

Abstract
SecA is an intensively studied mechanoenzyme that uses ATP hydrolysis to drive processive extrusion of secreted proteins through a protein-conducting channel in the cytoplasmic membrane of eubacteria. The ATPase motor of SecA is strongly homologous to that in DEAD-box RNA helicases. It remains unclear how local chemical events in its ATPase active site control the overall conformation of an ~100 kDa multidomain enzyme and drive protein transport. In this paper, we use biophysical methods to establish that a single electrostatic charge in the ATPase active site controls the global conformation of SecA. The enzyme undergoes an ATP-modulated endothermic conformational transition (ECT) believed to involve similar structural mechanics to the protein transport reaction. We have characterized the effects of an isosteric glutamate-to-glutamine mutation in the catalytic base, which mimics the immediate electrostatic consequences of ATP hydrolysis in the active site. Calorimetric studies demonstrate that this mutation facilitates the ECT in E. coli SecA and triggers it completely in B. subtilis SecA. Consistent with the substantial increase in entropy observed in the course of the ECT, hydrogen-deuterium exchange mass spectrometry demonstrates that it increases protein backbone dynamics in domain-domain interfaces at remote locations from the ATPase active site. The catalytic glutamate is one of ~250 charged amino acids in SecA, and yet neutralization of its sidechain charge is sufficient to trigger a global order-disorder transition in this 100 kDa enzyme. The intricate network of structural interactions mediating this effect couples local electrostatic changes during ATP hydrolysis to global conformational and dynamic changes in SecA. This network forms the foundation of the allosteric mechanochemistry that efficiently harnesses the chemical energy stored in ATP to drive complex mechanical processes.
Keywords: SecA, ATPase, thermodynamics, entropy, protein dynamics, allostery, hydrogen-deuterium exchange
Introduction
Following synthesis in the cytoplasm, secreted protein domains require translocation through cellular membranes by active transport. In bacteria, the Sec system provides a mechanism for post-translational translocation of proteins that function outside of the cytoplasm. Secreted “preproteins” contain a cleavable N-terminal hydrophobic extension, called a signal sequence 1. This extension targets them for delivery to SecA 2–4, a cytosolic ATPase that drives their extrusion through a heterotrimeric protein-conducting channel called SecYEG 5–7. SecA is a 100 kDa enzyme that contains six globular domains: an N-terminal pair of 20–30 kDa domains comprising its “DEAD-box” ATPase motor (described in detail below), an 18 kDa “preprotein crosslinking domain”, a 16 kDa α-helical scaffold domain (HSD), an 11 kDa α-helical wing domain (HWD), and a C-terminal 2.5 kDa Zn++-binding domain. SecA may partially insert into the pore of SecYEG as its ATPase drives preprotein extrusion 8,9. Each molecule of ATP hydrolyzed by SecA leads to translocation of 10–15 amino acids of preprotein 10,11. Based on mechanochemical studies of SecA and evolutionarily related ATPases 12–15, this translocation reaction seems likely to be driven by mechanical rearrangements of SecA’s domains coupled to its ATPase cycle. Such motions could processively push the preprotein itself through the pore of SecYEG while SecA remains bound to the surface of SecYEG 16,17, or alternatively, they could control insertion and retraction of SecA from SecYEG, which would drive transport if preprotein were bound to SecA during insertion but released during retraction 12,18,19. However, both of these models are speculative, and the mechanism by which SecA drives preprotein transport remains unclear despite extensive mechanistic 10,11,18–21, thermodynamic 13,22–27, and structural studies 12,16,28–32.
A fundamental question regarding SecA function is how hydrolysis of a single phosphodiester bond at its ATPase active site can control the functional conformational transitions of this large mechanoenzyme. The chemistry of the hydrolysis reaction is straightforward:
Extensive research has shown that catalysis of this reaction is critically dependent on electrostatic interactions with ATP and its Mg++ cofactor in the active site of ATPases 33,34. However, it remains unclear how these interactions and their variation during the hydrolysis reaction drive the large-scale conformational transitions that perform thermodynamic work in SecA and other ATP-driven mechanoenzymes.
The ATPase active site in SecA is located at the interface of domains called nucleotide-binding folds I and II (NBF-I and NBF-II) 12, which together comprise the DEAD-box ATPase motor. NBF-I and NBF-II are believed to function as an ATP-dependent mechanical clamp as they close around the nucleotide bound at their mutual interface 12,28. These domains bear the strongest structural and sequence similarity to the ATPase domains in superfamily I and II ATP-dependent helicases 14,35, especially those in the “DEAD-box” family of RNA helicases which perform a plethora of cellular functions 36–39. This family takes its name from a conserved asp-glu-ala-asp (DEAD) sequence that includes several catalytically vital residues 39,40. The glutamate in this sequence (E210 in E. coli SecA) is the catalytic base that activates water for hydrolytic attack on ATP 41. DEAD-box motor domains have more remote but clearly significant structural homology to a much wider variety of mechanically active ATPases including F1 ATPase 15,42, AAA ATPases, and ABC transporter proteins 43–46. The ATPase motor domains from these diverse families share a conserved core β-sheet topology as well as stereotyped Walker A and B sequence motifs 47 that participate in nucleotide binding. The final amino acid in the Walker B motif, an aspartate that ligates the Mg2+ cofactor of ATP, is the first amino acid in the DEAD sequence. The catalytic base is found at different sites in different functional families of F1-like ATPases but occurs at the same position adjacent to this aspartate in both the superfamily I and II helicases and the ABC transporters.
Studies of the structural mechanics of all of these F1-like mechanoenzymes suggest that their homologous motor domains generally function as ATP-dependent mechanical clamps 12,14,28,29,35,15,42,43–46. ATP binding in the interfacial active site stabilizes the closed state of the clamp via the chemical contacts made by the nucleotide to both of the flanking domains. Opening of the clamp, which prevents the nucleotide from simultaneously interacting with both domains, lowers affinity for the bound nucleotide. Therefore, ATP binding generally closes the clamp, while hydrolysis weakens interfacial contacts to permit opening of the clamp. These conformational changes are controlled by the structural interactions in the ATPase active site at the clamp interface, which includes an extensive network of electrostatic interactions between the flanking protein domains that also connects these domains to the bound nucleotide. In the case of SecA, these interactions must ultimately control the functional conformational changes throughout this 100 kDa ATPase. While interdomain interactions and protein-nucleotide interactions have been implicated in allosteric control of SecA 12,26,27, substantial uncertainty remains concerning the pathway of conformational communication and the mechanism by which this communication is controlled by ATP binding and hydrolysis.
At room temperature in the absence of nucleotide, the DEAD-box motor domains of SecA adopt a closed conformation resembling that found in the nucleotide-bound structures of homologous mechanoenzymes 14. However, at temperatures slightly above physiological, SecA undergoes a global conformational transition, called the endothermic conformational transition (ECT), that significantly reduces nucleotide-binding affinity 13,22,23,48,49. (Note that this widely used term implies directionality from the low-temperature conformational ground state to the high-temperature excited state, even though this transition represents a reversible equilibrium.) Based on the structural paradigms observed in related mechanoenzymes, this conformational transition is believed to open the ATPase active site at the interface between the DEAD-box motor domains (as schematized in Figure 9 below), and this hypothesis supported by mutagenesis studies 12. These studies furthermore demonstrate that the ECT couples opening of the DEAD-box motor domains to global changes in interdomain interactions throughout SecA 12. Nucleotide binding reverses the ECT, presumably via closure of the clamp formed by the DEAD-box motor domains 12,24. Therefore, the ECT is a global conformational transition allosterically controlled by nucleotide binding at the ATPase active site.
Given these properties, the ECT could emulate the ATP-driven conformational changes that mediate preprotein translocation through SecYEG. This model is attractive because otherwise SecA would have to possess a second distinct ATP-modulated conformational reaction cycle. Temperatures above physiological promote tight membrane association of SecA in vivo, which is consistent with the ECT gating functional interaction with SecYEG during protein transport 9,18,50. This observation suggests that SecA is intimately associated with SecYEG in the nucleotide-free state, which directly contradicts data showing that the non-hydrolyzable ATP analog AMP-PNP stabilizes interaction of SecA with the channel 18,51. However, AMP-PNP has been shown to have poor affinity for the ATPase active site in SecA 13,23 and many other mechanically active ATPases 52. In several cases, this analog has resulted in misleading mechanistic inferences because it lacks sufficient binding energy to induce the proper ATP-dependent conformational changes 53–55.
To elucidate the stereochemical and mechanistic effects of ATP binding to SecA, and to deepen understanding of the allosteric control of global conformation by its ATPase cycle, we sought to develop alternative methods to trap SecA in an ATP-bound conformation. In ABC transporters, mechanoejzymes sharing key ATP binding and hydrolysis motifs, this goal was achieved by mutagenesis of the catalytic glutamate to glutamine 43,44,56, which generally blocks ATP hydrolysis but preserves ATP-binding affinity for proteins belonging to this ATPase family. In this paper, we report that the E-to-Q mutation in the active site of either B. subtilis SecA (E208Q-BsSecA) or E. coli SecA (E210Q-EcSecA) has dramatically different thermodynamic effects than the equivalent mutation in ABC transporter motors. The single point-charge change caused by this isosteric mutation significantly perturbs thermodynamic couplings throughout SecA, demonstrating that an exquisitely balanced network of electrostatic interactions 33,34 enables small changes in active site charge to alter the global conformational state of a 100 kDa mechanoenzyme.
Methods
Buffers and reagent
Experiments were conducted in KEMT (50 mM KCl, 20 μM EDTA, 1.0 mM MgCl2, 25 mM Tris-Cl, pH 7.6) or TKM 23 (50 mM Tris-HCl, pH 8.0, 50 mM KCl, 5 mM MgCl2) buffers. MANT-labeled and unlabeled nucleotides were purchased from Invitrogen (Carlsbad, CA) and Sigma/Fluka (St. Louis, MO), respectively; concentrations were determined using ε260 = 15,400 M−1·m−1 for adenine and ε356 = 5,800 M−1·m−1 for MANT.
Protein biochemistry methods
Cloning, mutagenesis, and protein expression, purification, and crystallization methods are described in the online Supporting Information (SI). They are equivalent to those previously reported 12,13 except for use of an additional gel filtration column during purification. In brief, full-length proteins spanning the exact native sequences without additional residues were purified through DEAE Sepharose, SP Sepharose, Butyl Sepharose, and Sephacryl S300 columns prior to concentration to 5–10 mg/ml in KEMT buffer for enzyme assays and fluorimetry, TKM buffer for calorimetry, or 300 mM (NH4)2SO4, 1 mM DTT, 20 mM BES, pH 7.0 for crystallization. Protein concentration was determined by absorbance using ε280 = 51,480 M−1·m−1 or 76,000 M−1·m−1 for BsSecA or EcSecA, respectively, diluted into 6 M guanidinium-HCl, 20 mM sodium phosphate, pH 6.5.
Assays methods
ATPase and fluorescence anisotropy assays were conducted and analyzed as previously 13 described using the buffers indicated. Calorimetric methods are described in the SI. Samples for calorimetry were prepared by dialyzing 100–500 μl of protein stock for > 18 hours against 1 L of TKM buffer in either 3.5K or 10K Slide-A-Lyzer dialysis cassettes (Pierce, Rockford, IL). Buffer was changed 3 times with each step lasting > 3 hours. The final dialysis buffer was used to dissolve solid Mg-ADP for use in calorimetric experiments. Amide 1H/2H exchange rate measurements using mass spectrometry were conducted as described in Sharma et al. 57 with slight modifications described in the SI.
Results
The E-to-Q mutation in the catalytic base of SecA impairs ATPase activity
The basal ATPase activities of wild-type (WT) and mutant SecA enzymes from B. subtilis and E. coli were measured using the Malachite Green phosphate-release assay. Fig. 1 shows that the basal turnover rate of WT-BsSecA (kcat = 7.2 ± 0.6 min−1) is ~15-fold higher than that of WT-EcSecA (kcat = 0.480 ± 0.024 min−1), consistent with previous reports 13,41. In both enzymes, mutation of the catalytic base from glutamate to glutamine very strongly reduces the rate of ATP hydrolysis, to a level similar to background in comparable enzyme preparations (kcat = 0.24 ± 0.18 min−1 for BsSecA and kcat = 0.120 ± 0.006 min−1 for EcSecA), also consistent with previous work on the E. coli enzyme 41. Note that these rates represent upper limits for the ATPase velocities of the mutant enzymes. Even though our three-column purification procedure yields highly pure enzymes (Fig. 1B), the observed turnover rates of the mutants are so low that they could derive from trace contamination by an active ATPase (e.g., less than 0.1% by mass). It is extremely challenging to exclude this possibility with enzyme turnover rates on the order of 1 per 500 seconds.
Figure 1. The ATPase activity of SecA is reduced in the E-to-Q mutants.
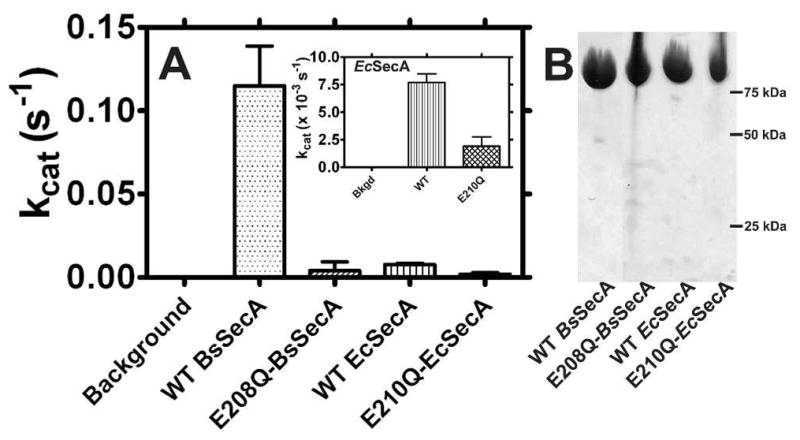
(A) ATP hydrolysis rates of WT and mutant SecAs were measured using the Malachite Green phosphate-release assay. Reactions at 25 °C containing 1–2 μM SecA and 2 mM Mg-ATP in KEMT buffer were monitored at 10–15 minute intervals for 30–90 minutes. The observed activity of E210Q-EcSecA is so low that it could be attributable to contaminating enzymes despite the high protein purity (see text). (B) Coomasie-blue stained SDS-PAGE gel of purified SecA variants.
The E-to-Q mutation reduces nucleotide affinity and accelerates binding and release rates
We used fluorescence anisotropy assays to determine the effects of the E-to-Q mutation on the affinity (Fig. 2) and kinetics (Figs. 3–4) of SecA’s interaction with MANT-labeled ADP and ATP analogues at 25 °C 13 (Table 1). Previous research has shown that MANT-labeled analogues closely track the binding properties of unlabeled adenine nucleotides 13, although they bind with ~5-fold higher affinity to EcSecA. Fig. 2 shows that the E-to-Q mutation reduces the affinity for Mg-MANT-ADP by ~20-fold in BsSecA (increasing the Kd from 160 ± 40 nM to 3.50 ± 0.14 μM) and by ~10-fold in EcSecA (increasing the Kd from 12 ± 3 nM to 120 ± 11 nM). The somewhat more severe defect in BsSecA compared to EcSecA suggest that the influence of the E-to-Q mutation may differ in some details between the enzymes even though it produces a similar biochemical phenotype.
Figure 2. Nucleotide-binding affinity is reduced to a different extent in E208Q-BsSecA vs. E210Q-EcSecA.

SecA variants were titrated onto a fixed concentration of MANT-labeled nucleotide at 25 °C in KEMT buffer (pH 7.6) while MANT fluorescence anisotropy was monitored at 352 nm. (A) WT (■) and E208Q (◆) BsSecA were titrated onto 900 nM Mg-MANT-ATP. (B) WT (□) and E210Q (◇) EcSecA were titrated onto 30 nM Mg-MANT-ATP. (C) WT (●) and E208Q (▲) BsSecA were titrated onto 900 nM Mg-MANT-ADP. (D) WT (○) and E210Q (△) EcSecA were titrated onto 30 nM Mg-MANT-ADP.
Figure 3. E-to-Q mutants of SecA do not kinetically trap ATP.
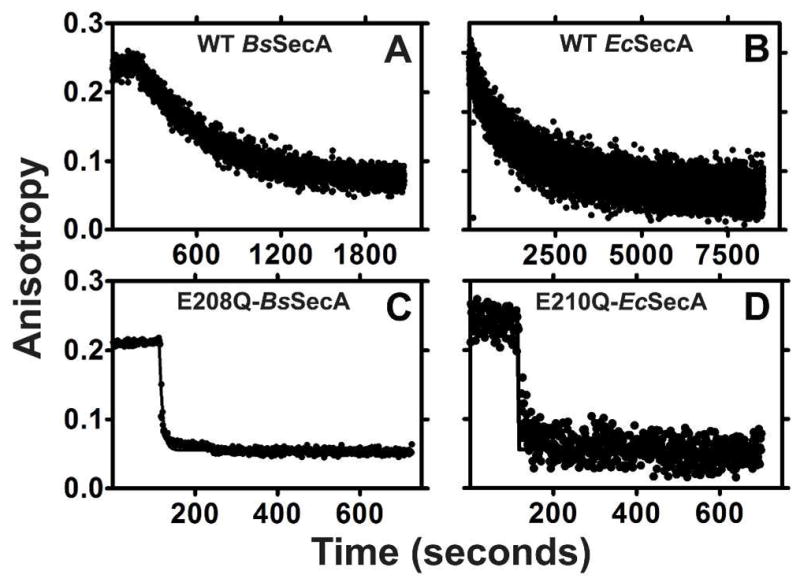
Competitive displacement of MANT-labeled nucleotides by unlabeled nucleotides was monitored kinetically using fluorescence anisotropy spectroscopy at 352 nm in KEMT buffer. WT-BsSecA (panel A), WT-EcSecA (panel B), E208Q-BsSecA (panel C), or E210Q-EcSecA (panel D) at 250–750 nM was equilibrated with 60 nM Mg-MANT-ATP in KEMT buffer (pH 7.6) at 5°C for 3–5 minutes before adding a > 100-fold excess of unlabeled Mg-ADP to initiate the exchange reaction.
Figure 4. Nucleotides bind to the E-to-Q mutants faster than to WT SecA.
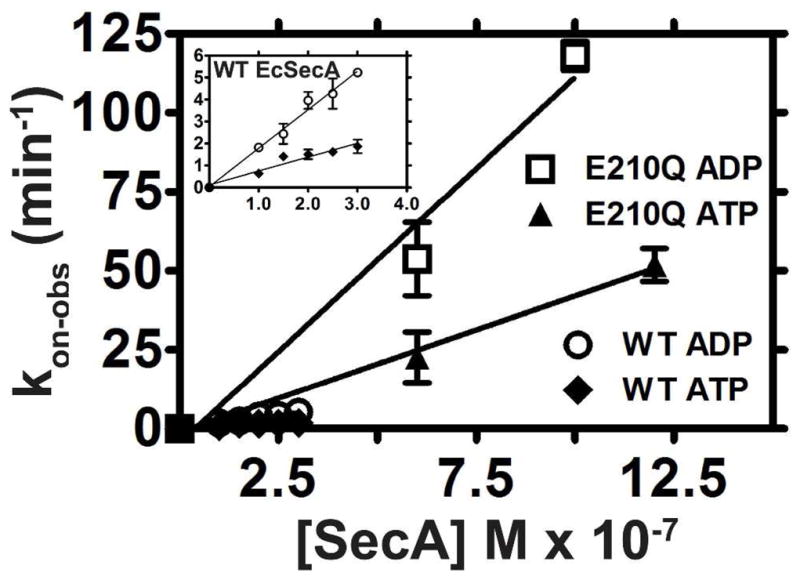
WT or E210Q EcSecA were added to a cuvette containing 60 nM MANT-labeled nucleotide in KEMT buffer (pH 7.6) at 25 °C, and binding was monitored using fluorescence anisotropy spectroscopy at 352 nm. The on-rate (kon) was calculated from the observed single-exponential binding curves as explained in the SI. The labels ADP and ATP refer to Mg-MANT-ADP and Mg-MANT-ATP, respectively. The inset shows a magnified view of the data for the WT enzyme.
Table 1.
Kinetic and thermodynamic parameters of nucleotide interactions with WT and mutant SecA.
| Parameter | Ligand | Method | Temp. | WT BsSecA | BsSecA E208Q | WT EcSecA | EcSecA E210Q |
|---|---|---|---|---|---|---|---|
| n | Mg-ADP | ITC | 25 °C | 0.89 | 0.92 | 0.93 | -- |
| ΔH° (kcal mol−1) | Mg-ADP | ITC | 25 °C | −13.00 ± 0.07 | −14.0 ± 0.1 | −20.0 ± 0.1 | -- |
| ΔS° (cal mol−1 K−1)a | Mg-ADP | ITC | 25 °C | −13.0 ± 0.3 | −24.0 ± 0.3 | −34.0 ± 0.4 | -- |
| ΔG° (kcal mol−1) | Mg-ADP | ITC | 25 °C | −9.20 ± 0.04 | −6.90 ± 0.02 | −9.80 ± 0.04 | -- |
| Kd (nM) | Mg-ADP | ITC | 25 °C | 180 ± 10 | 7700 ± 300 | 62 ± 4 | -- |
| kcat (min−1)b | Mg-ATP | M.G.c | 25 °C | 7.2 ± 0.6 | 0.24 ± 0.18d | 0.480 ± 0.024 | 0.120 ± 0.006d |
| Kd (nM) | Mg-MANT-ADP | Anisotropy | 25 °C | 160 ± 40e | 3500 ± 140 | 12 ± 3 | 120 ± 11 |
| Kd (nM) | Mg-MANT-ATP | Anisotropy | 25 °C | 190 ± 50f | 3800 ± 120 | 22 ± 7f | 120 ± 9 |
| Kd (nM) | Mg-MANT-ADP | Anisotropy | 5°C | 250 ± 30 | 460 ± 10 | 17 ± 7 | 16 ± 9 |
| koff (min−1) | Mg-MANT-ATP | Anisotropy | 5°C | 0.120 ± 0.003f | 32 ± 3 | 0.0400 ± 0.0004f | 5.6 ± 0.6 |
| koff (min−1) | Mg-MANT-ADP | Anisotropy | 25 °C | 1.1 ± 0.1 | -- | 0.070 ± 0.003 | -- |
| koff (min−1) | Mg-MANT-ATP | Anisotropy | 25 °C | 0.80 ± 0.03f | -- | 0.0500 ± 0.0007f | -- |
| kon (M−1 min−1) | Mg-MANT-ADP | Anisotropy | 25 °C | 11.0 ± 0.9 × 106 | -- | 18.0 ± 0.9×106 | 120.0 ± 0.9×106 |
| kon (M−1 min−1) | Mg-MANT-ATP | Anisotropy | 25 °C | 5.0 ± 0.6 × 106 | -- | 6.0 ± 0.6×106 | 40.0 ± 0.4 × 106 |
| Kd = koff/kon (nM) | Mg-MANT-ADP | Anisotropy | 25 °C | 100 ± 20 | -- | 4.0 ± 0.4 | -- |
| Kd = koff/kon (nM) | Mg-MANT-ATP | Anisotropy | 25 °C | 150 ± 20f | -- | 8 ± 1f | -- |
| Tm (°C) | none | DSC | N/A | 40.0 ± 0.1, 49.0 ± 0.1 | 27.0 ± 0.1, 41.0 ± 0.0 | 41.00 ± 0.03, 44.00 ± 0.05 | 39.0 ± 0.1, 44.0 ± 0.1 |
| Tm (°C) | Mg-ADP | DSC | N/A | 47.0 ± 0.4, 51.0 ± 0.4 | 32.00 ± 0.03, 41.00 ± 0.03 | 47.00 ± 0.03 | 41.0 ± 0.1, 44.0 ± 0.1 |
Calculated by the ORIGIN ITC module from ΔG° = ΔH° − TΔS°, with ΔG° being calculated from the optimized value of Kd from curve-fitting the ITC data.
Previous research has shown that kcat is equal to koff for Mg-ADP release 13, meaning that Mg-ADP release is rate limiting in the basal ATP hydrolysis cycle of SecA. The kcat values reported here for Mg-ADP do not match the koff values reported below for Mg-MANT-ADP because the fluorescently derivatized nucleotide binds to SecA tighter and is released more slowly than Mg-ADP itself.
Measured via Malachite Green phosphate-release assay calibrated with inorganic phosphate.
This level of ATP turnover may reflect the activity of contaminating enzymes. See text.
This value was obtained using 900 nM [Mg-MANT-ADP]. WT BsSecA showed evidence of a modest reduction in Kd value in experiments conducted at substantially lower nucleotide and therefore protein concentrations, suggesting that dissolution of the BsSecA dimer may increase nucleotide-binding affinity slightly.
Mg-MANT-ATP affinities and off-rates for the wild-type enzymes reflect the release rates of Mg-MANT-ADP.
Fig. 3 shows that the E-to-Q mutation in both enzymes produces substantially larger increases in the rate of Mg-MANT-ADP release (koff) than in its binding affinity. The mutant enzymes release this nucleotide so rapidly that it is difficult to quantify the exact rate at 25 °C, which means that it must be at least 100-fold faster than in the WT enzymes given the configuration of our experimental set up. (At 5 °C, a temperature at which nucleotide-exchange kinetics are substantially slower due to factors explained below, the mutation increases koff by 270-fold in BsSecA and 140-fold in EcSecA, as shown in Table 1.) Previous research 13 has demonstrated that the ratio of the measured off-rate for Mg-MANT-ADP release to the measured on-rate for its binding accurately recapitulates its Kd (i.e., koff/kon = Kd), indicating that binding is a well-behaved two-state process that is accurately monitored by the anisotropy of the fluorescently labeled nucleotide. Therefore, the disproportionately large increase in koff compared to Kd for Mg-MANT-ADP indicates that the mutation must accelerate its rate of binding in addition to its rate of release.
This inference is confirmed by direct measurements of the rate of Mg-MANT-ADP binding to the WT and mutant enzymes. While the rate was difficult to quantify accurately for E208Q-BsSecA due to its high velocity, it is clearly accelerated compared to the WT enzyme (data not shown). The rate of Mg-MANT-ADP binding to both WT and E208Q-EcSecA could be quantified (Fig. 4 and Table 1), showing that the mutation increases kon by 6.6-fold (from 18.0 ± 0.9 × 106 M−1 min−1 to 120.0 ± 0.9 × 106 M−1 min−1). The measured values of the on-rates match those expected from the measured values of Kd and koff for both WT and mutant enzymes (i.e., koff/kon = ~Kd for the measured values), supporting the accuracy of our kinetic measurements. The observed increases in both kon and koff in both enzymes suggest that the E-to-Q mutation substantially accelerates opening of the interface between NBF-I and NBF-II, which forms the ATPase active site 12,13.
Equivalent affinity (Fig. 2) and kinetic (Figs. 3–4) measurements can be done using Mg-MANT-ATP. Previously reported measurements of pre-steady-state kinetics have demonstrated that ATP is hydrolyzed very rapidly after binding to wild type SecA, while release of the ADP product occurs much more slowly 41 (and no rapidly released population is observed in kinetic experiments, ruling out nonproductive binding of ATP). Therefore, binding experiments conducted on WT enzymes with hydrolyzable ATP species report a combination of ATP binding rate and ADP release rate. However, equivalent measurements on the ATPase-deficient E-to-Q mutants yield accurate estimates of binding affinity because these enzymes release ATP substantially more rapidly than they hydrolyze it (Table 1). Both affinity and kinetic measurements with Mg-MANT-ATP show equivalent trends to those observed for Mg-MANT-ADP (Table 1), including a strong acceleration in binding rate and a net reduction in apparent binding affinity. The ATP analogue binds to both WT and E-to-Q variants of EcSecA and BsSecA ~3-fold slower than the ADP analogue. While there is a strong reduction in ATP-binding affinity in both mutant enzymes, there is again a larger reduction in the BsSecA compared to the EcSecA (~20-fold vs. ~5-fold), reinforcing the conclusion that the exact influence of the mutation may differ between the enzymes.
These results contrast sharply with those observed for the stereochemically equivalent mutation in the active sites of a wide variety of ABC transporter motors, which consistently kinetically trap ATP in the presence of the E-to-Q mutation (i.e., release it exceedingly slowly) 43,45. The equivalent mutation in SecA clearly does not kinetically trap ATP and instead substantially accelerates its release rate, resulting in a large reduction in net binding affinity. Furthermore, introducing the equivalent mutation into MJ0669, a model DEAD-box helicase from Methanococcus jannaschii, similarly fails to kinetically trap ATP (Fig. S1). Therefore, even though the motor domains of ABC transporters and the ATP-dependent helicases are homologous and both function as ATP-dependent mechanical clamps, there appear to be major differences in the role of electrostatic interactions in controlling protein conformation or dynamics 33,34. Therefore, we determined the crystal structure of E208Q-BsSecA to examine whether the mutation perturbs active-site stereochemistry.
Perturbations in active-site stereochemistry are unlikely to account for reduced nucleotide-binding affinity
The nucleotide-free B. subtilis mutant protein crystallized isomorphously with the previously reported structure of the WT enzyme, indicating no significant change in interdomain interactions when the protein is packed in the crystal lattice. The structure of the mutant crystal was solved using molecular replacement and refined to a free R-factor of 26.7% at 3.4 Å resolution (Table S1 and Fig. S2A). Electron density in the active site, which is the most stably ordered region of the structure, was clearly interpretable despite the limited resolution of the diffraction data (data not shown). While local stereochemistry is mostly preserved, the sidechains of Arg-489, Arg-517, and Arg-525 show shifts in conformation resulting in altered hydrogen-bonding (H-bonding) interactions in the active site in the presence of the E-to-Q mutation (Fig. S2A). Disruption or rearrangement of cooperative H-bonding interactions involving these Arg residues, which are all directly or indirectly involved in nucleotide binding, could potentially interfere with nucleotide binding.
To evaluate this possibility, we mutated Arg-489 to lysine 51,58 in WT-BsSecA and E208Q-BsSecA to change the stereochemistry of this residue’s H-bonding interactions while preserving net charge in the active site. The crystal structure of E208Q-R489K- BsSecA was solved and refined to a free R-factor of 26.9% at 3.3 Å resolution (Fig. S2B and Table S1). It shows no electron density for the sidechain of residue 489, indicating conformational disorder and confirming that the R489K mutation changes cooperative H-bonding interactions in the active site. However, this mutation does not significantly perturb Mg-MANT-ADP binding affinity either in the WT or the E208Q background (Fig S3A), showing that the detailed hydrogen-bonding interactions of Arg-489 are not an important determinant of nucleotide binding energy.
Active-site stereochemistry controls nucleotide-release rate
We evaluated next the influence of net electrostatic charge in the active site on nucleotide-binding affinity and kinetics, focusing on reversal of the charge difference caused by the E-to-Q mutation. Ser-211 is spatially adjacent to the catalytic glutamate E208 in the active site of BsSecA. Therefore, Ser-211 was mutated to aspartate or glutamate in both the WT and E208Q- BsSecA. These mutations, which modestly reduce Mg-MANT-ADP binding affinity in the WT background and modestly increase it in the presence of the E208Q mutation (data not shown), greatly accelerate Mg-MANT-ADP release rate in the WT background (Fig. S3B,D) and fail to reverse the accelerated release rate in the presence of the E208Q mutation (Fig. S3C,E). These experiments varying active-site electrostatic charge demonstrate that active-site stereochemistry is a much more important determinant of nucleotide release rate than net electrostatic interaction with the nucleotide. This rate was previously inferred to depend on global conformational fluctuations in SecA based on the parallel effects of temperature changes on release rate and protein conformation 13. Taken together, these observations suggest that perturbations in active-site stereochemistry, like those produced by the E-to-Q mutation, might change global conformational couplings within SecA.
Mg-MANT-ADP binding to the E-to-Q mutants is temperature-dependent
SecA is known to undergo an endothermic conformational transition (ECT) at physiological temperatures. This conformational change reduces nucleotide-binding affinity and accelerates nucleotide-release rate 13,59. Facilitation of this transition would be expected to reduce nucleotide-binding affinity, so such an effect could contribute to the defects in nucleotide binding observed in the E-to-Q mutants. Therefore, we measured the Mg-MANT-ADP binding affinities at temperatures from 5–35 °C for WT and E-to-Q mutants of B. subtilis and E. coli SecA (Fig. 5). The E-to-Q mutants both show dramatic changes in nucleotide-binding affinity in this temperature range, while the WT proteins show no significant variation except for a very slight increase in Kd above 33 °C due to an increasing population of protein in the domain-dissociated, low-affinity conformation produced by the ECT. Most strikingly, at 5 °C, E208Q-BsSecA binds Mg-MANT-ADP with comparable low-nanomolar affinity to the WT enzyme, so reducing temperature completely suppresses the defect in Mg-MANT-ADP-binding affinity. However, the Kd of the mutant begins to increase around 15°C and reaches the micromolar range at 25 °C. E210Q-EcSecA shows a qualitatively similar dependence of Kd on temperature, although the Kd only begins to increase above 20 °C. These data are consistent with the affinity measurements presented above showing a greater reduction in binding affinity for E208Q-BsSecA compared to E210Q-EcSecA at 25 °C. Moreover, the observed temperature-dependence of Mg-MANT-ADP-binding affinity supports the hypothesis that the E-to-Q mutation changes global conformational couplings within SecA and facilitates the ECT. Specifically, this dependence suggests that the mutation lowers the temperature at which the ECT occurs and that it thereby produces a reduction in Mg-MANT-ADP-binding affinity at temperatures at which the WT enzyme remains in the conformational ground state and retains maximal binding affinity (Fig. 5). To evaluate this hypothesis more directly, we undertook calorimetric studies of nucleotide binding and enzyme conformation.
Figure 5. Temperature dependence of Kd in WT and E-to-Q SecA.
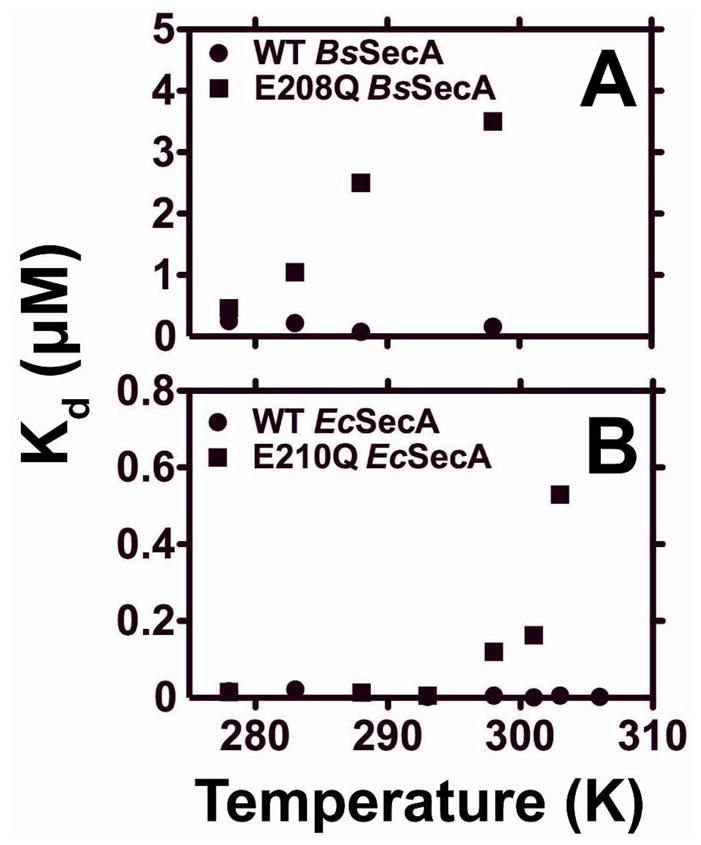
The Mg-MANT-ADP-binding affinity of WT (●) or mutant (■) variants of BsSecA (panel A) or EcSecA (panel B) were measured at temperatures from 5–30 °C in KEMT buffer (pH 7.6) using the fluorescence anisotropy assay shown in Fig. 2 with 900 nM Mg-MANT-ADP.
ITC shows Mg-ADP binding is coupled to a larger entropy loss in E208Q-BsSecA
We used isothermal titration calorimetry (ITC) to measure the enthalpy and entropy of Mg-ADP binding to WT-BsSecA, E208Q-BsSecA, and WT-EcSecA at 25 °C (Fig. 6 and Table 1). We were unable to obtain interpretable data for E210Q-EcSecA because this protein displayed irreproducible behavior in ITC experiments (as documented in Fig. S4). However, high quality ITC data was obtained for WT-EcSecA as well as both WT and E208Q-BsSecA. WT-EcSecA was reported to have ~5-fold lower affinity for Mg-ADP compared to the MANT-labeled analog based on competitive fluorescence binding experiments 13. Our ITC data confirm this inference for EcSecA and demonstrate that Mg-ADP binds to WT BsSecA with roughly comparable affinity either with or without MANT derivatization. Underivatized Mg-ADP binds to BsSecA ~3-fold more weakly than to EcSecA (180 ± 10 nM vs. 62 ± 4 nM), but this difference is physiological insignificant given the fact that cytosolic Mg-ADP concentration is at least 1,000-fold greater than the higher of these two Kd values and cytosolic Mg-ATP concentration is even higher in growing bacterial cells 60.
Figure 6. Isothermal titration calorimetry (ITC) reveals that large entropy losses accompany nucleotide binding to E208Q-BsSecA and WT-EcSecA.
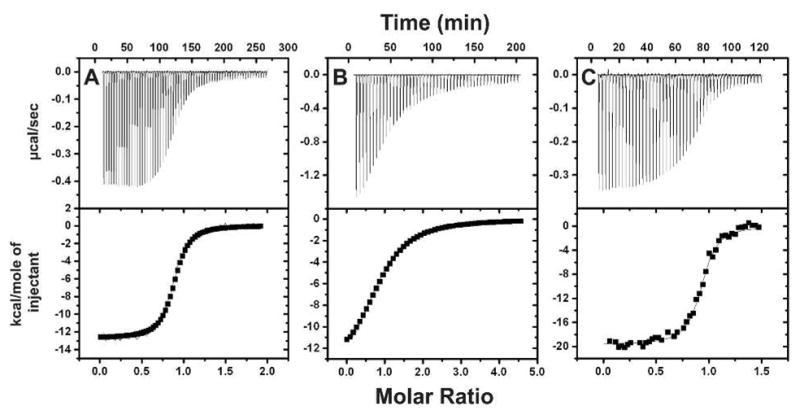
ITC experiments were performed at 25 °C in TKM buffer (pH 8.0). Mg-ADP was titrated onto 20 μM WT-BsSecA (panel A), 30 μM E208Q-BsSecA (panel B), or 10 μM WT-EcSecA (panel C). Table 1 gives the thermodynamic parameters inferred from curve-fitting these data.
The detailed thermodynamic data generated by the ITC experiments (Fig. 6 and Table 1) demonstrate that there is a much higher entropy loss during binding of Mg-ADP to E208Q-BsSecA compared to the WT enzyme at 25 °C (ΔS° of −24 vs. −13 cal mol−1 K−1), while there is a comparatively small change in binding enthalpy (−14 vs. −13 kcal mol−1). Thus, the E208Q mutant modestly increases the enthalpy gain upon binding, perhaps due to reduced electrostatic repulsion of the phosphates of ADP, while it doubles the unfavorable entropy of binding. The most straightforward explanation for this entropic effect, which accounts for the strong reduction in binding affinity produced by the E208Q mutation in BsSecA, is that the mutant enzyme is more disordered than the WT enzyme in the apo conformation but not the Mg-ADP-bound conformation. This explanation is consistent with the inference made above based on the temperature dependence of Mg-MANT-ADP-binding affinity that the E208Q mutation facilitates the ECT, causing it to be underway at 25 °C in the mutant enzyme but not the WT enzyme. The conformation produced by this transition, which greatly reduces Mg-ADP-binding affinity, is believed to be more dynamic than the low-temperature ground-state conformation 12,26. Therefore, because reversal of the ECT must precede Mg-ADP binding, the entropy loss involved in reversing the transition is coupled to Mg-ADP binding to E208Q-BsSecA. This entropy loss accounts for the lower net affinity of binding to E208Q-BsSecA compared to WT BsSecA, because the WT enzyme is in the conformational ground state at 25 °C and the ECT does not have to be reversed before it can bind Mg-ADP at this temperature.
The ITC experiments demonstrate that Mg-ADP binding to WT-EcSecA at 25 °C involves a substantially larger entropy loss (−34 cal mol−1 ° K−1) and a substantially larger enthalpy gain (−20 kcal mol−1) than binding to WT or even E208Q BsSecA (Fig. 6 and Table 1). These large entropy and enthalpy changes almost entirely compensate for one another, because the net binding affinities of the two enzymes differ by only ~10-fold (Table 1). The ITC experiments were conducted at a temperature well below the onset of the ECT in the apo form of either WT enzyme (see below), so the high entropic cost of Mg-ADP binding to EcSecA cannot come from coupling to the reversal of the ECT. It must reflect some other molecular process involving a reduction in dynamics, either a reduction in protein dynamics in the low-temperature ground-state conformation of the enzyme and/or a change in solvent dynamics upon nucleotide binding. Both processes typically involve strong enthalpy-entropy compensation of the kind observed in comparing Mg-ADP-binding to EcSecA vs. BsSecA. The process responsible for the much larger entropy loss upon Mg-ADP binding to EcSecA must be either absent or greatly reduced in magnitude in BsSecA, making it unlikely to be an essential feature of the mechanochemistry of the enzyme. NMR studies have suggested that helicase motifs V and VI in the ATPase active site have elevated dynamics in apo EcSecA 26,61, and immobilization of these segments during Mg-ADP binding to EcSecA could potentially account for the larger entropy loss observed during binding to this enzyme compared to BsSecA (Table 1). However, data presented below demonstrate that both of these segments adopt predominantly folded conformations in apo EcSecA at 25 °C on the timescale of amide hydrogen-deuterium exchange, making it unlikely that a simple unfolding transition in motifs V or VI is responsible for the much larger entropy loss upon Mg-ADP binding to EcSecA vs. BsSecA.
The E208Q mutation in BsSecA destabilizes the ECT by 13 °C
We used thermal titrations monitored by intrinsic tryptophan fluorescence spectroscopy 12 (Fig. S5) and differential scanning calorimetry (DSC – Figs. 7 & S6) to verify the hypothesis that the E-to-Q mutation facilitates the ECT in BsSecA. Previous research 23,24 demonstrated that WT-BsSecA undergoes two endothermic transitions between 20 °C and 60 °C. The first, with a midpoint of 40 °C, represents the ECT and is fully reversible, while the second, with a midpoint at 49 °C, involves the irreversible unfolding of some protein domain, probably NBF-I 23,24. Addition of Mg-ADP leads to a progressive increase in the temperature of the first transition due to inhibition of the ECT by nucleotide binding 23 (i.e., to 47 °C in the presence of sufficient Mg-ADP to saturate the active site). Our DSC data on WT-BsSecA recapitulate these earlier observations (Fig. 7A,B). Strikingly, as predicted based on the temperature-dependence of Mg-MANT-ADP-binding affinity, DSC analysis shows that the ECT is shifted to lower temperature in E208Q-BsSecA (Fig. 7C). The observed 13° downshift to 27 °C reflects a dramatic alteration in the global conformational properties of the apo enzyme in this temperature range based on a single isosteric point-mutation in the active site. The second endothermic transition in BsSecA is also shifted to lower temperature by ~8°, consistent with the previous interpretation that this transition involves unfolding of NBF-I (i.e., the domain containing the E208Q mutation). Addition of Mg-ADP to E208Q-BsSecA results in an increase of 5°C in the Tm of the first transition (i.e., shifting the ECT to 32 °C) as well as an increase in its enthalpy (Fig. 7D). However, both of these changes are substantially attenuated compared to those observed upon Mg-ADP binding to the WT enzyme, consistent with the data presented above showing that the E-to-Q mutation strongly perturbs nucleotide interactions at the high-affinity ATPase site in BsSecA. The Tm of the second transition in E208Q-BsSecA is essentially unchanged by the addition of Mg-ADP; this observation provides further evidence that nucleotide release effectively accompanies the ECT, because residual nucleotide binding to the Walker motifs in NBF-I after the occurrence of the ECT would be expected to inhibit its unfolding during the second transition and thereby increase the Tm of this transition.
Fig 7. Differential scanning calorimetry (DSC) of WT and E-to-Q BsSecA and EcSecA.
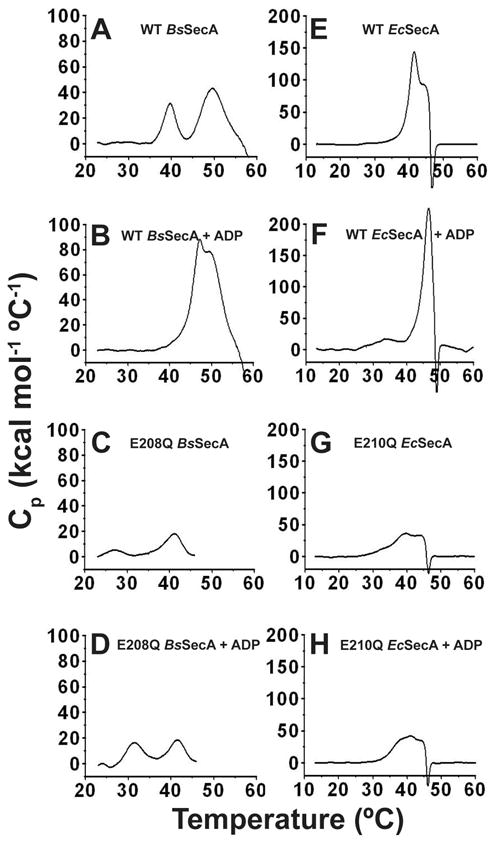
All DSC scans were conducted on the same instrument at 1 °C per minute in TKM buffer (pH 8.0). The same vertical scale is used here for all experiments conducted on enzymes from the same organism, while Fig. S6 shows the same data replotted with the vertical scale adjusted to enable clear visualization of the details of the transitions in each experiment. (A,B) 10 μM WT-BsSecA without (panel A) or with (panel B) 1 mM Mg-ADP. (C,D) 20 μM E208Q-BsSecA without (panel C) or with (panel D) 1 mM Mg-ADP. (E,F) 10 μM WT-EcSecA without (panel E) or with (panel F) 1 mM Mg-ADP. (G,H) 20 μM E210Q-EcSecA without (panel G) or with (panel H) 1 mM Mg-ADP. Protein aggregation following the second conformational transition, which is probably unfolding of NBF-I, likely contributes to the very large magnitude of Cp in some experiments. This phenomenon prevents reliable integration of enthalpy. However, it does not interfere with observation of the temperature and approximate magnitude of the initial transition corresponding to the ECT. The data shown here for WT BsSecA accurately recapitulates previously published results 23,24.
DSC analysis of EcSecA in the absence and presence of the E210Q mutation shows qualitatively similar trends (Fig. 7E-H). The mutation reduces the Tm of its ECT and substantially attenuates the increase in the Tm and enthalpy of this transition upon Mg-ADP binding (Fig. 7G-H vs. 7E-F). Notably, the single endothermic transition in WT-EcSecA is split into at least two sequential transitions by the E210Q mutation. This split makes the calorimetric behavior of E210Q-EcSecA more similar to that of WT BsSecA, although a broad shoulder on the low-temperature side of the first endothermic transition in E210Q-EcSecA indicates that several sequential endothermic sub-transitions may contribute to the ECT in this mutant enzyme. Critically, the onset of the earliest thermal transition in apo E210Q-EcSecA occurs at ~30 °C (Fig. 7G) compared to ~20 °C in apo E208Q-BsSecA (Fig. 7C). Therefore, while the E-to-Q mutation facilitates the ECT in both enzymes, it occurs at a significantly lower temperature when the mutation is introduced into BsSecA compared to EcSecA. This quantitative difference explains the substantially more severe impairment in the affinity of E208Q-BsSecA vs. E210Q-EcSecA in the initial nucleotide-binding experiments reported above, which were conducted at 25 °C. At this temperature, the ECT is in progress in E208Q-BsSecA but not in E210Q-EcSecA. Therefore, the qualitative effects of the E-to-Q mutation, which are fundamentally similar in both enzymes, were masked by the quantitative differences in their thermodynamics properties. The next section presents further evidence supporting the similarity of the functional conformational changes and the qualitative effects of the E-to-Q mutation in the catalytic base in both enzymes.
Hydrogen-deuterium exchange experiments to map the effect of the ECT on BsSecA dynamics
The DSC experiments reported above provide direct evidence showing that the E-to-Q mutation in the catalytic base of SecA perturbs the thermodynamics of the ECT. The observation that a single isosteric point-mutation triggers the ECT in the apo enzyme at 25 °C provided a unique opportunity to use hydrogen-deuterium exchange (HDX) measurements at a single temperature to characterize the global conformational changes in SecA during this transition that is likely to be the principal nucleotide-controlled process mediating polypeptide transport. HDX provides information on protein dynamics by quantitatively measuring the rate of exchange of backbone and sidechain amide hydrogen atoms for deuterium atoms from water 62,63. Because chemical exchange requires exposure of the amide group to solvent, the method is sensitive to conformational fluctuations breaking backbone hydrogen bonds (e.g., in regular protein secondary structures). Backbone HDX proceeds via two mechanisms, “EX1” involving transient conformational fluctuations exposing the backbone amides to solvent and “EX2” involving an enhanced population of a locally or globally unfolded protein conformation 62,63. An increase in HDX rate via either mechanism requires an increase in net backbone dynamics because even the EX2 mechanism involves substantial fractional residency in an unfolded conformation on the millisecond timescale of the HDX process. Therefore, we used mass spectrometric HDX measurements 63 to probe the conformational state of the BsSecA variants at 10 and 100 seconds at 25 °C, where the WT enzyme is in the ground-state conformation but the E208Q enzyme is undergoing the ECT. The data from these experiments are presented in Fig. 8A-B and Supporting Information file Kim-SecA-HDX-Data. xls, while the key conclusions are summarized schematically in Fig. 9.
Figure 8. Summary of results from hydrogen-deuterium exchange mass spectrometry (HDX-MS) experiments.
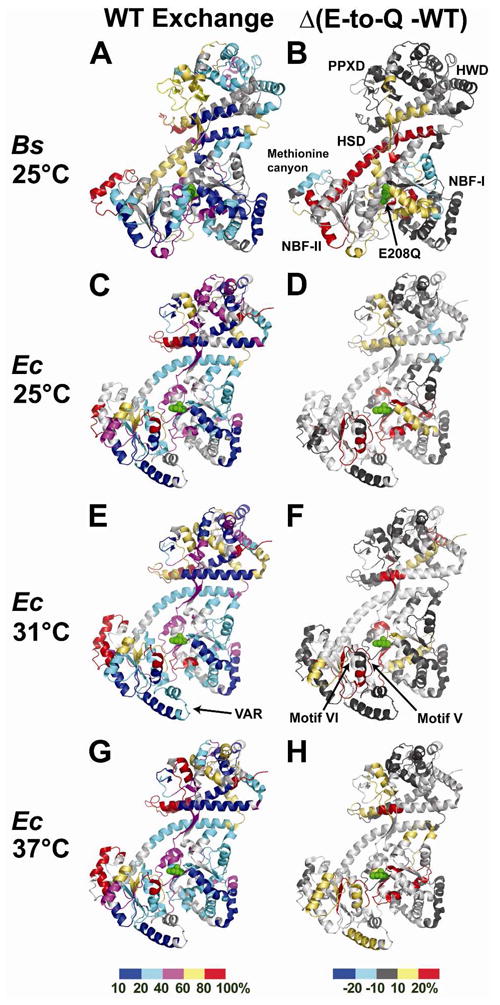
Average peptide 1H/2H exchange levels at the 10 second time point in TKM buffer (pH 8.0) are plotted onto ribbon diagrams of the corresponding WT enzymes (PDB id 1M74 for BsSecA and PDB id 2FSG for EcSecA). The catalytic glutamates are shown in green sphere representation. The panels on the left (A,C,E,G) show the absolute percent exchange of observable protons in peptides in the WT enzyme, while the panels on the right (B,D,F,H) show the difference in this value in the E-to-Q mutant minus that in the corresponding WT enzyme at the same time point; these latter images highlight the regions in which backbone dynamics are altered by the mutation. The exchange levels are color-coded as indicated in the bar at the bottom of each column (10–20% exchange in blue, 21–40% in cyan, 41–60% in magenta, 61–80% in yellow, and 81–100% in red for the absolute exchange levels shown in the left column, and ≥ 21% reduction in exchange in blue, 20–11% reduction in cyan, 10% reduction to 10% increase in dark gray, 11–20% increase in yellow, and ≥ 21% increase in red for the differences in exchange levels shown in the right column). Experiments were conducted on BsSecA at 25 °C (panels A and B) where the DSC data (Fig. 7A,C) indicate that the WT enzyme is in the conformational ground state but the E208Q mutant is undergoing the ECT. Experiments were conducted on EcSecA at 25 °C (panels C and D), 31 °C (panels E and F), and 37 °C (panels G and H). The DSC data on WT-EcSecA (Fig. 7E) indicate that this enzyme is in the conformational ground-state at all three temperatures, while the fine-structure in the broad DSC peak for the E210Q mutant (Fig. 7G) suggests that it sequentially occupies different conformational sub-states as it undergoes the ECT from 30–40 °C.
Figure 9. Schematic diagram summarizing conformational changes during the ECT and related features of the HDX data.
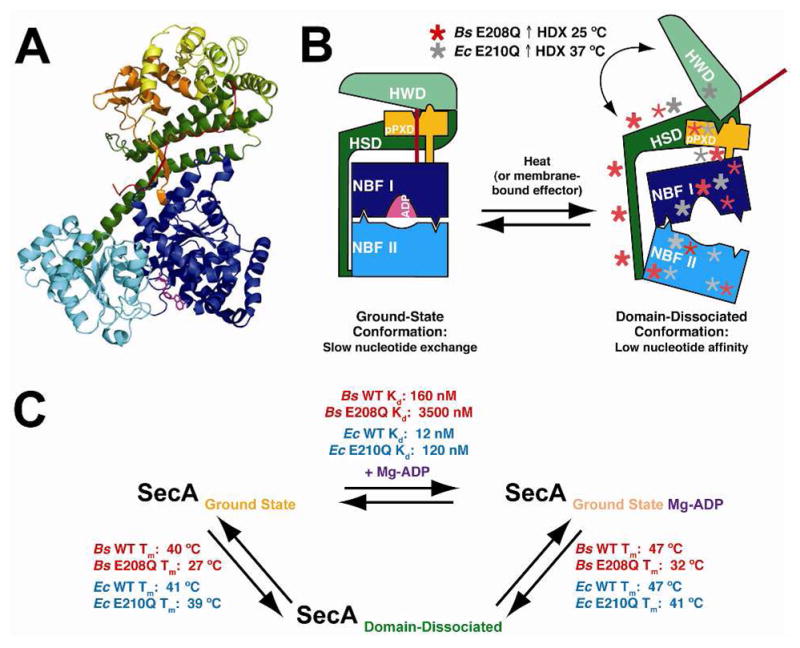
(A) A ribbon diagram of the BsSecA crystal structure (1M6N) bound to Mg-ADP (magenta) shows each domain colored separately to correspond to the labeled domains in panel B. (B) Schematic diagram showing key features of the endothermic conformational transition (ECT) between the ground state and the domain-dissociated state of SecA. The domain-dissociated state is marked with asterisks representing regions of increased mobility as measured by HDX in SecA mutants compared to the corresponding WT enzymes. Increases in BsSecA E208Q HDX are depicted in red, and increases in EcSecA E210Q HDX are shown in grey. Bold asterisks represent regions with large changes in HDX rates. (C) Schematic diagram showing equilibria between the major conformational states of SecA along with associated thermodynamic parameters.
In the absence of nucleotide, the WT protein shows modestly elevated amide proton exchange rates in the HWD and in local segments of the NBFs, more elevated dynamics in the PPXD and in the long central α-helix in the HSD, and highly elevated dynamics in the methionine canyon in NBF-II (Fig. 8A). The E208Q mutation strongly modulates amide proton exchange rates at sites throughout the protein, especially at interdomain interfaces (Figs. 8A-B & 9A-B). The most dramatic change involves strong acceleration of HDX rate throughout the long central α-helix in the HSD, indicating substantially greater backbone solvent exposure in this segment that serves as a physical scaffold organizing the interdomain interactions in the conformational ground-state of SecA. As explained above, this observation indicates that the E208Q mutation increases backbone dynamics in this protein segment, either via enhanced conformational fluctuation or enhanced population of an unfolded conformation. The mutation produces similarly strong enhancements in HDX rates in some segments near the mutation site in NBF-I, i.e., in one β-strand connecting this domain to the PPXD, and in the first α-helix in NBF-II, which lies on the opposite surface of this domain from the ATP-binding site. The mutation also modestly enhances HDX rates elsewhere in the HSD and in the interface between NBF-I and NBF-II. Therefore, the HDX data verify inferences from earlier mutational 12,49,64 and NMR 26 analyses that the ECT involves a global conformational change increasing the solvent-exposure and net backbone dynamics of interdomain interfaces throughout SecA, as summarized in Fig. 9. The observation that this transition is induced by an isosteric glutamine-to-glutamate mutation demonstrates an exquisitely sensitive coupling of active-site electrostatics 34 to global protein conformation.
While the E208Q mutation accelerates HDX rate in most regions of SecA, it modestly reduces the rate in the methionine canyon and on the distal surface of NBF-I relative to the ATPase active site. The observation of increased order at these sites suggest that they may interact with protein binding partners or other ligands specifically in the conformational state produced by the ECT. The coupling of interactions stabilizing local structure at these sites to a global conformational change generally increasing disorder elsewhere would enable such interactions to trigger the ECT and thereby coordinate the efficient progression of the conformational reaction cycle during the polypeptide transport reaction.
Equivalent dynamic changes in E210Q-EcSecA at slightly higher temperature
Qualitatively similar results to those obtained with BsSecA are observed in HDX experiments performed on EcSecA (Fig. 8C–H), suggesting that the fundamental conformational phenomena underlying SecA function are conserved between species even if the energetic consequences of the E-to-Q mutation in the catalytic base are slightly different. At 25 °C, 31 °C, and 37 °C, WT-EcSecA shows a similar overall HDX pattern as WT-BsSecA at 25 °C, although segments in the PPXD and HWD show some acceleration in HDX rate as temperature is raised (Fig. 8C vs. 8E vs. 8G). These observations are consistent with the protein adopting an equivalent conformation until the temperature nears the onset of the ECT, which has a midpoint of ~41 °C in EcSecA (Fig. 7E).
Although a prior NMR study showed enhanced mobility of the residues in helicase motifs V and VI in EcSecA in the absence of nucleotide 26,61, the HDX data support both of these motifs adopting stably folded conformations at 25 °C on the timescale of the amide-exchange process (Fig. 8C and Supporting Information file Kim-SecA-HDX-Data. xls). Two peptides are observed that include motif V (residues 501-512 65), one covering residues 500-512 and the other covering residues 502-512. These peptides respectively show ~60–80% exchange at 10 seconds and ~80–100% exchange at 100 seconds, which is similar to the levels observed for the equivalent peptide in BsSecA at 25 °C (50–60% at 10 seconds, 60–80% at 100 seconds). However, motif V adopts an extended conformation in which most residues do not participate in backbone H-bonds, so only a modest degree of protection from HDX is expected even in the folded structure. Two peptides are also observed that include motif VI (residues 563-579 65), one covering residues 550-586 and the other covering residues 572-586. The longer peptide, which includes the entire motif, shows ~25% exchange at 10 seconds and ~30% exchange at 100 seconds, while the shorter peptide shows ~35% exchange at 10 seconds and ~45% exchange at 100 seconds. These observed levels of exchange correspond roughly to the fraction of residues in these segments that do not participate in regular secondary structures and therefore would be expected to exchange even in the fully folded conformation. These HDX observations suggest that neither helicase motif V nor VI adopts a predominantly disordered conformation at 25 °C in WT EcSecA, although they do not exclude the possibility of transient unfolding in a minor fraction of the population.
HDX rates in E210Q-EcSecA show progressively greater changes compared to the WT protein as temperature is raised from 25°C (Fig. 8D) to 37 °C (Fig. 8H), consistent with the strong reduction in its nucleotide-binding affinity (Fig. 5B) and the onset of the ECT (Fig. 7G) in this temperature range in the mutant protein. While HDX data were not obtained from E210Q-EcSecA for the long central α-helix in the HSD, enhanced exchange rates are observed in the regions of the structure with which it interacts, with stronger effects being observed at 31 °C (Fig. 8F) and 37 °C (Fig. 8H) where the protein is undergoing the ECT. These data demonstrate that net backbone dynamics are enhanced at the higher temperatures at widely distributed sites in E210Q-EcSecA, including some residues in the cores of the HSD, NBF-I, and NBF-II. The overall pattern of the observed changes is similar to that observed in the HDX experiments on E208Q-BsSecA (Fig. 8B), as schematized in Fig. 9. Therefore, the E-to-Q mutation in the catalytic base facilitates the ECT in both enzymes, but it does so at a higher temperature in EcSecA compared to BsSecA.
In comparing apo E210Q-EcSecA to the WT enzyme, evidence is observed of local increases in HDX in some regions of the protein while other segments involved in the ECT remain relatively unperturbed. These observations suggest that the broad shoulder at 25–35 °C in DSC experiments on the mutant enzyme (Fig. 7G) may reflect a cascade of localized changes in protein dynamics sequentially affecting different regions of the mutant enzyme, rather than a simple two-state conformational transition. For example, segments of NBF-I proximal to the mutation site as well as motif V and an adjacent segment of NBF-II all show significant increases in HDX rates in the mutant enzyme compared to the WT enzyme even at 25 °C (Fig. 8D), well prior to the midpoint of the ECT in the mutant enzyme at ~39 °C (as monitored by DSC in Fig. 7G). In contrast, some regions in the PPXD and NBF-II that show enhanced exchange at the outset of the ECT at 37 °C in the mutant enzyme (Fig. 8H), are unperturbed at the intermediate temperature of 31 °C (Fig. 8F). These include one of the α-helices in the VAR segment 66 (shown at the very bottom of the structure in Fig. 8C-H). This segment contains a pair of α-helices that packs on the surface of NBF-II in SecA orthologs from Gram-negative eubacteria like E. coli, but it is absent in orthologs from Gram-positive eubacteria like B. subtilis. Notably, this segment exchanges more slowly than any other observed region of NBF-II in EcSecA (Fig. 8C,E,G), consistent with the proposal that it allosterically stabilizes the interactions between NBF-I and NBF-II that control nucleotide release, and it could therefore contribute to the lesser effect of the E-to-Q mutation in the catalytic base of EcSecA vs. BsSecA at 25 °C. While the HDX data on NBF-II shows that portions of its core as well as the VAR segment remain predominantly folded in E210Q-EcSecA at 37 °C, the coverage of NBF-II is incomplete, preventing assessment of the dynamics of many segments including helicase motif VI (which was not observed in any of the HDX experiments conducted on E210Q-EcSecA). Therefore, future experiments will be required to elucidate the conformational details accounting for the broad shoulder or pre-transition at 25–35 °C in DSC experiments on apo E210Q-EcSecA (Fig. 7G) and to determine whether it reflects sequential conformational transitions increasing the flexibility of local protein segments. However, even if it does, the HDX data in Fig. 8 show that the overall conformational state produced by these sequential transitions is very similar to that produced by the ostensibly discrete ECT in BsSecA.
Discussion
The thermodynamic studies reported in this paper substantially enhance understanding of the physicochemical factors controlling the function of a large mechanoenzyme mediating an exceedingly complex molecular process. These studies demonstrate that altering a single electrostatic charge in the ATPase active site of SecA can trigger a global conformational change in this 100 kDa mechanoenzyme. With more than 250 charged amino acid residues in SecA, the network of electrostatic interactions in the enzyme must be exquisitely balanced 26,27, and this balance is likely to play a central role in coupling transient electrostatic changes during ATP hydrolysis in the active site to the global conformational changes that drive processive polypeptide transport 33,34. These conformational changes alter cooperative atomic interactions between different domains 12,13,26 and possibly also between structural modules within individual domains (e.g., helicase motifs V/VI and the methionine canyon in NBF-II). The thermodynamic data presented in this paper show a substantial increase in entropy during the principal functional conformational transition in SecA (i.e., the ECT), while the HDX data support the interpretation that this increase derives from an enhancement in net backbone dynamics in many of its domains/modules. Thus, the conformational reaction cycle of SecA harnesses both the electrostatic and statistical properties of the protein polymer to orchestrate the efficient progression of an extremely complex physical process that is tightly coupled to the binding and hydrolysis of ATP.
The glutamate-to-glutamine mutation in the catalytic base emulates its change in electrostatic state during the initial step in the ATPase reaction in which this glutamate abstracts a proton from the hydrolytic water molecule to initiate a nucleophilic attack on the γ-phosphate of ATP 33,34. This proton-transfer event induces cleavage of the phosphoanhydride bond, which is rapidly followed by conformational rearrangement of the released inorganic phosphate and ADP in the active site. Subsequent events, including proton release from the sidechain of the catalytic glutamate and release of the cleaved inorganic phosphate from the active site, are likely to involve significant changes in electrostatic and other energetic interactions in the active site. The conformational effects of the E-to-Q mutation demonstrate that there is exquisitely sensitive coupling of the global conformational state of SecA to local electrostatic interactions at the catalytic center 33,34, and they are likely to reflect at least in part the influence on global protein conformation of the initial proton-abstraction event during ATP hydrolysis. Because electrostatic interactions can propagate through a protein structure very rapidly, the conformational couplings of the catalytic glutamate elucidated in our experiments can help drive SecA through the proper global conformational trajectory contemporaneously during the ATP hydrolysis reaction.
The HDX data in Fig. 8 demonstrate that the ECT involves a substantial increase in net backbone dynamics in widely distributed regions of SecA, and this increase is likely to be an important driving force for this critical functional conformational transition. For a reaction to be endothermic, it must involve an increase in enthalpy (i.e., input of heat) as well as entropy, because changing ΔG = ΔH − T·ΔS from positive to negative values by increasing temperature mathematically requires both ΔH > 0 and ΔS > 0. The widespread increase in conformational entropy, inferred from coordinated thermodynamic and HDX experiments on both BsSecA and EcSecA (Fig. 8), is likely to make a critical contribution to the entropy gain driving the ECT.
The delocalized nature of these changes is noteworthy because order-to-disorder transitions in helicase motifs V and VI have previously been hypothesized to be the entropic driver for the ECT 26. Our HDX data (Fig. 8) suggest that the entropic driver instead involves broadly distributed increases in protein dynamics throughout the multidomain protein structure, including most interdomain interfaces in SecA. This inference is supported by the observation that the E210Q mutation in EcSecA produces a significant increase in HDX rate and thus net backbone dynamics near both the mutation site in NBF-I and near helicase motif V in NBF-II even at 25 °C (Fig. 8C vs. 8D), which is well below the temperature of the ECT in E210Q-EcSecA. In the absence of Mg-ADP, the ECT in this protein variant has an apparent midpoint at ~39 °C following a broad pre-transition from 25–35 °C as monitored by DSC (Fig. 7G). Dynamic transitions already in progress at 25 °C in this protein variant are unlikely to represent the major entropic driver for a conformational transition centered at 39 °C. Therefore, the globally distributed increases in net backbone dynamics observed in the HDX studies of both BsSecA and EcSecA represent a more likely candidate to be the entropic driver controlling the progression of the ECT.
Nonetheless, the totality of our experimental data suggests that a number of distinct conformational transitions involving alterations in protein dynamics in different regions of SecA contribute to controlling the efficient progression of its conformational reaction cycle. It seems likely that some of these transitions can proceed independently, while others have varying degrees of thermodynamic interdependency. For example, the broad shoulder preceding the ECT in DSC experiments on E210Q-EcSecA (Fig. 7G) suggests that this mutant enzyme undergoes one or more endothermic conformational sub-transitions preceding the global change in interdomain interactions that occurs during the ECT (Fig. 8); presumably residue E210 in EcSecA strengthens the thermodynamic linkage between these sequential conformational transitions in the WT enzyme. Another example is provided by our earlier study characterizing the nucleotide-interaction properties of EcSecA 13, which demonstrated that different mutations have uncorrelated effects on Mg-ADP-release rate and the ECT. Therefore, the ECT must differ in at least some details from the conformational transition controlling Mg-ADP release. This latter transition could conceivably involve an order-to-disorder transition in helicase motifs V and VI uncoupled from global conformational changes 61; this possibility is supported by the observation that the E210Q mutation in EcSecA greatly accelerates Mg-MANT-ADP-release rate (Fig. 3) while also increasing the dynamics of helicase motif V (Fig. 8C vs. 8D) at 25 °C, well below the onset of the ECT in this enzyme (Fig. 7G). Notably, motifs V and VI both exhibit very high B-factors in crystal structures of some homologous helicases 67,68. However, the large entropy-enthalpy compensation that accompanies Mg-ADP binding to WT-EcSecA compared to WT-BsSecA at 25 °C (Table 1) is likely to derive from a dynamic transition involving different protein segments or alternatively dynamics on a faster timescale than that accessible to HDX measurements, because our data support both of these segments adopting a folded conformation in apo EcSecA at this temperature on the timescale of the HDX process. While future studies will be required to evaluate the specific structural hypotheses advanced here, the available thermodynamic and dynamic data provide strong evidence that SecA undergoes a variety of distinct conformational transitions that are likely to involve changes in protein dynamics.
Conclusions
Although the ECT strongly increases net protein dynamics at many sites, the HDX data suggest that it simultaneously involves a reduction in protein dynamics at specific sites (e.g., the methionine canyon in Fig. 8A vs. 8B), and these sites could participate in stabilizing structural interactions with ligands or other proteins that guide the progression of SecA’s conformational reaction cycle. In this manner, interacting structural modules undergoing sequential conformational transitions could control the progression of a large enzyme like SecA through a sequence of different conformational states each optimized to make different molecular contacts. A molecular trajectory of this kind, controlled by an exquisitely balanced network of electrostatic interactions connecting to the ATPase active site, offers clear advantages for efficient coupling of ATP binding and hydrolysis to a complex molecular process like processive extrusion of a polypeptide chain through an interacting channel. Having a cascade of distinct dynamic transitions underlie such a sequence of conformational changes would enable the inherent plasticity in protein structure, and the diverse physicochemical factors underlying this plasticity, to be exploited for efficient execution of a remarkably complex molecular process. Future research will be required to critically evaluate these mechanistic hypotheses that emerge from the integrated biophysical studies of SecA presented in the current paper. Please see the SI for further discussion.
Supplementary Material
Acknowledgments
The authors thank Y. Gelis and C. Kalodimos of Rutgers University and E. Folta-Stogniew of the Keck Biotechnology Resource Center at Yale University for use of ITC instruments as well as J. Maki and L. Gierasch for use of their DSC instrument. We are grateful to these individuals and C. Brouillette, V. Frasca, F. Forouhar, and P. Lobel for advice on data analysis and interpretation.
Funding Sources
This work was supported by NIH grants R01-GM58549 and R01-GM072867 (to JFH) and grant U54 GM094597 (to GTM and JFH). DK was supported by NIH Training Grant GM08281-17 and by the Departments of Biological Sciences and Biochemistry and Molecular Biophysics at Columbia University.
Footnotes
Supporting Information Statement
An expanded discussion, the crystallographic data table, and Figures S1–S5 are provided, as well as a file containing processed HDX data. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Devillers-Thiery A, Kindt T, Scheele G, Blobel G. Proc Natl Acad Sci U S A. 1975;72:5016. doi: 10.1073/pnas.72.12.5016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Oliver DB, Beckwith J. Journal of bacteriology. 1982;150:686. doi: 10.1128/jb.150.2.686-691.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bieker-Brady K, Silhavy TJ. EMBO J. 1992;11:3165. doi: 10.1002/j.1460-2075.1992.tb05393.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Huie JL, Silhavy TJ. Journal of bacteriology. 1995;177:3518. doi: 10.1128/jb.177.12.3518-3526.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Brundage L, Hendrick JP, Schiebel E, Driessen AJ, Wickner W. Cell. 1990;62:649. doi: 10.1016/0092-8674(90)90111-q. [DOI] [PubMed] [Google Scholar]
- 6.Driessen AJ. EMBO J. 1992;11:847. doi: 10.1002/j.1460-2075.1992.tb05122.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hanada M, Nishiyama KI, Mizushima S, Tokuda H. J Biol Chem. 1994;269:23625. [PubMed] [Google Scholar]
- 8.Eichler J, Brunner J, Wickner W. EMBO J. 1997;16:2188. doi: 10.1093/emboj/16.9.2188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Eichler J, Wickner W. Proc Natl Acad Sci U S A. 1997;94:5574. doi: 10.1073/pnas.94.11.5574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Schiebel E, Driessen AJ, Hartl FU, Wickner W. Cell. 1991;64:927. doi: 10.1016/0092-8674(91)90317-r. [DOI] [PubMed] [Google Scholar]
- 11.van der Wolk JP, de Wit JG, Driessen AJ. EMBO J. 1997;16:7297. doi: 10.1093/emboj/16.24.7297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hunt JF, Weinkauf S, Henry L, Fak JJ, McNicholas P, Oliver DB, Deisenhofer J. Science. 2002;297:2018. doi: 10.1126/science.1074424. [DOI] [PubMed] [Google Scholar]
- 13.Fak JJ, Itkin A, Ciobanu DD, Lin EC, Song XJ, Chou YT, Gierasch LM, Hunt JF. Biochemistry. 2004;43:7307. doi: 10.1021/bi0357208. [DOI] [PubMed] [Google Scholar]
- 14.Velankar SS, Soultanas P, Dillingham MS, Subramanya HS, Wigley DB. Cell. 1999;97:75. doi: 10.1016/s0092-8674(00)80716-3. [DOI] [PubMed] [Google Scholar]
- 15.Leslie AG, Abrahams JP, Braig K, Lutter R, Menz RI, Orriss GL, van Raaij MJ, Walker JE. Biochemical Society transactions. 1999;27:37. doi: 10.1042/bst0270037. [DOI] [PubMed] [Google Scholar]
- 16.Sharma V, Arockiasamy A, Ronning DR, Savva CG, Holzenburg A, Braunstein M, Jacobs WR, Jr, Sacchettini JC. Proc Natl Acad Sci U S A. 2003;100:2243. doi: 10.1073/pnas.0538077100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cavanaugh LF, Palmer AG, 3rd, Gierasch LM, Hunt JF. Nature structural & molecular biology. 2006;13:566. doi: 10.1038/nsmb0706-566. [DOI] [PubMed] [Google Scholar]
- 18.Economou A, Wickner W. Cell. 1994;78:835. doi: 10.1016/s0092-8674(94)90582-7. [DOI] [PubMed] [Google Scholar]
- 19.Rajapandi T, Oliver D. Biochem Biophys Res Commun. 1994;200:1477. doi: 10.1006/bbrc.1994.1617. [DOI] [PubMed] [Google Scholar]
- 20.Driessen AJ. Trends Biochem Sci. 1992;17:219. doi: 10.1016/0968-0004(92)90381-i. [DOI] [PubMed] [Google Scholar]
- 21.Or E, Boyd D, Gon S, Beckwith J, Rapoport T. J Biol Chem. 2005;280:9097. doi: 10.1074/jbc.M413947200. [DOI] [PubMed] [Google Scholar]
- 22.Ulbrandt ND, London E, Oliver DB. J Biol Chem. 1992;267:15184. [PubMed] [Google Scholar]
- 23.den Blaauwen T, Fekkes P, de Wit JG, Kuiper W, Driessen AJ. Biochemistry. 1996;35:11994. doi: 10.1021/bi9605088. [DOI] [PubMed] [Google Scholar]
- 24.den Blaauwen T, van der Wolk JP, van der Does C, van Wely KH, Driessen AJ. FEBS Lett. 1999;458:145. doi: 10.1016/s0014-5793(99)01139-4. [DOI] [PubMed] [Google Scholar]
- 25.Schmidt M, Ding H, Ramamurthy V, Mukerji I, Oliver D. J Biol Chem. 2000;275:15440. doi: 10.1074/jbc.M000605200. [DOI] [PubMed] [Google Scholar]
- 26.Keramisanou D, Biris N, Gelis I, Sianidis G, Karamanou S, Economou A, Kalodimos CG. Nature structural & molecular biology. 2006;13:594. doi: 10.1038/nsmb1108. [DOI] [PubMed] [Google Scholar]
- 27.Karamanou S, Gouridis G, Papanikou E, Sianidis G, Gelis I, Keramisanou D, Vrontou E, Kalodimos CG, Economou A. EMBO J. 2007;26:2904. doi: 10.1038/sj.emboj.7601721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Vrontou E, Karamanou S, Baud C, Sianidis G, Economou A. J Biol Chem. 2004;279:22490. doi: 10.1074/jbc.M401008200. [DOI] [PubMed] [Google Scholar]
- 29.Osborne AR, Clemons WM, Jr, Rapoport TA. Proc Natl Acad Sci U S A. 2004;101:10937. doi: 10.1073/pnas.0401742101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Vassylyev DG, Mori H, Vassylyeva MN, Tsukazaki T, Kimura Y, Tahirov TH, Ito K. J Mol Biol. 2006;364:248. doi: 10.1016/j.jmb.2006.09.061. [DOI] [PubMed] [Google Scholar]
- 31.Zimmer J, Li W, Rapoport TA. J Mol Biol. 2006;364:259. doi: 10.1016/j.jmb.2006.08.044. [DOI] [PubMed] [Google Scholar]
- 32.Zimmer J, Nam Y, Rapoport TA. Nature. 2008;455:936. doi: 10.1038/nature07335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jencks WP. Catalysis in chemistry and enzymology. Vol. 40. McGraw-Hill; New York: 1969. pp. 111–115. [Google Scholar]
- 34.Jencks WP. Advances in enzymology and related areas of molecular biology. 1980;51:75. doi: 10.1002/9780470122969.ch2. [DOI] [PubMed] [Google Scholar]
- 35.Story RM, Li H, Abelson JN. Proc Natl Acad Sci U S A. 2001;98:1465. doi: 10.1073/pnas.98.4.1465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Caruthers JM, McKay DB. Curr Opin Struct Biol. 2002;12:123. doi: 10.1016/s0959-440x(02)00298-1. [DOI] [PubMed] [Google Scholar]
- 37.Singleton MR, Wigley DB. Journal of bacteriology. 2002;184:1819. doi: 10.1128/JB.184.7.1819-1826.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.de la Cruz J, Kressler D, Linder P. Trends Biochem Sci. 1999;24:192. doi: 10.1016/s0968-0004(99)01376-6. [DOI] [PubMed] [Google Scholar]
- 39.Linder P, Lasko PF, Ashburner M, Leroy P, Nielsen PJ, Nishi K, Schnier J, Slonimski PP. Nature. 1989;337:121. doi: 10.1038/337121a0. [DOI] [PubMed] [Google Scholar]
- 40.Koonin EV, Gorbalenya AE. FEBS Lett. 1992;298:6. doi: 10.1016/0014-5793(92)80009-6. [DOI] [PubMed] [Google Scholar]
- 41.Zito CR, Antony E, Hunt JF, Oliver DB, Hingorani MM. J Biol Chem. 2005;280:14611. doi: 10.1074/jbc.M414224200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Abrahams JP, Leslie AG, Lutter R, Walker JE. Nature. 1994;370:621. doi: 10.1038/370621a0. [DOI] [PubMed] [Google Scholar]
- 43.Smith PC, Karpowich N, Millen L, Moody JE, Rosen J, Thomas PJ, Hunt JF. Mol Cell. 2002;10:139. doi: 10.1016/s1097-2765(02)00576-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Moody JE, Millen L, Binns D, Hunt JF, Thomas PJ. J Biol Chem. 2002;277:21111. doi: 10.1074/jbc.C200228200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chen J, Lu G, Lin J, Davidson AL, Quiocho FA. Mol Cell. 2003;12:651. doi: 10.1016/j.molcel.2003.08.004. [DOI] [PubMed] [Google Scholar]
- 46.Khare D, Oldham ML, Orelle C, Davidson AL, Chen J. Mol Cell. 2009;33:528. doi: 10.1016/j.molcel.2009.01.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Walker JE, Saraste M, Runswick MJ, Gay NJ. EMBO J. 1982;1:945. doi: 10.1002/j.1460-2075.1982.tb01276.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ramamurthy V, Oliver D. J Biol Chem. 1997;272:23239. doi: 10.1074/jbc.272.37.23239. [DOI] [PubMed] [Google Scholar]
- 49.Ding H, Mukerji I, Oliver D. Biochemistry. 2001;40:1835. doi: 10.1021/bi002058w. [DOI] [PubMed] [Google Scholar]
- 50.van der Does C, Manting EH, Kaufmann A, Lutz M, Driessen AJ. Biochemistry. 1998;37:201. doi: 10.1021/bi972105t. [DOI] [PubMed] [Google Scholar]
- 51.Economou A, Pogliano JA, Beckwith J, Oliver DB, Wickner W. Cell. 1995;83:1171. doi: 10.1016/0092-8674(95)90143-4. [DOI] [PubMed] [Google Scholar]
- 52.Weber J, Senior AE. Biochim Biophys Acta. 1997;1319:19. doi: 10.1016/s0005-2728(96)00121-1. [DOI] [PubMed] [Google Scholar]
- 53.Shimo-Kon R, Muneyuki E, Sakai H, Adachi K, Yoshida M, Kinosita K., Jr Biophys J. 98:1227. doi: 10.1016/j.bpj.2009.11.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Rye HS, Burston SG, Fenton WA, Beechem JM, Xu Z, Sigler PB, Horwich AL. Nature. 1997;388:792. doi: 10.1038/42047. [DOI] [PubMed] [Google Scholar]
- 55.Roseman AM, Chen S, White H, Braig K, Saibil HR. Cell. 1996;87:241. doi: 10.1016/s0092-8674(00)81342-2. [DOI] [PubMed] [Google Scholar]
- 56.Yuan YR, Blecker S, Martsinkevich O, Millen L, Thomas PJ, Hunt JF. J Biol Chem. 2001;276:32313. doi: 10.1074/jbc.M100758200. [DOI] [PubMed] [Google Scholar]
- 57.Sharma S, Zheng H, Huang YJ, Ertekin A, Hamuro Y, Rossi P, Tejero R, Acton TB, Xiao R, Jiang M, Zhao L, Ma LC, Swapna GV, Aramini JM, Montelione GT. Proteins. 2009 [Google Scholar]
- 58.Sianidis G, Karamanou S, Vrontou E, Boulias K, Repanas K, Kyrpides N, Politou AS, Economou A. EMBO J. 2001;20:961. doi: 10.1093/emboj/20.5.961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Natale P, Swaving J, van der Does C, de Keyzer J, Driessen AJ. J Biol Chem. 2004;279:13769. doi: 10.1074/jbc.M312892200. [DOI] [PubMed] [Google Scholar]
- 60.Bennett BD, Kimball EH, Gao M, Osterhout R, Van Dien SJ, Rabinowitz JD. Nature chemical biology. 2009;5:593. doi: 10.1038/nchembio.186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Chou YT, Swain JF, Gierasch LM. J Biol Chem. 2002;277:50985. doi: 10.1074/jbc.M209237200. [DOI] [PubMed] [Google Scholar]
- 62.Bai Y, Englander JJ, Mayne L, Milne JS, Englander SW. Methods Enzymol. 1995;259:344. doi: 10.1016/0076-6879(95)59051-x. [DOI] [PubMed] [Google Scholar]
- 63.Englander JJ, Del Mar C, Li W, Englander SW, Kim JS, Stranz DD, Hamuro Y, Woods VL., Jr Proc Natl Acad Sci U S A. 2003;100:7057. doi: 10.1073/pnas.1232301100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ding H, Mukerji I, Oliver D. Biochemistry. 2003;42:13468. doi: 10.1021/bi035099b. [DOI] [PubMed] [Google Scholar]
- 65.Papanikou E, Karamanou S, Economou A. Nature reviews Microbiology. 2007;5:839. doi: 10.1038/nrmicro1771. [DOI] [PubMed] [Google Scholar]
- 66.Das S, Grady LM, Michtavy J, Zhou Y, Cohan FM, Hingorani MM, Oliver DB. Journal of bacteriology. 2012;194:2205. doi: 10.1128/JB.00039-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Caruthers JM, Johnson ER, McKay DB. Proc Natl Acad Sci U S A. 2000;97:13080. doi: 10.1073/pnas.97.24.13080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Liu H, Rudolf J, Johnson KA, McMahon SA, Oke M, Carter L, McRobbie AM, Brown SE, Naismith JH, White MF. Cell. 2008;133:801. doi: 10.1016/j.cell.2008.04.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.