

Abstract
Advances in brain imaging technology in the past five years have contributed greatly to the understanding of Alzheimer’s disease (AD). Here, we review recent research related to amyloid imaging, new methods for magnetic resonance imaging analyses, and statistical methods. We also review research that evaluates AD risk factors and brain imaging, in the context of AD prediction and progression. We selected a variety of illustrative studies, describing how they advanced the field and are leading AD research in promising new directions.
Keywords: Alzheimer’s disease, amyloid, imaging, magnetic resonance imaging, methods, positron emission tomography, prediction, progression, risk factors
Alzheimer’s disease (AD) is the most prevalent neurodegenerative disorder worldwide. It is the sixth most common cause of death in the US. All of us are at risk, and there is no known cure. About 13.2 million Americans will be diagnosed with AD by the year 2050 [1]. Late onset AD, the more common form of the disease, is 58–74% heritable [2–4]. Apolipoprotein E ε4 allele (APOE4) is a long-standing known AD risk gene [5], that thus far increases late-onset AD risk by the greatest factor [6]. However, in the last three years, large multiple-cohort genome-wide association studies (GWAS) have found several new single nucleotide polymorphisms (SNPs) that each affect the lifetime odds ratios for developing AD by approximately ±0.10–0.20 [7–10]. However, much of the genetic risk for AD is still unexplained, probably because a large number of genetic variants, each with a small effect, combine with environmental factors to contribute to AD onset. As we will show later, neuroimaging can help reveal the function of these new AD risk variants and how they influence brain integrity over the lifespan.
By the time AD can be detected using standard clinical assessments, the brain typically has undergone widespread irreversible neuronal and synaptic loss [12]. This neurodegeneration may be promoted by long-standing toxic processes such as amyloid aggregation and neurofibrillary tangle formation (the hallmarks of AD) and inflammation. To avert the looming AD crisis, we must identify better ways to track and treat AD in its earliest stages, even in people who are presymptomatic. Neuroimaging is an ideal tool for this; in the past five years alone, it has helped to show how controllable lifestyle factors, including exercise, education, and dietary factors such as folate and iron levels, affect brain integrity and disease risk later in life [13–18]. Neuroimaging advances, such as in vivo mapping of amyloid plaques and tau neurofibrillary tangles, and increasingly sensitive techniques for detecting atrophy on magnetic resonance images (MRI) have also made it easier to track AD progression. Large-scale genetic and epidemiological analyses of brain images may also discover factors that promote and deter atrophy. Only in the last few years have we routinely seen analyses of hundreds to thousands of brain scans. The largest neuroimaging genetics study to date [19] offered ~99.92% power to detect genetic variants that affect as little as 1% of the variance in hippocampal volume. Given this new level of power to distinguish risk gene effects on the brain [20], whole new lines of discovery are possible. We now know how multiple AD risk-conferring genes affect brain function and structure [21], and we are beginning to discover networks of genetic risk factors that affect the brain simultaneously. Finally, a major line goal of AD imaging is to identify at-risk older adults who are most likely to decline cognitively. If a drug trial uses neuroimaging as an outcome measure, the time and cost of the trial will be greatly reduced if imminent decliners can be selected in advance. In this paper we note ways in which imaging help identify decliners, boosting power for AD-related clinical trials.
AMYLOID IMAGING
A major advance in AD research over the past 5 years has been the ability to assess levels of amyloid plaques and tau neurofibrillary tangles in the living brain. Before amyloid imaging was possible, AD could only be definitively diagnosed at autopsy based on characteristic microscopic features [22]. Some early pioneering studies linked postmortem pathology to in vivo patterns of glucose metabolism measured using fluorodeoxyglucose (FDG) positron emission tomography (PET) [23]. However, typically, the unpredictable interval between the patients’ last clinical assessments and their deaths make it difficult to compare clinical symptoms to AD pathology. By contrast, amyloid PET scans of the living brain make it possible to evaluate longitudinal changes in AD pathology, and to relate them to clinical changes assessed at the same time as the scans. A key goal has been to establish these PET measures as reliable biomarkers of clinical severity or disease risk. If a correlation is established with cognitive scores, or with future decline in those scores, the PET measures become more promising metrics to evaluate treatment and prevention efforts. Proof of concept for plaque and tangle PET imaging in living humans was established in 2002 [24] using 2-(1-(6-[(2-[18F]fluoroethyl)(methyl)amino]-2-naphthyl)ethylidene)malononitrile (also known as [18F]FDDNP). In 2004, the PET probe, N-methyl-[11C]2-(4-methylaminophenyl)-6-hydroxy benzothiazole (also known as Pittsburgh Compound B or [11C]PIB) was also introduced [25]. Like [18F]FDDNP, [11C]PIB allows PET imaging of amyloid plaques in the living brain. However, unlike [18F]FDDNP, [11C]PIB has a higher signal to noise ratio and is specific to senile amyloid plaques, while [18F]FDDNP signal reflects senile and diffuse amyloid plaques as well as neurofibrillary tangle load [26]. [11C]PIB has a shorter half-life, so scans must be performed very nearby, soon after probe creation. Depending on study goals, each probe may offer advantages. [18F]FDDNP is particularly useful for examining medial temporal lobe pathology, where neurofibrillary tangles are likely to predominate in preclinical AD [27], while [11C]PIB is useful for studies that focus specifically on amyloid deposition. Higher levels of both [11C]PIB and [18F]FDDNP are correlated with lower cerebrospinal fluid (CSF) levels of amyloid-β 42 (Aβ42) [28, 29], the main building block for amyloid plaques, and higher levels of CSF tau [29], the building block for neurofibrillary tangles. This provides converging evidence for the validity of these tools.
PET imaging of AD pathology has helped chart the trajectory of pathology as it spreads in the living brain. [18F]FDDNP has shown regional plaque and tangle load differences between cognitively intact older adults and those with mild cognitive impairment (MCI) [30]. Cognitive performance was linked to the plaque and tangle load, and the associations were displayed across the brain in 3D [31]. PET measures were correlated with subtle cognitive differences even in the normal elderly, and thus show promise for gauging presymptomatic disease progression. In a cross-sectional study, we found that those with lower cognitive ability had higher [18F]FDDNP signal in regions that approximated the classic Braak and Braak trajectory; as performance declined, pathology increased in lateral temporal, parietal, and frontal cortices [31] (Fig. 1). Cognitive performance was also related to [11C]PIB signal in normal elderly and MCI subjects. In one study, the relationship between amyloid load and episodic memory was mediated by hippocampal volume [32].
Fig. 1.
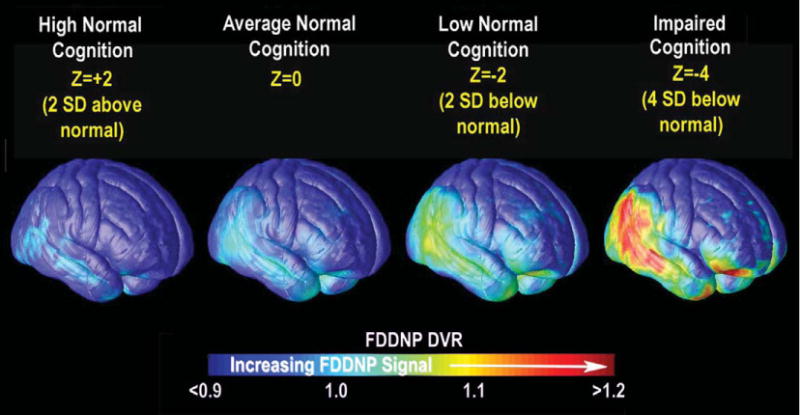
Spread of AD pathology mapped with PET. By regressing cortical measures of [18F]FDDNP signal (DVR) against cognitive scores in cross sectional data, we computed the pattern of pathology for subjects who were certain numbers of standard deviations above and below the norm for cognitive performance. Red indicates where greater predicted [18F]FDDNP signal was associated with poorer cognition based on a nonlinear spatially-varying model. Adapted from Braskie et al. [31] with permission from the authors and publishers.
Higher levels of [11C]PIB signal in the brain are heritable [33], associated with family history of AD [34], and similar in monozygotic twins discordant for AD [35]. APOE genotype, the strongest genetic risk factor for late onset AD, is related to both [11C]PIB and [18F]FDDNP signal in cognitively intact older adults [36, 37]. However, it does not fully explain the heritability of [11C]PIB signal [33], suggesting that amyloid imaging on a large scale may eventually uncover new genetic risk factors for late onset AD. Longitudinal [11C]PIB and structural MRI have also offered insight into disease progression, showing that amyloid deposition occurs at a constant slow rate while neurodegeneration accelerates; clinical symptoms were related more strongly to neurodegeneration than to amyloid deposition, which typically occurs before cognitive decline is evident [38]. However, [11C]PIB signal levels can still predict conversion from normal cognition and amnestic MCI to AD [39–41]. Additionally, [11C]PIB data were complementary to MRI data and together, both allowed for a more accurate AD diagnosis [42]. [11C]PIB has even been used to evaluate how an AD drug treatment (bapineuzumab) affects amyloid plaque load in vivo [43]. Other [11C]PIB studies explored the cognitive reserve hypothesis, which suggests that people with brains that better compensate for physiological deficits may accumulate more pathology before clinical symptoms are evident [44]. Education modulates the relationship between [11C]PIB signal and cognition, with greater plaque deposition relating to cognition less strongly in more highly educated adults [45, 46]. Amyloid imaging has also been useful for evaluating disruptions to the default mode network (DMN), a co-activated network of cortical brain regions that show more functional MRI activity at rest than during a directed task. The DMN shows deficits in AD patients [47], and DMN activity strength correlates with working memory performance [48]. Both [18F]FDDNP and [11C]PIB have shown that higher AD pathology is associated with lower DMN activity, even in cognitively intact older adults [49–53].
New PET amyloid probes such as [(18)F]3′-F-PiB (Flutemetamol) [54], (18)F-AV-45 (Florbetapir) [55], and (18)F-AV-1 (Florbetaben) [56] are now being investigated. These combine the specificity of [11C]PIB with the longer half-life of [18F]FDDNP, so the amyloid-specific probe can be created at a separate location and shipped. A promising lead, [18F]-THK523, also exists for in vivo imaging of neurofibrillary tangles, but has not yet been human-tested [57].
NEW METHODS FOR MRI ANALYSIS
Both greater ventricular volume [58–61] and lower medial temporal lobe structure volumes, especially of the hippocampus [60–65], predict cognitive decline and are associated with AD and genetic risk for AD. However, manual segmentation of these structures is time-consuming and is a limiting factor when a large study sample size is needed to obtain reliable results. Methods for fully automatic segmentation of subcortical structures have greatly improved in recent years. Large numbers of subjects can now be processed accurately and quickly, with high reproducibility. This allows researchers to detect subtle relationships more efficiently, while controlling for multiple confounds simultaneously. For example, the Enhancing NeuroImaging Genetics through Meta-Analysis (ENIGMA) project recently analyzed hippocampal volume using automated methods in 21,000 subjects scanned at over 20 sites worldwide [19]. This was the first study in history to identify new genetic variants that influence hippocampal volumes; clearly, traditional manual tracing would not have been feasible in such a sample size.
One new hippocampal segmentation method is a machine learning method based on “adaptive boosting”, in which an automated method learns from expert manual tracings; it can also learn from its mistakes. AdaBoost [66] is a meta-algorithm that combines weak classifiers (those that do not perform perfectly on their own but perform better than chance) such that regions with hippocampal segmentation errors receive more attention in subsequent iterations. This method has been extended to segment hippocampi of large numbers of AD and MCI patients automatically. The level of agreement between automated segmentations and manual ones was similar to agreement between two expert trained raters working independently. However, automated segmentation can be used to analyze hundreds of scans in a few hours [67]. The segmentation of MRI data from large samples, such as the Alzheimer’s Disease Neuroimaging Initiative (ADNI) cohort, has led to detailed calculations of sample sizes required to detect correlations between hippocampal structure, clinical assessments, and CSF biomarkers [68]. Work is still underway to determine which segmentation methods agree best with manual tracings. The ENIGMA consortium, which performed the largest-ever hippocampal volume analysis, concluded that the most accurate software depends on the dataset, but the methods correlate well with each other, as long as some manual quality control is performed [19]. Shape-based morphometry studies have attempted to evaluate measures of hippocampal structure more complex than volume, as subregional measures may better distinguish diagnostic groups or predict imminent decline. One study combined radial distance (the distance from the medial core of structure to each surface point) and multivariate tensor-based morphometry (mTBM; which uses spatial derivatives of the deformation maps used to register each brain image to a template image) [69] to study hippocampal structure in AD and MCI patients and normal controls [70]. This combines complementary information from radial distance, which measures hippocampal size in terms of the surface normal direction, and mTBM, which measures deformation within surfaces, to identify differences in hippocampal and lateral ventricle structure that relate to diagnosis (Fig. 2). Except for group differences between AD and MCI in the hippocampus (where mTBM performed the best), these combined methods detected diagnostic group differences at least as well as mTBM, TBM, or radial distance alone, allowing statistically significant differences to be detected in smaller samples [70]. This may be partly because TBM resolves more diffuse atrophy (such as in the temporal lobe as a whole) more sensitively than smaller-scale changes (such as in the hippocampus) [71].
Fig. 2.
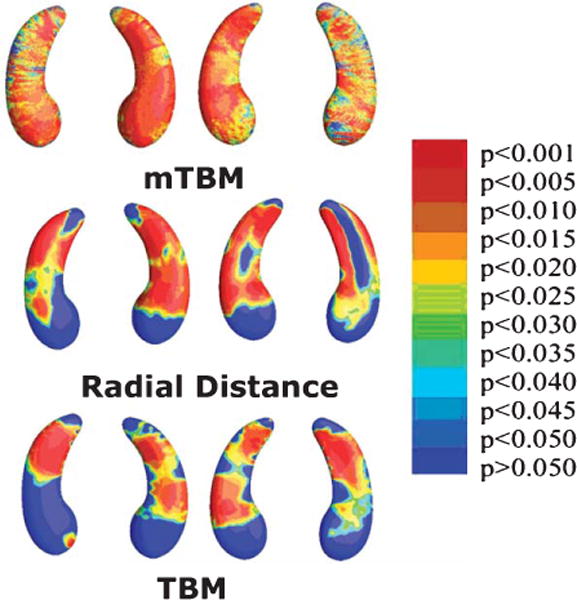
Mapping hippocampal correlates of tau proteins in the CSF. Here 3D maps show where alterations of hippocampal shape relate to levels of tau protein, measured in the CSF using lumbar puncture. To create these maps, we use a method called ‘multivariate tensor-based morphometry’ (mTBM; top row), as well as other methods to assess surface morphometry: radial distance maps (middle row) and standard TBM (bottom row). Non-blue colors show vertices with statistical differences, at the 0.05 level, uncorrected. Relationships were detected most sensitively with mTBM. Clearly, advanced mathematical methods can boost power to pick up associations between brain and CSF biomarkers, offering more detail than traditional measures of hippocampal volume. Adapted from Wang et al. [70] with permission of the authors and publishers.
Other methods target the ventricular system. Ventricular enlargement is the most prominent radiological marker of atrophy on brain MRI, albeit a relatively nonspecific one. Our laboratory recently created and validated an automated three-dimensional surface-meshing tool to automatically extract the lateral ventricles of the brain [72]. Previously, the narrowness of the inferior and posterior horns made it difficult for a computer program to label all regions of the ventricles consistently, but this new tool used surface models from several manually traced images, and aligned them to a new scan using a warping algorithm. By averaging several estimates of the segmentation, we obtained highly accurate and robust results. Such an approach, termed multi-atlas segmentation, decreased segmentation error and increased the statistical power to differentiate AD patients from controls [72]. When used to map ventricular changes in the ADNI cohort, baseline ventricular morphology was correlated with a cognitive decline over the following year, and was related to CSF Aβ42 levels [73]. This confirms earlier study findings that ventricular morphology is both AD-relevant and predictive of clinical symptoms. Such information is invaluable in AD prevention trials where an enriched sample is necessary to evaluate treatments using a reasonable sample size.
STATISTICAL METHODS
Recent publications have provided useful guidelines for the most efficient use of resources in future studies. By assessing changes in groups of subjects scanned longitudinally, several studies have empirically determined the best methods for boosting statistical power, often by calculating sample sizes needed to evaluate certain effects. For instance, in a large (n= 1,030) longitudinal TBM study, the ability to detect structural atrophy over time was enhanced by summarizing changes in statistically defined regions of interest derived from a training sample of 22 AD patients. Sample sizes needed to detect brain changes using the best MRI analysis methods were far lower than those needed to detect cognitive change using typical clinical tests [74]. In general, longer follow-up periods (such as 24 months) increased the power to detect change. Even so, many subjects drop out of longitudinal studies, so the added power of a long trial must be traded-off against the risk of losing too many subjects. The attrition rate for ADNI is only around 7% per year, but, in simulations, when more than 15–16% of subjects dropped out annually, a shorter time period (such as 12 months) provided more statistical power [75].
Cerebral glucose metabolism rate changes over one year are also easier to detect using statistically-defined regions of interest derived from a training subsample of the data [76]. Even so, for a statistically-designed region of interest to be approved by the FDA as a reasonable outcome measure in a clinical trial, some criteria would have to be determined to identify it from a subset of the data. Given the power of statistical brain maps and the range of methods to analyze them, detailed head-to-head comparisons with more standard volumetric measures are needed [77]. In related work, our lab used a Support Vector Machine algorithm to combine numerous types of biomarkers, including brain imaging, to classify subjects as AD, MCI, or healthy elderly controls [78]. MRI measures best identified AD subjects, but PET-FDG and CSF biomarkers (particularly Aβ42) better identified subjects as having MCI. This work also showed that if those most likely to decline can be identified, fewer subjects are required to detect a specific slowing of the atrophy rate over time in response to a given treatment [78]. Finally, many studies combine multiple biomarkers for predicting decline, but some of the biomarker data are often unavailable for some of the subjects. For instance, in ADNI, only half the subjects had FDG-PET and still fewer had amyloid imaging or lumbar puncture to assess CSF analytes. Sparse coding methods, an innovation from mathematics, are being used to address the problem of missing data in predictive models [79].
RISK FACTORS
Both genetic and environmental factors influence AD risk, and in recent years, many of these factors have also been shown to influence brain structure and function in regions relevant to AD. Numerous studies have demonstrated that possession of APOE4 is related to brain differences in healthy and cognitively impaired older adults [80]. In recent years, several GWAS have identified and confirmed new genetic risk factors for late onset AD [7–9, 81, 82]. Some top AD polymorphisms have since been associated with differences in brain structure [83] and function [84,85]. For instance, diffusion tensor imaging has shown that the allele C in the clusterin (CLU) gene at rs11136000 is associated with lower white matter integrity in healthy young adults in many regions affected in AD [11] (Fig. 3). At least one copy of this adverse allele is carried by approximately 84% of non-demented Caucasians [6], and the C allele increases the lifetime odds of AD by 1.14 in Caucasians [6]. This suggests a developmental vulnerability to AD in these young people. The CLU risk allele has also been associated with differences in brain function, assessed using functional MRI [84, 85]. Variants in other top AD-related genes such as PICALM, CR1, and BIN1 have been associated with AD-relevant measures such as hippocampal volume or gray matter density, entorhinal cortex thickness [86, 87]. Several new studies have also found that maternal versus paternal (or no) family history of AD may affect brain structure in ways that are important to AD risk [88–92]. Maternal family history may be useful to include in future genetic studies of AD, and AD family history in general may be useful for selecting people for clinical trials, especially when study efficiency relies on identifying those likely to decline cognitively.
Fig. 3.
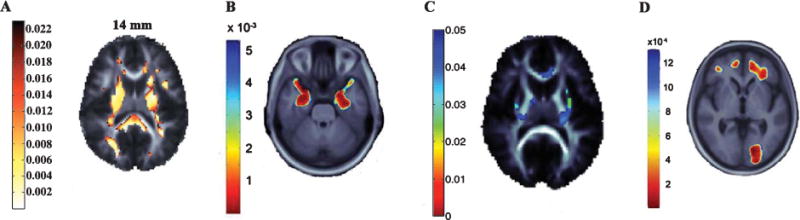
Commonly carried genes associated with brain atrophy on MRI and brain integrity on diffusion tensor imaging. Highlighted voxels in each panel represent p-values indicating the relationship between a genetic variant and a brain feature. A) The AD risk allele C at rs11136000 in the CLU gene was associated with lower diffusion tensor imaging fractional anisotropy (FA), a measure of white matter integrity, in healthy young adults [11]. B) rs10845840 in the GRIN2B glutamate receptor gene—important in learning and memory—is associated with medial temporal lobe structure in MCI patients [94]. C) The H63D polymorphism in the HFE gene was associated with white matter FA in healthy young adults [13]. D) Regional brain tissue volumes in healthy older adults was lower in those who carried the obesity-associated allele of rs3751812 in the FTO gene [99], in line with prior work showing higher brain atrophy in more obese people. Figures are adapted from the referenced publications with the permission of the authors and publishers.
Most of the newly discovered AD risk genes increase a person’s aggregate risk only mildly [6], so some tools aim to evaluate the effects of multiple genetic variants simultaneously. A few studies have used polygenic scores to assess the cumulative effect of certain AD risk variants on brain structure. In these studies, each subject is assigned a score based on the number of allele copies for certain previously identified risk variants, in some cases using odd ratios to weight the importance of different genes. This allows researchers to assess cumulative effects of several genetic risk factors on brain measures related to AD [86, 93]. In addition, several new tools are available that offer more flexibility for exploring genetic risk than a single weighted value can provide. For instance, our lab created a tool that allowed a genome-wide search for gene variants that are associated with brain structure [94]. This tool identified one gene variant, in the GRIN2B gene, that was associated with temporal lobe volume in AD and MCI patients and healthy older controls (Fig. 3). The protein encoded by GRIN2B is the N-methyl-D-aspartate (NMDA) glutamate receptor NR2B subunit, which is important in learning and memory and is already a target for AD research [94]. Another new tool allows for a voxelwise gene-wide association, which searches the whole brain, point-by-point, for evidence of gene effects [95]. Each gene may contain multiple SNPs, many of which may be highly correlated, so this method tests all SNPs in a gene at once, reducing the number of tests performed, and thus increasing statistical power to detect effects (as there is a less heavy correction for multiple comparisons).
Yet another tool tested genome-wide association at each voxel in the brain, studying only the most associated variant at each voxel to reduce the number of multiple comparisons [96]. An additional new tool to emerge accepts input from the user as to which SNPs will be evaluated, and controls for the effect of each SNP when evaluating each other SNP in the input list [97]. One recent study used penalized linear discriminant analysis to first identify a characteristic pattern of AD-related brain differences. We then used that imaging signature as a biomarker in a genome-wide association, performed using sparse reduced-rank regression, to identify genetic variants of interest [98]. Tools such as these help researchers to evaluate how multiple genetic variants together increase AD risk. They can also identify new genetic risk variants, bringing us closer to determining key mechanisms in the disease process.
Several studies have revealed how environmental risk factors for AD, such as cardiovascular risk, homocysteine levels, and insulin resistance affect brain structure and function in AD-relevant ways. For instance, obesity and a commonly-carried risk gene for obesity (FTO) (Fig. 3) have been linked to more atrophied brains and smaller hippocampi in older adults [15, 17, 99–101]. Along with increased body mass, higher blood pressure has also been associated with differences in functional brain patterns in AD-relevant regions during a memory task [102], and blood cholesterol levels have been linked to hippocampal and other brain gray matter volume [103, 104]. Higher systolic blood pressure has even been associated with more amyloid deposition in AD-relevant brain regions in healthy older adults [105]. High levels of homocysteine (a derivative of the amino acid methionine) elevate risk for cardiovascular disease [106] and AD [107]. They have been linked to greater white matter atrophy in a large sample of older adults, and in the patients with MCI alone [18]. This effect may relate in part to a commonly-carried variant in the folate pathway gene, MTHFR [108]. Using B vitamins to lower homocysteine levels may slow the rate of brain atrophy in MCI patients [109]. Higher peripheral insulin is also associated with less AD-related brain atrophy and dementia severity in early AD [110]; in cognitively intact older adults, insulin resistance was associated with hippocampal volume [111]. Finally, iron homeostasis is important for healthy brain functioning, and its disruption may lead to cognitive impairment [112]. Our laboratory found that levels of serum transferrin, a protein that transports iron in the body, and a variant in the hemochromatotic HFE gene were both associated with white matter integrity in young adults [13] (Fig. 3). Gaining a better understanding of how environmental risk factors affect brain structure aids AD research in at least three ways: 1) it provides focal points for treatment efforts, and a way to evaluate them even before clinical decline is evident; 2) it helps identify those most likely to decline cognitively, boosting statistical power for clinical trials; and 3) it uncovers possible factors that should be controlled for when examining other genetic and environmental effects.
PREDICTING COGNITIVE DECLINE OR CONVERSION TO AD
The ability to predict who will decline cognitively or develop AD over time is crucial to AD research. It provides a focus for treatment efforts, boosts power for clinical trials, and allows for further investigation of genetic and environmental factors that contribute to AD before clinical symptoms are evident and possibly before massive destruction of brain tissue has taken place. Predictors of cognitive decline in cognitively normal older adults include MRI measures of temporal and parietal structures [113] (particularly the CA1 of the hippocampus and the subiculum [114]) and the lateral ventricles [115]. Information from both MRI and CSF biomarkers was better for predicting cognitive decline than either measure alone [116]. Similarly, several other studies have found that multiple measures together best predict MCI patient conversion to AD. These measures typically included baseline cognitive tests [117–120], medial temporal lobe or hippocampal structure [117, 118, 120, 121], CSF biomarker levels [118], and cerebral glucose metabolism rates as measured by PET [119, 121]. Of studies that focused on individual biomarkers rather than combinations of them, two found that certain baseline MRI measures (medial temporal cortex or a structural abnormality score that reflect the degree of AD-like features) predict MCI conversion to AD somewhat better than CSF biomarker levels do [122, 123]. In fact, atrophy in a number of AD-related regions (such as the temporal and frontal lobes, temporoparietal cortex, anterior and posterior cingulate, and the precuneus) has been found to be greater in MCI patients who convert to AD within 3 years versus those who do not [124]. Other studies found that a smaller CA1 of the hippocampus or subiculum [125] and lower baseline right caudate volumes [126] were associated with increased conversion from MCI to AD. One study found that for those with moderate MCI, although hippocampal and ventricular volume predicted conversion to AD, baseline cognitive testing predicted it better [61]. Because brain structure typically changes before cognitive ability [12], the same study performed earlier before cognitive changes were evident may have shown different results.
DISEASE PROGRESSION
Understanding the progression of AD beginning in its presymptomatic phases is an essential goal for AD research for at least three reasons. First, determining in what order measurable biomarkers change helps clarify which factors help cause the disease and which may be incidental. Second, identifying the earliest biomarkers of change can be used to focus treatment efforts on those who do not yet have massive loss of brain tissue. Third, gaining a better understanding of the disease progression provides insight into the effects of treatment efforts or lifestyle factors at each disease stage. Several recent papers have used multiple types of biomarkers to assess the sequence of changes in early MCI and AD. In one study of amnestic MCI patients, hippocampal atrophy led to atrophy of the cingulum bundle and uncinate fasciculus, which was followed by glucose hypometabolism of the cingulate and subgenual cortices [127]. Another study assessed longitudinal amyloid deposition (using PIB PET scans) and ventricular expansion (using MRI) over 1 year. Ventricular expansion rates increased with worsening diagnosis and correlated with cognitive test scores, but amyloid deposition rates were similar among diagnostic groups. This suggests that amyloid deposition occurs at a constant slow rate, and that clinical symptoms relate to neurodegeneration rather than to amyloid deposition [38]. Other researchers found that low levels of CSF Aβ42 was correlated with brain and hippocampal atrophy rates and with the rate of ventricular expansion in cognitively normal older adults [128], demonstrating that the change in CSF Aβ42 precedes and can predict brain atrophy, even before AD symptoms are evident. Cross-sectionally, CSF Aβ42 correlated with whole brain volumes in nondemented subjects, but in AD, CSF tau and phosphorylated tau were higher in those with smaller brains; this suggests that Aβ toxicity may precede both clinical symptoms and changes in tau [129]. Other longitudinal studies focused on one imaging modality. Research showed that three years before an AD diagnosis, amnestic MCI patients showed brain loss largely in the medial temporal lobe (particularly anterior hippocampus, entorhinal cortex, and amygdala) compared with controls. By one year prior to diagnosis, this atrophy had spread to include the middle temporal gyrus and more posterior medial temporal lobe, as well as parts of the parietal lobe. By the time the patients progressed to AD, the temporal and parietal atrophy was more severe, and substantial frontal lobe atrophy had occurred as well [130]. This spread of atrophy roughly mirrors the pattern of neurofibrillary tangle progression noted by others [131], and is evidenced in cross-sectional research in which the temporal and parietal cortices, which are affected earlier [132], are also more severely atrophied compared with other cortical regions in AD versus MCI [133]. In MCI and AD patients, increased hippocampal volume loss rates over a one-year period were associated with lower CSF Aβ42 [134], and the atrophy rates increased with worsening diagnosis and cognitive decline [135]. Similarly, temporal lobe atrophy rates in MCI were associated with greater cognitive decline [136], and AD patients with higher CSF phosphorylated tau/Aβ42 ratios also had faster atrophy rates [136]. Others have found that hippocampal volumes are smaller in those with amnestic versus multi-domain MCI [137], and are good predictors of clinical diagnosis [138]. Together, these findings suggest that temporal lobe and hippocampal atrophy are excellent biomarkers for clinical and pathological aspects of AD, and that such atrophy may be an early sign of AD processes in asymptomatic older adults. In AD and MCI, it may serve as the most sensitive outcome measure for clinical trials [139].
Familial early onset AD (FAD), caused by rare autosomal dominant genetic mutations, also has offered insights into disease progression in recent years. The benefit of such studiesis that it is possible to determine, even in asymptomatic adults, who will develop AD, and approximately when. This precludes the need for lengthy prospective or retrospective studies to examine presymptomatic AD brain changes. Additionally, smaller initial sample sizes can be used because all of the mutation carriers will eventually develop AD as opposed to a much smaller fraction of older adults who are simply “at-risk” for AD based on environmental or genetic risk factors. Approximately 5.5 years prior to AD diagnosis, presymptomatic FAD mutation carriers had greater hippocampal atrophy rates than non-carriers, and smaller baseline hippocampi 3 years before diagnosis [140]. Additionally, FAD mutation carriers had lower white matter integrity in the columns of the fornix, even presymptomatically [141]. It is unclear to what extent FAD brain changes serve as a good model for late-onset AD, presymptomatically or otherwise. Expansion of such research to directly compare the two will be useful, because it may facilitate better treatments for those who are presymptomatic. It may also offer insights into general AD triggers and features of disease progression regardless of the specific genetic risk.
CONCLUSION
Improvements in brain scans and the toolstoanalyze them offer a wealth of opportunity in the field of AD research, allowing high-throughput analyses and new insights into disease processes. These developments have led to rapid discoveries regarding disease risk factors, how to predict AD onset, and how to monitor its progression. Such insights are essential for planning future studies as they identify sensitive biomarkers that most reliably reflect disease processes, allowing for more focused treatment and prevention efforts with the greatest statistical power to detect their effects.
Acknowledgments
This manuscript was supported, in part, by the National Institute on Aging (R01 AG040060-01), by the National Institute of Child Health and Human Development (R01 HD050735), and by the Northern California Institute for Research & Education/NIH (RC2 AG036535).
Footnotes
Authors’ disclosures available online (http://www.j-alz.com/disclosures/view.php?id=1335).
References
- 1.Hebert LE, Scherr PA, Bienias JL, Bennett DA, Evans DA. Alzheimer disease in the US population: Prevalence estimates using the 2000 census. Arch Neurol. 2003;60:1119–1122. doi: 10.1001/archneur.60.8.1119. [DOI] [PubMed] [Google Scholar]
- 2.Bergem AL, Engedal K, Kringlen E. The role of heredity in late-onset Alzheimer disease and vascular dementia. A twin study. Arch Gen Psychiatry. 1997;54:264–270. doi: 10.1001/archpsyc.1997.01830150090013. [DOI] [PubMed] [Google Scholar]
- 3.Gatz M, Pedersen NL, Berg S, Johansson B, Johansson K, Mortimer JA, Posner SF, Viitanen M, Winblad B, Ahlbom A. Heritability for Alzheimer’s disease: The study of dementia in Swedish twins. J Gerontol A Biol Sci Med Sci. 1997;52:M117–M125. doi: 10.1093/gerona/52a.2.m117. [DOI] [PubMed] [Google Scholar]
- 4.Gatz M, Reynolds CA, Fratiglioni L, Johansson B, Mortimer JA, Berg S, Fiske A, Pedersen NL. Role of genes and environments for explaining Alzheimer disease. Arch Gen Psychiatry. 2006;63:168–174. doi: 10.1001/archpsyc.63.2.168. [DOI] [PubMed] [Google Scholar]
- 5.Corder EH, Saunders AM, Strittmatter WJ, Schmechel DE, Gaskell PC, Small GW, Roses AD, Haines JL, Pericak-Vance MA. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science. 1993;261:921–923. doi: 10.1126/science.8346443. [DOI] [PubMed] [Google Scholar]
- 6.Bertram L, McQueen MB, Mullin K, Blacker D, Tanzi RE. Systematic meta-analyses of Alzheimer disease genetic association studies: The AlzGene database. Nat Genet. 2007;39:17–23. doi: 10.1038/ng1934. [DOI] [PubMed] [Google Scholar]
- 7.Harold D, Abraham R, Hollingworth P, Sims R, Gerrish A, Hamshere ML, Pahwa JS, Moskvina V, Dowzell K, Williams A, Jones N, Thomas C, Stretton A, Morgan AR, Lovestone S, Powell J, Proitsi P, Lupton MK, Brayne C, Rubinsztein DC, Gill M, Lawlor B, Lynch A, Morgan K, Brown KS, Passmore PA, Craig D, McGuinness B, Todd S, Holmes C, Mann D, Smith AD, Love S, Kehoe PG, Hardy J, Mead S, Fox N, Rossor M, Collinge J, Maier W, Jessen F, Schurmann B, van den Bussche H, Heuser I, Kornhuber J, Wiltfang J, Dichgans M, Frolich L, Hampel H, Hull M, Rujescu D, Goate AM, Kauwe JS, Cruchaga C, Nowotny P, Morris JC, Mayo K, Sleegers K, Bettens K, Engelborghs S, De Deyn PP, Van Broeckhoven C, Livingston G, Bass NJ, Gurling H, McQuillin A, Gwilliam R, Deloukas P, Al-Chalabi A, Shaw CE, Tsolaki M, Singleton AB, Guerreiro R, Muhleisen TW, Nothen MM, Moebus S, Jockel KH, Klopp N, Wichmann HE, Carrasquillo MM, Pankratz VS, Younkin SG, Holmans PA, O’Donovan M, Owen MJ, Williams J. Genome-wide association study identifies variants at CLU and PICALM associated with Alzheimer’s disease. Nat Genet. 2009;41:1088–1093. doi: 10.1038/ng.440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lambert JC, Heath S, Even G, Campion D, Sleegers K, Hiltunen M, Combarros O, Zelenika D, Bullido MJ, Tavernier B, Letenneur L, Bettens K, Berr C, Pasquier F, Fievet N, Barberger-Gateau P, Engelborghs S, De Deyn P, Mateo I, Franck A, Helisalmi S, Porcellini E, Hanon O, de Pancorbo MM, Lendon C, Dufouil C, Jaillard C, Leveillard T, Alvarez V, Bosco P, Mancuso M, Panza F, Nacmias B, Bossu P, Piccardi P, Annoni G, Seripa D, Galimberti D, Hannequin D, Licastro F, Soininen H, Ritchie K, Blanche H, Dartigues JF, Tzourio C, Gut I, Van Broeckhoven C, Alperovitch A, Lathrop M, Amouyel P. Genome-wide association study identifies variants at CLU and CR1 associated with Alzheimer’s disease. Nat Genet. 2009;41:1094–1099. doi: 10.1038/ng.439. [DOI] [PubMed] [Google Scholar]
- 9.Seshadri S, Fitzpatrick AL, Ikram MA, DeStefano AL, Gudnason V, Boada M, Bis JC, Smith AV, Carassquillo MM, Lambert JC, Harold D, Schrijvers EM, Ramirez-Lorca R, Debette S, Longstreth WT, Jr, Janssens AC, Pankratz VS, Dartigues JF, Hollingworth P, Aspelund T, Hernandez I, Beiser A, Kuller LH, Koudstaal PJ, Dickson DW, Tzourio C, Abraham R, Antunez C, Du Y, Rotter JI, Aulchenko YS, Harris TB, Petersen RC, Berr C, Owen MJ, Lopez-Arrieta J, Varadarajan BN, Becker JT, Rivadeneira F, Nalls MA, Graff-Radford NR, Campion D, Auerbach S, Rice K, Hofman A, Jonsson PV, Schmidt H, Lathrop M, Mosley TH, Au R, Psaty BM, Uitterlinden AG, Farrer LA, Lumley T, Ruiz A, Williams J, Amouyel P, Younkin SG, Wolf PA, Launer LJ, Lopez OL, van Duijn CM, Breteler MM. Genome-wide analysis of genetic loci associated with Alzheimer disease. JAMA. 2010;303:1832–1840. doi: 10.1001/jama.2010.574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ertekin-Taner N. Genetics of Alzheimer disease in the pre- and post-GWAS era. Alzheimers Res Ther. 2010;2:3. doi: 10.1186/alzrt26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Braskie MN, Jahanshad N, Stein JL, Barysheva M, McMahon KL, de Zubicaray GI, Martin NG, Wright MJ, Ringman JM, Toga AW, Thompson PM. Common Alzheimer’s disease risk variant within the CLU gene affects white matter microstructure in young adults. J Neurosci. 2011;31:6764–6770. doi: 10.1523/JNEUROSCI.5794-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jack CR, Jr, Knopman DS, Jagust WJ, Shaw LM, Aisen PS, Weiner MW, Petersen RC, Trojanowski JQ. Hypothetical model of dynamic biomarkers of the Alzheimer’s pathological cascade. Lancet Neurol. 2010;9:119–128. doi: 10.1016/S1474-4422(09)70299-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jahanshad N, Kohannim O, Hibar DP, Stein JL, McMahon KL, de Zubicaray GI, Medland SE, Montgomery GW, Whitfield JB, Martin NG, Wright MJ, Toga AW, Thompson PM. Brain structure in healthy adults is related to serum transferrin and the H63D polymorphism in the HFE gene. Proc Natl Acad Sci U S A. 2012;109:E851–E859. doi: 10.1073/pnas.1105543109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ho AJ, Raji CA, Becker JT, Lopez OL, Kuller LH, Hua X, Dinov ID, Stein JL, Rosano C, Toga AW, Thompson PM. The effects of physical activity, education, and body mass index on the aging brain. Hum Brain Mapp. 2011;32:1371–1382. doi: 10.1002/hbm.21113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ho AJ, Raji CA, Becker JT, Lopez OL, Kuller LH, Hua X, Lee S, Hibar D, Dinov ID, Stein JL, Jack CR, Jr, Weiner MW, Toga AW, Thompson PM. Obesity is linked with lower brain volume in 700 AD and MCI patients. Neurobiol Aging. 2010;31:1326–1339. doi: 10.1016/j.neurobiolaging.2010.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ho AJ, Raji CA, Parikshak N, Becker JT, Lopez OL, Kuller LH, Hua X, Leow AD, Toga AW, Thompson PM. Mapping effects of body mass index, insulinemia, and diabetes mellitus on brain structure in cognitively normal elders. Am Acad Neurol. 2009;722009:A171. [Google Scholar]
- 17.Ho AJ, Raji CA, Saharan P, DeGiorgio A, Madsen SK, Hibar DP, Stein JL, Becker JT, Lopez OL, Toga AW, Thompson PM. Hippocampal volume is related to body mass index in Alzheimer’s disease. Neuroreport. 2011;22:10–14. doi: 10.1097/wnr.0b013e3283412868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rajagopalan P, Hua X, Toga AW, Jack CR, Jr, Weiner MW, Thompson PM. Homocysteine effects on brain volumes mapped in 732 elderly individuals. Neuroreport. 2011;22:391–395. doi: 10.1097/WNR.0b013e328346bf85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Stein JL, Medland SE, Vasquez AA, Hibar DP, Senstad RE, Winkler AM, Toro R, Appel K, Bartecek R, Bergmann Ø, Bernard M, Brown AA, Cannon DM, Chakravarty M, Christoforou A, Domin M, Grimm O, Hollinshead M, Holmes AJ, Homuth G, Hottenga J-J, Langan C, Lopez LM, Hansell NK, Hwang KS, Kim S, Laje G, Lee PH, Liu X, Loth E, Lourdusamy A, Maniega SM, Mattingsdal M, Mohnke S, Nho K, Nugent AC, O’Brien C, Papmeyer M, Pütz B, Ramasamy A, Rasmussen J, Rijpkema M, Risacher SL, Roddey JC, Rose EJ, Ryten M, Shen L, Sprooten E, Strengman E, Teumer A, Trabzuni D, Turner J, van Eijk K, van Erp TGM, van Tol M-J, Wittfeld K, Wolf C, Woudstra S, Aleman A, Alhusaini S, Almasy L, Binder EB, Brohawn DG, Cantor RM, Carless MA, Corvin A, Czisch M, Curran JE, Davies G, de Almeida MAA, Delanty N, Depondt C, Duggirala R, Dyer TD, Erk S, Fagerness J, Fox PT, Freimer NB, Gill M, Göring HHH, Hagler DJ, Hoehn D, Holsboer F, Hoogman M, Hosten N, Jahanshad N, Johnson MP, Kasperaviciute D, Kent JWJ, Kochunov P, Lancaster JL, Lawrie SM, Liewald DC, Mandl R, Matarin M, Mattheisen M, Meisenzahl E, Melle I, Moses EK, Mühleisen TW, Nauck M, Nöthen MM, Olvera RL, Pandolfo M, Pike GB, Puls R, Reinvang I, Rentería ME, Rietschel M, Roffman JL, Royle NA, Rujescu D, Savitz J, Schnack HG, Schnell K, Seiferth N, Smith C, Steen VM, Hernández MCV, van den Heuvel M, van der Wee NJ, Van Haren NEM, Veltman JA, Völzke H, Walker R, Westlye LT, Whelan CD, Agartz I, Boomsma DI, Cavalleri GL, Dale AM, Djurovic S, Drevets WC, Hagoort P, Hall J, Heinz A, Jack CRJ, Foroud TM, Le Hellard S, Macciardi F, Montgomery GW, Poline JB, Porteous DJ, Sisodiya SM, Starr JM, Sussmann J, Toga AW, Veltman DJ, Walter H, Weiner MW, ADNI, EpiGenConsortium, IMAGENConsortium, SaguenayYouthStudyGroup. Bis JC, Ikram MA, Smith AV, Gudnason V, Tzourio C, Vernooij MW, Launer LJ, DeCarli C, Seshadri S, for TheChange-Consortium. Andreassen OA, Apostolova LG, Bastin ME, Blangero J, Brunner HG, Buckner RL, Cichon S, Coppola G, de Zubicaray GI, Deary IJ, Donohoe G, de Geus EJC, Espeseth T, Fernández G, Glahn DC, Grabe HJ, Hardy J, Hulshoff Pol HE, Jenkinson M, Kahn RS, McDonald C, McIntosh AM, McMahon FJ, McMahon KL, Meyer-Lindenberg A, Morris DW, Müller-Myhsok B, Nichols TE, Ophoff RA, Paus T, Pausova Z, Penninx BW, Potkin SG, Sämann PG, Saykin AJ, Schumann G, Smoller JW, Wardlaw JM, Weale ME, Martin NG, Franke B, Wright MJ, Thompson PM, ENIGMA Identification of common variants associated with human hippocampal and intracranial volumes. Nat Genet. 2012;44:552–561. doi: 10.1038/ng.2250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Thompson PM, Martin NG, Wright MJ. Imaging genomics. Curr Opin Neurol. 2010;23:368–373. doi: 10.1097/WCO.0b013e32833b764c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kohannim O, Hua X, Rajagopalan P, Hibar DP, Jahanshad N, Toga AW, Jack CR, Weiner MW, Thompson PM, ADNI Accelerated brain atrophy mapped in carriers of multiple AD risk genes: Empowering clinical trials. Organization for Human Brain Mapping; 10–14 June 2012; Beijing, China. 2012. http://ww4.aievolution.com/hbm1201/index.cfm?do=abs.viewAbs&abs=4982. [Google Scholar]
- 22.Thompson PM, Vinters HV. Pathologic lesions in neurodegenerative diseases. Prog Mol Biol Transl Sci. 2012;107:1–40. doi: 10.1016/B978-0-12-385883-2.00009-6. [DOI] [PubMed] [Google Scholar]
- 23.Mega MS, Chen SS, Thompson PM, Woods RP, Karaca TJ, Tiwari A, Vinters HV, Small GW, Toga AW. Mapping histology to metabolism: Coregistration of stained whole-brain sections to premortem PET in Alzheimer’s disease. Neuroimage. 1997;5:147–153. doi: 10.1006/nimg.1996.0255. [DOI] [PubMed] [Google Scholar]
- 24.Shoghi-Jadid K, Small GW, Agdeppa ED, Kepe V, Ercoli LM, Siddarth P, Read S, Satyamurthy N, Petric A, Huang SC, Barrio JR. Localization of neurofibrillary tangles and beta-amyloid plaques in the brains of living patients with Alzheimer disease. Am J Geriatr Psychiatry. 2002;10:24–35. [PubMed] [Google Scholar]
- 25.Klunk WE, Engler H, Nordberg A, Wang Y, Blomqvist G, Holt DP, Bergstrom M, Savitcheva I, Huang GF, Estrada S, Ausen B, Debnath ML, Barletta J, Price JC, Sandell J, Lopresti BJ, Wall A, Koivisto P, Antoni G, Mathis CA, Langstrom B. Imaging brain amyloid in Alzheimer’s disease with Pittsburgh Compound-B. Ann Neurol. 2004;55:306–319. doi: 10.1002/ana.20009. [DOI] [PubMed] [Google Scholar]
- 26.Vallabhajosula S. Positron emission tomography radiopharmaceuticals for imaging brain Beta-amyloid. Semin Nucl Med. 2011;41:283–299. doi: 10.1053/j.semnuclmed.2011.02.005. [DOI] [PubMed] [Google Scholar]
- 27.Braak H, Braak E. Frequency of stages of Alzheimer-related lesions in different age categories. Neurobiol Aging. 1997;18:351–357. doi: 10.1016/s0197-4580(97)00056-0. [DOI] [PubMed] [Google Scholar]
- 28.Fagan AM, Mintun MA, Mach RH, Lee SY, Dence CS, Shah AR, LaRossa GN, Spinner ML, Klunk WE, Mathis CA, DeKosky ST, Morris JC, Holtzman DM. Inverse relation between in vivo amyloid imaging load and cerebrospinal fluid Abeta42 in humans. Ann Neurol. 2006;59:512–519. doi: 10.1002/ana.20730. [DOI] [PubMed] [Google Scholar]
- 29.Tolboom N, van der Flier WM, Yaqub M, Boellaard R, Verwey NA, Blankenstein MA, Windhorst AD, Scheltens P, Lammertsma AA, van Berckel BN. Relationship of cerebrospinal fluid markers to 11C-PiB and 18F-FDDNP binding. J Nucl Med. 2009;50:1464–1470. doi: 10.2967/jnumed.109.064360. [DOI] [PubMed] [Google Scholar]
- 30.Small GW, Kepe V, Ercoli LM, Siddarth P, Bookheimer SY, Miller KJ, Lavretsky H, Burggren AC, Cole GM, Vinters HV, Thompson PM, Huang SC, Satyamurthy N, Phelps ME, Barrio JR. PET of brain amyloid and tau in mild cognitive impairment. N Engl J Med. 2006;355:2652–2663. doi: 10.1056/NEJMoa054625. [DOI] [PubMed] [Google Scholar]
- 31.Braskie MN, Klunder AD, Hayashi KM, Protas H, Kepe V, Miller KJ, Huang SC, Barrio JR, Ercoli LM, Siddarth P, Satyamurthy N, Liu J, Toga AW, Bookheimer SY, Small GW, Thompson PM. Plaque and tangle imaging and cognition in normal aging and Alzheimer’s disease. Neurobiol Aging. 2010;31:1669–1678. doi: 10.1016/j.neurobiolaging.2008.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mormino EC, Kluth JT, Madison CM, Rabinovici GD, Baker SL, Miller BL, Koeppe RA, Mathis CA, Weiner MW, Jagust WJ. Episodic memory loss is related to hippocampal-mediated beta-amyloid deposition in elderly subjects. Brain. 2009;132(Pt 5):1310–1323. doi: 10.1093/brain/awn320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hinrichs AL, Mintun MA, Head D, Fagan AM, Holtzman DM, Morris JC, Goate AM. Cortical binding of pittsburgh compound B, an endophenotype for genetic studies of Alzheimer’s disease. Biol Psychiatry. 2010;67:581–583. doi: 10.1016/j.biopsych.2009.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mosconi L, Rinne JO, Tsui WH, Berti V, Li Y, Wang H, Murray J, Scheinin N, Nagren K, Williams S, Glodzik L, De Santi S, Vallabhajosula S, de Leon MJ. Increased fibrillar amyloid-{beta} burden in normal individuals with a family history of late-onset Alzheimer’s. Proc Natl Acad Sci U S A. 2010;107:5949–5954. doi: 10.1073/pnas.0914141107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Scheinin NM, Aalto S, Kaprio J, Koskenvuo M, Raiha I, Rokka J, Hinkka-Yli-Salomaki S, Rinne JO. Early detection of Alzheimer disease: (1)(1)C-PiB PET in twins discordant for cognitive impairment. Neurology. 2011;77:453–460. doi: 10.1212/WNL.0b013e318225118e. [DOI] [PubMed] [Google Scholar]
- 36.Reiman EM, Chen K, Liu X, Bandy D, Yu M, Lee W, Ayutyanont N, Keppler J, Reeder SA, Langbaum JB, Alexander GE, Klunk WE, Mathis CA, Price JC, Aizenstein HJ, DeKosky ST, Caselli RJ. Fibrillar amyloid-beta burden in cognitively normal people at 3 levels of genetic risk for Alzheimer’s disease. Proc Natl Acad Sci U S A. 2009;106:6820–6825. doi: 10.1073/pnas.0900345106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Small GW, Siddarth P, Burggren AC, Kepe V, Ercoli LM, Miller KJ, Lavretsky H, Thompson PM, Cole GM, Huang SC, Phelps ME, Bookheimer SY, Barrio JR. Influence of cognitive status, age, and APOE-4 genetic risk on brain FDDNP positron-emission tomography imaging in persons without dementia. Arch Gen Psychiatry. 2009;66:81–87. doi: 10.1001/archgenpsychiatry.2008.516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jack CR, Jr, Lowe VJ, Weigand SD, Wiste HJ, Senjem ML, Knopman DS, Shiung MM, Gunter JL, Boeve BF, Kemp BJ, Weiner M, Petersen RC. Serial PIB and MRI in normal, mild cognitive impairment and Alzheimer’s disease: Implications for sequence of pathological events in Alzheimer’s disease. Brain. 2009;132(Pt 5):1355–1365. doi: 10.1093/brain/awp062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wolk DA, Price JC, Saxton JA, Snitz BE, James JA, Lopez OL, Aizenstein HJ, Cohen AD, Weissfeld LA, Mathis CA, Klunk WE, De-Kosky ST. Amyloid imaging in mild cognitive impairment subtypes. Ann Neurol. 2009;65:557–568. doi: 10.1002/ana.21598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Morris JC, Roe CM, Grant EA, Head D, Storandt M, Goate AM, Fagan AM, Holtzman DM, Mintun MA. Pittsburgh compound B imaging and prediction of progression from cognitive normality to symptomatic Alzheimer disease. Arch Neurol. 2009;66:1469–1475. doi: 10.1001/archneurol.2009.269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Villemagne VL, Pike KE, Chetelat G, Ellis KA, Mulligan RS, Bourgeat P, Ackermann U, Jones G, Szoeke C, Salvado O, Martins R, O’Keefe G, Mathis CA, Klunk WE, Ames D, Masters CL, Rowe CC. Longitudinal assessment of Abeta and cognition in aging and Alzheimer disease. Ann Neurol. 2011;69:181–192. doi: 10.1002/ana.22248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jack CR, Jr, Lowe VJ, Senjem ML, Weigand SD, Kemp BJ, Shiung MM, Knopman DS, Boeve BF, Klunk WE, Mathis CA, Petersen RC. 11C PiB and structural MRI provide complementary information in imaging of Alzheimer’s disease and amnestic mild cognitive impairment. Brain. 2008;131(Pt 3):665–680. doi: 10.1093/brain/awm336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rinne JO, Brooks DJ, Rossor MN, Fox NC, Bullock R, Klunk WE, Mathis CA, Blennow K, Barakos J, Okello AA, Rodriguez Martinez de Liano S, Liu E, Koller M, Gregg KM, Schenk D, Black R, Grundman M. 11C-PiB PET assessment of change in fibrillar amyloid-beta load in patients with Alzheimer’s disease treated with bap-ineuzumab: A phase 2, double-blind, placebo-controlled, ascending-dose study. Lancet Neurol. 2010;9:363–372. doi: 10.1016/S1474-4422(10)70043-0. [DOI] [PubMed] [Google Scholar]
- 44.Stern Y. What is cognitive reserve? Theory and research application of the reserve concept. J Int Neuropsychol Soc. 2002;8:448–460. [PubMed] [Google Scholar]
- 45.Kemppainen NM, Aalto S, Karrasch M, Nagren K, Savisto N, Oikonen V, Viitanen M, Parkkola R, Rinne JO. Cognitive reserve hypothesis: Pittsburgh Compound B and fluorodeoxyglucose positron emission tomography in relation to education in mild Alzheimer’s disease. Ann Neurol. 2008;63:112–118. doi: 10.1002/ana.21212. [DOI] [PubMed] [Google Scholar]
- 46.Rentz DM, Locascio JJ, Becker JA, Moran EK, Eng E, Buckner RL, Sperling RA, Johnson KA. Cognition, reserve, and amyloid deposition in normal aging. Ann Neurol. 2010;67:353–364. doi: 10.1002/ana.21904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Greicius MD, Srivastava G, Reiss AL, Menon V. Default-mode network activity distinguishes Alzheimer’s disease from healthy aging: Evidence from functional MRI. Proc Natl Acad Sci U S A. 2004;101:4637–4642. doi: 10.1073/pnas.0308627101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hampson M, Driesen NR, Skudlarski P, Gore JC, Constable RT. Brain connectivity related to working memory performance. J Neurosci. 2006;26:13338–13343. doi: 10.1523/JNEUROSCI.3408-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Shin J, Kepe V, Small GW, Phelps ME, Barrio JR. Multimodal imaging of Alzheimer pathophysiology in the brain’s default mode network. Int J Alzheimers Dis. 2011;2011:687945. doi: 10.4061/2011/687945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sheline YI, Raichle ME, Snyder AZ, Morris JC, Head D, Wang S, Mintun MA. Amyloid plaques disrupt resting state default mode network connectivity in cognitively normal elderly. Biol Psychiatry. 2010;67:584–587. doi: 10.1016/j.biopsych.2009.08.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Mormino EC, Smiljic A, Hayenga AO, Onami SH, Greicius MD, Rabinovici GD, Janabi M, Baker SL, Yen IV, Madison CM, Miller BL, Jagust WJ. Relationships between beta-amyloid and functional connectivity in different components of the default mode network in aging. Cereb Cortex. 2011;21:2399–2407. doi: 10.1093/cercor/bhr025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hedden T, Van Dijk KR, Becker JA, Mehta A, Sperling RA, Johnson KA, Buckner RL. Disruption of functional connectivity in clinically normal older adults harboring amyloid burden. J Neurosci. 2009;29:12686–12694. doi: 10.1523/JNEUROSCI.3189-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sperling RA, Laviolette PS, O’Keefe K, O’Brien J, Rentz DM, Pihlajamaki M, Marshall G, Hyman BT, Selkoe DJ, Hedden T, Buckner RL, Becker JA, Johnson KA. Amyloid deposition is associated with impaired default network function in older persons without dementia. Neuron. 2009;63:178–188. doi: 10.1016/j.neuron.2009.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Nelissen N, Van Laere K, Thurfjell L, Owenius R, Vandenbulcke M, Koole M, Bormans G, Brooks DJ, Vandenberghe R. Phase 1 study of the Pittsburgh compound B derivative 18F-flutemetamol in healthy volunteers and patients with probable Alzheimer disease. J Nucl Med. 2009;50:1251–1259. doi: 10.2967/jnumed.109.063305. [DOI] [PubMed] [Google Scholar]
- 55.Choi SR, Golding G, Zhuang Z, Zhang W, Lim N, Hefti F, Benedum TE, Kilbourn MR, Skovronsky D, Kung HF. Preclinical properties of 18F-AV-45: A PET agent for Abeta plaques in the brain. J Nucl Med. 2009;50:1887–1894. doi: 10.2967/jnumed.109.065284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Rowe CC, Ackerman U, Browne W, Mulligan R, Pike KL, O’Keefe G, Tochon-Danguy H, Chan G, Berlangieri SU, Jones G, Dickinson-Rowe KL, Kung HP, Zhang W, Kung MP, Skovronsky D, Dyrks T, Holl G, Krause S, Friebe M, Lehman L, Lindemann S, Dinkelborg LM, Masters CL, Villemagne VL. Imaging of amyloid beta in Alzheimer’s disease with 18F-BAY94-9172, a novel PET tracer: Proof of mechanism. Lancet Neurol. 2008;7:129–135. doi: 10.1016/S1474-4422(08)70001-2. [DOI] [PubMed] [Google Scholar]
- 57.Fodero-Tavoletti MT, Okamura N, Furumoto S, Mulligan RS, Connor AR, McLean CA, Cao D, Rigopoulos A, Cartwright GA, O’Keefe G, Gong S, Adlard PA, Barnham KJ, Rowe CC, Masters CL, Kudo Y, Cappai R, Yanai K, Villemagne VL. 18F-THK523: A novel in vivo tau imaging ligand for Alzheimer’s disease. Brain. 2011;134(Pt 4):1089–1100. doi: 10.1093/brain/awr038. [DOI] [PubMed] [Google Scholar]
- 58.Vemuri P, Wiste HJ, Weigand SD, Knopman DS, Trojanowski JQ, Shaw LM, Bernstein MA, Aisen PS, Weiner M, Petersen RC, Jack CR., Jr Serial MRI and CSF biomarkers in normal aging, MCI, and AD. Neurology. 2010;75:143–151. doi: 10.1212/WNL.0b013e3181e7ca82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Chou YY, Lepore N, Saharan P, Madsen SK, Hua X, Jack CR, Shaw LM, Trojanowski JQ, Weiner MW, Toga AW, Thompson PM. Ventricular maps in 804 ADNI subjects: Correlations with CSF biomarkers and clinical decline. Neurobiol Aging. 2010;31:1386–1400. doi: 10.1016/j.neurobiolaging.2010.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kovacevic S, Rafii MS, Brewer JB. High-throughput, fully automated volumetry for prediction of MMSE and CDR decline in mild cognitive impairment. Alzheimer Dis Assoc Disord. 2009;23:139–145. doi: 10.1097/WAD.0b013e318192e745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Fleisher AS, Sun S, Taylor C, Ward CP, Gamst AC, Petersen RC, Jack CR, Jr, Aisen PS, Thal LJ. Volumetric MRI vs clinical predictors of Alzheimer disease in mild cognitive impairment. Neurology. 2008;70:191–199. doi: 10.1212/01.wnl.0000287091.57376.65. [DOI] [PubMed] [Google Scholar]
- 62.Risacher SL, Saykin AJ, West JD, Shen L, Firpi HA, McDonald BC. Baseline MRI predictors of conversion from MCI to probable AD in the ADNI cohort. Curr Alzheimer Res. 2009;6:347–361. doi: 10.2174/156720509788929273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Galluzzi S, Geroldi C, Ghidoni R, Paghera B, Amicucci G, Bonetti M, Zanetti O, Cotelli M, Gennarelli M, Frisoni GB. The new Alzheimer’s criteria in a naturalistic series of patients with mild cognitive impairment. J Neurol. 2010;257:2004–2014. doi: 10.1007/s00415-010-5650-0. [DOI] [PubMed] [Google Scholar]
- 64.Apostolova LG, Thompson PM, Green AE, Hwang KS, Zoumalan C, Jack CR, Jr, Harvey DJ, Petersen RC, Thal LJ, Aisen PS, Toga AW, Cummings JL, Decarli CS. 3D comparison of low, intermediate, and advanced hippocampal atrophy in MCI. Hum Brain Mapp. 2010;31:786–797. doi: 10.1002/hbm.20905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Hua X, Leow AD, Parikshak N, Lee S, Chiang MC, Toga AW, Jack CR, Jr, Weiner MW, Thompson PM. Tensor-based morphometry as a neuroimaging biomarker for Alzheimer’s disease: An MRI study of 676 AD, MCI, and normal subjects. Neuroimage. 2008;43:458–469. doi: 10.1016/j.neuroimage.2008.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Freund Y, Schapire R. A decision-theoretic generalization of online learning and an application to boosting. J Comp Syst Sci. 1997;55:119–139. [Google Scholar]
- 67.Morra JH, Tu Z, Apostolova LG, Green AE, Avedissian C, Madsen SK, Parikshak N, Hua X, Toga AW, Jack CR, Jr, Weiner MW, Thompson PM. Validation of a fully automated 3D hippocampal segmentation method using subjects with Alzheimer’s disease mild cognitive impairment, and elderly controls. Neuroimage. 2008;43:59–68. doi: 10.1016/j.neuroimage.2008.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Morra JH, Tu Z, Apostolova LG, Green AE, Avedissian C, Madsen SK, Parikshak N, Hua X, Toga AW, Jack CR, Jr, Schuff N, Weiner MW, Thompson PM. Automated 3D mapping of hippocampal atrophy and its clinical correlates in 400 subjects with Alzheimer’s disease, mild cognitive impairment, and elderly controls. Human Brain Mapp. 2009;30:2766–2788. doi: 10.1002/hbm.20708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Wang Y, Wang Y, Toga AW, Thompson PM. Hippocampal morphometry in AD with surface fluid registration and multivariate tensor-based morphometry. Organization for Human Brain Mapping; 6–10 June 2010; Barcelona, Spain. 2010. [Google Scholar]
- 70.Wang Y, Song Y, Rajagopalan P, An T, Liu K, Chou YY, Gutman B, Toga AW, Thompson PM. Surface-based TBM boosts power to detect disease effects on the brain: An N= 804 ADNI study. Neuroimage. 2011;56:1993–2010. doi: 10.1016/j.neuroimage.2011.03.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Hua X, Leow AD, Lee S, Klunder AD, Toga AW, Lepore N, Chou YY, Brun C, Chiang MC, Barysheva M, Jack CR, Jr, Bernstein MA, Britson PJ, Ward CP, Whitwell JL, Borowski B, Fleisher AS, Fox NC, Boyes RG, Barnes J, Harvey D, Kornak J, Schuff N, Boreta L, Alexander GE, Weiner MW, Thompson PM, Alzheimer’s Disease Neuroimaging, I 3D characterization of brain atrophy in Alzheimer’s disease and mild cognitive impairment using tensor-based morphometry. Neuroimage. 2008;41:19–34. doi: 10.1016/j.neuroimage.2008.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Chou YY, Lepore N, de Zubicaray GI, Carmichael OT, Becker JT, Toga AW, Thompson PM. Automated ventricular mapping with multi-atlas fluid image alignment reveals genetic effects in Alzheimer’s disease. Neuroimage. 2008;40:615–630. doi: 10.1016/j.neuroimage.2007.11.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Chou YY, Lepore N, Saharan P, Madsen SK, Hua X, Jack CR, Shaw LM, Trojanowski JQ, Weiner MW, Toga AW, Thompson PM. Ventricular maps in 804 ADNI subjects: Correlations with CSF biomarkers and clinical decline. Neurobiol Aging. 2010;31:1386–1400. doi: 10.1016/j.neurobiolaging.2010.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Hua X, Gutman B, Boyle CP, Rajagopalan P, Leow AD, Yanovsky I, Kumar AR, Toga AW, Jack CR, Jr, Schuff N, Alexander GE, Chen K, Reiman EM, Weiner MW, Thompson PM, the Alzheimer’s Disease Neuroimaging, Initiative Accurate measurement of brain changes in longitudinal MRI scans using tensor-based morphometry. Neuroimage. 2011;57:5–14. doi: 10.1016/j.neuroimage.2011.01.079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Hua X, Lee S, Hibar DP, Yanovsky I, Leow AD, Toga AW, Jack CR, Jr, Bernstein MA, Reiman EM, Harvey DJ, Kornak J, Schuff N, Alexander GE, Weiner MW, Thompson PM. Mapping Alzheimer’s disease progression in 1309 MRI scans: Power estimates for different inter-scan intervals. Neuroimage. 2010;51:63–75. doi: 10.1016/j.neuroimage.2010.01.104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Chen K, Langbaum JB, Fleisher AS, Ayutyanont N, Reschke C, Lee W, Liu X, Bandy D, Alexander GE, Thompson PM, Foster NL, Harvey DJ, de Leon MJ, Koeppe RA, Jagust WJ, Weiner MW, Reiman EM. Twelve-month metabolic declines in probable Alzheimer’s disease and amnestic mild cognitive impairment assessed using an empirically pre-defined statistical region-of-interest: Findings from the Alzheimer’s Disease Neuroimaging Initiative. Neuroimage. 2010;51:654–664. doi: 10.1016/j.neuroimage.2010.02.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Wyman BT, Harvey DJ, Crawford K, Bernstein MA, Carmichael O, Cole PE, Crane P, DeCarli C, Fox NC, Gunter J, Hill D, Killiany R, Pachai C, Schwarz A, Schuff N, Senjem M, Suhy J, Thompson PM, Weiner MW, Jack CR. Standardization of analysis sets for reporting results from ADNI MRI data. Alzheimers Dement. 2012 doi: 10.1016/j.jalz.2012.06.004. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Kohannim O, Hua X, Hibar DP, Lee S, Chou YY, Toga AW, Jack CR, Jr, Weiner MW, Thompson PM. Boosting power for clinical trials using classifiers based on multiple biomarkers. Neurobiol Aging. 2010;31:1429–1442. doi: 10.1016/j.neurobiolaging.2010.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Yuan L, Wang Y, Thompson PM, Narayan VA, Ye J, for the Alzheimer’s Disease Neuroimaging Initiative Multi-source feature learning for joint analysis of incomplete multiple heterogeneous neuroimaging data. Neuroimage. 2012 doi: 10.1016/j.neuroimage.2012.03.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Scarmeas N, Stern Y. Imaging studies and APOE genotype in persons at risk for Alzheimer’s disease. Curr Psychiatry Rep. 2006;8:11–17. doi: 10.1007/s11920-006-0076-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Naj AC, Jun G, Beecham GW, Wang LS, Vardarajan BN, Buros J, Gallins PJ, Buxbaum JD, Jarvik GP, Crane PK, Larson EB, Bird TD, Boeve BF, Graff-Radford NR, De Jager PL, Evans D, Schneider JA, Carrasquillo MM, Ertekin-Taner N, Younkin SG, Cruchaga C, Kauwe JS, Nowotny P, Kramer P, Hardy J, Huentelman MJ, Myers AJ, Barmada MM, Demirci FY, Baldwin CT, Green RC, Rogaeva E, George-Hyslop PS, Arnold SE, Barber R, Beach T, Bigio EH, Bowen JD, Boxer A, Burke JR, Cairns NJ, Carlson CS, Carney RM, Carroll SL, Chui HC, Clark DG, Corneveaux J, Cotman CW, Cummings JL, Decarli C, Dekosky ST, Diaz-Arrastia R, Dick M, Dickson DW, Ellis WG, Faber KM, Fallon KB, Farlow MR, Ferris S, Frosch MP, Galasko DR, Ganguli M, Gearing M, Geschwind DH, Ghetti B, Gilbert JR, Gilman S, Giordani B, Glass JD, Growdon JH, Hamilton RL, Harrell LE, Head E, Honig LS, Hulette CM, Hyman BT, Jicha GA, Jin LW, Johnson N, Karlawish J, Karydas A, Kaye JA, Kim R, Koo EH, Kowall NW, Lah JJ, Levey AI, Lieberman AP, Lopez OL, Mack WJ, Marson DC, Martiniuk F, Mash DC, Masliah E, McCormick WC, McCurry SM, McDavid AN, McKee AC, Mesulam M, Miller BL, Miller CA, Miller JW, Parisi JE, Perl DP, Peskind E, Petersen RC, Poon WW, Quinn JF, Rajbhandary RA, Raskind M, Reisberg B, Ringman JM, Roberson ED, Rosenberg RN, Sano M, Schneider LS, Seeley W, Shelanski ML, Slifer MA, Smith CD, Sonnen JA, Spina S, Stern RA, Tanzi RE, Trojanowski JQ, Troncoso JC, Van Deerlin VM, Vinters HV, Vonsattel JP, Weintraub S, Welsh-Bohmer KA, Williamson J, Woltjer RL, Cantwell LB, Dombroski BA, Beekly D, Lunetta KL, Martin ER, Kamboh MI, Saykin AJ, Reiman EM, Bennett DA, Morris JC, Montine TJ, Goate AM, Blacker D, Tsuang DW, Hakonarson H, Kukull WA, Foroud TM, Haines JL, Mayeux R, Pericak-Vance MA, Farrer LA, Schellenberg GD. Common variants at MS4A4/MS4A6E, CD2AP, CD33 and EPHA1 are associated with late-onset Alzheimer’s disease. Nat Genet. 2011;43:436–441. doi: 10.1038/ng.801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Hollingworth P, Harold D, Sims R, Gerrish A, Lambert JC, Carrasquillo MM, Abraham R, Hamshere ML, Pahwa JS, Moskvina V, Dowzell K, Jones N, Stretton A, Thomas C, Richards A, Ivanov D, Widdowson C, Chapman J, Lovestone S, Powell J, Proitsi P, Lupton MK, Brayne C, Rubinsztein DC, Gill M, Lawlor B, Lynch A, Brown KS, Passmore PA, Craig D, McGuinness B, Todd S, Holmes C, Mann D, Smith AD, Beaumont H, Warden D, Wilcock G, Love S, Kehoe PG, Hooper NM, Vardy ER, Hardy J, Mead S, Fox NC, Rossor M, Collinge J, Maier W, Jessen F, Ruther E, Schurmann B, Heun R, Kolsch H, van den Bussche H, Heuser I, Kornhuber J, Wiltfang J, Dichgans M, Frolich L, Hampel H, Gallacher J, Hull M, Rujescu D, Giegling I, Goate AM, Kauwe JS, Cruchaga C, Nowotny P, Morris JC, Mayo K, Sleegers K, Bettens K, Engelborghs S, De Deyn PP, Van Broeckhoven C, Livingston G, Bass NJ, Gurling H, McQuillin A, Gwilliam R, Deloukas P, Al-Chalabi A, Shaw CE, Tsolaki M, Singleton AB, Guerreiro R, Muhleisen TW, Nothen MM, Moebus S, Jockel KH, Klopp N, Wichmann HE, Pankratz VS, Sando SB, Aasly JO, Barcikowska M, Wszolek ZK, Dickson DW, Graff-Radford NR, Petersen RC, van Duijn CM, Breteler MM, Ikram MA, Destefano AL, Fitzpatrick AL, Lopez O, Launer LJ, Seshadri S, Berr C, Campion D, Epelbaum J, Dartigues JF, Tzourio C, Alperovitch A, Lathrop M, Feulner TM, Friedrich P, Riehle C, Krawczak M, Schreiber S, Mayhaus M, Nicolhaus S, Wagenpfeil S, Steinberg S, Stefansson H, Stefansson K, Snaedal J, Bjornsson S, Jonsson PV, Chouraki V, Genier-Boley B, Hiltunen M, Soininen H, Combarros O, Zelenika D, Delepine M, Bullido MJ, Pasquier F, Mateo I, Frank-Garcia A, Porcellini E, Hanon O, Coto E, Alvarez V, Bosco P, Siciliano G, Mancuso M, Panza F, Solfrizzi V, Nacmias B, Sorbi S, Bossu P, Piccardi P, Arosio B, Annoni G, Seripa D, Pilotto A, Scarpini E, Galimberti D, Brice A, Hannequin D, Licastro F, Jones L, Holmans PA, Jonsson T, Riemenschneider M, Morgan K, Younkin SG, Owen MJ, O’Donovan M, Amouyel P, Williams J. Common variants at ABCA7, MS4A6A/MS4A4E, EPHA1, CD33 and CD2AP are associated with Alzheimer’s disease. Nat Genet. 2011;43:429–435. doi: 10.1038/ng.803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Braskie MN, Ringman JM, Thompson PM. Neuroimaging measures as endophenotypes in Alzheimer’s disease. Int J Alzheimers Dis. 2011;2011:490140. doi: 10.4061/2011/490140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Erk S, Meyer-Lindenberg A, Opitz von Boberfeld C, Esslinger C, Schnell K, Kirsch P, Mattheisen M, Muhleisen TW, Cichon S, Witt SH, Rietschel M, Nothen MM, Walter H. Hippocampal function in healthy carriers of the CLU Alzheimer’s disease risk variant. J Neurosci. 2011;31:18180–18184. doi: 10.1523/JNEUROSCI.4960-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Lancaster TM, Baird A, Wolf C, Jackson MC, Johnston SJ, Donev R, Thome J, Linden DE. Neural hyperactivation in carriers of the Alzheimer’s risk variant on the clusterin gene. Eur Neuropsychopharmacol. 2011;21:880–884. doi: 10.1016/j.euroneuro.2011.02.001. [DOI] [PubMed] [Google Scholar]
- 86.Biffi A, Anderson CD, Desikan RS, Sabuncu M, Cortellini L, Schmansky N, Salat D, Rosand J. Genetic variation and neuroimaging measures in Alzheimer disease. Arch Neurol. 2010;67:677–685. doi: 10.1001/archneurol.2010.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Bralten J, Franke B, Arias-Vasquez A, Heister A, Brunner HG, Fernandez G, Rijpkema M. CR1 genotype is associated with entorhinal cortex volume in young healthy adults. Neurobiol Aging. 2011;32:2106 e2107–e2111. doi: 10.1016/j.neurobiolaging.2011.05.017. [DOI] [PubMed] [Google Scholar]
- 88.Andrawis JP, Hwang KS, Green AE, Kotlerman J, Elashoff D, Morra JH, Cummings JL, Toga AW, Thompson PM, Apostolova LG. Effects of ApoE4 and maternal history of dementia on hippocampal atrophy. Neurobiol Aging. 2010;33:856–866. doi: 10.1016/j.neurobiolaging.2010.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Honea RA, Swerdlow RH, Vidoni ED, Burns JM. Progressive regional atrophy in normal adults with a maternal history of Alzheimer disease. Neurology. 2011;76:822–829. doi: 10.1212/WNL.0b013e31820e7b74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Mosconi L, Glodzik L, Mistur R, McHugh P, Rich KE, Javier E, Williams S, Pirraglia E, De Santi S, Mehta PD, Zinkowski R, Blennow K, Pratico D, de Leon MJ. Oxidative stress and amyloid-beta pathology in normal individuals with a maternal history of Alzheimer’s. Biol Psychiatry. 2010;68:913–921. doi: 10.1016/j.biopsych.2010.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Mosconi L, Mistur R, Switalski R, Brys M, Glodzik L, Rich K, Pirraglia E, Tsui W, De Santi S, de Leon MJ. Declining brain glucose metabolism in normal individuals with a maternal history of Alzheimer disease. Neurology. 2009;72:513–520. doi: 10.1212/01.wnl.0000333247.51383.43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Honea RA, Swerdlow RH, Vidoni ED, Goodwin J, Burns JM. Reduced gray matter volume in normal adults with a maternal family history of Alzheimer disease. Neurology. 2010;74:113–120. doi: 10.1212/WNL.0b013e3181c918cb. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Sabuncu MR, Buckner RL, Smoller JW, Lee PH, Fischl B, Sperling RA. The association between a polygenic Alzheimer score and cortical thickness in clinically normal subjects. Cereb Cortex. 2011 doi: 10.1093/cercor/bhr348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Stein JL, Hua X, Morra JH, Lee S, Hibar DP, Ho AJ, Leow AD, Toga AW, Sul JH, Kang HM, Eskin E, Saykin AJ, Shen L, Foroud T, Pankratz N, Huentelman MJ, Craig DW, Gerber JD, Allen AN, Corneveaux JJ, Stephan DA, Webster J, DeChairo BM, Potkin SG, Jack CR, Jr, Weiner MW, Thompson PM. Genome-wide analysis reveals novel genes influencing temporal lobe structure with relevance to neurodegeneration in Alzheimer’s disease. Neuroimage. 2010;51:542–554. doi: 10.1016/j.neuroimage.2010.02.068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Hibar DP, Stein JL, Kohannim O, Jahanshad N, Saykin AJ, Jack CR, Weiner MW, Toga AW, Thompson PM, Alzheimer’s Disease Neuroimaging, Initiative Voxelwise gene-wide association study (vGene WAS): Multivariate gene-based association testing in 731 elderly subjects. NeuroImage. 2011;56:1875–1891. doi: 10.1016/j.neuroimage.2011.03.077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Stein JL, Hua X, Lee S, Ho AJ, Leow AD, Toga AW, Saykin AJ, Shen L, Foroud T, Pankratz N, Huentelman MJ, Craig DW, Gerber JD, Allen AN, Corneveaux JJ, Dechairo BM, Potkin SG, Weiner MW, Thompson P, Alzheimer’s Disease Neuroimaging, Initiative Voxelwise genome-wide association study (vGWAS) Neuroimage. 2010;53:1160–1174. doi: 10.1016/j.neuroimage.2010.02.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Kohannim O, Jahanshad N, Braskie MN, Stein JL, Chiang MC, Reese AH, Toga AW, McMahon KL, de Zubicaray GI, Medland SE, Montgomery GW, Martin NG, Wright MJ, Thompson PM. Predicting white matter integrity from multiple common genetic variants. Neuropsychopharmacology. 2012 doi: 10.1038/npp.2012.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Vounou M, Janousova E, Wolz R, Stein JL, Thompson PM, Rueckert D, Montana G, Alzheimer’s Disease Neuroimaging, Initiative Sparse reduced-rank regression detects genetic associations with voxel-wise longitudinal phenotypes in Alzheimer’s disease. Neuroimage. 2012;60:700–716. doi: 10.1016/j.neuroimage.2011.12.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Ho AJ, Stein JL, Hua X, Lee S, Hibar DP, Leow AD, Dinov ID, Toga AW, Saykin AJ, Shen L, Foroud T, Pankratz N, Huentelman MJ, Craig DW, Gerber JD, Allen AN, Corneveaux JJ, Stephan DA, DeCarli CS, DeChairo BM, Potkin SG, Jack CR, Jr, Weiner MW, Raji CA, Lopez OL, Becker JT, Carmichael OT, Thompson PM. A commonly carried allele of the obesity-related FTO gene is associated with reduced brain volume in the healthy elderly. Proc Natl Acad Sci U S A. 2010;107:8404–8409. doi: 10.1073/pnas.0910878107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Raji CA, Ho AJ, Parikshak NN, Becker JT, Lopez OL, Kuller LH, Hua X, Leow AD, Toga AW, Thompson PM. Brain structure and obesity. Hum Brain Mapp. 2010;31:353–364. doi: 10.1002/hbm.20870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Walther K, Birdsill AC, Glisky EL, Ryan L. Structural brain differences and cognitive functioning related to body mass index in older females. Hum Brain Mapp. 2010;31:1052–1064. doi: 10.1002/hbm.20916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Braskie MN, Small GW, Bookheimer SY. Vascular health risks and fMRI activation during a memory task in older adults. Neurobiol Aging. 2010;31:1532–1542. doi: 10.1016/j.neurobiolaging.2008.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Ward MA, Bendlin BB, McLaren DG, Hess TM, Gallagher CL, Kastman EK, Rowley HA, Asthana S, Carlsson CM, Sager MA, Johnson SC. Low HDL cholesterol is associated with lower gray matter volume in cognitively healthy adults. Front Aging Neurosci. 2010;2:29. doi: 10.3389/fnagi.2010.00029. pii. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Wolf H, Hensel A, Arendt T, Kivipelto M, Winblad B, Gertz HJ. Serum lipids and hippocampal volume: The link to Alzheimer’s disease? Ann Neurol. 2004;56:745–748. doi: 10.1002/ana.20289. [DOI] [PubMed] [Google Scholar]
- 105.Langbaum JB, Chen K, Launer LJ, Fleisher AS, Lee W, Liu X, Protas HD, Reeder SA, Bandy D, Yu M, Caselli RJ, Reiman EM. Blood pressure is associated with higher brain amyloid burden and lower glucose metabolism in healthy late middle-age persons. Neurobiol Aging. 2012;33:827 e11–e19. doi: 10.1016/j.neurobiolaging.2011.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.El Oudi M, Bouguerra C, Aouni Z, Mazigh C, Bellaaj R, Machghoul S. Homocysteine and inflammatory biomarkers plasma levels, and severity of acute coronary syndrome. Ann Biol Clin (Paris) 2011;69:175–180. doi: 10.1684/abc.2011.0533. [DOI] [PubMed] [Google Scholar]
- 107.Seshadri S, Beiser A, Selhub J, Jacques PF, Rosenberg IH, D’Agostino RB, Wilson PW, Wolf PA. Plasma homocysteine as a risk factor for dementia and Alzheimer’s disease. N Engl J Med. 2002;346:476–483. doi: 10.1056/NEJMoa011613. [DOI] [PubMed] [Google Scholar]
- 108.Rajagopalan P, Jahanshad N, Stein JL, Toga AW, Jack CR, Weiner MW, Thompson PM. Commonly-carried variant in the folate pathway gene, MTHFR, may partly account for homocysteine related brain atrophy. Soc Neurosci Abstr. 2011 Program No. 551.22/D7. [Google Scholar]
- 109.Smith AD, Smith SM, de Jager CA, Whitbread P, Johnston C, Agacinski G, Oulhaj A, Bradley KM, Jacoby R, Refsum H. Homocysteine-lowering by B vitamins slows the rate of accelerated brain atrophy in mild cognitive impairment: A randomized controlled trial. PLoS One. 2010;5:e12244. doi: 10.1371/journal.pone.0012244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Burns JM, Donnelly JE, Anderson HS, Mayo MS, Spencer-Gardner L, Thomas G, Cronk BB, Haddad Z, Klima D, Hansen D, Brooks WM. Peripheral insulin and brain structure in early Alzheimer disease. Neurology. 2007;69:1094–1104. doi: 10.1212/01.wnl.0000276952.91704.af. [DOI] [PubMed] [Google Scholar]
- 111.Rasgon NL, Kenna HA, Wroolie TE, Kelley R, Silverman D, Brooks J, Williams KE, Powers BN, Hallmayer J, Reiss A. Insulin resistance and hippocampal volume in women at risk for Alzheimer’s disease. Neurobiol Aging. 2011;32:1942–1948. doi: 10.1016/j.neurobiolaging.2009.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Bartzokis G. Alzheimer’s disease as homeostatic responses to age-related myelin breakdown. Neurobiol Aging. 2009;32:1341–1371. doi: 10.1016/j.neurobiolaging.2009.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Chiang GC, Insel PS, Tosun D, Schuff N, Truran-Sacrey D, Raptentsetsang S, Jack CR, Jr, Weiner MW. Identifying cognitively healthy elderly individuals with subsequent memory decline by using automated MR temporoparietal volumes. Radiology. 2011;259:844–851. doi: 10.1148/radiol.11101637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Apostolova LG, Mosconi L, Thompson PM, Green AE, Hwang KS, Ramirez A, Mistur R, Tsui WH, de Leon MJ. Subregional hippocampal atrophy predicts Alzheimer’s dementia in the cognitively normal. Neurobiol Aging. 2010;31:1077–1088. doi: 10.1016/j.neurobiolaging.2008.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Carmichael OT, Kuller LH, Lopez OL, Thompson PM, Dutton RA, Lu A, Lee SE, Lee JY, Aizenstein HJ, Meltzer CC, Liu Y, Toga AW, Becker JT. Ventricular volume and dementia progression in the Cardiovascular Health Study. Neurobiol Aging. 2007;28:389–397. doi: 10.1016/j.neurobiolaging.2006.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Nettiksimmons J, Harvey D, Brewer J, Carmichael O, DeCarli C, Jack CR, Jr, Petersen R, Shaw LM, Trojanowski JQ, Weiner MW, Beckett L. Subtypes based on cerebrospinal fluid and magnetic resonance imaging markers in normal elderly predict cognitive decline. Neurobiol Aging. 2010;31:1419–1428. doi: 10.1016/j.neurobiolaging.2010.04.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Gomar JJ, Bobes-Bascaran MT, Conejero-Goldberg C, Davies P, Goldberg TE. Utility of combinations of biomarkers, cognitive markers, and risk factors to predict conversion from mild cognitive impairment to Alzheimer disease in patients in the Alzheimer’s disease neuroimaging initiative. Arch Gen Psychiatry. 2011;68:961–969. doi: 10.1001/archgenpsychiatry.2011.96. [DOI] [PubMed] [Google Scholar]
- 118.Heister D, Brewer JB, Magda S, Blennow K, McEvoy LK. Predicting MCI outcome with clinically available MRI and CSF biomarkers. Neurology. 2011;77:1619–1628. doi: 10.1212/WNL.0b013e3182343314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Landau SM, Harvey D, Madison CM, Reiman EM, Foster NL, Aisen PS, Petersen RC, Shaw LM, Trojanowski JQ, Jack CR, Jr, Weiner MW, Jagust WJ. Comparing predictors of conversion and decline in mild cognitive impairment. Neurology. 2010;75:230–238. doi: 10.1212/WNL.0b013e3181e8e8b8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Devanand DP, Liu X, Tabert MH, Pradhaban G, Cuasay K, Bell K, de Leon MJ, Doty RL, Stern Y, Pelton GH. Combining early markers strongly predicts conversion from mild cognitive impairment to Alzheimer’s disease. Biol Psychiatry. 2008;64:871–879. doi: 10.1016/j.biopsych.2008.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Chen K, Ayutyanont N, Langbaum JB, Fleisher AS, Reschke C, Lee W, Liu X, Bandy D, Alexander GE, Thompson PM, Shaw L, Trojanowski JQ, Jack CR, Jr, Landau SM, Foster NL, Harvey DJ, Weiner MW, Koeppe RA, Jagust WJ, Reiman EM. Characterizing Alzheimer’s disease using a hypometabolic convergence index. Neuroimage. 2011;56:52–60. doi: 10.1016/j.neuroimage.2011.01.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Vemuri P, Wiste HJ, Weigand SD, Shaw LM, Trojanowski JQ, Weiner MW, Knopman DS, Petersen RC, Jack CR., Jr MRI and CSF biomarkers in normal, MCI, and AD subjects: Predicting future clinical change. Neurology. 2009;73:294–301. doi: 10.1212/WNL.0b013e3181af79fb. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Desikan RS, Cabral HJ, Settecase F, Hess CP, Dillon WP, Glastonbury CM, Weiner MW, Schmansky NJ, Salat DH, Fischl B. Automated MRI measures predict progression to Alzheimer’s disease. Neurobiol Aging. 2010;31:1364–1374. doi: 10.1016/j.neurobiolaging.2010.04.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Whitwell JL, Shiung MM, Przybelski SA, Weigand SD, Knopman DS, Boeve BF, Petersen RC, Jack CR., Jr MRI patterns of atrophy associated with progression to AD in amnestic mild cognitive impairment. Neurology. 2008;70:512–520. doi: 10.1212/01.wnl.0000280575.77437.a2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Apostolova LG, Dutton RA, Dinov ID, Hayashi KM, Toga AW, Cummings JL, Thompson PM. Conversion of mild cognitive impairment to Alzheimer disease predicted by hippocampal atrophy maps. Arch Neurol. 2006;63:693–699. doi: 10.1001/archneur.63.5.693. [DOI] [PubMed] [Google Scholar]
- 126.Madsen SK, Ho AJ, Hua X, Saharan PS, Toga AW, Jack CR, Jr, Weiner MW, Thompson PM. 3D maps localize caudate nucleus atrophy in 400 Alzheimer’s disease, mild cognitive impairment, and healthy elderly subjects. Neurobiol Aging. 2010;31:1312–1325. doi: 10.1016/j.neurobiolaging.2010.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Villain N, Fouquet M, Baron JC, Mezenge F, Landeau B, de La Sayette V, Viader F, Eustache F, Desgranges B, Chetelat G. Sequential relationships between grey matter and white matter atrophy and brain metabolic abnormalities in early Alzheimer’s disease. Brain. 2010;133:3301–3314. doi: 10.1093/brain/awq203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Schott JM, Bartlett JW, Fox NC, Barnes J. Increased brain atrophy rates in cognitively normal older adults with low cerebrospinal fluid Abeta1-42. Ann Neurol. 2010;68:825–834. doi: 10.1002/ana.22315. [DOI] [PubMed] [Google Scholar]
- 129.Fagan AM, Head D, Shah AR, Marcus D, Mintun M, Morris JC, Holtzman DM. Decreased cerebrospinal fluid Abeta(42) correlates with brain atrophy in cognitively normal elderly. Ann Neurol. 2009;65:176–183. doi: 10.1002/ana.21559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Whitwell JL, Przybelski SA, Weigand SD, Knopman DS, Boeve BF, Petersen RC, Jack CR., Jr 3D maps from multiple MRI illustrate changing atrophy patterns as subjects progress from mild cognitive impairment to Alzheimer’s disease. Brain. 2007;130(Pt 7):1777–1786. doi: 10.1093/brain/awm112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Braak H, Braak E. Staging of Alzheimer’s disease-related neurofibrillary changes. Neurobiol Aging. 1995;16:271–278. doi: 10.1016/0197-4580(95)00021-6. discussion 278–284. [DOI] [PubMed] [Google Scholar]
- 132.Frisoni GB, Prestia A, Rasser PE, Bonetti M, Thompson PM. In vivo mapping of incremental cortical atrophy from incipient to overt Alzheimer’s disease. J Neurol. 2009;256:916–924. doi: 10.1007/s00415-009-5040-7. [DOI] [PubMed] [Google Scholar]
- 133.Apostolova LG, Steiner CA, Akopyan GG, Dutton RA, Hayashi KM, Toga AW, Cummings JL, Thompson PM. Three-dimensional gray matter atrophy mapping in mild cognitive impairment and mild Alzheimer disease. Arch Neurol. 2007;64:1489–1495. doi: 10.1001/archneur.64.10.1489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Schuff N, Woerner N, Boreta L, Kornfield T, Shaw LM, Trojanowski JQ, Thompson PM, Jack CR, Jr, Weiner MW. MRI of hippocampal volume loss in early Alzheimer’s disease in relation to ApoE genotype and biomarkers. Brain. 2009;132(Pt 4):1067–1077. doi: 10.1093/brain/awp007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Morra JH, Tu Z, Apostolova LG, Green AE, Avedissian C, Madsen SK, Parikshak N, Toga AW, Jack CR, Jr, Schuff N, Weiner MW, Thompson PM. Automated mapping of hippocampal atrophy in 1-year repeat MRI data from 490 subjects with Alzheimer’s disease, mild cognitive impairment, and elderly controls. Neuroimage. 2009;45(Suppl 1):S3–S15. doi: 10.1016/j.neuroimage.2008.10.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Leow AD, Yanovsky I, Parikshak N, Hua X, Lee S, Toga AW, Jack CR, Jr, Bernstein MA, Britson PJ, Gunter JL, Ward CP, Borowski B, Shaw LM, Trojanowski JQ, Fleisher AS, Harvey D, Kornak J, Schuff N, Alexander GE, Weiner MW, Thompson PM. Alzheimer’s disease neuroimaging initiative: A one-year follow up study using tensor-based morphometry correlating degenerative rates, biomarkers and cognition. Neuroimage. 2009;45:645–655. doi: 10.1016/j.neuroimage.2009.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Becker JT, Davis SW, Hayashi KM, Meltzer CC, Toga AW, Lopez OL, Thompson PM. Three-dimensional patterns of hippocampal atrophy in mild cognitive impairment. Arch Neurol. 2006;63:97–101. doi: 10.1001/archneur.63.1.97. [DOI] [PubMed] [Google Scholar]
- 138.Apostolova LG, Dinov ID, Dutton RA, Hayashi KM, Toga AW, Cummings JL, Thompson PM. 3D comparison of hippocampal atrophy in amnestic mild cognitive impairment and Alzheimer’s disease. Brain. 2006;129(Pt 11):2867–2873. doi: 10.1093/brain/awl274. [DOI] [PubMed] [Google Scholar]
- 139.Holland D, Brewer JB, Hagler DJ, Fennema-Notestine C, Dale AM. Subregional neuroanatomical change as a biomarker for Alzheimer’s disease. Proc Natl Acad Sci U S A. 2009;106:20954–20959. doi: 10.1073/pnas.0906053106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Ridha BH, Barnes J, Bartlett JW, Godbolt A, Pepple T, Rossor MN, Fox NC. Tracking atrophy progression in familial Alzheimer’s disease: A serial MRI study. Lancet Neurol. 2006;5:828–834. doi: 10.1016/S1474-4422(06)70550-6. [DOI] [PubMed] [Google Scholar]
- 141.Ringman JM, O’Neill J, Geschwind D, Medina L, Apostolova LG, Rodriguez Y, Schaffer B, Varpetian A, Tseng B, Ortiz F, Fitten J, Cummings JL, Bartzokis G. Diffusion tensor imaging in preclinical and presymptomatic carriers of familial Alzheimer’s disease mutations. Brain. 2007;130(Pt 7):1767–1776. doi: 10.1093/brain/awm102. [DOI] [PubMed] [Google Scholar]