

Background: The Ebola VP30 is required for viral transcription and can exist in phosphorylated (inactive) and dephosphorylated (active) forms.
Results: VP30 is dephosphorylated by PP1; the small PP1-targeting molecule 1E7-03 inhibits VP30 dephosphorylation and viral transcription, thereby blocking viral replication.
Conclusion: PP1 plays an important role in Ebola virus transcription.
Significance: Targeting PP1 is a feasible approach for inhibition of Ebola virus.
Keywords: Ebola Virus, Phosphoprotein Phosphatase 1 (PP1), Small Molecule, Transcription, Viral Replication
Abstract
The filovirus Ebola (EBOV) causes the most severe hemorrhagic fever known. The EBOV RNA-dependent polymerase complex includes a filovirus-specific VP30, which is critical for the transcriptional but not replication activity of EBOV polymerase; to support transcription, VP30 must be in a dephosphorylated form. Here we show that EBOV VP30 is phosphorylated not only at the N-terminal serine clusters identified previously but also at the threonine residues at positions 143 and 146. We also show that host cell protein phosphatase 1 (PP1) controls VP30 dephosphorylation because expression of a PP1-binding peptide cdNIPP1 increased VP30 phosphorylation. Moreover, targeting PP1 mRNA by shRNA resulted in the overexpression of SIPP1, a cytoplasm-shuttling regulatory subunit of PP1, and increased EBOV transcription, suggesting that cytoplasmic accumulation of PP1 induces EBOV transcription. Furthermore, we developed a small molecule compound, 1E7-03, that targeted a non-catalytic site of PP1 and increased VP30 dephosphorylation. The compound inhibited the transcription but increased replication of the viral genome and completely suppressed replication of EBOV in cultured cells. Finally, mutations of Thr143 and Thr146 of VP30 significantly inhibited EBOV transcription and strongly induced VP30 phosphorylation in the N-terminal Ser residues 29–46, suggesting a novel mechanism of regulation of VP30 phosphorylation. Our findings suggest that targeting PP1 with small molecules is a feasible approach to achieve dysregulation of the EBOV polymerase activity. This novel approach may be used for the development of antivirals against EBOV and other filovirus species.
Introduction
EBOV,4 along with the closely related Sudan, Bundibugyo, Taï Forest, Reston, Marburg, and Ravn viruses, is a member of the family Filoviridae and a biosafety level 4 pathogen, which causes the most severe hemorrhagic fevers known in humans and non-human primates, with a mortality rate in humans of up to 90% (1). The latest outbreak of EBOV started several months ago in Guinea and spread to Liberia and Sierra Leone (2). Currently, there are no approved treatments against filoviruses. Thus, there is an urgent need for universal treatments against all diverse species and strains of filoviruses.
The ribonucleoprotein complex of EBOV consists of nucleoprotein (NP), phosphoprotein (VP35), and the large subunit of polymerase (L). In addition, it also includes the VP30 protein (reviewed in Ref. 3). The polymerase complex can mediate both the transcription of individual genes and replication of the whole genome. In the transcription mode, the polymerase sequentially transcribes each gene, starting at the 3′-proximal gene, initiating the transcription at the gene-start signal and terminating at the gene-end signal. In the replication mode, the polymerase uses the genomic RNA to produce a complementary strand of anti-genome and subsequently generates a new genomic RNA strand using antigenome as a template. Although the exact mechanism of the polymerase switch between the transcription and replication modes is unknown, several studies pointed to VP30 as a transcription activation factor unique for filoviruses (4–6).
EBOV VP30 protein was shown to be phosphorylated at two serine clusters at positions 29–31 and 42–46 and at a threonine in position 52, located close to the RNA-binding domain (7). Phosphorylation of VP30 blocks the ability of the viral polymerase to function during transcription but not genome replication (5–7). VP30 that was expressed and phosphorylated in cultured cells and then immunoprecipitated was dephosphorylated in vitro by added catalytic subunits of protein phosphatase 1, 2A, or 2C (PP1, -2A, or -2C) (7). However, the identity of the phosphatase that might dephosphorylate VP30 in cultured cells and control EBOV transcription remained unknown.
The PP1 holoenzyme consists of a constant catalytic subunit (PP1α, PP1β/δ, or PP1γ) and a variable regulatory subunit that determines the localization, activity, and substrate specificity of the phosphatase (8). Major regulatory subunits of PP1, such as NIPP1 (nuclear inhibitor of PP1) or PNUTS (phosphatase nuclear targeting subunit), bind the PP1 catalytic subunit with nanomolar affinity (8). The binding occurs through one or a combination of short binding motifs, such as the well established RVXF motif and the recently identified SILK, MyPhoNE, SpiDoC, and iDoHA motifs (9). Engagements of different combinations of these motifs define both the composition of PP1 holoenzymes and their unique specificity for different substrates in various cell compartments.
We previously showed that PP1 activates human immunodeficiency virus 1 (HIV-1) transcription and that expression of NIPP1 or a short central domain of NIPP1 (cdNIPP1) inhibits HIV-1 transcription and replication (10–12). We recently developed a small molecule, 1H4, that targets the RVXF-binding pocket of PP1 and efficiently inhibits HIV-1 (13).
In the present study, we determined whether PP1 regulates VP30 phosphorylation and whether PP1 can be therapeutically targeted for EBOV inhibition. We analyzed EBOV VP30 phosphorylation in cultured cells and in EBOV virions using high-resolution mass spectrometry that resulted in identification of the novel Thr143 and Thr146 phosphorylation sites in addition to the previously identified phosphorylation at the N-terminal Ser clusters in positions 29–31 and 42–46. We next determined whether PP1 controls VP30 phosphorylation by expressing its inhibitor cdNIPP1 or using PP1-targeting shRNA.
These experiments showed that expression of PP1-inhibitory cdNIPP increases VP30 phosphorylation and that overexpression of SIPP1, a splicing factor that interacts with PQBP1 (polyglutamine tract-binding protein 1) and PP1 and shuttles PP1 to cytoplasm (14), decreases it. Next, we developed about 300 small molecule compounds based on the 1H4 structure (13) that were targeted to PP1. We showed that the non-toxic compound 1E7-03 increases EBOV VP30 phosphorylation and effectively suppresses replication of EBOV particles in cell cultures. Furthermore, we analyzed the effect of 1E7-03 on EBOV transcription and replication using the mini-genome system and showed that the molecule reduces the transcription of EBOV genes but not replication of EBOV genome. Finally, we analyzed the role of the identified VP30 Thr143 and Thr146 phosphorylation sites. We found that disabling of these sites increases VP30 phosphorylation within the serine clusters in residues 29–31 and 42–46 and inhibits EBOV transcription. Our study demonstrates a novel role of PP1 in regulation of VP30 phosphorylation and suggests a new approach to inhibit EBOV by targeting PP1 by the identified small molecules that can be further developed as an EBOV-inhibitory drug.
EXPERIMENTAL PROCEDURES
Design of the 1H4 Derivative Library
The Enamine stock collection, which included about 2 million small molecule compounds at the time the study was initiated, was used for the virtual screening. After excluding the compounds with reactive groups, the remaining 600,000 compounds were virtually docked to PP1 as we described recently (13). About 300 unique compounds that were identified in the virtual screening were further evaluated biologically for inhibition of HIV-1 and the lack of toxicity. As a result, a single compound, 1H4, inhibiting HIV-1 transcription was identified. The compound competed with the RVXF-containing peptide for the binding to PP1, which effectively blocked the interaction between HIV-1 Tat and PP1 (13). To improve binding potency and reduce cytotoxicity, we designed multiple 1H4 derivatives, as described under “Results.”
Analysis of the PP1-targeted Compounds' Cytotoxicity
Vero-E6 cells and 293T cells were grown to confluence in 96-well tissue culture plates. Each compound (1E7, 1E7-03, and 1E7-04) was serially diluted in cell medium and added to the cell monolayer in triplicates. The plates were incubated for 1 or 3 days at 37 °C and 5% CO2. After incubation, the effect of each compound or DMSO control on the cell viability was determined using Cell Titer-Glo reagent (Promega) according to the manufacturer's protocol. Non-treated control wells were assumed to represent 100% cell viability and were used to calculate the percentage viability of cells treated with the compounds.
In Vitro Binding of Recombinant PP1 to the Immobilized Compound 1E7
100 nmol of 1E7 analog with an additional NH2 group were coupled with N-hydroxysuccinimide-activated Sepharose (GE Healthcare) according to the manufacturer's protocol. N-hydroxysuccinimide-activated Sepharose beads that were blocked with Tris-HCl buffer served as a control. 10 units of recombinant protein PP1 (New England Biolabs) were incubated for 2 h at 4 °C with the beads containing immobilized 1E7 in 50 mm HEPES, pH 7.5, 100 mm NaCl, 2 mm DTT, 0.01% Nonidet P-40; washed three times; and then trypsinized overnight at 37 °C. Solution above the beads was collected, dried, and dissolved in 0.1% trifluoroacetic acid (TFA), and the peptides were purified using C18 Ziptips (Millipore) according to the manufacturer's protocol.
Plasmids
The expression vectors for cdNIPP1, amino acids 143–224, fused to eGFP, or its inactive mutant V201A/F203A were described previously (15). The plasmid pCEZ-VP30 expressing EBOV VP30 (16) was kindly provided by Dr. Yoshihiro Kawaoka (University of Wisconsin). The EBOV VP30 coding sequence (amino acids 1–246) was amplified by PCR and subcloned in EcoRI and BamHI sites of PrecisionShuttle mammalian vector pCMV6-AC-IRES-GFP-Puro (Origene) in frame with Myc and DDK (FLAG) sequences.
Analysis of VP30 Phosphorylation
Phosphorylation of EBOV VP30 in cells was analyzed as described previously (17). Briefly, FLAG-tagged VP30 was expressed in 293T cells or Vero-E6 cells. The cells were pulsed with [32P]orthophosphate for 2 h with or without the addition of okadaic acid or 1E7-03. VP30 was immunoprecipitated with anti-FLAG antibodies (Sigma) and resolved on SDS-PAGE. VP30 expression was analyzed by immunoblotting, and VP30 phosphorylation was analyzed using a Packard Cyclone PhosphorImager (PerkinElmer Life Sciences).
Mass Spectrometry Analysis
FLAG-tagged VP30 was expressed in 293T cells and immunoprecipitated with anti-FLAG antibodies. Recombinant EBOV expressing enhanced green fluorescent protein (EBOV-eGFP) (18) was propagated in Vero-E6 cells, purified on a sucrose gradient, lysed with 10% SDS, and heat-inactivated at 95 °C for 10 min. Recombinant FLAG-tagged VP30 or proteins of the purified virus were resolved on 10% SDS-PAGE. The gel was stained with SimpleBlue safe stain (Invitrogen), washed with water, and cut into pieces. Gel pieces were crushed into smaller pieces, dehydrated with acetonitrile (MP Biochemicals), and rehydrated with 50 mm ammonium bicarbonate for 5 min. Dehydration and rehydration were repeated three times. Reduction and alkylation of the cysteine residues was conducted by treating the samples with 10 mm DTT for 1 h at 60 °C followed by 50 mm iodoacetamide in the dark at room temperature for 30 min. Trypsin Gold (Promega) reconstituted in 50 mm ammonium bicarbonate was added to the gel pieces at a final concentration 10 ng/μl, and the mixture was incubated at 37 °C overnight. Eluted peptides were collected and dried on a Savant SpeedVac concentrator (ThermoFisher). Samples were reconstituted in 0.1% TFA and purified using C18 Ziptips (Millipore) according to the manufacturer's protocol. Peptides were eluted with 80% acetonitrile containing 0.1% TFA and dried on a SpeedVac. For the liquid chromatography-mass spectrometry (LC-MS) procedure, samples were reconstituted in 40 μl of 0.1% formic acid. Next, samples were separated by reversed-phase liquid chromatography (HPLC), using microcapillary column C18, coupled in line with nanospray and tandem mass spectrometer ThermoFisher LTQ Orbitrap XL. The LC gradient was run for 60 min from 2 to 80% of acetonitrile containing 0.1% formic acid at a flow rate of 400 nl/min. One Fourier transform MS scan and three data-dependent Fourier transform MS/MS scans on major multicharged MS peaks with resolution 30,000 were performed in a single measurement block. The normalized collision-induced dissociation energy was 35%. The MS/MS spectra were analyzed against the NCBI human protein database using Proteome Discover version 1.2 software (ThermoFisher) with the SEQUEST (Scripps Research Institute) search engine. Because the FLAG-tagged VP30 was not present in the database due to the modifications (the addition of FLAG and Myc sequences), its sequence was manually added to the database. Only peptides having X-correlation (Xcorr) cut-offs of 1.0 for [M + 2H]2+, 1.5 for [M + 3H]3+, and higher charge state were considered. These SEQUEST criteria thresholds resulted in less than 1% false discovery rate.
PP1α Knockdown Cell Lines
Vero-E6 cells were infected with pan-PP1 shRNA (human) lentiviral particles and a control shRNA lentiviral particle (Santa Cruz Biotechnology, Inc.). About 2 × 104 cells were inoculated at 1.1 pfu/1,000 cells. Stable clones were selected in the presence of 5 μg/ml puromycin. Two clones, which demonstrated a significant decrease in PP1α mRNA expression, were selected and used for further studies.
Analysis of PP1α and PP1 Regulatory Subunit mRNA Expression
Total RNA was extracted using TRIzol reagent (Invitrogen). 100 ng of total RNA was reverse-transcribed to cDNA with hexamers and oligo(dT) primers using the SuperscriptTM RT-PCR kit (Invitrogen). Real-time PCR analysis was conducted on a Roche LightCycler 480 detection system (Roche Applied Science) with SYBR Green. 45 cycles of denaturation were performed with primers for GAPDH, PP1α, SIPP1, Sds22, or PNUTS with 10 s of denaturation at 95 °C, 10 s of annealing at 60 °C, and 10 s of extension at 72 °C; sequences of the primers can be provided upon request. Mean Cp values and expression levels were determined using ΔΔCp analysis with GAPDH as a reference. Unpaired t test was used to evaluate statistical significance.
Experiments with EBOV
Experiments with live EBOV-eGFP were performed in BSL-4 facilities of the Galveston National Laboratory and Robert E. Shope Laboratory (University of Texas Medical Branch). To measure titers of EBOV-eGFP in supernatants of infected Vero-E6 cells, aliquots were taken every 24 h, frozen, and titrated in Vero-E6 cell monolayers under 0.9% methylcellulose/minimum Eagle's medium overlay. After 3–4 days at 37 °C, fluorescent viral plaques were counted under a UV microscope.
Experiments with Respiratory Syncytial Virus (RSV)
To measure titers of RSV in HEp-2 cell supernatants, aliquots were taken on days 4 and 6 postinfection, frozen, and titrated in HEp-2 cell monolayers under a 0.9% methylcellulose/minimum Eagle's medium overlay. After 4 days at 37 °C, viral plaques were detected using monoclonal antibodies 1243 and 1129 against RSV F protein (kindly provided by Dr. Peter Collins, National Institutes of Health), followed by incubation with a secondary HRP-labeled anti-mouse IgG (KPL) and stained with the chromogenic 4CN two-component peroxidase substrate system (KPL).
Construction of the Bicistronic Minigenome
To generate the bicistronic minigenome, we used the monocistronic minigenome construct (19) previously modified by replacement of the chloramphenicol gene with the firefly luciferase gene (20), kindly provided by Dr. Elke Mühlberger (19). The NsiI restriction endonuclease site was generated downstream of the firefly luciferase gene by insertion of a G nucleotide using the QuikChange site-directed mutagenesis kit (Stratagene). The Renilla luciferase gene was PCR-amplified with the EBOV L transcriptional termination signal (gene-end) added downstream the open reading frame and flanking NsiI restriction endonuclease sites upstream of the open reading frame and downstream of the gene-end signal and inserted into the NsiI site of the minigenome plasmid. Thereafter, a C nucleotide was introduced to create a BglII restriction endonuclease site upstream of the Renilla luciferase open reading frame as above, for subsequent insertion of the authentic stem-loop structure required for transcription (4). To insert the stem-loop structure, the NP-VP35 gene junction PCR product (nucleotides 2,690–3,128 in the EBOV genome), incorporating the non-coding parts of the genes and the intergenic sequence that form the stem-loop structure, with NotI and BglII sites introduced at the termini during amplification, was inserted into the minigenome using the respective restriction enzyme sites.
Construction of the Mutated VP30 Plasmids
The EBOV VP30 was reported to contain two clusters of serine residues that can be phosphorylated: serines 29, 30, and 31 and serines 42, 44, and 46 (7). To generate constructs encoding permanently “phosphorylated” (VP30phos) or “dephosphorylated” (VP30dephos) forms of VP30 protein, Ser codons in pCEZ-VP30 were replaced with either Asp (GAT in amino acid positions 29, 30, 31, 42, 44, and 46) or Ala (GCA, GCA, GCC, GCA, GCC, and GCA in positions 29, 30, 31, 42, 44, and 46), respectively, using the QuikChange site-directed mutagenesis kit (Stratagene); primer sequences can be provided upon request.
To generate plasmids pCEZ-VP30143D, pCEZ-VP30146D, pCEZ-VP30143D146D, and pCEZ-VP30143A146A with the newly identified sites of phosphorylation in positions 143 and 146 mutagenized, the two Thr codons were replaced with the Asp codon GAC (amino acid position 143) and Asp codon GAT (position 146) or Ala codon GCC (position 143) and Ala codon GCA (position 146) using standard molecular biological methods; primer sequences can be provided upon request. To generate constructs expressing mutated forms of VP30 for analysis of their phosphorylation, the VP30 ORFs from the pCEZ-VP30dephos, pCEZ-VP30143D146D, and pCEZ-VP30143A146A plasmids were cloned in pCMV6-AC-IRES-GFP-Puro vector (OriGene). An additional construct, VP30 S29–46A T143A,T146A, was obtained from the VP30 S29–46A construct by PCR mutagenesis using the same pair of primers as for generation of the pCEZ-VP30143A146A plasmid.
Minigenome Experiments
The plasmids pCEZ-NP, pCEZ-VP35, pCEZ-VP30, pCEZ-L, and pC-T7 (16) were kindly provided by Dr. Yoshihiro Kawaoka. For minigenome experiments, the following amounts of plasmids were used: pCEZ-NP, 0.25 μg; pCEZ-VP35, 0.25 μg; pCEZ-VP30 or its mutants, 0.15 μg; pCEZ-L, 2.0 μg; pC-T7, 0.5 μg; mono- or bicistronic minigenome, 0.5 μg. The plasmids were transfected in 293T cells with Mirus transfection reagent (Mirus Bio). In 48 h, transcription was measured by a dual luciferase reporter assay (Promega), and RNA was isolated for the analysis of replication of the minigenome by Northern blotting. RNA samples were separated in 1.5% agarose (Fisher) with 0.66 m formaldehyde (Fisher) and transferred to Protran BA 85 nitrocellulose membranes (GE Healthcare), UV-immobilized, and hybridized overnight with a luciferase-specific RNA probe of negative polarity, labeled with digoxigenin (Roche Applied Science). RNA bands were visualized by incubation of the membranes with anti-digoxigenin antibodies labeled with alkaline phosphatase (Roche Applied Science), followed by incubation with ECF substrate (GE Healthcare), and quantified with a Typhoon Tryo PhosphorImager.
RESULTS
Analysis of VP30 Phosphorylation by Mass Spectrometry
We used mass spectrometry to analyze phosphorylation of vector-expressed VP30 and also VP30 in EBOV viral particles. To analyze the vector-expressed protein, FLAG-tagged VP30 was expressed in 293T cells, immunoprecipitated, and resolved by SDS-PAGE (Fig. 1A, lane 2). The high resolution LC-MS/MS analysis of tryptic peptides extracted from the gel identified the continuous VP30 protein sequence with the exception of the C-terminal part, although the C-terminal Myc and FLAG tags were detected (65% coverage; Fig. 1B, thin lines). Analysis of post-translational modifications indicated phosphorylation of Ser29 and Ser30 (Fig. 1, B and C) and also Thr143 and Thr146 (Fig. 1, B and D). We next analyzed whether VP30 packaged in EBOV particles is also phosphorylated. Proteins from purified EBOV particles were resolved on 10% SDS-PAGE (Fig. 1E) and in-gel trypsinized, and the extracted peptides were analyzed by high resolution LS-MS/MS. The continuous VP30 sequence was identified with the exception of the C-terminal part (49% coverage; Fig. 1B, thick lines). Analysis of post-translational modifications indicated the phosphorylation of Ser42 (Fig. 1F) and also Thr63, Thr96, Thr143, and Thr146 (not shown; outlined in Fig. 1B). These data confirmed the previously reported phosphorylation of VP30 in the serine clusters of Ser29–Ser31 and Ser42–Ser46 (7) and also indicated that Thr63, Thr96, Thr143, and Thr146 might also be phosphorylated. The phosphorylated Thr143 and Thr146 were identified both in the VP30 expressed in cultured cells and that packaged in EBOV (Fig. 1B).
FIGURE 1.

EBOV VP30 has two clusters of phosphorylation. A, purification of FLAG-tagged VP30 for MS/MS analysis. FLAG-tagged VP30-expressing 293T cells were treated with 0.1 μm okadaic acid for 3 h. VP30 was immunoprecipitated, resolved on 10% SDS-PAGE, and stained with colloidal Coomassie Blue. Lane 1, molecular weight markers; lane 2, VP30; lane 3, mock-transfected control. The position of VP30 is shown by an arrow. B, MS/MS analysis of virion-associated and recombinant VP30, the SEQUEST search results. VP30 was in-gel-digested with trypsin, and the eluted peptides were subjected to MS analysis on a Thermo LTQ Orbitrap XL mass spectrometer. Peptides identified with high, median, and low probability are underscored with green, red, and blue lines, respectively; the thin and thick lines depict data for recombinant and virion-associated VP30, respectively. Peptides that were found to be phosphorylated in virion-associated and recombinant VP30 are overscored with brown and black lines, respectively. The positions of phosphorylated amino acids in recombinant and virion-associated VP30 are indicated with black and brown arrows, respectively. Note that threonines 143 and 146 were found to be phosphorylated in both virion-associated and recombinant VP30. C, MS/MS spectrum of the VP30 peptide 29–36. The colored peaks indicate matched MS/MS fragments. Green indicates precursors, as outlined in the figure; blue and red indicate y and a ions, respectively. The spectrum gives positive identification of SSSRENYR peptide with the indicated phosphorylation sites. D, MS/MS spectrum of the VP30 peptide 142–149. The colored peaks indicate matched MS/MS fragments (as described in C). The spectrum gives positive identification of the ITLLTLIK peptide with the indicated phosphorylation sites. E, electrophoretic separation of the purified EBOV for MS/MS analysis. EBOV was resolved in 10% SDS-PAGE and stained with colloidal Coomassie Blue. Lane 1, molecular weight markers; lane 2, proteins of purified inactivated EBOV. F, MS/MS analysis of the peptide 41–49 from the EBOV-incorporated VP30. The colored peaks indicate matched MS/MS fragments (as described in C). The spectrum gives positive identification of QSESASQVR with the indicated phosphorylation site.
EBOV VP30 Is Dephosphorylated by PP1
To confirm that VP30 is phosphorylated in cultured cells, FLAG-tagged VP30 was expressed in 293T cells, and the cells were treated with [32P]orthophosphate and also with 100 nm okadaic acid. Treatment with okadaic acid induced VP30 phosphorylation (Fig. 2A, compare lanes 2 and 3). However, okadaic acid is known to preferentially inhibit PP2A over PP1 (21). Thus, to determine whether VP30 is dephosphorylated by PP1 in cultured cells, PP1-binding peptide, cdNIPP (10), was co-expressed with FLAG-tagged VP30 in 293T cells. Expression of cdNIPP significantly increased VP30 phosphorylation as compared with cdNIPP inactive mutant that does not bind PP1 (10) (Fig. 2B), suggesting that PP1 dephosphorylates VP30 in cultured cells.
FIGURE 2.

EBOV VP30 is dephosphorylated by PP1. A, phosphorylation of EBOV VP30 is increased in the presence of okadaic acid. FLAG-tagged VP30 was expressed in 293T cells and metabolically labeled with [32P]orthophosphate with or without 0.1 μm okadaic acid (OA). VP30 was immunoprecipitated with anti-FLAG antibodies, resolved on 10% SDS-PAGE, and either exposed to a PhosphorImager screen (top) or probed for VP30 with anti-FLAG antibodies (bottom). The positions of VP30 are shown by arrows. B, PP1 inhibition induces VP30 phosphorylation: phosphorylation of EBOV VP30 in the presence of WT or the mutated non-active form of cdNIPP1. Top, FLAG-tagged VP30 and cdNIPP1-eGFP (WT or mutant) were expressed in 293T cells and metabolically labeled with [32P]orthophosphate. VP30 was immunoprecipitated, resolved on 10% SDS-PAGE, and either exposed to a PhosphorImager screen (top) or probed for VP30 with anti-FLAG antibodies (bottom). Bottom, averages of five independent experiments ± S.D. (error bars). The p value was calculated by Student's paired t test. C, expression of PP1α mRNA is reduced in PP1 KD cells. Concentrations of PP1 RNA in Vero-E6 cells stably transduced with vectors expressing control non-targeting shRNA or PP1α-targeting shRNA were determined by real-time RT-PCR with 18 S ribosomal RNA as an internal control. Shown are means of triplicate values ± S.D. The p value was calculated by Student's paired t test. D, VP30 phosphorylation is reduced in PP1 KD cells. FLAG-tagged VP30 was expressed in Vero-E6 cells untreated (WT) or stably transduced with non-targeting shRNA (Sh) or PP1α-targeting shRNA (KD). The cells were metabolically labeled with [32P]orthophosphate, and VP30 was immunoprecipitated with anti-FLAG antibodies, resolved on 10% SDS-PAGE, and either exposed to a PhosphorImager screen (top) or probed for VP30 with anti-FLAG antibodies (bottom). E, expression of SIPP1 is increased in PP1 KD Vero-E6 cells. Concentrations of the regulatory subunits of PP1 SIPP1, Sds22, and PNUTS were determined by real-time RT-PCR with 18 S ribosomal RNA as an internal control. Shown are means of triplicate values ± S.D. F, EBOV minigenome. Top, the EBOV genome; overlapping of some genes is shown; the EBOV-specific transcriptional gene-start and gene-end signals are highlighted in green and red, respectively. Left, the EBOV monocistronic minigenome used in the present experiment and the bicistronic minigenome used in the experiments shown in Fig. 7. The EBOV leader sequence, which includes the promoter, non-coding sequence, and gene-start, and the EBOV trailer sequence, including the gene-end and non-coding sequence, flank the firefly luciferase ORF in the monocistronic minigenome and flank the firefly and the Renilla luciferase ORFs in the bicistronic minigenome. Right, the support plasmids encoding the EBOV polymerase complex and T7 polymerase. Transcription was analyzed by luminometry; in addition, in the experiments shown in Fig. 7C, the minigenome was used for analysis of replication by Northern blotting. G, EBOV transcription is increased in PP1 KD cells. Transcription of the monocystronic minigenome in Vero-E6 cells stably transduced with vector expressing PP1α-targeting shRNA: the mean luciferase signals based on triplicate values ± S.E. The p value was calculated by Student's paired t test. WB, Western blot.
To further analyze the involvement of PP1 in VP30 dephosphorylation and therefore in EBOV transcription, we generated a PP1α knockdown (PP1 KD) Vero-E6 stable cell line. Cells expressing PP1α-targeting shRNA expressed about 2.5 times less PP1α mRNA compared with the parental cells or a control cell line that expressed a non-targeting shRNA (Fig. 2C). Unexpectedly, analysis of VP30 phosphorylation showed a significantly reduced VP30 phosphorylation in PP1 KD cells compared with parental or control shRNA-expressing cells (Fig. 2D). Because PP1 functions as a heterodimer, we analyzed PP1 distribution in the cells by quantifying expression of its cytoplasmic and nuclear regulatory subunits. Specifically, we analyzed the expression of SIPP1, a nucleocytoplasmic shuttling subunit of PP1 that accumulates in the cytoplasm in UV or X-irradiated cells (14); PNUTS, a major nuclear subunit of PP1 (22); and Sds22, a subunit of PP1 that shuttles PP1 to the nucleus (23). SIPP1 mRNA expression was significantly increased in PP1 KD cells, whereas the expressions of Sds22 and PNUTS mRNAs were not altered (Fig. 2E). Thus, knockdown of PP1α leads to a significant increase in PP1 holoenzyme in cytoplasm, most likely as a compensation for the knockout of the catalytic subunit. Therefore, the observed reduction in VP30 phosphorylation is probably caused by the increased concentration of PP1 holoenzyme in the cytoplasm.
Because disabling the transcriptional activity of EBOV polymerase will prevent synthesis of the viral proteins and virus replication, it cannot be easily analyzed in virus-infected cells. We therefore analyzed the effects of PP1 KD on EBOV transcription by using the previously developed EBOV minigenome system (Fig. 2F) (19, 24). The system includes the monocistronic minigenome, under control of the T7 promoter containing the firefly luciferase reporter gene under control of EBOV-specific transcription initiation (gene-start) and termination (gene-end) signals, and plasmids expressing the viral proteins involved in transcription and replication (L, NP, VP35, and VP30) under the control of the chicken β-actin promoter. Importantly, without the VP30 transcription activation factor, it is still fully capable of replication but not transcription (19). The T7 polymerase, required to drive the minigenome system, was provided by a co-expressed plasmid. The transcriptional activity was analyzed by the luciferase assay. Consistently with the reduced phosphorylation of VP30 in PP1 KD cells (Fig. 2D), the EBOV transcription was significantly increased (Fig. 2G). Taken together, these results clearly indicate that PP1 is involved in regulation of EBOV by controlling the VP30 phosphorylation.
Development of PP1-targeted Small Molecules
We previously developed PP1-targeted small molecule 1H4 (13). To improve the binding potency and reduce cytotoxicity of the compound, we designed multiple 1H4 derivatives. To enhance the interaction with Asp242, Lys260, and Gln292 residues of PP1, the 4th position of the 1,2,3,4-tetrahydroacridine heterocycle was varied by including aromatic hydrocarbons with different functional groups. To provide more rich hydrogen bonding with Tyr255, Arg261, and Cys291 and van der Waals interactions with Phe257, Met290, and Phe293 of PP1 and to facilitate docking into the RVXF-accommodating groove, the 9-carboxylic tail was modified by extending it with aromatic rings while retaining the flexibility. In total, 42 compounds were generated. Based on the ability of the compounds to inhibit HIV-1, which is sensitive to PP1 inhibition (25), three compounds were selected for the current study: 1E7, 1E7-03, and 1E7-04 (Fig. 3A). To confirm that the 1E7 family of compounds interacts with PP1, an analog of 1E7 was synthesized with an unsubstituted terminal NH2 group that was used to couple the compound to N-hydroxysuccinimide-Sepharose beads. Mass spectrometer analysis of the purified enzyme incubated with the 1E7 analog immobilized on the beads or the control beads showed a specific association of PP1 to the beads with the immobilized 1E7 analog (Fig. 3B).
FIGURE 3.

Effect of the PP1-targeting compounds on EBOV replication and VP30 phosphorylation. A, chemical structures of the compounds 1E7, 1E7-03, and 1E7-04. B, binding of PP1 to 1E7 analog in vitro. An analog of 1E7 containing an additional NH2 group was coupled with N-hydroxysuccinimide-Sepharose and incubated with PP1. The beads were treated with trypsin, and the eluted peptides were analyzed by nano-LC connected to an LTQ XL Orbitrap mass spectrometer. The PP1 peptide EIFLSQPILLELEAPLK, z = 2, m/z = 977.07 Da, was detected by SEQUEST with high confidence. The bar represents the LC signal amplitude for this mass with tolerance 0.03 Da. C, multistep growth kinetics of EBOV-eGFP in Vero-E6 cells, which were treated with the indicated compounds 30 min prior to the infection with EBOV-eGFP at MOI 0.01 pfu/cell. Following 1-h-long adsorption, media were removed, and fresh media containing the original concentrations of the molecules were added. For the samples in which no virus was detected, the values 2-fold below the limit of detection were assigned. Shown are mean EBOV-eGFP titers ± S.E. in the medium, based on triplicate monolayers. D, induction of VP30 phosphorylation by 1E7-03. 293T cells expressing FLAG-tagged VP30 were treated with 0.1 μm okadaic acid (OA) or 10 μm 1E7-03 and labeled with [32P]orthophosphate. VP30 was immunoprecipitated, resolved on 10% SDS-PAGE, and either exposed to a PhosphorImager screen (top) or probed for VP30 with anti-FLAG antibodies (bottom). The ImageQuant software was used to quantify the PhosphorImager data, which were normalized by dividing phosphorylation values by protein values. The bars show averages of three independent experiments ± S.D. (error bars). The p values were calculated by Student's paired t test. E, induction of VP30 phosphorylation in Vero-E6 cells. Vero-E6 cells expressing FLAG-tagged VP30 were treated with 0.1 μm okadaic acid (OA) and labeled with [32P]orthophosphate. VP30 was immunoprecipitated, resolved on 10% SDS-PAGE, and either exposed to a PhosphorImager screen (top) or probed for VP30 with anti-FLAG antibodies (bottom). The bars show unadjusted PhosphorImager mean values ± S.D. The p value was calculated by Student's paired t test. F, comparison of 1E7-03-induced phosphorylation of VP30 in Vero-E6 and 293T cells; the experiment was performed similarly to that shown in E. WB, Western blot.
Comparison of Antiviral Activities of Individual Compounds of the 1E7 Family
The antiviral effects of the compounds against EBOV-eGFP, which is an infectious virus with replication characteristics identical to those of WT EBOV in cultured cells (18), were tested in Vero-E6 cells. Treatments with 1E7, 1E7-03, or 1E7-04 for 30 min prior to the infection with a multiplicity of infection (MOI) of 0.01 pfu/cell resulted in a dose-dependent inhibition of the virus replication (Fig. 3C). For example, the addition of 1E7-03 at 10 μm resulted in the reduction of the viral titers on days 2 and 3 by 200- and 147-fold, respectively.
Cytotoxicity of the PP1-targeted Compounds
To test the cytotoxicity of 1E7, 1E7-03, and 1E7-04, Vero-E6 and 293T cells were treated with the compounds at different concentrations, and cell viability was analyzed on days 1 and 3 (Fig. 4). 1E7-04 significantly reduced the viability of both the Vero-E6 and 293T cells at an IC50 of 10.6 and 32.5 μm, respectively, at day 3. In contrast, treatment with 1E7 had no effect on cell viability at day 1 and slightly decreased viability of Vero-E6 but not 293T cells at the greatest concentration tested, 73 μm, at day 3. Treatment with 1E7-03 resulted in cell viability similar to that in cells treated with 1E7 except that the reduction of viability of Vero-E6 on day 3 was even less (Fig. 4).
FIGURE 4.

Viability of Vero-E6 and 293T cells treated with the indicated compounds at the indicated concentrations on days 1 and 3. The values represent mean percentages of cells with viability of untreated cells considered to be 100% based on triplicate monolayers ± S.D. (error bars). Note that the error bars for Vero-E6 cells are not seen due to their small size.
1E7-03 Increases Phosphorylation of VP30
We next analyzed the effect of 1E7-03 on EBOV VP30 phosphorylation using FLAG-tagged VP30 expressed in 293T cells. We observed that treatment of cells with 1E7-03 at 10 μm concentration increased phosphorylation of VP30 to a level comparable with that in cells treated by okadaic acid (Fig. 3D). VP30 expressed in Vero-E6 cells was also phosphorylated, and the phosphorylation was increased in the cells treated with okadaic acid (Fig. 3E). We also compared VP30 phosphorylation in Vero-E6 and 293T cells. VP30 phosphorylation in the presence of okadaic acid or 10 μm 1E7-03 in Vero-E6 cells was comparable with that in the presence of 10 μm 1E7-03 in 293T cells (Fig. 3F).
The Leading Compound 1E7-03 Effectively Inhibits Replication of EBOV
Focusing on the non-cytotoxic compound 1E7-03, cells pretreated with 1E7-03 at 3 μm and infected with MOI 0.001 pfu/cell did not display any visible eGFP fluorescence up to 5 days postinfection (Fig. 5A, middle). In time-of-addition experiments, 10 μm 1E7-03 was added to the cells at 24 h prior to the infection, during the infection, and 24 h or 48 h postinfection with EBOV-eGFP at MOI 0.01 pfu/cell. Adding the compound 24 h prior to the infection resulted in a lack of any detectable virus in the medium up to 3 days postinfection and very low viral titers on the subsequent days. For example, on days 4 and 6 postinfection, the relative reduction of the viral titers was 2,750-fold and 5,537-fold, respectively (Fig. 5B). Adding the compound at 0, 24, or 48 h postinfection resulted in a reduction of the viral titers at day 4 by 77-, 33-, and 79-fold, respectively, and at day 6 by 384-, 221-, and 31-fold, respectively (Fig. 5B). Analysis of 10 μm 1E7-03 compound stability in aqueous solution showed a decrease by 50% after 3 h of incubation at 37 °C (Fig. 5C). Consequently, we tested the effect of 1E7-03 addition at a 3 μm dose every 24 h, with the first dose administered 24 h prior to or during infection with MOI 0.01 pfu/cell. We found that each treatment regimen resulted in a dramatic reduction in virus replication. Adding the compound starting at 24 h prior to infection resulted in the most effective inhibition. For example, no virus was detected up to 3 days postinfection, equating to a >68,000-fold reduction of the viral titers; on days 4, 5, and 6, the relative reductions of the viral titers were 25,000-, 136,000-, and 147,000-fold, respectively. When treatment started at 0 h (i.e. concurrent with infection), the relative reductions of the viral titers on days 4, 5, and 6 were 794-, 1,080-, and 858-fold, respectively (Figs. 5D and 6). To check whether the ability of 1E7-03 to inhibit EBOV replication was specific to this virus and not due to an intrinsic toxicity that might not be easily detected by viability tests, we analyzed the replication of a non-related RSV in the presence of 10 μm 1E7-03. The compound did not affect replication of RSV (Fig. 5E), suggesting that it specifically targets replication of EBOV.
FIGURE 5.

Effect of the small molecules on replication of EBOV-eGFP in Vero-E6 cells. A, cells on day 5 postinfection with EBOV-eGFP at MOI 0.001 pfu/cell: untreated (left) or treated with 1E7-03 (3 μm) 30 min prior to infection under UV (left, middle) or bright field (BF) microscopy (right). B, effect of the time of 1E7-03 addition on multistep growth kinetics of EBOV at MOI 0.01 pfu/ml. 1E7-03 was added at 10 μm 24 h prior to, during, or 24 or 48 h after infection. After 1-h-long adsorption, the medium was removed, cells were washed with phosphate-buffered saline, and fresh medium was added; in the case of treatment, 24 h prior to the infection or during the infection with 1E7-03, fresh medium contained 10 μm 1E7-03. C, concentration of 1E7-03 in water solution. Freshly dissolved 1E7-03 at a 1 μm concentration was aliquoted in 50-μl samples. Each sample was incubated at 37 °C for the indicated time and frozen until analyzed. The samples were analyzed by electrospray ionization-MS, and the amplitudes of peaks m/z = 504.2123 Da, z = 1 were recorded. D, effect of adding the 1E7-03 compound at 3 μm every 24 h starting at the indicated time. B and D, mean EBOV-eGFP titers ± S.E. (error bars) in the medium, based on triplicate monolayers. The dotted lines indicate the limit of detection. E, the 1E7-03 compound has no effect on replication of RSV. HEp-2 cells were infected with RSV at MOI 0.1 pfu/ml and treated with 1E7-03 at 3 μm daily, as described for EBOV (D). Medium aliquots were collected daily and flash-frozen; RSV was titrated in HEp-2 monolayers, and on day 2, plaques were immunostained using mouse monoclonal antibodies specific for the F protein of the virus and counted.
FIGURE 6.

UV microscopy of Vero-E6 cells, which were infected with EBOV-eGFP at 0.01 pfu/cell and treated daily with 1E7-03 at 3 μm starting 24 h prior to the infection or during infection. Days 5 and 8 postinfection are shown. No eGFP-positive cells were observed in 1E7-03-treated monolayers on days 1–4, which are therefore not shown.
1E7-03 Reduces the Transcription but Not Replication of EBOV Genome
The increase in VP30 phosphorylation was expected to disable the transcriptional but not the replication activity of EBOV polymerase. We therefore analyzed the effects of 1E7-03 on transcription and replication by using the EBOV minigenome system described above (Fig. 2F). Because VP30 is required for reinitiation of transcription of downstream genes (6), we also generated a bicistronic minigenome (Fig. 2F). The second transcriptional unit, a Renilla luciferase gene, was flanked by the EBOV-specific gene-start and gene-end transcriptional signals and placed immediately downstream of the firefly luciferase transcriptional unit. Previous studies demonstrated that filovirus non-translated regions upstream of each open reading frame form stem-loop structures, which include transcriptional gene-start signals (26–28). These stem-loop structures were shown to be important for VP30-mediated transcription (4). We therefore also inserted the authentic EBOV Zaire NP-VP35 gene junction (nucleotides 2,690–3,128), which includes the non-coding parts of the genes and the intergenic sequence, and which forms the stem-loop structure (nucleotides 3,029–3,045), required for transcription (4) (data not shown). As a control, a phosphorylation mimicking VP30 (VP30phos), which abrogates the transcription but not replication of the mini-genome (7), was created by replacing serines at positions 29, 30, 31, 42, 44, and 46 (7) with aspartic acid residues. A dephosphorylated form, VP30dephos, which promotes transcription, was created by replacing the serine clusters with alanines (7). The construct, along with the plasmids expressing NP, VP35, L, and VP30 as well as the T7 polymerase-expressing plasmid, was transfected to 293T cells, which resulted in an effective transcription of both the firefly luciferase gene (transcriptional unit 1) and the Renilla luciferase gene (transcriptional unit 2). We analyzed the effects of 1E7-03, VP30dephos, and VP30phos on transcription of the monocistronic (Fig. 7A) or the bicistronic (Fig. 7B) minigenomes and replication of the monocistronic minigenome (Fig. 7C). This was achieved by transfecting 293T cells with the mono- or bicistronic minigenome; plasmids NP, VP35, L, and one of the following plasmids: VP30, VP30dephos, and VP30phos; and the T7 plasmid. In addition, the cells were also treated with 3 μm 1E7-03 where indicated. On day 2 posttransfection, the cells were harvested, and transcriptional activities of the monocistronic and bicistronic minigenomes were analyzed by a single or dual luciferase assay, respectively. To quantify replication of the minigenome, total RNA was isolated from collected cells, and the positive-sense anti-genome was analyzed by Northern blotting by hybridizing with the digoxigenin-labeled RNA probes of negative polarity. Bands were detected by incubation with alkaline phosphatase-labeled anti-digoxigenin antibodies, followed by incubation with the substrate and phosphorimaging (Fig. 7C). The following effects were observed. Consistent with the previous data (7), expression of VP30 was required for transcription because the absence of VP30 abrogated the luciferase signal (Fig. 7, A and B, column 8 versus column 2). Expression of VP30phos abrogated the transcription (Fig. 7, A and B, column 4 versus column 2), whereas expression of VP30dephos had no inhibitory effect on transcription (Fig. 7, A and B, column 5 versus column 2), as reported previously (7). VP30phos increased genome replication (Fig. 7C, lane 7 versus lane 5); the inhibition of EBOV transcription and increase in genome replication by the phosphorylated VP30 was recently demonstrated by Biedenkopf et al. (5). The VP30-dependent regulation of EBOV transcription/replication balance appears to be similar to the regulation of the transcription/replication balance of RSV, for which the transcription anti-termination factor, M2-1, promotes transcription, and the negative regulator of transcription, M2-2, shifts the balance toward replication (29, 30). However, to our knowledge, no phosphorylation-dependent mechanism of regulation of transcription/replication has been identified for RSV so far. Treatment of cells with 1E7-03 reduced transcription (Fig. 7, A and B, column 3 versus column 2) but increased replication (Fig. 7C, lane 4 versus lane 5; the quantitative data are provided in the figure legend). These results are consistent with the observed increase in phosphorylation of VP30 in the presence of 1E7-03 (Fig. 3D) and the observed abrogation of transcription in the presence of VP30phos. The effects of VP30 and their mutated forms as well as 1E7-03 were similar for both the upstream and the downstream genes (Fig. 7B, top and bottom, respectively). Thus, 1E7-03 prevents VP30 dephosphorylation by PP1 and prevents the effective transcription of the viral genes, which in turn prevents the effective replication of the viral particles.
FIGURE 7.

Effect of 1E7-03 on the transcription and replication of EBOV genome. A and B, analysis of transcription of the monocistronic (A) and bicistronic (B) minigenome by the luciferase assay: mean values based on triplicate samples ± S.E. (error bars). Shown is statistical significance of the 1E7-03-mediated reduction of the transcription (Student's unpaired t test), lane 3 versus lane 2 (A and B). A, p < 0.01; B, gene 1, p = 0.03; gene 2, p = 0.05. C, analysis of replication of the monocistronic minigenome by Northern blotting; the EBOV miniantigenome bands were quantified by a PhosphorImager. Please note that irrelevant lanes between lanes 6 and 7 were removed. The following values were obtained after background subtractions (lanes 1–7): 228, 1,439, 6,788, 33,881, 25,696, 23,621, and 52,504. The experiment was repeated with results essentially similar to those shown in the figure.
VP30 Phosphorylation at Thr143 and Thr146 Inhibits EBOV Transcription
We next characterized the effects of Thr143 and Thr146, which we found to be phosphorylated in VP30 expressed in cultured cells or packaged in EBOV virions (Fig. 1). We generated a set of VP30 mutants, in which the threonine residues were mutated to asparagine residues to mimic phosphorylation (T143D,T146D) or to alanine to mimic their dephosphorylation (T143A,T146A). To exclude the effect of phosphorylation at the N-terminal serine residues (7), we also generated mutants in which the N-terminal serine clusters (Ser29, Ser30, Ser31, Ser42, Ser44, and Ser46) were replaced with alanines to make them permanently dephosphorylated, with or without the mutations T143A,T146A. To analyze the effect of the Thr143 and Thr146 mutations on VP30 phosphorylation, the FLAG-tagged mutated proteins were expressed in 293T cells. Unexpectedly, not only T143D,T146D but also T143A,T146A mutations led to a significant increase in VP30 phosphorylation (Fig. 8A, lanes 3 and 4). This effect was comparable with the increase of VP30 phosphorylation induced by 1E7-03 treatment (Fig. 8A, lane 5). The increase in VP30 phosphorylation appeared to be due to the phosphorylation of the N-terminal serine clusters because substitution of serines 29, 30, 31, 42, 44, and 46 to alanines (the S29–46A mutant) prevented this increased phosphorylation (Fig. 8A, lanes 6 and 7). Similarly, treatment with 1E7-03 had no effect on phosphorylation of VP30 S29–46A and VP30 S29–46A T143A,T146A mutants (Fig. 8A, lanes 8 and 9). Thus, phosphorylation of Thr143 and Thr146 affected the phosphorylation of the N-terminal serine cluster.
FIGURE 8.
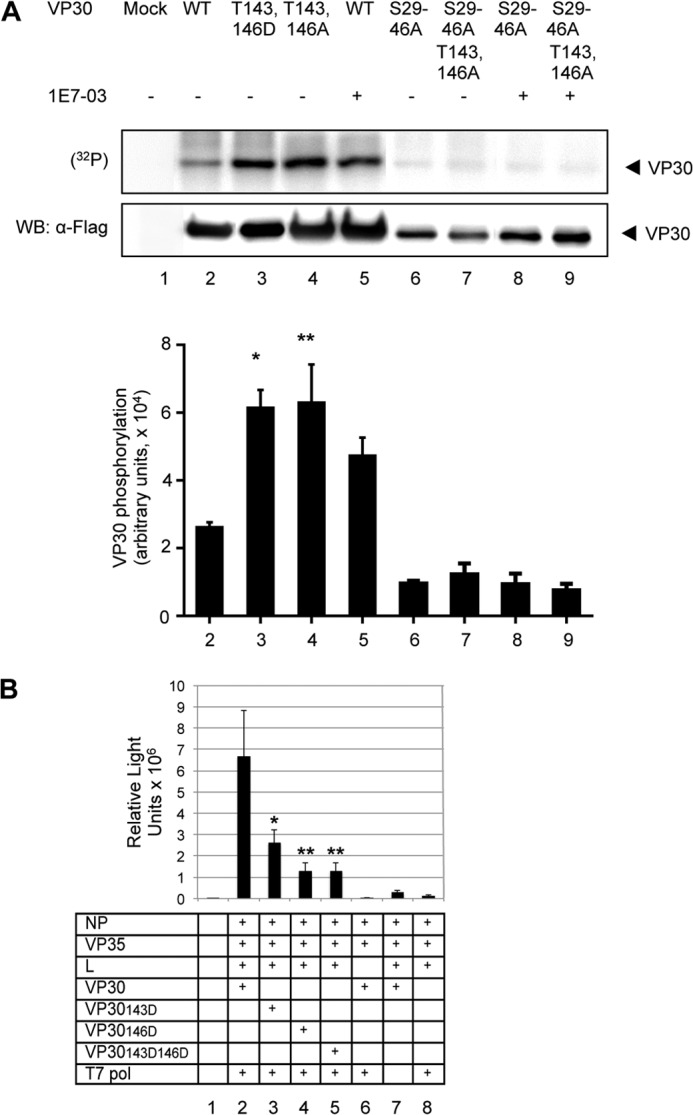
Role of Thr143 and Thr146 in EBOV transcription. A, mutations of Thr143 and Thr146 increase VP30 phosphorylation. FLAG-tagged VP30 (WT) and its mutated forms T143D,T146D, VP30 T143A,T146A, VP30 S29–46A, and VP30 S29–46A T143A,T146A were expressed in 293T cells followed by treatment of cells with 10 μm 1E7-03. The cells were metabolically labeled with [32P]orthophosphate. VP30 was immunoprecipitated with anti-FLAG antibodies, resolved on 10% SDS-PAGE, and either exposed to a PhosphorImager screen (top) or probed for VP30 with anti-FLAG antibodies (middle). The quantitative phosphorylation data were normalized to VP30 expression by dividing phosphorylation values by protein values; mean values ± S.D. (error bars) are indicated (bottom). The p values were calculated by Student's paired t test. B, mutations in VP30 Thr143 and Thr146 residues reduce EBOV transcription: transcription of the monocistronic minigenome in the presence of VP30 in which threonines in position 143 and/or 146 were substituted with aspartic acids to mimic permanent phosphorylation. Mean luciferase signals are based on six samples ± S.E. The p values were calculated by Student's paired t test; comparison with non-mutated VP30: *, p = 0.028, **, p = 0.006.
We next validated the functional importance of Thr143 and Thr146. We generated three VP30 mutants in which threonine residues were replaced with aspartic acid to mimic phosphorylation, VP30143D, VP30146D, and VP30143D146D, and analyzed the effects of the mutations on transcription using the EBOV minigenome system described above. Each of the two individual mutations or their combination significantly reduced the level of EBOV transcription. Specifically, T143D reduced the transcription by 61%, T146D by 81%, and their combination by 81% (Fig. 8B). Thus, phosphorylation of VP30 Thr143 and Thr146 inhibits EBOV transcription, suggesting that these amino acid residues play a regulatory role in the transcription/replication balance.
DISCUSSION
The present study demonstrated multiple novel findings. First, using high resolution mass spectrometry, we identified new phosphorylation sites in the VP30 component of the EBOV polymerase complex and demonstrated their role in regulation of EBOV transcription/replication balance. Second, we provided evidence that dephosphorylation of VP30 is controlled by PP1. Third, we designed a small molecule compound, 1E7-03, which effectively binds PP1, induces VP30 phosphorylation, and effectively inhibits replication of EBOV particles. We also demonstrated that 1E7-03 shifts the transcription/replication balance of the EBOV polymerase toward replication, suggesting that its antiviral effect results from the imbalance of EBOV polymerase it causes.
Previously, plasmid-expressed VP30 was shown to be phosphorylated in cultured cells, and its phosphorylation was significantly decreased with alanine mutations of serines 29–31 and serines 42, 44, and 46, suggesting phosphorylation of these residues by cellular kinases (7). In our study, phosphorylation on Ser29 and Ser30 in VP30 expressed in cultured cells and Ser42 in VP30 incorporated in EBOV virions were demonstrated directly by mass spectrometry analysis. This is the first study in which phosphorylation of virion-incorporated VP30 was conducted. Although we observed different residues being phosphorylated in the N-terminal serine clusters in the recombinant VP30 and the VP30 incorporated in EBOV virions, we cannot exclude the possibility that additional residues in these clusters were also phosphorylated but not detected in our MS analysis. Furthermore, the levels of phosphorylation of various sites may be different, as demonstrated for several viruses, including RSV (31), and detection of sites with a lesser level of phosphorylation may be complicated. Further detailed phosphopeptide mapping analysis is needed to analyze VP30 phosphorylation. Also, the fact that EBOV was assembled in Vero-E6 cells and VP30 was expressed in 293T cells may have led to the differences in the N-terminal serine cluster phosphorylation. In addition, VP30 Thr143 and Thr146 residues were found to be phosphorylated both in cultured cells and virions, and Thr63 and Thr96 were phosphorylated in VP30 expressed in cultured cells. Our data complement the previous findings by extending the number of VP30 residues that may be phosphorylated. We further analyzed the phosphorylation and functional importance of Thr143 and Thr146; the role of the other potential sites of phosphorylation remains to be analyzed.
Previously published studies demonstrated that VP30 expressed in cultured cells is effectively dephosphorylated in vitro by added purified catalytic subunits of PP1 or PP2A, and, somewhat less effectively, by PP2C, suggesting that in vivo, phosphorylation of VP30 may be controlled by PP1 or PP2A (7). The present study specifically investigated the role of PP1 in phosphorylation of VP30. Treatment of cultured cells with high concentrations of okadaic acid (IC50 ∼100 nm) over a 24–36-h-long period was previously shown to inhibit EBOV transcription (7). Okadaic acid inhibits PP2A at low nanomolar IC50 and PP1 at high nanomolar IC50 (11, 21). However, at high concentrations (100 nm and above), okadaic acid blocks cellular transcription by promoting phosphorylation of cellular RNA polymerase II (32). Thus, a long term incubation of cells with high concentration of okadaic acid is likely to produce artifacts due to its strong cellular effects. We showed that short, 2-h-long, treatment with 100 nm okadaic acid induced VP30 phosphorylation, which has not been noted before. Moreover, we used a specific PP1 inhibitor, cdNIPP1, which we previously demonstrated to be non-toxic when stably expressed in 293T cells (10), to analyze the role of PP1 in VP30 phosphorylation. Expression of cdNIPP1 increased phosphorylation of VP30, suggesting that it is controlled by PP1. Thus, our study specifically demonstrates for the first time that PP1 is involved in dephosphorylation of VP30 under physiological conditions. The PP1α knockdown in cultured cells led to up-regulation of the PP1 cytoplasmic shuttling regulatory subunit, SIPP1, and reduction of VP30 phosphorylation. Because SIPP1 has high affinity to PP1 (33), the increased expression of SIPP1 will translate to the higher levels of SIPP1/PP1 holoenzyme in the cytoplasm. This effect also coincided with the increase in EBOV transcription, suggesting that SIPP1 is likely to target PP1 to VP30. Because PP1 exists as a heterodimer or even a heterotrimer (PP1/Sds22/inhibitor-3) (23), and some of its catalytic subunits are interchangeable, complete PP1 knockout is difficult to achieve. The use of PP1-targeting peptides, such as cdNIPP1, is clearly a more straightforward approach. Furthermore, in an early study in vivo, NIPP1 was shown to co-purify with PP1β/δ from rat liver extracts (34). The recent crystal structure of the NIPP1-PP1 complex was determined for PP1α (35). Thus, cdNIPP1 is likely to bind equally well PP1α and PP1β/δ. Whether both of these catalytic subunits are involved in VP30 dephosphorylation remains to be determined. Nevertheless, our PP1 KD experiments clearly indicate that PP1 plays a role in EBOV transcription. Further studies are needed to confirm the role of PP1 holoenzyme in VP30 dephosphorylation and to identify the regulatory cytoplasmic PP1 subunit that may target PP1 to VP30. Also, we cannot fully exclude the possibility that PP2A may also participate in dephosphorylation of certain residues of VP30.
Either alanine or aspartic acid mutation in Thr143 and Thr146 led to a strong induction of VP30 phosphorylation, which apparently took place in the N-terminal serine clusters because their mutations prevented the induction of phosphorylation, similar to the phenomenon known as “cascade phosphorylation” described for vesicular stomatitis virus (36) and RSV (37). Consistent with that, the phosphorylation mimic mutations strongly inhibited transcription of the EBOV genome. It appears that targeted phosphorylation of Thr143 and/or Thr146 has a strong regulatory effect on VP30 phosphorylation. Identifying a kinase that targets these two residues would be important for the development of antivirals against filoviruses and may represent a novel regulatory pathway that may be targeted for the development of antivirals.
When our study was in progress, Biedenkopf et al. (5) reported that phosphorylation of VP30 shifts the transcription/replication balance of the EBOV polymerase toward replication. Whereas the study of Biedenkopf et al. used strand-specific quantitative PCR, our study used Northern blotting, which is a more direct method and which complements the study of Biedenkopf et al. (5). VP30 proteins of filoviruses do not have any analogs among other non-segmented negative strand viruses, which typically have three proteins: nucleoprotein, phosphoprotein (its filovirus analog is called VP35), and L. To our best knowledge, RSV and pneumonia virus of mice are the only other non-segmented negative strand viruses, which have proteins (M2-1 and M2-2) with functions somewhat similar to filovirus VP30 (30, 38, 39). However, the M2-1-regulated transcription/replication balance is controlled by phosphorylation of the P protein, which interferes with its interaction with M2-1, resulting in the reduction of transcription elongation (40). The other important difference includes the effects of these factors on transcription. The RSV M2-1 protein serves as a transcription elongation factor whose absence results in a premature termination of transcription (39), and M2-2 mediates a regulatory switch from transcription to replication (30). In contrast, EBOV VP30 is involved in initiation of transcription, and its absence results in a lack of transcription but not the premature termination of transcription (4, 19).
Importantly, the small molecule compound generated in the study affected the EBOV transcription/replication balance the same way as permanent phosphorylation of VP30 (i.e. reducing the transcription and increasing replication of the viral genome). Martinez et al. (41) demonstrated indirectly that the dephosphorylated, but not the phosphorylated, form reinitiates EBOV transcription based on the ability of the permanently phosphorylated and dephosphorylated forms of VP30 to support recovery of the virus. In contrast to the study by Martinez et al. (41), this study used a bicistronic minigenome to provide direct evidence that dephosphorylated form of VP30 is required not only for initiation but also for reinitiation of transcription.
Our study demonstrated that 1E7-03 strongly inhibits replication of EBOV and provides evidence that the molecule increases phosphorylation of the viral VP30. Because the transcription of EBOV, and probably other filoviruses, is critically dependent on VP30 dephosphorylation, the molecule prevents the effective transcription but increases replication of the EBOV genome. As a result of the transcription/replication imbalance, viral replication is effectively prevented (Fig. 9A).
FIGURE 9.

A model of the effects of 1E7-03 on EBOV. A, during the EBOV life cycle, VP30 exists in two modes: active (dephosphorylated), in which it promotes transcription of the viral genome by the viral polymerase complex, and inactive (phosphorylated), when the polymerase switches to the replication mode. The 1E7-03 compound prevents dephosphorylation of VP30 by PP1, thereby skewing the transcription/replication balance toward replication and reducing transcription, which restricts viral growth. The viral genome of the negative polarity is shown in black with the seven genes depicted as thick bars; the seven mRNAs are depicted in red, and the anti-genomes of the positive polarity are depicted in blue. B, comparison of the phosphorylation sites of VP30 of various species of filoviruses. The phosphorylated serine (S) residues of EBOV (7) and Marburg virus (48) and the putative phosphorylated residues of additional filovirus species are highlighted in red. Shown in the order of PubMed accession numbers are the VP30 N-terminal 60 amino acids of EBOV, Q05323; Sudan virus (SUDV), AY729654.1; Bundibugyo virus (BDBV), YP_003815438.1; Taï Forest virus (TAFV), FJ217162.1; Marburg virus (MARV), P35258; and Ravn virus (RAVV), DQ447649.1.
The small molecule preventing the effective dephosphorylation of VP30 presented here, which provides the complete inhibition of EBOV in vitro, represents a novel approach for the development of antivirals against EBOV, which are urgently needed to combat the devastating disease caused by the virus. The present study suggests that 1E7-03 can be used as a starting point for the development of a highly effective drug against EBOV. A remarkable similarity in the phosphorylation sites at the N termini of the VP30 proteins of multiple filovirus species (Fig. 9B) suggests that the approach is feasible for development of a pan-filoviral drug.
The study demonstrates that with repeated treatments, 1E7 effectively suppresses concentration of EBOV at 3 μm concentration (Fig. 5D). Our recent study showed that mice injected with 25 mg/kg 1E7-03 accumulated 90 μm plasma concentration of the compound up to 8 h (25). Thus, concentrations of 10 and 3 μm, which effectively suppress replication of EBOV in vitro (Fig. 4, B and D), will be achieved by administering the compound at ∼3 and 1 mg/kg, respectively. These doses are comparable with the active doses of the anti-influenza drug oseltamivir, when administered to children (2 mg/kg) (42), and the anti-influenza drug zanamivir tested in cynomolgus macaques (10–20 mg/kg) (43). Importantly, our leading compound 1E7-03 is non-toxic, presumably because it targets only one PP1 non-catalytic site and probably prevents the interaction of PP1 holoenzyme with EBOV VP30, whereas the host cell PP1 regulatory subunits typically bind PP1 through several interacting sites with a much higher affinity (44). Several lines of evidence suggest that sequestering of PP1 per se does not result in a general toxic effect, unlike the toxic effects of PP1 inhibitors, such as microcystin and okadaic acid, that bind to the active site of PP1 with global PP1 inhibition and cannot be used as therapeutics (45). First, our recent study showed that PP1-targeted compound 1H4 did not prevent the interaction of PP1 with major regulatory subunits, such as NIPP1 or PNUTS (13). Second, we recently demonstrated that high doses of 1E7-03 administered to mice by injections do not result in a toxic effect (25). Third, a stable expression of PP1-sequestering cdNIPP1, although inhibitory for HIV-1, did not compromise cell growth (10). Fourth, viable cell lines stably expressing cdNIPP1 (10) and transgenic mice in which genetic PP1 inhibition through induced expression of inhibitor-1 (I-1) resulted in ∼70% reduction of the PP1 activity (46) were developed. Therefore, it is unlikely that the compound interferes with interactions of PP1 with its cellular regulatory subunits and its cellular substrates. Although direct targeting of a RNA virus frequently results in selection for drug-resistant mutants (47), targeting of an interaction of a viral protein with a cellular component described here is unlikely to have such effect. Thus, targeting of PP1 for inhibition of its interaction with filovirus VP30 may represent a viable approach for development of an antiviral molecule.
Acknowledgments
We thank Dr. Elke Mühlberger (Boston University) for providing the EBOV minigenome and for the useful advice; Dr. Yoshihiro Kawaoka (University of Wisconsin) for providing the plasmids expressing EBOV NP, VP35, L, VP30, and the T7 polymerase; and Dr. Mathieu Bollen and Dr. Monique Beullens (University of Leuven, Belgium) for cdNIPP1-expressing vectors. We thank Dr. Chandrasekhar Yallampalli (University of Texas Medical Branch) for kindly providing access to the Typhoon Tryo PhosphorImager. We thank Dr. Krishna Narayanan and Dr. Cheng Huang (University of Texas Medical Branch) for valuable suggestions in Northern blot analysis. We thank Anand Soorneedi for technical assistance. We also thank Dr. Michelle Meyer for reading the manuscript.
This work was supported, in whole or in part, by National Institutes of Health Grants 1 U19 AI109664-01 (to A. B.), 1SC1GM082325, 1P50HL118006-01, 8G12MD007597, and 1 U19 AI109664-01(to S. N.). This work was also supported by a departmental start-up grant from the University of Texas Medical Branch (to A. B.) and by District of Columbia Developmental Center for AIDS Research Grant P30AI087714 (to S. N.).
- EBOV
- Ebola virus
- NP
- nucleoprotein
- PP
- protein phosphatase
- eGFP
- enhanced GFP
- RSV
- respiratory syncytial virus
- KD
- knockdown
- MOI
- multiplicity of infection
- S29–46A
- mutation of serines 29, 30, 31, 42, 44, and 46 to alanine.
REFERENCES
- 1. World Health Organization (1978) Ebola haemmorhagic fever in Zaire, 1976. Bull. World Health Organ. 56, 271–293 [PMC free article] [PubMed] [Google Scholar]
- 2. Centers for Disease Control and Prevention (2014) Outbreak of Ebola in Guinea, Liberia, and Sierra Leone, Centers for Disease Control and Prevention, Atlanta [Google Scholar]
- 3. Kuhn J. H. (2008) Filoviruses, 1st Ed., pp. 190–251, Springer, New York [Google Scholar]
- 4. Weik M., Modrof J., Klenk H. D., Becker S., Mühlberger E. (2002) Ebola virus VP30-mediated transcription is regulated by RNA secondary structure formation. J. Virol. 76, 8532–8539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Biedenkopf N., Hartlieb B., Hoenen T., Becker S. (2013) Phosphorylation of Ebola virus VP30 influences the composition of the viral nucleocapsid complex: impact on viral transcription and replication. J. Biol. Chem. 288, 11165–11174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Martínez M. J., Biedenkopf N., Volchkova V., Hartlieb B., Alazard-Dany N., Reynard O., Becker S., Volchkov V. (2008) Role of Ebola virus VP30 in transcription reinitiation. J. Virol. 82, 12569–12573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Modrof J., Mühlberger E., Klenk H. D., Becker S. (2002) Phosphorylation of VP30 impairs Ebola virus transcription. J. Biol. Chem. 277, 33099–33104 [DOI] [PubMed] [Google Scholar]
- 8. Bollen M., Peti W., Ragusa M. J., Beullens M. (2010) The extended PP1 toolkit: designed to create specificity. Trends Biochem. Sci. 35, 450–458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Heroes E., Lesage B., Görnemann J., Beullens M., Van Meervelt L., Bollen M. (2013) The PP1 binding code: a molecular-Lego strategy that governs specificity. FEBS J. 280, 584–595 [DOI] [PubMed] [Google Scholar]
- 10. Ammosova T., Yedavalli V. R., Niu X., Jerebtsova M., Van Eynde A., Beullens M., Bollen M., Jeang K. T., Nekhai S. (2011) Expression of a protein phosphatase 1 inhibitor, cdNIPP1, increases CDK9 threonine 186 phosphorylation and inhibits HIV-1 transcription. J. Biol. Chem. 286, 3798–3804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ammosova T., Jerebtsova M., Beullens M., Lesage B., Jackson A., Kashanchi F., Southerland W., Gordeuk V. R., Bollen M., Nekhai S. (2005) Nuclear targeting of protein phosphatase-1 by HIV-1 Tat protein. J. Biol. Chem. 280, 36364–36371 [DOI] [PubMed] [Google Scholar]
- 12. Ammosova T., Jerebtsova M., Beullens M., Voloshin Y., Ray P. E., Kumar A., Bollen M., Nekhai S. (2003) Nuclear protein phosphatase-1 regulates HIV-1 transcription. J. Biol. Chem. 278, 32189–32194 [DOI] [PubMed] [Google Scholar]
- 13. Ammosova T., Platonov M., Yedavalli V. R., Obukhov Y., Gordeuk V. R., Jeang K. T., Kovalskyy D., Nekhai S. (2012) Small molecules targeted to a non-catalytic “RVxF” binding site of protein phosphatase-1 inhibit HIV-1. PLoS One 7, e39481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Llorian M., Beullens M., Lesage B., Nicolaescu E., Beke L., Landuyt W., Ortiz J. M., Bollen M. (2005) Nucleocytoplasmic shuttling of the splicing factor SIPP1. J. Biol. Chem. 280, 38862–38869 [DOI] [PubMed] [Google Scholar]
- 15. Beullens M., Vulsteke V., Van Eynde A., Jagiello I., Stalmans W., Bollen M. (2000) The C-terminus of NIPP1 (nuclear inhibitor of protein phosphatase-1) contains a novel binding site for protein phosphatase-1 that is controlled by tyrosine phosphorylation and RNA binding. Biochem. J. 352, 651–658 [PMC free article] [PubMed] [Google Scholar]
- 16. Neumann G., Feldmann H., Watanabe S., Lukashevich I., Kawaoka Y. (2002) Reverse genetics demonstrates that proteolytic processing of the Ebola virus glycoprotein is not essential for replication in cell culture. J. Virol. 76, 406–410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ammosova T., Obukhov Y., Kotelkin A., Breuer D., Beullens M., Gordeuk V. R., Bollen M., Nekhai S. (2011) Protein phosphatase-1 activates CDK9 by dephosphorylating Ser175. PLoS One 6, e18985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Towner J. S., Paragas J., Dover J. E., Gupta M., Goldsmith C. S., Huggins J. W., Nichol S. T. (2005) Generation of eGFP expressing recombinant Zaire ebolavirus for analysis of early pathogenesis events and high-throughput antiviral drug screening. Virology 332, 20–27 [DOI] [PubMed] [Google Scholar]
- 19. Mühlberger E., Weik M., Volchkov V. E., Klenk H. D., Becker S. (1999) Comparison of the transcription and replication strategies of marburg virus and Ebola virus by using artificial replication systems. J. Virol. 73, 2333–2342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Watanabe S., Noda T., Halfmann P., Jasenosky L., Kawaoka Y. (2007) Ebola virus (EBOV) VP24 inhibits transcription and replication of the EBOV genome. J. Infect. Dis. 196, S284–S290 [DOI] [PubMed] [Google Scholar]
- 21. Ammosova T., Washington K., Debebe Z., Brady J., Nekhai S. (2005) Dephosphorylation of CDK9 by protein phosphatase 2A and protein phosphatase-1 in Tat-activated HIV-1 transcription. Retrovirology 2, 47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Allen P. B., Kwon Y. G., Nairn A. C., Greengard P. (1998) Isolation and characterization of PNUTS, a putative protein phosphatase 1 nuclear targeting subunit. J. Biol. Chem. 273, 4089–4095 [DOI] [PubMed] [Google Scholar]
- 23. Lesage B., Beullens M., Pedelini L., Garcia-Gimeno M. A., Waelkens E., Sanz P., Bollen M. (2007) A complex of catalytically inactive protein phosphatase-1 sandwiched between Sds22 and inhibitor-3. Biochemistry 46, 8909–8919 [DOI] [PubMed] [Google Scholar]
- 24. Mühlberger E., Lötfering B., Klenk H. D., Becker S. (1998) Three of the four nucleocapsid proteins of Marburg virus, NP, VP35, and L, are sufficient to mediate replication and transcription of Marburg virus-specific monocistronic minigenomes. J. Virol. 72, 8756–8764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ammosova T., Platonov M., Ivanov A., Kont Y. S., Kumari N., Kehn-Hall K., Jerebtsova M., Kulkarni A. K., Üren A., Kovalskyy D., Nekhai S. (2014) 1E7–03, a small molecule targeting host protein phosphatase-1, inhibits HIV-1 transcription. Br. J. Pharmacol. 10.1111/bph.12863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Sanchez A., Kiley M. P., Holloway B. P., Auperin D. D. (1993) Sequence analysis of the Ebola virus genome: organization, genetic elements, and comparison with the genome of Marburg virus. Virus Res. 29, 215–240 [DOI] [PubMed] [Google Scholar]
- 27. Bukreyev A. A., Belanov E. F., Blinov V. M., Netesov S. V. (1995) Complete nucleotide sequences of Marburg virus genes 5 and 6 encoding VP30 and VP24 proteins. Biochem. Mol. Biol. Int. 35, 605–613 [PubMed] [Google Scholar]
- 28. Mühlberger E., Trommer S., Funke C., Volchkov V., Klenk H. D., Becker S. (1996) Termini of all mRNA species of Marburg virus: sequence and secondary structure. Virology 223, 376–380 [DOI] [PubMed] [Google Scholar]
- 29. Collins P. L., Hill M. G., Camargo E., Grosfeld H., Chanock R. M., Murphy B. R. (1995) Production of infectious human respiratory syncytial virus from cloned cDNA confirms an essential role for the transcription elongation factor from the 5′ proximal open reading frame of the M2 mRNA in gene expression and provides a capability for vaccine development. Proc. Natl. Acad. Sci. U.S.A. 92, 11563–11567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Bermingham A., Collins P. L. (1999) The M2-2 protein of human respiratory syncytial virus is a regulatory factor involved in the balance between RNA replication and transcription. Proc. Natl. Acad. Sci. U.S.A. 96, 11259–11264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Asenjo A., Rodríguez L., Villanueva N. (2005) Determination of phosphorylated residues from human respiratory syncytial virus P protein that are dynamically dephosphorylated by cellular phosphatases: a possible role for serine 54. J. Gen. Virol. 86, 1109–1120 [DOI] [PubMed] [Google Scholar]
- 32. Washington K., Ammosova T., Beullens M., Jerebtsova M., Kumar A., Bollen M., Nekhai S. (2002) Protein phosphatase-1 dephosphorylates the C-terminal domain of RNA polymerase-II. J. Biol. Chem. 277, 40442–40448 [DOI] [PubMed] [Google Scholar]
- 33. Llorian M., Beullens M., Andrés I., Ortiz J. M., Bollen M. (2004) SIPP1, a novel pre-mRNA splicing factor and interactor of protein phosphatase-1. Biochem. J. 378, 229–238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Jagiello I., Beullens M., Stalmans W., Bollen M. (1995) Subunit structure and regulation of protein phosphatase-1 in rat liver nuclei. J. Biol. Chem. 270, 17257–17263 [DOI] [PubMed] [Google Scholar]
- 35. O'Connell N., Nichols S. R., Heroes E., Beullens M., Bollen M., Peti W., Page R. (2012) The molecular basis for substrate specificity of the nuclear NIPP1:PP1 holoenzyme. Structure 20, 1746–1756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Barik S., Banerjee A. K. (1992) Sequential phosphorylation of the phosphoprotein of vesicular stomatitis virus by cellular and viral protein kinases is essential for transcription activation. J. Virol. 66, 1109–1118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Barik S., McLean T., Dupuy L. C. (1995) Phosphorylation of Ser232 directly regulates the transcriptional activity of the P protein of human respiratory syncytial virus: phosphorylation of Ser237 may play an accessory role. Virology 213, 405–412 [DOI] [PubMed] [Google Scholar]
- 38. Dibben O., Thorpe L. C., Easton A. J. (2008) Roles of the PVM M2–1, M2–2 and P gene ORF 2 (P-2) proteins in viral replication. Virus Res. 131, 47–53 [DOI] [PubMed] [Google Scholar]
- 39. Collins P. L., Hill M. G., Cristina J., Grosfeld H. (1996) Transcription elongation factor of respiratory syncytial virus, a nonsegmented negative-strand RNA virus. Proc. Natl. Acad. Sci. U.S.A. 93, 81–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Asenjo A., Calvo E., Villanueva N. (2006) Phosphorylation of human respiratory syncytial virus P protein at threonine 108 controls its interaction with the M2–1 protein in the viral RNA polymerase complex. J. Gen. Virol. 87, 3637–3642 [DOI] [PubMed] [Google Scholar]
- 41. Martinez M. J., Volchkova V. A., Raoul H., Alazard-Dany N., Reynard O., Volchkov V. E. (2011) Role of VP30 phosphorylation in the Ebola virus replication cycle. J. Infect. Dis. 204, S934–S940 [DOI] [PubMed] [Google Scholar]
- 42. Sugaya N., Ohashi Y. (2010) Long-acting neuraminidase inhibitor laninamivir octanoate (CS-8958) versus oseltamivir as treatment for children with influenza virus infection. Antimicrob. Agents Chemother. 54, 2575–2582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Stittelaar K. J., Tisdale M., van Amerongen G., van Lavieren R. F., Pistoor F., Simon J., Osterhaus A. D. (2008) Evaluation of intravenous zanamivir against experimental influenza A (H5N1) virus infection in cynomolgus macaques. Antiviral Res. 80, 225–228 [DOI] [PubMed] [Google Scholar]
- 44. Bollen M., Beullens M. (2002) Signaling by protein phosphatases in the nucleus. Trends Cell Biol. 12, 138–145 [DOI] [PubMed] [Google Scholar]
- 45. Fujiki H., Suganuma M. (2009) Carcinogenic aspects of protein phosphatase 1 and 2A inhibitors. Prog. Mol. Subcell. Biol. 46, 221–254 [DOI] [PubMed] [Google Scholar]
- 46. Genoux D., Haditsch U., Knobloch M., Michalon A., Storm D., Mansuy I. M. (2002) Protein phosphatase 1 is a molecular constraint on learning and memory. Nature 418, 970–975 [DOI] [PubMed] [Google Scholar]
- 47. McKimm-Breschkin J. L. (2013) Influenza neuraminidase inhibitors: antiviral action and mechanisms of resistance. Influenza Other Respir. Viruses 7, 25–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Modrof J., Möritz C., Kolesnikova L., Konakova T., Hartlieb B., Randolf A., Mühlberger E., Becker S. (2001) Phosphorylation of Marburg virus VP30 at serines 40 and 42 is critical for its interaction with NP inclusions. Virology 287, 171–182 [DOI] [PubMed] [Google Scholar]