

Abstract
Retinoic acid receptor-related orphan nuclear receptor alpha (RORα) is a potent tumor suppressor that reduces cell proliferation and inhibits tumor growth. However, the molecular mechanism by which it inhibits cell proliferation remains unknown. We demonstrate a noncanonical nuclear receptor pathway in which RORα binds to E2F1 to inhibit cell cycle progression. We showed that RORα bound to the heptad repeat and marked box region of E2F1 and suppressed E2F1-regulated transcription in epithelial cells. Binding of RORα inhibited E2F1 acetylation and its DNA-binding activity by recruiting histone deacetylase 1 (HDAC1) to the protein complexes. Knockdown of HDAC1 or inhibition of HDAC activity at least partially rescued transcription factor activity of E2F1 that was repressed by RORα. Importantly, RORα levels were increased in mammary ducts compared to terminal end buds and inversely correlated with expression of E2F1 target genes and cell proliferation. Silencing RORα in mammary epithelial cells significantly enhanced cell proliferation in the ductal epithelial cells and promoted side branching of the mammary ducts. These results reveal a novel link between RORα and E2F1 in regulating cell cycle progression and mammary tissue morphogenesis.
INTRODUCTION
Nuclear receptors, a family of ligand-dependent transcription factors, regulate gene expression by directly binding to the cis response elements in the regulatory regions (1, 2). Retinoic acid receptor-related orphan nuclear receptor alpha (RORα) is an orphan nuclear receptor and plays critical roles in many physiological processes, including cell differentiation, metabolism, inflammation, transformation, and circadian rhythm (3–11). The canonical way that RORα regulates gene expression is through ROR response elements in the regulatory regions of the target genes (12, 13). RORα recruits a number of transcriptional coactivators and corepressors to the target genes, including steroid receptor coactivator 1 (14) and p300 (15), nuclear receptor corepressor, and silencing mediator for retinoic acid and thyroid hormone receptors (16). These transcriptional cofactors modulate the chromatin structure to induce or repress transcription. Unlike the canonical pathway of nuclear receptor to regulate gene expression, a number of recent studies show that RORα acts as a transcriptional cofactor, binding to β-catenin or p53 to regulate target gene expression or modulate protein stability (7, 17).
Spatial and temporal regulation of cell proliferation is crucial for normal tissue development and maintenance of the differentiated state (18). For instance, mammary epithelial cells in the mature ducts remain growth arrested, while cells in terminal end buds are highly proliferative (19, 20). The different cell proliferation status in terminal end buds and ducts is important for mammary gland branching morphogenesis and maintenance of the ductal structure (21, 22). Enhancing cell proliferation in mammary epithelial cells in the long run is sufficient to induce tumorigenesis (23, 24). We and others have shown that RORα expression is significantly downregulated during tumor development and progression (7, 25), and exogenous RORα reduces cell proliferation and inhibits tumor growth. But the molecular mechanism by which RORα inhibits cell proliferation remains to be determined.
Cell proliferation is controlled by a series of cell cycle regulators, such as transcription factor E2Fs, cyclin E, cyclin-dependent kinases, and cell cycle brakes pRB and p27 (26–31). E2F1 is a member of the E2F transcription factor family and plays important roles in regulating G1/S transition, DNA synthesis, and apoptosis (32–35). E2F1 activity is regulated by a number of proteins through protein-protein interaction (36–38). For example, binding of pRb inhibits E2F1 activity at G1 phase, and phosphorylation of pRB releases it from E2F1 and leads to E2F1 activation (39). E2F1 activity is also regulated by a number of covalent modifications, such as acetylation and phosphorylation (40, 41). Acetylation of lysine residues 117, 120, and 125 by the acetyltransferase complex CBP/p/CAF enhances E2F1 DNA binding ability and stabilizes E2F1 (41).
Using gene expression profile analysis, we showed that transcription of E2F1 target genes was suppressed by RORα. Coimmunoprecipitation (co-IP) and in vitro binding data demonstrated that RORα bound to E2F1 and enhanced the interaction between E2F1 and histone deacetylase 1 (HDAC1), which, in turn, reduced E2F1 acetylation and inhibited its DNA-binding activity. Importantly, high levels of RORα were associated with reduced cell proliferation and repression of E2F1 target genes during mammary branch morphogenesis. Silencing RORα in the mammary epithelial cells enhanced cell proliferation in the ductal epithelial cells and promoted side branching. These results identified a noncanonical pathway of RORα to regulate gene expression and mammary gland development, in which RORα binds to E2F1 to inhibit E2F1-dependent cell cycle progression.
MATERIALS AND METHODS
Antibodies and reagents.
The 5-ethynyl-2′-deoxyuridine (EdU) staining kit was from Invitrogen. Matrigel (laminin-rich extracellular matrix [lrECM]) and type I collagen were from BD Bioscience. RORα cDNA was purchased from Open Biosystems. E2F1 and DP1 cDNA clones were purchased from Addgene. E2F1-Luc luciferase reporter vector was purchased from Signosis, Inc. Plasmids carrying short hairpin RNA (shRNA) against RORα (shRORα plasmids) and shHDAC1 plasmids (a kind gift from Zhou P. Binhua) were purchased from Sigma. HDAC inhibitor trichostatin A (TSA) was purchased from Sigma. Antibodies against the following antibodies were obtained: RORα, E2F1, and lamin A/C (Santa Cruz); Flag (Sigma); Ki67 (Vector Laboratories); acetyllysine (Millipore); and HDAC1 (Affinity BioReagents).
Cell culture and virus preparation.
HMT-3522 S1 cells (hereafter referred to as S1 cells; a kind gift from Mina J. Bissell) were maintained in Dulbecco modified Eagle medium (DMEM)–F-12 medium containing insulin at 2.5 × 10−7 g/ml, transferrin at 1.0 × 10−5 g/ml, sodium selenite at 2.6 × 10−9 g/ml, 1.0 × 10−10 M estradiol, 1.4 × 10−6 M hydrocortisone, prolactin at 5.0 × 10−6 μg/ml, and epidermal growth factor (EGF) at 1.0 × 10−8 ng/ml. MDA-MB 231 cells (ATCC) were propagated in DMEM–F-12 (Sigma) with 10% fetal bovine serum (Invitrogen). MDA-MB 157 cells (ATCC) were propagated in DMEM (Sigma) with 10% fetal bovine serum (Invitrogen). Three-dimensional (3D) lrECM on-top cultures were prepared by trypsinization of cells from tissue culture flasks, seeding of single cells on top of Engelbreth-Holm-Swarm (EHS) tumor extract (Matrigel; BD Biosciences), and addition of medium containing 5% EHS. MDA-MB 157 cells and MDA-MB 231 cells were seeded at 1.4 × 104 per cm2. MDA-MB 157 cells and MDA-MB 231 cells were maintained in H14 medium with 1% fetal bovine serum.
Flag-tagged RORα cDNA was cloned into plasmid pCDH1 and generated expression vector pCDH1-RORAl-Flag. HEK293 FT cells were transfected with pCDH1 or shRNA vector (Sigma) plus packaging lentivector using Lipofectamine (Invitrogen). Cancer cells were infected with lentivirus and selected by puromycin 48 h after infection.
Microarray analysis.
Control and RORα-expressing MDA-MB 231 and MDA-MB 157 cells were isolated from 3D cultures with phosphate-buffered saline (PBS)-EDTA (5 mM EDTA, 1 mM NaVO4, 1.5 mM NaF in PBS) on ice as previously described (42). Purified total cellular RNA was extracted using an RNeasy minikit with on-column DNase digestion (Qiagen). RNA was quantified by measuring optical density at 260 nm, and quality was verified by agarose gel electrophoresis. Affymetrix microarray analysis was performed using the Affymetrix HuGene-1.0 high-throughput array (HTA) GeneChip system. Preprocessing, normalization, and filtering were performed using R Bioconductor (43). Gene set enrichment analysis was performed with GSEA v2.07 (http://www.broadinstitute.org/gsea/index.jsp) (44). E2F1 target gene expression levels in terminal end buds and mammary ducts were derived from a published microarray data set (Gene Expression Omnibus accession number GSE2988). Unsupervised hierarchical clustering analysis was performed with Cluster (uncentered correlation, average linkage), and results were visualized with TreeView (http://www.eisenlab.org/eisen/?page_id=42).
Luciferase reporter assay.
A luciferase reporter vector containing four E2F response elements (TTTCGCGC) was purchased from Signosis. MDA-MB 231 cells were transfected with pCDNA plus pE2F1-Luc luciferase vector (1:5). A luciferase vector pE2F1-Luc stable expression cell clone was selected by G418 (8.0 × 10−4 g/ml) treatment. Cell lysates were collected for the luciferase assay 48 h after infection with pCDH1-RORAl-Flag.
Immunohistochemistry.
Mouse mammary gland sections and xenograft tumor sections were deparaffinized and hydrated from xylene, 100% ethanol, 95% ethanol, 85% ethanol, and 70% ethanol to PBS solution. Endogenous peroxidase was blocked by incubation with 3% H2O2 for 20 min. At the antigen retrieval step, slides were steamed in citrate sodium buffer for 30 min. Slides were incubated with antibodies at 4°C overnight, and then the sections were incubated with goat anti-rabbit IgG conjugated with horseradish peroxidase at room temperature for 60 min. After diaminobenzidine (DAB) staining, images were taken by Nikon and scored blindly.
RT-PCR.
Total RNA was extracted from cells using TRIzol reagent (Invitrogen). cDNA was synthesized using the SuperScript First Strand Synthesis kit (Invitrogen) from 0.5- to 1.0-μg RNA samples. cDNA synthesis was performed with the SuperScript III First-Strand Synthesis System according to the manufacturer's instructions. Quantitative reverse transcription-PCRs (RT-PCRs) were carried out using SYBR green PCR master mix reagents on an ABI 7500 Fast real-time PCR system (Applied Biosystems). Thermal cycling was conducted at 95°C for 30 s, followed by 40 cycles of amplification at 95°C for 5 s, 55°C for 30 s, and 72°C for 15 s. The relative quantification of gene expression for each sample was analyzed by the threshold cycle (CT) method. The following primers were used for amplification: for CDC6, 5′-TGGGCGATGACAACCTATGC-3′ and 5′-TGGCTAGTTCTCTTTTGCTAGGA-3′; for ESPL1, 5′-CCGCCTTGAAGGAGTTCCTG-3′ and 5′-GGGGTAGACACTAAGTAGCCAT-3′; for MCM4, 5′-GACGTAGAGGCGAGGATTCC-3′ and 5′-GAGTGCCGTATGTCAGTGGT-3′; for POLE2, 5′-GAATGCAGTCAGTCTGTTGATGA-3′ and 5′-AACCTATCACCGGAGGAGTAAAT-3′; and for 18S rRNA, 5′-ACCTGGTTGATCCTGCCAGT-3′ and 5′-CTGACCGGGTTGGTTTTGAT-3′.
Coimmunoprecipitation assay.
Cells were lysed with 400 μl of ice-cold hypotonic gentle lysis buffer (10 mM Tris-HCl [pH 7.5], 10 mM NaCl, 2 mM EDTA, 0.5% Triton X-100, 1× protease inhibitor cocktail set I [Calbiochem]) and incubated on ice for 10 min. After sonication, the protein complexes were pulled down with anti-Flag M2 affinity gel (Sigma), monoclonal antihemagglutinin (anti-HA)-agarose (Sigma), or anti-E2F antibody (Santa Cruz).
ChIP assay.
Vector control and RORα-expressing MDA-MB 231 cells were cross-linked using formaldehyde for the chromatin immunoprecipitation (ChIP) assay. The ChIP assay was performed based on the Upstate Biotechnology ChIP protocol, with a few modifications. After formaldehyde cross-linking, nuclei were isolated with a nuclear isolation kit (Sigma) and resuspended in ChIP lysis buffer (1% SDS, 10 mM EDTA, 50 mM Tris-HCl [pH 8.0]) containing protease inhibitor cocktail. Protein-DNA complexes were immunoprecipitated as per the Upstate protocol. Isolated DNA was then analyzed by RT-PCR using the following primers: CDC6 promoter, 5′-ACTACAGCCAATCAGAATCGAGGC-3′ and 5′-TCCTCTTCTTTCCACCTCCTCAGT-3′, and POLE2 promoter, 5′-GGAGACCAAGCAGGGATCTT-3′ and 5′-GCAACTTGAAGGCGGAGAG-3′.
GST pulldown assay.
Full-length E2F1 gene and deletion mutants (M1 to M5) were cloned into pGEX-4T-1 plasmid. Full-length RORα1, the DBD (containing the DNA-binding domain and hinge domain), or ligand-binding domain (LBD) was cloned into the pGEX-4T-1 plasmid. Glutathione S-transferase (GST) fusion protein expression was induced in Escherichia coli by 0.1 mM isopropyl-β-d-thiogalactopyranoside (IPTG) at 19°C overnight. Bacteria were lysed in radioimmunoprecipitation assay (RIPA) buffer (50 mM Tris, 150 mM NaCl, 0.5% sodium deoxycholate, 1% NP-40) with protease inhibitors and purified using Pierce glutathione-agarose (Thermo Scientific). Purified GST-fused proteins were confirmed by Coomassie staining. HEK293 FT cells were transfected with pCDH1-RORα1-flag or pCDH1-E2F1-flag plasmid using FuGENE HD transfection reagent (Promega). Cells were lysed in RIPA buffer with protease inhibitors 36 h after transfection. For the pulldown assay, GST-fused protein binding beads incubated with HEK293 FT cells at 4°C for 4 h and then washed with RIPA buffer three times for immunoblotting analysis.
Flow cytometry analysis.
Vector control and RORα-expressing MDA-MB 231 cells were starved with serum for 24 h and then trypsinized for flow cytometry analysis. shRORα-expressing and vector control HMT-3522 S1 cells were trypsinized for flow cytometry analysis after starvation with EGF for 24 h. Cells were washed with 5 ml of ice-cold PBS, suspended in 500 μl of PBS, and then immediately fixed with 5 ml of cold 70% ethyl alcohol (EtOH). Fixed cells were incubated with 200 μl of propidium iodide solution (0.2 mg/ml) and 20 μl of RNase (10 mg/ml) at 37°C for 45 min. Flow cytometry analysis was performed at the fluorescence-activated cell sorting (FACS) facility at the School of Medicine, University of Kentucky.
Mammary fat pad clearance and mammary epithelium cell transplantation.
Primary mammary organoids were isolated from 6- to 7-week-old female FVB mice (Friend virus B-outbred colony of Swiss mice) with collagenase-trypsin solution (0.2% trypsin, 0.2% collagenase type IV, 5% fetal calf serum, 5 μg/ml of insulin, and 50 μg/ml of gentamicin in 50 ml of DMEM–F-12) as previously described (45). The isolated mammary organoids were infected with lentivirus containing shControl or shRORα before transplantation. Three-week-old female FVB mice were anesthetized, and the inguinal fat pads were cleared by surgically removing the developing mammary epithelium. A small pocket was formed in the cleared fat pad and shControl-infected mammary organoids were placed in the pocket; shRORα-infected mammary organoids were transplanted on the contralateral side using the same procedure. Five weeks after surgery, the outgrowth of transplants was analyzed by performing whole mounts.
Whole-mount staining of mammary glands.
The fourth mammary glands were freshly dissected, spread on a glass slide, and fixed in Carnoy's fixative (6 parts ethanol, 3 parts chloroform, and 1 part glacial acetic acid) overnight. After being washed in 70% ethanol for 15 min, glands were rinsed in distilled water for 5 min and then stained in carmine alum stain solution (0.2% [wt/vol] carmine, 0.5% [wt/vol] aluminum potassium sulfate) for several hours to overnight. Slides were washed with 70% ethanol, followed by 95% and 100% ethanol. For long-term storage and photographic documentation, glands were cleared in xylene and mounted with Cytoseal 60 (Thermo Scientific) mounting medium. Whole mounts were photographed using a Nikon microscope.
Statistical analysis.
Experiments were repeated at least three times. Results were reported as means ± standard errors of the means (SEM); significance of difference was assessed by an independent Student t test. A P value of <0.05 represented statistical significance, and a P value of <0.01 represented high statistical significance.
RESULTS
RORα inhibits cell proliferation and expression of the E2F1 target genes.
We showed previously that downregulation of RORα in breast cancer is associated with enhanced cell proliferation and aggressive phenotypes of breast cancer cells in 3D culture (25). Expression of RORα in T4-2 cells inhibits tumor growth, suggesting that RORα may modulate cell proliferation (25). Introducing exogenous RORα in breast cancer cell lines MDA-MB 157 and MDA-MB 231 (Fig. 1A) significantly inhibited cell proliferation in 3D culture (Fig. 1B). Ki67 staining showed that expression of RORα also inhibited tumor growth and suppressed proliferation of breast cancer cells in vivo (Fig. 1C and D).
FIG 1.

RORα inhibits cell proliferation and expression of the E2F1-targeted genes. (A) RORα expression was assessed by Western blotting in breast cancer cells infected with lentivirus containing the RORα-expressing construct. (B) Cell proliferation in control and RORα-expressing breast cancer cells was analyzed by EdU staining. Cells were cultured in 3D Matrigel. RORα expression inhibited cell proliferation in MDA-MB 157 and MDA-MB 231 cells (n = 3; *, P < 0.05; **, P < 0.01). (C) Control or RORα-expressing T4 cells or MDA-MB 231 cells were injected into the mammary fat pad in SCID mice. Restoring RORα expression in T4 cells or MDA-MB 231 cells inhibited tumor growth (n = 6; *, P < 0.05; **, P < 0.01). (D) Cell proliferation was examined by Ki67 staining in tumors formed by control or RORα-expressing T4-2 and MDA-MB 231 cells. The bar graph shows the Ki67 positivity ratio (n = 3; **, P < 0.01). (E) Gene set enrichment analysis (GSEA) identified the gene set “CELL CYCLE_KEGG” to be enriched among genes that show a reduced expression in RORα-expressing cells (P = 0.094). (F) GSEA analysis showed that the gene set “E2F1_Q6_01” had reduced expression in RORα-expressing breast cancer cells. (G) Heat map showing that E2F1 target genes were downregulated in RORα-expressing breast cancer cells compared with control cells. (H) Cell cycle progression in control and RORα-expressing cells was examined by flow cytometry analysis. The percentage of cells in G1 phase was significantly increased, while that of cells in S phase was significantly decreased in RORα-expressing MDA-MB 231 cells (n = 3; *, P < 0.05). (I) The top portion shows silencing efficiency of shRORα in S1 cells, examined by Western blotting. The bottom portion shows cell proliferation in control and shRORα-expressing S1 cells, analyzed by EdU staining. Cells were cultured in 3D Matrigel. Knockdown of RORα expression increased cell proliferation in S1 cells (n = 2; *, P < 0.05). (J) Cell cycle progression was analyzed by flow cytometry in control and RORα-silenced S1 cells. The percentage of cells in G1 phase was significantly decreased, while the percentages of cells in S phase and G2 phase were significantly increased in RORα-silenced cells compared with control cells (n = 3; *, P < 0.05).
To understand how RORα inhibits cell proliferation, we performed microarray analysis in control and RORα-expressing breast cancer cells. Hundreds of genes were identified to be differentially expressed between the control and the RORα-expressing cells. Interestingly, most of these genes do not contain ROR response elements in their regulatory regions, suggesting that RORα modulates their expression indirectly. Using gene set enrichment analysis (GSEA), we found that genes related to the KEGG cell cycle pathway (Fig. 1E) and E2F1-targeted genes were downregulated in RORα-expressing breast cancer cells (Fig. 1F and G). Flow cytometry analysis showed that ectopic expression of RORα in MDA-MB 231 cells significantly increased the number of cells at G0/G1 phase and reduced the number of cells in S phase (Fig. 1H). Silencing RORα in nonmalignant S1 cells inhibited cell proliferation (Fig. 1I) and reduced the percentage of cells in G1 phase (P < 0.05), while the number of cells in S phase and G2 phase was significantly increased in RORα-silenced cells compared to control cells (P < 0.05) (Fig. 1J). These results suggest that RORα inhibits cell proliferation by inducing G1/S arrest in mammary epithelial cells.
To confirm that RORα inhibits E2F1-targeted genes, a couple of genes that regulate cell cycle progression and DNA replication were analyzed by RT-PCR. The results showed that mRNA levels of CDC6 (P < 0.01), ESPL1 (P < 0.01), MCM4 (P < 0.01), and POLE2 (P < 0.01) were significantly downregulated in RORα-expressing MDA-MB 231 cells compared with control cells (P < 0.01) (Fig. 2A). Similar results were also observed with MDA-MB 157 cells (Fig. 2A). Silencing RORα in S1 cells enhanced the expression of CDC6 (P < 0.01), MCM4 (P < 0.01), and POLE2 (P < 0.05) (Fig. 2B). Eln, a gene that is not regulated by E2F1, was used as a negative control in real-time RT-PCR (data not shown). To examine whether RORα directly regulates E2F1 transcriptional activity, luciferase assays were performed with MDA-MB 231 cells that were stably transfected with the E2F1 response report vector (E2F1-Luc). Introducing exogenous RORα significantly inhibited the luciferase activity driven by E2F1 (P < 0.01) (Fig. 2C). These results indicate that RORα suppressed E2F1-regulated transcription.
FIG 2.

RORα inhibits expression of E2F1-targeted genes. (A) mRNA levels of E2F1-targeted genes CDC6, ESPL1, MCM4, and POLE2 in control and RORα-expressing MDA-MB 231 cells and MDA-MB 157 cells were analyzed by real-time RT-PCR. Expression of these E2F1 target genes was significantly downregulated in RORα-expressing MDA-MB 231 cells and MDA-MB 157 cells compared to control cells (n = 4; *, P < 0.05; **, P < 0.01). (B) Real-time RT-PCR results showed that mRNA levels of CDC6, ESPL1, MCM4, and POLE2 were upregulated in RORα knockdown S1 cells compared with shRNA control S1 cells (n = 4; *, P < 0.05; **, P < 0.01). (C) Luciferase analysis was performed for MDA-MB 231 cells that were stably transfected with control (ctrl-Luc) or E2F1 response luciferase reporter vector (E2F1-Luc). RORα inhibited E2F1-driven reporter activity (n = 3; **, P < 0.01). (D) E2F1 protein levels in control and RORα-expressing MDA-MB 231 cells were examined by Western blotting. (E and F) Chromatin immunoprecipitation results showed that overexpression of RORα significantly inhibited binding of E2F1 to the promoter regions of CDC6 (E) and POLE2 (F). Levels of AcH3 and AcH4 in the promoter regions of CDC6 and POLE2 were also reduced in RORα-expressing cells (n = 3; *, P < 0.05; **, P < 0.01). The ChIP data were normalized to input DNA, and the results represent the relative changes in RORα group compared to the control group.
We found that RORα expression had little effect on E2F1 protein levels in MDA-MB 231 cells (Fig. 2D). To understand how RORα inhibits E2F1 activity, we performed chromatin immunoprecipitation experiments. The E2F1-chromatin complexes were pulled down with anti-E2F1 antibody from the control and RORα-expressing MDA-MB 231 cells. Enrichment of specific DNA fragments was measured by quantitative PCR. Ectopic expression of RORα significantly inhibited binding of E2F1 to the promoter regions of CDC6 (P < 0.01) and POLE2 (P < 0.05) (Fig. 2E and F). Levels of acetylated histone H3 (AcH3) and H4 (AcH4) in the promoter regions of CDC6 and POLE2 were also significantly reduced upon RORα expression (Fig. 2E and F). Thus, RORα inhibits binding of E2F1 to the targeted genes and suppresses E2F1-induced chromatin remodeling.
RORα binds to E2F1 through the heptad repeat and marked box domain.
During cell cycle progression, a number of coregulators, such as pRb, Sp1, and KAP1, dynamically bind to E2F1 and modulate its activity (36–38). We hypothesized that RORα suppresses E2F1 transcription factor activity through a similar mechanism. Coimmunoprecipitation experiments were performed to determine whether RORα binds to E2F1 in epithelial cells. RORα and/or HA-E2F1 vector was cotransfected in HEK293 cells, and the protein complexes were immunoprecipitated by anti-HA antibody. A significant amount of RORα was pulled down in the HA-E2F1-expressing cells but not in the control cells, indicating that RORα binds to E2F1 in HEK293 cells (Fig. 3A). The interaction between RORα and E2F1 was confirmed in MDA-MB 231 cells (Fig. 3B). We also showed interaction between endogenous RORα and E2F1 in S1 cells by coimmunoprecipitation using E2F1 antibody (Fig. 3C). In addition, confocal analysis showed that RORα was partially colocalized with E2F1 in the nuclei of MDA-MB 231 cells (data not shown).
FIG 3.

RORα binds to the heptad repeat and marked box region of E2F1. (A) RORα was coimmunoprecipitated with E2F1 in HEK293 cells. The cells were transfected with RORα and/or E2F1 expression vectors. (B) RORα was coimmunoprecipitated with E2F1 in MDA-MB 231 cells. The cells were transfected with RORα and/or E2F1 expression vectors. (C) Binding of endogenous RORα and E2F1 was detected in S1 cells by coimmunoprecipitation experiments. (D) Binding of GST-tagged RORα to E2F1was detected by in vitro GST pulldown assays. DBD, DBD and hinge domain of RORα; LBD, LBD domain of RORα. The DBD and hinge region interacted with E2F1. (E) Binding of GST-tagged E2F1 to RORα was detected by in vitro GST pulldown assays. Deletion of the heptad repeat and marked box region (amino acid 195 to 317) of E2F1 significantly reduced binding of E2F1 to RORα.
RORα exhibits a typical nuclear receptor domain structure consisting of three major domains: a DNA-binding domain (DBD), hinge, and ligand-binding domain (LBD). To understand which domain of RORα is required for E2F1 binding, we performed an in vitro GST pulldown assay. The results showed that the DBD-and-hinge region was able to interact with E2F1 (Fig. 3D). E2F1 contains a cyclin A-binding domain, helix-loop-helix DNA-binding domain, heptad repeat, marked box, and transactivation domain (Fig. 3E). To further map the regions in E2F1 involved in RORα-E2F1complex formation, in vitro GST pulldown assays were performed with a panel of GST-tagged E2F1 mutants. We found that deletion of the heptad repeat and marked box domain (amino acid 195 to 317) in E2F1 dramatically reduced the ability of E2F1 to bind with RORα, indicating that this region is required for RORα-E2F1 interaction. The heptad repeat domain mediates the heterodimer formation of E2F1 and DP1. Interestingly, binding of RORα had no effect on formation of E2F1-DP1 dimerization (Fig. 4A and B). The results of in vivo and in vitro binding assays indicate that RORα directly interacts with E2F1 to inhibit its activity, but how binding of RORα inhibits E2F1 transcriptional activity remained to be determined.
FIG 4.

RORα reduces E2F1 acetylation by recruiting HDAC1. (A) Coimmunoprecipitation experiments were performed with HEK293 cells cotransfected with RORα, Flag-tagged E2F1, and HA-tagged DP1. (B) A binding assay showed that binding of Flag-tagged DP1 to GST-tagged E2F1 was not inhibited by RORα. (C) Lysine acetylation of endogenous E2F1 was inhibited by RORα in MDA-MB 231 cells. (D) RORα and E2F1 both interacted with HDAC1 and were examined by immunoprecipitation experiments performed with HEK293 cells transfected with HDAC1, RORα, and E2F1. (E) A luciferase reporter assay showed that inhibition of HDAC1 activity (TSA treatment or knockdown of HDAC1 by shHDAC1-containing lentivirus) rescued E2F1 activity in RORα-expressing MDA-MB 231 cells (n = 3; **, P < 0.01). (F) Immunoprecipitation experiment results showed that knockdown of HDAC1 rescued E2F1 acetylation in RORα-expressing MDA-MB 231 cells. (G) EdU staining results showed that silencing HDAC1 partially rescued cell proliferation in RORα-expressing MDA-MB 231 cells (n = 3; *, P < 0.05).
RORα recruits HDAC1 to E2F1 and reduces its acetylation.
It has been shown that covalent modifications, such as acetylation, regulate binding of E2F1 to chromatin and E2F1-dependent transcription (40, 41). To determine whether binding of RORα inhibits E2F1 activity through modulation of the covalent modifications, we checked the acetylation levels on endogenous E2F1 protein in the control and the RORα-expressing MDA-MB 231 cells using immunoprecipitation. RORα expression significantly inhibited the lysine acetylation on E2F1 (Fig. 4C). Since HDACs play a critical role in reducing protein acetylation (46, 47), we asked whether RORα reduces E2F1 acetylation by recruiting HDACs to the protein complexes. We examined interaction between RORα and endogenous HDAC1 in HEK293 cells using coimmunoprecipitation, and the data showed that HDAC1 bound to RORα (data not shown). To determine whether RORα recruits HDAC1 to E2F1, coimmunoprecipitation experiments were performed in HDAC1-, E2F1-, and/or RORα-expressing HEK293 cells. Expression of RORα dramatically enhanced interaction between HDAC1 and E2F1, whereas a weak association between E2F1 and HDAC1 was detected in the absence of RORα (Fig. 4D). These results indicate that RORα recruits HDAC1 to E2F1, which, in turn, reduces acetylated lysine levels in E2F1.
To assess whether RORα inhibits E2F1 by recruiting HDAC1 to E2F1, we performed rescue experiments with MDA-MB 231 cells containing the E2F1-Luc reporter construct. Expression of RORα significantly inhibits the luciferase activity in these cells (Fig. 2C and 4E). Inhibiting HDAC1 with TSA or shRNA against HDAC1 enhanced E2F1 transcription activity that was suppressed by RORα (Fig. 4E). Furthermore, silencing HDAC1 partially rescued E2F1 acetylation (Fig. 4F) and cell proliferation (Fig. 4G) in RORα-expressing MDA-MB 231 cells. These data indicate that HDAC1 is required for the inhibitory activity of RORα on E2F1.
RORα reversely correlated with cell proliferation and E2F1 target gene expression in mammary glands.
The terminal end bud, a bulbous invasive structure at the tip of growing ducts (48–50), has much higher cell proliferation than does the mature duct (19, 20). This spatial regulation of cell proliferation plays important roles in normal mammary branching morphogenesis. To understand the roles of the RORα-E2F1 axis in spatial regulation of cell proliferation in mammary tissue, we analyzed the expression of RORα and E2F1-targeted genes in a published microarray data set generated from mouse terminal end bud and mature duct tissues (51). RORα mRNA levels were significantly reduced in the terminal end buds compared to duct glands, while expression of E2F1-targeted genes was significantly upregulated in the terminal end bulb (Fig. 5A to C). Immunohistochemistry analysis confirmed that RORα expression was reduced in terminal end buds compared to ductal epithelial cells in mouse mammary glands. There were more Ki67-positive cells in the terminal end buds than in ducts (Fig. 5D), which is consistent with the previous finding that cells in terminal end buds are highly proliferative (19, 20). A reverse correlation of RORα with cell proliferation and expression of the E2F1 target genes in terminal end buds and ductal epithelial cells suggests potential roles for RORα in regulating mammary branching morphogenesis.
FIG 5.

RORα levels are reversely correlated with cell proliferation and E2F1 target expression in mammary glands. (A) Heat map showing that E2F1-targeted genes were activated in terminal end buds (TEBs) compared to mammary ducts (DUC). (B) RORα mRNA levels were lower in TEBs than in DUC (*, P < 0.05). (C) Quantification data showing that expression levels of E2F1 target genes were significantly reduced in terminal end buds (**, P < 0.01). A bar graph represents the sum of the mRNA levels of E2F1 target genes in the microarray data generated from ductal and TEB epithelial cells. In panels B and C, values on the y axis are mRNA level (log2). (D) Immunohistochemistry analysis of RORα and Ki67 expression in mammary glands derived from 5-week-old female mice. Ki67-positive cells are enriched in terminal end buds, while intensive RORα staining was detected in mammary ducts (scale bar, 20 μm). (E) Expression of Ki67 was analyzed in the mammary ducts and terminal end buds derived from transplanted mammary glands. The primary epithelial cells were infected with lentivirus containing control shRNA or shRORα constructs before the transplantation (scale bar, 40 μm). (F) Bar graph showing the ratio of Ki67 cell in shControl and shRORα-expressing ductal structure and terminal end buds of mammary glands. Increased cell proliferation was detected in RORα knockdown mammary gland epithelial cells compared with that in the control group (n = 5; **, P < 0.01). (G) Whole-mount staining of transplanted mammary glands harvested 5 weeks following transplantation (scale bar, 400 μm). (H) Quantification of whole-mount staining results showed that RORα-silenced mammary glands had increased side branching (n = 3; *, P < 0.05).
Next, we performed mammary gland transplant experiments to examine the function of RORα in spatial regulation of cell proliferation during branching morphogenesis. Primary mammary organoids were isolated from FVB mice and infected with lentivirus containing control shRNA or shRORα. The infected mammary gland organoids were transplanted into 3-week-old FVB mice with cleared fat pads. Five weeks following transplantation, the transplanted mammary glands were taken out for immunohistochemistry analysis and whole-mount staining. Ki67 staining showed that knockdown of RORα significantly enhanced cell proliferation in the ductal epithelial cells (Fig. 5E and F) but had little effect on cell proliferation in the terminal end buds (Fig. 5E and F). We also found that RORα-silenced mammary glands had increased side branching (Fig. 5G and H), suggesting that RORα-induced growth arrest is important for normal mammary branch morphogenesis.
DISCUSSION
A function for RORα in regulating cell proliferation has been reported previously (7, 25), but how RORα inhibits cell proliferation in normal tissue development and disease progression remains unclear. In the present study, we found that RORα bound to E2F1 as a cofactor and inhibited E2F1 activity by recruiting HDAC1 to the RORα-E2F1 complex. These findings identify a noncanonical pathway of RORα to suppress E2F1-targeted gene and cell cycle progression.
Cross talk between E2F and other transcription factors has been reported previously (52–56). For instance, interaction between aryl hydrocarbon receptor (AHR) and E2F1 has been detected in mouse hepatoma cells, and binding of AHR suppresses E2F1-mediated apoptosis by modulating E2F1 transcriptional activity (54, 55, 57). We showed that binding of RORα reduced acetylation levels on E2F1 and inhibits its DNA binding activity. Lysine acetylation is an important posttranslational modification that regulates E2F1 activity. It has been reported that lysine residues 117, 120, and 125 on E2F1 are acetylated by the acetyltransferase complex CBP/p/CAF and that the acetyl group can be removed by HDAC proteins. Acetylation of E2F1 enhances its DNA binding ability and increases protein stability (41). It has been shown that Rb, mSin3B, Ebp1, and RbAp48 recruit class I HDACs (HDAC 1, HDAC2, and HDAC3) to E2Fs and modulate E2F target gene expression (58–61). Our data showed that RORα enhanced interaction between E2F1 and HDAC1; reducing HDAC1 expression or inhibiting its activity rescued E2F1 activity suppressed by RORα. However, microarray data showed that the majority, but not all, of E2F1-targeted genes are inhibited by RORα, suggesting that some E2F1-targeted gene may be dominantly regulated by other transcription factors or epigenetically silenced. These results identify a novel pathway by which nuclear receptor modulates E2F1 transcriptional activity.
Cell proliferation is tightly regulated during branching morphogenesis, a fundamental developmental process that shapes the formation of mammary glands (21, 22). Steroid hormones, local growth factors, and their receptors play important roles in mammary gland branching morphogenesis. For example, deletion of insulin-like growth factor I receptor in mammary epithelial cells inhibits cell proliferation in terminal end buds and impairs mammary branching morphogenesis in mice (19). We found that RORα expression was reversely correlated with cell proliferation in terminal end bud and mammary ducts, suggesting RORα as the inhibitor of cell proliferation during branch morphogenesis. In fact, knockdown of RORα increased E2F1 target gene expression and cell proliferation in the mammary ducts and enhanced side branching in the transplanted mammary gland. Therefore, increased RORα expression in mammary ducts is crucial to maintain the ductal structure by inhibiting cell proliferation (Fig. 6). In addition, it has been reported that Wnt signaling has been identified as a downstream target of progesterone to induce side-branch morphogenesis (62, 63). RORα has been shown to bind to β-catenin and inhibits the Wnt pathway during colon cancer development (7). Thus, the Wnt/β-catenin pathway is also a potential target of RORα to regulate branching morphogenesis.
FIG 6.
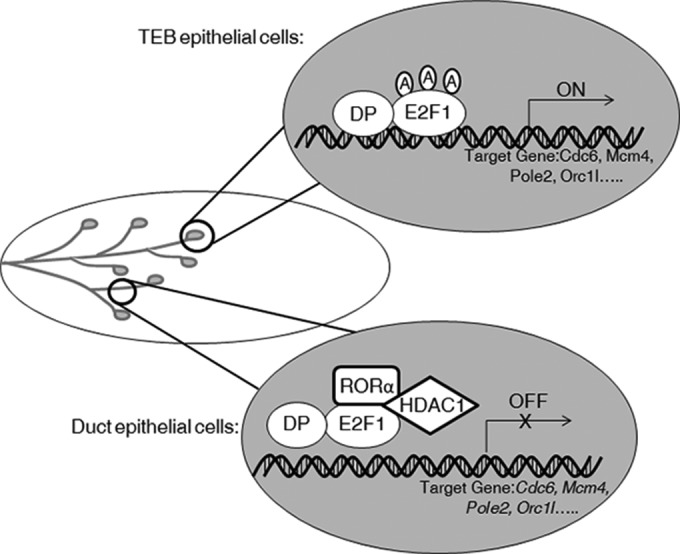
RORα suppresses E2F1 transcriptional activity by enhancing HDAC1-E2F1 interaction in ductal epithelial cells during mammary gland morphogenesis.
Uncontrolled cell proliferation is a hallmark of cancer development and progression, and dysregulation of the pRB/E2F pathway plays a critical role in the enhanced cell proliferation in cancer tissue. Mutation or deletion of pRB has been detected in many cancers (64–67). However, it has been shown that in human breast cancer patients, only about 20 to 30% of breast tumors are pRB deficient and less than 10% of tumors show inactivated or mutated pRB (64, 65). These results suggest that other proteins or pathways inhibit expression or activation of E2F1 during breast cancer development. Our previous study shows that 70% of human breast cancers have reduced RORα expression (25). Ectopic expression of RORα in breast cancer cells reduces cell proliferation and inhibits and tumor growth. Thus, aberrant activation of the E2F1 pathway and cell cycle progression in breast cancer are at least partially due to reduced RORα expression. Further investigation of the connection of RORα with E2F1 activation and cell proliferation in breast cancer tissue may lead to discovery of a potential pathway to inhibit breast cancer progression.
ACKNOWLEDGMENTS
This study was supported by grants from the ACS (IRG 85-001-22 to R. Xu) and AHA (12SDG8600000 to R. Xu). This work was also supported by the National Center for Research Resources and the National Center for Advancing Translational Sciences, National Institutes of Health (grant UL1TR000117).
The content of this article is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
Footnotes
Published ahead of print 2 June 2014
REFERENCES
- 1.Evans RM. 1988. The steroid and thyroid hormone receptor superfamily. Science 240:889–895. 10.1126/science.3283939 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Olefsky JM. 2001. Nuclear receptor minireview series. J. Biol. Chem. 276:36863–36864. 10.1074/jbc.R100047200 [DOI] [PubMed] [Google Scholar]
- 3.Delerive P, Monte D, Dubois G, Trottein F, Fruchart-Najib J, Mariani J, Fruchart JC, Staels B. 2001. The orphan nuclear receptor ROR alpha is a negative regulator of the inflammatory response. EMBO Rep. 2:42–48. 10.1093/embo-reports/kve007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dussault I, Fawcett D, Matthyssen A, Bader JA, Giguere V. 1998. Orphan nuclear receptor ROR alpha-deficient mice display the cerebellar defects of staggerer. Mech. Dev. 70:147–153. 10.1016/S0925-4773(97)00187-1 [DOI] [PubMed] [Google Scholar]
- 5.Dzhagalov I, Giguere V, He YW. 2004. Lymphocyte development and function in the absence of retinoic acid-related orphan receptor alpha. J. Immunol. 173:2952–2959. 10.4049/jimmunol.173.5.2952 [DOI] [PubMed] [Google Scholar]
- 6.Lau P, Nixon SJ, Parton RG, Muscat GE. 2004. RORalpha regulates the expression of genes involved in lipid homeostasis in skeletal muscle cells: caveolin-3 and CPT-1 are direct targets of ROR. J. Biol. Chem. 279:36828–36840. 10.1074/jbc.M404927200 [DOI] [PubMed] [Google Scholar]
- 7.Lee JM, Kim IS, Kim H, Lee JS, Kim K, Yim HY, Jeong J, Kim JH, Kim JY, Lee H, Seo SB, Kim H, Rosenfeld MG, Kim KI, Baek SH. 2010. RORalpha attenuates Wnt/beta-catenin signaling by PKCalpha-dependent phosphorylation in colon cancer. Mol. Cell 37:183–195. 10.1016/j.molcel.2009.12.022 [DOI] [PubMed] [Google Scholar]
- 8.Sato TK, Panda S, Miraglia LJ, Reyes TM, Rudic RD, McNamara P, Naik KA, FitzGerald GA, Kay SA, Hogenesch JB. 2004. A functional genomics strategy reveals Rora as a component of the mammalian circadian clock. Neuron 43:527–537. 10.1016/j.neuron.2004.07.018 [DOI] [PubMed] [Google Scholar]
- 9.Steinmayr M, Andre E, Conquet F, Rondi-Reig L, Delhaye-Bouchaud N, Auclair N, Daniel H, Crepel F, Mariani J, Sotelo C, Becker-Andre M. 1998. staggerer phenotype in retinoid-related orphan receptor alpha-deficient mice. Proc. Natl. Acad. Sci. U. S. A. 95:3960–3965. 10.1073/pnas.95.7.3960 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhu Y, McAvoy S, Kuhn R, Smith DI. 2006. RORA, a large common fragile site gene, is involved in cellular stress response. Oncogene 25:2901–2908. 10.1038/sj.onc.1209314 [DOI] [PubMed] [Google Scholar]
- 11.Solt LA, Burris TP. 2012. Action of RORs and their ligands in (patho)physiology. Trends Endocrinol. Metab. 23:619–627. 10.1016/j.tem.2012.05.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jetten AM. 2009. Retinoid-related orphan receptors (RORs): critical roles in development, immunity, circadian rhythm, and cellular metabolism. Nucl. Recept. Signal. 7:e003. 10.1621/nrs.07003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jetten AM, Kurebayashi S, Ueda E. 2001. The ROR nuclear orphan receptor subfamily: critical regulators of multiple biological processes. Prog. Nucleic Acid Res. Mol. Biol. 69:205–247. 10.1016/S0079-6603(01)69048-2 [DOI] [PubMed] [Google Scholar]
- 14.Gold DA, Baek SH, Schork NJ, Rose DW, Larsen DD, Sachs BD, Rosenfeld MG, Hamilton BA. 2003. RORalpha coordinates reciprocal signaling in cerebellar development through sonic hedgehog and calcium-dependent pathways. Neuron 40:1119–1131. 10.1016/S0896-6273(03)00769-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lau P, Bailey P, Dowhan DH, Muscat GE. 1999. Exogenous expression of a dominant negative RORalpha1 vector in muscle cells impairs differentiation: RORalpha1 directly interacts with p300 and myoD. Nucleic Acids Res. 27:411–420. 10.1093/nar/27.2.411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Harding HP, Atkins GB, Jaffe AB, Seo WJ, Lazar MA. 1997. Transcriptional activation and repression by RORalpha, an orphan nuclear receptor required for cerebellar development. Mol. Endocrinol. 11:1737–1746. 10.1210/mend.11.11.0002 [DOI] [PubMed] [Google Scholar]
- 17.Kim H, Lee JM, Lee G, Bhin J, Oh SK, Kim K, Pyo KE, Lee JS, Yim HY, Kim KI, Hwang D, Chung J, Baek SH. 2011. DNA damage-induced RORalpha is crucial for p53 stabilization and increased apoptosis. Mol. Cell 44:797–810. 10.1016/j.molcel.2011.09.023 [DOI] [PubMed] [Google Scholar]
- 18.Huang S, Ingber DE. 1999. The structural and mechanical complexity of cell-growth control. Nat. Cell Biol. 1:E131–E138. 10.1038/13043 [DOI] [PubMed] [Google Scholar]
- 19.Bonnette SG, Hadsell DL. 2001. Targeted disruption of the IGF-I receptor gene decreases cellular proliferation in mammary terminal end buds. Endocrinology 142:4937–4945. 10.1210/endo.142.11.8500 [DOI] [PubMed] [Google Scholar]
- 20.Russo J, Russo IH. 1980. Influence of differentiation and cell kinetics on the susceptibility of the rat mammary gland to carcinogenesis. Cancer Res. 40:2677–2687 [PubMed] [Google Scholar]
- 21.Williams JM, Daniel CW. 1983. Mammary ductal elongation: differentiation of myoepithelium and basal lamina during branching morphogenesis. Dev. Biol. 97:274–290. 10.1016/0012-1606(83)90086-6 [DOI] [PubMed] [Google Scholar]
- 22.Brantley DM, Chen CL, Muraoka RS, Bushdid PB, Bradberry JL, Kittrell F, Medina D, Matrisian LM, Kerr LD, Yull FE. 2001. Nuclear factor-kappaB (NF-kappaB) regulates proliferation and branching in mouse mammary epithelium. Mol. Biol. Cell 12:1445–1455. 10.1091/mbc.12.5.1445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cohen SM, Ellwein LB. 1990. Cell proliferation in carcinogenesis. Science 249:1007–1011. 10.1126/science.2204108 [DOI] [PubMed] [Google Scholar]
- 24.Mommers EC, van Diest PJ, Leonhart AM, Meijer CJ, Baak JP. 1999. Balance of cell proliferation and apoptosis in breast carcinogenesis. Breast Cancer Res. Treat. 58:163–169. 10.1023/A:1006396103777 [DOI] [PubMed] [Google Scholar]
- 25.Xiong G, Wang C, Evers BM, Zhou BP, Xu R. 2012. RORalpha suppresses breast tumor invasion by inducing SEMA3F expression. Cancer Res. 72:1728–1739. 10.1158/0008-5472.CAN-11-2762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Koff A, Giordano A, Desai D, Yamashita K, Harper JW, Elledge S, Nishimoto T, Morgan DO, Franza BR, Roberts JM. 1992. Formation and activation of a cyclin E-cdk2 complex during the G1 phase of the human cell cycle. Science 257:1689–1694. 10.1126/science.1388288 [DOI] [PubMed] [Google Scholar]
- 27.Graña X, Reddy EP. 1995. Cell cycle control in mammalian cells: role of cyclins, cyclin dependent kinases (CDKs), growth suppressor genes and cyclin-dependent kinase inhibitors (CKIs). Oncogene 11:211–219 [PubMed] [Google Scholar]
- 28.Ren B, Cam H, Takahashi Y, Volkert T, Terragni J, Young RA, Dynlacht BD. 2002. E2F integrates cell cycle progression with DNA repair, replication, and G(2)/M checkpoints. Genes Dev. 16:245–256. 10.1101/gad.949802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Harrington EA, Bruce JL, Harlow E, Dyson N. 1998. pRB plays an essential role in cell cycle arrest induced by DNA damage. Proc. Natl. Acad. Sci. U. S. A. 95:11945–11950. 10.1073/pnas.95.20.11945 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Coats S, Flanagan WM, Nourse J, Roberts JM. 1996. Requirement of p27Kip1 for restriction point control of the fibroblast cell cycle. Science 272:877–880. 10.1126/science.272.5263.877 [DOI] [PubMed] [Google Scholar]
- 31.Date DA, Burrows AC, Venere M, Jackson MW, Summers MK. 2013. Coordinated regulation of p31(Comet) and Mad2 expression is required for cellular proliferation. Cell Cycle 12:3824–3832. 10.4161/cc.26811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.DeGregori J, Kowalik T, Nevins JR. 1995. Cellular targets for activation by the E2F1 transcription factor include DNA synthesis- and G1/S-regulatory genes. Mol. Cell. Biol. 15:4215–4224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.DeGregori J, Leone G, Miron A, Jakoi L, Nevins JR. 1997. Distinct roles for E2F proteins in cell growth control and apoptosis. Proc. Natl. Acad. Sci. U. S. A. 94:7245–7250. 10.1073/pnas.94.14.7245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wu L, Timmers C, Maiti B, Saavedra HI, Sang L, Chong GT, Nuckolls F, Giangrande P, Wright FA, Field SJ, Greenberg ME, Orkin S, Nevins JR, Robinson ML, Leone G. 2001. The E2F1-3 transcription factors are essential for cellular proliferation. Nature 414:457–462. 10.1038/35106593 [DOI] [PubMed] [Google Scholar]
- 35.Wu X, Levine AJ. 1994. p53 and E2F-1 cooperate to mediate apoptosis. Proc. Natl. Acad. Sci. U. S. A. 91:3602–3606. 10.1073/pnas.91.9.3602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lin SY, Black AR, Kostic D, Pajovic S, Hoover CN, Azizkhan JC. 1996. Cell cycle-regulated association of E2F1 and Sp1 is related to their functional interaction. Mol. Cell. Biol. 16:1668–1675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dick FA, Dyson N. 2003. pRB contains an E2F1-specific binding domain that allows E2F1-induced apoptosis to be regulated separately from other E2F activities. Mol. Cell 12:639–649. 10.1016/S1097-2765(03)00344-7 [DOI] [PubMed] [Google Scholar]
- 38.Wang C, Rauscher FJ, III, Cress WD, Chen J. 2007. Regulation of E2F1 function by the nuclear corepressor KAP1. J. Biol. Chem. 282:29902–29909. 10.1074/jbc.M704757200 [DOI] [PubMed] [Google Scholar]
- 39.Weinberg RA. 1995. The retinoblastoma protein and cell cycle control. Cell 81:323–330. 10.1016/0092-8674(95)90385-2 [DOI] [PubMed] [Google Scholar]
- 40.Ivanova IA, Nakrieko KA, Dagnino L. 2009. Phosphorylation by p38 MAP kinase is required for E2F1 degradation and keratinocyte differentiation. Oncogene 28:52–62. 10.1038/onc.2008.354 [DOI] [PubMed] [Google Scholar]
- 41.Martínez-Balbás MA, Bauer UM, Nielsen SJ, Brehm A, Kouzarides T. 2000. Regulation of E2F1 activity by acetylation. EMBO J. 19:662–671. 10.1093/emboj/19.4.662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lee GY, Kenny PA, Lee EH, Bissell MJ. 2007. Three-dimensional culture models of normal and malignant breast epithelial cells. Nat. Methods 4:359–365. 10.1038/nmeth1015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gentleman RC, Carey VJ, Bates DM, Bolstad B, Dettling M, Dudoit S, Ellis B, Gautier L, Ge Y, Gentry J, Hornik K, Hothorn T, Huber W, Iacus S, Irizarry R, Leisch F, Li C, Maechler M, Rossini AJ, Sawitzki G, Smith C, Smyth G, Tierney L, Yang JY, Zhang J. 2004. Bioconductor: open software development for computational biology and bioinformatics. Genome Biol. 5:R80. 10.1186/gb-2004-5-10-r80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, Mesirov JP. 2005. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. U. S. A. 102:15545–15550. 10.1073/pnas.0506580102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Fata JE, Mori H, Ewald AJ, Zhang H, Yao E, Werb Z, Bissell MJ. 2007. The MAPK(ERK-1,2) pathway integrates distinct and antagonistic signals from TGFalpha and FGF7 in morphogenesis of mouse mammary epithelium. Dev. Biol. 306:193–207. 10.1016/j.ydbio.2007.03.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Glozak MA, Sengupta N, Zhang X, Seto E. 2005. Acetylation and deacetylation of non-histone proteins. Gene 363:15–23. 10.1016/j.gene.2005.09.010 [DOI] [PubMed] [Google Scholar]
- 47.Marks P, Rifkind RA, Richon VM, Breslow R, Miller T, Kelly WK. 2001. Histone deacetylases and cancer: causes and therapies. Nat. Rev. Cancer 1:194–202. 10.1038/35106079 [DOI] [PubMed] [Google Scholar]
- 48.Kenney NJ, Smith GH, Lawrence E, Barrett JC, Salomon DS. 2001. Identification of stem cell units in the terminal end bud and duct of the mouse mammary gland. J. Biomed. Biotechnol. 1:133–143. 10.1155/S1110724301000304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Moraes RC, Zhang X, Harrington N, Fung JY, Wu MF, Hilsenbeck SG, Allred DC, Lewis MT. 2007. Constitutive activation of smoothened (SMO) in mammary glands of transgenic mice leads to increased proliferation, altered differentiation and ductal dysplasia. Development 134:1231–1242. 10.1242/dev.02797 [DOI] [PubMed] [Google Scholar]
- 50.Watson CJ, Khaled WT. 2008. Mammary development in the embryo and adult: a journey of morphogenesis and commitment. Development 135:995–1003. 10.1242/dev.005439 [DOI] [PubMed] [Google Scholar]
- 51.Kouros-Mehr H, Werb Z. 2006. Candidate regulators of mammary branching morphogenesis identified by genome-wide transcript analysis. Dev. Dyn. 235:3404–3412. 10.1002/dvdy.20978 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Schug TT, Berry DC, Shaw NS, Travis SN, Noy N. 2007. Opposing effects of retinoic acid on cell growth result from alternate activation of two different nuclear receptors. Cell 129:723–733. 10.1016/j.cell.2007.02.050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Balk SP, Knudsen KE. 2008. AR, the cell cycle, and prostate cancer. Nucl. Recept. Signal. 6:e001. 10.1621/nrs.06001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Marlowe JL, Knudsen ES, Schwemberger S, Puga A. 2004. The aryl hydrocarbon receptor displaces p300 from E2F-dependent promoters and represses S phase-specific gene expression. J. Biol. Chem. 279:29013–29022. 10.1074/jbc.M404315200 [DOI] [PubMed] [Google Scholar]
- 55.Marlowe JL, Puga A. 2005. Aryl hydrocarbon receptor, cell cycle regulation, toxicity, and tumorigenesis. J. Cell. Biochem. 96:1174–1184. 10.1002/jcb.20656 [DOI] [PubMed] [Google Scholar]
- 56.Nguyen-Vu T, Vedin LL, Liu K, Jonsson P, Lin JZ, Candelaria NR, Candelaria LP, Addanki S, Williams C, Gustafsson JA, Steffensen KR, Lin CY. 2013. Liver x receptor ligands disrupt breast cancer cell proliferation through an E2F-mediated mechanism. Breast Cancer Res. 15:R51. 10.1186/bcr3443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Marlowe JL, Fan Y, Chang X, Peng L, Knudsen ES, Xia Y, Puga A. 2008. The aryl hydrocarbon receptor binds to E2F1 and inhibits E2F1-induced apoptosis. Mol. Biol. Cell 19:3263–3271. 10.1091/mbc.E08-04-0359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Magnaghi-Jaulin L, Groisman R, Naguibneva I, Robin P, Lorain S, Le Villain JP, Troalen F, Trouche D, Harel-Bellan A. 1998. Retinoblastoma protein represses transcription by recruiting a histone deacetylase. Nature 391:601–605. 10.1038/35410 [DOI] [PubMed] [Google Scholar]
- 59.Rayman JB, Takahashi Y, Indjeian VB, Dannenberg JH, Catchpole S, Watson RJ, te Riele H, Dynlacht BD. 2002. E2F mediates cell cycle-dependent transcriptional repression in vivo by recruitment of an HDAC1/mSin3B corepressor complex. Genes Dev. 16:933–947. 10.1101/gad.969202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zhang Y, Woodford N, Xia X, Hamburger AW. 2003. Repression of E2F1-mediated transcription by the ErbB3 binding protein Ebp1 involves histone deacetylases. Nucleic Acids Res. 31:2168–2177. 10.1093/nar/gkg318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Nicolas E, Ait-Si-Ali S, Trouche D. 2001. The histone deacetylase HDAC3 targets RbAp48 to the retinoblastoma protein. Nucleic Acids Res. 29:3131–3136. 10.1093/nar/29.15.3131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Robinson GW, Hennighausen L, Johnson PF. 2000. Side-branching in the mammary gland: the progesterone-Wnt connection. Genes Dev. 14:889–894 [PubMed] [Google Scholar]
- 63.Brisken C, Heineman A, Chavarria T, Elenbaas B, Tan J, Dey SK, McMahon JA, McMahon AP, Weinberg RA. 2000. Essential function of Wnt-4 in mammary gland development downstream of progesterone signaling. Genes Dev. 14:650–654 [PMC free article] [PubMed] [Google Scholar]
- 64.Borg A, Zhang QX, Alm P, Olsson H, Sellberg G. 1992. The retinoblastoma gene in breast cancer: allele loss is not correlated with loss of gene protein expression. Cancer Res. 52:2991–2994 [PubMed] [Google Scholar]
- 65.Wakasugi E, Kobayashi T, Tamaki Y, Nakano Y, Ito Y, Miyashiro I, Komoike Y, Miyazaki M, Takeda T, Monden T, Monden M. 1997. Analysis of phosphorylation of pRB and its regulatory proteins in breast cancer. J. Clin. Pathol. 50:407–412. 10.1136/jcp.50.5.407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Cote RJ, Dunn MD, Chatterjee SJ, Stein JP, Shi SR, Tran QC, Hu SX, Xu HJ, Groshen S, Taylor CR, Skinner DG, Benedict WF. 1998. Elevated and absent pRb expression is associated with bladder cancer progression and has cooperative effects with p53. Cancer Res. 58:1090–1094 [PubMed] [Google Scholar]
- 67.He J, Olson JJ, James CD. 1995. Lack of p16INK4 or retinoblastoma protein (pRb), or amplification-associated overexpression of cdk4 is observed in distinct subsets of malignant glial tumors and cell lines. Cancer Res. 55:4833–4836 [PubMed] [Google Scholar]