

Abstract
This review focuses upon the development, scope, and utility of the highly versatile chemoselective alkoxyamine-based ‘neoglycosylation’ reaction first described by Peri and Dumy. The fundamentals of neoglycosylation and the subsequent development of a ‘neoglycorandomization’ platform to afford differentially-glycosylated libraries of plant-based natural products, microbial-based natural products, and small molecule-based drugs for drug discovery applications are discussed.
Section 1 – Introduction
Carbohydrates are pervasive in nature where they serve a wide range of vital structural and functional roles. Simple carbohydrate units such as monosaccharides are integral to fundamental metabolism while carbohydrate polymers and saccharide conjugation contribute to cell membrane/wall structural integrity, cellular communication and signaling mechanisms, fundamental protein folding/function and the activity of small molecules. The dramatic functional diversity of carbohydrates derives from their inherent structural diversity by virtue of the multiple stereocenters, functional group substitutions and regio-/stereochemical connections offered by each of hundreds of naturally-occurring saccharide units.1–3 Thus, the natural combinatorial potential of carbohydrates far surpasses that of proteins and nucleic acids and, from a biosynthetic perspective, requires a far more extensive network of carbohydrate precursors and assembly machinery.4–7
This inherent carbohydrate structural diversity presents a monumental challenge in terms of reagent synthesis for the study or exploitation of carbohydrate function. Chief among these are selective and divergent protecting group strategies and selective anomeric activation methods for regio-/sterochemical control of carbohydrate coupling or conjugation.8–9 Within this context, chemoselective glycosylation reactions are advantageous in minimizing the number synthetic steps to achieve carbohydrate reagents for biological study. Specifically, the use of a single chemoselective carbohydrate coupling reaction minimally eliminates four essential steps in each conventional glycoside bond-forming reaction – selective functional group protection of both the donor and acceptor, anomeric activation of the saccharide donor, the key coupling reaction, and global deprotection (Scheme 1). Therefore, the strength of chemoselective glycosylation lies in the ability to rapidly generate glycodiverse libraries via a one-step divergent process.
Scheme 1.
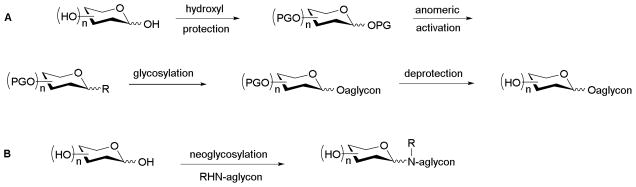
Comparison of conventional glycosylation strategy (A) to neoglycosylation (B)
While there exists a range of convenient chemoselective carbohydrate conjugation methods,10 this review focuses upon the scope and utility of the alkoxyamine-based ‘neoglycosylation’ reaction first described Peri and Dumy.11 Specifically, this review discusses the fundamentals of neoglycosylation and the subsequent development of a ‘neoglycorandomization’ platform to afford differentially-glycosylated libraries of plant-based natural products, microbial-based natural products, and small molecule-based drugs for drug discovery applications.
Section 2 - Chemical Aspects of Neoglycosylation
An interest to rapidly construct homogeneous glycoproteins (i.e., glycodiverse proteins with a constant peptide domain) served as early inspiration for neoglycosylation. Initial forays toward chemoselective glycosylation using Schiff base formation (i.e., selective coupling between aldehydes and amines) involved approaches including coupling aminooxy-appended peptides with reducing sugars12,13 and aminooxy-appended oligosaccharides to existing glycopeptides (Scheme 2).14
Scheme 2.
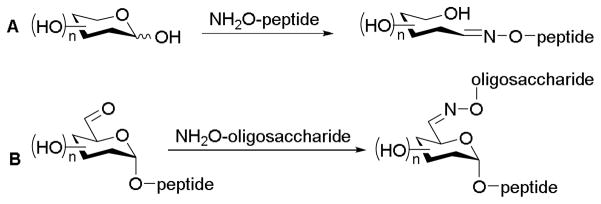
Chemoselective glycosylation between aminooxy-coupled compounds and aldehyde-containing sugars. (A) Anomeric-coupled glycopeptides reported by Mutter;12 (B) C6-coupled glycopeptides reported by Bertozzi.14
A primary drawback of these early pioneering strategies stemmed from the production of linear, non-cyclic carbohydrate oximes which lacked the desired conformational similarity to native cyclic carbohydrate conjugates. Peri et al. developed an effective method of chemoselective glycosylation between N,O-dialkylhydroxyamines and hexoses to rectify this deficiency.11 Through the use of alkoxyamines as chemoselective ‘handles,’ aglycons could selectively couple to a non-reducing, unprotected sugar with the final product population fully existing as cyclized saccharides referred to ‘neoglycosides’. The chemoselectivity of the neoglycosylation reaction derives from the paired reactivity of the nucleophilic alkoxyamine and the open chain aldehyde form of non-reducing aldoses, the latter of which is in equilibrium with its respective α - and β-hemiacetal isomers.
Importantly, neoglycosylation reactions are typically regiospecific due to both an infrequency of aldehyde groups in target acceptors and the use of excess carbohydrate in the reaction mixture. After dehydration of the tetrahedral intermediate, the reactive electrophilic iminium ion sets the stage for intramolecular cyclization to afford, in most cases, the corresponding thermodynamically-favored cyclic N-glycoside (Scheme 3). Configuration of the anomeric stereocenter is determined by attack on the iminium carbon at the Re or Si face resulting in the corresponding anomers. Furanoside and pyranoside isomerism also occurs in this mechanistic step depending on attack on the iminium carbon by the C4 or C5 hydroxyl group, respectively. The basics regarding alkoxyamine acceptor generation, and neoglycoside reaction conditions as well as additional insights regarding the propensity for neoglycoside cyclization and anomeric stereoselectivites are within the remainder of this section.
Scheme 3.

Mechanism of the neoglycosylation reaction involving reducing sugars and alkoxyamines (R1NHOR2).11
Acceptor synthesis
For neoglycosylation to be a viable alternative to conventional O-glycosylation, aglycon installation of the chemoselective handle must be a relatively simple and concise process. Although different means have been used to attach a variety of alkoxyamine groups, a simple two-step reductive amination strategy is perhaps among the most common (Scheme 4). This commonly involves condensation between an alkoxyamine (such as methoxyamine) and an aglycon aldehyde or ketone in the presence of weak base (e.g., triethylamine, pyridine) followed by reduction of the corresponding imine in a polar solvent (Table 1). With aglycons lacking a convenient reductive amination connection point, further synthetic manipulation may be necessary to prepare a pharmacophore aglycon for coupling as exemplified via the use of tethered handles for betulinic acid neoglycosides15 or alternative strategies as required for chlorambucil neoglycosides.16
Scheme 4.
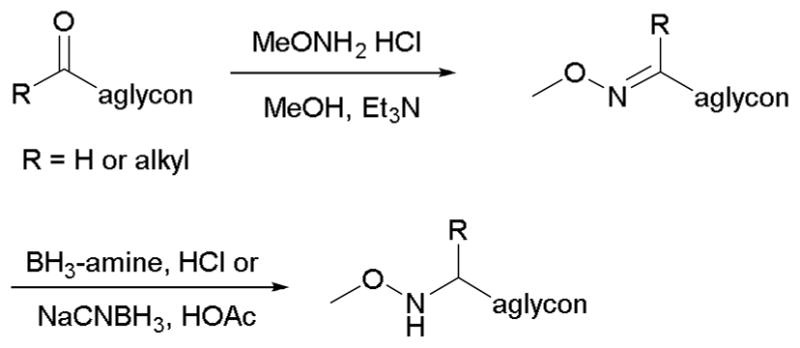
Typical methoxyamine handle installation on an aglycon.
Table 1.
Representative reductive amination handle installation conditions
| means of handle attachment | reducing agent | solvent and conditions | rxn time | yield | ref |
|---|---|---|---|---|---|
| aldehyde | NaCNBH3 | HOAc, r.t. | 1 h | 80% | 11 |
| aldehyde | BH3-Et3N | HCl/EtOH, 0°C | 5 m | 65% | 16 |
| ketone | BH3- tBuNH2 | HCl,/EtOH/ dioxane, 0 °C | 2.5 h | 69% | 19 |
| ketone | NaCNBH3 | HOAc/DCE/ MeOH, r.t. | 90 m | 88% | 24 |
| glycyl linker | BH3-Me3N | HCl/EtOH, 0 °C | 5 h | 85% | 15 |
General reaction conditions
Reaction conditions for neoglycoside formation can vary but require acidic conditions (a pH of ~4–5.5)11 using either an organic acid17 or aqueous buffer.18 This pH range is to favour both nucleophilic attack by the alkoxyamine and subsequent dehydration of the tetrahedral intermediate. Polar aprotic solvents (DMSO, DMF) are frequently used due to greater hexose solubility; though in a methanolic system reactions still occur in a similar timeframe (appx. 24 h) with comparable yields. Other significant conditions that promote increased yields and lower reaction times include heating to 40 ºC and using an excess of reducing sugar (2–3 equivalents).15,16,19–26 Although not systematically assessed, empirical studies suggest reactions are favoured under higher concentrations. Other reaction condition optimization include the use of microwave or nucleophilic catalysts as discussed later in this review. Table 2 summarizes representative published neoglycosylation conditions used for peptide and small molecule aglycons.
Table 2.
Neoglycosylation conditions by aglycon
| neoaglycon | solvent | [neoaglycon] mM | equivalents of sugar | reaction temp (°C) | time (h) | yield (%)a | ref |
|---|---|---|---|---|---|---|---|
| LysAlaLys | 0.1 M NaOAc (pH 4) | 20 | 1.2 | rt–60 | 48–144 | 56b | 11 |
| methyl D-Glc | 1:2 HOAc/DMF | n/rc | n/r | rt | 4–6 | 72b | 17 |
| tri- to nonapeptide | 0.1 M NaOAc (pH 4 or 5.1) | n/r | 75 | 40–45 | 24–48 | 60–85 | 55–57 |
| peptoidd | 6:1 0.1 M NaOAc/MeOH (pH 4) | 6.3 or 8.0 | 100–200 | 40 | 0.17–5 | 81–94 | 58 |
| betulinic acide | 6:1 MeOH/CH2Cl2f | 80 | 1–3 | 40 | 48 | 33b | 15 |
| betulinic acidg | 6:1 MeOH/CH2Cl2f | 80 | 3 | 40 | 48 | 40b | 15 |
| calicheamicin | MeOH, HOAc (1.5 eq.) | 90 | 4 | 40 | 20–48 | 49b | 22 |
| chlorambucil | MeOH, HOAc (1.5 eq.) | 90 | 2 | 40 | 3–48 | 63b | 16 |
| colchicine | 3:1 DMF/HOAc | 90–100 | 2 | 40 | 24 | 51b | 20 |
| cyclopamine | 8:1 MeOH/HOAc | 90–100 | 3 | 40 | 4–28 | 49b | 24 |
| digitoxin | 3:1 DMF/HOAc | 90 | 2 | 40 | 48 | n/r | 19 |
| doxycycline | 4:1 DMF/TFA | 60 | 1.2–2 | 40 | 12 | 11–50 | 26 |
| fluorescein | 3:1 DMF/HOAc | 100 | 2–3 | 45 | 48 | 35–65 | - - -h |
| podophylltotoxin | 3:1 DMF/HOAc | 90 | 10 | 40 | 36 | <1b | - - -h |
| vancomycini | 2.5% TFA in DMSO | 40 | 10 | 40 | 24–72 | 14–30 | 21 |
| vancomycinj | 3:1 DMSO/HOAc | 20 | 5–10 | 40 | 24–48 | 31–95 | 25 |
| warfarin | 3:1 DMF/HOAc | 150 | 2 | 50 | 24–48 | 41b | 23 |
Isolated yields.
Average yield.
Not reported.
Microwave irradiation.
Ester glycyl linker.
Additional acid not required due to aglycon carboxylic acid group.
Amide glycyl linker.
Unpublished results.
Monosaccharide library.
Disaccharide library.
Neoglycosylation is compatible with a diverse array of carbohydrates.15,16,19–26 These include hexoses, pentoses, tetroses, D- and L-sugars, N-acetyl sugars, azido sugars, deoxysugars (2-,3-,4-, and 6-positions), fluorosugars, alkoxy- and acyloxysugars, glycuronic acids, disaccharides, and saccharides from natural products (e.g., digitoxose, noviose). In most cases, ketoses and oligosaccharides are poor donors, the latter possibly due to solubility issues. Aminosugars present a challenge due to potential competition between the alkoxylamine and sugar amine for the reactive aldose. However, this can be circumvented by using N-acyl- protected or azidosugars followed by deprotection or reduction, respectively.26
Cyclic versus acyclic products
The use of N,O-disubstituted hydroxylamine-containing acceptors in the neoglycosylation reaction typically favors formation of cyclic neoglycosides. Peri reported that both O-methyl and O-benzyl groups exclusively provided the products in cyclic form, largely as neopyranosides except for a small percentage (i.e., 20% or less) of furanoside with D-galactose and D-mannose analogs.11 Additionally, Langenhan and coworkers found that replacing the original methoxyamine handle of digitoxin neoglycosides19 with other alkoxyamines (e.g., Et, i-Pr, allyl, Bn) had no noticeable effect on the configuration of the neoglycosyl products.27 However, use of a bulky t-butoxyamine handle in the same study inhibited neoglycoside formation. Likewise, in compounds employing ‘inverse’ handles (i.e., N-methyl groups and O-linked aglycons), cyclical neoglycosides were also observed.11 In contrast to alkoxyamine-based conjugates, Goff and Thorson reported mixtures of linear and cyclized neoglycosides using hydroxyamines and acyl hydrazides as the chemoselective ligation moieties.16 They found that hexoses tended to assemble mostly as closed rings, pentoses were apt to exist as a greater ratio of open to closed than hexoses, and tetroses were configured fully in the open chain. The presence of linear saccharides using hydroxyamine was also reported by Dondoni28–30 and Goti.31,32 Glycosyl acyl hydrazides however, have widely been represented in the literature as existing as fully cyclic.33–39 These same reports have exclusively used hexoses and hexose variants (e.g., deoxy, N-acetyl) as the sugar component as exemplified by the work of Bendiak and coworkers.40 This trend can be rationalized using the description of the hydrolysis mechanism proposed by Nitz and co-workers.41
The ultimate formation of linear and/or cyclic neoglycosides is dictated by both the nature of the chemoselective handle and the reducing sugar employed. With respect to the handle, intramolecular ring closure to afford a final neutral species is driven via the initial charged iminium ion intermediate formed from reaction of a reducing sugar and an N-alkoxyamine. In contrast, simple deprotonation of the corresponding iminium ion intermediate formed from reaction of a reducing sugar and a N-hydroxyamine (or acyl hydrazide) satisfies the electronic requirements without intramolecular cyclization. The second factor in the trend of cyclic vs. acyclic neoglycosides is due to the type of saccharide used in conjugation with hydroxylamine and acyl hydrazide handles. With respect to the saccharide, propensity for cyclization parallels carbohydrate electronegativity. Specifically, relatively electron-poor sugars (e.g., hexoses) generally have a higher propensity for cyclization in the context of neoglycosylation than electron-rich sugars (e.g., pentoses and tetroses) and this also correlates to the ease by which protonation of their linear form can occur.41 This trend was also confirmed by Godula and Bertozzi, who reported that acyl hydrazide coupling to hexoses favoured cyclic products while pentose-/tetrose-based products trended toward linear neoglycosides.42
Anomeric stereoselectivity
Thermodynamics is a major driving force in determining anomeric stereoselectivity. Peri observed anomeric diastereoselectivities in excess of 96%, with C2-equatorial glycosides (e.g., gluco-, galacto-) preferring the β-anomer and C2-axial glycosides (e.g., manno-) with the α-configuration - likely due to a thermodynamic equilibrium between the open iminium intermediate and closed ring form.11 As with conventional glycosylation reactions, formation of the β-anomer (typically equatorial in D-hexoses) provides lowered 1,3-diaxial strain countered against the stability of the α-anomer (typically axial in D-hexoses) due to delocalization of the n-σ* anomeric orbital group. In neoglycoside libraries reported by Thorson and coworkers,15,16,19–26 there was typically a strong bias toward the β-anomer with the use of a methoxyamine handle. According to Perrin, the presence of electron-withdrawing groups near the anomeric center of protonated N-glycosides enhances the anomeric effect.43–44 The low electron-withdrawing nature of alkoxyamine handles typically employed may enhance the impact of aglycon sterics upon anomeric stereoselectivity. This trend holds for aglycons with a significant amount of bulk near the point of glycosylation,15,19–26 although the effect may be diminished when attachment occurs on a more linear substrate.16 While many neoglycosides favor formation of a single anomer, it is important to note that any anomeric mixtures observed are in dynamic solution equilibrium. Although some anomeric mixtures may be resolved chromatographically, the separated anomers typically redistribute back into the original diastereomeric mixtures. This has been put forth as a potential advantage in the context of bioactivity where binding of one anomer to a biological target would help drive the equilibrium toward the bioactive anomer.
Structural and functional relationship to O-glycosides
Aside from the unnatural alkoxyamine linkage, evidence from spectroscopic data and computational analysis strongly suggests that neoglycosides are structurally similar to conventional O-glycosides. Using 1D-NMR chemical shift data and 1H NMR coupling constants, model neohexosides and neopentosides were found to adopt a pyranoside structure with D-sugars typically in the 4C1 conformation and L-glycosides as 1C4.11,15,16,19–26,43–49 Tetroses, as alkoxyamine neoglycosides, typically exist as furanosides.16,46 In their work with (1→6) methoxyamine-linked disaccharides, Peri and coworkers ascertained that the conformational behavior of these neoglycosides was similar to the naturally-occurring congeners gentiobiose and allolactose.18 Using ab initio and molecular mechanics and dynamics calculations in comparison with NMR analysis, they found that only slight differences in conformation existed, likely arising from variance between glycosidic bond length (i.e., C-O vs. C-N) and bond angle (i.e., C-O-C vs. C-N-C). Langenhan et al. reported that the torsion angles of a digitoxigenin neo-D-glucoside, based upon X-ray crystallography, were within the range of those displayed by 23 other cardiac O-glycosides.19 Furthermore, the same study determined the neoglycosides to be completely stable for > 1 month in a 1:1 DMSO/buffer solution at basic and neutral pH, but to slowly hydrolyze under acidic conditions. Enzymatically, model neoglycosides are resistant to glycosidase hydrolysis, unlike their O-glycoside counterparts.50 Finally, direct comparisons of the bioactivities of lipid A neoglycoside antagonists (inflammation)51 or steroidal neoglycosides (anticancer)52 and their O-glycoside counterparts revealed nearly identical bioactivity among all comparator pairings supporting the contention that neoglycosides are good functional mimetics of O-glycosides.
Section 3 – Methods Development (Oligosaccharide and Peptide Applications)
The use of N,O-substituted hydroxylamines in chemoselective glycosylation was first suggested by Peri and coworkers11 as a means of overcoming the linear amine-glycosyl conjugation products (see Figure 1 for structures). Their initial peptide model was based upon installation of a Lys γ-amine N-methyl-O-acetylhydroxylamine handle. This early work revealed aglycons bearing the requisite handle attached via either the alkoxyamine nitrogen or oxygen to function as a substrates for chemoselective conjugation to D-glucose (1), lactose, maltotriose, or D-GalNAc in 24% to 95% yield. Additionally, this study illustrated successful ligation with a variety of N,O-disubstituted handles including those with alkyl and benzyl substituents. Peri and Nicotra expanded the utility of neoglycosylation to form disaccharide and trisaccharide analogs (2).17 Specifically, installation of an alkoxyamine at the 6-position of suitably protected D-glucose followed by conjugation with aldohexoses using either organic (i.e., DMF/AcOH) or aqueous (i.e., acetate buffer) solvent systems revealed 6-N-methoxyiminoglucose (or its precursor 6-aldehyde) as useful sequential building blocks in successive conjugations. Similar methodology was used in an attempt to prepare lipid A antagonist 3 where successful neoglycoside formation was accomplished using a glycosyl bromide rather than a free reducing sugar as the donor.51 The resulting β-(1→6) neoglycoside analog of E. coli lipid A was found to have similar activity as the conventional O-glycoside version. Although the number of synthetic steps to make either the O- glycoside-based and neoglycoside-based antagonists were similar, the neoglycoside had greater solubility in aqueous buffers and was therefore advantageous in biological assays.
Figure 1.

More recently, Peri used neoglycosylation to develop a ligand of the GTPase Ras to elucidate the Ras-substrate binding interface.53 An N-sulfonamidylglucoside previously identified as a Ras ligand, lacked aqueous solubility needed for proper NMR characterization. In this study, a three step process from easily-obtained starting materials led to neoglucoside variant 4, which displayed improved aqueous solubility and long-term stability. Further variation of the sugar revealed that, although binding to Ras was predominately driven via aromatic side chains, the nature of sugar also influenced binding specificity, affinity, and the inhibitory potential.54
Carrasco and coworkers studied the utility of neoglycosylation for creating alkoxyamino side chain-bearing amino acid building blocks orthogonal to peptide coupling (see Figure 2). Specifically, processes using Boc- and Fmoc-based peptide syntheses were used to create an O-methylaminohomoserine handle, which was incorporated into peptides of 3 to 9 residues.55–57 D-Glucose (5) and lactose were coupled to these peptides over 24 h with yields of 60–85%.55 They also found that amino acids based on threonine (6), serine, and homoserine, bearing N-methoxyamine side chains were effective acceptors for D-glucose ligation.57 An extension of this work included the examination of microwave irradiation upon neoglycosylation. When 100 molar-fold excesses of sugar to N-methylaminooxypeptoids were used in the presence of microwaves, reaction times were cut from 12 h to 10–30 minutes in the production of 7.58 Notably, this process appeared to offer chemoselectivity for the N,O-disubstituted handle over free amines (i.e., α-amine) as corresponding α-oxime formation was not detected.
Figure 2.

More recently, André-Miral and coworkers described the use of neoglycosylation in creating β-(1→3) disaccharides 8 via a subsequent glycosynthase-catalyzed glycosylation of a neoglycoside-comprised acceptor (Figure 2).59 Specifically, ligation of N-alkyl-O-benzylhydroxylamines to D-glucose in a THF/AcOH solvent mix yielded exclusively the β-anomers in yields of 43–89%. The resulting enzymatic coupling, using a mutant bacterial glycosynthase, produced varying ratios of β-(1→2) and β-(1→3) disaccharides dependent on the N-alkyl group, with yields of 74–87%. This study is comparable to that of Gantt et al.60 Their work describes glycosylation of a number of small molecule-bearing alkoxyamines by the highly permissive glycosyltransferase OleD and evolved OleD variant ASP. They found that N,O-disubstituted alkoxyamine nucleophiles are recognized by certain enzymes. Complimentary to these approaches, Dasgupta and Nitz describe a method of using N,O-dimethylhydroxylamine as an anomeric protecting group orthogonal to acetate, benzyl, TBDPS, and benzylidene that can be removed using N-chlorosuccinimide in THF/water without affecting disaccharide bonds.61
Section 4 – Natural Product and Small Molecule Drug Discovery Applications (Neoglycorandomization)
Neoglycorandomization is a complementary strategy to enzymatic glycorandomization where the fundamental goal of both is to provide focused libraries of glycosylated target molecules (natural products or small molecule-based drugs) that differ solely via their attached sugars.2 Initially, both strategies were focused upon using these technologies to probe sugar SAR of naturally glycosylated natural product-based drugs. However, the broad applicability of the neoglycosylation reaction and the development, via directed evolution, of highly permissive glycosyltransferases,60,62,63 enabled the expansion of these platforms toward non-glycosylated target molecules. While the neoglycosylation reaction serves as the enabling conjugation reaction, neoglycorandomization conceptually differs from the work highlighted in the prior sections of this review via its focus upon the generation of larger combinatorial neoglycoside-based libraries. An important corollary of neoglycosylation in drug discovery is simple, effective, and efficient reaction setup and purification. Individual reactions can be accomplished in microreaction vials (e.g., 4 mL) on mg scale. Using stir “fleas” and a controlled heating block, dozens of reactions can be conducted manually in parallel, dried via speedvac and subsequently rapidly purified usually with solid-phase extraction cartridges and follow-up HPLC clean-up as necessary – processes all clearly amendable to existing automation. The major long-term goal of this work is to systematically study the impact of differential glycosylation upon bioactive small molecule/drug pharmacological properties and neoglycorandomization presents an excellent tool to facilitate such studies. Table 3 summarizes the structures relevant to neoglycorandomization examples briefly highlighted in alphabetical order (based upon aglycon) herein.
Table 3.
Neoglycosides of natural products and small molecules.
| compound (type) |
neoglycosidea | natural form of glycosylation |
activity (parent) |
activity (neoglycoside) |
library size |
ref |
|---|---|---|---|---|---|---|
| betulinic acid (triterpenoid) |
8 X = O or NH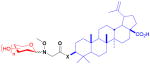
|
n/ab | apoptosis | anticancer or anti-HIV-1 | 32 (ester) 5 (amide) |
15 |
| calicheamicin (enediyne antibiotic) |
9 R = H or monosaccharide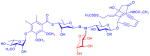
|
aryltri- or tetrasaccharide | anticancer (DNA cleavage) | anticancer | 2 | 22 |
| chlorambucil (nitrogen mustard) |
10 |
n/a | anticancer (DNA alkylation) | anticancer | 63c | 16 |
| colchicine (alkaloid) |
11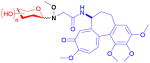
|
n/a | tubulin polymeri- zation inhibitor | anticancer | 58 | 20 |
| cyclopamine (alkaloid) |
12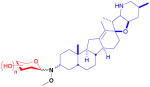
|
n/a | inhibition of Hedgehog pathway | anticancer | 16 (3R) 16 (3S) |
24 |
| digitoxin (steroid) |
13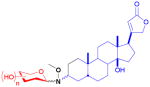
|
tridigitoxoside | cardiac glycoside | anticancer | 39 (3R) 39(3S) |
19 |
| doxycycline (tetracycline) |
14 |
n/a | inhibition of bacterial protein synthesis | antibiotic | 37 | 26 |
| vancomycin (nonribosomal peptide) | 15 R = OH 16 R = N(OMe)sugar 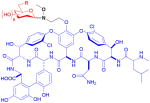
|
disaccharide | inhibition of bacterial cell wall synthesis | antibiotic | 8 (15) 8 (16) |
21 25 |
| warfarin (coumarin) |
17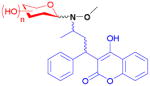
|
n/a | anticoagulant | anticancer | 38 | 23 |
Saccharide depicted in red, handle in black, aglycon in blue.
Not naturally glycosylated.
Library consists of 54 neoglycosides attached by a methoxyamine handle, five by hydroxyamine, and four by hydrazide
Betulinic acid
Betulinic acid, a triterpenoid isolated from birch tree bark, has a broad range of biological activities including apoptosis induction, antivirus, and antiparasitic effects. While not a naturally-glycosylated compound, betulinic acid is structurally related to the saponin family of glycosides,64 making it an attractive target for study.15 Installation of the methoxyamine handle reminiscent to that described for digitoxin neoglycosylation19 was unsuccessful, likely due to the hindered dimethyl group at C4. Rather, an N-methoxyglycine group was appended via a three-step scheme at the C3 alcohol to make 8 (see Table 3). Because the carboxylic acid group in betulinic acid acted as an effective proton source, the usual DMF/AcOH solvent system could be replaced with more volatile MeOH/CH2Cl2. A library of 32 unique neoglycosides was produced and studied for cytotoxicity against a panel of seven carcinomas. Compared to parent, none of the analogs had improved anticancer activity. However, evaluation of cytoprotective effects on CEM-SS lymphocytes against HIV-1 revealed that the majority of the neoglycosides had activity greater than the parent, with seven displaying greater than 10-fold improvement over betulinic acid without toxicity to the host cell. A subgroup of neoglycosides with the handle attached to the aglycon via an amide rather than ester was also evaluated, revealing the nature of the handle to influence activity (i.e., increased cytotoxicity in general). Even more interesting was the bifurcation of activity according to carbohydrate. Those neoglycosides with greater anticancer activity had diminished anti-HIV activity. A similar trend was observed with more cytoprotective neoglycosides having negligible cytotoxicities. Along with these assays, the library was screened for suppression of the glial fibrillary acidic protein (GFAP) based upon the prior identification of the parent betulinic acid as inhibiting production of a mutant form of GFAP which gives rise to the fatal neurodegenerative disorder Alexander disease. Luciferase- and ELISA-based assays revealed that both the ester-linked D-altroside and D-xyloside neoglycosides were able to decrease the amount of both wild type and mutant GFAP production to at least 60% of control without significantly affecting GFAP-producing astrocyte viability at concentrations ≤15 μM.65,66
Calicheamicin
Neoglycosylation is dependent on the presence of a secondary alkoxyamine handle to form the proper selective bond with reducing sugars. While this functional group is not typically common in naturally-occurring metabolites, two members of the enediyne antitumor antibiotics (calicheamicin and esperamicin)67,68 contain a rare N,O-disaccharide bond that contributes to their ability to bind DNA. To explore the potential of this natural neoglycoside handle, two calicheamicin variants, the trisaccharide-containing α3I and its N-acetyl analog, were tested as aglycons in single-step neoglycosylation reactions with D-ribose in methanol using an equivalent of acetic acid.22 This study revealed production of the corresponding neoglycosides, in 33% and 64% yields respectively, where molecular modeling based upon the NMR structure of calicheamicin,69 suggested a minimal adverse effect by the appended ribose upon DNA-binding. Consistent with this model, the corresponding neoglycosides (9) retained activity in both an in vitro DNA cleavage assay70 and in cytotoxicity assays against a panel of five cancer cell lines. Although the GI50 values of the calicheamicin neoglycosides were slightly higher than the parental congeners, the two neoglycosides still displayed potent cytoxicity (i.e., GI50 values of 5 to 190 nM). This study further illustrates the accommodation of the neoglycosylation handle to include a range of alkoxy substitutions (including O-glycosides). In addition, this work sets the stage for potential alternative avenues for calicheamicin conjugation in the context of antibody drug conjugates where existing conjugates under clinical investigation still suffer from off target side effects likely due to premature release of the toxic payload.71,72
Chlorambucil
Chlorambucil is a nitrogen mustard-based DNA alkylating agent that is used clinically for chronic lymphocytic leukemia, but with potentially toxic side effects. A D-glucoside analog was found to be actively transported into cells which is distinct the diffusion dependent mechanism of the parent drug,73 thereby suggesting glycosylation might be beneficial to improving the drug’s properties. Goff and Thorson described a five-step alteration of the chlorambucil butanoic acid group to install the ligation handle with only one required chromatography step.16,74 A 54-member library was created (10, see Table 3) and assayed for antiproliferative activity (i.e., GI50) activity against a panel of ten cancer cell lines. Nineteen neoglycosides had GI50 values in the nanomolar range in at least one cell line with six of the nineteen in three or more cell lines. The D-threoside and D-glucuronolactonide analogs had 8- and 6-fold average improvements over the parent across all ten cell lines tested. Structure-activity analysis found that those neoglycosides that formed furanosides tended to have greater potency and, of that group, those with 2,3-trans-dihydroxyl orientation were more potent than 2,3-cis. The nature of the N-methoxyamine handle was also examined, in part based upon lessons learned from the impact of linker composition on the corresponding betulinic acid neoglycoside library.15 N-hydroxyamino- and N-acylhydrazine-based groups were alternatively explored based on reports that they produced similar chemoselective glycosylation results.40,75 In the same inhibition assay, these variants were markedly less active compared to the corresponding N-methoxyamine-based neoglycosides likely due to the propensity of the furanosides (i.e., glucuronolactonide), pentosides, or tetrosides to adopt open-chain forms. Consistent with this, further scrutiny revealed the N-hydroxyamino-D-riboside to adopt a 1:1 open-chain nitrone to cyclized product ratio while the D-fucoside led to cyclized conformer. Peracetlyation of the N-hydroxyamino-D-riboside mixture in this study invoked cyclization which, upon global deprotection, reverted back to the mixture of closed and open ring forms. This is consistent with the prior discussion (see section 2) pertaining to the influence of both the nature of the chemoselective handle (i.e., N,O-disubstituted vs. N-substituted) and the reducing sugar (i.e., electron-rich vs. electron-poor) in dictating ring closure. This finding also implicates the use of hydroxyamine handles to potentially serve as an intermediate to ‘capture’ alternative alkyl subsitituents en route to alternative alkoxy handle substitutions such as those described by Langenhan and coworkers.27
Colchicine
Colchicine is a plant-based alkaloid that inhibits tubulin polymerization and, like betulinic acid, is a non-glycosylated natural product. Using an eight-step process, the methoxyamine handle was installed at the colchicine C19 acetamide methyl carbon and ligated with an unprecedented number of 71 unique sugars by Ahmed et al.20,76 Cytotoxicity screening of the 58 resultant neoglycosides (11) identified 15 with IC50 values of <1 μM in at least one cell line, with some analogs equipotent to the parent. Interestingly, among this set were analogs that displayed divergent mechanisms. Specifically, some functioned mechanistically as the parent (a tubulin desstabilizer), others functioned mechanistically as paclitaxel (a tubulin stabilizer), while a few neoglycosides displayed tubulin-independent cytotoxicity. Additionally, many of the hits displayed in vitro cell line specificities distinct from parent natural product.
Cyclopamine
Cyclopamine is an inhibitor of the developmental Hedgehog (Hh) pathway, conserved in higher organisms, and has recently been studied as a non-cytotoxic means of controlling unchecked cell division and cancer stem cell growth.77 Installation of the methoxyamine handle at C3 was accomplished in four steps from parent followed by separation of the (3R)- and (3S)-diastereomers and neoglycosylation of both with an identical set of 16 sugars.24 Four of the 16 unique sugars improved inhibition of cancer cell growth over the parent from 4- to 12-fold, where all four were non-metabolic sugars. This was similarly observed in the chlorambucil study.16 In addition, it was found that the stereocenter at C3 (12) did not have an effect towards GI50 values, though stereochemistry at that position had previously been reported to influence Hh inhibition with other semi-synthetic analogs.78 Neoglycosylation was also found to improve cyclopamine solubility, a noted deficiency of the parent natural product.
Digitoxin
The first natural product neoglycorandomization study was that by Langenhan et al. to study the effect of altering the tridigitoxose glycon of digitoxin on the anticancer activity of the drug.19,79,80 A one-step process was used to hydrolyze the trisaccharide tail of digitoxin while oxidizing the 3-hydroxyl group to the requisite ketone en route to handle installation. Reductive amination of the methoxyimine with borane t-butylamine complex provided a 1:1 diastereomeric mixture of the aglycon. A glycodiverse library of commercially-available sugars was used to couple with both the (3R)- and (3S)-diastereomers of the aglycon (13). Subsequent cytotoxicty assay against ten carcinoma cell lines revealed that, while the (3S)-variants had negligible cytotoxicity (IC50 >25 μM), six of the 39 (3R)-compounds had either notable potency (i.e., IC50 of 18–200 nM as compared to the parent digitoxin with an average IC50 of 440 nM) across most cell lines or showed cell line selectivity. As a crude measure of potential cardiotoxicity, the six best hits were subsequently tested for anti-Na+/K+ ATPase activity and surprisingly found to be >4- fold less inhibitory than the parent digitoxin (where the IC50 of neoglycoside hits could not be reached due to solubility limitations). The subsequent notable activity of two best hits, the L-riboside and L-xyloside, in the NCI 60 panel cell line screen prompted further study in the NCI hollow fiber assay, the outcome of which was noted among the best ever observed in this assay (unpublished results). These two hits also displayed impressive in vivo efficacy in a non-small cell lung (A549) xenograft model (unpublished results) and subsequent comparison of the in vitro cytotoxicity of two lead neoglycosides to their corresponding O-glycosides revealed equipotency.52 Importantly, these lead compounds and aspects of the steroidal glycosylation platform developed served as a basis for a completely new class of drug leads referred to extracellular drug conjugates. Distinct from conventional antibody drug conjugates, which require internalization and hydrolytic release of a drug cargo for activity, EDCs act extracellularly and function as intact antibody-drug conjugates.81,82
Additional studies by Langenhan and coworkers on the digitoxin scaffold27 found that the alkoxy group of the ligation handle also could influence the cytotoxic properties of digitoxin neoglycosides. Using L-ribose and L-xylose (based upon the studies described above), it was found that digitoxin neoglycosides with longer (e.g., ethyl, allyl) or bulkier (e.g., t-butyl, benzyl) alkoxyamine handles tended to have reduced activity when compared to the prior methyl analogs.27 However, they concluded that the glycoside structure had a larger influence on cytotoxicity than the alkoxygroup.83 This model was also used to explore the impact of nucleophilic catalysts to speed the neoglycosylation reaction.84 A subsequent study to assess the impact of the length of the digitoxose saccharide chain for both O- and MeON-glycosides of digitoxin towards cytotoxicity revealed the monodigitoxoside to be the best in both formats.85
Doxycycline
Doxycycline is a semi-synthetic broad-spectrum tetracycline antibiotic and, like that of many other antibiotics, strains of tetracycline-resistant bacteria are becoming increasingly common.86 The latest variant of the tetracycline class, tigecycline, incorporates a glycyl linker at C9 of 9-aminodoxycycline reminiscent to that used in neoglycosylation strategies toward betulinic acid,15 colchicine,20 and vancomycin.21 A four-step scheme was used to nitrate and attach methoxyglycine to the doxycycline scaffold followed by neoglycosylation with 31 different sugars (14, see Table 3).26 To extend the study of neoglycosylation, aminosugars were also used, which marks the first instance of these sugars being systematically employed in neoglycosylation. To overcome the incompatibility issue between neoglycosylation and aminosugars, azidosugars were conjugated with the aglycon then reduced via hydrogenolysis. Of the 37 resulting analogs, the 2′-amino-α-D-neoglucoside was found to have antibiotic activity rivaling that of parent against tetracycline-resistant and tetracycline-sensitive E. coli, with four others having similar activity as parent against a tetracycline-sensitive E. coli strain.
Vancomycin
Vancomycin, a glycosylated natural product of considerable value in antibiotic treatment, was modified via neoglycosylation by Griffith et al. to identify potential candidates with activity against vancomycin-resistant Enterococci (VRE).21 Based on the structure of the related teicoplanin antibiotic, the natural disaccharide of vancomycin was replaced with 2-,3-,4-, or 6-N-decanoyl or biphenoyl D-glucose using a methoxyaminoethyl tether at the phenol of the vancomycin aglycon (15). Resulting testing against 15 VRE strains of varying resistance found that alteration of the sugar moiety improved activity against VRE strains with the 3- and 4-acylated sugars identified as the best sugars in this context.87
Peltier-Pain et al. subsequently created a group of disaccharide analogs and studied the effect of alteration of the distal sugar moiety.25 Using the emerging technique of reverse glycosyltransferase-catalyzed reactions,63 the C6′-N-methoxyamino-β-D-glucosyl vancomycin was enzymatically generated via a single pot two enzyme (OleD TDP16/GtfE) process from vancomycin aglycon and the corresponding para-nitrophenyl O-glycoside donor. A variety of sugars, including those from natural glycopeptides, were installed using neoglycosylation with yields of 31–95% (16). Assays against methicillin- or vancomycin-resistant bacterial strains indicated that, while these analogs displayed decreased activity over vancomycin, they functioned via the same mechanism of action as the parent vancomycin.
Warfarin
The anticoagulant warfarin was neoglycosylated at position 11 in three steps from the parent drug.23,88 To obtain more enantiopure products (warfarin is produced as a racemate), an efficient method of separating the enantiomers using a chiral ketal protecting group was also developed. All 38 neoglycosides were found to be ineffective inhibitors (up to 70-fold decrease) of the vitamin K epoxide reductase complex 1, the target of the parent drug which is involved in blood coagulation. Notably, many library members (17) had up to a 100-fold increase in cytotoxicity over warfarin when tested against an eight-member carcinoma panel. This dramatic reversal of activity demonstrates the potential for neoglycosylation to reveal new potential applications for otherwise well-studied drugs.
Section 5 – Conclusion and future prospects
The published precedent illustrates the power of neoglycosylation as an enabling tool for the discovery of novel bioactive probes and potential early stage leads. Specifically, the body of work summarized within this review illustrates the neoglycosylation reaction to be highly versatile and amenable to a range of solvent conditions, aglycon/sugar functionality, alkoxyamine handle variation and high throughput synthesis platforms. This versatility, in conjunction with the notable combinatorial potential of carbohydrates and the ability to rapidly vary the nature of the neoglycosylation handle alkoxy substitution, offers a nearly limitless access to new structural (and potentially functional) diversity where creative new strategies for alkoxyamine handle installation and/or development of new ‘carboselective’ handles are expected to offer strategic advances. Moreover, the biological evaluation of neoglycosides and neoglycorandomized libraries reported to date highlights the potential for this technology to improve in vitro properties (solubility, potency) and, in some cases, alter the fundamental mechanism of action of a parent compound. In vivo studies published to date also indicate neoglycosides to be stable when delivered via IP or IV injection and to display improved properties (PK, PD and efficacy). Preliminary data from the study of new neoglycorandomized libraries yet to be published (including those based upon brefeldin, doxorubicin, podophyllotoxin, taxol, perillyl alcohol and topotecan for cancer; macrolides and nucleosides for antibiotics/immunosuppression; parthenolide for cancer/immunosuppression; and fluorophores for high throughput sugar transport assays89) are consistent with published precedent and promise to continue to deliver new exciting revelations and bioactive entities.
Acknowledgments
The authors would like to thank both past and current Thorson group members and collaborators that have contributed to neoglycorandomization development and the biological evaluation of corresponding neoglycorandomized libraries/library members. We are also grateful for financial support of this extensive effort provided, in part, by the National Institutes of Health grants CA84374 and AI52218 (to J.S.T.), the National Center for Advancing Translational Sciences (UL1TR000117), the University of Kentucky College of Pharmacy and the University of Kentucky Markey Cancer Center.
Contributor Information
Randal D. Goff, Email: rgoff@wwcc.wy.edu.
Jon. S. Thorson, Email: jsthorson@uky.edu.
Notes and references
- 1.Lin C-I, McCarty RM, Liu H-w. Chem Soc Rev. 2013;42:4377–4407. doi: 10.1039/c2cs35438a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gantt RW, Peltier-Pain P, Thorson JS. Nat Prod Rep. 2011;28:1811–1853. doi: 10.1039/c1np00045d. [DOI] [PubMed] [Google Scholar]
- 3.Singh S, Phillips GN, Jr, Thorson JS. Nat Prod Rep. 2012;29:1201–1237. doi: 10.1039/c2np20039b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hudak JE, Bertozzi CR. Chem Biol. 2014;21:16–37. doi: 10.1016/j.chembiol.2013.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.de Paz JL, Seeberger PH. Methods Mol Biol. 2012;808:1–12. doi: 10.1007/978-1-61779-373-8_1. [DOI] [PubMed] [Google Scholar]
- 6.Bertozzi CR, Freeze HH, Varki A, Esko JD. In: Essentials of Glycobiology. 2. Varki A, editor. 2009. pp. 719–732. [PubMed] [Google Scholar]
- 7.Thibodeaux CJ, Melancon CE, III, Liu H-w. Angew Chem, Int Ed. 2008;47:9814–9859. doi: 10.1002/anie.200801204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Boons G-J, editor. Carbohydrate Chemistry. Blackie Academic & Professional; New York: 1998. [Google Scholar]
- 9.Hanessian S, editor. Preparative Carbohydrate Chemistry. Marcel Dekker; New York: 1997. [Google Scholar]
- 10.Langenhan JM, Griffith BR, Thorson JS. J Nat Prod. 2005;68:1696–1711. doi: 10.1021/np0502084. [DOI] [PubMed] [Google Scholar]
- 11.Peri F, Dumy P, Mutter M. Tetrahedron. 1998;54:12269–12278. [Google Scholar]
- 12.Cervigni SE, Dumy P, Mutter M. Angew Chem Int Ed. 1996;35:1230–1232. [Google Scholar]
- 13.Zhao Y, Kent SBH, Chait BT. Proc Natl Acad Sci USA. 1997;94:1629–1633. doi: 10.1073/pnas.94.5.1629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rodriguez EC, Winans KA, King DS, Bertozzi CR. J Am Chem Soc. 1997;119:9905–9906. [Google Scholar]
- 15.Goff RD, Thorson JS. Org Lett. 2009;11:461–464. doi: 10.1021/ol8025704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Goff RD, Thorson JS. J Med Chem. 2010;53:8129–8139. doi: 10.1021/jm101024j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Peri F, Deutman A, La Ferla B, Nicotra F. Chem Commun. 2002:1504–1505. doi: 10.1039/b203605c. [DOI] [PubMed] [Google Scholar]
- 18.Peri F, Jiménez-Barbero J, Garcia-Aparício V, Tvaroška I, Nicotra F. Chem Eur J. 2004;10:1433–1444. doi: 10.1002/chem.200305587. [DOI] [PubMed] [Google Scholar]
- 19.Langenhan JM, Peters NR, Guzei IA, Hoffman MA, Thorson JS. Proc Nat Acad Sci USA. 2005;102:12305–12310. doi: 10.1073/pnas.0503270102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ahmed A, Peters NR, Fitzgerald MK, Watson JA, Jr, Hoffmann FM, Thorson JS. J Am Chem Soc. 2006;128:14224–14225. doi: 10.1021/ja064686s. [DOI] [PubMed] [Google Scholar]
- 21.Griffith BR, Krepel C, Fu X, Blanchard S, Ahmed A, Edmiston CE, Thorson JS. J Am Chem Soc. 2007;129:8150–8155. doi: 10.1021/ja068602r. [DOI] [PubMed] [Google Scholar]
- 22.Goff RD, Singh S, Thorson JS. ChemMedChem. 2011;6:774–776. doi: 10.1002/cmdc.201100028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Peltier-Pain P, Timmons CS, Grandemange A, Benoit E, Thorson JS. ChemMedChem. 2011;6:1347–1350. doi: 10.1002/cmdc.201100178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Goff RD, Thorson JS. Org Lett. 2012;14:2454–2457. doi: 10.1021/ol300703z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Peliter-Pain P, Marchillo K, Zhou M, Andes DR, Thorson JS. Org Lett. 2012;14:5086–5089. doi: 10.1021/ol3023374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhang J, Ponomareva LV, Marchillo K, Zhou M, Andes DR, Thorson JS. J Nat Prod. 2013;76:1627–1636. doi: 10.1021/np4003096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Langenhan JM, Engle JM, Slevin LK, Fay LR, Lucker RW, Smith KR, Endo MM. Bioorg Med Chem Lett. 2008;18:670–673. doi: 10.1016/j.bmcl.2007.11.058. [DOI] [PubMed] [Google Scholar]
- 28.Dondoni A, Perrone D. Tetrahedron Lett. 1999;40:9375–9378. [Google Scholar]
- 29.Dondoni A, Giovannini PP, Perrone D. J Org Chem. 2002;67:7203–7214. doi: 10.1021/jo020252d. [DOI] [PubMed] [Google Scholar]
- 30.Dondoni A, Perrone D. Tetrahedron. 2003;59:4261–4273. [Google Scholar]
- 31.Cicchi S, Corsi M, Marradi M, Goti A. Tetrahedron Lett. 2002;43:2741–2743. [Google Scholar]
- 32.Cicchi S, Marradi M, Corsi M, Faggi C, Goti A. Eur J Org Chem. 2003:4152–4160. [Google Scholar]
- 33.Leteux C, Childs RA, Chai W, Stoll MS, Kogelberg H, Geizi T. Glycobiology. 1998;8:227–236. doi: 10.1093/glycob/8.3.227. [DOI] [PubMed] [Google Scholar]
- 34.Augé J, Germain-Lubin NJ. Carbohydr Chem. 2000;19:379–392. [Google Scholar]
- 35.Peluso S, Ufret ML, O’Reilly MK, Imperiali B. Chem Biol. 2002;9:1323–1328. doi: 10.1016/s1074-5521(02)00281-8. [DOI] [PubMed] [Google Scholar]
- 36.Guillaumie F, Thomas ORT, Jensen KJ. Bioconjugate Chem. 2002;13:285–294. doi: 10.1021/bc0155364. [DOI] [PubMed] [Google Scholar]
- 37.Flinn NS, Quibell M, Monk TP, Ramjee MK, Urch CJ. Bioconjugate Chem. 2005;16:722–728. doi: 10.1021/bc050041q. [DOI] [PubMed] [Google Scholar]
- 38.Chuang YJ, Zhou X, Pan Z, Turchi C. Biochem Biophys Res Commun. 2009;389:22–27. doi: 10.1016/j.bbrc.2009.08.079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mallevre F, Roget A, Minon T, Kervella Y, Ropartz D, Ralet MC, Canut H, Livache T. Bioconj Chem. 2013;24:1264. doi: 10.1021/bc300667b. [DOI] [PubMed] [Google Scholar]
- 40.Bendiak B. Carbohydr Res. 1997;304:85–90. doi: 10.1016/s0008-6215(97)00213-9. [DOI] [PubMed] [Google Scholar]
- 41.Gundmundsdottir AV, Paul CE, Nitz M. Carbohydr Res. 2009;344:278–284. doi: 10.1016/j.carres.2008.11.007. [DOI] [PubMed] [Google Scholar]
- 42.Godula K, Bertozzi CR. J Am Chem Soc. 2010;132:9963–9965. doi: 10.1021/ja103009d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Perrin CL, Armstrong KB. J Am Chem Soc. 1993;115:6825–6834. [Google Scholar]
- 44.Perrin CL. Tetrahedron. 1995;51:11901–11935. [Google Scholar]
- 45.Perrin CL, Kuperman J. J Am Chem Soc. 2003;125:8846–8851. doi: 10.1021/ja035782l. [DOI] [PubMed] [Google Scholar]
- 46.Serianni AS, Barker R. J Org Chem. 1984;49:3292–3300. [Google Scholar]
- 47.Agrawal PK. Phytochemistry. 1992;31:3307–3330. doi: 10.1016/0031-9422(92)83678-r. [DOI] [PubMed] [Google Scholar]
- 48.Benesi AJ, Falzone CJ, Banerjee S, Farber GK. Carbohydr Res. 1994;258:27–33. [Google Scholar]
- 49.Pretsch E, Bühlmann P, Afolter C. Structure Determination of Organic Compounds. 3. Springer; Berlin: 2000. [Google Scholar]
- 50.Iqbal A, Chibli H, Hamilton CJ. Carbohydr Res. 2013;377:1–3. doi: 10.1016/j.carres.2013.05.006. [DOI] [PubMed] [Google Scholar]
- 51.Peri F, Marinzi C, Barath M, Granucci F, Urbano M, Nicotra F. Bioorg Med Chem. 2006;14:190–199. doi: 10.1016/j.bmc.2005.08.047. [DOI] [PubMed] [Google Scholar]
- 52.Hutchinson CR, Shekhani MS, Prudent JR. 8,361,973. US Patent. 2011 Dec 29;
- 53.Palmioli A, Sacco E, Abraham S, Thomas CJ, Di Domizio A, De Giola L, Gaponenko V, Vanoni M, Peri F. Bioorg Med Chem Lett. 2009;19:4217–4222. doi: 10.1016/j.bmcl.2009.05.107. [DOI] [PubMed] [Google Scholar]
- 54.Sacco E, Abraham SJ, Palmioli A, Damore G, Bargna A, Mazzoleni E, Gaponenko V, Vanoni M, Peri F. Med Chem Commun. 2011;2:396. [Google Scholar]
- 55.Carrasco MR, Nguyen MJ, Burnell DR, MacLaren MD, Hengel SM. Tetrahedron Lett. 2002;43:5727–5729. [Google Scholar]
- 56.Carrasco MR, Brown RT, Serafimova IM, Silva O. J Org Chem. 2003;68:195–197. doi: 10.1021/jo026641p. [DOI] [PubMed] [Google Scholar]
- 57.Carrasco MR, Brown RT. J Org Chem. 2003;68:8853–8858. doi: 10.1021/jo034984x. [DOI] [PubMed] [Google Scholar]
- 58.Seo J, Michaelian N, Owens SC, Dashner ST, Wong AJ, Barron AE, Carrasco MR. Org Lett. 2009;11:5210–5213. doi: 10.1021/ol9021468. [DOI] [PubMed] [Google Scholar]
- 59.Teze D, Dion M, Daligault F, Tran V, André-Miral C, Tellier C. Bioorg Med Chem Lett. 2013;23:448–451. doi: 10.1016/j.bmcl.2012.11.065. [DOI] [PubMed] [Google Scholar]
- 60.Gantt RW, Goff RD, Williams GJ, Thorson JS. Angew Chem, Int Ed. 2008;47:8889–8892. doi: 10.1002/anie.200803508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Dasgupta D, Nitz M. J Org Chem. 2011;76:1918–1921. doi: 10.1021/jo102372m. [DOI] [PubMed] [Google Scholar]
- 62.Williams GJ, Zhang C, Thorson JS. Nature Chem Biol. 2007;3:657–662. doi: 10.1038/nchembio.2007.28. [DOI] [PubMed] [Google Scholar]
- 63.Gantt RW, Peltier-Pain P, Cournoyer WJ, Thorson JS. Nature Chem Biol. 2011;7:685–691. doi: 10.1038/nchembio.638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Vincken JP, Heng L, de Groot A, Gruppen H. Phytochemistry. 2007;68:275–297. doi: 10.1016/j.phytochem.2006.10.008. [DOI] [PubMed] [Google Scholar]
- 65.Messing A, Cho W, Thorson JS, Goff RD. 8,372,808. US Patent. 2013 Feb 12;
- 66.Goff RD, Thorson JS. 8,653,043. US Patent. 2014 Feb 18;
- 67.Lee MD, Dunne TS, Chang CC, Siegel MM, Morton GO, Ellestad GA, McGahren WJ, Borders DB. J Am Chem Soc. 1992;114:985–997. [Google Scholar]
- 68.Konishi M, Ohkuma H, Saitoh K, Kawaguchi H, Golik J, Dubay G, Groenewold G, Krishnan B, Doyle TW. J Antibiot. 1985;38:1605. doi: 10.7164/antibiotics.38.1605. [DOI] [PubMed] [Google Scholar]
- 69.Kumar RA, Ikemoto N, Patel DJ. J Mol Biol. 1997;265:187–201. doi: 10.1006/jmbi.1996.0718. [DOI] [PubMed] [Google Scholar]
- 70.Biggins JB, Prudent JR, Marshall DJ, Ruppen M, Thorson JS. Proc Natl Acad Sci. 2000;97:13537–13542. doi: 10.1073/pnas.240460997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Ricart AD. Clin Cancer Res. 2011;17:6417–6427. doi: 10.1158/1078-0432.CCR-11-0486. [DOI] [PubMed] [Google Scholar]
- 72.Trail PA. Antibodies. 2013;2:113–129. [Google Scholar]
- 73.Veyhl M, Wagner K, Volk C, Gorboulev V, Baumgarten K, Weber WM, Schaper M, Bertam B, Wiessler M, Keopsell H. Proc Natl Acad Sci USA. 1998;95:2914–2919. doi: 10.1073/pnas.95.6.2914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Thorson JS, Goff RD. 8,552,176. US Patent. 2013 Oct 8;
- 75.Merino P, Franco S, Merchan FL, Tejero T. Synth Commun. 1997;27:3529–3537. [Google Scholar]
- 76.Thorson JS, Ahmed A. 8,232,254. US Patent. 2012 Jul 31;
- 77.Heretsch P, Tzagkaroulaki L, Giannis A. Angew Chem Int Ed. 2010;49:3418–3427. doi: 10.1002/anie.200906967. [DOI] [PubMed] [Google Scholar]
- 78.Tremblay MR, Nevalainen M, Nair SJ, Porter JR, Castro AC, Behnke ML, Yu LC, Hagel M, White K, Faia K, Grenier L, Campbell MJ, Cushing J, Woodward CN, Hoyt J, Foley MA, Read MA, Sydor JR, Tong JK, Palombella VJ, McGovern K, Adams J. J Med Chem. 2008;51:6646–6649. doi: 10.1021/jm8008508. [DOI] [PubMed] [Google Scholar]
- 79.Thorson JS, Langenhan JM. 7,754,874. US Patent. 2010 Jul 13;
- 80.Thorson JS, Langenhan JM. 8,344,133. US Patent. 2013 Jan 1;
- 81.Hutchinson CR, Prudent JR, Thorson JS. 8,470,980. US Patent. 2013 Jun 25;
- 82.Sweeny L, Hartman YE, Zinn KR, Prudent JR, Marshall DJ, Shekhani MS, Rosenthal EL. Oral Oncol. 2013;49:991–997. doi: 10.1016/j.oraloncology.2013.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Langenhan JM, Endo MM, Engle JM, Fukumoto LL, Rogalsky DR, Slevin LK, Fay LR, Lucker RW, Rohlfing JR, Smith KR, Tjaden AE, Werner HM. Carbohydr Res. 2011;346:2663–2676. doi: 10.1016/j.carres.2011.09.019. [DOI] [PubMed] [Google Scholar]
- 84.Loskot SA, Zhang J, Langenhan JM. J Org Chem. 2013;78:12189–12193. doi: 10.1021/jo401688p. [DOI] [PubMed] [Google Scholar]
- 85.Iyer AKV, Zhou M, Azad N, Elbaz H, Wang L, Rogalsky DK, Rojanasakul Y, O’Doherty GA, Langenhan JM. ACS Med Chem Lett. 2010;1:326. doi: 10.1021/ml1000933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Sun C, Hunt DK, Clark RB, Lofland D, O’Brien WJ, Plamodon L, Xiao X. J Med Chem. 2011;54:3704–3731. doi: 10.1021/jm1015395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Thorson JS, Griffith BR. 8,236,926. US Patent. 2012 Aug 7;
- 88.Thorson JS, Timmons SC. 8,278,436. US Patent. 2012 Oct 2;
- 89.Thorson JS, Fitzgibbon JR. 8,211,654. US Patent. 2012 Jul 3;