-fold with a coefficient of variation of ≤0.6 across all experiments were defined as being differentially regulated.
Identification of CpxR binding sites.
Putative CpxR binding sites were identified in the promoter regions of the differentially regulated genes using Clone Manager Professional software (Sci-Ed Software). The search was limited to 300 bp preceding the ATG start codon for each gene and used the published CpxR consensus binding site (i.e., GTAAN6GTAA), with an allowance for limited mismatches without gap insertion (51). To estimate the frequency of CpxR binding sites in the V. cholerae genome, we randomly selected 25 genes (15 from the large chromosome and 10 from the small chromosome) using the random-number generator function in Microsoft Excel. The promoters of these genes were then searched as described above for the presence of the CpxR consensus binding site.
Analysis of Cpx expression on agar plates.
A chromosomal cpxP-lacZ reporter was used to examine the expression of the Cpx system in a panel of various RND efflux mutants. Overnight cultures of the reporter strains were diluted 100-fold into LB broth and grown at 37°C with shaking for 1 h. The cultures were then normalized to an OD600 of 0.1; equal volumes of the cultures were spun down, and the pellet was resuspended in an equal volume of phosphate-buffered saline (PBS). The resuspended cultures were then diluted 1,000-fold before 2 μl was inoculated onto LB agar plates containing 160 μg/ml X-gal. As a positive control, the same cultures were also inoculated onto LB agar plates containing 160 μg/ml X-gal and 500 μM CuCl2. The inoculated plates were incubated overnight at 37°C before being photographed using a digital camera.
Antimicrobial susceptibility tests.
Antimicrobial susceptibility tests were performed as previously reported (31) using the following antibiotic disks purchased from Becton, Dickinson (Franklin Lakes, NJ, USA): ampicillin (10 μg/ml), tetracycline (10 μg/ml), gentamicin (10 μg/ml), erythromycin (15 μg/ml), and polymyxin B (300 μg/ml). Test compounds that were not commercially available were prepared by spotting 20 μl of concentrated stock onto 6-mm blank disks from Becton, Dickinson (i.e., 1 M CuCl2, 10% sarcosyl, and 10% deoxycholate). LB agar plates were inoculated with a lawn of the test strains using overnight cultures before the disks were aseptically placed on the surfaces of the agar plates. The plates were then inoculated overnight at 37°C before the zones of growth inhibition were measured in mm using a ruler. The Student t test was used to determine statistical significance for the zones of growth inhibition relative to that of the WT.
Plating efficiency.
Overnight cultures of the tested V. cholerae strains were diluted 100-fold into LB broth or LB broth containing 2 M KCl before being incubated at 37°C with shaking for 1 h. The cultures were then normalized to an OD600 of 0.25, at which time equal aliquots of each strain were spun down in a microcentrifuge and the resulting cell pellets resuspended in PBS. Serial dilutions of the cultures were then inoculated onto LB agar and thiosulfate-citrate-bile sucrose (TCBS) agar plates using an Eddy Jet 2 spiral plater (IUL Instruments). The agar plates were then incubated overnight at 37°C before the viable cells were enumerated using a Flash & Go automatic colony counter (IUL Instruments).
Cholera toxin quantitation and TcpA Western blotting.
CT production was determined by the GM1 enzyme-linked immunosorbent assay (ELISA) as previously described (30) using purified CT (Sigma) as a standard. TcpA production was determined by Western immunoblotting as previously described (30) using polyclonal rabbit antisera against TcpA that was graciously provided by Jun Zhu (University of Pennsylvania). Immunoreactive proteins on the Western blots were visualized using the SuperSignal West Pico chemiluminescence detection kit (Pierce Biotechnology).
Microarray data accession number.
The microarray data have been deposited in NCBI's Gene Expression Omnibus database and are available through accession number GSE55037.
RESULTS
Identification of CpxR-regulated genes.
To gain a better understanding of the genetic basis of the V. cholerae Cpx response, we used microarrays to identify CpxR-regulated genes. This was done by defining the effect of the cpxA* mutation on the V. cholerae transcriptome. The cpxA* mutation inactivates the CpxA phosphatase activity, which results in accumulation of CpxR in its activated form (i.e., CpxR∼P). Analysis of the microarray revealed that the levels of 25 transcripts were changed ≥2-fold in response to the constitutive activation of CpxR; 22 genes were upregulated, and three genes were downregulated (Table 2). We hypothesized that if CpxR directly regulated the expression of these genes, then their respective promoters would likely contain a CpxR consensus binding sequence. We therefore searched the putative promoter regions of the 25 genes for sequence similarity to the published CpxR consensus binding site (GTAAN6GTAA) (51). Eighteen of the genes (72%) contained a putative CpxR consensus sequence in their respective promoters; all but one of the genes was upregulated by CpxR. An analysis of 25 randomly selected promoters identified two genes (8%) containing CpxR consensus binding sequences. Seven genes did not contain CpxR binding sites and were likely regulated in an indirect manner. The preferential location of CpxR consensus binding sites in the promoters of positivity-regulated genes suggests that CpxR functions primarily as a transcriptional activator in V. cholerae.
TABLE 2.
CpxR-regulated genes
| Gene categorya | Open reading frame | Expression ratio (SD) relative to expression in the cpxA* mutant | CpxR binding site? | Gene | Gene product |
|---|---|---|---|---|---|
| Cellular processes | vc2691 | 67.0 (16.8) | Yesb | cpxP | Periplasmic protein CpxP, putative |
| Cell envelope protein | vc1854 | 0.5 (0.1) | No | ompT | OmpT porin |
| Metabolism | vca0151 | 5.2 (2.3) | Yes | Oxidoreductase, putative | |
| vca0249 | 2.3 (0.3) | Yes | Cytochrome b561, putative | ||
| vca0538 | 10.4 (3.3) | Yesc | Cytochrome b561 | ||
| Regulatory function proteins | vc0166 | 3.2 (0.7) | Yes (2 sites) | vexR | Transcriptional regulator, TetR family |
| vc2692 | 8.1 (1.7) | Yesb | cpxR | Transcriptional regulator CpxR | |
| Transport and binding proteins | vc0042 | 0.5 (0.2) | Yes | trkH | Potassium uptake protein |
| vc0164 | 2.4 (0.2) | Yesd | vexB | RND efflux pump VexB | |
| vc0165 | 2.2 (0.3) | Yesd | vexA | Membrane fusion protein VexA | |
| vc0913 | 14.8 (2.4) | Yes | vexG | Membrane fusion proteins VexG | |
| vc0914 | 12.4 (2.2) | Yese | vexH | RND efflux pump VexH | |
| vc2436 | 2.1 (0.3) | Yes | tolC | Outer membrane pore protein TolC | |
| vca0576 | 4.2 (2.2) | Yes | hutA | Heme transport protein HutA | |
| vca0603 | 2.6 (1.4) | No | ABC transporter substrate-binding protein | ||
| vca0782 | 4.3 (1.9) | Yes | ABC transporter, ATP-binding protein | ||
| Hypothetical and conserved hypothetical proteins | vc0191 | 2.0 (0.9) | Yes | ||
| vc0806 | 0.5 (0.1) | No | |||
| vc0915 | 2.4 (1.1) | No | |||
| vc0938 | 5.5 (1.3) | No | |||
| vca0126 | 2.0 (0.5) | No | |||
| vca0139 | 51.1 (25.4) | Yes (3 sites) | |||
| vca0162 | 3.0 (1.2) | No | |||
| vca0539 | 11.2 (2.0) | Yesc | |||
| vca0781 | 3.1 (1.2) | Yesf |
Gene categories were derived from the work of Heidelberg et al. (46).
The CpxR consensus binding site was located in the intergenic region between the divergently transcribed cpxP and cpxR genes.
vca0538 and vca0539 are in an operon that contains three CpxR consensus binding sites located upstream of vca0538.
vc0166, vc0165, and vc0164 are in an operon with two CpxR consensus binding site located upstream of vc0166.
vc0913 and vc0914 are in an operon with a CpxR consensus binding site located upstream of vc0913.
vca0782 and vca0781 are in an operon with a CpxR consensus binding site located upstream of vca0782.
Transcripts for cpxP and cpxR, which are known to be regulated by CpxR, were in the list of upregulated genes, lending credence to our approach. CpxP is located next to cpxR and is expressed from a divergent promoter. A CpxR consensus binding sequence is located in the intergenic region separating these two genes, a location that is consistent with CpxR's known regulation of its own expression as well as the divergently transcribed cpxP gene (40, 52). In contrast to that in E. coli, the Cpx response in V. cholerae did not appear to activate genes involved in alleviating membrane stress (e.g., dsbA, degP, and fkpA) resulting from misfolded membrane proteins (51, 53, 54), a finding that may reflect functional differences between the Cpx responses in the Vibrionaceae and the Enterobacteriaceae.
Notably, 11 of the 25 CpxR-regulated genes were involved in maintaining the permeability barrier of the cell (Table 2). Five of the upregulated genes encoded the production of two broad-spectrum RND efflux systems: VexAB, VexGH, and their cognate outer membrane pore component, TolC. Furthermore, a putative CpxR consensus binding sequence was found in the promoter regions of vexRAB, vexGH, and tolC, suggesting that CpxR regulates their expression (Fig. 1). One of the downregulated genes was ompT, which encodes a ToxR-regulated porin that possesses a large-diameter pore. Repression of ompT is associated with decreased susceptibility to low-molecular-weight antimicrobial compounds, such as bile salts (55–57).
FIG 1.

CpxR consensus binding sites in the vexRAB, vexGH, and tolC promoters. Putative CpxR consensus binding sequences in the promoter regions of the vexRAB, vexGH, and tolC genes are indicated by gray boxes. The start codon for each gene is marked by boldface. Numbering is relative to the start codon for each gene.
CpxR is a positive regulator of the V. cholerae RND efflux systems.
The expression of the vexRAB and vexGH RND efflux systems increased in the cpxA* mutant (Table 2). This finding, combined with the presence of CpxR consensus binding sequences in the vexRAB and vexGH promoters (Fig. 1), suggested that CpxR likely regulates the expression of these two RND efflux systems. To test this hypothesis and to validate the microarray results, we quantified vexRAB and vexGH expression in the WT, in the cpxA* mutant (which constitutively activates CpxR), and in a cpxR deletion mutant during growth in LB broth and during growth under AKI conditions. These growth conditions were selected, as they represent conditions where the RND efflux systems have been shown to contribute to both antimicrobial resistance and virulence factor production (10). We measured reporter expression at two time points (2 and 4 h postinoculation for the LB cultures and 3.5 and 6.5 h postinoculation for the AKI cultures) to control for potential growth-dependent effects on expression.
With the activation of CpxR, in the cpxA* background, vexRAB expression was significantly increased relative to that in the WT under AKI growth conditions. In contrast, the absence of CpxR in the ΔcpxR background did not influence vexRAB expression under either condition (Fig. 2A and B). Thus, CpxR does not regulate the basal-level expression of vexRAB but can enhance vexRAB expression under conditions where the Cpx system is activated. The expression level of vexGH was increased during growth in LB broth and under AKI conditions in the cpxA* background (Fig. 2C and D). However, the basal level of vexGH expression under AKI conditions appeared to be much lower than was observed in LB broth. Unlike with vexRAB deletion, cpxR deletion resulted in a significant reduction in vexGH expression during growth in LB broth (Fig. 2C) but not during growth under AKI conditions (Fig. 2D), implying that CpxR positively affects the basal-level expression of vexGH during growth in LB broth but not under AKI conditions. Collectively, these observations indicate that there are medium-dependent differences in the levels of activation of the Cpx system and that the Cpx system is likely inactive during growth under virulence gene-inducing conditions. These findings validate the microarray results and support the conclusion that CpxR is a positive regulator of the vexRAB and vexGH RND efflux systems.
FIG 2.
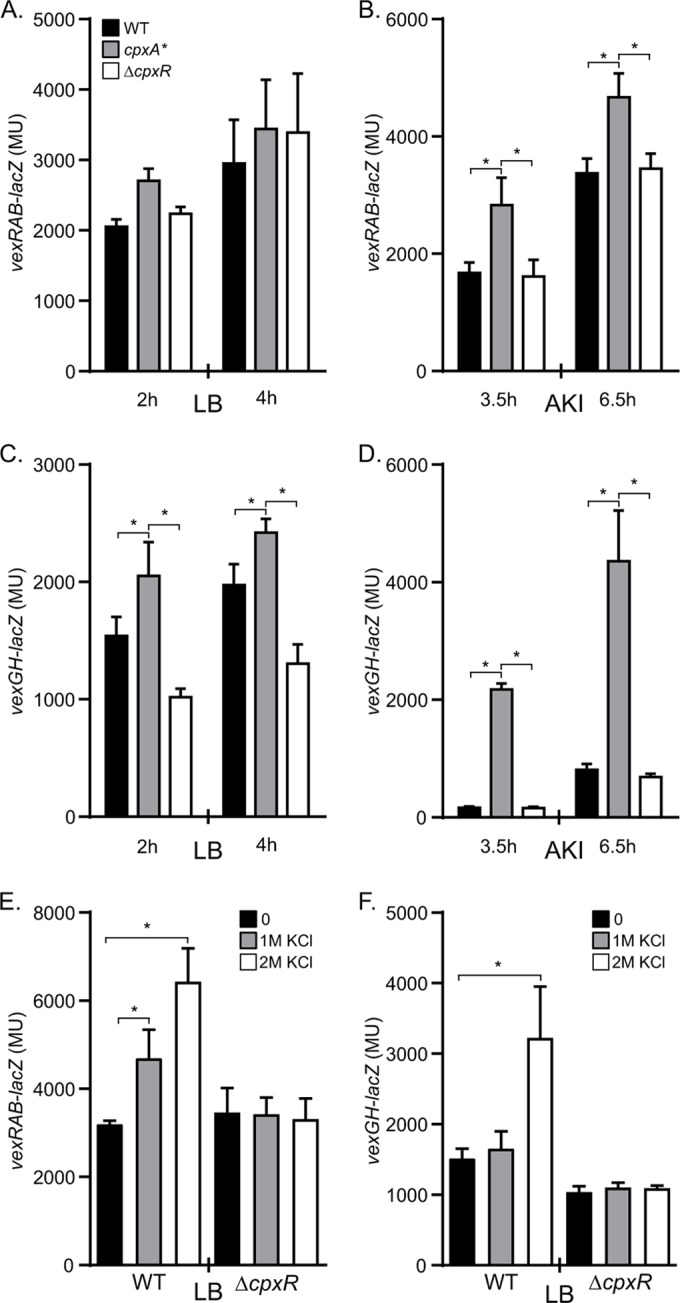
Expression of vexRAB and vexGH in V. cholerae cpx mutants. V. cholerae strains bearing either a vexRAB-lacZ (A, B, and E) or a vexGH-lacZ (C, D, and F) reporter were grown in LB broth (A and C), under AKI conditions (B and D), or in LB broth containing KCl at the indicated concentrations (E and F). Culture aliquots were taken at 2 h and 4 h (A and C), 3.5 and 6.5 h (B and D), or at 4 h (E and F) and assayed for β-galactosidase activity. The presented data are the means ± standard deviations (SD) of results from three independent experiments. Statistical significance was determined by analysis of variance (ANOVA) with the Tukey-Kramer multiple-comparison test (A, B, C, and D). (E and F) Values were compared to those with 0 KCl. *, P < 0.001.
Since vexRAB and vexGH appeared to be positively regulated by CpxR, we predicted that their expression should increase with activation of the Cpx system. We therefore quantified vexRAB and vexGH expression in the WT and a ΔcpxR mutant following growth in LB broth containing various concentrations of KCl, a known inducer of the V. cholerae Cpx system (36). The results showed a KCl concentration-dependent increase in vexRAB expression. Growth in 1 M KCl resulted in a 1.5-fold increase in vexRAB-lacZ expression, while growth in 2 M KCl caused a 2-fold increase in vexRAB-lacZ expression relative to that after growth in LB broth without KCl (Fig. 2E). This phenotype was found to be dependent on CpxR, as deletion of cpxR abolished the KCl-dependent induction of vexRAB (Fig. 2E). The expression of vexGH was also induced by KCl, as evidenced by a 2-fold increase in vexGH-lacZ expression during growth in 2 M KCl relative to that of the LB broth-grown control (Fig. 2F). The induction of vexGH by KCl was also dependent on CpxR, as deletion of cpxR abolished the KCl-dependent increase in vexGH expression (Fig. 2F). Together, these results confirm that the data obtained with the cpxA* mutant reflects activated CpxR and provides additional evidence to support the conclusion that the vexRAB and vexGH RND efflux systems are positively regulated by the Cpx system in a CpxR-dependent manner.
Activation of the Cpx response enhances V. cholerae resistance to antimicrobial compounds.
Since both the vexRAB and vexGH RND efflux systems were upregulated in the cpxA* mutant (Fig. 2), we predicted that the cpxA* mutant would exhibit enhanced resistance to antimicrobial compounds that are substrates for these two RND efflux systems (e.g., bile salts and other detergent-like molecules). To test this hypothesis, we calculated the plating efficiency of CpxR-activated V. cholerae on TCBS agar. Growth of V. cholerae on TCBS agar is dependent upon the expression of the RND efflux systems, which provide resistance to bile salts and other detergent-like compounds that are present in this medium (9, 30, 31, 58, 59). In these experiments, we compared the plating efficiencies of the cpxA* and ΔcpxR strains to that of the WT. We also compared the plating efficiencies of the WT and the ΔcpxR mutant grown in LB broth with and without 2 M KCl. The results showed that there was a 4.4-fold increase in the recovery of the cpxA* mutant relative to that of the WT, while the recovery of the ΔcpxR mutant was not significantly different from that of the WT (Fig. 3). Activation of the Cpx system by growth in 2 M KCl resulted in a 3-fold increase in the recovery of the KCl-grown cells relative to the recovery of cells grown in LB broth without KCl. Growth of the ΔcpxR mutant in 2 M KCl did not have a significant effect on its recovery, which confirmed that this phenotype was dependent on cpxR (Fig. 3). In conjunction with the previous data (Fig. 2), these results provide further evidence to support the idea that activation of the Cpx system induces vexRAB and vexGH expression in a CpxR-dependent manner, thereby enhancing antimicrobial resistance.
FIG 3.

Effect of CpxR activation on the recovery of Vibrio cholerae on TCBS agar. The V. cholerae WT, cpxA* mutant, and ΔcpxR mutant were cultured in LB broth or LB broth containing 2 M KCl before aliquots were diluted in PBS and plated in duplicate onto LB agar and TCBS agar plates for enumeration. The plates were incubated overnight at 37°C before the resulting colonies were counted. The recovery ratio for each mutant was calculated by the following equation: (the number of mutant colonies on TCBS/the number of mutant colonies on LB agar)/(the number of WT colonies on TCBS/the number of WT colonies on LB agar). The values are the means of results of three independent experiments ± SD. Statistical significance relative to WT values was determined by ANOVA with the Tukey-Kramer multiple-comparison test. *, P < 0.001.
Next, the effect of the Cpx system on V. cholerae antimicrobial susceptibility was investigated by examining the resistance of the WT, cpxA*, and ΔcpxR strains to antimicrobial agents by disk diffusion assays. The results showed that the cpxA* mutant exhibited increased resistance to ampicillin but not to other tested antimicrobial compounds (see Table S1 in the supplemental material). Since VexAB and VexGH are the only two RND efflux systems that contribute to ampicillin resistance, this finding is consistent with the idea that both of these RND efflux systems are upregulated in the cpxA* mutant. It is not surprising that differences in susceptibility were not observed for the other RND-dependent antimicrobial substrates, as the contributions of VexAB and VexGH to resistance to these substrates is masked due to redundancy among the six RND efflux systems (9, 30, 31). There was no significant difference in susceptibility to any of the compounds tested between the WT and the ΔcpxR mutant strain, suggesting that the tested xenobiotics did not function to activate the expression of the Cpx response. This was verified by plating V. cholerae containing a cpxP-lacZ reporter on LB agar containing subinhibitory amounts of these xenobiotics; no differences in expression of the reporter in the presence or absence of any of the compounds were noted (data not shown), confirming the supposition that the Cpx system was not activated in response to these antimicrobial compounds. Based on these observations, we conclude that the Cpx system is not required for V. cholerae's intrinsic resistance to xenobiotic antimicrobial compounds, but its activation was able to enhance the pathogen's resistance to antimicrobials through increased expression of vexRAB and vexGH.
Ectopic expression of cpxR activates vexRAB and vexGH expression.
The observation that the vexRAB and vexGH promoters contain CpxR consensus binding sequences (Fig. 1) and were upregulated in a cpxA* background (Fig. 2) suggested that CpxR was a positive regulator of vexRAB and vexGH. To confirm this hypothesis, we expressed cpxR from the arabinose-inducible promoter in pBAD33 in V. cholerae harboring either the vexRAB-lacZ or the vexGH-lacZ reporter. This resulted in a dramatic arabinose-dose-dependent increase in both vexRAB and vexGH expression (Fig. 4). In contrast, cpxR expression did not affect the expression of any other RND efflux system (see Fig. S1 in the supplemental material), confirming that CpxR was specific for vexRAB and vexGH. These results confirmed our hypothesis that CpxR is a positive regulator of the vexRAB and vexGH RND efflux systems. Taken together, these observations are consistent with the hypothesis that CpxR functions as an activator at the vexRAB and vexGH promoters.
Mutation of vexB and vexH activate the Cpx system.
Our collective findings revealed that the VexAB and VexGH RND efflux systems are components of the Cpx response. This suggested the possibility that active efflux by the RND efflux systems might function to suppress the Cpx response. To test this hypothesis, we used a chromosomal cpxP-lacZ reporter as an indicator of the activation state of the Cpx system; cpxP expression is regulated by CpxR (36). We compared levels of cpxP expression in the WT and JB485, a strain lacking all six RND efflux systems (9). JB485 produced dark-blue colonies on LB agar–X-gal plates, whereas the WT and the JB485 ΔcpxR mutant yielded white colonies (Fig. 5A). Control cultures grown on agar plates containing 500 μM CuCl2 (a Cpx inducer) showed induction of cpxP in the WT but not in the cpxR mutants, validating the idea that the cpxP-lacZ construct faithfully reports the activation state of the Cpx system in each strain. Thus, the absence of RND efflux activity induces the Cpx system, a finding consistent with the idea that the RND efflux systems normally suppress the activity of the Cpx system in V. cholerae.
FIG 5.
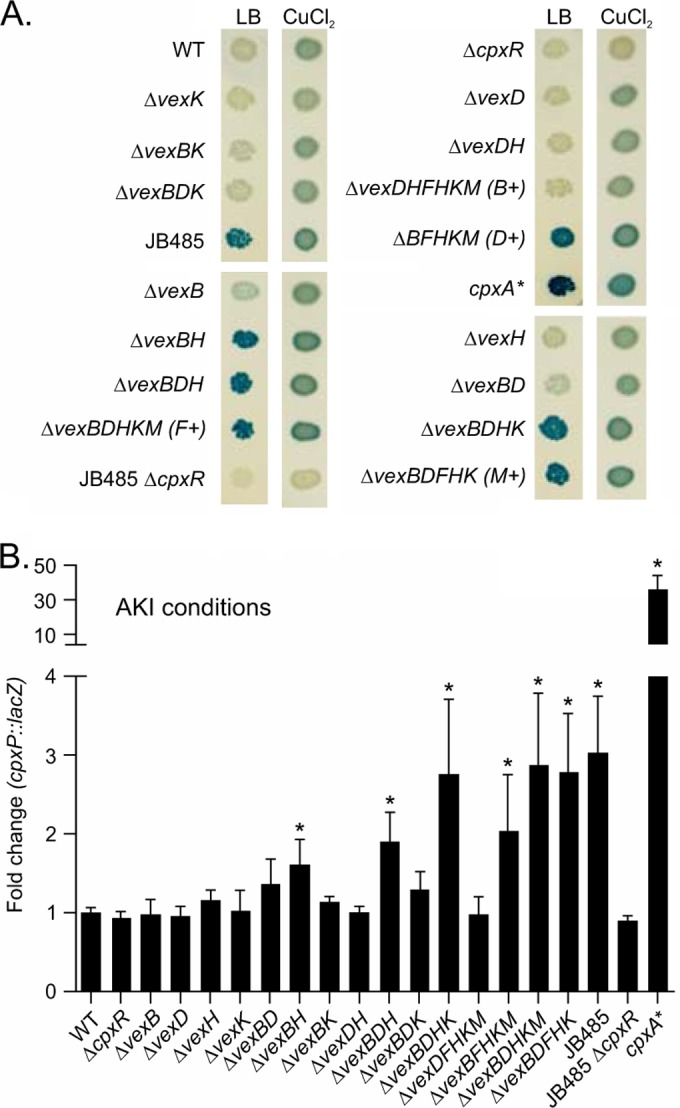
Induction of the Cpx system in V. cholerae RND efflux mutants. Expression of a chromosomal cpxP-lacZ reporter in V. cholerae strains with the indicated characteristics. (A) The strains were inoculated onto the surfaces of LB agar–X-gal plates with and without 500 μM CuCl2 before the plates were incubated overnight at 37°C and photographed. (B) The same V. cholerae cpxP-lacZ fusion strains were cultured under AKI conditions for 5 h, after which culture aliquots were collected and their β-galactosidase activities assayed. The fold change was calculated as the β-galactosidase activity (measured in Miller units) in the mutant divided by that in the WT. The bars represent the means ± SD of results from three independent experiments. Statistical relevance to WT values was calculated using a one-way ANOVA with Dunnett's post hoc test. *, P < 0.001.
Previous studies have shown that four of the six RND systems (vexB, vexD, vexH, and vexK) contribute to the in vitro resistance of V. cholerae to antimicrobial compounds with both distinct and redundant roles (9, 30, 31). We therefore sought to determine which of the RND efflux systems contributes to Cpx suppression by examining cpxP expression in a panel of strains that included both single and multiple RND efflux mutants. For these experiments, the cpxP-lacZ reporter was introduced into the chromosome of each of the RND mutants. Expression of cpxP was then examined on LB agar–X-gal plates and LB agar–X-gal plates containing CuCl2 (as a positive control). LB agar was used to screen for cpxP expression because previous studies have shown that the Cpx system was poorly expressed in LB broth (36). Among the four single RND efflux mutants (vexB, vexD, vexH, and vexK mutants), only the vexB mutant produced light-blue colonies on LB agar–X-gal (Fig. 5A); the vexH mutant produced colonies that appeared white to the naked eye but were discernible as faint blue under magnification, while the vexD and vexK mutant colonies were white. These observations suggest that cpxP expression is activated in the absence of either vexB or vexH (albeit to a very low level in the absence of vexH) but that the absence of either vexD or vexK does not induce cpxP expression. Furthermore, a vexBDK triple mutant produced colonies that were similar in color to the vexB single mutant, supporting the idea that vexD and vexK do not influence cpxP expression under these conditions. When the vexB and vexH deletions were combined, the resulting strain produced dark-blue colonies similar to those of JB485 (Fig. 5A), consistent with the hypothesis that these two RND efflux systems are functionally redundant for this phenotype. We hypothesized that if Cpx activation was a result of efflux activity provided by vexGH and/or vexRAB, then a strain lacking all five RND efflux systems except vexRAB should suppress expression of the Cpx system relative to that in JB485. Indeed, this was what we observed with the ΔvexDFHKM (vexB+) strain, which produced faint-blue colonies that were similar in color to those of the ΔvexH mutant (Fig. 5A). We conclude that efflux activity provided by the VexAB and VexGH RND efflux systems maintains the Cpx system in a suppressed state during growth of V. cholerae on LB agar.
We also examined whether the RND efflux systems influence the expression of the Cpx system during growth under virulence-inducing conditions. For these experiments, each RND mutant was grown under AKI conditions before being assayed for cpxP expression. There was no difference in cpxP expression among any of the single-deletion mutants (Fig. 5B). However, when a vexB mutant was combined with a vexH deletion, we observed an approximately 2-fold increase in cpxP expression. The vexBK and vexDH mutations did not appear to affect cpxP expression. This suggested that during growth under AKI conditions, vexB and vexH function in a redundant manner to limit the activation of the Cpx system. There was an increase in cpxP expression in the vexBDHK mutant relative to that in the vexBDH mutant, suggesting that the VexIJK RND efflux system contributed to the suppression of the Cpx system. The expression of cpxP in the vexBDHKM (vexF+) and vexBDFHK (vexM+) mutants was similar to that in the vexBDHK mutant. Expression of cpxP in the vexDFHKM (vexB+) mutant was similar to that in the WT, confirming the idea that the VexAB RND efflux system is sufficient to complement the absence of the five other RND efflux systems. The increase in cpxP expression in JB485 relative to that in the vexBFHKM (vexD+) mutant suggests that the VexCD RND efflux system contributes to the suppression of the Cpx system during growth under AKI conditions. These findings are reminiscent of our previous observations regarding CT and TCP production in RND efflux-deficient V. cholerae, where all six RND systems were required for high-level CT and TCP production (30). The concordance of these observations raises the intriguing possibility that the factor responsible for activation of the Cpx system during AKI growth may also contribute, by a Cpx-independent mechanism, to the virulence attenuation observed in JB485 (9).
CpxR contributes to vexRAB and vexGH expression in RND efflux-negative V. cholerae.
The above data suggested that the Cpx system was activated by loss of the RND efflux systems (Fig. 5). Based on this and the finding that CpxR activated vexRAB and vexGH expression (Fig. 2 and 4), we hypothesized that the expression of vexRAB and vexGH in JB485 would be upregulated in a CpxR-dependent manner. To test this, we quantified vexRAB and vexGH expression in the WT, JB485, and JB485 ΔcpxR strains during growth under AKI conditions. AKI conditions were selected, as they showed the most dramatic effect on the expression of vexRAB and vexGH (Fig. 2). The results showed a significant increase in the expression of both vexRAB and vexGH in strain JB485 relative to that in the WT at 3.5 and 6.5 h (Fig. 6A and B). The level of vexRAB expression in JB485 ΔcpxR was reduced relative to that in JB485 but was still greater than in the WT (Fig. 6A). This suggests that CpxR contributes to the induction of vexRAB in JB485 but that additional factors also contribute to vexRAB upregulation. In contrast to what occurred in the vexRAB mutant, the expression level of vexGH returned to WT levels in the JB485 ΔcpxR mutant, indicating that CpxR was responsible for the upregulation of vexGH in JB485 (Fig. 6B). Together, these results provide additional evidence supporting the conclusion that the loss of RND efflux activity results in CpxR activation and that CpxR functions as a positive regulator of vexRAB and vexGH.
FIG 6.
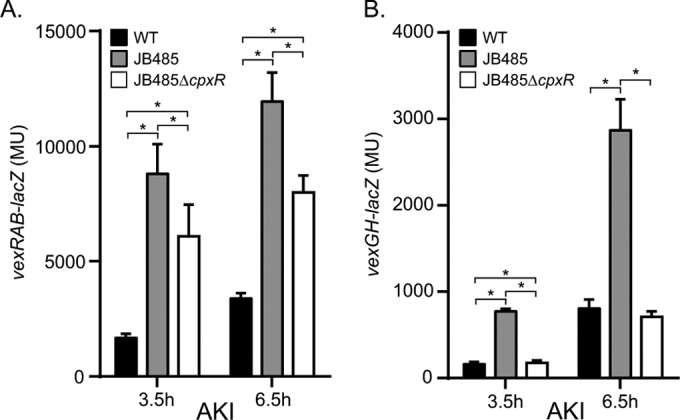
CpxR activates vexRAB and vexGH expression in the absence of RND efflux activity. V. cholerae strains containing a vexRAB (A) or vexGH (B) reporter plasmid were grown under AKI growth conditions. Aliquots were taken at 3.5 and 6.5 h and assayed for β-galactosidase activity. The presented data are the means ± SD of results from three independent experiments. Statistical significance was determined by ANOVA with the Tukey-Kramer multiple-comparison test. *, P < 0.001.
The Cpx system does not affect CT or TCP production.
The RND efflux systems were shown to be required for the production of both CT and TCP in V. cholerae (9). The mechanism by which the RND systems repressed CT and TCP production, while still unknown, was linked to repression of the ToxR regulon (9). The upregulation of the Cpx system in the RND null mutant suggested the possibility that CpxR functions to repress CT and TCP production. We investigated this hypothesis by quantifying CT and TCP production in the WT, cpxA*, ΔcpxR, JB485, and JB485 ΔcpxR strains. If cpxR was responsible for attenuated CT and TCP production, then its deletion in the RND-negative background should result in increased CT and TCP production. Likewise, CT and TCP production should be decreased in the WT by the cpxA* mutation due to constitutive activation of the Cpx system. Our results showed that there was no significant difference in CT or TCP production between the WT, cpxA*, and ΔcpxR strains or between JB485 and JB485 ΔcpxR (Fig. S2). This indicated that the defect in virulence factor production in JB485 was not a result of the activation of the Cpx system. These results also suggested that the Cpx system does not function to regulate virulence factor production in V. cholerae, a result that is consistent with previous findings showing that the Cpx system was dispensable for V. cholerae colonization of the infant mouse small intestine (36).
DISCUSSION
Bacteria have evolved overlapping mechanisms to sense and respond to stress that can result from exposure to toxic molecules, from adhesion to abiotic surfaces, or from misfolded proteins. The Cpx two-component system represents one such system that in E. coli mitigates envelope stress resulting from protein misfolding (34). V. cholerae also encodes a Cpx two-component system that shares a conserved genetic organization with the E. coli Cpx system (36). However, there are differences in the amino acid sequences of the sensor domains of the V. cholerae and E. coli CpxA and CpxP proteins, and the V. cholerae Cpx system does not respond to stimuli that activate the E. coli Cpx system (36). Thus, the Cpx systems may play different roles in the physiology of these two related Gammaproteobacteria.
Our characterization of the V. cholerae CpxR transcriptome supports the idea that there are differences between the V. cholerae and E. coli Cpx regulons. One key difference that we found is that several V. cholerae genes involved in protein fate (e.g., degP and dsbA) either were not detected as being regulated or were found to be repressed by CpxR (Table 2). This finding is similar to findings for Haemophilus ducreyi, where degP was also noted to be absent from the Cpx regulon (60). Together, these observations suggest that the physiological role for Cpx systems may not be universally conserved among Gram-negative bacteria. Although degP and dsbA appear to be regulated independently of CpxR in V. cholerae, the activation of the Cpx system in V. cholerae dsbC and dsbD mutants (36), which likely results in misfolded cell envelope proteins, suggests that the Cpx system can respond to misfolded proteins. The mechanism by which this occurs remains to be determined but may involve an alternate sigma factor (sigma E) (rpoE), which has been shown to functionally overlap the Cpx system in responding to extracytoplasmic stress in E. coli (53, 61).
The list of V. cholerae CpxR-regulated genes included a number of genes that mediate uptake and efflux of low-molecular-weight antimicrobial compounds. This included the ompT porin (which was repressed) and the vexRAB, vexGH, and tolC genes (which were upregulated). The latter loci encode the production of two broad-spectrum RND efflux systems that contribute to V. cholerae resistance to multiple antimicrobial compounds and pathogenesis (9, 10, 31). Several lines of evidence suggest that the vexRAB and vexGH operons are regulated by CpxR: the promoters of both operons contain CpxR consensus binding sequences (Fig. 1), both operons were upregulated in the cpxA* mutant (Fig. 2), ectopic cpxR expression activated their expression in V. cholerae (Fig. 4), and KCl (a Cpx system activator) induced their expression in the WT but not in a ΔcpxR mutant (Fig. 2). This conclusion is buttressed by the observations that the cpxA* mutant exhibited increased resistance to ampicillin (an antibiotic substrate of the VexAB and VexGH RND systems) and exhibited a growth advantage on TCBS agar (see Table S1 in the supplemental material and Fig. 3) and that induction of the Cpx system with 2 M KCl provided a growth advantage for the WT on TCBS but not for a ΔcpxR mutant (Fig. 3). While our data strongly support the conclusion that CpxR is an activator of these two RND efflux systems, CpxR was not required for V. cholerae's intrinsic resistance to antimicrobial compounds (Fig. 3 and Table S1). This suggests that the Cpx system may contribute to xenobiotic resistance under Cpx-inducing conditions by further increasing the expression of vexRAB and vexGH; however, the Cpx system is not required for V. cholerae's intrinsic resistance to xenobiotics.
Besides finding that the V. cholerae Cpx response promotes the expression of RND efflux systems, we found that inactivation of RND efflux stimulates the Cpx response. Mutation of vexB and/or vexH resulted in activation of the V. cholerae Cpx system. It is interesting to note that this phenotype was also observed with the deletion of RND efflux systems in H. ducreyi and Klebsiella pneumoniae (62, 63), which suggests that the genetic linkage between the Cpx system and the efflux systems is not unique to V. cholerae. The mechanism(s) by which the RND efflux systems modulate the activity of the Cpx system is not known. However, since RND efflux systems function in small-molecule export, we speculate that this phenotype is the result of the intracellular accumulation of an endogenously produced small molecule in the RND efflux mutants. Recent studies suggest that a natural function of RND efflux systems may be to remove metabolic waste from within the cell (64), which is consistent with this hypothesis. In E. coli, there is evidence to suggest that in the absence of RND-mediated efflux, metabolites accumulate in the cell and activate the expression of the MAR, Bae, and Cpx stress response systems (29, 43, 64–67). These regulatory systems then activate the expression of the acrAB, acrD, and mdtABCD RND efflux systems and other stress-mitigating genes. Although V. cholerae appears to lack the MAR and Bae systems, we propose that a similar mechanism occurs in V. cholerae. We speculate that an as-yet-unidentified cellular metabolite accumulates in the absence of RND efflux and activates the Cpx system. Our observation that different RND efflux pumps were required to suppress the Cpx system during growth on LB agar versus growth under AKI conditions suggests that the Cpx system may respond to the accumulation of multiple different endogenous molecules. Determining the identities of such effector molecules is a key challenge for future studies.
We previously proposed that a small molecule accumulated in V. cholerae in the absence of RND-mediated efflux and repressed CT and TCP production during growth under AKI conditions (30). This proposal was supported by the facts that V. cholerae virulence factor production was dependent upon all six RND efflux systems and that there was functional redundancy among the RND systems for this phenotype (9, 30, 32). The similarity of these results to the Cpx data, along with the inclusion of the ToxR-regulated ompT in the list of CpxR-responsive genes, suggests that attenuation of CT and TCP production in the RND-deficient strain may be due to the activation of the Cpx system. Although the Cpx system has been linked to virulence in other pathogens (68–73), our data indicate that the Cpx system does not affect V. cholerae virulence factor production. This was evidenced by the fact that cpxR deletion, constitutive activation of the Cpx system (i.e., cpxA*), or chemical activation of the Cpx system (i.e., with CuCl2) did not affect CT or TCP production. This conclusion is further supported by a previous study which showed that the Cpx system was dispensable for V. cholerae intestinal colonization (36).
While our findings strongly suggest that the Cpx system does not exert a significant influence over the expression of the ToxR-regulated virulence factors CT and TCP, we cannot exclude the possibility that the Cpx system plays a role in the regulation of other genes in vivo. Late in infection, V. cholerae is thought to encounter growth conditions conducive to the induction of the Cpx system, including high cell density, nutrient limitation, and the likely accumulation of metabolic waste/by-products (74, 75). The finding that both the vexAB and vexGH RND efflux systems are upregulated late during infection in humans and animals (74–77) is consistent with the hypothesis that the Cpx system is also induced late in infection. Thus, it is tempting to speculate that the Cpx system may contribute to late gene expression during infection. Late induced genes contribute to important phenotypes. This includes genes that contribute to V. cholerae survival in the environment and genes that contribute to the hyperinfectious phenotype associated with human-shed vibrios and is thought to be a key factor responsible for the epidemic spread of cholera (75, 78).
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by National Institutes of Health (NIH) awards R01AI091845 (J.E.B.) and R37AI042347 (M.K.W.). M.K.W. is an HHMI investigator. D.L.T. was supported by NIH training grant T32AI049820.
Footnotes
Published ahead of print 5 May 2014
Supplemental material for this article may be found at http://dx.doi.org/10.1128/IAI.00025-14.
REFERENCES
- 1.WHO. 2012. Cholera annual report 2011. Wkly. Epidemiol. Re. 87:289–304 [Google Scholar]
- 2.Reidl J, Klose KE. 2002. Vibrio cholerae and cholera: out of the water and into the host. FEMS Microbiol. Rev. 26:125–139. 10.1111/j.1574-6976.2002.tb00605.x [DOI] [PubMed] [Google Scholar]
- 3.Betley MJ, Miller VL, Mekalanos JJ. 1986. Genetics of bacterial enterotoxins. Annu. Rev. Microbiol. 40:577–605. 10.1146/annurev.mi.40.100186.003045 [DOI] [PubMed] [Google Scholar]
- 4.Taylor RK, Miller VL, Furlong DB, Mekalanos JJ. 1987. Use of phoA gene fusions to identify a pilus colonization factor coordinately regulated with cholera toxin. Proc. Natl. Acad. Sci. U. S. A. 84:2833–2837. 10.1073/pnas.84.9.2833 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Miller VL, Taylor RK, Mekalanos JJ. 1987. Cholera toxin transcriptional activator ToxR is a transmembrane DNA binding protein. Cell 48:271–279. 10.1016/0092-8674(87)90430-2 [DOI] [PubMed] [Google Scholar]
- 6.Herrington DA, Hall RH, Losonsky G, Mekalanos JJ, Taylor RK, Levine MM. 1988. Toxin, toxin-coregulated pili, and the toxR regulon are essential for Vibrio cholerae pathogenesis in humans. J. Exp. Med. 168:1487–1492. 10.1084/jem.168.4.1487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tacket CO, Taylor RK, Losonsky G, Lim Y, Nataro JP, Kaper JB, Levine MM. 1998. Investigation of the roles of toxin-coregulated pili and mannose-sensitive hemagglutinin pili in the pathogenesis of Vibrio cholerae O139 infection. Infect. Immun. 66:692–695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Thelin KH, Taylor RK. 1996. Toxin-coregulated pilus, but not mannose-sensitive hemagglutinin, is required for colonization by Vibrio cholerae O1 El Tor biotype and O139 strains. Infect. Immun. 64:2853–2856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bina XR, Provenzano D, Nguyen N, Bina JE. 2008. Vibrio cholerae RND family efflux systems are required for antimicrobial resistance, optimal virulence factor production, and colonization of the infant mouse small intestine. Infect. Immun. 76:3595–3605. 10.1128/IAI.01620-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Taylor DL, Bina XR, Bina JE. 2012. Vibrio cholerae vexH encodes a multiple drug efflux pump that contributes to the production of cholera toxin and the toxin co-regulated pilus. PLoS One 7:e38208. 10.1371/journal.pone.0038208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tikhonova EB, Zgurskaya HI. 2004. AcrA, AcrB, and TolC of Escherichia coli form a stable intermembrane multidrug efflux complex. J. Biol. Chem. 279:32116–32124. 10.1074/jbc.M402230200 [DOI] [PubMed] [Google Scholar]
- 12.Zgurskaya HI, Nikaido H. 1999. Bypassing the periplasm: reconstitution of the AcrAB multidrug efflux pump of Escherichia coli. Proc. Natl. Acad. Sci. U. S. A. 96:7190–7195. 10.1073/pnas.96.13.7190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Koronakis V, Sharff A, Koronakis E, Luisi B, Hughes C. 2000. Crystal structure of the bacterial membrane protein TolC central to multidrug efflux and protein export. Nature 405:914–919. 10.1038/35016007 [DOI] [PubMed] [Google Scholar]
- 14.Akama H, Matsuura T, Kashiwagi S, Yoneyama H, Narita S, Tsukihara T, Nakagawa A, Nakae T. 2004. Crystal structure of the membrane fusion protein, MexA, of the multidrug transporter in Pseudomonas aeruginosa. J. Biol. Chem. 279:25939–25942. 10.1074/jbc.C400164200 [DOI] [PubMed] [Google Scholar]
- 15.Eswaran J, Koronakis E, Higgins MK, Hughes C, Koronakis V. 2004. Three's company: component structures bring a closer view of tripartite drug efflux pumps. Curr. Opin. Struct. Biol. 14:741–747. 10.1016/j.sbi.2004.10.003 [DOI] [PubMed] [Google Scholar]
- 16.Saier MH, Jr, Paulsen IT. 2001. Phylogeny of multidrug transporters. Semin. Cell Dev. Biol. 12:205–213. 10.1006/scdb.2000.0246 [DOI] [PubMed] [Google Scholar]
- 17.Van Bambeke F, Glupczynski Y, Plesiat P, Pechere JC, Tulkens PM. 2003. Antibiotic efflux pumps in prokaryotic cells: occurrence, impact on resistance and strategies for the future of antimicrobial therapy. J. Antimicrob. Chemother. 51:1055–1065. 10.1093/jac/dkg224 [DOI] [PubMed] [Google Scholar]
- 18.Martinez JL. 2009. The role of natural environments in the evolution of resistance traits in pathogenic bacteria. Proc. Biol. Sci. 276:2521–2530. 10.1098/rspb.2009.0320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Alonso A, Sanchez P, Martinez JL. 2001. Environmental selection of antibiotic resistance genes. Environ. Microbiol. 3:1–9. 10.1046/j.1462-2920.2001.00161.x [DOI] [PubMed] [Google Scholar]
- 20.Alvarez-Ortega C, Olivares J, Martinez JL. 2013. RND multidrug efflux pumps: what are they good for? Front. Microbiol. 4:7. 10.3389/fmicb.2013.00007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Evans K, Passador L, Srikumar R, Tsang E, Nezezon J, Poole K. 1998. Influence of the MexAB-OprM multidrug efflux system on quorum sensing in Pseudomonas aeruginosa. J. Bacteriol. 180:5443–5447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kohler T, van Delden C, Curty LK, Hamzehpour MM, Pechere JC. 2001. Overexpression of the MexEF-OprN multidrug efflux system affects cell-to-cell signaling in Pseudomonas aeruginosa. J. Bacteriol. 183:5213–5222. 10.1128/JB.183.18.5213-5222.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Martinez JL, Delgado-Iribarren A, Baquero F. 1990. Mechanisms of iron acquisition and bacterial virulence. FEMS Microbiol. Rev. 6:45–56 [DOI] [PubMed] [Google Scholar]
- 24.Maggiorani Valecillos A, Rodríquez Palenzuela P, López-Solanilla E. 2006. The role of several multidrug resistance systems in Erwinia chrysanthemi pathogenesis. Mol. Plant Microbe Interact. 19:607–613. 10.1094/MPMI-19-0607 [DOI] [PubMed] [Google Scholar]
- 25.Cox JS, Chen B, McNeil M, Jacobs WR., Jr 1999. Complex lipid determines tissue-specific replication of Mycobacterium tuberculosis in mice. Nature 402:79–83. 10.1038/47042 [DOI] [PubMed] [Google Scholar]
- 26.Tahlan K, Wilson R, Kastrinsky DB, Arora K, Nair V, Fischer E, Barnes SW, Walker JR, Alland D, Barry CE, III, Boshoff HI. 2012. SQ109 targets MmpL3, a membrane transporter of trehalose monomycolate involved in mycolic acid donation to the cell wall core of Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 56:1797–1809. 10.1128/AAC.05708-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Piddock LJ. 2006. Multidrug-resistance efflux pumps—not just for resistance. Nat. Rev. Microbiol. 4:629–636. 10.1038/nrmicro1464 [DOI] [PubMed] [Google Scholar]
- 28.Nies DH. 2003. Efflux-mediated heavy metal resistance in prokaryotes. FEMS Microbiol. Rev. 27:313–339. 10.1016/S0168-6445(03)00048-2 [DOI] [PubMed] [Google Scholar]
- 29.Ruiz C, Levy SB. 2014. Regulation of acrAB expression by cellular metabolites in Escherichia coli. J. Antimicrob. Chemother. 69:390–399. 10.1093/jac/dkt352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Taylor DL, Bina XR, Bina JE. 2012. Vibrio cholerae VexH encodes a multiple drug efflux pump that contributes to the production of cholera toxin and the toxin co-regulated pilus. PLoS One 7:e38208. 10.1371/journal.pone.0038208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bina JE, Provenzano D, Wang C, Bina XR, Mekalanos JJ. 2006. Characterization of the Vibrio cholerae vexAB and vexCD efflux systems. Arch. Microbiol. 186:171–181. 10.1007/s00203-006-0133-5 [DOI] [PubMed] [Google Scholar]
- 32.Bina XR, Bina JE. 2010. The cyclic dipeptide cyclo(Phe-Pro) inhibits cholera toxin and toxin-coregulated pilus production in O1 El Tor Vibrio cholerae. J. Bacteriol. 192:3829–3832. 10.1128/JB.00191-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Buelow DR, Raivio TL. 2010. Three (and more) component regulatory systems—auxiliary regulators of bacterial histidine kinases. Mol. Microbiol. 75:547–566. 10.1111/j.1365-2958.2009.06982.x [DOI] [PubMed] [Google Scholar]
- 34.Vogt SL, Raivio TL. 2012. Just scratching the surface: an expanding view of the Cpx envelope stress response. FEMS Microbiol. Lett. 326:2–11. 10.1111/j.1574-6968.2011.02406.x [DOI] [PubMed] [Google Scholar]
- 35.Raivio TL, Popkin DL, Silhavy TJ. 1999. The Cpx envelope stress response is controlled by amplification and feedback inhibition. J. Bacteriol. 181:5263–5272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Slamti L, Waldor MK. 2009. Genetic analysis of activation of the Vibrio cholerae Cpx pathway. J. Bacteriol. 191:5044–5056. 10.1128/JB.00406-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Raivio TL, Silhavy TJ. 1997. Transduction of envelope stress in Escherichia coli by the Cpx two-component system. J. Bacteriol. 179:7724–7733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fleischer R, Heermann R, Jung K, Hunke S. 2007. Purification, reconstitution, and characterization of the CpxRAP envelope stress system of Escherichia coli. J. Biol. Chem. 282:8583–8593. 10.1074/jbc.M605785200 [DOI] [PubMed] [Google Scholar]
- 39.Raivio TL, Silhavy TJ. 2001. Periplasmic stress and ECF sigma factors. Annu. Rev. Microbiol. 55:591–624. 10.1146/annurev.micro.55.1.591 [DOI] [PubMed] [Google Scholar]
- 40.Raivio TL, Silhavy TJ. 1999. The sigmaE and Cpx regulatory pathways: overlapping but distinct envelope stress responses. Curr. Opin. Microbiol. 2:159–165. 10.1016/S1369-5274(99)80028-9 [DOI] [PubMed] [Google Scholar]
- 41.Prigent-Combaret C, Brombacher E, Vidal O, Ambert A, Lejeune P, Landini P, Dorel C. 2001. Complex regulatory network controls initial adhesion and biofilm formation in Escherichia coli via regulation of the csgD gene. J. Bacteriol. 183:7213–7223. 10.1128/JB.183.24.7213-7223.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mathur J, Waldor MK. 2004. The Vibrio cholerae ToxR-regulated porin OmpU confers resistance to antimicrobial peptides. Infect. Immun. 72:3577–3583. 10.1128/IAI.72.6.3577-3583.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rosner JL, Martin RG. 2013. Reduction of cellular stress by TolC-dependent efflux pumps in Escherichia coli indicated by BaeSR and CpxARP activation of spy in efflux mutants. J. Bacteriol. 195:1042–1050. 10.1128/JB.01996-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Koronakis V, Eswaran J, Hughes C. 2004. Structure and function of TolC: the bacterial exit duct for proteins and drugs. Annu. Rev. Biochem. 73:467–489. 10.1146/annurev.biochem.73.011303.074104 [DOI] [PubMed] [Google Scholar]
- 45.Baba T, Ara T, Hasegawa M, Takai Y, Okumura Y, Baba M, Datsenko KA, Tomita M, Wanner BL, Mori H. 2006. Construction of Escherichia coli K-12 in-frame, single-gene knockout mutants: the Keio collection. Mol. Syst. Biol. 2:2006.0008. 10.1038/msb4100050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Heidelberg JF, Eisen JA, Nelson WC, Clayton RA, Gwinn ML, Dodson RJ, Haft DH, Hickey EK, Peterson JD, Umayam L, Gill SR, Nelson KE, Read TD, Tettelin H, Richardson D, Ermolaeva MD, Vamathevan J, Bass S, Qin H, Dragoi I, Sellers P, McDonald L, Utterback T, Fleishmann RD, Nierman WC, White O, Salzberg SL, Smith HO, Colwell RR, Mekalanos JJ, Venter JC, Fraser CM. 2000. DNA sequence of both chromosomes of the cholera pathogen Vibrio cholerae. Nature 406:477–483. 10.1038/35020000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Iwanaga M, Yamamoto K, Higa N, Ichinose Y, Nakasone N, Tanabe M. 1986. Culture conditions for stimulating cholera toxin production by Vibrio cholerae O1 El Tor. Microbiol. Immunol. 30:1075–1083. 10.1111/j.1348-0421.1986.tb03037.x [DOI] [PubMed] [Google Scholar]
- 48.Bina XR, Taylor DL, Vikram A, Ante VM, Bina JE. 2013. Vibrio cholerae ToxR downregulates virulence factor production in response to cyclo(Phe-Pro). mBio 4(5):e00366–13. 10.1128/mBio.00366-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Miller JH. 1972. Experiments in molecular genetics. Cold Spring Harbor Laboratory, Cold Spring Harbor, NY [Google Scholar]
- 50.Ding Y, Davis BM, Waldor MK. 2004. Hfq is essential for Vibrio cholerae virulence and downregulates sigma expression. Mol. Microbiol. 53:345–354. 10.1111/j.1365-2958.2004.04142.x [DOI] [PubMed] [Google Scholar]
- 51.Pogliano J, Lynch AS, Belin D, Lin EC, Beckwith J. 1997. Regulation of Escherichia coli cell envelope proteins involved in protein folding and degradation by the Cpx two-component system. Genes Dev. 11:1169–1182. 10.1101/gad.11.9.1169 [DOI] [PubMed] [Google Scholar]
- 52.Danese PN, Silhavy TJ. 1998. CpxP, a stress-combative member of the Cpx regulon. J. Bacteriol. 180:831–839 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.De Wulf P, McGuire AM, Liu X, Lin EC. 2002. Genome-wide profiling of promoter recognition by the two-component response regulator CpxR-P in Escherichia coli. J. Biol. Chem. 277:26652–26661. 10.1074/jbc.M203487200 [DOI] [PubMed] [Google Scholar]
- 54.Danese PN, Silhavy TJ. 1997. The sigma(E) and the Cpx signal transduction systems control the synthesis of periplasmic protein-folding enzymes in Escherichia coli. Genes Dev. 11:1183–1193. 10.1101/gad.11.9.1183 [DOI] [PubMed] [Google Scholar]
- 55.Provenzano D, Klose KE. 2000. Altered expression of the ToxR-regulated porins OmpU and OmpT diminishes Vibrio cholerae bile resistance, virulence factor expression, and intestinal colonization. Proc. Natl. Acad. Sci. U. S. A. 97:10220–10224. 10.1073/pnas.170219997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Provenzano D, Lauriano CM, Klose KE. 2001. Characterization of the role of the ToxR-modulated outer membrane porins OmpU and OmpT in Vibrio cholerae virulence. J. Bacteriol. 183:3652–3662. 10.1128/JB.183.12.3652-3662.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Simonet VC, Basle A, Klose KE, Delcour AH. 2003. The Vibrio cholerae porins OmpU and OmpT have distinct channel properties. J. Biol. Chem. 278:17539–17545. 10.1074/jbc.M301202200 [DOI] [PubMed] [Google Scholar]
- 58.Kobayashi T, Enomoto S, Sakazaki R, Kuwahara S. 1963. A new selective isolation medium for the Vibrio group; on a modified Nakanishi's medium (TCBS agar medium). Nihon Saikingaku Zasshi 18:387–392 (In Japanese.) 10.3412/jsb.18.387 [DOI] [PubMed] [Google Scholar]
- 59.Nakanishi Y. 1963. An isolation agar medium for cholerae and enteropathogenic halophilic vibrios. Mod. Media 9:246 [Google Scholar]
- 60.Labandeira-Rey M, Brautigam CA, Hansen EJ. 2010. Characterization of the CpxRA regulon in Haemophilus ducreyi. Infect. Immun. 78:4779–4791. 10.1128/IAI.00678-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kovacikova G, Skorupski K. 2002. The alternative sigma factor sigma(E) plays an important role in intestinal survival and virulence in Vibrio cholerae. Infect. Immun. 70:5355–5362. 10.1128/IAI.70.10.5355-5362.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Srinivasan VB, Rajamohan G. 2013. KpnEF, a new member of the Klebsiella pneumoniae cell envelope stress response regulon, is an SMR-type efflux pump involved in broad-spectrum antimicrobial resistance. Antimicrob. Agents Chemother. 57:4449–4462. 10.1128/AAC.02284-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Rinker SD, Trombley MP, Gu X, Fortney KR, Bauer ME. 2011. Deletion of mtrC in Haemophilus ducreyi increases sensitivity to human antimicrobial peptides and activates the CpxRA regulon. Infect. Immun. 79:2324–2334. 10.1128/IAI.01316-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Helling RB, Janes BK, Kimball H, Tran T, Bundesmann M, Check P, Phelan D, Miller C. 2002. Toxic waste disposal in Escherichia coli. J. Bacteriol. 184:3699–3703. 10.1128/JB.184.13.3699-3703.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Rosner JL, Martin RG. 2009. An excretory function for the Escherichia coli outer membrane pore TolC: upregulation of marA and soxS transcription and Rob activity due to metabolites accumulated in tolC mutants. J. Bacteriol. 191:5283–5292. 10.1128/JB.00507-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Bury-Mone S, Nomane Y, Reymond N, Barbet R, Jacquet E, Imbeaud S, Jacq A, Bouloc P. 2009. Global analysis of extracytoplasmic stress signaling in Escherichia coli. PLoS Genet. 5:e1000651. 10.1371/journal.pgen.1000651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Hirakawa H, Inazumi Y, Masaki T, Hirata T, Yamaguchi A. 2005. Indole induces the expression of multidrug exporter genes in Escherichia coli. Mol. Microbiol. 55:1113–1126. 10.1111/j.1365-2958.2004.04449.x [DOI] [PubMed] [Google Scholar]
- 68.Gangaiah D, Zhang X, Fortney KR, Baker B, Liu Y, Munson RS, Jr, Spinola SM. 2013. Activation of CpxRA in Haemophilus ducreyi primarily inhibits the expression of its targets, including major virulence determinants. J. Bacteriol. 195:3486–3502. 10.1128/JB.00372-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Debnath I, Norton JP, Barber AE, Ott EM, Dhakal BK, Kulesus RR, Mulvey MA. 2013. The Cpx stress response system potentiates the fitness and virulence of uropathogenic Escherichia coli. Infect. Immun. 81:1450–1459. 10.1128/IAI.01213-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Leuko S, Raivio TL. 2012. Mutations that impact the enteropathogenic Escherichia coli Cpx envelope stress response attenuate virulence in Galleria mellonella. Infect. Immun. 80:3077–3085. 10.1128/IAI.00081-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Herbert Tran EE, Goodrich-Blair H. 2009. CpxRA contributes to Xenorhabdus nematophila virulence through regulation of lrhA and modulation of insect immunity. Appl. Environ. Microbiol. 75:3998–4006. 10.1128/AEM.02657-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Humphreys S, Rowley G, Stevenson A, Anjum MF, Woodward MJ, Gilbert S, Kormanec J, Roberts M. 2004. Role of the two-component regulator CpxAR in the virulence of Salmonella enterica serotype Typhimurium. Infect. Immun. 72:4654–4661. 10.1128/IAI.72.8.4654-4661.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Gal-Mor O, Segal G. 2003. Identification of CpxR as a positive regulator of icm and dot virulence genes of Legionella pneumophila. J. Bacteriol. 185:4908–4919. 10.1128/JB.185.16.4908-4919.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Bina J, Zhu J, Dziejman M, Faruque S, Calderwood S, Mekalanos J. 2003. ToxR regulon of Vibrio cholerae and its expression in vibrios shed by cholera patients. Proc. Natl. Acad. Sci. U. S. A. 100:2801–2806. 10.1073/pnas.2628026100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Merrell DS, Butler SM, Qadri F, Dolganov NA, Alam A, Cohen MB, Calderwood SB, Schoolnik GK, Camilli A. 2002. Host-induced epidemic spread of the cholera bacterium. Nature 417:642–645. 10.1038/nature00778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Lombardo MJ, Michalski J, Martinez-Wilson H, Morin C, Hilton T, Osorio CG, Nataro JP, Tacket CO, Camilli A, Kaper JB. 2007. An in vivo expression technology screen for Vibrio cholerae genes expressed in human volunteers. Proc. Natl. Acad. Sci. U. S. A. 104:18229–18234. 10.1073/pnas.0705636104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Xu Q, Dziejman M, Mekalanos JJ. 2003. Determination of the transcriptome of Vibrio cholerae during intraintestinal growth and midexponential phase in vitro. Proc. Natl. Acad. Sci. U. S. A. 100:1286–1291. 10.1073/pnas.0337479100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Schild S, Tamayo R, Nelson EJ, Qadri F, Calderwood SB, Camilli A. 2007. Genes induced late in infection increase fitness of Vibrio cholerae after release into the environment. Cell Host Microbe 2:264–277. 10.1016/j.chom.2007.09.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Linn T, St Pierre R. 1990. Improved vector system for constructing transcriptional fusions that ensures independent translation of lacZ. J. Bacteriol. 172:1077–1084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Guzman LM, Belin D, Carson MJ, Beckwith J. 1995. Tight regulation, modulation, and high-level expression by vectors containing the arabinose PBAD promoter. J. Bacteriol. 177:4121–4130 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.