

Abstract
Mixed-lineage kinase 3 (MLK3) activates mitogen-activated protein kinase (MAPK) signaling pathways and has important functions in migration, invasion, proliferation, tumorigenesis, and apoptosis. We investigated the role of the E3 ligase carboxyl terminus of Hsc70-interacting protein (CHIP) in the regulation of MLK3 protein levels. We show that CHIP interacts with MLK3 and, together with the E2 ubiquitin-conjugating enzyme UbcH5 (UbcH5a, -b, -c, or -d), ubiquitinates MLK3 in vitro. CHIP or Hsp70 overexpression promoted endogenous MLK3 ubiquitination and induced a decline in MLK3 protein levels in cells with Hsp90 inhibition. Furthermore, CHIP overexpression caused a proteasome-dependent reduction in exogenous MLK3 protein. Geldanamycin (GA), heat shock, and osmotic shock treatments also reduced the level of MLK3 protein via a CHIP-dependent mechanism. In addition, CHIP depletion in ovarian cancer SKOV3 cells increased cell invasion, and the enhancement of invasiveness was abrogated by small interfering RNA (siRNA)-mediated knockdown of MLK3. Thus, CHIP modulates MLK3 protein levels in response to GA and stress stimuli, and CHIP-dependent regulation of MLK3 is required for suppression of SKOV3 ovarian cancer cell invasion.
INTRODUCTION
Mixed-lineage kinase 3 (MLK3) is a mitogen-activated protein kinase (MAPK) kinase kinase (MAP3K) that has regulatory roles in diverse biological processes such as proliferation, migration, differentiation, invasion, and neuronal cell apoptosis (1–9). MLK3 activates the c-Jun N-terminal kinase (JNK) and p38 MAPK signaling pathways, and in certain cellular contexts, MLK3 has a kinase-independent function in the activation of extracellular signal-regulated kinase (ERK) MAPK signaling (1, 10–12). Aberrant levels of total and/or activated MLK3 protein have been observed in ovarian and breast cancer cells in comparison to nonneoplastic ovarian and breast epithelial cells (5, 6). Moreover, a critical function for MLK3 in breast and ovarian cancer cell invasion was recently identified (5, 6). The regulation of MLK3 by phosphorylation, dimerization, and interaction with Rho GTPases has been the focus of numerous studies; however, the mechanism(s) that controls the level of total MLK3 protein in cells remains poorly understood (13–16).
We previously identified that in SKOV3 ovarian cancer cells, the proinflammatory cytokines tumor necrosis factor alpha (TNF-α) and interleukin-1β (IL-1β) induced the activation of MLK3 and that TNF-α also stimulated MLK3 ubiquitination (17). TNF-α-dependent activation of MLK3 is facilitated by TNF receptor-associated factor 2 (TRAF2) and TRAF6, which interact with MLK3 and are recruited to the TNF receptor (17, 18). Recently, it was demonstrated that TRAF6 promotes K63-linked polyubiquitination of MLK3 in vitro (19). Furthermore, MLK3 activation in response to IL-1β is dependent on TRAF6 binding and K63-linked polyubiquitination (17, 19). MLK3 also undergoes K48-linked polyubiquitination; however, the role of this modification in MLK3 protein function and turnover has not been elucidated (17).
MLK3 interacts with the chaperone protein Hsp90 and the cochaperone protein p50cdc37, which participate in the folding and stabilization of signaling proteins involved in proliferation, apoptosis, and survival (20, 21). The dissociation of Hsp90 from its client proteins can be triggered by specific stimuli or by exposure to Hsp90 ATPase inhibitors such as geldanamycin (GA) (22, 23). Treatment of cancer cell lines with GA causes a reduction in the level of endogenous MLK3 protein and an inhibition of JNK signaling, which suggests that the Hsp90-MLK3 interaction is important for MLK3 function and stability (21, 24). The disruption of Hsp90 chaperone-client interactions can lead to ubiquitination and proteasomal degradation of the client proteins via Hsp70-dependent recruitment of the carboxyl terminus of Hsc70-interacting protein (CHIP) E3 ubiquitin ligase (20, 22, 23). CHIP is a Ubox E3 protein that mediates cytosolic protein polyubiquitination and targets Hsp70-bound proteins for degradation by the 26S proteasome, thereby coupling the chaperone and ubiquitin-proteasome systems (25–28). In this study, we investigated the role of CHIP in the ubiquitination and degradation of MLK3 in response to GA, heat shock, and osmotic shock. We show that GA, heat shock, and osmotic shock promoted a decline in the level of MLK3 protein that is dependent on CHIP. Furthermore, CHIP mediates MLK3 polyubiquitination and proteasome-dependent degradation, and CHIP-dependent regulation of MLK3 is required for suppression of SKOV3 ovarian cancer cell invasion.
MATERIALS AND METHODS
Cell culture.
Human SKOV3 and TOV21G ovarian cancer and human embryonic kidney (HEK293) cells were purchased from the American Type Culture Collection (ATCC) (Manassas, VA). HEY1B ovarian cancer cells were a gift from Douglas Leaman (The University of Toledo). SKOV3, HEY1B, and HEK293 cells were cultured at 37°C in Dulbecco's modified Eagle's medium (DMEM) (Mediatech, Herndon, VA), which contained 10% fetal bovine serum (FBS) (HyClone, Logan, UT). TOV21G cells were cultured in medium 199 (Mediatech, Inc.) with 10% MCDB 105 (Sigma-Aldrich, St. Louis, MO) and 15% FBS. All tissue culture media were supplemented with 2 mM l-glutamine, 25 μg/ml streptomycin, and 25 IU penicillin (Mediatech, Inc.). Cells were cultured in a humidified atmosphere with 5% CO2 at 37°C.
Plasmid and siRNA transfections.
The following mammalian expression constructs for expression of human CHIP, MLK3, Hsp70, and Hsp90 were used in this study: pCMV-FLAG-CHIP, pCMV-FLAG-MLK3, pCMV-GST-MLK3, pCMV-GFP-Hsp70, pCMV-HA-Hsp90, and pCMV-His-MLK3. The constructs that were used for the expression of human MLK3 and CHIP in Escherichia coli were pCMV-His-MLK3 and p-CMV-GST-CHIP. Transient transfections were performed by using PolyJet (SignaGen Laboratories, Rockville, MD), and small interfering RNA (siRNA) knockdown was performed by using Lipofectamine 2000 (Invitrogen, Carlsbad, CA) or GeneMute (SignaGen Laboratories) according to the manufacturer's instructions. The nontarget and human CHIP siRNAs were obtained from Santa Cruz Technologies (Santa Cruz, CA). The human MLK3 siRNA was previously described (1).
Cell treatments.
Cells were either left untreated, treated with vehicle, or treated with a final concentration of 10 μM GA (InvivoGen, San Diego, CA), 250 mM sorbitol, 50 μM cycloheximide (Thermo Fisher Scientific, Rockford, IL), 20 ng/ml TNF-α (Life Technologies, Grand Island, NY), or 10 μM MG132 (Selleckchem, Houston, TX) for the indicated time periods. For heat shock treatments, cells were cultured at 37°C (control) or 42°C for the indicated time periods. The cells were harvested immediately after the treatments, and whole-cell extracts were prepared.
Quantitative real-time PCR.
Total RNA was prepared from cells by using TRIzol reagent (Invitrogen) according to the manufacturer's instructions. Two micrograms of RNA was reverse transcribed into cDNA, and real-time PCR (RT-PCR) was performed with Ssofast EvaGreen Supermix (Bio-Rad Laboratories, Hercules, CA). The MLK3 and β-actin primers used for RT-PCR were described previously (5). Statistical analysis was performed by using Student's t test, and a P value of <0.05 was considered statistically significant.
Recombinant MLK3 and CHIP protein purification from bacterial and mammalian cells.
Mammalian His-CHIP, glutathione S-transferase (GST)–CHIP, and His-MLK3 plasmids were expressed in E. coli BL21 cells. The cells were lysed in bacterial protein extraction reagent (B-PER) (Thermo Fisher Scientific) with a complement of protease inhibitors (1 μM leupeptin, 1 μM aprotinin, 1 mM benzamidine, and 1 mM phenylmethylsulfonyl fluoride [PMSF]). Cell lysates were subjected to His pulldown with Ni-nitrilotriacetic acid (NTA)-agarose or GST pulldown with glutathione (GSH)-agarose beads. The GST fusion or His-tagged proteins were eluted from the agarose beads with 10 mM glutathione (GST fusion proteins) or 0.5 M imidazole (His-tagged proteins) and stored at −20°C.
His-MLK3 was purified from HEK293 cells for in vitro ubiquitination assays. Cells transfected with His-MLK3 were lysed in immunoprecipitation (IP) lysis buffer (50 mM Tris-HCl [pH 7.5], 1% Triton X-100, 150 mM NaCl, 0.1% 2-mercaptoethanol, 50 mM NaF, 1 mM Na3VO4, 1 μM leupeptin, 1 μM aprotinin, 1 mM PMSF, and 1 mM benzamidine), and His-MLK3 was purified on Ni-NTA-agarose beads and eluted off the beads with 0.5 M imidazole.
CHIP/MLK3 in vitro binding assay.
Equal quantities of His-MLK3 and GST-CHIP (purified from E. coli BL21) were incubated together in IP lysis buffer, and GST pulldown assays were performed. His-MLK3 and GST-CHIP protein levels were assessed via immunoblotting with anti-CHIP and anti-MLK3 antibodies.
Immunoprecipitations and immunoblotting.
HEK293 cells were transfected with appropriate plasmids to express human MLK3, Hsp70, Hsp90, or CHIP. At 24 h posttransfection, the cells were lysed in IP lysis buffer. FLAG immunoprecipitations or GST pulldowns were performed and subjected to immunoblotting with anti-GST and anti-FLAG antibodies (Agilent, Santa Clara, CA). Antibodies against MLK3 (C-20), GST (Z-5), JNK1 (C-17), Hsp90 (H-114), Raf-1 (C-20), B-Raf (F-3), β-actin (C-4), and CHIP (H-231) were obtained from Santa Cruz Biotechnology (Santa Cruz, CA). The activation state phosphorylated JNK (p-JNK) (Thr183/Tyr185), phosphorylated MLK3 (p-MLK3) (Thr277/Ser281), phosphorylated ERK (p-ERK) (Thr202/Tyr204), and phosphorylated p38 (p-p38) (Thr180/Tyr182) antibodies were obtained from Cell Signaling Technology (Beverly, MA). The K63 ubiquitin (D7A11) and K48 ubiquitin (D9D5) antibodies were also obtained from Cell Signaling Technology. Other antibodies that were used in this study include Hsp70 (W-27; Thermo Scientific, Freemont, CA), green fluorescent protein (GFP; Novus Biologicals, Littleton, CO), and antiubiquitin (BD Biosciences, San Jose, CA) antibodies. Densitometric analysis of immunoblots was performed using Image J software (National Institutes of Health).
Analysis of endogenous MLK3 ubiquitination.
SKOV3 cells were treated with 10 μM GA for different time intervals and then harvested in IP lysis buffer. N-Ethylmaleimide was added to the IP lysis buffer at a final concentration of 10 mM just prior to cell lysis. Immunoprecipitations were performed with anti-K48 ubiquitin or anti-K63 ubiquitin antibodies, and the immunoprecipitates were immunoblotted with an anti-MLK3 antibody. To determine the effect of CHIP or Hsp70 overexpression on endogenous MLK3 ubiquitination, HEK293 cells were transfected with FLAG-CHIP or GFP-Hsp70, and 24 h later, the cells were treated with 5 μM GA or dimethyl sulfoxide (DMSO) for 1 h. Endogenous ubiquitinated MLK3 was immunoprecipitated from cell lysates (prepared with IP lysis buffer as described above) with antiubiquitin antibody, and the immunoprecipitates were immunoblotted with anti-MLK3 antibody.
In vitro ubiquitination assay.
Specific E2s from an E2 screening kit (UBPBio, Aurora, CO) were incubated together with purified human His-CHIP, ubiquitin, E1, and His-MLK3 in ATP ubiquitination buffer according to the manufacturer's instructions. The reactions were carried out for 1 h at 30°C and stopped by the addition of 1× SDS sample buffer to the mixtures.
Invasion assays.
SKOV3 cell invasion assays were performed with Transwell membranes coated with Matrigel as described previously (5). Cells (2.5 × 105) were seeded onto the upper chamber and incubated for 16 h. The invaded cells were stained, and the number of cells per field of view was counted by using an inverted light microscope. The average of 6 fields of view was obtained per experiment. Data are presented as means, with n representing 4 independent experiments. Comparison of 2 groups was performed by using Student's t test. A P value of <0.05 was considered statistically significant.
RESULTS
GA enhances MLK3 K48-linked ubiquitination and reduces the level of endogenous MLK3 protein.
GA treatment results in a decline in the level of endogenous MLK3 protein in MCF-7 breast cancer cells, which we postulated might be due to ubiquitin/proteasome-mediated degradation of MLK3 (21). To determine whether GA also elicits a decline in MLK3 levels in ovarian cancer cells, SKOV3 ovarian cancer cells were treated with GA, and MLK3 protein levels were analyzed by immunoblotting. In SKOV3 cells treated with GA for 6 h, the amount of MLK3 protein was reduced to 53% of the amount in untreated cells (Fig. 1A). A further decline in the amount of MLK3 protein was observed after 8 and 10 h, and by 12 h, the amount of MLK3 protein was reduced to 15% of the amount in untreated cells (Fig. 1A). The level of phosphorylated, active MLK3 in cytoplasmic extracts was also reduced by GA treatment (see Fig. S1 in the supplemental material). GA induces Hsp70 expression through the heat shock response and has a minimal effect on Hsp90 protein (29–31). Consistent with this, SKOV3 cells treated with GA for 4 to 12 h had a higher level of Hsp70 protein than did untreated cells, whereas Hsp90 protein levels were not affected (Fig. 1A). Furthermore, GA treatment reduced the level of active, phosphorylated ERK (p-ERK) and active, phosphorylated JNK (p-JNK). The level of activated, phosphorylated p38 (p-p38) was increased by 6 h and 8 h and then declined after 10 h and 12 h of GA treatment. The total levels of ERK, JNK, and p38 or β-actin were not affected by GA treatment (Fig. 1A). Because B-Raf and Raf-1 MAP3Ks are also major regulators of ERK signaling, the effect of GA on the level of these proteins was also analyzed. It was previously demonstrated that wild-type (WT) B-Raf does not require Hsp90 for stability and is unaffected by Hsp90 inhibition (32). In contrast, the B-Raf V600E mutant and Raf-1 require Hsp90 for stability, and GA treatment reduces the levels of these proteins in cells (32, 33). As expected, in SKOV3 cells which express wild-type B-Raf, GA did not elicit a reduction in the level of B-Raf protein (Fig. 1A) (34). The level of Raf-1, also an Hsp90 client, declined only after 10 h of GA treatment (Fig. 1A). A similar GA-mediated reduction in MLK3 protein levels was observed in HEY1B and TOV21G ovarian cancer cell lines, which express wild-type B-Raf (see Fig. S2A in the supplemental material) (34). Taken together, these results indicate that the GA-dependent decline in MLK3 protein levels is closely correlated with an increase in Hsp70 protein levels and a deactivation of ERK and JNK signaling.
FIG 1.

GA stimulates MLK3 ubiquitination and reduces the level of endogenous MLK3 protein. (A) SKOV3 cells were treated with GA (10 μM) for the indicated time periods, and whole-cell extracts were immunoblotted with the indicated antibodies. The MLK3 band intensity for each time point was normalized to the band intensity for β-actin and expressed as a percentage of the value for untreated cells. (B) SKOV3 cells were treated as described above for panel A. MLK3 mRNA transcript levels were analyzed by quantitative RT-PCR, normalized to the β-actin transcript levels, and expressed as a percentage of the value for control, untreated cells. The values represent the means ± standard deviations (n = 3). No statistically significant differences in mRNA levels were observed between the GA-treated and untreated cells. A P value of <0.05 was considered statistically significant. (C) SKOV3 cells were treated with cycloheximide (CHX) (50 μM) alone or cycloheximide (50 μM) and GA (10 μM) for the indicated time periods. The MLK3 band intensity was determined for each time point as described above for panel A. (D, top) Endogenous MLK3 was immunoprecipitated (IP) from cell lysates with anti-K63 ubiquitin (K63-Ub) or anti-K48 ubiquitin (K48-Ub) antibodies, and the immunoprecipitates were immunoblotted (IB) with anti-MLK3 antibody. (Bottom) The cell lysates were also immunoblotted with anti-MLK3 and anti-β-actin antibodies. (E) HEK293 cells were treated with vehicle or MG132 (10 μM) for the indicated time periods. MLK3 and β-actin protein levels were assessed by immunoblot analysis with anti-MLK3 and anti-β-actin antibodies. (F) SKOV3 cells were untreated or treated with MG132 (10 μM) for 2 h and then treated with GA (10 μM) for 6 h. Immunoblotting of cell extracts was performed as described for panel E.
GA treatment reduced MLK3 protein levels but did not reduce the level of MLK3 mRNA in SKOV3, HEY1B, and TOV21G cells (Fig. 1B; see also Fig. S2B in the supplemental material). Furthermore, the MLK3 protein half-life was analyzed by using the protein synthesis inhibitor cycloheximide, and it was observed that after a 10-h cycloheximide treatment, the MLK3 protein level was reduced to 52% of the amount present in untreated SKOV3 cells (Fig. 1C, left). In comparison, SKOV3 cells treated with both cycloheximide and GA had only 5% of MLK3 protein remaining after 10 h (Fig. 1C, right). These results indicate that GA substantially reduces the half-life of MLK3 protein in SKOV3 cells.
We speculated that if GA treatment could induce ubiquitin/proteasome-dependent degradation of MLK3, then elevated levels of ubiquitinated MLK3 should be evident in GA-treated cells. To test this, K48- or K63-linked ubiquitinated proteins were immunoprecipitated from untreated and GA-treated SKOV3 cells, and the immunoprecipitates were subjected to immunoblotting with anti-MLK3 antibody. MLK3 conjugated with K48-linked ubiquitin was detected in the immunoprecipitations (Fig. 1D, right), whereas minimal K63-linked ubiquitinated MLK3 was detected (Fig. 1D, left). The level of K48-linked ubiquitinated MLK3 was highest in cells treated with GA for 6 h and 8 h, and this correlated with a decline in MLK3 total protein levels in the cell lysates (Fig. 1D, right and bottom). These results support the hypothesis that GA promotes the degradation of MLK3 protein via a mechanism that involves K48-linked ubiquitination.
A common fate for K48-linked ubiquitinated proteins is degradation by the 26S proteasome (28). To determine whether the degradation of endogenous MLK3 protein is proteasome dependent, HEK293 cells were treated with the proteasome inhibitor MG132, and MLK3 protein was analyzed in cell extracts by immunoblotting. MG132 treatments for 2 and 4 h resulted in elevated endogenous MLK3 protein levels in comparison to those in untreated cells or cells treated with MG132 for 1 h (Fig. 1E). These results suggest that proteasomal degradation is a major mechanism by which the level of endogenous MLK3 protein is regulated. Furthermore, the GA-mediated reduction of MLK3 protein levels was blocked by MG132 treatment in SKOV3, HEY1B, and TOV21G cells, which indicates that GA-induced degradation of MLK3 is also proteasome dependent (Fig. 1F; see also Fig. S2 in the supplemental material).
Heat shock or osmotic shock causes a decline in the level of endogenous MLK3 protein.
To determine whether stress stimuli could also cause a reduction in the level of endogenous MLK3 protein, SKOV3 cells were exposed to heat shock or osmotic shock (sorbitol), and MLK3 protein levels were analyzed by immunoblotting. A reduced level of endogenous MLK3 protein but not MLK3 mRNA was observed in cells exposed to heat shock (2 to 10 h) or osmotic shock (6 to 10 h) in comparison to untreated cells (Fig. 2A to C). After a 2-h exposure to heat stress, the MLK3 protein level was reduced to 30% of the amount present in untreated cells and declined further after 4, 6, 8, and 10 h (Fig. 2A). SKOV3 cells exposed to osmotic shock for 6 h or 8 h had MLK3 protein levels that were 24% and 8% of the amount observed in untreated cells, respectively. Heat and osmotic stresses had minimal effects on B-Raf and Raf1 protein levels (Fig. 2A and B). The Hsp70 protein level was elevated after 2 h of exposure to osmotic or heat stresses and increased further over the 10-h time courses (Fig. 2A and B). Both heat shock and sorbitol treatments resulted in substantial reductions in the levels of p-ERK, p-JNK, and p-p38 but not total ERK, JNK, p38, or β-actin (Fig. 2A and B). In comparison to untreated cells, SKOV3 cells treated with cycloheximide had 73% of the MLK3 protein remaining after a 6-h treatment (Fig. 2D, left); however, cells treated with cycloheximide and sorbitol for 6 h (Fig. 2D, middle) or with cycloheximide and heat shock for 6 h (Fig. 2D, right) had only 16% and 1% of the total MLK3 protein remaining, respectively. These novel findings indicate that heat shock and osmotic shock induce Hsp70 protein expression, reduce the half-life of MLK3 protein, and inactivate ERK, JNK, and p38 MAPK signaling. MLK3 activates both JNK and p38 signaling pathways and is critical for sorbitol-induced activation of JNK but not for heat shock-mediated activation of JNK (see Fig. S3 in the supplemental material). Possibly, other MAP3Ks function in activating JNK in response to heat shock in ovarian cancer cells. Interestingly, treatment of SKOV3 cells with TNF-α, a stress stimulus that activates MLK3, did not result in a decline of MLK3 protein or an induction of Hsp70 protein levels. This result indicates that TNF-dependent regulation of MLK3 occurs via a mechanism that is different from those of the other stimuli tested, which all trigger MLK3 degradation (see Fig. S4 in the supplemental material).
FIG 2.

Osmotic and heat stresses reduce the level of endogenous MLK3 protein. (A and B) SKOV3 cells were exposed to heat shock (42°C) (A) or 0.25 M sorbitol (B) for the indicated time periods, and whole-cell extracts were immunoblotted with the indicated antibodies. (C) SKOV3 cells were exposed to heat shock or sorbitol as described above for panels A and B. The MLK3 mRNA transcript levels were analyzed by quantitative RT-PCR, normalized to β-actin transcript levels, and expressed as a percentage of the levels in control, untreated cells. The values represent the means ± standard deviations (n = 3). No statistically significant differences in mRNA levels were observed between the untreated and heat shock- or sorbitol-treated cells. A P value of <0.05 was considered statistically significant. (D) SKOV3 cells were treated with cycloheximide (CHX) (left), cycloheximide plus heat shock (right), or cycloheximide plus sorbitol (middle) for the indicated time periods. Whole-cell extracts were analyzed by immunoblotting with the indicated antibodies. The MLK3 band intensity for each time point was normalized to the band intensity for β-actin and expressed as a percentage of the value for untreated cells.
MLK3 interacts with Hsp70.
GA and heat shock both activate the heat shock response and induce Hsp70 transcription (35). Hsp70 binds to damaged proteins and recruits E3 ligases that promote protein ubiquitination and degradation (36). We postulated that stress or GA-induced destabilization of MLK3 could promote an interaction between Hsp70 and MLK3, which might lead to MLK3 degradation via an Hsp70-coupled E3 ligase. The MLK3-Hsp90 interaction was previously characterized; however, an interaction between Hsp70 and MLK3 has not been demonstrated (21). To test whether MLK3 interacts with Hsp70, GFP-Hsp70 was overexpressed together with FLAG-MLK3 in HEK293 cells. The cells were treated with DMSO or GA for 3 h, and FLAG-MLK3 immunoprecipitates were immunoblotted with anti-GFP antibody. GFP-Hsp70 was detected in FLAG-MLK3 immunoprecipitates in both the DMSO- and the GA-treated cells, which confirms the presence of MLK3-Hsp70 complexes in HEK293 cells (Fig. 3A). The dynamics of the formation of endogenous Hsp70-MLK3 and Hsp90-MLK3 complexes were investigated in GA-treated cells. In untreated SKOV3 cells, endogenous MLK3 was coimmunoprecipitated with endogenous Hsp90 (Fig. 3B). The MLK3-Hsp90 interaction was lost in cells treated with GA for time intervals ranging from 2 to 8 h (Fig. 3B), even though total Hsp90 protein levels remained constant throughout the GA time course (Fig. 3B). In contrast, Hsp70 was detected in the MLK3 immunoprecipitates in cells treated with GA for 4 h, 6 h, and 8 h, and this was accompanied by an increase in the level of Hsp70 and a decline in the level of MLK3 protein in the cell lysates (Fig. 3B). These results strongly suggest that GA disrupts the Hsp90-MLK3 interaction and that the subsequent binding of Hsp70 to MLK3 leads to a decline in MLK3 protein levels.
FIG 3.
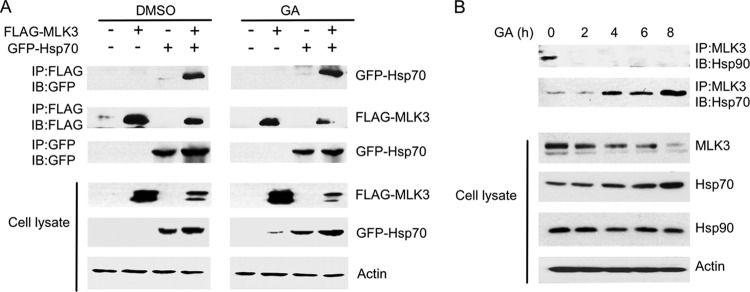
GA stimulates an association between MLK3 and Hsp70. (A) HEK293 cells were transfected with the indicated combinations of FLAG-MLK3 and GFP-Hsp70 WT plasmids. The cells were treated with DMSO or GA (5 μM) for 3 h. FLAG-MLK3 immunoprecipitates were immunoblotted with anti-GFP and anti-FLAG antibodies. GFP-Hsp70 immunoprecipitates were also immunoblotted with anti-GFP antibody. Whole-cell extracts were immunoblotted with anti-FLAG, anti-GFP, and anti-β-actin antibodies. (B) SKOV3 cells were treated with GA (10 μM) for the indicated time periods, and endogenous MLK3 immunoprecipitates were immunoblotted with anti-Hsp90 and anti-Hsp70 antibodies. Whole-cell extracts were also immunoblotted with anti-MLK3, anti-Hsp70, anti-Hsp90, and anti-β-actin antibodies, as indicated.
CHIP interacts with MLK3.
The E3 ligase(s) involved in ubiquitination and targeting of MLK3 for proteasomal degradation has not been identified. We reasoned that CHIP is a good candidate E3 ligase for this process because CHIP couples the chaperone and proteasome systems and facilitates ubiquitination of stress-activated proteins (28, 37). To determine whether CHIP associates with MLK3 in cells, GST-MLK3 and FLAG-CHIP were co-overexpressed in HEK293 cells. GST pulldowns were performed and immunoblotted with anti-FLAG antibody. FLAG-CHIP was present in GST-MLK3 pulldowns, which indicates that FLAG-CHIP is associated with GST-MLK3 in HEK293 cells (Fig. 4A). The level of GST-MLK3 was lower in cells that had coexpressed FLAG-CHIP, which is in agreement with a function for CHIP in promoting MLK3 degradation (Fig. 4A).
FIG 4.

CHIP interacts with MLK3 and promotes a proteasome-dependent decline in MLK3 protein levels. (A) HEK293 cells were transfected with combinations of GST-MLK3 and FLAG-CHIP plasmids and treated with GA for 3 h, as indicated. The GST-MLK3 pulldowns and the whole-cell extracts were immunoblotted with anti-FLAG and anti-GST antibodies. (B) Purified, recombinant His-MLK3 and GST-CHIP proteins were combined in an in vitro binding assay. (Left) GST pulldowns were performed and subjected to immunoblotting with anti-MLK3 and anti-CHIP antibodies. (Right) The GST-CHIP and His-MLK3 protein preparations and whole-cell extract (WCE) were immunoblotted with anti-Hsp70, anti-GST, or anti-His antibodies. (C) HEK293 cells were transfected with FLAG-CHIP and/or GST-MLK3 and treated with vehicle (left) or MG132 (10 μM) (right) for 3 h. GST-MLK3 and FLAG-CHIP protein levels were assessed by immunoblot analysis with anti-GST and anti-FLAG antibodies.
To determine if MLK3 interacts directly with CHIP, recombinant His-MLK3 and GST-CHIP purified from E. coli were incubated together in lysis buffer, and GST pulldowns were performed. His-MLK3 was detected only in the GST pulldowns that contained GST-CHIP (Fig. 4B, left). Because it is possible that the presence of Hsp70 in the purified CHIP and MLK3 protein preparations serves to bridge the interaction between CHIP and MLK3, immunoblotting for Hsp70 was performed with samples of each preparation. Hsp70 protein was not detected in the His-MLK3 or GST-CHIP preparations (Fig. 4B, right). These results suggest that MLK3 interacts directly with CHIP in vitro.
Next, we wished to determine whether CHIP overexpression could lead to a proteasome-dependent reduction in MLK3 protein levels. FLAG-CHIP and GST-MLK3 were co-overexpressed in HEK293 cells. The cells were either untreated or treated with MG132, and the level of GST-MLK3 protein in cell extracts was analyzed by immunoblotting. Cells with FLAG-CHIP overexpression had a dramatically reduced level of GST-MLK3. However, FLAG-CHIP had a minimal effect on the GST-MLK3 protein in cells that were pretreated with MG132 (Fig. 4C, right). These results indicate that CHIP modulates the level of exogenous MLK3 by a mechanism that involves proteasome-dependent degradation.
CHIP/Hsp70 complexes promote ubiquitination and proteasome-dependent degradation of protein substrates (27, 37, 38). Thus, the effect of CHIP and Hsp70 on endogenous MLK3 ubiquitination and protein levels was analyzed. FLAG-CHIP and/or GFP-Hsp70 WT overexpression had a minimal effect on the level of endogenous total or ubiquitinated MLK3 protein in HEK293 cells (Fig. 5A). We speculated that MLK3 binding to Hsp90 might stabilize MLK3 and prevent its ubiquitination by CHIP. To test this possibility, the dissociation of endogenous MLK3 from Hsp90 was induced by pretreatment of cells with GA for 1 h. In the GA-treated cells, FLAG-CHIP and/or GFP-Hsp70 overexpression induced MLK3 ubiquitination (Fig. 5A, bottom) and reduced the level of MLK3 protein (Fig. 5A, top). These results confirm that CHIP and Hsp70 promote MLK3 ubiquitination and modulate the level of endogenous MLK3 protein. We postulated that CHIP-induced degradation of exogenous MLK3 occurs because there is insufficient endogenous Hsp90 to stabilize the exogenous MLK3. To investigate this possibility, hemagglutinin (HA)-Hsp90 was overexpressed with FLAG-MLK3 and FLAG-CHIP or GFP-Hsp70. Indeed, the FLAG-CHIP- or GFP-Hsp70-induced degradation of MLK3 was blocked by elevating the level of Hsp90 (Fig. 5B and C).
FIG 5.

Hsp90 inhibits the CHIP- and Hsp70-dependent decline in MLK3 protein levels. (A) HEK293 cells were transfected with the indicated combinations of GFP-Hsp70 and FLAG-CHIP plasmids and treated with vehicle or GA (5 μM) for 1 h. (Top) Whole-cell extracts were immunoblotted with anti-MLK3, anti-β-actin, anti-FLAG, and anti-GFP antibodies. (Bottom) Immunoprecipitations were performed with antiubiquitin antibody, and ubiquitinated MLK3 was detected by immunoblot analysis with anti-MLK3 antibody. (B) Cell extracts from HEK293 cells expressing combinations of FLAG-MLK3, HA-Hsp90, and GFP-Hsp70 were immunoblotted with anti-FLAG, anti-GFP, anti-HA, and anti-β-actin antibodies. (C) Cell extracts from HEK293 cells expressing combinations of FLAG-MLK3, HA-Hsp90, and FLAG-CHIP were immunoblotted as described above for panel B.
CHIP promotes ubiquitination of MLK3.
Next, we wished to test if CHIP could facilitate ubiquitination of MLK3 in vitro. Ubiquitination assays were performed with purified His-CHIP and His-MLK3 together with E1, E2 (UbcH5a or -b), and ubiquitin (Fig. 6A and B). The E2s UbcH5a and UbcH5b were selected for the in vitro reactions because they function with CHIP to mediate polyubquitination of other substrates (39, 40). In vitro ubiquitination of His-MLK3 and His-CHIP autoubiquitination were observed in reaction mixtures that contained E1, E2, His-CHIP, His-MLK3, and ubiquitin (Fig. 6A and B). No detectable levels of His-MLK3 ubiquitination or His-CHIP autoubquitination were observed in control reaction mixtures that lacked E1, E2, His-CHIP, or ubiquitin (Fig. 6A and B). To the best of our knowledge, these results are the first demonstration that CHIP ubiquitinates MLK3 in vitro. To examine whether other E2 enzymes could function with CHIP to facilitate MLK3 ubiquitination, the E2 enzymes UbcH5c, UbcH5d, Ubc13/Uev1a plus Ube2W, Ubc13/Uev1a plus Cdc34, UbcH8, and UbcH9 were tested in the ubiquitination assay. Ubc13/Uev1a functions with RING-type E3s such as TRAF6 and promotes MLK3 ubiquitination (19). The CHIP-Ubc13/Uev1a complex generates unanchored K63 polyubiquitin chains, whereas the CHIP-UbcH5a complex forms polyubiquitin chains (K48 or K63) anchored to CHIP and to substrates (39). Thus, it has been proposed that a priming ubiquitination of the substrate by another E2 enzyme may be necessary for CHIP-Ubc13/Uev1a polyubiquitination to occur (39). Hence, for the analysis of CHIP-mediated in vitro ubiquitination of MLK3 by Ubc13/Uev1a, we combined Ubc13/Uev1a with either Ube2W or Cdc34, both of which can monoubiquitinate substrates (40). All members of the UbcH5 family that were tested (UbcH5a, -b, -c, and -d) facilitated His-MLK3 ubiquitination by His-CHIP (Fig. 6A to C). None of the other E2 enzymes tested (UbcH8, UbcH9, Ubc13/Uev1a/Ube2W, or Ubc13/Uev1a/Cdc34) could function with CHIP to promote MLK3 ubiquitination (Fig. 6C). Previous studies demonstrated that UbcH5a, -b, and -c and Ubc13/Uev1a/Ube2W mediate His-CHIP autoubiquitination and that UbcH8 and UbcH9 facilitate His-CHIP monoubiquitination (40). We observed similar results for His-CHIP autoubiquitination with UbcH5a, -b, -c, and -d and Ubc13/Uev1a/Ube2W but not with Ubc13/Uev1a/Cdc34 or UbcH9 (Fig. 6C). Furthermore, UbcH8 promoted only monoubiquitination of His-CHIP (Fig. 6C).
FIG 6.

CHIP ubiquitinates MLK3 in vitro. (A) In vitro ubiquitination reactions were performed with purified E1, UbcH5a, ubiquitin, His-CHIP, and His-MLK3 proteins. The samples were subjected to SDS-PAGE and immunoblotting with anti-MLK3 (top) and anti-CHIP (bottom) antibodies. (B) Reactions and immunoblotting were carried out as described above for panel A, except that UbcH5b was used instead of UbcH5a. (C) Reactions and immunoblotting were carried out as described above for panel A with the following E2 enzymes: UbcH5a, -b, -c, and -d; Ubc13/Uev1a plus Ube2W (13/2W); Ubc13/Uev1a plus Cdc34 (13/Cdc34); UbcH8; and UbcH9.
CHIP is required for the GA-, heat shock-, and sorbitol-induced decline in MLK3 protein levels.
Our results suggest that CHIP is a key regulator of endogenous and exogenous MLK3 protein levels. Next, we sought to determine whether CHIP mediates the stress-induced decline of endogenous MLK3 protein levels. The marked decline in endogenous MLK3 protein levels that was observed in GA-treated (6 h) SKOV3 cells (Fig. 1A) was much less pronounced in cells with CHIP siRNA knockdown (Fig. 7A). Similar results were observed in HEY1B and TOV21G ovarian cancer cells (see Fig. S2 in the supplemental material). For nonspecific siRNA-transfected cells exposed to heat shock for 60 min, the MLK3 protein level was reduced to 0.06% of that in the untreated cells (Fig. 7B). In contrast, the MLK3 protein level was not reduced in the CHIP siRNA knockdown cells exposed to heat shock for 60 min in comparison to the cells that were not exposed to heat shock (Fig. 7B). Similarly, nonspecific siRNA-transfected cells treated with sorbitol for 4 h exhibited a decline in MLK3 protein levels to 0.19% of the level in untreated cells, whereas no decline in MLK3 levels was observed in CHIP siRNA-transfected cells treated with sorbitol (Fig. 7C). These results indicate that CHIP is required for the degradation of MLK3 induced by GA, osmotic shock, and heat shock. In addition, JNK activation induced by heat shock and osmotic shock was enhanced in CHIP knockdown cells in comparison to cells transfected with nonspecific siRNA, which suggests that CHIP may suppress JNK signaling in response to stress stimuli (Fig. 7B and C).
FIG 7.
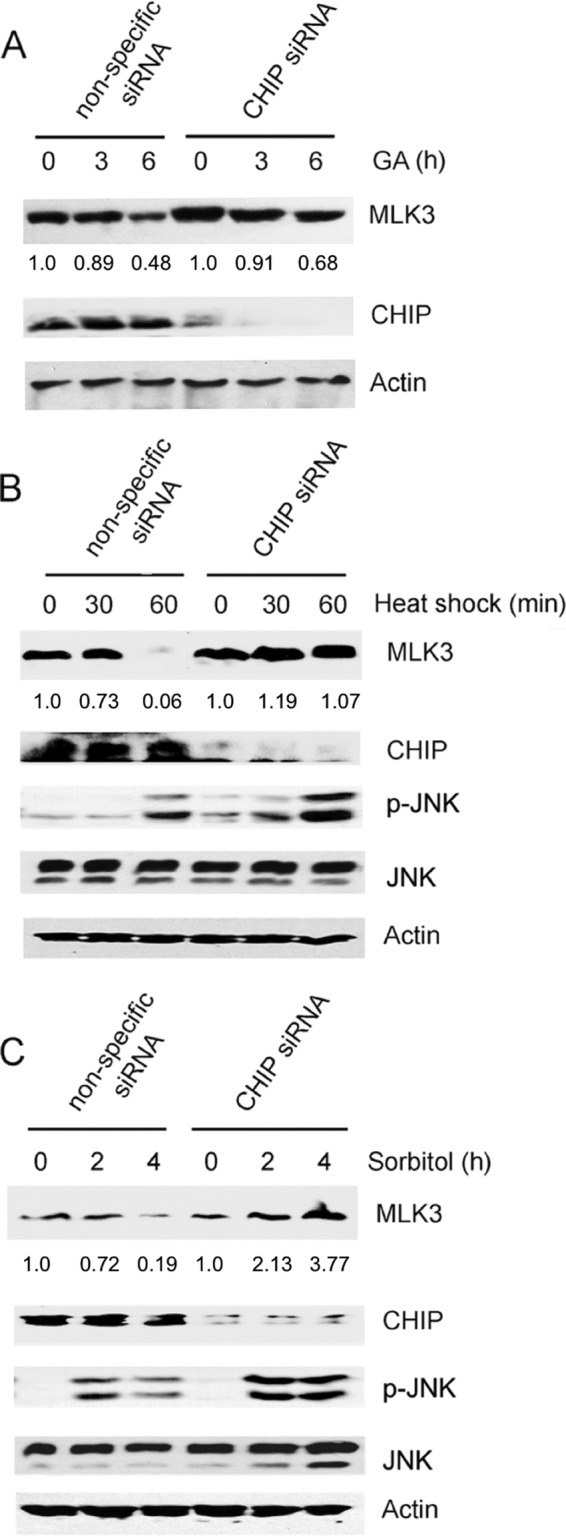
GA- and stress-induced degradation of MLK3 requires CHIP. (A) SKOV3 cells were transfected with nonspecific or CHIP siRNA oligonucleotides and then treated with GA (10 μM) for 3 and 6 h. Whole-cell extracts were immunoblotted with anti-MLK3, anti-CHIP, and anti-β-actin antibodies. The MLK3 band intensity for each time point was normalized to the band intensity for β-actin and expressed as a percentage of the value for untreated cells. (B) HEK293 cells were transfected with CHIP and nonspecific siRNA oligonucleotides as described above for panel A and then subjected to a 42°C heat shock for 30 or 60 min. The cell lysates were immunoblotted with the indicated antibodies. The MLK3 band intensity for each time point was analyzed as described above for panel A. (C) HEK293 cells were transfected with CHIP and nonspecific siRNA oligonucleotides as described above for panel A and then treated with 0.25 M sorbitol for 2 or 4 h. The cell lysates were immunoblotted with the indicated antibodies. The MLK3 band intensity for each time point was analyzed as described above for panel A.
MLK3 is required for invasion of CHIP knockdown SKOV3 cells.
MLK3 has an important function in ovarian and breast cancer cell invasion, and we postulated that CHIP-dependent regulation of the MLK3 protein level might be critical for cell invasion and establishment of a malignant phenotype (5, 6). To investigate the function of CHIP in MLK3-dependent regulation of ovarian cancer cell invasion, SKOV3 cells with MLK3 and/or CHIP siRNA knockdown were subjected to Matrigel invasion assays. CHIP and MLK3 knockdown in SKOV3 cells was verified by immunoblotting (Fig. 8, bottom). Invasion was reduced by approximately 42% in cells with MLK3 knockdown in comparison to cells transfected with nonspecific siRNA, which is consistent with a requirement for MLK3 in ovarian cancer cell invasion, as previously reported (Fig. 8, top) (5). In contrast, SKOV3 cells with CHIP knockdown had a 2.6-fold increase in invasion in comparison to cells transfected with nonspecific siRNA, which indicates that CHIP has a suppressive effect on ovarian cancer cell invasion (Fig. 8, top). Cells with combined knockdown of CHIP and MLK3 had only a 1.7-fold increase in invasion; thus, CHIP-dependent suppression of ovarian cancer cell invasion may be mediated, in part, by MLK3 (Fig. 8, top).
FIG 8.
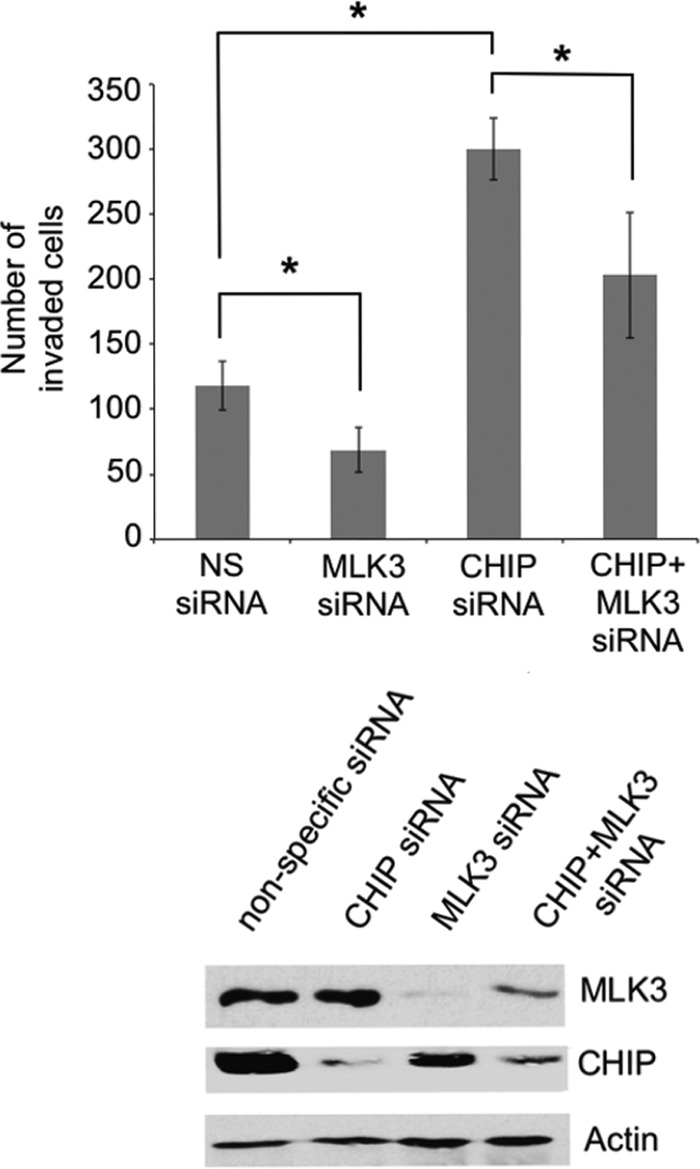
MLK3 is required for CHIP-mediated suppression of cell invasion. SKOV3 cells were transfected with nonspecific, CHIP, MLK3, or CHIP plus MLK3 siRNA oligonucleotides. Cell invasion was assessed by using Transwell membranes coated with Matrigel. (Top) The cells that invaded were stained and counted. The values represent the means ± standard deviations (*, P < 0.05 relative to the nonspecific siRNA control, determined by unpaired Student's t test) (n = 4). (Bottom) Whole-cell extracts were prepared from each set of transfected cells for analysis by immunoblotting with the indicated antibodies.
DISCUSSION
Environmental stresses induce protein damage and destabilization. The cytotoxicity associated with the accumulation of damaged proteins is avoided by targeting these proteins for degradation by the proteasome (36, 41). In a proposed model, Hsp70 recognizes damaged proteins and recruits CHIP, which ubiquitinates these proteins and targets them for degradation by the 26S proteasome (36). Our results show that in response to GA or stress stimuli, the endogenous MLK3 protein level is reduced by a CHIP-dependent mechanism. Hsp70 and CHIP interact with MLK3 and promote MLK3 ubiquitination, and CHIP-mediated regulation of MLK3 protein levels is proteasome dependent. We propose that extended exposure to stress stimuli promotes Hsp90-MLK3 dissociation and that Hsp70 subsequently binds to MLK3. This binding results in the recruitment of CHIP (via the Hsp70-CHIP interaction) and ubiquitination of MLK3. Hence, MLK3 binding to Hsp70 could be a critical step that leads to CHIP-mediated MLK3 ubiquitination and proteasomal degradation in response to specific stress stimuli (Fig. 9).
FIG 9.
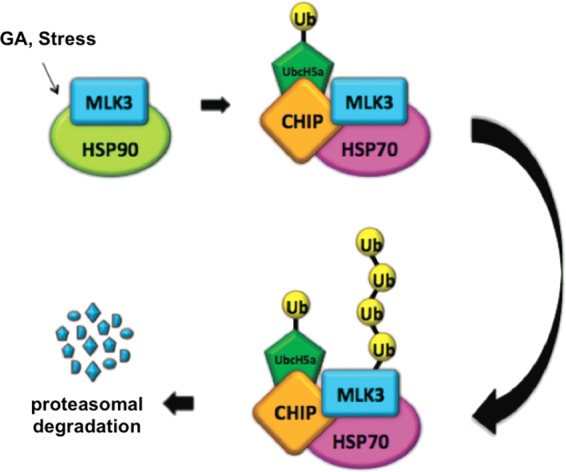
Proposed model for CHIP-mediated ubiquitination and degradation of MLK3. GA or stress stimuli promote MLK3-Hsp90 dissociation, and MLK3 subsequently interacts with Hsp70. Hsp70 recruits CHIP, which, together with E1, E2 (UbcH5), and ubiquitin, promotes polyubiquitination of MLK3. Polyubiquitinated MLK3 undergoes proteasome-mediated degradation.
TRAF6, together with the E2 enzyme Ubc13/Uev1a, promotes MLK3 ubiquitination (19). We observed that CHIP-mediated ubiquitination of MLK3 was accomplished by the UbcH5 family of E2 enzymes but not by other CHIP-E2 complexes tested. The CHIP-Ubc13/Uev1a complex facilitated CHIP autoubiquitination but not ubiquitination of MLK3. Thus, we conclude that MLK3 is not an optimal substrate for CHIP-Ubc13/Uev1a in vitro.
The FLAG-CHIP- or GFP-Hsp70-dependent decline in endogenous MLK3 levels was observed only in cells that were treated with the Hsp90 inhibitor GA. These results highlight the important function of Hsp90 in MLK3 stabilization and suggest that only unbound MLK3 is ubiquitinated by CHIP. The level of GST-MLK3 was substantially reduced (in the absence of GA treatment) in cells with co-overexpression of FLAG-CHIP and GST-MLK3. We postulate that endogenous Hsp90, despite its relatively high level of expression in many cell types, is insufficient to fully stabilize all of the exogenous GST-MLK3. Thus, any GST-MLK3 that is not bound to Hsp90 is destabilized, ubiquitinated by FLAG-CHIP, and targeted for proteasome-dependent degradation. In agreement with this hypothesis, co-overexpression of HA-Hsp90 together with FLAG-CHIP or GFP-Hsp70 prevented the decline of GST-MLK3 protein.
CHIP knockdown or overexpression had a minimal effect on MLK3 protein levels in cells that were not exposed to stresses. This finding suggests that CHIP-mediated regulation of endogenous MLK3 is stimulus dependent. It is intriguing to speculate that, like GA, heat and osmotic stresses also disrupt the Hsp90-MLK3 interaction and cause unfolding or destabilization of MLK3. However, further investigation of Hsp90-MLK3 binding in response to these stimuli is needed to demonstrate this conclusively. Possibly, the turnover of MLK3 protein in cells that are not exposed to stresses is regulated by other E3 ligases. For instance, in Caenorhabditis elegans, the E3 ligase RPM-1 is required for regulation of basal MLK1 levels, and this regulation is important for axon regeneration (42).
GA and heat shock both induce the heat shock response, which elevates Hsp70 expression levels and induces proteasome-mediated protein degradation (30, 31). We observed that osmotic stress also elevated Hsp70 and reduced MLK3 protein levels, which might indicate that the modulation of MLK3 by osmotic stress occurs via an Hsp70-dependent mechanism. Sorbitol-mediated osmotic shock in keratinocytes elevates Hsp70 expression levels after 4 h, and this increase is blocked by pretreatment of cells with the p38 MAPK inhibitor PD169316 (43). Because the p38 MAPK pathway is critical for the cellular response to changes in osmolarity, it is plausible that p38-dependent regulation of Hsp70 is involved in the establishment of stress tolerance (43, 44).
Our results indicate that in HEK293 cells, JNK is activated by osmotic and heat stresses. CHIP knockdown cells had impaired heat shock- or osmotic shock-induced MLK3 protein degradation, and JNK activation was enhanced. Possibly, CHIP knockdown resulted in the upregulation of MAP3Ks such as MLK3, ASK1, or DLK and/or inhibited MAPK phosphatases, all of which could have contributed to the enhanced JNK activation (38, 45).
Failure of the ubiquitin proteasome system and the resulting accumulation of misfolded proteins are characteristic of degenerative diseases such as Huntington's, Parkinson's, and Alzheimer's diseases (46). CHIP knockdown in breast cancer cells substantially elevates levels of cell invasion (47). Similarly, we observed that SKOV3 cells with CHIP knockdown exhibited increased cell invasion. The mechanism of CHIP-dependent regulation of invasion is not well understood; however, it is possible that the loss of CHIP causes the accumulation of signaling proteins such as MLK3, TRAF2, Raf-1, and ErbB2, which are key regulators of invasion (47–49). The accumulation of TRAF2 in CHIP knockdown cells might also increase cell invasion by enhancing MLK3 activation (5, 17). The enhancement of invasiveness in CHIP knockdown SKOV3 cells was reduced by depletion of MLK3, which underscores the crucial function of MLK3 in invasion and the importance of CHIP-dependent regulation of MLK3 in ovarian cancer cells. It will be interesting to determine if CHIP or Hsp90 is deregulated in ovarian cancer cells and if its levels correlate with the invasive capacity of these cells.
The loss of MLK3 in response to stress stimuli may seem counterintuitive, as MLK3 is a stress-activated kinase; however, MLK3 also has other important kinase-dependent and kinase-independent biological functions. For instance, in neuronal cells, a complex of Rac-MLK3-JNK anchored by the POSH scaffold protein elicits apoptosis in response to nerve growth factor (NGF) deprivation, whereas in some epithelial cancer cells, MLK3 has a role in proliferation, migration, and tumor cell invasion (1, 5–7, 50, 51). One possibility is that MLK3, due to its proapoptotic function in some cell types, is downregulated as part of a stress-induced attenuation of apoptosis that occurs as a mechanism to cope with exposures to environmental stresses (35). However, whether MLK3 is a major contributor to stress-induced apoptosis in SKOV3 cells remains to be determined. Another possibility is that after prolonged stress exposure, MLK3 protein is severely damaged such that proper refolding cannot be achieved, and the misfolded protein is targeted for degradation by the protein quality control system.
The investigation of the effect of stresses on MLK3 protein levels has been limited; however, a few interesting studies have shed some light on this topic. Chen et al. demonstrated that exogenous MLK3 undergoes proteolytic cleavage at a site within the kinase domain in response to phorbol-12-myristate-13-acetate (PMA) and TNF (52). This cleavage produces a C-terminal fragment that heterodimerizes with full-length MLK3 and reduces MLK3 activation loop phosphorylation and MLK3-dependent activation of JNK. Furthermore, production of the C-terminal fragment is blocked by proteasome inhibition (52). A previous study by Gonda et al. showed that the Drosophila melanogaster MLK Slipper (Slpr) is phosphorylated in response to heat stress on a serine that is within a MAPK PXSP consensus sequence (53). Expression of a phosphomimetic PXEP mutant conferred an advantage to the mutant flies over the control flies in their ability to tolerate thermostress, and those authors suggested that phosphorylation of this serine residue in response to heat stress may be critical for JNK signaling and an optimal stress response (53). Mammalian MLK3 is phosphorylated by JNK, and this phosphorylation induces the redistribution of MLK3 to a Triton-soluble fraction and enhances MLK3 activation (54). Moreover, Wang et al. also described a potential stabilizing effect of JNK phosphorylation on MLK3 protein (55). Thus, phosphorylation of MLK3 by JNK seems to positively regulate MLK3 stability and function. It remains to be determined if there is a pool of mammalian MLK3 that is phosphorylated in response to thermostress and if this phosphorylation affects MLK3 stability or blocks its ubiquitination by CHIP. Interestingly, we observed a low level of MLK3 protein in SKOV3 cells even after 8 h of GA or stress treatments. Because CHIP regulates cytoplasmic protein degradation, MLK3 protein localized to the plasma membrane may escape ubiquitination by CHIP. It is intriguing to speculate that in response to GA or stress stimuli, a pool of functional MLK3 localized at the plasma membrane might participate in the cellular stress response.
Our results provide the first demonstration that CHIP promotes the ubiquitination and proteasome-dependent degradation of MLK3. Furthermore, we have identified that CHIP-mediated regulation of MLK3 is critical for the suppression of ovarian cancer cell invasion. These findings are a significant advance toward understanding the mechanisms that regulate MLK3 protein degradation in human ovarian cancer cells. A thorough understanding of how MLK3 protein is regulated in neoplastic and nonneoplastic cells will be essential for the identification of new strategies to target MLK3 in cancer.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by National Institutes of Health grant GM102831-01 and a deArce-Koch Memorial Endowment Fund award (The University of Toledo) to D.N.C.
We declare that we have no conflicts of interest.
Footnotes
Published ahead of print 9 June 2014
Supplemental material for this article may be found at http://dx.doi.org/10.1128/MCB.00296-14.
REFERENCES
- 1.Chadee DN, Kyriakis JM. 2004. MLK3 is required for mitogen activation of B-Raf, ERK and cell proliferation. Nat. Cell Biol. 6:770–776. 10.1038/ncb1152 [DOI] [PubMed] [Google Scholar]
- 2.Kovalenko PL, Kunovska L, Chen J, Gallo KA, Basson MD. 2012. Loss of MLK3 signaling impedes ulcer healing by modulating MAPK signaling in mouse intestinal mucosa. Am. J. Physiol. Gastrointest. Liver Physiol. 303:G951–G960. 10.1152/ajpgi.00158.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zou W, Greenblatt MB, Shim JH, Kant S, Zhai B, Lotinun S, Brady N, Hu DZ, Gygi SP, Baron R, Davis RJ, Jones D, Glimcher LH. 2011. MLK3 regulates bone development downstream of the faciogenital dysplasia protein FGD1 in mice. J. Clin. Invest. 121:4383–4392. 10.1172/JCI59041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mishra P, Senthivinayagam S, Rangasamy V, Sondarva G, Rana B. 2010. Mixed lineage kinase-3/JNK1 axis promotes migration of human gastric cancer cells following gastrin stimulation. Mol. Endocrinol. 24:598–607. 10.1210/me.2009-0387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhan Y, Abi Saab WF, Modi N, Stewart AM, Liu J, Chadee DN. 2012. Mixed lineage kinase 3 is required for matrix metalloproteinase expression and invasion in ovarian cancer cells. Exp. Cell Res. 318:1641–1648. 10.1016/j.yexcr.2012.05.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chen J, Miller EM, Gallo KA. 2010. MLK3 is critical for breast cancer cell migration and promotes a malignant phenotype in mammary epithelial cells. Oncogene 29:4399–4411. 10.1038/onc.2010.198 [DOI] [PubMed] [Google Scholar]
- 7.Chen J, Gallo KA. 2012. MLK3 regulates paxillin phosphorylation in chemokine-mediated breast cancer cell migration and invasion to drive metastasis. Cancer Res. 72:4130–4140. 10.1158/0008-5472.CAN-12-0655 [DOI] [PubMed] [Google Scholar]
- 8.Swenson-Fields KI, Sandquist JC, Rossol-Allison J, Blat IC, Wennerberg K, Burridge K, Means AR. 2008. MLK3 limits activated Galphaq signaling to Rho by binding to p63RhoGEF. Mol. Cell 32:43–56. 10.1016/j.molcel.2008.09.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Xu Z, Maroney AC, Dobrzanski P, Kukekov NV, Greene LA. 2001. The MLK family mediates c-Jun N-terminal kinase activation in neuronal apoptosis. Mol. Cell. Biol. 21:4713–4724. 10.1128/MCB.21.14.4713-4724.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gallo KA, Mark MR, Scadden DT, Wang Z, Gu Q, Godowski PJ. 1994. Identification and characterization of SPRK, a novel src-homology 3 domain-containing proline-rich kinase with serine/threonine kinase activity. J. Biol. Chem. 269:15092–15100 [PubMed] [Google Scholar]
- 11.Rana A, Gallo K, Godowski P, Hirai S, Ohno S, Zon L, Kyriakis JM, Avruch J. 1996. The mixed lineage kinase SPRK phosphorylates and activates the stress-activated protein kinase activator, SEK-1. J. Biol. Chem. 271:19025–19028. 10.1074/jbc.271.32.19025 [DOI] [PubMed] [Google Scholar]
- 12.Gallo KA, Johnson GL. 2002. Mixed-lineage kinase control of JNK and p38 MAPK pathways. Nat. Rev. Mol. Cell Biol. 3:663–672. 10.1038/nrm906 [DOI] [PubMed] [Google Scholar]
- 13.Du Y, Bock BC, Schachter KA, Chao M, Gallo KA. 2005. Cdc42 induces activation loop phosphorylation and membrane targeting of mixed lineage kinase 3. J. Biol. Chem. 280:42984–42993. 10.1074/jbc.M502671200 [DOI] [PubMed] [Google Scholar]
- 14.Vacratsis PO, Gallo KA. 2000. Zipper-mediated oligomerization of the mixed lineage kinase SPRK/MLK-3 is not required for its activation by the GTPase cdc 42 but is necessary for its activation of the JNK pathway. Monomeric SPRK L410P does not catalyze the activating phosphorylation of Thr258 of murine mitogen-activated protein kinase kinase 4. J. Biol. Chem. 275:27893–27900. 10.1074/jbc.M002858200 [DOI] [PubMed] [Google Scholar]
- 15.Leung IW, Lassam N. 1998. Dimerization via tandem leucine zippers is essential for the activation of the mitogen-activated protein kinase kinase kinase, MLK-3. J. Biol. Chem. 273:32408–32415. 10.1074/jbc.273.49.32408 [DOI] [PubMed] [Google Scholar]
- 16.Leung IW, Lassam N. 2001. The kinase activation loop is the key to mixed lineage kinase-3 activation via both autophosphorylation and hematopoietic progenitor kinase 1 phosphorylation. J. Biol. Chem. 276:1961–1967. 10.1074/jbc.M004092200 [DOI] [PubMed] [Google Scholar]
- 17.Korchnak AC, Zhan Y, Aguilar MT, Chadee DN. 2009. Cytokine-induced activation of mixed lineage kinase 3 requires TRAF2 and TRAF6. Cell. Signal. 21:1620–1625. 10.1016/j.cellsig.2009.06.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sondarva G, Kundu CN, Mehrotra S, Mishra R, Rangasamy V, Sathyanarayana P, Ray RS, Rana B, Rana A. 2010. TRAF2-MLK3 interaction is essential for TNF-alpha-induced MLK3 activation. Cell Res. 20:89–98. 10.1038/cr.2009.125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Humphrey RK, Yu SMA, Bellary A, Gonuguntla S, Yebra M, Jhala US. 2013. Lysine 63-linked ubiquitination modulates mixed lineage kinase-3 interaction with JIP1 scaffold protein in cytokine-induced pancreatic beta cell death. J. Biol. Chem. 288:2428–2440. 10.1074/jbc.M112.425884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Whitesell L, Lindquist SL. 2005. HSP90 and the chaperoning of cancer. Nat. Rev. Cancer 5:761–772. 10.1038/nrc1716 [DOI] [PubMed] [Google Scholar]
- 21.Zhang H, Wu W, Du Y, Santos SJ, Conrad SE, Watson JT, Grammatikakis N, Gallo KA. 2004. Hsp90/p50cdc37 is required for mixed-lineage kinase (MLK) 3 signaling. J. Biol. Chem. 279:19457–19463. 10.1074/jbc.M311377200 [DOI] [PubMed] [Google Scholar]
- 22.Roe SM, Prodromou C, O'Brien R, Ladbury JE, Piper PW, Pearl LH. 1999. Structural basis for inhibition of the Hsp90 molecular chaperone by the antitumor antibiotics radicicol and geldanamycin. J. Med. Chem. 42:260–266. 10.1021/jm980403y [DOI] [PubMed] [Google Scholar]
- 23.Stebbins CE, Russo AA, Schneider C, Rosen N, Hartl FU, Pavletich NP. 1997. Crystal structure of an Hsp90-geldanamycin complex: targeting of a protein chaperone by an antitumor agent. Cell 89:239–250. 10.1016/S0092-8674(00)80203-2 [DOI] [PubMed] [Google Scholar]
- 24.Haupt A, Joberty G, Bantscheff M, Frohlich H, Stehr H, Schweiger MR, Fischer A, Kerick M, Boerno ST, Dahl A, Lappe M, Lehrach H, Gonzalez C, Drewes G, Lange BMH. 2012. Hsp90 inhibition differentially destabilises MAP kinase and TGF-beta signalling components in cancer cells revealed by kinase-targeted chemoproteomics. BMC Cancer 12:38. 10.1186/1471-2407-12-38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jiang J, Ballinger CA, Wu Y, Dai Q, Cyr DM, Hohfeld J, Patterson C. 2001. CHIP is a U-box-dependent E3 ubiquitin ligase: identification of Hsc70 as a target for ubiquitylation. J. Biol. Chem. 276:42938–42944. 10.1074/jbc.M101968200 [DOI] [PubMed] [Google Scholar]
- 26.Hatakeyama S, Yada M, Matsumoto M, Ishida N, Nakayama KI. 2001. U box proteins as a new family of ubiquitin-protein ligases. J. Biol. Chem. 276:33111–33120. 10.1074/jbc.M102755200 [DOI] [PubMed] [Google Scholar]
- 27.Ballinger CA, Connell P, Wu Y, Hu Z, Thompson LJ, Yin LY, Patterson C. 1999. Identification of CHIP, a novel tetratricopeptide repeat-containing protein that interacts with heat shock proteins and negatively regulates chaperone functions. Mol. Cell. Biol. 19:4535–4545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.McDonough H, Patterson C. 2003. CHIP: a link between the chaperone and proteasome systems. Cell Stress Chaperones 8:303–308. 10.1379/1466-1268(2003)008<0303:CALBTC>2.0.CO;2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yun BG, Matts RL. 2005. Differential effects of Hsp90 inhibition on protein kinases regulating signal transduction pathways required for myoblast differentiation. Exp. Cell Res. 307:212–223. 10.1016/j.yexcr.2005.03.003 [DOI] [PubMed] [Google Scholar]
- 30.Zou J, Guo Y, Guettouche T, Smith DF, Voellmy R. 1998. Repression of heat shock transcription factor HSF1 activation by HSP90 (HSP90 complex) that forms a stress-sensitive complex with HSF1. Cell 94:471–480. 10.1016/S0092-8674(00)81588-3 [DOI] [PubMed] [Google Scholar]
- 31.Sittler A, Lurz R, Lueder G, Priller J, Lehrach H, Hayer-Hartl MK, Hartl FU, Wanker EE. 2001. Geldanamycin activates a heat shock response and inhibits huntingtin aggregation in a cell culture model of Huntington's disease. Hum. Mol. Genet. 10:1307–1315. 10.1093/hmg/10.12.1307 [DOI] [PubMed] [Google Scholar]
- 32.Grbovic OM, Basso AD, Sawai A, Ye Q, Friedlander P, Solit D, Rosen N. 2006. V600E B-Raf requires the Hsp90 chaperone for stability and is degraded in response to Hsp90 inhibitors. Proc. Natl. Acad. Sci. U. S. A. 103:57–62. 10.1073/pnas.0609973103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Schulte TW, Blagosklonny MV, Romanova L, Mushinski JF, Monia BP, Johnston JF, Nguyen P, Trepel J, Neckers LM. 1996. Destabilization of Raf-1 by geldanamycin leads to disruption of the Raf-1-MEK-mitogen-activated protein kinase signalling pathway. Mol. Cell. Biol. 16:5839–5845 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Anglesio MS, Wiegand KC, Melnyk N, Chow C, Salamanca C, Prentice LM, Senz J, Yang W, Spillman MA, Cochrane DR, Shumansky K, Shah SP, Kalloger SE, Huntsman DG. 2013. Type-specific cell line models for type-specific ovarian cancer research. PLoS One 8:e72162. 10.1371/journal.pone.0072162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dai Q, Zhang C, Wu Y, McDonough H, Whaley RA, Godfrey V, Li HH, Madamanchi N, Xu W, Neckers L, Cyr D, Patterson C. 2003. CHIP activates HSF1 and confers protection against apoptosis and cellular stress. EMBO J. 22:5446–5458. 10.1093/emboj/cdg529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pratt WB, Morishima Y, Peng HM, Osawa Y. 2010. Proposal for a role of the Hsp90/Hsp70-based chaperone machinery in making triage decisions when proteins undergo oxidative and toxic damage. Exp. Biol. Med. (Maywood) 235:278–289. 10.1258/ebm.2009.009250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hwang JR, Zhang C, Patterson C. 2005. C-terminus of heat shock protein 70-interacting protein facilitates degradation of apoptosis signal-regulating kinase 1 and inhibits apoptosis signal-regulating kinase 1-dependent apoptosis. Cell Stress Chaperones 10:147–156. 10.1379/CSC-90R.1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Daviau A, Proulx R, Robitaille K, Di Fruscio M, Tanguay RM, Landry J, Patterson C, Durocher Y, Blouin R. 2006. Down-regulation of the mixed-lineage dual leucine zipper-bearing kinase by heat shock protein 70 and its co-chaperone CHIP. J. Biol. Chem. 281:31467–31477. 10.1074/jbc.M607612200 [DOI] [PubMed] [Google Scholar]
- 39.Windheim M, Peggie M, Cohen P. 2008. Two different classes of E2 ubiquitin-conjugating enzymes are required for the mono-ubiquitination of proteins and elongation by polyubiquitin chains with a specific topology. Biochem. J. 409:723–729. 10.1042/BJ20071338 [DOI] [PubMed] [Google Scholar]
- 40.Soss SE, Yue Y, Dhe-Paganon S, Chazin WJ. 2011. E2 conjugating enzyme selectivity and requirements for function of the E3 ubiquitin ligase CHIP. J. Biol. Chem. 286:21277–21286. 10.1074/jbc.M111.224006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Burkewitz K, Choe K, Strange K. 2011. Hypertonic stress induces rapid and widespread protein damage in C. elegans. Am. J. Physiol. Cell Physiol. 301:C566–C576. 10.1152/ajpcell.00030.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nix P, Hisamoto N, Matsumoto K, Bastiani M. 2011. Axon regeneration requires coordinate activation of p38 and JNK MAPK pathways. Proc. Natl. Acad. Sci. U. S. A. 108:10738–10743. 10.1073/pnas.1104830108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Garmyn M, Mammone T, Pupe A, Declercq L, Maes D. 2000. Human keratinocytes respond to osmotic stress by p38 MAP kinase regulated induction of HSP70 and HSP27. J. Invest. Dermatol. 115:549. [DOI] [PubMed] [Google Scholar]
- 44.Garmyn M, Mammone T, Pupe A, Gan D, Declercq L, Maes D. 2001. Human keratinocytes respond to osmotic stress by p38 map kinase regulated induction of HSP70 and HSP27. J. Invest. Dermatol. 117:1290–1295. 10.1046/j.0022-202x.2001.01553.x [DOI] [PubMed] [Google Scholar]
- 45.Gao Y, Han C, Huang H, Xin Y, Xu Y, Luo L, Yin Z. 2010. Heat shock protein 70 together with its co-chaperone CHIP inhibits TNF-alpha induced apoptosis by promoting proteasomal degradation of apoptosis signal-regulating kinase1. Apoptosis 15:822–833. 10.1007/s10495-010-0495-7 [DOI] [PubMed] [Google Scholar]
- 46.Menzies FM, Moreau K, Rubinsztein DC. 2011. Protein misfolding disorders and macroautophagy. Curr. Opin. Cell Biol. 23:190–197. 10.1016/j.ceb.2010.10.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Jang KW, Lee KH, Kim SH, Jin T, Choi EY, Jeon HJ, Kim E, Han YS, Chung JH. 2011. Ubiquitin ligase CHIP induces TRAF2 proteasomal degradation and NF-kappaB inactivation to regulate breast cancer cell invasion. J. Cell. Biochem. 112:3612–3620. 10.1002/jcb.23292 [DOI] [PubMed] [Google Scholar]
- 48.Xu W, Marcu M, Yuan X, Mimnaugh E, Patterson C, Neckers L. 2002. Chaperone-dependent E3 ubiquitin ligase CHIP mediates a degradative pathway for c-ErbB2/Neu. Proc. Natl. Acad. Sci. U. S. A. 99:12847–12852. 10.1073/pnas.202365899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Demand J, Alberti S, Patterson C, Hohfeld J. 2001. Cooperation of a ubiquitin domain protein and an E3 ubiquitin ligase during chaperone/proteasome coupling. Curr. Biol. 11:1569–1577. 10.1016/S0960-9822(01)00487-0 [DOI] [PubMed] [Google Scholar]
- 50.Zhan Y, Modi N, Stewart AM, Hieronimus RI, Liu J, Gutmann DH, Chadee DN. 2011. Regulation of mixed lineage kinase 3 is required for neurofibromatosis-2-mediated growth suppression in human cancer. Oncogene 30:781–789. 10.1038/onc.2010.453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Xu Z, Kukekov NV, Greene LA. 2003. POSH acts as a scaffold for a multiprotein complex that mediates JNK activation in apoptosis. EMBO J. 22:252–261. 10.1093/emboj/cdg021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Liou GY, Zhang H, Miller EM, Seibold SA, Chen W, Gallo KA. 2010. Induced, selective proteolysis of MLK3 negatively regulates MLK3/JNK signalling. Biochem. J. 427:435–443. 10.1042/BJ20091077 [DOI] [PubMed] [Google Scholar]
- 53.Gonda RL, Garlena RA, Stronach B. 2012. Drosophila heat shock response requires the JNK pathway and phosphorylation of mixed lineage kinase at a conserved serine-proline motif. PLoS One 7:e42369. 10.1371/journal.pone.0042369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Schachter KA, Du Y, Lin A, Gallo KA. 2006. Dynamic positive feedback phosphorylation of mixed lineage kinase 3 by JNK reversibly regulates its distribution to Triton-soluble domains. J. Biol. Chem. 281:19134–19144. 10.1074/jbc.M603324200 [DOI] [PubMed] [Google Scholar]
- 55.Wang C, Tao Y, Wang Y, Xu Z. 2010. Regulation of the protein stability of POSH and MLK family. Protein Cell 1:871–878. 10.1007/s13238-010-0111-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.