

Abstract
When the NIH Mouse Models of Human Cancer Consortium (MMCC) initiated the Prostate Steering Committee 15 years ago, there were no genetically engineered mouse (GEM) models of prostate cancer (PCa). Today, a PubMed search for “prostate cancer mouse model” yields 3,200 publications and this list continues to grow. The first generation of GEM utilized the newly discovered and characterized probasin (PB) promoter driving viral oncogenes such as SV40 large T antigen to yield the LADY and TRAMP models. As the PCa research field has matured, the second generation of models has incorporated the single and multiple molecular changes observed in human disease, such as loss of PTEN and over-expression of Myc. Application of these models has revealed that mice are particularly resistant to developing invasive PCa, and once they achieve invasive disease, the PCa rarely resembles human disease. Nevertheless, these models and their application have provided vital information on human PCa progression. The aim of this review is to provide a brief primer on mouse and human prostate histology and pathology, provide descriptions of mouse models, as well as attempt to answer the age old question: Which GEM model of PCa is the best for my research question?
Keywords: Mouse models, prostate cancer, TRAMP, LADY, PTEN, Myc
I. Introduction: Modeling Human Prostate Cancer in Mice
Prostate cancer (PCa) is the most commonly diagnosed cancer in the United States[1] and carries a lifetime risk of one in six[2]. Due to the increase in early detection through prostate specific antigen (PSA) screening, mortality from PCa is decreasing; however the PSA test has recently come under increased scrutiny, as it may contribute to overtreatment for low-risk PCa[3]. On the other side of the spectrum, PCa patients with metastatic disease, whose five year disease specific survival rate has remained at about 30% from 1973[2,4], continue to fare poorly due to lack of curative therapeutics for advanced and metastatic disease. Therefore, it is important that basic and translational research continue in an effort to better understand the molecular events that contribute to indolent versus aggressive disease, as well as develop novel targeting strategies for advanced disease. In fact, a focus on distinguishing indolent disease from aggressive PCa, as well as understanding mechanisms of progression to metastasis, has been a focus of PCa researchers for many years. However, it was only with the advent of genetically engineered mice (GEM), that identifying specific molecular alterations related to PCa in a physiological context became a reality. In an effort to develop models of human cancers, the Mouse Models of Human Cancer Consortium (MMCC) was formed in 1999 by the National Institutes of Health (NIH). The PCa subset of the NIH MMCC is comprised of PCa researchers working with GEM as well as associated human and mouse pathologists. As a subset of the PCa working group, the Prostate Cancer Steering Committee, now the Prostate Pathology Committee (PPC), evolved in order to provide a consensus on observed mouse pathologies.
Modeling PCa with GEM is complicated by two facts. While human males have a one in six lifetime risk of developing PCa[2], mice very rarely develop spontaneous PCa[5], suggesting there are fundamental differences between human and mouse prostate biology and tumorigenesis. Also, the murine prostate is not a single organ as in the human, but rather is divided into four distinct lobes. This raises debate over which lobe(s) of the mouse prostate is the most representative of human prostate, as well as concerns over the lack of similarity of murine stroma surrounding the lobes, in comparison to human stroma. The advantages, (TGFβ RII ng GEM models of PCa is that the disease progresses in a shorter-lived, immunocompetent host within a genetically homogenous animal population, allowing for well-controlled, temporal observations on the effect of gene manipulations and drug treatments.
Fifteen years of studying various GEM has revealed that no single mouse model encompasses the entire spectrum of human PCa progression faithfully. PCa is usually a slowly developing cancer occurring late in life, and modeling these features in mice is counterproductive to experimental design. Nevertheless, several initial criteria are important to consider. A mouse model of PCa should reproducibly recapitulate one or more phases of disease progression. Murine PCa (mPCa) should originate within epithelial cells of the prostate. Ideally, the model should progress to an invasive adenocarcinoma, but prostatic intraepithelial neoplasia (PIN), the precursor lesion to adenocarcinoma, can also be informative. As most human PCa responds to androgen deprivation therapy (ADT), the tumor should be androgen dependent (AD) and respond to castration (preferably, not because expression of the transgene responsible for tumorigenesis is driven by an androgen-dependent promoter). In order to be considered a model of progression to “castrate resistant” prostate cancer (CRPC), experimental evidence of tumor regression in the GEM, must be followed by subsequent failure and continued outgrowth of the tumor. Failure of ADT and emergence of CRPC in humans is usually associated with expression of nuclear androgen receptor (AR) since CRPC remains dependent on AR signaling[6]. If mPCa does not respond to castration or is AR negative, the tumor should be classified as androgen independent (AI), and this behavior may be associated with molecular mechanisms comparable to human AI disease. Often, this phenotype in mice involves neuroendocrine (NE) differentiation. Although pure NE or “small cell carcinomas” are rare in humans as a primary PCa, treated PCa patients frequently develop NE differentiation and frank NE carcinomas sometimes constitute these recurrent/resistant tumors[7]. Finally, in the mouse models the tumors should achieve metastasis, ideally to bone, the most common site of metastasis observed in human PCa patients[8]. Rare bone metastases have been reported in some GEM, but visceral metastasis are more common and also valuable.
II. Human versus Mouse
II.A. Prostate anatomy
The human prostate sits at the base of the bladder and encircles the urethra. It is traditionally divided into four zones and right or left: the anterior fibromuscular stroma zone, the periurethral transition zone (TZ), the peripheral zone (PZ), and the central zone (CZ). In fact, the prostate is composed of multiple glands in each of these “zones” with ductal conduits to the urethra. All of these glands are invested in a contiguous fibromuscular stroma which abuts the lower pelvic soft tissues such that there is neither a true capsule surface nor investment with peritoneal lining. Conversely, the mouse prostate exists as four separate lobes with conduits to the urethra but which abut the peritoneal space and are invested within a peritoneal lining. The anterior, dorsal, lateral, and ventral prostate lobes are paired right and left in the mouse and each has a distinct anatomy and histology. Detailed descriptions of individual lobe histology are summarized in Suwa et al[5]. Although there have been studies suggesting that specific mouse lobes or mPCa lesions in specific lobes are more representative of human prostatic zones and PCa, the PPC concluded that it is premature to assume one mouse lobe is more relevant to human prostate and PCa than another[9,10].
At the microscopic level, the mouse and human prostate become more similar. Here, fibromuscular stroma, which is much more pronounced in human than mouse, encircles glands of epithelium[9]. The epithelial cell compartment is comprised of two cell layers, consisting of basal and terminally differentiated luminal cells, the later which secrete prostatic secretions in the luminal space. In addition, there are populations of epithelial cell precursors as well as scattered neuroendocrine cells. In the mouse, basal cells give rise to luminal and neuroendocrine cells during prostatic development[11], but in the adult mouse, basal to luminal cell differentiation following castration and testosterone re-administration either does not occur[12,13] or it is a slow and rare event[14] mediated by rare bipotential progenitors[15]. Interestingly, GEM studies suggest that the PCa tumor cell of origin in mPCa can be luminal, basal, or neuroendocrine[13-17].
II.B. Prostate pathology
For an extensive review of human and mouse pathology, please see Shappell et al[9] and Ittmann et al[10], which elegantly summarize human and mouse pathology, as well as present the consensus findings of the MMHC PPC. Definitions of mouse pathologies are summarized in Table 1, and mouse versus human PCa progression is illustrated in Figure 1 with histology provided in Figure 2.
Table 1. Mouse pathology definitions.
| Mouse Pathology | Characteristics |
|---|---|
| Hyperplasia | Proliferation of normal cells |
| PIN | Proliferation of atypical cells, contained within glands |
| Adenocarcinoma | Destructive proliferation with glandular differentiation , invading into stroma, adjacent glands |
| Neuroendocrine carcinoma | Destructive proliferation characterized by rosette formation, lacking cytoplasm; confirmed by synaptophysin or chromogranin staining |
| Sarcomatoid carcinoma | Destructive proliferation of atypical spindle cells |
Figure 1. Schematic representation of PCa progression.
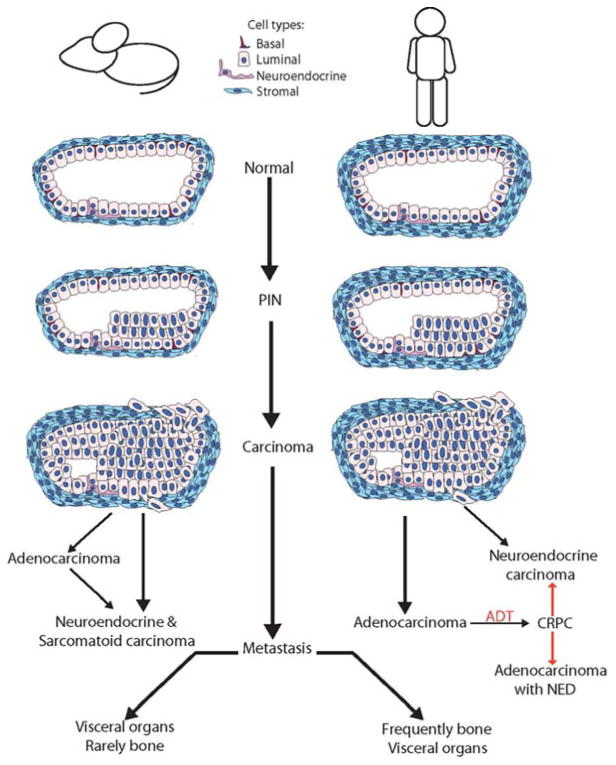
In the mouse, the prostate gland is surrounded by a thin stromal layer, while human prostate glands are surrounded by thick fibromuscular stroma. The precursor lesion to PCa is PIN, which is highlighted by the proliferation of atypical epithelial cells within the glands. PIN progresses to carcinoma. Carcinoma is characterized by invasion into the stroma by atypical epithelial cells and loss of basal cells. In humans, almost all PCa is adenocarcinoma. Systemic treatment includes ADT, and advanced PCa patients will frequently progress to CRPC, which has been associated with development of NE differentiation. These heavily treated patients can progress to NE tumors, which occur very rarely at primary diagnosis. Conversely, in mPCa, models often develop NE differentiation and NE tumors. Sarcomatoid tumors (rare in humans) are also seen as primary mPCa. Mouse models of adenocarcinoma may also progress to NE or sarcomatoid tumors following surgical castration, a proxy for ADT. Metastatic carcinoma, characterized by the dissemination and colonization of tumor cells in distant sites shows a predilection for specific sites. In human PCa, metastatic lesions are found predominantly in bone with NE cancers more common in visceral organs. mPCa, however, homes largely to lymph nodes and visceral organs with very rare examples of mouse PCa bone involvement.
Figure 2. Human versus mouse PIN and PCa.
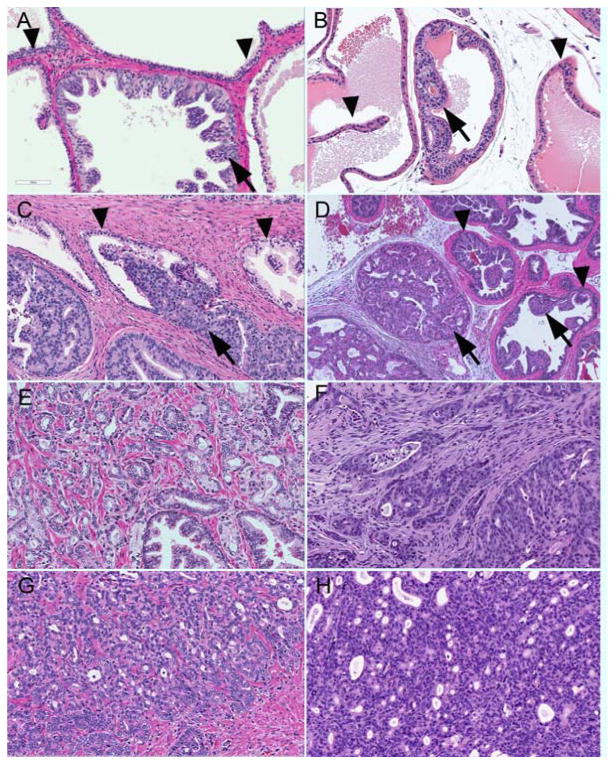
A. Human low grade PIN with minimal papillary growth with atypical nuclei (arrows), and clear in situ location indicated by the basal cells layer (triangles). B. Mouse low grade PIN with cribriform architecture (arrows) also with a layer of basal cells (triangles) present. HGPIN in human (C) and mouse (D) with attenuation, basal cells (triangles), and proliferation of atypical nuclei within glands (arrows). PCa can progress into low grade acinar patterns of adenocarcinoma (human Gleason pattern 3+3 E; Pten mouse D) or high grade with minimal or absent glands spaces (human Gleason pattern 4+5 F; Pten mouse G) Basal cells are absent, and the stroma is reactive to the invasive tumor (tumor associated stroma).
II.B.1. Hyperplasia
In the human, benign prostatic hyperplasia (BPH) is not a precursor lesion to PCa, and BPH patients do not have an increased incidence of PCa[18]. The PZ is the main zone of origin for PCa, while BPH arises solely in the TZ[9]. Because there is no consensus on which lobes of the mouse prostate most accurately recapitulate the human prostate zones[9,10], mPCa can begin as hyperplasia. Epithelial hyperplasia in the mouse can be diffuse or focal, have small areas of nuclear atypia, and tufting, and papillary or cribiform changes are not uncommon[9,10].
II.B.2. PIN
PIN in humans is characterized by the focal proliferation of atypical cells and appears to be the result of a clonal expansion of a single transformed cell within the gland or duct. Although most PIN lesions remain indolent, high grade PIN (HGPIN) on biopsy is associated with a high risk for subsequent adenocarcinoma, and it appears that at least some HGPIN lesions can progress to adenocarcinoma[19]. HGPIN phenotypes with similarities to uncommon PCa subtypes have been recorded, but are often not identified in conjunction with these cancers[20].
Similarly, mouse PIN (mPIN), where atypical cells within established glands become more pronounced over time, can progress to carcinoma[9,10]. There have been several proposed PIN grading schemes for SV40 and non-SV40 based models[21,22]. For example, Park et al proposed lesions of mPIN I-IV, where mPIN I lesions have one or two layers of atypical cells which progresses through mPIN IV lesions which fill the entire lumen and may bulge into the fibromuscular sheath, resulting in discontinuous smooth muscle actin (SMA) and laminin[22]. In contrast to human PIN grading, the authors did not use cytologic criteria (dysplasia), but instead use extent criteria. Nevertheless, the criteria are easily applied and therefore useful in reporting and comparing disease in GEM. The main distinction between epithelial hyperplasia and mPIN, is that mPIN arises in discrete foci (clonal expansions) and shows some evidence of nuclear atypia/dysplasia. Experimentally, it can be demonstrated to be progressive, while hyperplasia in GEM is more diffuse (poly or non-clonal) and does not progress. For a comparison of human PIN and mPIN, please see Figure 2.
II.B.3. Carcinoma
In human PCa, progression of HGPIN to carcinoma is defined by the presence of epithelial invasion into stroma. When HGPIN lesions progress to PCa, the majority (99%) of cases are adenocarcinoma, while the remaining 1% is comprised of NE or small cell carcinoma and even rarer subtypes such as sarcomatoid carcinoma, which are discussed in more depth by Humphrey[23]. Adenocarcinomas are an AD PCa and are graded using the Gleason pattern scale, in which a pathologist scores the level of disorganization (1 normal glandular structure to 5 very abnormal) of the primary/dominant area of tumor as well as any secondary areas of the tumor. The Gleason grade is the sum of these two scores and this is used in conjunction with the tumor stage for prognostic purposes. NE and sarcomatoid carcinomas have distinct histologic phenotypes, are often scored as Gleason pattern 5, and are usually androgen independent (AI) limiting the utility of hormonal therapy. In addition, they typically do not respond well to chemotherapy and radiation[24-26].
Unlike human PCa, where the majority of tumors are adenocarcinomas, GEM tumors exhibit adenocarcinomas, NE, and sarcomatoid carcinomas. Murine adenocarcinomas often have squamous or mucinous differentiation which are uncommon in human PCa[10]. Additionally, many aggressive GEM models progress to sarcomatoid carcinoma tumors[10], suggesting an inherent difference between human and mouse PCa progression. Some publications have referred to these tumors as adenocarcinomas which have undergone epithelial to mesenchymal transition (EMT)[27-30].
mPCa can be broken down into microinvasive and invasive carcinoma, which is determined by the amount of infiltration and destructive growth. Invasive carcinoma consists of a neoplastic proliferation outside of the ducts and may invade contiguous local structures within and beyond the prostate. Meanwhile microinvasive carcinoma is always seen in the setting of an mPIN lesion, but with individual cells or small nests of cells invading beyond the basal layer and basement membrane of the gland space. Both invasive and microinvasive mPCa is usually associated with reactive stroma changes, whereas human PCa typically elicits very little stromal reaction, at least when evaluated by histology alone[10]. It should be noted, however, that when reactive stroma is observed in human PCa patients by additional immunohistochemistry (IHC), it correlates with poor prognosis[31,32], and stroma derived from PCa patients (Carcinoma Associated Fibroblasts, CAF, [33]), can transform non-tumorigenic immortalized epithelial cell lines, clearly suggesting an important role for the stroma in human PCa. For a comparison of human and mouse PCa progression, see Figure 1.
II.B.4. Metastasis
About 15% of PCa patients present with metastatic disease, and 20-30% of patients treated with definitive local therapy will progress to metastatic disease[34]. Of these metastatic patients, 90% suffer from skeletal metastases which are largely osteoblastic (bone forming)[8] . Of the 90% of patients suffering from bone metastasis, this is the sole site of metastasis for 86% of them[35]. Conversely, NE tumors exhibit metastatic tropism to visceral organs (bladder, lymph nodes, liver, adrenal gland), brain, and spinal cord but also generate lytic bone lesions[25]. Patients presenting with NE PCa frequently present at an advanced stage with symptoms due to metastases.
There are limited GEM which develop distal metastasis. These metastatic lesions tend to occur in visceral organs, such as lymph nodes and lungs, and are largely NE and sarcomatoid carcinomas. Although GEM models of PCa can exhibit bone metastasis, the frequency is low and some descriptions were later found to be direct extension and invasion from the prostate rather than true hematogenous bone metastases[10,36-38]. The relative paucity in which GEM models of PCa develop distal metastases may be explained by the observation that prostate tumor burden can be extensive, thus not providing time for mice to develop overt metastases.
II.B.5. Castrate Resistant Prostate Cancer
Because PCa usually depends on androgen receptor (AR) signaling, advanced PCa is treated by ADT, and while successful for a time, nearly all patients relapse and progress to CRPC[39]. Although initially termed AI disease because it no longer responds to first generation ADT, subsequent studies of CRPC have revealed that AR signaling continues through a variety of mechanisms including increased AR expression, AR splice variants (ARv), ligand-independent activation, activating point mutations in the AR, and increased androgen and dihydrotestosterone (DHT) synthesis by the adrenal glands and by the tumor[6]. The continued dependence of CRPC tumors on AR signaling allows patients to respond to high affinity anti-androgens such as MDV3100 (enzalutamide)[40] and agents targeting the androgen biosynthesis pathway such as abiraterone[41]. Another method by which tumors can escape ADT is by undergoing NE differentiation, whereby cells acquire NE markers such synaptophysin and chromogranin. Focal NE differentiation is common in human PCa[7] and heavily ADT treated adenocarcinoma patients can develop frank NE tumors[24]. These patients therefore, have progressed from CRPC to true AI disease. For a comparison of NE and NE differentiation in human and mouse, see Figure 3. A much less common response to treatment is the progression to sarcomatoid carcinoma, but this is rare in human PCa patients[26].
Figure 3. Neuroendocrine features in the human and mouse prostate.
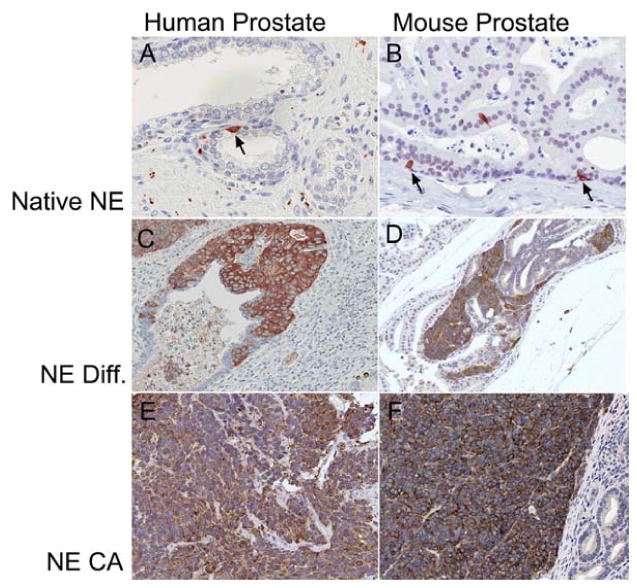
Synaptophysin staining denotes areas of NE features. In the normal human (A) and mouse (B) prostate, NE cells are rare. PCa in humans often has focal NE differentiation (C) and this is also seen in some of the mouse modes that initially appear to have adenocarcinomas (Pten;Δp53 mouse D). NE differentiation occurs in areas of clearly glandular differentiation without “small cell” features. Conversely, true NE tumors, usually with a characteristic solid growth pattern of “small cells” in both the human (E) and the TRAMP mouse (F) express synaptophysin globally in the tumor.
Modeling ADT in GEM can be achieved by murine orchiectomy (surgical castration) or treatment with pharmacological inhibitors of AR or androgen biosynthesis. In order to model CRPC in GEM, regression of the established tumor after ADT followed by subsequent regrowth/recurrence of the tumor should be demonstrated. It is important to note that commonly used promoters to drive transgene expression (such as the probasin [PB] and Nkx3.1 promoters) are themselves androgen regulated[42-46]. Therefore, tumor regression following ADT in mice utilizing these promoters should be interpreted cautiously. In addition, the mechanism that causes the outgrowth of the tumor following ADT is important. The ideal GEM model would, therefore, show progression to CRPC through mechanisms utilized by human PCa, such as continued AR signaling, NE differentiation, or the rare progression to AI disease via development of NE tumors. Although not every GEM model has reported castration studies, the majority of models tested progress to be AI, meaning they either never regress or rebound rapidly following castration by the development of AR negative NE or sarcomatoid carcinomas.
III. Experimental Considerations for GEM Studies
There are several considerations to make when selecting the appropriate mPCa model. First and foremost, the model must be representative of the disease state under examination. Models of PIN and early invasion, of which there are many, are appropriate for prevention and early detection studies of PCa. There are fewer models which progress to adenocarcinoma and metastasis, which can be used to examine novel therapies and metastasis promoters. Models of AI and CRPC are limited and some remain poorly defined. Therefore, picking a model can be challenging if the focus is metastatic or castrate resistant disease, where models are limited and penetrance is low. Again, selecting an appropriate promoter to drive the transgene or Cre is important. As previously mentioned, utilizing an androgen dependent promoter to drive transgene expression, such as one driven by PB, may not be appropriate for ADT studies. Another concern is the utility of the model. Ideally, a model should have high penetrance of phenotype, consistent latency and progression. Although the best model may include three genetic events driven by a prostate specific Cre, the breeding of four alleles into a single mouse is time consuming and requires a large breeding program, which is not practical for most investigators, particularly if the penetrance of the phenotype is limited.
Additionally, caution should be used in selecting the background of mouse strains for proposed studies. For example, microarray comparison of five commonly used mouse strains has demonstrated that 932 genes have strain-specific gene expression differences, which can account for varying tumor penetrance on different backgrounds[47]. These strain differences have been reported to result in dramatic differences incidence of NE tumors[48]. Also, some strains of mice develop spontaneous lung neoplasm which can be mistaken for metastases unless appropriate IHC analysis is performed[10]. Similarly, the method of gene deletion targeting can affect the result. Different groups have observed different results knocking out the same gene using the same Cre, and this may be attributable to strain differences, as well as excision of different exons during homologous recombination with Cre. These concerns will be discussed as appropriate in the sections below. In an attempt to aid investigators, select mouse models are summarized in Table 2, including the most severe phenotype, penetrance, and multiple reports, if available.
Table 2. Selected mouse models of PCa.
Mouse models were selected based on their usefulness to address specific aspects of PCa. Time, penetrance, and pathology are reported as in referenced manuscript. These and additional models are reviewed in greater detail within the review. Path: additional pathology reports published on the model, outside of the primary authors, if available. Abbreviations: C57: C57Bl/6; AEH atypical epithelial hyperplasia; PIN prostatic intraepithelial neoplasia; NE Neuroendocrine; NED NE differentiation; AD adenocarcinoma; SC sarcomatoid carcinoma; PI post induction; AI Androgen Independent; CRPC Castrate Resistant PCa; SQD: squamous differentiation CXT castration
| Model | Driver | Event | Time: Penetrance, Pathology | Ref | Path. | Model of: |
|---|---|---|---|---|---|---|
| TRAMP | PB | SV40 early region | 8-12 weeks: hyperplasia 18-24 weeks: PIN 28 weeks: AD 100% lymph, 67% lung met Castration: progress to 100% NE |
[38] [53] |
FVB: NE[48] C57: AEH [48] |
Visceral metastasis, NE (FVB), NED to NE following ADT (C57) |
| LADY | LPB | SV40 Tag | 10-15 weeks: dysplasia (12T-7f, s) 15-22 weeks: carcinoma 25-30 weeks: dysplasia (12T-10) 33 weeks: metastatic carcinoma 90% |
[57] | Visceral metastasis, NE | |
| 12T-7s; PB-Hepsin | 50% Metastatic carcinoma (17% bone metastasis) | [58] | NE bone mets [10] | NE, NE bone metastasis | ||
| 12T-7s; β-catenin | 18 weeks: AD with NED | [64] | AD & NED [10] | AD with NED | ||
| Pten+/- | Pten+/- | 6-30 weeks: hyperplasia, dysplasia 6-22 weeks: 62.5% hyperplasia, 37.5% PIN 40-65 weeks: 25% PIN, 6.25% AD |
[80] [82] [84] |
PIN to PCa progression | ||
| Pten+/-; Nkx3.1-/- | 26-52 weeks: 60% HGPIN 52 weeks: 100% HGPIN >52 weeks: 84% adenocarcinoma, 25% lymph node mets |
[89] [94] |
HGPIN to AD to metastasis progression | |||
| Pten flox/flox | PB-Cre4 | Pten flox/flox (exon 4,5) | 9 weeks: HGPIN 17-26 weeks: 100% carcinoma No metastasis up to 130 |
[83,1 02] | HGPIN to carcinoma | |
| Pten flox/flox (exon 5) | 4 weeks: hyperplasia 6 weeks: 100% PIN 9-29 weeks: 100% AD 12-29 weeks: 45% lung met Castration |
[95] | PCa progression, AI disease | |||
| Ptenflox/flox; p53flox/flox |
11 weeks: 100% AD 28 weeks: death, no mets |
[102] | SC [10,27] | SC (by 30 weeks) | ||
| Ptenflox/flox; Smad4flox/flox |
7 weeks: low-grade PIN 11 weeks: invasive PCa 15 weeks: highly aggressive PCa with stromal reaction 32 weeks: death (bladder obstruct) 100% lymph node, 12% lung mets |
[113] | AD[10] | PCa progression, visceral metastasis | ||
| Ptenflox/flox; p53flox/flox; Smad4flox/flox | 17 weeks: death 12.5% bone metastasis | [37] | Bone metastasis | |||
| Nkx3.1-Cre ERT2 | Ptenflox/flox | 26-39 weeks PI: PIN 39-52:PIN III, IV 25% >69 weeks: no progression Castration (17 weeks PI): initial recession >69 weeks: 100% microinvasive AD with areas of poorly differentiated AD, 14% squamous metaplasia |
[106, 107] | HGPIN AD[10] Post CXT: AD+ SQD [10] |
Compartment specific tumor cell of origin, CRPC | |
| CK14-CreERT2 CK8-CreERT2 |
Ptenflox/flox | 13 weeks PI: 36% PIN 26 weeks PI: 97% PIN 13 weeks PI: 100% PIN/early cancer 26 weeks PI: 100% PIN/AD |
[13] | PIN [10] AD[10] |
Compartment specific tumor cell of origin, AI | |
| Hi-Myc | ARR2PB | Myc | >13 weeks: PIN >26 weeks: AD |
[132] | AD[10] | PIN to AD |
| Myc, PB-hepsin | 13 weeks: PIN 20 weeks: 80% AD >26 weeks: 100% high grade AD |
[36] | PIN to AD | |||
| Myc, IκBa +/- | 26 weeks: 100% AD Castration: AR+ proliferating AD |
[134] | PCa progression to CRPC | |||
| Z-Myc | PB-Cre4 | z-Myc; Ptenflox/+; p53flox/flox | 10-24 weeks: AD with lymph node metastasis | [137] | PCa,all lobes [10] | PCa progression without NED, heterogenous PCa |
IV. Mouse Models of Prostate Cancer
IV.A. 1st generation mouse models: Oncogenic “sledgehammer” driven mouse models
Early modeling of mPCa focused on eliciting a tumor in the prostate, through whatever means necessary, including expression of ectopic oncogenes. The Simian Virus 40 (SV40) early region, which is comprised of large tumor T antigen (Tag) and small t antigen was the first oncogene targeted to the murine prostate for this purpose. Although Tag was originally identified to bind and inhibit the p53 and Rb tumor suppressors, while small t antigen binds the phosphatase pp2A, it is now recognized as targeting many other intracellular proteins involved in multiple aspects of transformation[49], making SV40 early region an oncogenic “sledgehammer.”
IV.A.1. TRAMP
The first reported GEM of PCa used the prostate steroid binding protein (C3-1) promoter driving the expression of the SV40 early region which resulted in prostatic and mammary gland adenocarcinoma and rare lung metastasis[50]. Since this C3-1 promoter was not specific for the prostate, a prostate specific construct was developed using the PB promoter[42]. Using the small PB promoter (-286/+28 b.p.) yielded variable expression of SV40 early region and varying phenotypes in the founder animals[51], but selecting a line with higher levels of transgene expression in the ventral and dorsal lobes, yielded the TRansgenic Adenocarcinoma Mouse Prostate (TRAMP) model. This model is characterized by rapid progression to prostatic neoplasia at 28 weeks, with 100% penetrance of lymph node metastasis and 67% pulmonary metastasis[38]. The authors also reported bone metastasis on the FVB background, but not the C57Bl/6[38], as well as differences in survival and tumor origin (dorsolateral v lateral, respectively)[21]. Further studies of TRAMP mice have revealed that the androgen-dependence of the tumors is variable, i.e. some animals (20%) show a dramatic loss of genitourinary (GU) volume following castration while others maintain a weight similar to or greater than intact transgenic animals[52]. Of the castrated TRAMP mice which progressed, their disease was poorly differentiated and more metastatic, as opposed to the better differentiated tumors in intact animals[52]. These AI tumors are 100% synaptophysin positive, and metastasis are 67% positive, suggesting these tumors are NE[53]. TRAMP mice also exhibit Phyllodes-like lesions with varying degrees depending on the mouse background[48,53]. These tumors are now recognized to be “small cell” or NE carcinomas[48]. Extensive analysis of TRAMP mice has also revealed that they harbor AR single base substitution within their tumors, and the incidence of AR mutation increases with castration[54]. As one of the first models, TRAMP has been utilized to validate genes involved in PCa progression[55], as well as chemopreventative approaches and novel therapeutics[56].
IV.A.2. LADY
In the LADY model, the large PB promoter (LPB) drives expression of SV40 Large T-antigen, Tag, (a deletion construct prevents the expression of the small t antigen). Although similar targeting strategies were employed to generate these models, due to the lack of small t antigen, the LADY is not a TRAMP. Amongst the eleven lines that were generated, phenotypes varied, likely due to variable transgene expression. The lines were termed 12 after the LPB fragment, estimated to be 12 kb, T for the Tag transgene, and numbered subsequently to 11. Collectively, these lines are referred to as LADY. The most aggressive mouse line developed from 12T-7 which had multiple transgenes on two chromosomes. Offspring of this founder were divided into the fast, 12T-7f, and slow, 12T-7s, growing lines. Each progressed to develop mPIN with limited local invasive adenocarcinoma by 15-22 weeks[57]. Although this line grows tumors very quickly, it never achieves metastatic disease in intact mice, so it is an excellent system to interrogate genes involved in promoting metastasis. To that end, the 12T-7s line, when crossed with PB-hepsin, drives metastasis to the liver, lung, and bone[58]. These metastasis are all NE cancers that affect 50% of the animals, 33% of which had bone metastases, resulting in a 17% penetrance of bone metastasis in the model overall[58].
Conversely, the mouse line with the slowest progression to dysplasia was the 12T-10 line[57]. The 12T-10 line faithfully recapitulates HGPIN with limited stromal involvement, as seen in human HGPIN and PCa, and then progresses from invasive NE carcinoma around 33 weeks of age and eventually with a 100% penetrance[59]. Although bone metastasis are rare, older 12T-10 mice (48-52 weeks) exhibit frequent metastasis to lymph nodes, lungs, and liver[59]. Studies using the LADY model have indicated that micronutrients (vitamin E, selenium, and lycopene) can reduce PCa incidence[60,61]. Coupling LADY with loss of Transforming growth factor β receptor type II (TGFβ RII), which is lost during PCa progression[62], drives more invasive mPCa and increases visceral metastasis[63]. All of these tumors are NE cancers.
Interestingly, activation of β-catenin in the 12T-7s shifts the pathology of the mPIN towards adenocarcinoma with focal NED without apparent NE cancer[64], suggesting that the Wnt/β-catenin pathway may control adenocarcinoma and NED development. β-catenin stabilization alone is sufficient to drive the formation of mPIN[65] . Other studies which have utilized PB-Cre4 to inactivated APC have yielded adenocarcinoma with areas of squamous metaplasia[66], which the PCC has classified as predominantly intracystic carcinoma[10].
IV.A.3. Considerations for utilizing the SV40 Tag models
The major advantage of all the Tag based models is they are easy to generate, have high penetrance, well characterized progression, develop metastases, and they have been used in numerous studies. However, there are several concerns with use of the SV40 Tag models, perhaps most obviously that Tag is an exogenous oncogene which does not exist in human PCa. However, Zhou et al have shown that simultaneous PB-Cre4 deletion of p53 and Rb results in highly aggressive and metastatic carcinomas, which are AI and express NE markers[67], whereas inactivation of Rb alone via a modified Tag fragment yields only microinvasive adenocarcinoma[68]. This may be due to the timing of tumor suppressor inactivation. Meanwhile, Vinall et al showed that heterozygous knock-in of a common tumor associated p53 mutation (R270H) was sufficient to generate HGmPIN and these sometimes progressed to non-NE sarcomatoid carcinomas. The authors proposed that the mutant p53 was both dominant negative and also might have a gain of function[30]. In human PCa, loss of Rb is believed to occur early in 60% of patients[69], while the loss of p53 by mutation or deletion occurs in 20-40% of more advanced and metastatic disease[70,71]. Thus, the Tag models recapitulate the loss of Rb/p53 seen in human PCa but these mouse models develop AR negative NE cancers rather than the AR positive adenocarcinoma seen in humans.
Another concern is that the Tag models mainly produce NE cancer and other carcinoma subtypes, which are rare in the human population. For example, in TRAMP, subsequent studies have shown that the FVB background produces NE cancer, while only 20% of the BL/6 produce NE tumors[48]. Castration of TRAMP animals on a mixed background results in 100% progression to NE tumors[53]. Others have suggested, following extensive analysis of TRAMP tumors on both backgrounds, that TRAMP tumors do not meet the criteria for adenocarcinoma, and instead these were classified as atypical epithelial hyperplasia[48]. The positive aspect of these observations, however, is that patients do undergo NE differentiation as they progress to CRPC and some CRPC patients will develop NE tumors[24,25]. These clinical results should bring renewed interest in mouse models that develop NE cancer. Therefore, TRAMP studies may identify novel pathways critical for these late-stage tumors. Additionally, accepting that TRAMP produces mice with mostly NE tumors allowed Qi et al to observe that loss of Siah2 prevented formation of NE tumors and instead maintained them as atypical epithelial hyperplasia[72].
IV.A.4. Development of prostate specific Cre recombinases
Early studies of prostate tumorigenesis in the mouse were severely limited by the lack of a prostate-specific promoter to “drive” the expression of Cre recombinase, relying instead on global knockouts. Many genes of interest in PCa are either embryonic lethal or cause other neoplasias, which result in mouse death prior to development of PCa. Moreover, any prostate phenotype in a global knockout mouse should be interpreted with caution, as it may be representative of a developmental phenotype. The identification of rat PB as an androgen responsive prostate specific promoter[42,45,73] provided the groundwork to generate the first prostate specific Cre, PB-Cre[74]. However, the low expression of PB prompted the subsequent refinement of PB into ARR2PB[46], resulting in the development of the PB-Cre4 mouse, which expresses high levels of Cre in the luminal cells of the mouse prostate[75].
IV.B. 2nd generation mouse models: Models based on human genetic lesions
Following the successes and limitations of the Tag mouse models and the advent of PB-Cre4, researchers began targeting genetic lesions observed in human PCa patients to murine prostates to develop new and more relevant mouse models of PCa. These new models incorporate single or multiple genetic losses and gains, as well as traditional and novel Cre recombinase targeting strategies.
IV.B.1. Pten
Phosphatase and tensin homolog deleted on chromosome ten (PTEN) is frequently lost in a variety of human cancers, and it is considered a significant tumor suppressor [76]. The primary role of PTEN is to de-phosphorylate phosphatidylinositol-3,4,5-trisphosphate (PtdIns(3,4,5) P3), the accumulation of which activates AKT and PDK1, resulting in cell proliferation and survival[77]. Extensive research efforts by numerous groups have demonstrated that PTEN plays numerous roles, including supporting cell metabolism, polarity, motility, cancer “stem-ness,” and stromal-epithelial interactions[76]. In human PCa, PTEN deletions occur in approximately 23% of HGPIN, 69% of localized PCa[78], and 86% of metastatic CRPC[79].
IV.B.1.a. Pten heterozygous knockout studies
The homozygous knockout of Pten in the mouse is embryonic lethal approximately at embryonic day (ED) 3.5-9.5 [80-82]. Heterozygous Pten knockout (Pten+/-) mice develop lymphomas, dysplastic intestinal polyps, endometrial complex atypical hyperplasia, PIN, and thyroid neoplasms, but some tumors (skin, breast, brain) commonly associated with PTEN deletion in humans are largely absent from these mice[82]. Considering the variability in the onset of embryonic lethality, it is perhaps unsurprising that Pten+/- mice have a spectrum of prostate phenotypes. Di Cristifano et al reported seeing that 70% of mice had hyperplasia and dysplasia between 6-30 weeks[80]. Lowering Pten expression even further by introducing a hypomorphic allele (Pten+/hyp) was reported to promote the progression from hyperplasia to PIN as compared to the Pten+/- controls[83]. Podsypanina noted hyperplasia in 62.5% of mice and PIN in 37.5% at 6-22 weeks with invasion rarely seen[82], while Stambolic reported that in older mice aged 40-65 weeks, the most common histology was PIN (25%), with one case of adenocarcinoma (6.25%)[84].
IV.B.1.b. Applications of the Pten heterozygous knockout
Due to the slow progression of PCa in Pten+/- against a background of severe health issues, some of these studies combined Pten+/- mice with other genes lost in PCa to accelerate disease progression. For example, decreased expression of p27Kip1 in human PCa correlates with increased recurrence following radical prostatectomy, independent of stage and grade[85], but p27Kip1 loss in a mouse results in only prostate hyperplasia[86]. Once coupled with a heterozygous Pten deletion, however, these mice rapidly (13-22 weeks with 100% penetrance versus 39-69 weeks with 50% penetrance ) develop PIN and about 25% of mice develop invasive PCa[87]. It should be noted that all Pten +/-; p27Kip1 -/- die by 22 weeks due to intestinal occlusions[87].
Similarly, NKX3.1 plays a critical role in urogenital development and function[43,88] and is frequently lost early in human PIN and PCa samples[89,90]. However, Nkx3.1 loss alone is not sufficient to induce PCa or HGPIN in mice. Nkx3.1-/- mice develop hyperplasia and some dysplasia[43,44,91,92], while mice older than a year or generated by a PSA-Cre driven conditional knockout develop PIN[92,93]. Combining Nkx3.1-/- with the Pten+/- accelerated the incidence and progression to HGPIN/early carcinoma with 60% of Pten+/-; Nkx3.1-/- mice exhibiting HGPIN at 26 weeks versus 10% of the Pten+/- with 100% penetrance of HGPIN by 52 weeks[89]. Surgical castration of these animals at 24 weeks resulted in partial regression of the lesions and decreased expression of AR[94]. Later studies would determine that aging Pten+/-; Nkx3.1+/- mice beyond a year allowed the HGPIN lesions to progress to invasive adenocarcinoma in 84% of the animals examined[94].
IV.B.1.c. Targeted deletion of Pten
Due to the health problems afflicting the Pten+/- mice and the embryonic lethality of Pten KO mice, subsequent studies have focused on targeting Pten deletion in an organ-specific manner. Two separate groups generated conditional Pten knockout mice, which have yielded different results. The targeting vector used by Trotman et al excised exons 4 and 5[83], while Wang et al targeted exon 5 on mice of a different background[95]. Wang et al reported hyperplasia at 4 weeks, PIN at 6 weeks, and full adenocarcinoma with 100% penetrance at 9-29 weeks[95]. Mice castrated at 16 weeks, when adenocarcinomas are established, respond to surgical castration, as observed by an increase in apoptosis[95]. In animals aged 11-17 weeks with established adenocarcinoma, castration did extend the survival time of castrated versus intact animals, however castrated animals maintained prostates 5-10 fold larger than WT controls after 11 weeks of castration. In these castrated animals, IHC AR levels were diminished[95], contrary to human CRPC[96-98]. Decreased levels of AR are consistent with other studies with castrated Pten-driven tumors[99,100], although some did observe increased NE differentiation[99]. Subsequent studies would determine that castration or genetic ablation of the AR at any age in the Ptenflox/flox does not prevent tumor formation[101], suggesting that these tumors are AI from the start. Additionally, Wang et al also observed metastasis to the lymph nodes and lung at 12-29 weeks in 45% of the animals[95], as did an additional group employing the PB-Cre4[99].
While Trotman et al had also observed invasive PCa by 42 weeks, the neoplastic lesions observed in the lungs were not consistent morphologically or molecularly with PCa metastases[83]. Additionally, the studies of Trotman et al demonstrated that Pten dose was critically important for tumor promotion, where a mouse with one allele of Pten deleted had a slower progression to PCa than a mouse with a Pten hypomorphic allele; which in turn exhibited a slower progression to invasive carcinoma compared to a mouse which had both alleles of Pten deleted in the prostate[83]. Further studies utilizing the same Ptenflox/flox mouse, demonstrated disease progression from HGPIN at 9 weeks to invasive cancer at 28-42 weeks, but the mice never developed metastatic disease even up to 130 weeks of aging[102]. The conditional knockout of Pten on a mixed strain background consistently progresses to invasive cancer, although the penetrance and timeframe vary between different studies [83,95,99,100,102,103]. Moreover, the development of metastatic lesions has only been reported in two studies, highlighting the critical importance of age-matched littermate controls for all experiments. Interestingly, a constitutive activation of Akt in the prostate only results in PIN and does not progress at any time point examined[104], confirming that Pten loss has additional consequences beyond constitutive activation of AKT.
IV.B.1.d. Applications of the conditional Pten knockout model
Pten conditional knockout mice have been used for several application-based studies, which generally have focused on determining if a specific gene of interest is involved in promoting or repressing mPCa progression. Some of these studies have generated additional and improved PCa mouse models, while others have focused on molecular events of particular interest in human PCa.
Alternative promoters
Several studies have focused on driving Pten loss with alternate promoters. Driving Pten loss with the human PSA promoter results in 100% incidence of adenocarcinoma and carcinoma by 56 weeks[103,105]. This is a much slower progression to adenocarcinoma than the PB-Cre4 driven Pten loss, and is likely due to the lower levels of transgene expression from the PSA promoter[103]. An inducible Pten knockout has also been developed by crossing the Ptenflox/flox animal with the Nkx3.1-Cre ERT2 mouse[106], which yields temporal control over Pten loss, while additionally ablating one allele of Nkx3.1, as Cre recombinase was “knocked-in” to the endogenous Nkx3.1 locus. These animals, following inductions with tamoxifen at 2 months, slowly develop HGPIN (PIN III, IV) with microinvasion[107]. This model is perhaps the best model of CRPC in mice because following castration, tumors regress, but then continue to progress to microinvasive adenocarcinoma while maintaining nuclear AR[107]. This study suggests AR-mediated signaling remains active in these mice, much as it does in human patients[96]. Combining castration (a proxy for ADT) with clinically used inhibitors of AKT (MK2206) and mTOR (MK8669) significantly reduced tumor burden in these mice, suggesting this combination may be useful in human patients[107]. Interestingly, the authors note some animal tumors exhibit squamous metaplasia and carcinoma[107], which has been observed in other Pten models as well. An activated BRAF mutant (BRAFV600E)[108] bred into this model decreased the time required to develop adenocarcinoma and promoted metastasis to lymph nodes and lung but made these tumors castrate-resistant (AR levels following castrations were not shown)[109]. Analysis of animals by the PPC determined that after 30 weeks following tamoxifen induction these tumors had progressed to sarcomatoid carcinomas[10]. Subsequent studies by Aytes et al have also coupled Nkx3.1-CreERT2; Ptenflox/flox mice with an oncogenic Ras (KrasLSL-G12D/+)[110]. Nkx3.1-Cre ERT2;Ptenflox/flox;KrasLSL-G12D/+ mice also develop aggressive adenocarcinomas with metastasis (100% penetrance with metastasis to mostly lung and liver) [110] with noted focal intestinal metaplasia[10]. Although there were no bone metastases observed in these compound mice, disseminated tumor cells were isolated from the bone marrow[110], suggesting that these tumor cells are capable of homing to bone.
Another promoter strategy has been designed to determine the tumor cell of origin by targeting deletions to the luminal or basal compartment. In the mouse, basal cells give rise to luminal and neuroendocrine cells during prostatic development[11], but in the adult mouse, basal to luminal cell differentiation was reported not to exist[12,13]. However, recent reports have demonstrated that basal to luminal cell differentiation is a slow and rare event[14] mediated by rare bipotential progenitors[15]. Nevertheless, both basal and luminal cells have been reported to be the tumor cell of origin for PCa[13,106,111]. Up until the report by Choi et al, all of the Pten deletions in the prostate were targeted to the luminal compartment. Indeed, deletion of Pten by an inducible basal CK14-CreERT2 or luminal CK8-CreERT2 both gave rise to PIN and adenocarcinoma[13]. Subsequent studies by Lu et al demonstrated that basal-derived Pten knockout lesions progressed more rapidly and were more aggressive than luminal-derived lesions[14]. Interestingly, both basal and luminal derived lesions continued to grow under castrated conditions, and demonstrated loss of AR expression, particularly notable in the basal derived tumors[14]. Conversely, other groups have reported that the luminal-derived tumors (Nkx3.1-CreERT2 and CK8-Cre ERT2) progress more rapidly[13,15], which raises additional questions about the contributions of mouse background and Cre penetrance to observed phenotypes. The neuroendocrine compartment can also give rise to PCa as exhibited by targeting SV40 T antigen expression with the cryptidin-2 promoter[16,17].
Additional gene alterations
Another interesting approach has been to complement previous observations from human PCa and apply them to this model. Based on the success of the Tag model, an obvious gene of interest was Trp53, which encodes the tumor suppressor p53. While tissue specific knockout of p53 results in no observable phenotype before or after 75 weeks, coupling it with a tissue specific Pten knockout results in very aggressive PCa with onset at 11 weeks and 100% penetrance of invasive adenocarcinoma resulting in death by 28 weeks with no observed metastasis[102]. The progression of these mice, described extensively by Martin et al, demonstrated that the adenocarcinomas progressed to sarcomatoid carcinomas, and occasionally there were areas of basal carcinoma and prostatic urothelial carcinoma[27]. The PPC, following examination of slides from Chen et al, also found the 30 week animals to display feature of a sarcomatoid carcinoma[10].
Interestingly, coupling the PB-Cre4 Ptenflox/flox; p53flox/flox mouse with telomerase loss then reactivation results in lethal aggressive adenocarcinoma with bone metastasis observed in 25% of the mice by 24 weeks of age[37]. Upon examination of samples provided by Ding et al, the PPC determined that these tumors represented adenocarcinomas and sarcomatoid carcinomas, with the sarcomatoid component leading to bone invasion[10]. Regardless, examination of the metastatic samples indicated that 11% had lost Smad4[37], which is lost during PCa progression mostly by promoter methylation[112]. It was previously shown that Smad4 knockout on the background of a prostate specific Pten deletion drives progression to invasive disease by 15 weeks of age with observed lymph node and lung metastasis (100% and 12%, respectively)[113]. In fact, when Ding et al generated a triple prostate specific knockout of Pten, Smad4, and p53, the survival time dropped to 17 weeks and had 12.5% mice with bone metastasis[37].
A relatively recent discovery in human PCa has been the gene fusion event between the androgen-regulated TMPRSS2 promoter and ETS family members (ETV1, ERG, ETV4)[114-116]. Interestingly, PB driven ERG results in PIN[117], as does ARR2PB driven over-expression of ETV1 or ERG[116,118] or no phenotype over the lifetime of the mouse[119]. Similarly, driving TMPRSS2-ERG with ARR2PB promoter yielded no histological phenotype up to 60 weeks[120], and similar negative results were reported following “knocking-in” the ETV1 or EGR into the TMPRSS2 locus[121]. However, when ERG is over-expressed on a Pten+/- background, within 26 weeks, all mice develop invasive PCa[119]. Combining the ERG or ETV1 knock in into the TMPRESS2 locus with the PB-Cre4 Ptenflox/flox mouse yields invasive adenocarcinoma or invasive carcinoma with lymph node metastasis, respectively[121]. This is particularly relevant to human PCa because patient samples frequently have both the TMPRSS2-ERG gene fusion and PTEN loss[119,120], and patients who lack these genetic alternations have a diminished chance of biochemical failure following ADT[122].
Other successful combinations of Pten loss include pairing it with over-expression of fibroblast growth factor 8, isoform b (FGF8b) so that the bigenic animals progress from adenocarcinoma to lymph node metastasis[123]. Loss of Gata3 coupled with Pten loss results in decreased time to invasion versus Pten loss alone[124]. Over-expression of Bmi1 with a Pten haplo-insufficiency drives invasive adenocarcinoma[125]. Coupling PB-Cre4 Ptenflox/flox with an activated K-RAS (K-rasG12D/WT)[126], which alone cannot induce PCa, results in rapid progression to carcinoma with heterogeneous AR expression, reduced AR target gene expression, and 100% penetrance of lung and liver metastasis[29]. Interestingly, examination of bone marrow aspirates from these mice revealed cells expressing PB-Cre4, Pten deletion, and the activated K-Ras, however neither imaging nor histology studies were able to observe bone metastasis[29], suggesting that if these PCa cells are targeting the bone, they are failing to colonize successfully.
Novel drug therapies and imaging studies
Studies utilizing the well characterized Pten model have provided translational and pre-clinical data which one day may inform future approaches to treating PCa. For example, treating conditional Pten knockout mice with MDV3100 and PI3 kinase inhibitors in combination with surgical castration significantly reduced tumor volume[127]. Studies with the Pten mouse also has revealed that concomitant targeting of AR and mTOR with Rapamycin may one day be a successful targeting strategy for androgen dependent disease in humans[100]. In addition, the Pten model has been used for imaging studies[100] and bioluminescent imaging studies (BLI)[99,128].
IV.B.1.e. Concerns with Pten knockout model
The phenotypes reported in the Pten knockout model are variable in nature, and these variations may be explained by the use of differing targeting vectors as well as by differing strain background of mice utilized. Indeed, when the PB-Cre4 and Ptenflox/flox[95] mice are back crossed into a C57/Bl6 background, these mice do not progress beyond mouse PIN with microinvasion and castration does not result in continued proliferation, relapse, or NE differentiation[128]. Some studies of the PB-Cre4 Ptenflox/flox mice have observed metastasis, while others have failed to do so. It is advisable that this model be employed in studies designed to identify metastasis promoting pathways, as opposed to therapeutic studies designed to inhibit disease progression. Additionally, the histology of the Pten model has been problematic with reports of squamous differentiation, mucinous metaplasia, and outright sarcomatoid carcinoma in some models[10]. Similarly, based on the studies of Wang et al and Mulholland et al, PB-Cre4; Ptenflox/flox tumors are AI[95,101]. Therefore, while there are aspects of PCa progression which these Pten-based compound models recapitulate faithfully, most of them fail to achieve CRPC and bone lesions, which are the major clinical concerns for advanced PCa patients.
IV.B.2. Myc
C-Myc is a proto-oncogene which plays numerous roles in the cell cytoplasm and nucleus. Myc is over-expressed at an mRNA and protein level in human PCa samples and Myc over-expression increases with increasing tumor stage in PCa (76% positive at PIN, 82% at carcinoma), suggesting that Myc over-expression is an early event in human PCa[129,130]. Myc over-expression can happen through a variety of mechanisms, including gene amplification, rearrangement, activation of the Wnt/β-catenin pathway, loss of FOXP3, and germline Myc promoter variation[131]. Targeting Myc over-expression to the mouse prostate, therefore, is a valid model of human PCa.
IV.B.2.a. Prostate-specific Myc over-expression
In terms of mouse modeling, the first studies to evaluate the consequence of Myc over-expression in the mouse prostate were performed by Ellwood-Yen et al. In these studies, Myc was driven by the small PB promoter that gives weak expression or the more potent ARR2PB promoter, yielding the Lo-Myc and Hi-My mice, respectively[132]. Hi-Myc mice progress from mouse PIN at 13 weeks to adenocarcinoma with invasion by 26 weeks, while Lo-Myc mice progress slower with both PIN and adenocarcinoma onset being delayed by about 30 weeks, consistent with lower transgene expression[132]. With the expression of the transgene and onset of PIN, Nkx3.1 expression decreases[133], recapitulating the loss of Nkx3.1 observed in human PIN cases. Hi-Myc PIN or adenocarcinoma lesions regress in response to castration but they do not become CR. This highlights a concern of using an androgen regulated promoter to drive the transgene for castration studies. Neither of these mouse strains gives rise to metastatic disease[132].
IV.B.2.b. Applications of the Myc model
The majority of studies have coupled the Hi-Myc model with other genes of interest in human PCa. For example, coupling the Hi-Myc model with the PB-hepsin mouse accelerated progression to adenocarcinoma by 12 weeks versus 24 weeks[36]. Interestingly, constitutive activation of the NF-κB pathway in the Hi-Myc model results in adenocarcinoma which is no longer sensitive to castration, suggesting that NF-κB may play a role in CRPC[134]. In this case, NF-κB continues to drive AR activity and maintain AR target gene expression, including the PB-Myc.
The Hi-Myc model has also been combined with the Pten pathway by generating bigenic animals over-expressing Myc and activated AKT or ablated Pten. In human PCa samples, Myc amplifications associate with alterations in the PI3 kinase pathway[135], and prostate specific knockout of Pten or activation of Akt coupled with the Hi-Myc model accelerates the progression to adenocarcinoma (13 weeks and 16-20 weeks, respectively)[135]. The PPC does note there is a component of intestinal metaplasia in this model as well[10]. Interestingly, the over-expression of Myc in these animals rendered mTOR inhibition ineffective, suggesting mTOR inhibitors may be contraindicated in PCa patients that exhibit Myc overexpression[135].
In an attempt to divorce Myc oncogene overexpression from the control of the androgen dependence of the ARR2PB promoter fragment, Kim et al employed an alternative targeting strategy designed by Roh et al. Roh et al designed the Z-Myc mouse, which expresses LacZ and maintains the Myc transgene silent, until recombination occurs[136]. Combining the Z-Myc transgene with PB-Cre4; Ptenflox/+; p53flox/+ results in invasive PCa in all lobes of the mouse prostate with an overall penetrance of 100% in animals aged 33-46 weeks[137]. Interestingly, Z-Myc; Ptenflox/+; p53flox/flox have highly aggressive adenocarcinoma starting at 10 weeks with lymph node metastasis, while a similar geneotype, c-Myc; Ptenflox/flox; p53flox/+ develops adenocarcinoma only[137]. These studies also showed that Pten was lost earlier than p53 and that the lesions are heterogeneous, much as is the case in human PCa[137].
IV.B.2.c. Concerns with Myc model
The major concern with the Myc models is that they mostly do not continue to progress following castration or develop metastasis. However, due to the early onset of Myc amplification during PCa progression[129-131], this is perhaps not surprising, and poises the Myc model to be an excellent starting point for additional genetic alterations which drive PCa progression. Additionally, the oncogenic activation strategy in the Z-Myc mouse[136] is an excellent approach towards avoiding the pitfalls of using AR-dependent (yet prostate-specific) promoters during castration studies.
Additional GEM
Unfortunately, not all GEM yield mPCa or mPIN, and this sections will cover some of these models because they make powerful statements about the biology of mouse tumorigenesis that can extrapolated to human PCa. For an extensive review of additional mouse models please see[9,10,138-140].
IV.B.3. The role of AR in PCa
The functional role of AR in human PCa is complicated. By and large, AR is a marker for (and major determinant of) the acquisition of a mature and differentiated luminal epithelium in the prostate. However, aberrations of AR expression, such as expression of AR variants (ARv), and changes in cofactor binding are common in PCa and contribute to the ability of PCa to escape conventional ADT[6]. Conversely, tumors which are truly AI, such as NE carcinoma, are associated with poor clinical outcome[141] because they often do not express AR and therefore are not treated with ADT[25]. Although pure NE tumors are rare, focal NED is common in human PCa and heavily ADT treated adenocarcinoma patients can develop NE tumors[7]. Our work has shown that NE secretions from the mouse prostate NE tumor, NE-10 (established from the Lady 12-T10 line[57]), can support the growth of LNCaP xenografts in castrated mice, indicating that secreted factors by the NE cancer can support continued AR signaling in CRPC[142]. The NE secreted proteins bombasin and gastrin releasing peptide were identified as two of these secreted factors and shown to activate the NF-κB pathway in the LNCaP tumors[142]. Since the constitutive activation of the NF-κB pathway in the Hi-Myc model results in adenocarcinoma which is CRPC, this suggests that NE secretions activate the NF-κB to induce CRPC[134].
IV.B.3.a. AR in GEM
There have been several GEM models which alter AR signaling in the mouse prostate. The first simply over-expressed AR under the control of the PB promoter and resulted in mPIN in mice older than a 52 weeks[143], suggesting that higher levels of AR activity alone are not sufficient to drive invasion. Similarly, expression of human AR with naturally occurring N terminal glutamine polymorphisms resulted in altered AR-target gene expression but no prostate phenotype was observed[144]. Targeting human AR over-expression to the mouse prostate using the Osr1 promoter, however, results in mPIN in 50% of animals and adenocarcinoma in 5% of animals by 52 weeks of age[145]. Due to the targeting strategy utilizing a prostate-specific but constitutively active promoter, the expression of the transgene (here, AR) remains constant in intact or castrated animals, suggesting this may be an appropriate model or targeting strategy for modeling CRPC in mice[145]. Very recent studies have generated the first GEM with an ARv. Liu et al coupled ARv567es, which occurs frequently in human CRPC and is missing exons 5, 6, and 7[146,147], with the AAR2PB promoter[148]. These mice progress from hyperplasia (16-20 weeks), to mPIN (30-40 weeks), and adenocarcinoma (52 weeks)[148]. These mice respond to castration at 16 weeks by maintaining nuclear ARv567es expression and losing full length AR nuclear localization, while mice castrated at a year developed more aggressive adenocarcinoma than their sham-castrated counterparts, suggesting this may be a novel model of CRPC[148].
IV.B.4. The role of stroma in PCa
GEM models of mPCa frequently exhibit reactive stroma, as exhibited by a dramatic increase in stromal proliferation adjacent to PIN and cancer. In humans, reactive stroma in PCa is less histologically obvious[10]. However, human PCa patients with confirmed reactive stroma have poor clinical outcome[31,32], and CAFs isolated from human PCa patients can support transformation of immortalized cell lines in tissue recombination experiments[33]. There are several stromal factors which have been shown to contribute to PCa progression, including AR and Transforming Growth Factor B (TGFβ).
IV.B.4.a. Models of stromal PCa tumorigenesis
Stromal AR expression plays a critical role in the development of the prostate[149] and supports PCa progression[150]. It is therefore not surprising that loss of AR in the stroma represses PIN formation in Pten+/- mice[151]. Conversely, activation of TGFβ Receptor II (TGFβ RII) in GEM stroma is sufficient to drive the formation of mPIN in the epithelium[152] and eventually adenocarcinoma[153]. These two GEM stromal models highlight the importance of the stroma on tumor development and progression. The majority of the work in stromal tumorigenesis, however, has focused on human-derived cell lines, CAFs, and tissue recombination assays, described here[33,150,154] .
V. Discussion
The past fifteen years of GEM modeling of PCa has revealed that even if the molecular changes observed in human PCa are precisely engineered in the mouse, the mPCa generated differ both in phenotype and behavior from human PCa. GEM mPCa models tend to progress spontaneously from adenocarcinoma to NE carcinomas and sarcomatoid carcinomas whereas these changes are most common in human PCa patients after therapy and recurrence [25,26]. What are the mechanisms for spontaneous NE or sarcomatoid/EMT differentiation in GEM and are these the same as the mechanisms for therapeutic resistance and disease recurrence in men with PCa? Although NE differentiation and the development of NE carcinoma following PCa treatment have both been reported previously[7,24,25], there is renewed interest in these observations due to the sequential analysis of PCa progression in humans. Genetic analysis of NE tumors by Beltran et al revealed 40% of NE tumors harbor amplifications and over-expression of Aurora kinase and N-Myc[155]. More recent studies performed have demonstrated that 60% of hormone-naïve PCa tumors, which will later progress to NE tumors, already harbor these genetic changes[156]. Conversely, these amplifications only occur in 5% of the entire PCa population[156]. Perhaps the most significant observation from these studies is that there was 100% concordance between ERG rearrangement and Aurora kinase in the NE metastatic samples, suggesting that adenocarcinoma gives rise to the NE tumor component[156]. Because of the success of second generation anti-androgens, it is likely that more NED and NE tumors will be observed in humans. Therefore, due to the progression of TRAMP and LADY 12T-10 line to a NE phenotype, these models may be appropriate for studying NE differentiation in human PCa.
There are other critical pre-clinical questions which GEM models have yet to address in detail. One of the methods by which PCa becomes castrate-resistant is by the expression of ARvs. Although there are TRAMP AR mutants, these are single base substitutions, and not large deletions or rearrangements[54]. Murine ARvs have been reported in the Myc-CaP cell line[157], but to date these mutations have not been translated into a GEM. The translation of human ARvs into GEM has only recently occurred with Liu et al generating an AAR2PB driven ARv567es mouse [148], and subsequent studies utilizing this model coupled with other genetic lesions, such as Pten loss or Myc over-expression, will address the role of ARvs in PCa progression to CRPC in GEM, and perhaps generate a more faithful model of PCa progression.
Similarly, the failure of our GEM models of PCa to metastasize to bone is striking. In human PCa patients, 90% of PCa metastases occur in bone[35], and the majority have strong expression of nuclear AR[158]. In the limited GEM models that develop metastatic bone lesions, the penetrance varies from “rare” up to about 17%. Moreover, these bone lesions tend to be NE or sarcomatoid in nature. The possibilities that account for this could be as simple as different bone environments between humans and mice or differing levels of circulating androgens.
As we are entering the 3rd generation of GEM models of PCa, new tools and ideas are being applied to established models. Application of basal versus luminal promoters, as described by Choi et al[13], may provide additional insights into compartment-specific drivers of human PCa progression. Focusing on global changes in the cell, such as reactivation of telomerase, changes in epigenetic regulation, and the role of stroma in PCa progression are also becoming more prominent in GEM models. Finally, the ability to activate oncogenes in a prostate-specific manner but independent of androgen may yield better models of CRPC[136,145]. The next generations of mouse PCa models, therefore, will continue to incorporate the genetic alterations observed in human PCa patients, but should also include the global, environmental, and dietary considerations of human PCa.
Acknowledgments
When selecting GEM models for this review, we have focused on selecting models which have been well-characterized, frequently used, and of particular use to the PCa research field. Additionally, we have included certain models for their particular significance in utilizing novel human PCa lesions or unique approaches to tumorigenesis. Our hope is that this review will serve as a tool in selecting which model may be appropriate for a given research study. We would like to thank all researchers whose work we have cited, those whose contributions were omitted in the interest of space constraints, and those whose omission was unintentional. The brevity of this review is greatly attributable to the excellent and exhaustive reviews on mouse models[55,138-140], mouse PCa pathology[9,10], and human PCa progression[139] published previously.
MMG was supported by the VUMC Integrated Biological Systems Training in Oncology training grant (2 T32 CA119925-06). DJD was supported by an NIH Pathway to Independence award (1 K99 CA172122), the VUMC Multidisciplinary Training Grant in Molecular Endocrinology (5 T32 DK007563-21), the VUMC Integrated Biological Systems Training in Oncology training grant (1 T32 CA119925), and the American Cancer Society Great Lakes Division-Michigan Cancer Research Fund Postdoctoral Fellowship. XY was supported by Department of Defense (DOD) PC111074. RJJ was supported by the DOD Prostate Cancer Research Program (PCRP) (W81XWH-10-1-0236). ZC was supported by the National Institute on Minority Health and Health Disparities (NIMHD) grants MD 5 R01 004038, G12 MD 007586, and 5 U54 CA 163069. AB was supported by NCI (U01 CA141582), the National Center for Research Resources (K26 RR024037) and by a grant from the University of California Davis, School of Medicine, Department of Pathology, Advisory Research Committee. RJM was supported by the NCI (R0-CA076142-14) and NIDDK (R01-DK055748-13).
Contributor Information
Magdalena M. Grabowska, Email: magda.grabowska@vanderbilt.edu.
David J. DeGraff, Email: david.degraff@vanderbilt.edu.
Xiuping Yu, Email: xiuping.yu@vanderbilt.edu.
Ren Jie Jin, Email: renjie.jin@vanderbilt.edu.
Zhenbang Chen, Email: zchen@mmc.edu.
Alexander D. Borowsky, Email: adborowsky@ucdavis.edu.
Robert J. Matusik, Email: robert.matusik@vanderbilt.edu.
References
- 1.Howlader N, Noone A, Krapcho M, Garshell J, Neyman N, Altekruse S, Kosary C, Yu M, Ruhl J, Tatalovich Z, Cho H, Mariotto A, Lewis D, Chen H, Feuer E. National Cancer Institute; [accessed June 2, 2013]. SEER Cancer Statistics Review, 1975-2010. Available from URL: http://seer.cancer.gov/csr/1975_2010/ [Google Scholar]
- 2.(SEER) SEaER: SEER Stat Fact Sheets: Prostate. [accessed May, 2013]; [Google Scholar]
- 3.Miller AB. New Data on Prostate-Cancer Mortality after PSA Screening. N Engl J Med. 2012;366:1047–48. doi: 10.1056/NEJMe1200185. [DOI] [PubMed] [Google Scholar]
- 4.Stanford J, Stephenson R, Coyle L, Cerhan J, Correa R, Eley J, Gilliland F, Hankey B, Kolonel L, Kosary C, Ross R, Severson R. Prostate Cancer Trends, 1973-1995. Bethesda, MD: SEER Program, National Cancer Instittue; 1999. Survival. [Google Scholar]
- 5.Suwa T, Nyska A, Haseman JK, Mahler JF, Maronpot RR. Spontaneous Lesions in Control B6C3F1 Mice and Recommended Sectioning of Male Accessory Sex Organs. Toxicol Pathol. 2002;30:228–34. doi: 10.1080/019262302753559560. [DOI] [PubMed] [Google Scholar]
- 6.Vis AN, Schröder FH. Key targets of hormonal treatment of prostate cancer. Part 1: the androgen receptor and steroidogenic pathways. BJU Int. 2009;104:438–48. doi: 10.1111/j.1464-410X.2009.08695.x. [DOI] [PubMed] [Google Scholar]
- 7.Sun Y, Niu J, Huang J. Neuroendocrine differentiation in prostate cancer. American Journal of Translation Research. 2009;1:148–62. [PMC free article] [PubMed] [Google Scholar]
- 8.Morrissey C, Vessella RL. The role of tumor microenvironment in prostate cancer bone metastasis. J Cell Biochem. 2007;101:873–86. doi: 10.1002/jcb.21214. [DOI] [PubMed] [Google Scholar]
- 9.Shappell SB, Thomas GV, Roberts RL, Herbert R, Ittmann MM, Rubin MA, Humphrey PA, Sundberg JP, Rozengurt N, Barrios R, Ward JM, Cardiff RD. Prostate Pathology of Genetically Engineered Mice: Definitions and Classification. The Consensus Report from the Bar Harbor Meeting of the Mouse Models of Human Cancer Consortium Prostate Pathology Committee. Cancer Res. 2004;64:2270–305. doi: 10.1158/0008-5472.can-03-0946. [DOI] [PubMed] [Google Scholar]
- 10.Ittmann M, Huang J, Radaelli E, Martin P, Signoretti S, Sullivan R, Simons BW, Ward JM, Robinson BD, Chu GC, Loda M, Thomas G, Borowsky A, Cardiff RD. Animal Models of Human Prostate Cancer: The Consensus Report of the New York Meeting of the Mouse Models of Human Cancers Consortium Prostate Pathology Committee. Cancer Res. 2013;73:2718–36. doi: 10.1158/0008-5472.CAN-12-4213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ousset M, Van Keymeulen A, Bouvencourt G, Sharma N, Achouri Y, Simons BD, Blanpain C. Multipotent and unipotent progenitors contribute to prostate postnatal development. Nature Cell Bioliogy. 2012;14:1131–38. doi: 10.1038/ncb2600. [DOI] [PubMed] [Google Scholar]
- 12.Liu J, Pascal LE, Isharwal S, Metzger D, Ramos Garcia R, Pilch J, Kasper S, Williams K, Basse PH, Nelson JB, Chambon P, Wang Z. Regenerated Luminal Epithelial Cells Are Derived from Preexisting Luminal Epithelial Cells in Adult Mouse Prostate. Mol Endocrinol. 2011;25:1849–57. doi: 10.1210/me.2011-1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Choi N, Zhang B, Zhang L, Ittmann M, Xin L. Adult Murine Prostate Basal and Luminal Cells Are Self-Sustained Lineages that Can Both Serve as Targets for Prostate Cancer Initiation. Cancer Cell. 2012;21:253–65. doi: 10.1016/j.ccr.2012.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lu TL, Huang YF, You LR, Chao NC, Su FY, Chang JL, Chen CM. Conditionally Ablated Pten in Prostate Basal Cells Promotes Basal-to-Luminal Differentiation and Causes Invasive Prostate Cancer in Mice. The American Journal of Pathology. 2013;182:975–91. doi: 10.1016/j.ajpath.2012.11.025. [DOI] [PubMed] [Google Scholar]
- 15.Wang ZA, Mitrofanova A, Bergren SK, Abate-Shen C, Cardiff RD, Califano A, Shen MM. Lineage analysis of basal epithelial cells reveals their unexpected plasticity and supports a cell-of-origin model for prostate cancer heterogeneity. Nat Cell Biol. 2013;15:274–83. doi: 10.1038/ncb2697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hu Y, Ippolito JE, Garabedian EM, Humphrey PA, Gordon JI. Molecular Characterization of a Metastatic Neuroendocrine Cell Cancer Arising in the Prostates of Transgenic Mice. J Biol Chem. 2002;277:44462–74. doi: 10.1074/jbc.M205784200. [DOI] [PubMed] [Google Scholar]
- 17.Garabedian EM, Humphrey PA, Gordon JI. A transgenic mouse model of metastatic prostate cancer originating from neuroendocrine cells. Proc Natl Acad Sci U S A. 1998;95:15382–87. doi: 10.1073/pnas.95.26.15382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chokkalingam AP, Nyrén O, Johansson JE, Gridley G, McLaughlin JK, Adami HO, Hsing AW. Prostate carcinoma risk subsequent to diagnosis of benign prostatic hyperplasia. Cancer. 2003;98:1727–34. doi: 10.1002/cncr.11710. [DOI] [PubMed] [Google Scholar]
- 19.Bostwick DG, Liu L, Brawer MK, Qian J. High-grade prostatic intraepithelial neoplasia. Rev Urol. 2004;6:171–79. [PMC free article] [PubMed] [Google Scholar]
- 20.Reyes A, Swanson PE, Carbone JM, Humphrey PA. Unusual Histologic Types of High-Grade Prostatic Intraepithelial Neoplasia. The American Journal of Surgical Pathology. 1997;21:1215–22. doi: 10.1097/00000478-199710000-00013. [DOI] [PubMed] [Google Scholar]
- 21.Gingrich JR, Barrios RJ, Foster BA, Greenberg NM. Pathologic progression of autochthonous prostate cancer in the TRAMP model. Prostate Cancer and Prostatic Disease. 1999;2:70–75. doi: 10.1038/sj.pcan.4500296. [DOI] [PubMed] [Google Scholar]
- 22.Park JH, Walls JE, Galvez JJ, Kim M, Abate-Shen C, Shen MM, Cardiff RD. Prostatic Intraepithelial Neoplasia in Genetically Engineered Mice. The American Journal of Pathology. 2002;161:727–35. doi: 10.1016/S0002-9440(10)64228-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Humphrey PA. Histological variants of prostatic carcinoma and their significance. Histopathology. 2012;60:59–74. doi: 10.1111/j.1365-2559.2011.04039.x. [DOI] [PubMed] [Google Scholar]
- 24.Wang W, Epstein JI. Small Cell Carcinoma of the Prostate: A Morphologic and Immunohistochemical Study of 95 Cases. The American Journal of Surgical Pathology. 2008;32:65–71. doi: 10.1097/PAS.0b013e318058a96b. [DOI] [PubMed] [Google Scholar]
- 25.Palmgren JS, Karavadia SS, Wakefield MR. Unusual and Underappreciated: Small Cell Carcinoma of the Prostate. Semin Oncol. 2007;34:22–29. doi: 10.1053/j.seminoncol.2006.10.026. [DOI] [PubMed] [Google Scholar]
- 26.Hansel DE, Epstein JI. Sarcomatoid carcinoma of the prostate: a study of 42 cases. Am J Surg Pathol. 2006;30:1316–21. doi: 10.1097/01.pas.0000209838.92842.bf. [DOI] [PubMed] [Google Scholar]
- 27.Martin P, Liu YN, Pierce R, Abou-Kheir W, Casey O, Seng V, Camacho D, Simpson RM, Kelly K. Prostate Epithelial Pten/TP53 Loss Leads to Transformation of Multipotential Progenitors and Epithelial to Mesenchymal Transition. The American Journal of Pathology. 2011;179:422–35. doi: 10.1016/j.ajpath.2011.03.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Acevedo VD, Gangula RD, Freeman KW, Li R, Zhang Y, Wang F, Ayala GE, Peterson LE, Ittmann M, Spencer DM. Inducible FGFR-1 Activation Leads to Irreversible Prostate Adenocarcinoma and an Epithelial-to-Mesenchymal Transition. Cancer Cell. 2007;12:559–71. doi: 10.1016/j.ccr.2007.11.004. [DOI] [PubMed] [Google Scholar]
- 29.Mulholland DJ, Kobayashi N, Ruscetti M, Zhi A, Tran LM, Huang J, Gleave M, Wu H. Pten Loss and RAS/MAPK Activation Cooperate to Promote EMT and Metastasis Initiated from Prostate Cancer Stem/Progenitor Cells. Cancer Res. 2012;72:1878–89. doi: 10.1158/0008-5472.CAN-11-3132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Vinall RL, Chen JQ, Hubbard NE, Sulaimon SS, Shen MM, DeVere White RW, Borowsky AD. Initiation of prostate cancer in mice by Tp53R270H: evidence for an alternative molecular progression. Dis Model Mech. 2012;5:914–20. doi: 10.1242/dmm.008995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ayala GE, Muezzinoglu B, Hammerich KH, Frolov A, Liu H, Scardino PT, Li R, Sayeeduddin M, Ittmann MM, Kadmon D, Miles BJ, Wheeler TM, Rowley DR. Determining Prostate Cancer-Specific Death through Quantification of Stromogenic Carcinoma Area in Prostatectomy Specimens. The American Journal of Pathology. 2011;178:79–87. doi: 10.1016/j.ajpath.2010.09.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ayala G, Tuxhorn JA, Wheeler TM, Frolov A, Scardino PT, Ohori M, Wheeler M, Spitler J, Rowley DR. Reactive Stroma as a Predictor of Biochemical-Free Recurrence in Prostate Cancer. Clin Cancer Res. 2003;9:4792–801. [PubMed] [Google Scholar]
- 33.Hayward SW, Wang Y, Cao M, Hom YK, Zhang B, Grossfeld GD, Sudilovsky D, Cunha GR. Malignant Transformation in a Nontumorigenic Human Prostatic Epithelial Cell Line. Cancer Res. 2001;61:8135–42. [PubMed] [Google Scholar]
- 34.Small EJ, Schellhammer PF, Higano CS, Redfern CH, Nemunaitis JJ, Valone FH, Verjee SS, Jones LA, Hershberg RM. Placebo-Controlled Phase III Trial of Immunologic Therapy with Sipuleucel-T (APC8015) in Patients with Metastatic, Asymptomatic Hormone Refractory Prostate Cancer. J Clin Oncol. 2006;24:3089–94. doi: 10.1200/JCO.2005.04.5252. [DOI] [PubMed] [Google Scholar]
- 35.Hess KR, Varadhachary GR, Taylor SH, Wei W, Raber MN, Lenzi R, Abbruzzese JL. Metastatic patterns in adenocarcinoma. Cancer. 2006;106:1624–33. doi: 10.1002/cncr.21778. [DOI] [PubMed] [Google Scholar]
- 36.Nandana S, Ellwood-Yen K, Sawyers C, Wills M, Weidow B, Case T, Vasioukhin V, Matusik R. Hepsin cooperates with MYC in the progression of adenocarcinoma in a prostate cancer mouse model. The Prostate. 2010;70:591–600. doi: 10.1002/pros.21093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ding Z, Wu CJ, Jaskelioff M, Ivanova E, Kost-Alimova M, Protopopov A, Chu Gerald C, Wang G, Lu X, Labrot EmmaS, Hu J, Wang W, Xiao Y, Zhang H, Zhang J, Zhang J, Gan B, Perry Samuel R, Jiang S, Li L, Horner JW, Wang YA, Chin L, DePinho Ronald A. Telomerase Reactivation following Telomere Dysfunction Yields Murine Prostate Tumors with Bone Metastases. Cell. 2012;148:896–907. doi: 10.1016/j.cell.2012.01.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gingrich JR, Barrios RJ, Morton RA, Boyce BF, DeMayo FJ, Finegold MJ, Angelopoulou R, Rosen JM, Greenberg NM. Metastatic Prostate Cancer in a Transgenic Mouse. Cancer Res. 1996;56:4096–102. [PubMed] [Google Scholar]
- 39.Prostate Cancer Trialists' Collaborative G: Maximum androgen blockade in advanced prostate cancer: an overview of 22 randomised trials with 3283 deaths in 5710 patients. The Lancet. 1995;346:265–69. [PubMed] [Google Scholar]
- 40.Scher HI, Fizazi K, Saad F, Taplin ME, Sternberg CN, Miller K, de Wit R, Mulders P, Chi KN, Shore ND, Armstrong AJ, Flaig TW, Fléchon A, Mainwaring P, Fleming M, Hainsworth JD, Hirmand M, Selby B, Seely L, de Bono JS. Increased Survival with Enzalutamide in Prostate Cancer after Chemotherapy. N Engl J Med. 2012;367:1187–97. doi: 10.1056/NEJMoa1207506. [DOI] [PubMed] [Google Scholar]
- 41.Fizazi K, Scher HI, Molina A, Logothetis CJ, Chi KN, Jones RJ, Staffurth JN, North S, Vogelzang NJ, Saad F, Mainwaring P, Harland S, Goodman OB, Sternberg CN, Li JH, Kheoh T, Haqq CM, de Bono JS. Abiraterone acetate for treatment of metastatic castration-resistant prostate cancer: final overall survival analysis of the COU-AA-301 randomised, double-blind, placebo-controlled phase 3 study. The Lancet Oncology. 2012;13:983–92. doi: 10.1016/S1470-2045(12)70379-0. [DOI] [PubMed] [Google Scholar]
- 42.Greenberg NM, DeMayo FJ, Sheppard PC, Barrios R, Lebovitz R, Finegold M, Angelopoulou R, Dodd JG, Duckworth ML, Rosen JM. The rat probasin gene promoter directs hormonally and developmentally regulated expression of a heterologous gene specifically to the prostate in transgenic mice. Mol Endocrinol. 1994;8:230–9. doi: 10.1210/mend.8.2.8170479. [DOI] [PubMed] [Google Scholar]
- 43.Bhatia-Gaur R, Donjacour AA, Sciavolino PJ, Kim M, Desai N, Young P, Norton CR, Gridley T, Cardiff RD, Cunha GR, Abate-Shen C, Shen MM. Roles for Nkx3.1 in prostate development and cancer. Genes Dev. 1999;13:966–77. doi: 10.1101/gad.13.8.966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tanaka M, Komuro I, Inagaki H, Jenkins NA, Copeland NG, Izumo S. Nkx3.1, a murine homolog of Drosophila bagpipe, regulates epithelial ductal branching and proliferation of the prostate and palatine glands. Dev Dyn. 2000;219:248–60. doi: 10.1002/1097-0177(2000)9999:9999<::aid-dvdy1054>3.3.co;2-5. [DOI] [PubMed] [Google Scholar]
- 45.Rennie PS, Bruchovsky N, Leco KJ, Sheppard PC, McQueen SA, Cheng H, Snoek R, Hamel A, Bock ME, MacDonald BS. Characterization of two cis-acting DNA elements involved in the androgen regulation of the probasin gene. Mol Endocrinol. 1993;7:23–36. doi: 10.1210/mend.7.1.8446105. [DOI] [PubMed] [Google Scholar]
- 46.Zhang J, Thomas TZ, Kasper S, Matusik RJ. A Small Composite Probasin Promoter Confers High Levels of Prostate-Specific Gene Expression through Regulation by Androgens and Glucocorticoids in Vitro and in Vivo. Endocrinology. 2000;141:4698–710. doi: 10.1210/endo.141.12.7837. [DOI] [PubMed] [Google Scholar]
- 47.Bianchi-Frias D, Pritchard C, Mecham B, Coleman I, Nelson P. Genetic background influences murine prostate gene expression: implications for cancer phenotypes. Genome Biol. 2007;8:R117. doi: 10.1186/gb-2007-8-6-r117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chiaverotti T, Couto SS, Donjacour A, Mao JH, Nagase H, Cardiff RD, Cunha GR, Balmain A. Dissociation of Epithelial and Neuroendocrine Carcinoma Lineages in the Transgenic Adenocarcinoma of Mouse Prostate Model of Prostate Cancer. The American journal of pathology. 2008;172:236–46. doi: 10.2353/ajpath.2008.070602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ahuja D, Saenz-Robles MT, Pipas JM. SV40 large T antigen targets multiple cellular pathways to elicit cellular transformation. Oncogene. 2005;24:7729–45. doi: 10.1038/sj.onc.1209046. [DOI] [PubMed] [Google Scholar]
- 50.Maroulakou IG, Anver M, Garrett L, Green JE. Prostate and mammary adenocarcinoma in transgenic mice carrying a rat C3(1) simian virus 40 large tumor antigen fusion gene. Proc Natl Acad Sci U S A. 1994;91:11236–40. doi: 10.1073/pnas.91.23.11236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Greenberg NM, DeMayo F, Finegold MJ, Medina D, Tilley WD, Aspinall JO, Cunha GR, Donjacour AA, Matusik RJ, Rosen JM. Prostate cancer in a transgenic mouse. Proc Natl Acad Sci U S A. 1995;92:3439–43. doi: 10.1073/pnas.92.8.3439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gingrich JR, Barrios RJ, Kattan MW, Nahm HS, Finegold MJ, Greenberg NM. Androgen-independent Prostate Cancer Progression in the TRAMP Model. Cancer Res. 1997;57:4687–91. [PubMed] [Google Scholar]
- 53.Kaplan-Lefko PJ, Chen TM, Ittmann MM, Barrios RJ, Ayala GE, Huss WJ, Maddison LA, Foster BA, Greenberg NM. Pathobiology of autochthonous prostate cancer in a pre-clinical transgenic mouse model. The Prostate. 2003;55:219–37. doi: 10.1002/pros.10215. [DOI] [PubMed] [Google Scholar]
- 54.Han G, Foster BA, Mistry S, Buchanan G, Harris JM, Tilley WD, Greenberg NM. Hormone Status Selects for Spontaneous Somatic Androgen Receptor Variants That Demonstrate Specific Ligand and Cofactor Dependent Activities in Autochthonous Prostate Cancer. J Biol Chem. 2001;276:11204–13. doi: 10.1074/jbc.M008207200. [DOI] [PubMed] [Google Scholar]
- 55.Irshad S, Abate-Shen C. Modeling prostate cancer in mice: something old, something new, something premalignant, something metastatic. Cancer Metastasis Rev. 2012:1–14. doi: 10.1007/s10555-012-9409-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ahmad I, Sansom OJ, Leung HY. The role of murine models of prostate cancer in drug target discovery and validation. Expert Opinion on Drug Discovery. 2009;4:879–88. doi: 10.1517/17460440903049308. [DOI] [PubMed] [Google Scholar]
- 57.Kasper S, Sheppard PC, Yan Y, Pettigrew N, Borowsky AD, Prins G, Dodd JG, Duckworth ML, Matusik RJ. Development, progression, and androgen-dependence of prostate tumors in probasin-large T antigen transgenic mice: a model for prostate cancer. Lab Invest. 1998;78:i–xv. [PubMed] [Google Scholar]
- 58.Klezovitch O, Chevillet J, Mirosevich J, Roberts RL, Matusik RJ, Vasioukhin V. Hepsin promotes prostate cancer progression and metastasis. Cancer Cell. 2004;6:185–95. doi: 10.1016/j.ccr.2004.07.008. [DOI] [PubMed] [Google Scholar]
- 59.Masumori N, Thomas TZ, Chaurand P, Case T, Paul M, Kasper S, Caprioli RM, Tsukamoto T, Shappell SB, Matusik RJ. A Probasin-Large T Antigen Transgenic Mouse Line Develops Prostate Adenocarcinoma and Neuroendocrine Carcinoma with Metastatic Potential. Cancer Res. 2001;61:2239–49. [PubMed] [Google Scholar]
- 60.Palmer J, Venkateswaran V, Fleshner NE, Klotz LH, Cox ME. The impact of diet and micronutrient supplements on the expression of neuroendocrine markers in murine Lady transgenic prostate. The Prostate. 2008;68:345–53. doi: 10.1002/pros.20692. [DOI] [PubMed] [Google Scholar]
- 61.Venkateswaran V, Klotz LH, Ramani M, Sugar LM, Jacob LE, Nam RK, Fleshner NE. A Combination of Micronutrients Is Beneficial in Reducing the Incidence of Prostate Cancer and Increasing Survival in the Lady Transgenic Model. Cancer Prev Res (Phila Pa) 2009;2:473–83. doi: 10.1158/1940-6207.CAPR-08-0124. [DOI] [PubMed] [Google Scholar]
- 62.Guo Y, Jacobs SC, Kyprianou N. Down-regulation of protein and mRNA expression for transforming growth factor-β (TGF-β1) type I and type II receptors in human prostate cancer. Int J Cancer. 1997;71:573–79. doi: 10.1002/(sici)1097-0215(19970516)71:4<573::aid-ijc11>3.0.co;2-d. [DOI] [PubMed] [Google Scholar]
- 63.Tu WH, Thomas T, Masumori N, Bhowmick N, Gorska A, Shyr Y, Kasper S, Case T, Roberts R, Shappell S, Moses H, Matusik R. The loss of TGF-β signaling promotes prostate cancer metastasis. Neoplasia. 2003;5:267–77. doi: 10.1016/S1476-5586(03)80058-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Yu X, Wang Y, DeGraff DJ, Wills ML, Matusik RJ. Wnt/Beta-catenin activation promotes prostate tumor progression in a mouse model. Oncogene. 2011;30:1868–79. doi: 10.1038/onc.2010.560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Gounari F, Signoretti S, Bronson R, Klein L, Sellers WR, Kum J, Siermann A, Taketo MM, Boehme Hv, Khazaie K. Stabilization of b-catenin induces lesions reminiscent of prostatic intraepithelial neoplasia, but terminal squamous transdiferentiation of other secretory epithelia. Oncogene. 2002;21:4099–107. doi: 10.1038/sj.onc.1205562. [DOI] [PubMed] [Google Scholar]
- 66.Bruxvoort KJ, Charbonneau HM, Giambernardi TA, Goolsby JC, Qian CN, Zylstra CR, Robinson DR, Roy-Burman P, Shaw AK, Buckner-Berghuis BD, Sigler RE, Resau JH, Sullivan R, Bushman W, Williams BO. Inactivation of Apc in the Mouse Prostate Causes Prostate Carcinoma. Cancer Res. 2007;67:2490–96. doi: 10.1158/0008-5472.CAN-06-3028. [DOI] [PubMed] [Google Scholar]
- 67.Zhou Z, Flesken-Nikitin A, Corney DC, Wang W, Goodrich DW, Roy-Burman P, Nikitin AY. Synergy of p53 and Rb Deficiency in a Conditional Mouse Model for Metastatic Prostate Cancer. Cancer Res. 2006;66:7889–98. doi: 10.1158/0008-5472.CAN-06-0486. [DOI] [PubMed] [Google Scholar]
- 68.Hill R, Song Y, Cardiff RD, Van Dyke T. Heterogeneous Tumor Evolution Initiated by Loss of pRb Function in a Preclinical Prostate Cancer Model. Cancer Res. 2005;65:10243–54. doi: 10.1158/0008-5472.CAN-05-1579. [DOI] [PubMed] [Google Scholar]
- 69.Phillips SM, Barton CM, Lee SJ, Morton DG, Wallace DM, Lemoine NR, Neoptolemos JP. Loss of the retinoblastoma susceptibility gene (RB1) is a frequent and early event in prostatic tumorigenesis. Br J Cancer. 1994;70:1252–57. doi: 10.1038/bjc.1994.482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Bookstein R, MacGrogan D, Hilsenbeck SG, Sharkey F, Allred DC. p53 Is Mutated in a Subset of Advanced-Stage Prostate Cancers. Cancer Res. 1993;53:3369–73. [PubMed] [Google Scholar]
- 71.Saric T, Brkanac Z, Troyer DA, Padalecki SS, Sarosdy M, Williams K, Abadesco L, Leach RJ, O'Connell P. Genetic pattern of prostate cancer progression. Int J Cancer. 1999;81:219–24. doi: 10.1002/(sici)1097-0215(19990412)81:2<219::aid-ijc9>3.0.co;2-3. [DOI] [PubMed] [Google Scholar]
- 72.Qi J, Nakayama K, Cardiff RD, Borowsky AD, Kaul K, Williams R, Krajewski S, Mercola D, Carpenter PM, Bowtell D. Ronai ZeA: Siah2-Dependent Concerted Activity of HIF and FoxA2 Regulates Formation of Neuroendocrine Phenotype and Neuroendocrine Prostate Tumors. Cancer Cell. 2010;18:23–38. doi: 10.1016/j.ccr.2010.05.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Spence AM, Sheppard PC, Davie JR, Matuo Y, Nishi N, McKeehan WL, Dodd JG, Matusik RJ. Regulation of a bifunctional mRNA results in synthesis of secreted and nuclear probasin. Proc Natl Acad Sci U S A. 1989;86:7843–47. doi: 10.1073/pnas.86.20.7843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Maddison LA, Nahm H, DeMayo F, Greenberg NM. Prostate specific expression of Cre recombinase in transgenic mice. Genesis. 2000;26:154–56. doi: 10.1002/(sici)1526-968x(200002)26:2<154::aid-gene18>3.0.co;2-2. [DOI] [PubMed] [Google Scholar]
- 75.Wu X, Wu J, Huang J, Powell WC, Zhang J, Matusik RJ, Sangiorgi FO, Maxson RE, Sucov HM, Roy-Burman P. Generation of a prostate epithelial cell-specific Cre transgenic mouse model for tissue-specific gene ablation. Mech Dev. 2001;101:61–69. doi: 10.1016/s0925-4773(00)00551-7. [DOI] [PubMed] [Google Scholar]
- 76.Song MS, Salmena L, Pandolfi PP. The functions and regulation of the PTEN tumour suppressor. Nature Reviews in Molecular and Cell Biology. 2012;13:283–96. doi: 10.1038/nrm3330. [DOI] [PubMed] [Google Scholar]
- 77.Cantley LC, Neel BG. New insights into tumor suppression: PTEN suppresses tumor formation by restraining the phosphoinositide 3-kinase/AKT pathway. Proc Natl Acad Sci U S A. 1999;96:4240–45. doi: 10.1073/pnas.96.8.4240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Yoshimoto M, Cutz JC, Nuin PAS, Joshua AM, Bayani J, Evans AJ, Zielenska M, Squire JA. Interphase FISH analysis of PTEN in histologic sections shows genomic deletions in 68% of primary prostate cancer and 23% of high-grade prostatic intra-epithelial neoplasias. Cancer Genet Cytogenet. 2006;169:128–37. doi: 10.1016/j.cancergencyto.2006.04.003. [DOI] [PubMed] [Google Scholar]
- 79.Holcomb IN, Young JM, Coleman IM, Salari K, Grove DI, Hsu L, True LD, Roudier MP, Morrissey CM, Higano CS, Nelson PS, Vessella RL, Trask BJ. Comparative Analyses of Chromosome Alterations in Soft-Tissue Metastases within and across Patients with Castration-Resistant Prostate Cancer. Cancer Res. 2009;69:7793–802. doi: 10.1158/0008-5472.CAN-08-3810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Cristofano AD, Pesce B, Cordon-Cardo C, Pandolfi PP. Pten is essential for embryonic development and tumour suppression. Nat Genet. 1998;19:348–55. doi: 10.1038/1235. [DOI] [PubMed] [Google Scholar]
- 81.Suzuki A, de la Pompa JL, Stambolic V, Elia AJ, Sasaki T, Barrantes IdB, Ho A, Wakeham A, ltie A, Khoo W, Fukumoto M, Mak TW. High cancer susceptibility and embryonic lethality associated with mutation of the PTEN tumor suppressor gene in mice. Current biology : CB. 1998;8:1169–78. doi: 10.1016/s0960-9822(07)00488-5. [DOI] [PubMed] [Google Scholar]
- 82.Podsypanina K, Ellenson LH, Nemes A, Gu J, Tamura M, Yamada KM, Cordon-Cardo C, Catoretti G, Fisher PE, Parsons R. Mutation of Pten/Mmac1 in mice causes neoplasia in multiple organ systems. Proc Natl Acad Sci U S A. 1999;96:1563–68. doi: 10.1073/pnas.96.4.1563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Trotman LC, Niki M, Dotan ZA, Koutcher JA, Di Cristofano A, Xiao A, Khoo AS, Roy-Burman P, Greenberg NM, Dyke TV, Cordon-Cardo C, Pandolfi PP. Pten Dose Dictates Cancer Progression in the Prostate. PLoS Biol. 2003;1:e59. doi: 10.1371/journal.pbio.0000059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Stambolic V, Tsao MS, Macpherson D, Suzuki A, Chapman WB, Mak TW. High Incidence of Breast and Endometrial Neoplasia Resembling Human Cowden Syndrome in pten+/- Mice. Cancer Res. 2000;60:3605–11. [PubMed] [Google Scholar]
- 85.Vis AN, Noordzij MA, Fitoz K, Wildhagen MF, Schroder FH, van der Kwast TH. Prognostic value of cell cycle proteins p27(kip1) and MIB-1, and the cell adhesion protein CD44s in surgically treated patients with prostate cancer. The Journal of Urology. 2000;164:2156–61. [PubMed] [Google Scholar]
- 86.Cordon-Cardo C, Koff A, Drobnjak M, Capodieci P, Osman I, Millard SS, Gaudin PB, Fazzari M, Zhang ZF, Massague J, Scher HI. Distinct Altered Patterns of p27KIP1 Gene Expression in Benign Prostatic Hyperplasia and Prostatic Carcinoma. J Natl Cancer Inst. 1998;90:1284–91. doi: 10.1093/jnci/90.17.1284. [DOI] [PubMed] [Google Scholar]
- 87.Di Cristofano A, De Acetis M, Koff A, Cordon-Cardo C, Pandolfi PP. Pten and p27KIP1 cooperate in prostate cancer tumor suppression in the mouse. Nat Genet. 2001;27:222–24. doi: 10.1038/84879. [DOI] [PubMed] [Google Scholar]
- 88.Sciavolino PJ, Abrams EW, Yang L, Austenberg LP, Shen MM, Abate-Shen C. Tissue-specific expression of murine Nkx3.1 in the male urogenital system. Dev Dyn. 1997;209:127–38. doi: 10.1002/(SICI)1097-0177(199705)209:1<127::AID-AJA12>3.0.CO;2-Z. [DOI] [PubMed] [Google Scholar]
- 89.Kim MJ, Cardiff RD, Desai N, Banach-Petrosky WA, Parsons R, Shen MM, Abate-Shen C. Cooperativity of Nkx3.1 and Pten loss of function in a mouse model of prostate carcinogenesis. Proc Natl Acad Sci U S A. 2002;99:2884–89. doi: 10.1073/pnas.042688999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Bowen C, Bubendorf L, Voeller HJ, Slack R, Willi N, Sauter G, Gasser TC, Koivisto P, Lack EE, Kononen J, Kallioniemi OP, Gelmann EP. Loss of NKX3.1 Expression in Human Prostate Cancers Correlates with Tumor Progression1,2. Cancer Res. 2000;60:6111–15. [PubMed] [Google Scholar]
- 91.Schneider A, Brand T, Zweigerdt R, Arnold HH. Targeted disruption of the Nkx3.1 gene in mice results in morphogenetic defects of minor salivary glands: parallels to glandular duct morphogenesis in prostate. Mech Dev. 2000;95:163–74. doi: 10.1016/s0925-4773(00)00355-5. [DOI] [PubMed] [Google Scholar]
- 92.Abdulkadir SA, Magee JA, Peters TJ, Kaleem Z, Naughton CK, Humphrey PA, Milbrandt J. Conditional Loss of Nkx3.1 in Adult Mice Induces Prostatic Intraepithelial Neoplasia. Mol Cell Biol. 2002;22:1495–503. doi: 10.1128/mcb.22.5.1495-1503.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Kim MJ, Bhatia-Gaur R, Banach-Petrosky WA, Desai N, Wang Y, Hayward SW, Cunha GR, Cardiff RD, Shen MM, Abate-Shen C. Nkx3.1 Mutant Mice Recapitulate Early Stages of Prostate Carcinogenesis. Cancer Res. 2002;62:2999–3004. [PubMed] [Google Scholar]
- 94.Abate-Shen C, Banach-Petrosky WA, Sun X, Economides KD, Desai N, Gregg JP, Borowsky AD, Cardiff RD, Shen MM. Nkx3.1; Pten Mutant Mice Develop Invasive Prostate Adenocarcinoma and Lymph Node Metastases. Cancer Res. 2003;63:3886–90. [PubMed] [Google Scholar]
- 95.Wang S, Gao J, Lei Q, Rozengurt N, Pritchard C, Jiao J, Thomas GV, Li G, Roy-Burman P, Nelson PS, Liu X, Wu H. Prostate-specific deletion of the murine Pten tumor suppressor gene leads to metastatic prostate cancer. Cancer Cell. 2003;4:209–21. doi: 10.1016/s1535-6108(03)00215-0. [DOI] [PubMed] [Google Scholar]
- 96.Waltering KK, Urbanucci A, Visakorpi T. Androgen receptor (AR) aberrations in castration-resistant prostate cancer. Mol Cell Endocrinol. 2012;360:38–43. doi: 10.1016/j.mce.2011.12.019. [DOI] [PubMed] [Google Scholar]
- 97.Holzbeierlein J, Lal P, LaTulippe E, Smith A, Satagopan J, Zhang L, Ryan C, Smith S, Scher H, Scardino P, Reuter V, Gerald WL. Gene Expression Analysis of Human Prostate Carcinoma during Hormonal Therapy Identifies Androgen-Responsive Genes and Mechanisms of Therapy Resistance. The American Journal of Pathology. 2004;164:217–27. doi: 10.1016/S0002-9440(10)63112-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Visakorpi T, Hyytinen E, Koivisto P, Tanner M, Keinanen R, Palmberg C, Palotie A, Tammela T, Isola J, Kallioniemi OP. In vivo amplification of the androgen receptor gene and progression of human prostate cancer. Nat Genet. 1995;9:401–06. doi: 10.1038/ng0495-401. [DOI] [PubMed] [Google Scholar]
- 99.Liao CP, Zhong C, Saribekyan G, Bading J, Park R, Conti PS, Moats R, Berns A, Shi W, Zhou Z, Nikitin AY, Roy-Burman P. Mouse Models of Prostate Adenocarcinoma with the Capacity to Monitor Spontaneous Carcinogenesis by Bioluminescence or Fluorescence. Cancer Res. 2007;67:7525–33. doi: 10.1158/0008-5472.CAN-07-0668. [DOI] [PubMed] [Google Scholar]
- 100.Zhang W, Zhu J, Efferson CL, Ware C, Tammam J, Angagaw M, Laskey J, Bettano KA, Kasibhatla S, Reilly JF, Sur C, Majumder PK. Inhibition of Tumor Growth Progression by Antiandrogens and mTOR Inhibitor in a Pten-Deficient Mouse Model of Prostate Cancer. Cancer Res. 2009;69:7466–72. doi: 10.1158/0008-5472.CAN-08-4385. [DOI] [PubMed] [Google Scholar]
- 101.Mulholland David J, Tran Linh M, Li Y, Cai H, Morim A, Wang S, Plaisier S, Garraway Isla P, Huang J, Graeber Thomas G, Wu H. Cell Autonomous Role of PTEN in Regulating Castration-Resistant Prostate Cancer Growth. Cancer Cell. 2011;19:792–804. doi: 10.1016/j.ccr.2011.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Chen Z, Trotman LC, Shaffer D, Lin HK, Dotan ZA, Niki M, Koutcher JA, Scher HI, Ludwig T, Gerald W, Cordon-Cardo C, Pandolfi PP. Crucial role of p53-dependent cellular senescence in suppression of Pten-deficient tumorigenesis. Nature. 2005;436:725–30. doi: 10.1038/nature03918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Ma X, Ziel-van der Made AC, Autar B, van der Korput HA, Vermeij M, van Duijn P, Cleutjens KB, de Krijger R, Krimpenfort P, Berns A, van der Kwast TH, Trapman J. Targeted Biallelic Inactivation of Pten in the Mouse Prostate Leads to Prostate Cancer Accompanied by Increased Epithelial Cell Proliferation but not by Reduced Apoptosis. Cancer Res. 2005;65:5730–39. doi: 10.1158/0008-5472.CAN-04-4519. [DOI] [PubMed] [Google Scholar]
- 104.Majumder PK, Febbo PG, Bikoff R, Berger R, Xue Q, McMahon LM, Manola J, Brugarolas J, McDonnell TJ, Golub TR, Loda M, Lane HA, Sellers WR. mTOR inhibition reverses Akt-dependent prostate intraepithelial neoplasia through regulation of apoptotic and HIF-1-dependent pathways. Nat Med. 2004;10:594–601. doi: 10.1038/nm1052. [DOI] [PubMed] [Google Scholar]
- 105.Ratnacaram CK, Teletin M, Jiang M, Meng X, Chambon P, Metzger D. Temporally controlled ablation of PTEN in adult mouse prostate epithelium generates a model of invasive prostatic adenocarcinoma. Proc Natl Acad Sci U S A. 2008;105:2521–26. doi: 10.1073/pnas.0712021105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Wang X, Julio MKd, Economides KD, Walker D, Yu H, Halili MV, Hu YP, Price SM, Abate-Shen C, Shen MM. A luminal epithelial stem cell that is a cell of origin for prostate cancer. Nature. 2009;461:495–500. doi: 10.1038/nature08361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Floc'h N, Kinkade CW, Kobayashi T, Aytes A, Lefebvre C, Mitrofanova A, Cardiff RD, Califano A, Shen MM, Abate-Shen C. Dual Targeting of the Akt/mTOR Signaling Pathway Inhibits Castration-Resistant Prostate Cancer in a Genetically Engineered Mouse Model. Cancer Res. 2012;72:4483–93. doi: 10.1158/0008-5472.CAN-12-0283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Dankort D, Filenova E, Collado M, Serrano M, Jones K, McMahon M. A new mouse model to explore the initiation, progression, and therapy of BRAFV600E-induced lung tumors. Genes Dev. 2007;21:379–84. doi: 10.1101/gad.1516407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Wang J, Kobayashi T, Floc'h N, Kinkade CW, Aytes A, Dankort D, Lefebvre C, Mitrofanova A, Cardiff RD, McMahon M, Califano A, Shen MM, Abate-Shen C. B-Raf Activation Cooperates with PTEN Loss to Drive c-Myc Expression in Advanced Prostate Cancer. Cancer Res. 2012;72:4765–76. doi: 10.1158/0008-5472.CAN-12-0820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Aytes A, Mitrofanova A, Kinkade CW, Lefebvre C, Lei M, Phelan V, LeKaye HC, Koutcher JA, Cardiff RD, Califano A, Shen MM, Abate-Shen C. ETV4 promotes metastasis in response to activation of PI3-kinase and Ras signaling in a mouse model of advanced prostate cancer. Proc Natl Acad Sci U S A. 2013;110:E3506–E15. doi: 10.1073/pnas.1303558110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Goldstein AS, Huang J, Guo C, Garraway IP, Witte ON. Identification of a Cell of Origin for Human Prostate Cancer. Science. 2010;329:568–71. doi: 10.1126/science.1189992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Aitchison AA, Veerakumarasivam A, Vias M, Kumar R, Hamdy FC, Neal DE, Mills IG. Promoter methylation correlates with reduced Smad4 expression in advanced prostate cancer. The Prostate. 2008;68:661–74. doi: 10.1002/pros.20730. [DOI] [PubMed] [Google Scholar]
- 113.Ding Z, Wu CJ, Chu GC, Xiao Y, Ho D, Zhang J, Perry SR, Labrot ES, Wu X, Lis R, Hoshida Y, Hiller D, Hu B, Jiang S, Zheng H, Stegh AH, Scott KL, Signoretti S, Bardeesy N, Wang YA, Hill DE, Golub TR, Stampfer MJ, Wong WH, Loda M, Mucci L, Chin L, DePinho RA. SMAD4-dependent barrier constrains prostate cancer growth and metastatic progression. Nature. 2011;470:269–73. doi: 10.1038/nature09677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Tomlins SA, Rhodes DR, Perner S, Dhanasekaran SM, Mehra R, Sun XW, Varambally S, Cao X, Tchinda J, Kuefer R, Lee C, Montie JE, Shah RB, Pienta KJ, Rubin MA, Chinnaiyan AM. Recurrent Fusion of TMPRSS2 and ETS Transcription Factor Genes in Prostate Cancer. Science. 2005;310:644–48. doi: 10.1126/science.1117679. [DOI] [PubMed] [Google Scholar]
- 115.Tomlins SA, Mehra R, Rhodes DR, Smith LR, Roulston D, Helgeson BE, Cao X, Wei JT, Rubin MA, Shah RB, Chinnaiyan AM. TMPRSS2:ETV4 Gene Fusions Define a Third Molecular Subtype of Prostate Cancer. Cancer Res. 2006;66:3396–400. doi: 10.1158/0008-5472.CAN-06-0168. [DOI] [PubMed] [Google Scholar]
- 116.Tomlins SA, Laxman B, Dhanasekaran SM, Helgeson BE, Cao X, Morris DS, Menon A, Jing X, Cao Q, Han B, Yu J, Wang L, Montie JE, Rubin MA, Pienta KJ, Roulston D, Shah RB, Varambally S, Mehra R, Chinnaiyan AM. Distinct classes of chromosomal rearrangements create oncogenic ETS gene fusions in prostate cancer. Nature. 2007;448:595–99. doi: 10.1038/nature06024. [DOI] [PubMed] [Google Scholar]
- 117.Klezovitch O, Risk M, Coleman I, Lucas JM, Null M, True LD, Nelson PS, Vasioukhin V. A causal role for ERG in neoplastic transformation of prostate epithelium. Proc Natl Acad Sci U S A. 2008;105:2105–10. doi: 10.1073/pnas.0711711105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Tomlins SA, Laxman B, Varambally S, Cao X, Yu J, Helgeson BE, Cao Q, Prensner JR, Rubin MA, Shah RB, Mehra R, Chinnaiyan AM. The role of the TMPRSS2-ERG gene fusion in prostate cancer. Neoplasia. 2008;10:177–88. doi: 10.1593/neo.07822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Carver BS, Tran J, Gopalan A, Chen Z, Shaikh S, Carracedo A, Alimonti A, Nardella C, Varmeh S, Scardino PT, Cordon-Cardo C, Gerald W, Pandolfi PP. Aberrant ERG expression cooperates with loss of PTEN to promote cancer progression in the prostate. Nat Genet. 2009;41:619–24. doi: 10.1038/ng.370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.King JC, Xu J, Wongvipat J, Hieronymus H, Carver BS, Leung DH, Taylor BS, Sander C, Cardiff RD, Couto SS, Gerald WL, Sawyers CL. Cooperativity of TMPRSS2-ERG with PI3-kinase pathway activation in prostate oncogenesis. Nat Genet. 2009;41:524–26. doi: 10.1038/ng.371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Baena E, Shao Z, Linn DE, Glass K, Hamblen MJ, Fujiwara Y, Kim J, Nguyen M, Zhang X, Godinho FJ, Bronson RT, Mucci LA, Loda M, Yuan GC, Orkin SH, Li Z. ETV1 directs androgen metabolism and confers aggressive prostate cancer in targeted mice and patients. Genes Dev. 2013;27:683–98. doi: 10.1101/gad.211011.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Yoshimoto M, Joshua AM, Cunha IW, Coudry RA, Fonseca FP, Ludkovski O, Zielenska M, Soares FA, Squire JA. Absence of TMPRSS2:ERG fusions and PTEN losses in prostate cancer is associated with a favorable outcome. Mod Pathol. 2008;21:1451–60. doi: 10.1038/modpathol.2008.96. [DOI] [PubMed] [Google Scholar]
- 123.Zhong C, Saribekyan G, Liao CP, Cohen MB, Roy-Burman P. Cooperation between FGF8b Overexpression and PTEN Deficiency in Prostate Tumorigenesis. Cancer Res. 2006;66:2188–94. doi: 10.1158/0008-5472.CAN-05-3440. [DOI] [PubMed] [Google Scholar]
- 124.Nguyen AHT, Tremblay M, Haigh K, Koumakpayi IlH, Paquet Mn, Pandolfi PP, Mes-Masson AM, Saad F, Haigh JJ, Bouchard M. Gata3 antagonizes cancer progression in Pten-deficient prostates. Hum Mol Genet. 2013;22:2400–10. doi: 10.1093/hmg/ddt088. [DOI] [PubMed] [Google Scholar]
- 125.Nacerddine K, Beaudry JB, Ginjala V, Westerman B, Mattiroli F, Song JY, van der Poel H, Ponz OB, xE, Pritchard C, Cornelissen-Steijger P, Zevenhoven J, Tanger E, Sixma TK, Ganesan S, van Lohuizen M. Akt-mediated phosphorylation of Bmi1 modulates its oncogenic potential, E3 ligase activity, and DNA damage repair activity in mouse prostate cancer. The Journal of Clinical Investigation. 2012;122:1920–32. doi: 10.1172/JCI57477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Jackson EL, Willis N, Mercer K, Bronson RT, Crowley D, Montoya R, Jacks T, Tuveson DA. Analysis of lung tumor initiation and progression using conditional expression of oncogenic K-ras. Genes Dev. 2001;15:3243–48. doi: 10.1101/gad.943001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Carver Brett S, Chapinski C, Wongvipat J, Hieronymus H, Chen Y, Chandarlapaty S, Arora Vivek K, Le C, Koutcher J, Scher H, Scardino Peter T, Rosen N, Sawyers Charles L. Reciprocal Feedback Regulation of PI3K and Androgen Receptor Signaling in PTEN-Deficient Prostate Cancer. Cancer Cell. 2011;19:575–86. doi: 10.1016/j.ccr.2011.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Svensson RU, Haverkamp JM, Thedens DR, Cohen MB, Ratliff TL, Henry MD. Slow Disease Progression in a C57BL/6 Pten-Deficient Mouse Model of Prostate Cancer. The American Journal of Pathology. 2011;179:502–12. doi: 10.1016/j.ajpath.2011.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Gurel B, Iwata T, Koh CM, Jenkins RB, Lan F, Van Dang C, Hicks JL, Morgan J, Cornish TC, Sutcliffe S, Isaacs WB, Luo J, De Marzo AM. Nuclear MYC protein overexpression is an early alteration in human prostate carcinogenesis. Mod Pathol. 2008;21:1156–67. doi: 10.1038/modpathol.2008.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Fleming WH, Hamel A, MacDonald R, Ramsey E, Pettigrew NM, Johnston B, Dodd JG, Matusik RJ. Expression of the c-myc Protooncogene in Human Prostatic Carcinoma and Benign Prostatic Hyperplasia. Cancer Res. 1986;46:1535–38. [PubMed] [Google Scholar]
- 131.Koh CM, Bieberich CJ, Dang CV, Nelson WG, Yegnasubramanian S, De Marzo AM. MYC and Prostate Cancer. Genes & Cancer. 2010;1:617–28. doi: 10.1177/1947601910379132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Ellwood-Yen K, Graeber TG, Wongvipat J, Iruela-Arispe ML, Zhang J, Matusik R, Thomas GV, Sawyers CL. Myc-driven murine prostate cancer shares molecular features with human prostate tumors. Cancer Cell. 2003;4:223–38. doi: 10.1016/s1535-6108(03)00197-1. [DOI] [PubMed] [Google Scholar]
- 133.Iwata T, Schultz D, Hicks J, Hubbard GK, Mutton LN, Lotan TL, Bethel C, Lotz MT, Yegnasubramanian S, Nelson WG, Dang CV, Xu M, Anele U, Koh CM, Bieberich CJ, De Marzo AM. MYC Overexpression Induces Prostatic Intraepithelial Neoplasia and Loss of Nkx3.1 in Mouse Luminal Epithelial Cells. PLoS ONE. 2010;5:e9427. doi: 10.1371/journal.pone.0009427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Jin RJ, Lho Y, Connelly L, Wang Y, Yu X, Saint Jean L, Case TC, Ellwood-Yen K, Sawyers CL, Bhowmick NA, Blackwell TS, Yull FE, Matusik RJ. The Nuclear Factor-KappaB Pathway Controls the Progression of Prostate Cancer to Androgen-Independent Growth. Cancer Res. 2008;68:6762–69. doi: 10.1158/0008-5472.CAN-08-0107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Clegg NJ, Couto SS, Wongvipat J, Hieronymus H, Carver BS, Taylor BS, Ellwood-Yen K, Gerald WL, Sander C, Sawyers CL. MYC Cooperates with AKT in Prostate Tumorigenesis and Alters Sensitivity to mTOR Inhibitors. PLoS ONE. 2011;6:e17449. doi: 10.1371/journal.pone.0017449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Roh M, Kim J, Song C, Wills M, Abdulkadir SA. Transgenic mice for Cre-inducible overexpression of the oncogenes c-MYC and Pim-1 in multiple tissues. Genesis. 2006;44:447–53. doi: 10.1002/dvg.20235. [DOI] [PubMed] [Google Scholar]
- 137.Kim J, Roh M, Doubinskaia I, Algarroba GN, Eltoum IEA, Abdulkadir SA. A mouse model of heterogeneous, c-MYC-initiated prostate cancer with loss of Pten and p53. Oncogene. 2012;31:322–32. doi: 10.1038/onc.2011.236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Hensley P, Kyprianou N. Modeling prostate cancer in mice: limitations and opportunities. J Androl. 2012;33:133–44. doi: 10.2164/jandrol.111.013987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Shen MM, Abate-Shen C. Molecular genetics of prostate cancer: new prospects for old challenges. Genes Dev. 2010;24:1967–2000. doi: 10.1101/gad.1965810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Jeet V, Russell P, Khatri A. Modeling prostate cancer: a perspective on transgenic mouse models. Cancer Metastasis Rev. 2010;29:123–42. doi: 10.1007/s10555-010-9212-9. [DOI] [PubMed] [Google Scholar]
- 141.Marcus DM, Goodman M, Jani AB, Osunkoya AO, Rossi PJ. A comprehensive review of incidence and survival in patients with rare histological variants of prostate cancer in the United States from 1973 to 2008. Prostate Cancer and Prostatic Disease. 2012;15:283–88. doi: 10.1038/pcan.2012.4. [DOI] [PubMed] [Google Scholar]
- 142.Jin RJ, Wang Y, Masumori N, Ishii K, Tsukamoto T, Shappell SB, Hayward SW, Kasper S, Matusik RJ. NE-10 Neuroendocrine Cancer Promotes the LNCaP Xenograft Growth in Castrated Mice. Cancer Res. 2004;64:5489–95. doi: 10.1158/0008-5472.CAN-03-3117. [DOI] [PubMed] [Google Scholar]
- 143.Stanbrough M, Leav I, Kwan PWL, Bubley GJ, Balk SP. Prostatic intraepithelial neoplasia in mice expressing an androgen receptor transgene in prostate epithelium. Proc Natl Acad Sci U S A. 2001;98:10823–28. doi: 10.1073/pnas.191235898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Albertelli MA, Scheller A, Brogley M, Robins DM. Replacing the Mouse Androgen Receptor with Human Alleles Demonstrates Glutamine Tract Length-Dependent Effects on Physiology and Tumorigenesis in Mice. Mol Endocrinol. 2006;20:1248–60. doi: 10.1210/me.2006-0021. [DOI] [PubMed] [Google Scholar]
- 145.Zhu C, Luong R, Zhuo M, Johnson DT, McKenney JK, Cunha GR, Sun Z. Conditional Expression of the Androgen Receptor Induces Oncogenic Transformation of the Mouse Prostate. J Biol Chem. 2011;286:33478–88. doi: 10.1074/jbc.M111.269894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Zhang X, Morrissey C, Sun S, Ketchandji M, Nelson PS, True LD, Vakar-Lopez F, Vessella RL, Plymate SR. Androgen Receptor Variants Occur Frequently in Castration Resistant Prostate Cancer Metastases. PLoS ONE. 2011;6:e27970. doi: 10.1371/journal.pone.0027970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Sun S, Sprenger CCT, Vessella RL, Haugk K, Soriano K, Mostaghel EA, Page ST, Coleman IM, Nguyen HM, Sun H, Nelson PS, Plymate SR. Castration resistance in human prostate cancer is conferred by a frequently occurring androgen receptor splice variant. The Journal of Clinical Investigation. 2010;120:2715–30. doi: 10.1172/JCI41824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Liu G, Sprenger C, Sun S, Epilepsia KS, Haugk K, Zhang X, Coleman I, Nelson PS, Plymate S. AR Variant ARv567es Induces Carcinogenesis in a Novel Transgenic Mouse Model of Prostate Cancer. Neoplasia (N Y) 2013;15:1009–17. doi: 10.1593/neo.13784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Cunha GR. Mesenchymal-epithelial interactions: past, present, and future. Differentiation. 2008;76:578–86. doi: 10.1111/j.1432-0436.2008.00290.x. [DOI] [PubMed] [Google Scholar]
- 150.Ricke EA, Williams K, Lee YF, Couto S, Wang Y, Hayward SW, Cunha GR, Ricke WA. Androgen hormone action in prostatic carcinogenesis: stromal androgen receptors mediate prostate cancer progression, malignant transformation and metastasis. Carcinogenesis. 2012;33:1391–98. doi: 10.1093/carcin/bgs153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Lai KP, Yamashita S, Huang CK, Yeh S, Chang C. Loss of stromal androgen receptor leads to suppressed prostate tumourigenesis via modulation of pro-inflammatory cytokines/chemokines. EMBO Mol Med. 2012;4:791–807. doi: 10.1002/emmm.201101140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Bhowmick N, Chytil A, Plieth D, Gorska A, Dumont N, Shappell S, Washington M, Neilson E, Moses H. TGF-beta signaling in fibroblasts modulates the oncogenic potential of adjacent epithelia. Science. 2004;303:848–51. doi: 10.1126/science.1090922. [DOI] [PubMed] [Google Scholar]
- 153.Li X, Placencio V, Iturregui JM, Uwamariya C, Sharif-Afshar AR, Koyama T, Hayward SW, Bhowmick NA. Prostate tumor progression is mediated by a paracrine TGF-[beta]/Wnt3a signaling axis. Oncogene. 2008;27:7118–30. doi: 10.1038/onc.2008.293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Franco OE, Hayward SW. Chapter Nine - Targeting the Tumor Stroma as a Novel Therapeutic Approach for Prostate Cancer. In: Keiran SMS, editor. Advances in Pharmacology. Academic Press; 2012. pp. 267–313. [DOI] [PubMed] [Google Scholar]
- 155.Beltran H, Rickman DS, Park K, Chae SS, Sboner A, MacDonald TY, Wang Y, Sheikh KL, Terry Sp, Tagawa ST, Dhir R, Nelson JB, de la Taille A, Allory Y, Gerstein MB, Perner S, Pienta KJ, Chinnaiyan AM, Wang Y, Collins CC, Gleave ME, Demichelis F, Nanus DM, Rubin MA. Molecular Characterization of Neuroendocrine Prostate Cancer and Identification of New Drug Targets. Cancer Discovery. 2011;1:487–95. doi: 10.1158/2159-8290.CD-11-0130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Mosquera JM, Beltran H, Park K, MacDonald TY, Robinson BD, Tagawa ST, Perner S, Bismar TA, Erbersdobler A, Dhir R, Nelson JB, Nanus DM, Rubin MA. Concurrent AURKA and MYCN gene amplifications are harbingers of lethal treatment-related neuroendocrine prostate cancer. Neoplasia. 2013;15:1–10. doi: 10.1593/neo.121550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Watson PA, Chen YF, Balbas MD, Wongvipat J, Socci ND, Viale A, Kim K, Sawyers CL. Constitutively active androgen receptor splice variants expressed in castration-resistant prostate cancer require full-length androgen receptor. Proc Natl Acad Sci U S A. 2010;107:16759–65. doi: 10.1073/pnas.1012443107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Crnalic S, Hörnberg E, Wikström P, Lerner UH, Tieva Å, Svensson O, Widmark A, Bergh A. Nuclear androgen receptor staining in bone metastases is related to a poor outcome in prostate cancer patients. Endocr Relat Cancer. 2010;17:885–95. doi: 10.1677/ERC-10-0059. [DOI] [PubMed] [Google Scholar]