

Abstract
Whole-genome duplications are a widespread feature of plant genome evolution, having been detected in all flowering plant lineages. Despite the prevalence of these events, the extent to which duplicated genes (homeolog gene pairs) functionally diverge (neofunctionalization) is unclear. We present a genome-wide analysis of molecular evolution and regulatory neofunctionalization in maize (Zea mays L.). We demonstrate that 13% of all homeolog gene pairs in maize are regulatory neofunctionalized in leaves, and that regulatory neofunctionalized genes experience enhanced purifying selection. We show that significantly more genes have been regulatory neofunctionalized in foliar leaves than in husk leaves and that both leaf types have experienced selection for distinct functional roles. Furthermore, we demonstrate that biased subgenome expression dominance occurs only in the presence of regulatory neofunctionalization and that in nonregulatory neofunctionalized genes subgenome dominance is progressively acquired during development. Taken together, our study reveals several novel insights into the evolution of maize, genes, and gene expression, and provides a general model for gene evolution following whole-genome duplication in plants.
Whole-genome duplications are a dominant feature of plant genome evolution and have been detected in all angiosperm lineages (Adams and Wendel 2005; Soltis et al. 2009). Post-duplication, many duplicated genes are lost in a process known as fractionation, with this loss often showing a bias toward one of the subgenomes (Thomas et al. 2006; Woodhouse et al. 2010). However, some duplicated genes are retained in the genome as homeolog gene pairs. If retained, the individual genes in a homeolog gene pair may have identical functions, they may partition and share the original gene function (subfunctionalization), or they may diverge and develop novel functions (neofunctionalization) (Ohno 1970; Lynch and Conery 2000; Moore and Purugganan 2005; Freeling 2008; McGrath and Lynch 2012). Neofunctionalization has previously been categorized as regulatory neofunctionalizaton (R-NF) or coding neofunctionalization (C-NF) (Moore and Purugganan 2005). C-NF provides novel protein function via a gain-of-function mutation in the open reading frame of a gene. In contrast, R-NF results from expression divergence, which provides extant protein function in novel temporal or spatial environments. Thus, both R-NF and C-NF of homeolog gene pairs have long been proposed as a major source of evolutionary innovation (Ohno 1970; Moore and Purugganan 2005). While neofunctionalization in plants has been studied for specific genes, or at a genome scale in Arabidopsis thaliana and Glycine (Blanc and Wolfe 2004; Duarte et al. 2006; Erdmann et al. 2010; Liu et al. 2011; Guo et al. 2013; Roulin et al. 2013), it is unknown to what extent genome-wide neofunctionalization is important in other species, or to what extent R-NF and C-NF interact and interdepend.
The grass lineage provides an excellent system to study genome-wide neofunctionalization events because of the contrasting evolutionary trajectories of the closely related species maize (Zea mays L.) and sorghum (Sorghum bicolor) (Swigonova et al. 2004a). Maize underwent a recent whole-genome duplication event between 5 and 12 million years ago (Swigonova et al. 2004a). Since that time, rather than remain tetraploid, multiple chromosomal breakage and fusion events have combined the duplicated chromosomes into a diploid genome containing 10 novel mosaic chromosomes (Wei et al. 2007; Schnable et al. 2011). In contrast, the sorghum genome has retained the ancestral nonduplicated state (Swigonova et al. 2004a). Comparison of the maize genome to that of sorghum has thus facilitated the reconstruction of the two duplicated subgenomes of maize, based on biased fractionation (Paterson et al. 2009; Schnable et al. 2011). This analysis has identified 3228 homeolog gene pairs that have been retained post-duplication (Schnable et al. 2011). Given that maize has ∼32,000 genes (Schnable et al. 2009), homeolog gene pairs thus account for >20% of gene content. Despite constituting a large fraction of the maize genome, it is not yet known to what extent homeolog gene pairs have diverged in expression (R-NF) since the whole-genome duplication event.
Here we exploit a novel transcriptional data set from two maize leaf types (foliar leaf blade and husk leaf sheath) to investigate R-NF of genes in maize leaves following whole-genome duplication. Foliar leaves are the primary source tissue in maize and develop from the main shoot apical meristem. In contrast, husk leaves develop from axillary meristems and function to surround and protect the developing female inflorescence (the ear) (Iltis 2000). We provide evidence for widespread R-NF of retained homeolog genes in maize and show that significantly more genes have been R-NF in foliar leaves than husk leaves, suggesting that since genome duplication, gene-expression patterns in foliar leaves have been under greater selection than those in husk leaves. In addition, functional annotations of R-NF genes are consistent with the biological role of the leaves in which those genes are expressed. Moreover, we demonstrate that R-NF genes are under strong purifying selection and that biased subgenome expression dominance occurs only in the presence of R-NF. We further show that relaxed purifying selection occurs in the absence of R-NF, and is further enhanced by expression dominance. Taken together, these data provide a general model for gene evolution following whole-genome duplication, and provide evidence that regulatory neofunctionalization has played an important role in maize leaf evolution.
Results
Signatures of R-NF are apparent in over 400 homeolog gene pairs that are expressed during leaf development
To assess the evolutionary trajectory of homeolog gene pairs in maize we analyzed a recently published data set, which took advantage of biased fractionation of one ancestral genome in order to reconstruct the maize subgenomes (Schnable et al. 2011). This data set was utilized alongside a transcriptome data set from precisely typed equivalent stages of foliar and husk leaf development (Wang et al. 2013). A total of 2607 of the 3228 homeolog gene pairs that are present in the maize genome are expressed (i.e., gene expression >0) during early development in both foliar and husk leaves. To identify R-NF genes within this data set we defined a set of conservative and statistically rigorous criteria (see Methods). All genes were assigned a four-letter code, which is obtained from the statistically significant gene-expression behavior of the homeolog gene pair during early leaf development in both foliar and husk leaves (Fig. 1; see Methods for a detailed explanation).
Figure 1.

Identification and quantification of regulatory neofunctionalized genes. (A) Genes expressed in foliar (F) and husk (H) leaves were allocated to one of three expression profiles, ascending (A), neutral (N), and descending (D), based on the relative expression levels at three stages of early leaf development: plastochron (P) 1/2, P3/4, and P5 (see Methods for an explanation of profile classification). (B) Cartoon of how an example ancestral gene, with expression allocated to profile A in both F and H, could be manifested after whole-genome duplication. Blue background refers to the homeolog from subgenome-1, and orange to the homeolog from subgenome-2. Continuous line refers to the expression profile (A, N, or D) in F, whereas a dashed line refers to the expression profile in H. Four-letter expression code is made up of homeolog-1 F expression profile, homeolog-2 F expression profile, homeolog-1 H expression profile, homeolog-2 H expression profile. If the expression profile of homeolog-1 matches the expression profile of homeolog-2 in F, and the expression profile of homeolog-1 matches the expression profile of homeolog-2 in H, the gene pair is assumed to have undergone zero expression changes since duplication, and is thus nonregulatory neofunctionalized (non-R-NF). If the expression profile of homeolog-1 matches the expression profile of homeolog-2 in H, but the expression profiles in F are not the same, assuming only one expression change has occurred, this change must have been in F, and thus the gene pair is said to have been regulatory neofunctionalized (R-NF) in F only. If the expression profile of homeolog-1 matches the expression profile of homeolog-2 in F, but the expression profiles in H are not the same, assuming only one change has occurred, this change must have been in H, and thus the gene pair is said to have been R-NF in H only. If the expression profile of homeolog-1 does not match that of homeolog-2 in F, and if the expression profile of homeolog-1 does not match that of homeolog-2 in H, the gene pair is assumed to have undergone at least two changes in expression, and is thus R-NF in both F and H. (C) Results of dividing homeolog gene pairs using the method described in B. Numbers refer to homeolog gene pairs that have expression profiles consistent with the labeled category. A total of 2607 homeolog gene pairs are expressed in both foliar and husk leaf samples.
It is most parsimonious to assume that gene expression in the diploid parental species (assuming an allotetraploid origin) is identical, and that immediately after genome duplication, both genes of a homeolog gene pair have the same expression profiles in all tissues. Therefore, identical expression profiles in the transcriptome data indicate that a homeolog gene pair has not undergone R-NF. Correspondingly, homeolog gene pairs that have nonidentical expression profiles represent R-NF events. In the simplest case, where one gene of a homeolog gene pair has changed expression profile in either foliar or husk leaves, then the most parsimonious explanation is that there has been one change in expression since genome duplication (Fig. 1B). If both genes in a homeolog gene pair differ in expression profile in both leaf types, then there have been at least two changes in expression since genome duplication (Fig. 1B).
Using the above criteria, 2195 (84%) of the expressed homeolog gene pairs (n = 2607) have not changed expression profiles in leaf development since whole-genome duplication, 341 (13%) have undergone one change, and 71 (3%) have undergone two or more changes. Within the two-change category, 13 homeolog gene pairs have palindromic expression profiles (DNND, ANNA, NAAN) (Supplemental Table 1). These palindromic expression profiles may represent examples of subfunctionalized homeolog gene pairs in the context of early foliar and husk leaf development. These results are consistent with the previous finding in Arabidopsis, that divergent expression patterns were more frequently the result of regulatory neofunctionalization than subfunctionalization (Liu et al. 2011). Taken together, these data indicate that 16% of expressed homeolog gene pairs, and 13% of all homeolog-gene pairs, have undergone R-NF in the context of maize leaf development (Fig. 1C).
R-NF genes are more likely to have altered expression patterns in foliar leaves than in husk leaves
To determine whether R-NF is associated equally with altered foliar and husk leaf expression patterns, homeolog gene pairs exhibiting one change in expression profile were categorized into those that provide evidence for R-NF in foliar leaves and those that provide evidence for R-NF in husk leaves. Assuming that the observed expression code is the result of one change in expression profile, it follows that the ancestral pre-whole-genome duplication profile could have been that of either subgenome-1 or subgenome-2. For example, for the expression code AANA, the ancestral expression profile could have been either AA or NA. Post-duplication this would have become AAAA or NANA, and in both cases the single change to generate AANA must occur in the foliar leaf (Fig. 1B). Partitioning the homeolog gene pairs from the one change category in this way revealed that significantly more homeolog gene pairs are R-NF in foliar leaves than in husk leaves (n = 205 and n = 136, respectively, P = 1.87 × 10−5) (Fig. 1C).
Functional annotations of R-NF homeolog genes support selection for distinct biological roles in foliar and husk leaves
The unequal distribution of R-NF genes between foliar and husk leaves suggests that foliar leaves have been subject to greater selection since the recent whole-genome duplication event. If this is the case, R-NF genes should enhance the role of the foliar leaf as the primary source tissue. To determine whether specific functional annotations are overrepresented in the homeolog gene pair data set we independently analyzed the occurrence of annotation terms derived from gene ontology (GO) terms, MaizeCyc pathways, MapMan terms, and Pfam domains relative to all maize genes as well as relative to just homeolog genes (Supplemental Tables 2,3, respectively; see Methods). In the 2195 non-R-NF genes there are a total of 133 significantly overrepresented annotation terms relative to all annotated maize genes (P ≤ 0.05), while in the 412 R-NF genes there are 56, 7, and 20 overrepresented annotation terms in foliar, husk, and both leaves, respectively (Supplemental Table 2). Notably, foliar-specific R-NF genes are overrepresented for terms concerning photosynthesis, cell-wall precursor synthesis, and hormone metabolism (Supplemental Table 2). R-NF of photosynthesis-related genes in foliar leaves indicates that selection has acted on photosynthesis in this tissue type. As foliar leaves are the primary photosynthetic tissue, this selection may have been for enhanced photosynthetic productivity and/or efficiency since the whole-genome duplication event.
Although fewer in number than those in foliar leaves, husk-specific R-NF genes also provide evidence of selection for specific functions with overrepresented annotations including responses to stress and proline metabolism (Supplemental Table 2). Proline is accumulated by plants in response to osmotic stress, and has previously been found to be the major osmotic regulator accumulated in the husk leaves of maize (Xu et al. 2011). R-NF of stress-response genes in husk leaves indicates selection on these leaves for improved stress tolerance.
Of the 20 annotation terms overrepresented in those homeolog gene pairs R-NF in both foliar and husk leaves, there are eight concerning nitrogen metabolism, plus those related to cell-wall biosynthesis and transport (Supplemental Table 2). The overrepresentation of genes involved in nitrogen metabolism is consistent with previous findings showing that genes involved in amino acid biosynthesis and protein catabolism have been under artificial selection since the domestication of maize (Wright et al. 2005). Interestingly, the only annotation terms that are consistently enriched across all categories are those concerned with the regulation of transcription. Thus, although there is a distinct partitioning of R-NF genes in both foliar and husk leaves that is consistent with leaf function, in each category there are significantly more R-NF transcriptional regulators then would be expected given the number of such genes in the maize genome (or the number of such genes in the list of homeologs). This novel finding provides additional insight to previous observations that transcription factors are preferentially retained following whole-genome duplications (Blanc and Wolfe 2004; Freeling 2009). The identity of the sequence regions that are putative targets of these transcription factors are difficult to define, and we were unable to find direct evidence on a genome-wide scale of a link between R-NF transcription factors and the presence or absence of motifs in the promoter regions of differentially expressed genes.
Subgenome-1 homeologs are more likely to be R-NF than subgenome-2
The two ancestral subgenomes of maize have recently been reconstructed (Schnable et al. 2011). Assuming that the expression profiles of non-R-NF homeolog gene pairs are an unbiased sample of the underlying distribution of all gene-expression profiles, the most likely ancestral profile for R-NF homeolog gene pairs can be inferred using a Bayesian model comparison approach (see Methods). This approach revealed that of the homeolog gene pairs exhibiting one change in expression profile, significantly more genes are R-NF from subgenome-1 than from subgenome-2 (n = 197 and n = 144, P = 0.004). However, there was no association between subgenome and leaf type (P = 0.321, χ2 test for independence). Therefore, although there are more R-NF genes in foliar leaves and more R-NF genes from subgenome-1, there is no evidence that subgenome-1 genes are preferentially R-NF in foliar leaves or vice versa.
Biased subgenome expression dominance is only observed in R-NF homeolog gene pairs
As the originating subgenome of each homeolog gene has been previously identified we sought to determine whether R-NF impacted on subgenome expression dominance (Schnable et al. 2011). In previous analyses of this kind a homeolog gene was said to dominate if its expression was at least double that of its homeologous pair (Schnable et al. 2011). To improve both the specificity and sensitivity of expression dominance classification, statistically significant (P ≤ 0.05) differential expression of homeolog gene pairs was used rather than the twofold method utilized previously. This analysis revealed that for non-R-NF genes there is no detectable bias in subgenome expression dominance, with both subgenomes having equal numbers of dominant homeologs at all stages of development in both foliar and husk leaves (Fig. 2A). Interestingly, the proportion of genes that exhibit dominance in either subgenome is higher at later stages of development in both foliar and husk leaves (Fig. 2A). This indicates that in non-R-NF homeolog gene pairs, subgenome dominance is progressively acquired during plant development.
Figure 2.

Expression dominance in homeolog gene pairs during leaf development. (A) Nonregulatory neofunctionalized homeologs. (B) Regulatory neofunctionalized in only foliar leaves. (C) Regulatory neofunctionalized in only husk leaves. (D) Regulatory neofunctionalized in foliar and husk leaves. X-axis labels refer to distinct developmental stages in both foliar and husk leaf development. (P) Plastochron; (I) immature leaf; (E) fully expanded leaf (Wang et al. 2013).
In contrast, R-NF genes exhibit a consistent subgenome dominance bias, and do not exhibit the same progressive increase in dominance during development as non-R-NF genes (Fig. 2B–D). In virtually all cases, the proportion of homeolog gene pairs that exhibit subgenome-1 dominance is larger than the proportion that exhibit subgenome-2 dominance (Fig. 2B–D). Notably, the earlier primordia samples exhibit stronger subgenome-1 dominance bias than later stages of leaf development (Fig. 2B,C). This indicates that in R-NF homeolog gene pairs, biased subgenome dominance is already established in the earliest leaf primordia stages and becomes progressively less pronounced during leaf development (Fig. 2B,C). This is in contrast to non-R-NF genes where subgenome dominance is progressively acquired.
R-NF homeolog gene pairs are under stronger purifying selection and have more similar evolutionary rates than non-R-NF gene pairs
To determine whether R-NF genes exhibit differential rates of coding sequence evolution, each homeolog gene pair was aligned to its sorghum ortholog and Ka/Ks ratios were calculated. This analysis revealed that, in general, homeolog gene pairs are under strong purifying selection (Fig. 3A). Moreover, comparing R-NF homeolog gene pairs with all homeolog gene pairs revealed that R-NF homeolog gene pairs are under significantly higher purifying selection than non-R-NF pairs (Fig. 3A).
Figure 3.

Selection acts differentially on regulatory neofunctionalized genes. (A) Mean Ka/Ks ratio of each homeolog plotted for each category. A ratio of <1 indicates negative (purifying selection), whereas >1 indicates positive selection. P-values were calculated for all R-NF homeolog gene pairs (n = 412) and each category of R-NF homeolog gene pairs using Monte Carlo resampling tests. (F) Foliar; (H) Husk; (F&H) Foliar and Husk. All R-NF categories show significantly greater purifying selection than non-R-NF homeolog gene pairs. (B) Mean magnitude of the difference in Ka/Ks ratios between homeolog pairs. The greater the difference in Ka/Ks the more selection has been relaxed on one gene in the pair. The closer the ratio is to zero the more similar the selection acting upon both homeologs. P-values were calculated using Monte Carlo resampling tests. R-NF homeolog gene pairs show a significantly smaller difference to non-R-NF homeolog gene pairs. Significance levels are indicated by asterisks above the bars. (*) P < 0.05; (**) P < 0.01; (***) P < 0.001; (n.s.) not significant. Error bars are standard errors.
While R-NF homeolog gene pairs are under greater purifying selection, it may be that the R-NF homeolog in a gene pair is under relaxed purifying selection relative to the non-R-NF homeolog. To test this hypothesis the magnitude in the difference in Ka/Ks ratio of both homeologs in each gene pair was calculated. This revealed that R-NF genes have on average a significantly smaller difference in the magnitude of Ka/Ks than non-R-NF homeolog gene pairs (Fig. 3B). When analyzed by category this difference is significant for those homeolog gene pairs R-NF in the foliar and in both the foliar and husk, but not for those R-NF in just the husk (Fig. 3B). Taken together these data reveal that R-NF homeolog gene pairs experience stronger purifying selection and have evolutionary rates that are more similar to each other than non-R-NF gene pairs. Thus, in non-R-NF homeolog gene pairs one member of the gene pair is more likely to experience reduced purifying selection relative to its homeologous pair.
Dominant homeolog genes exhibit stronger purifying selection than their nondominant gene pair
To determine whether subgenome expression dominance accounts for the greater difference in Ka/Ks between non-R-NF homeolog gene pairs, the Ka/Ks ratio of the homeolog of each gene pair that dominates expression was compared with its nondominant partner. This revealed that nondominant homeologs are on average under significantly reduced purifying selection relative to their dominant counterparts. This observation is consistent for dominant and nondominant homeolog gene pairs at all stages of leaf development in both foliar and husk leaves (Fig. 4). Our findings are consistent with previous reports that revealed that low-expressed genes tend to evolve more rapidly than high-expressed genes (Drummond and Wilke 2008). However, our results demonstrate that this phenomenon also applies to recently duplicated genes. Nondominant homeolog genes are therefore a potential source of protein sequence novelty, and are thus strong candidates for coding-sequence neofunctionalization.
Figure 4.

Nondominant homeolog genes experience relaxed purifying selection. Ka/Ks ratios were compared for those non-R-NF homeolog gene pairs that exhibit subgenome dominance. The mean Ka/Ks ratio of nondominant homeolog genes (those expressed at significantly lower levels than their homeolog pair) was significantly higher than the dominant homeolog (those expressed at significantly higher levels than their homeolog pair). P-values were calculated using Wilcoxon signed-rank tests. Significance levels are indicated by asterisks above the bars. (***) P < 0.001. Error bars are standard errors.
Discussion
Duplication followed by neofunctionalization has long been proposed as a source of evolutionary novelty in genome evolution. Moreover, whole-genome duplications are a principal theme of plant evolution and have occurred in the evolution of all angiosperm lineages (Adams and Wendel 2005; Soltis et al. 2009). In maize, a recent whole-genome duplication (5–12 MYA) followed by multiple gene losses has sculpted the contemporary maize genome such that duplicated homeolog gene pairs comprise >20% of all genes. In this work we used maize as a model system to analyze the evolutionary paths taken by homeolog gene pairs following whole-genome duplication. Our work reveals that regulatory neofunctionalization (R-NF) is an important feature of maize evolution at the gene, genome, tissue, and developmental levels.
From our analysis and previous published work it is clear that there are broadly four possible fates for a duplicated gene pair if both copies are retained following a whole-genome duplication (Ohno 1970; Lynch and Conery 2000; Moore and Purugganan 2005; Freeling 2008; McGrath and Lynch 2012): (1) Both duplicates can maintain the same expression pattern and protein function; (2) the original gene function can be partitioned between the duplicate genes (subfunctionalization); (3) one or both duplicates can diverge in gene expression (regulatory neofunctionalization); (4) one or both duplicates can diverge in protein function (coding neofunctionalization) (Fig. 5). On a genome-wide scale it is currently difficult to accurately diagnose changes in protein function (though an attempt has recently been made using known protein interaction partners in Arabidopsis thaliana [Guo et al. 2013]), but changes in gene expression are readily determined. Previous studies of neofunctionalization in Arabidopsis thaliana have relied on microarray experiments to assess divergent expression of specific subsets of genes, and are thus not representative of the whole genome (Blanc and Wolfe 2004; Duarte et al. 2006). More recently, RNA sequencing has been used to assess expression divergence of duplicate genes in soybean (Glycine max L.) (Roulin et al. 2013). In our work, RNA sequencing allowed us to characterize R-NF at a genome-wide scale, revealing that ∼13% of homeolog gene pairs in the maize genome have undergone R-NF. As this estimate was generated only in the context of leaf development, it likely represents an underestimation of the extent of R-NF in maize genome evolution. There is some discussion over whether the maize whole-genome duplication was the result of an autotetraploid or allotetraploid event (Gaut and Doebley 1997; Swigonova et al. 2004a,b). Given the more likely scenario of an allotetraploid origin (Gaut and Doebley 1997), it is possible that some of the divergence in expression that we have classified as regulatory neofunctionalization (R-NF) preceded the whole-genome duplication in the two progenitors of the contemporary maize genome. In this context it is pertinent to note that differential expression of some genes has been found between diploid cotton genomes (pre-allotetraploidization) (Rapp et al. 2009; Flagel and Wendel 2010; Yoo et al. 2013). However, it is our contention that it is more parsimonious to assume that the majority of genes have the same expression profile at the point of genome duplication. In support of this assumption is the fact that 84% of genes in our data set are non-R-NF, and while this may represent convergence, it is more likely that 16% of homeolog gene pairs have changed expression than 84% of genes have converged in expression. Moreover, regardless of when this divergence occurred, it does not change the central finding of our work; that distinct selection signatures and patterns of subgenome dominance are hallmarks of homeolog gene pairs that have diverged in gene expression.
Figure 5.

A model for gene evolution following whole-genome duplication in maize. Once retained, a duplicated gene pair can either diverge in expression profile or maintain the original expression profile. Those pairs that maintain expression profiles are likely to either share the original gene function or undergo coding neofunctionalization. Those gene pairs that diverge in expression are likely regulatory neofunctionalized.
Expression dominance bias toward the less fractionated subgenome has previously been observed in both maize and Brassica rapa (Schnable et al. 2011; Cheng et al. 2012) but is not observed in more ancient plant tetraploids (Blanc and Wolfe 2004; Li et al. 2006). Similarly, recent work in cotton has demonstrated that expression dominance between homeologs occurs immediately following tetraploidization and increases over time (Adams and Wendel 2013; Yoo et al. 2013). Interestingly, we find that when defined using statistically significant differential expression testing, expression dominance is a dynamic feature of homeolog gene pairs in maize. Those gene pairs that have not undergone R-NF show no consistent expression bias toward either subgenome, and the proportion of gene pairs exhibiting either subgenome-1 (less fractionated) or subgenome-2 (more fractionated) dominance increases over the course of leaf development. In contrast, subgenome-1 expression dominance bias is consistently observed in R-NF homeolog gene pairs, with no apparent developmental progression in dominance. This indicates that in maize, the previously reported expression dominance bias toward the less fractionated subgenome occurs alongside R-NF of homeolog gene pairs (Schnable et al. 2011).
We find that non-R-NF homeolog genes are under reduced purifying selection, and of those non-R-NF homeolog gene pairs that exhibit subgenome dominance, the nondominant homeolog is consistently found to be under reduced purifying selection relative to the dominant homeolog. In light of this, it is interesting to note that only 10 genes in the maize genome show evidence of being under positive selection, and nine of these are non-R-NF (Supplemental Table 4). The fact that we find so few genes to be under positive selection supports the previous finding that positive selection on coding regions does not seem to play a significant role in post-genome duplication C-NF (Duarte et al. 2006). While positive selection has been shown to drive neofunctionalization in some duplicates (Zhang et al. 1998; Shiu et al. 2006; Conant and Wolfe 2008), our analysis suggests that following whole-genome duplication, reduced purifying selection is a more important evolutionary mechanism for altering protein function in plants. In support of our findings, relaxed purifying selection has previously been associated with functional divergence of duplicate genes within both plant and non-plant systems (Kondrashov et al. 2002; Zhang 2003; Flagel and Wendel 2009; Shan et al. 2009). In addition, it has previously been shown that reduced purifying selection is more important than positive selection in the maintenance of duplicated genes in the human genome (Nguyen et al. 2008). Collectively, our results point to a model for gene evolution following whole-genome duplication in maize whereby the emergence of novel protein function occurs through the relaxation of purifying selection in genes that have not undergone R-NF (Fig. 5). It remains possible that C-NF could follow R-NF; however, our data indicate that C-NF is more likely to occur in the absence of R-NF.
Our findings also shed new light on the selective pressures acting on maize since the whole-genome duplication event. Specifically, we show that foliar leaves have experienced selection for photosynthetic productivity/efficiency and that husk leaves have experienced selection for stress tolerance. Though the time interval of the analysis is defined by the genome duplication event ∼5–12 MYA, our findings are strongly consistent with artificial selection as a result of domestication ∼7000–12,000 yr ago (Matsuoka et al. 2002; Wright et al. 2005). Taken together, our results provide evidence that R-NF has played a major role in the evolution of maize, sculpting its genes, genome, tissues, and developmental processes, and provide a general model for gene evolution following whole-genome duplication in plants.
Methods
Transcript quantification and expression profile allocation
Maize leaf development transcriptome data sets were obtained from a previous study (Wang et al. 2013). Paired end reads were subject to quality-based trimming using the FASTX-Toolkit (Goecks et al. 2010) setting the phred quality threshold at 20 and discarding reads <21 nucleotides in length. Transcripts were quantified using RSEM (Li and Dewey 2011) (using default parameters) and the predicted coding sequences from version 5b of the maize genome. RSEM is specifically designed to enable accurate expression quantification between highly similar gene sequences. The mean Ks between maize homeolog genes is 0.38; thus, on average, a 90-bp paired end read (180 bp) will have ∼20 distinguishing SNPs, enabling RSEM to accurately discriminate between reads uniquely originating from each homeolog. For differential gene expression analysis, expected transcript counts originating from the same gene locus were summed and all possible pairwise comparisons between biologically replicated samples were performed using DESeq (Anders and Huber 2010). Expression values were normalized by DESeq using the default method, and in all cases differentially expressed genes were identified as those genes with a Benjamini-Hochberg corrected P-value ≤ 0.05 (Benjamini and Hochberg 1995). Genes were assigned to three expression profiles: ascending (A), neutral (N), and descending (D). The ascending expression code included those genes that increased significantly from either P1/2 to P3/4 (but not necessarily P3/4 to P5), from P3/4 to P5 (but not necessarily from P1/2 to P3/4), and those genes that increased significantly across both steps (P ≤ 0.05). There could not be a significant decrease between developmental stages, and thus in all cases the overall expression pattern is one of increasing expression. The same applies inversely to the descending category. In the neutral profile there could not be a significant difference between any of the samples. Genes that had significantly higher (or lower) expression in P3/4 than both P5 and P1/2 for both leaf types were assigned to “peak” (or “trough”) profiles. The number of genes assigned to these profiles was low (Foliar “P3/4 Peak” profile n = 34, Foliar “P3/4 Trough” profile n = 10, Husk “P3/4 Peak” profile n = 27, Husk “P3/4 Trough” profile n = 8), and hence statistical analysis using these profiles was not possible (Wang et al. 2013). Thus, they were excluded from this work.
While transcriptome data was collected at five distinct developmental stages (P1/2, P3/4, P5, I, E), for expression profile analysis we used only those stages that were directly equivalent between foliar and husk samples (Wang et al. 2013). FP1/2 is equivalent to HP1/2, FP3/4 is equivalent to HP3/4, and FP5 is equivalent to HP5. However, HI is not the equivalent developmental stage to FI and HE is not the equivalent developmental stage to FE. Thus, in order to provide a directly comparable developmental series we did not construct profiles including these later samples. A four-letter expression code was then generated by taking the expression profile of the subgenome-1 homeolog in both foliar and husk leaves, followed by the expression profile of the subgenome-2 homeolog in both foliar and husk leaves. Together this forms a four-letter expression code that describes the statistically significant behavior of a gene pairs expression (Fig. 1B). For example, for a hypothetical homeolog gene pair, if the subgenome-1 homeolog had an ascending (A) expression profile in both foliar and husk leaves, and the subgenome-2 homeolog had a descending (D) expression profile in foliar leaves and a neutral (N) expression profile in husk leaves, then the expression code for this homeolog gene pair would be AADN ([foliar profile subgenome-1][Husk profile subgenome-1][Foliar profile subgenome-2][Husk profile subgenome-2]).
Enrichment analysis
Enrichment analyses were carried out for Gene Ontology (GO) http://www.geneontology.org/ terms, MaizeCyc pathways http://maizecyc.maizegdb.org/, MapMan terms http://www.mapman.gabipd.org/ and Pfam domains http://pfam.sanger.ac.uk/. Analysis was performed relative to all maize genes (Supplemental Table 2), and relative to just the set of known homeolog genes (Supplemental Table 3). P-values were obtained by approximating Wallenius’ noncentral hypergeometric distribution. GOseq (Young et al. 2010) was used to compensate for overdetection of differential expression for long and highly expressed transcripts. The resulting P-values were subject to multiple hypothesis test correction to correct for Type I family-wise error using the Benjamini-Hochberg method. Significantly enriched annotation terms were identified as those that obtained a corrected P-value of ≤0.05.
Calculation of Ka/Ks ratios
Maize gene sequences were obtained from the B73 reference genome (Schnable et al. 2009) and sorghum from the published Sorghum bicolor genome (Paterson et al. 2009). For each homeolog gene pair the longest representative gene models were selected and the amino acid sequences were aligned to the Sorghum bicolor ortholog using MergeAlign (Collingridge and Kelly 2012). The amino-acid sequences in the three-sequence multiple sequence alignment were then replaced with their corresponding coding sequences. Each aligned homeolog gene was then compared independently with its Sorghum bicolor ortholog and the ratio of nonsynonymous (protein changed) substitution rate per nonsynonymous site (Ka) to synonymous (protein unchanged) substitution rate per synonymous site (Ks) was calculated using KaKs_Calculator Toolbox 2.0 (Wang et al. 2010). Ka/Ks values for homeolog genes in each category were averaged and standard errors calculated (Fig. 3).
Monte Carlo resampling tests
To account for differences in sample sizes and avoid requirement for distributional assumptions, Monte Carlo resampling tests were used to assess the significance of differences in Ka/Ks ratios between groups. Here, the mean Ka/Ks ratio of the test group was compared to a reference group of the same size that was randomly selected with replacement from the whole pool of homeolog genes. For each test group the process was repeated 10,000 times and the proportion of times that the mean of the test group was greater than the reference group recorded. A proportion ≥0.95 indicates a significantly greater difference in the test group than the overall population of homeolog genes, whereas a proportion of ≤0.05 indicates a significantly smaller difference in the test group.
Ancestral profile inference by Bayesian model comparison
To determine whether the homeolog from subgenome-1 or subgenome-2 has been neofunctionalized it is necessary to know the ancestral gene expression profile. As this data is unobtainable, ancestral profiles must be inferred. Assuming that the expression profiles of non-neofunctionalized homeolog gene pairs are an unbiased sample of the global distribution of gene expression profiles, we can infer the probability of observing an ancestral gene expression profile
 |
where Ai is the ancestral gene expression profile in question and Ni is the number of times that the same profile is observed in all of the homeolog genes present in this analysis. We evaluate the probability of observing an expression profile as inversely proportional to the number of changes between the observed and ancestral gene expression profile such that
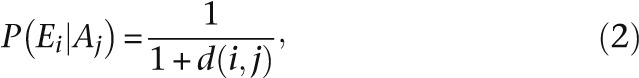 |
where P(Ei|Aj) is the probability of observing expression profile Ei given ancestral profile Aj and d(i,j) is the Hamming distance between the observed and ancestral profile. We can thus compute the posterior probability of each ancestral profile for any observed profile using Bayes rule and the law of total probability:
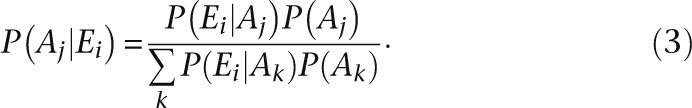 |
For the purposes of all downstream analyses we selected the maximum likelihood ancestral profile.
Acknowledgments
T.E.H. was supported by the Newton Abraham Scholarship and the BBSRC funded Oxford Interdisciplinary Bioscience Doctoral Training Partnership. S.K. is a Leverhulme Trust Career Development Fellow. J.A.L. is supported by an ERC Advanced Investigator Award (EDIP). We thank Jim Fouracre and Dmitry Filatov for their discussion and advice on the manuscript, and Dr. James Schnable for the provision of homeolog gene pair lists.
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.172684.114.
References
- Adams KL, Wendel JF. 2005. Polyploidy and genome evolution in plants. Curr Opin Plant Biol 8: 135–141 [DOI] [PubMed] [Google Scholar]
- Adams KL, Wendel JF. 2013. Dynamics of duplicated gene expression in Polyploid Cotton. In Polyploid and hybrid genomics (ed. Chen ZJ and Birchler JA). Wiley, Oxford, UK. [Google Scholar]
- Anders S, Huber W. 2010. Differential expression analysis for sequence count data. Genome Biol 11: R106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benjamini Y, Hochberg Y. 1995. Controlling the false discovery rate: a practical and powerful approach to mulitple testing. JR Stat Soc 57: 289–300 [Google Scholar]
- Blanc G, Wolfe KH. 2004. Functional divergence of duplicated genes formed by polyploidy during Arabidopsis evolution. Plant Cell 16: 1679–1691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng F, Wu J, Fang L, Sun S, Liu B, Lin K, Bonnema G, Wang X. 2012. Biased gene fractionation and dominant gene expression among the subgenomes of Brassica rapa. PLoS ONE 7: e36442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collingridge PW, Kelly S. 2012. MergeAlign: improving multiple sequence alignment performance by dynamic reconstruction of consensus multiple sequence alignments. BMC Bioinformatics 13: 117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conant GC, Wolfe KH. 2008. Turning a hobby into a job: how duplicated genes find new functions. Nat Rev Genet 9: 938–950 [DOI] [PubMed] [Google Scholar]
- Drummond DA, Wilke CO. 2008. Mistranslation-induced protein misfolding as a dominant constraint on coding-sequence evolution. Cell 134: 341–352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duarte JM, Cui L, Wall PK, Zhang Q, Zhang X, Leebens-Mack J, Ma H, Altman N, dePamphilis CW. 2006. Expression pattern shifts following duplication indicative of subfunctionalization and neofunctionalization in regulatory genes of Arabidopsis. Mol Biol Evol 23: 469–478 [DOI] [PubMed] [Google Scholar]
- Erdmann R, Gramzow L, Melzer R, Theissen G, Becker A. 2010. GORDITA (AGL63) is a young paralog of the Arabidopsis thaliana Bsister MADS box gene ABS (TT16) that has undergone neofunctionalization. Plant J 63: 914–924 [DOI] [PubMed] [Google Scholar]
- Flagel LE, Wendel JF. 2009. Gene duplication and evolutionary novelty in plants. New Phytol 183: 557–564 [DOI] [PubMed] [Google Scholar]
- Flagel LE, Wendel JF. 2010. Evolutionary rate variation, genomic dominance and duplicate gene expression evolution during allotetraploid cotton speciation. New Phytol 186: 184–193 [DOI] [PubMed] [Google Scholar]
- Freeling M 2008. The evolutionary position of subfunctionalization, downgraded. Genome Dyn 4: 25–40 [DOI] [PubMed] [Google Scholar]
- Freeling M 2009. Bias in plant gene content following different sorts of duplication: tandem, whole-genome, segmental, or by transposition. Annu Rev Plant Biol 60: 433–453 [DOI] [PubMed] [Google Scholar]
- Gaut BS, Doebley JF. 1997. DNA sequence evidence for the segmental allotetraploid origin of maize. Proc Natl Acad Sci 94: 6809–6814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goecks J, Nekrutenko A, Taylor J. 2010. Galaxy: a comprehensive approach for supporting accessible, reproducible, and transparent computational research in the life sciences. Genome Biol 11: R86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo H, Lee T-H, Wang X, Paterson AH. 2013. Function relaxation followed by diversifying selection after whole genome duplication in flowering plants. Plant Physiol 162: 769–778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iltis HH 2000. Homeotic sexual translocations and the origin of maize (Zea Mays, Poaceae): a new look at an old problem. Econ Bot 54: 7–42 [Google Scholar]
- Kondrashov FA, Rogozin IB, Wolf YI, Koonin E V. 2002. Selection in the evolution of gene duplications. Genome Biol 3: RESEARCH0008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li B, Dewey CN. 2011. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics 12: 323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L, Wang X, Stolc V, Li X, Zhang D, Su N, Tongprasit W, Li S, Cheng Z, Wang J, et al. 2006. Genome-wide transcription analyses in rice using tiling microarrays. Nat Genet 38: 124–129 [DOI] [PubMed] [Google Scholar]
- Liu S-L, Baute GJ, Adams KL. 2011. Organ and cell type-specific complementary expression patterns and regulatory neofunctionalization between duplicated genes in Arabidopsis thaliana. Genome Biol Evol 3: 1419–1436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch M, Conery JS. 2000. The evolutionary fate and consequences of duplicate genes. Science 290: 1151–1155 [DOI] [PubMed] [Google Scholar]
- Matsuoka Y, Vigouroux Y, Goodman MM, Sanchez GJ, Buckler E, Doebley J. 2002. A single domestication for maize shown by multilocus microsatellite genotyping. Proc Natl Acad Sci 99: 6080–6084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGrath C, Lynch M. 2012. Evolutionary significance of whole-genome duplication. In Polyploidy and genome evolution (ed. Soltis PS and Soltis DE), pp. 1–20. Springer, Berlin. [Google Scholar]
- Moore RC, Purugganan MD. 2005. The evolutionary dynamics of plant duplicate genes. Curr Opin Plant Biol 8: 122–128 [DOI] [PubMed] [Google Scholar]
- Nguyen D-Q, Webber C, Hehir-Kwa J, Pfundt R, Veltman J, Ponting CP. 2008. Reduced purifying selection prevails over positive selection in human copy number variant evolution. Genome Res 18: 1711–1723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohno S. 1970. Evolution by gene duplication. Springer, Berlin. [Google Scholar]
- Paterson AH, Bowers JE, Bruggmann R, Dubchak I, Grimwood J, Gundlach H, Haberer G, Hellsten U, Mitros T, Poliakov A, et al. 2009. The Sorghum bicolor genome and the diversification of grasses. Nature 457: 551–556 [DOI] [PubMed] [Google Scholar]
- Rapp RA, Udall JA, Wendel JF. 2009. Genomic expression dominance in allopolyploids. BMC Biol 7: 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roulin A, Auer PL, Libault M, Schlueter J, Farmer A, May G, Stacey G, Doerge RW, Jackson SA. 2013. The fate of duplicated genes in a polyploid plant genome. Plant J 73: 143–153 [DOI] [PubMed] [Google Scholar]
- Schnable PS, Ware D, Fulton RS, Stein JC, Wei F, Pasternak S, Liang C, Zhang J, Fulton L, Graves TA, et al. 2009. The B73 maize genome: complexity, diversity, and dynamics. Science 326: 1112–1115 [DOI] [PubMed] [Google Scholar]
- Schnable JC, Springer NM, Freeling M. 2011. Differentiation of the maize subgenomes by genome dominance and both ancient and ongoing gene loss. Proc Natl Acad Sci 108: 4069–4074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shan H, Zahn L, Guindon S, Wall PK, Kong H, Ma H, DePamphilis CW, Leebens-Mack J. 2009. Evolution of plant MADS box transcription factors: evidence for shifts in selection associated with early angiosperm diversification and concerted gene duplications. Mol Biol Evol 26: 2229–2244 [DOI] [PubMed] [Google Scholar]
- Shiu S-H, Byrnes JK, Pan R, Zhang P, Li W-H. 2006. Role of positive selection in the retention of duplicate genes in mammalian genomes. Proc Natl Acad Sci 103: 2232–2236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soltis DE, Albert VA, Leebens-Mack J, Bell CD, Paterson AH, Zheng C, Sankoff D, Depamphilis CW, Wall PK, Soltis PS. 2009. Polyploidy and angiosperm diversification. Am J Bot 96: 336–348 [DOI] [PubMed] [Google Scholar]
- Swigonova Z, Lai J, Ma J, Ramakrishna W, Llaca V, Bennetzen JL, Messing J. 2004a. Close split of sorghum and maize genome progenitors. Genome Res 14: 1916–1923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swigonova Z, Lai J, Ma J, Ramakrishna W, Llaca V, Bennetzen JL, Messing J. 2004b. On the tetraploid origin of the maize genome. Comp Funct Genomics 5: 281–284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas BC, Pedersen B, Freeling M. 2006. Following tetraploidy in an Arabidopsis ancestor, genes were removed preferentially from one homeolog leaving clusters enriched in dose-sensitive genes. Genome Res 16: 934–946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang D, Zhang Y, Zhang Z, Zhu J, Yu J. 2010. KaKs_Calculator 2.0: a toolkit incorporating gamma-series methods and sliding window strategies. Genomics Proteomics Bioinformatics 8: 77–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang P, Kelly S, Fouracre JP, Langdale JA. 2013. Genome-wide transcript analysis of early maize leaf development reveals gene cohorts associated with the differentiation of C4 Kranz anatomy. Plant J 75: 656–670 [DOI] [PubMed] [Google Scholar]
- Wei F, Coe E, Nelson W, Bharti AK, Engler F, Butler E, Kim H, Goicoechea JL, Chen M, Lee S, et al. 2007. Physical and genetic structure of the maize genome reflects its complex evolutionary history. PLoS Genet 3: e123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodhouse MR, Schnable JC, Pedersen BS, Lyons E, Lisch D, Subramaniam S, Freeling M. 2010. Following tetraploidy in maize, a short deletion mechanism removed genes preferentially from one of the two homologs. PLoS Biol 8: e1000409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright SI, Bi IV, Schroeder SG, Yamasaki M, Doebley JF, McMullen MD, Gaut BS. 2005. The effects of artificial selection on the maize genome. Science 308: 1310–1314 [DOI] [PubMed] [Google Scholar]
- Xu H, Lu Y, Tong S, Song F. 2011. Lipid peroxidation, antioxidant enzyme activity and osmotic adjustment changes in husk leaves of maize in black soils region of Northeast China. Ann Surg Oncol 6: 3098–3102 [Google Scholar]
- Yoo M-J, Szadkowski E, Wendel JF. 2013. Homoeolog expression bias and expression level dominance in allopolyploid cotton. Heredity 110: 171–180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young MD, Wakefield MJ, Smyth GK, Oshlack A. 2010. Gene ontology analysis for RNA-seq: accounting for selection bias. Genome Biol 11: R14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J 2003. Evolution by gene duplication: an update. Trends Ecol Evol 18: 292–298 [Google Scholar]
- Zhang J, Rosenberg HF, Nei M. 1998. Positive Darwinian selection after gene duplication in primate ribonuclease genes. Proc Natl Acad Sci 95: 3708–3713 [DOI] [PMC free article] [PubMed] [Google Scholar]