

Abstract
RecA is central to maintaining genome integrity in bacterial cells. Despite the near-ubiquitous conservation of RecA in eubacteria, the pathways that facilitate RecA loading and repair center assembly have remained poorly understood in Bacillus subtilis. Here, we show that RecA rapidly colocalizes with the DNA polymerase complex (replisome) immediately following DNA damage or damage-independent replication fork arrest. In Escherichia coli, the RecFOR and RecBCD pathways serve to load RecA and the choice between these two pathways depends on the type of damage under repair. We found in B. subtilis that the rapid localization of RecA to repair centers is strictly dependent on RecO and RecR in response to all types of damage examined, including a site-specific double-stranded break and damage-independent replication fork arrest. Furthermore, we provide evidence that, although RecF is not required for RecA repair center formation in vivo, RecF does increase the efficiency of repair center assembly, suggesting that RecF may influence the initial stages of RecA nucleation or filament extension. We further identify single-stranded DNA binding protein (SSB) as an additional component important for RecA repair center assembly. Truncation of the SSB C terminus impairs the ability of B. subtilis to form repair centers in response to damage and damage-independent fork arrest. With these results, we conclude that the SSB-dependent recruitment of RecOR to the replisome is necessary for loading and organizing RecA into repair centers in response to DNA damage and replication fork arrest.
INTRODUCTION
DNA repair pathways are critical for genome maintenance in all living cells (for a review, see reference 1). Both prokaryotic and eukaryotic organisms are constantly exposed to endogenous and exogenous sources of DNA damage that compromise genome integrity. In bacterial cells, RecA is central to genome integrity and is important for strand exchange during homologous recombination, stabilizing stalled replication forks, and induction of the SOS transcriptional response to DNA damage (for reviews, see references 2, 3, and 4).
In the Gram-positive bacterium Bacillus subtilis, the process that recruits RecA to excess single-stranded DNA (ssDNA) has remained unclear (5, 6). In B. subtilis and Escherichia coli, RecA fused to green fluorescent protein (GFP) forms foci in response to DNA damage (5–12). The visualization of assembled RecA-GFP foci (also referred to as repair centers) provides an in vivo assay to understand the events that lead to RecA loading within a living cell.
In E. coli, RecA is loaded through two major pathways: RecBCD and RecFOR (for reviews, see references 3, 13, 14, and 15). During double-strand break repair, double-stranded ends are processed by the RecBCD helicase-nuclease pathway (16). RecBCD generates a 3′ ssDNA extension after encountering a Chi site (17). Following end processing, RecBCD physically loads RecA onto the 3′ ssDNA extension (18, 19). The second loading pathway in E. coli is the RecFOR pathway (20). In this pathway, RecO and RecR form a complex that is required for RecA loading onto single-stranded DNA binding protein (SSB)-coated ssDNA in vitro. The third component, RecF, can accelerate this reaction, particularly on gapped DNA substrates (21–23). In E. coli, RecA also colocalizes to blocked forks and it was shown that RecF and RecO are important for efficient binding of RecA to stalled forks (8, 24). Collectively, these studies show that RecA in E. coli is loaded by the RecBCD and RecFOR pathways in vitro and in vivo.
In B. subtilis, the AddAB helicase-nuclease enzyme is required for double-stranded end processing and is homologous to E. coli RecBCD (25, 26). Like RecBCD, AddAB generates a 3′ ssDNA extension suitable for RecA binding and filament formation (25, 26). In contrast to RecBCD, B. subtilis AddAB has not been shown to physically bind or directly load RecA. B. subtilis also has the RecFOR pathway (27). A notable difference is that B. subtilis RecO is necessary and sufficient to load RecA onto SSB-coated ssDNA in vitro (28). Prior work showed that RecA-GFP foci were largely independent of recO and recR and that defects in AddAB do not substantially reduce RecA-GFP focus formation in vivo (5, 29). Therefore, in B. subtilis and other AddAB-containing bacteria, the process responsible for loading RecA in response to DNA breaks, strand gaps, and damage-independent replication fork arrest has remained unknown. This is surprising considering that AddAB exists in place of RecBCD in most bacterial organisms (30).
Here we report the proteins that are required for RecA to establish repair centers in response to endogenous DNA damage, exogenous sources of DNA damage, and damage-independent replication fork arrest. We use RecA-GFP focus formation as a proxy for RecA loading in vivo. Importantly, we found that RecA mediator proteins RecO and RecR are necessary for RecA to load and organize into repair centers in response to endogenous damage and to all sources of exogenous DNA damage and replication fork arrest tested. These results support the model that RecOR is critical for RecA filament nucleation on ssDNA in B. subtilis and show that AddAB does not direct loading and is dependent on RecOR for RecA loading following end processing. We found that RecF, another RecA mediator protein, did not influence RecA repair center formation in response to endogenous damage. However, in response to exogenous damage, RecA repair center formation was delayed in recF-deficient cells, showing that RecF participates in RecA nucleation or enhancement of filament extension. We also found that truncation of the C-terminal 35 amino acids of SSB prevents the DNA damage-dependent increase in assembly of RecA repair centers, supporting a model where SSB recruits RecA-mediator proteins to ssDNA for nucleation and assembly of RecA filaments. With these results, we conclude that RecA loading in B. subtilis is dependent on a single loading pathway, providing novel insight into the mechanism used to establish RecA repair centers in bacteria with the AddAB end processing pathway.
MATERIALS AND METHODS
Bacteriological methods.
The bacterial plasmids and strains are listed in Table S1 and Table S2 in the supplemental material, respectively. The following concentrations of compounds were used: 5 μg/ml chloramphenicol (Cat), 100 μg/ml spectinomycin (Spc), 12.5 μg/ml tetracycline (Tet), 0.5 μg/ml erythromycin (Erm), 5 μM IPTG (isopropyl-β-d-thiogalactopyranoside), and 0.125% xylose for recO and recR (31, 32). The concentrations of DNA-damaging agents are listed in the appropriate figure legends.
Live-cell microscopy.
Microscopy of live cells was completed as previously described (33–35; for a review, see reference 36). Briefly, a starting culture was inoculated in 1× defined S750 minimal media supplemented with 2% glucose at a starting optical density at 600 nm (OD600) of 0.05 and allowed to grow for three doublings (35, 36). Cultures were split, and one culture was challenged with the indicated damaging agent, while the other was left untreated as a control and grown for the indicated times. Following incubation, 200-μl aliquots of cells were stained with the vital membrane stain FM 4-64 [N-(3-triethylammoniumpropyl)-4-(p-diethylaminophenyl-hexatrienyl) pyridinium dibromide] (1 μg/ml) or 10 μM TMA-DPH [1-(4-(trimethylamino)phenyl)-6-phenylhexa-1,3,5-triene] and placed onto 1% agarose pads made with 1× Spizizen's salts. Cells were imaged with an Olympus BX61 microscope using an Olympus 100× oil immersion 1.45-numerical aperture (NA) total internal reflection fluorescence microscopy (TIRFM) objective lens (36). Imaging for each strain was performed independently a minimum of three times.
Compound synthesis.
All small-molecule compounds used in this study were synthesized by the Vahlteich Medicinal Chemistry Core, University of Michigan. 6-(p-Hydroxyphenylazo)-uracil (compound 3; HPUra) was synthesized by known methods (37) along with the novel silylated ether analogue compound 5 and the known analogue 6-(phenylhydrazino)-uracil (compound 6) (37). Complete experimental details are provided in the supplemental material. Stock solutions (20 mM) of assayed compounds were prepared in dimethyl sulfoxide (DMSO) or 50 mM KOH in the case of HPUra, stored in the dark, and kept for no more than 4 weeks. The compounds were added at 162 μM except when indicated otherwise. DMSO or KOH was used as a control.
MMC survival assay.
In general, sensitivity to DNA damage was determined as described previously (38–40). Briefly, strains were grown with the appropriate antibiotic overnight at 30°C. Single isolates were grown in 6 ml of LB in the dark to a final OD600 of 0.4 to 0.5. Five 1-ml aliquots of culture were centrifuged for 2 min at 10,000 rpm, and the supernatants were removed. Cells were resuspended in 1 ml of 0.85% saline solution containing various mitomycin C (MMC) concentrations (see Fig. 5). After 30 min, cells were pelleted, supernatant was aspirated, and cells were resuspended in 100 μl 0.85% saline solution. The suspension was serial diluted followed by plating on LB agar plates and was grown overnight at 30°C. Percent survival was calculated by taking the mean survival of an indicated treatment relative to the mean survival of untreated cultures.
FIG 5.

SSB is in the recOR pathway. (A) Immunoblot for RecA and DnaN in ssb3+ and ssbΔ35 cells. A 25-μg volume of soluble protein extract was applied per lane and separated via SDS-PAGE, followed by detection with affinity-purified RecA antisera in a 1:200 dilution and a 1:2,000 dilution for anti-DnaN antiserum. (B) Killing curve of cells with the indicated genetic backgrounds over a range of 0 to 0.3 μM MMC. The error bars represent the standard errors of the means (SEM).
Replication inhibition assays.
Cultures were grown in S750 minimal medium at 30°C to an OD600 of 0.35. Using the dnaB134 allele, replication initiation was inhibited while ongoing replication was completed by shifting cultures to 45°C for 1 h. Upon downshift to 30°C, aliquots of culture were removed at specific time points and added to an equal volume of ice-cold methanol prior to centrifugation and genomic DNA purification. When used, HPUra was added to cultures at a final concentration of 162 μM 20 min after the temperature downshift, with cells harvested at 60 min. Genomic DNA was sequenced on the Illumina HiSeq-2000 platform, generating 50-base single-end reads. Library preparation and sequencing were performed by the University of Michigan DNA Sequencing Core. Sequence data were aligned to the PY79 reference genome (accession number CP006881 [41]) using bwa aln with default parameters followed by bwa samse with the “-n” parameter set to 1 (42). For coverage calculation, the genome was split into 1,000-nucleotide-wide windows and coverage of each window was normalized per million reads mapped to the entire genome. Fold enrichment for each sample was calculated by dividing normalized coverage for each window by the normalized coverage of the same window for genomic DNA harvested immediately after the temperature downshift. Resulting enrichment values were then offset to achieve equal baseline values for the samples.
RESULTS
RecA colocalizes to the replisome in response to endogenous and exogenous DNA damage.
Previous work in E. coli and B. subtilis examined the ability of RecA-GFP to organize into foci (5–12, 43). Prior experiments have tested RecA-GFP as the only source of RecA in vivo (6, 8, 9, 38) GFP-RecA was expressed ectopically as the sole source of RecA in cells or in merodiploid cells with the native recA allele intact (5, 12). Here, we examined RecA-GFP as the only source of intracellular RecA, expressed at its native locus and under the control of its native promoter (6). The fusion allele was fully functional at low levels of damage (see Fig. S1 and results in the supplemental material). Unless otherwise stated, the experiments described below were performed under conditions of DNA damage where the recA-GFP allele was fully functional.
One current model suggests that RecA predominantly localizes away from the replisome in response to DNA damage and that replication is not required for repair center formation (44), while other work shows that replication is required for RecA-GFP to form foci (6) and that these foci form at the midcell (6, 8, 12). To differentiate between these two models, we constructed a strain with fluorescent fusions to RecA and the replisome marker DnaX (see Materials and Methods). Imaging of this strain showed that in untreated cells, ∼75% of RecA foci colocalized with DnaX (replisome) foci. This observation, in combination with the results of prior work (6), leads us to suggest that RecA initiates filament formation to establish repair centers at the replication fork followed by a search for homologous DNA. Overall, this result suggests that the bulk of the endogenous lesions occurring under the growth conditions used here elicit recruitment of RecA to the replisome (Fig. 1A).
FIG 1.

RecA colocalizes to the replisome. (A) Percentage of RecA-GFP foci colocalized with DnaX-mCherry during the indicated treatments. (B and C) Representative micrographs of RecA-GFP and DnaX-mCherry in cells challenged with phleomycin (B) and HPUra (C). The white bar represents 4 μm; membranes were stained with TMA-DPH.
With this result, we asked if different forms of DNA damage cause RecA to differentially localize to or away from the replisome. Our imaging results showed that following challenge with mitomycin C (MMC), an agent that forms mostly monoadducts and interstrand cross-links (45), ∼65% of RecA foci colocalized with replisomes. Similarly, cells challenged with phleomycin, a DNA-break-inducing peptide (46), showed that ∼68% of RecA colocalized with replisomes. We found it striking that the majority of RecA foci that assembled in response to MMC and phleomycin colocalized with the replisome, supporting earlier work showing that replication fork progression was important for RecA repair center assembly in response to a site-specific double-stranded break (DSB) (6). Following challenge with UV, a treatment that would result in DNA strand gaps and replication-stalling thymidine dimers, we found that foci formed immediately (within 5 min) and that ∼84% of RecA were colocalized with replisomes (Fig. 1). With these results, we conclude that the majority of RecA repair centers localized to the replisome in response to endogenous and exogenous sources of DNA damage, including phleomycin-generated DNA breaks and MMC-generated inter- and intrastrand cross-links, two types of damage that require homologous recombination for repair.
RecA colocalizes to the replisome in response to damage-independent fork arrest.
The compound HPUra has been widely used as a tool to rapidly block replication fork progression in B. subtilis and other Gram-positive bacteria (see, e.g., references 10, 37, 47, 48, 49, 50, and 51). HPUra is a replication-specific class III DNA polymerase inhibitor which blocks replication within minutes (52). To study the RecA response to damage-independent replication fork arrest, we chose to use this compound because it has been well characterized, but with the limitation that HPUra is not commercially available.
We began by synthesizing HPUra (see the supplemental material). In prior literature, the hydrazine congener H2-HPUra (compound 4) is described as the compound that inhibits DNA synthesis (see, e.g., references 37, 47, 50, and 51). We show the scheme for the attempted synthesis of compound 4 and related congeners in Fig. 2A. Briefly, we found that HPUra (compound 3) is readily synthesized as described previously (37). However, numerous attempts to reduce HPUra to H2-HPUra using sodium dithionite as described previously (50), or slight variations thereof, did not provide compound 4 but recovered only compound 3. This led us to the conclusion that either compound 3 is active as an inhibitor of DNA synthesis or compound 3 is reduced within the cell to produce compound 4. A detailed description of the synthesis and structure determination for compound 3 and congeners is provided in the supplemental material (see Fig. S2 to S12 in the supplemental material).
FIG 2.

Synthesis and activity of HPUra. (A) The synthesis scheme for HPUra (compound 3). (B) Log2 (fold enrichment) values of read coverage in 1,000-nucleotide-wide windows plotted versus PY79 genome position. The plot is centered on the origin of replication. Because the HPUra stock was dissolved in 50 mM KOH, the untreated control cells harvested at 60 min were subjected to a volume of 50 mM KOH at 20 min equal to that used with the treated cells harvested at 60 min. For HPUra, cells were treated at 20 min followed by harvesting of chromosomal DNA and analysis at the 60-min time point.
To test compound 3 for DNA synthesis inhibition, we synchronized replication initiation using a temperature-sensitive initiation mutant as described previously (6). We analyzed untreated cells 20 and 60 min after replication initiation and determined the position of replication forks by quantitative Illumina sequencing as described previously (53). During HPUra treatment, cells were challenged with HPUra 20 min after replication initiation followed by analysis of DNA content at 60 min. We show that replication forks in untreated cells progressed throughout the 60-min time course, while DNA replication in the HPUra-treated cells arrested immediately upon HPUra addition, showing the same DNA content as the untreated control at 20 min. With these results, we conclude that HPUra (compound 3) provides the desired effect of rapidly arresting DNA synthesis in vivo (Fig. 2B).
It has been shown that treatment of B. subtilis cells with HPUra blocks DNA synthesis and triggers replication stress as measured by RecA-GFP focus formation in most cells (54). We found that following HPUra treatment, RecA-GFP formed foci in most cells as rapidly as we could prepare the sample for imaging. We found that 76% of cells had RecA-GFP foci at 140 s posttreatment (n = 74) and that ∼94% of cells had RecA-GFP foci 5 min after HPUra treatment, demonstrating the rapid assembly of RecA foci following damage-independent replication fork arrest (Fig. 1A and C). We performed colocalization with RecA and DnaX and found that ∼93% of RecA foci (n = 181) were colocalized with replisomes following addition of HPUra to the growth medium (Fig. 1C). These results show that damage-independent fork arrest caused RecA to localize to nearly all replisomes in B. subtilis. This work also clarifies the method for HPUra synthesis (see the supplemental material).
RecA-GFP focus formation is dependent on recO and recR.
Having established that RecA-GFP localized to the replisome in most cells in response to endogenous and exogenous sources of damage, we asked which gene product(s) is required for RecA loading and hence for RecA-GFP focus formation in vivo. We predicted that recO might contribute to RecA-GFP focus formation since B. subtilis RecO helps nucleate formation of RecA onto SSB-coated ssDNA in vitro (28). To test recO in this capacity, we imaged and quantified RecA-GFP foci in B. subtilis cells bearing a recO disruption (recO::cat). In the absence of recO, RecA-GFP foci formed in less than 2.1% of cells left untreated or following exogenous DNA damage (Fig. 3). In addition, recO is necessary for damage-independent RecA-GFP filament formation in response to fork arrest. This result could be explained by release of the GFP moiety via proteolytic cleavage from RecA in a recO mutant. However, we found that RecA-GFP remained intact in the recO::cat background (see Fig. S13A in the supplemental material). We also complemented the recO mutant allele via ectopic expression (see Fig. S13B). Taking the results together, we found that, regardless of the type of damage, RecO is necessary for assembly of RecA repair centers in B. subtilis (Fig. 3).
FIG 3.

The recOR genes are necessary for RecA-GFP focus formation. A bar graph shows the percentage of cells with RecA-GFP foci in a wild-type (WT) strain (recA-gfp) and in isogenic strains lacking recO, recR, or recF following treatment with mitomycin C (40 nM), phleomycin (0.4 μM), HPUra (40 nM), and UV (40 J/m2). Error bars indicate the 95% confidence intervals.
Although B. subtilis RecO and RecR do not form a complex, they do function in the same genetic pathway (28). We performed the same experiment in a strain where the recR gene was replaced by a cat cassette and obtained results nearly identical to those obtained in recO-deficient cells (Fig. 3). Under all conditions examined, we found that RecA-GFP formed foci in 4.5% or less of cells deficient for recR. These results show that both recO and recR are necessary for RecA to form foci in response to DNA damage and damage-independent fork arrest (please see Discussion). This result was not due to proteolytic cleavage of RecA-GFP, and this phenotype was complemented by ectopic recR expression (see Fig. S13B in the supplemental material and data not shown). We interpret these results to mean that the RecOR pathway is necessary for RecA loading in vivo.
RecF is the third component of the RecFOR pathway, which in E. coli is critical for the repair of DNA gap substrates in vivo. This damage is the product of a single-stranded gap in otherwise double-stranded DNA following UV or mitomycin C exposure. To test the role of RecF in RecA-GFP repair center formation, we replaced the recF locus with a cat marker (recF::cat) and, interestingly, we found that the percentage of cells with RecA-GFP foci was mostly unaffected in untreated cells lacking recF (wild-type cells left untreated, 12.4% ± 2.6%; recF::cat cells left untreated, 9.2% ± 2.1%; recO::cat cells, 0.7% ± 0.7%; recR::cat strains left untreated, <2.5% ± 1.1%) (Fig. 3). We did, however, discover that in recF::cat cells, the percentage of cells with RecA-GFP foci failed to increase in response to exogenously introduced DNA damage and damage-independent replication fork arrest (Fig. 3). We conclude that the RecOR pathway is necessary for RecA-GFP foci to form in response to endogenous and low levels of exogenous sources of damage.
RecF affects the efficiency of RecA focus assembly.
We were concerned that the defects we observed in RecA-GFP focus assembly in the recFOR-deficient backgrounds might reflect poor efficiency at lower doses of DNA damage. Thus, we performed a time course of RecA-GFP focus assembly experiments with increases in the doses of phleomycin (3 μM) and MMC (300 nM) relative to the experiments represented in Fig. 3 (Fig. 4A). We show that the percentage of cells with RecA-GFP foci (solid lines) increased steadily, yet rapidly, until most cells had visible RecA foci (Fig. 4A; see also Fig. S14 in the supplemental material). Under the new conditions, with an increase in the DNA damage dose, we found that recO and recR were still required for assembly of RecA-GFP foci, even with more time to support filament formation (see Fig. S15 and S16). We also complemented the defect by ectopic expression of recO or recR (see Fig. S17). Interestingly, RecA-GFP foci were partially restored in the recF::cat strain at higher doses of damage (see Fig. S16). This observation suggests that RecF may be important for reducing the lag time of RecA-GFP nucleation in vivo.
FIG 4.

RecF is important for efficient RecA-GFP focus formation. (A) Results of a time course experiment representing the percentages of cells with RecA-GFP foci challenged with 3 μM phleomycin (black) or 300 nM MMC (gray) and the percentages of recF-deficient cells challenged with 3 μM phleomycin (black dashed) or 300 nM MMC (gray dashed). (B) An immunoblot for RecA and SSB in cells with (+) or without (−) recF. A 25-μg volume of soluble protein extract was separated via SDS-PAGE, followed by detection with affinity-purified RecA antisera in a 1:200 dilution and a 1:1,000 dilution for anti-SSB antiserum. (C) Results of a time course experiment for RecA-GFP focus formation following expression of I-SceI with 0.5% xylose in S750 minimal medium supplemented with 1% arabinose as described previously (6). For panels A and C, the error bars represent the 95% confidence intervals, and the number of cells scored (n) is >312 for each time point shown.
To further address the role of RecF, we performed a time course of RecA-GFP focus formation in recF-deficient cells (dashed lines) in response to MMC and phleomycin (Fig. 4A). We found that during the 60-min time course, RecA-GFP repair centers were considerably delayed in formation compared with the wild-type control. The delay in RecA-GFP repair center assembly was not due to a decrease in RecA-GFP protein levels since RecA protein abundance was unchanged in recF-deficient cells (Fig. 4B). With this result, we suggest that RecF function is distinct from that of RecOR and that RecOR is important for both initial RecA nucleation and steady filament growth, as suggested by in vitro experiments (55). Furthermore, our work shows that B. subtilis RecF contributes to RecA loading (foci) in response to many different types of lesions, including DNA breaks.
The results reported above showed that recOR is required for RecA to organize into repair centers in response to all DNA damage and damage-independent fork arrest and that RecF is important for efficient RecA-GFP organization. However, we had not yet tested the requirements of RecFOR in RecA loading following a double-stranded break (DSB). Therefore, we monitored RecA-GFP loading in response to an I-SceI endonuclease-catalyzed DSB as described previously (6, 38). We found over a 120-min time course that RecA-GFP foci failed to form in cells deficient for recO or recR. In contrast, after 120 min postexpression of I-SceI, nearly 70% of wild-type cells showed RecA-GFP repair centers (Fig. 4C). In recF-deficient cells, we continued to observe a delay in RecA-GFP focus formation. With these results, we conclude that recOR is necessary for RecA-GFP focus formation in response to replication fork-blocking lesions, damage-independent fork arrest (HPUra), and DSBs. Furthermore, we conclude that RecF was an important contributor to efficient RecA-GFP repair center formation under all conditions tested, including DSBs.
The SSB C terminus contributes to the DNA damage-induced assembly of RecA-GFP foci.
In both B. subtilis and E. coli, RecO is an SSB partner (20, 56, 57). Because we showed above that RecOR is necessary for RecA to assemble into repair centers and these repair centers primarily localize to the replisome, we asked if SSB contributes to this response. In E. coli, the SSB C terminus mediates interaction with its binding partners through the last two residues (PF), which are required for SSB interaction (for a review, see reference 57). Furthermore, in E. coli, the PF residues are essential and mutation of just the proline (ssb113) causes temperature-sensitive growth and UV sensitivity (58–60). Such a strong phenotype limits the ability to study E. coli SSB binding partners in vivo. Interestingly, ssb alleles in B. subtilis encoding truncations of the C-terminal 35 amino acids are viable (56, 61), providing a system to understand the effect of the SSB C terminus on DNA replication and repair in vivo (56, 61). To this end, we integrated the ssbΔ35 allele, which uses an IPTG-regulated promoter to drive gene expression of the essential downstream gene rpsR, as well as the corresponding wild-type (ssb3+) control into an isogenic strain background carrying the recA-gfp allele (56, 61). In the ssb3+ strain, RecA-GFP foci formed in ∼15% of cells and showed virtually the same response to MMC, phleomycin, HPUra, and UV as the wild-type ssb strain (Table 1; compare with Fig. 3) (6, 38).
TABLE 1.
Truncation of the SSB C-terminal tail eliminates RecA filament formation in response to DNA damage and replication fork stress
| B. subtilis strain relevant genotype | % filament formation under indicated conditiona |
||||
|---|---|---|---|---|---|
| Exponential growth | 40 nM MMC | 0.3 μM phleomycin | 40 μM HPUra | 40 J/m2 UV | |
| ssb3+ | 15.0 ± 3.0 | 44.3 ± 2.5 | 46.4 ± 5.4 | 87.3 ± 3.8 | 86.5 ± 3.5 |
| ssbΔ35 | 20.0 ± 3.1 | 21.5 ± 3.7 | 26.7 ± 4.0 | 21.6 ± 3.9 | 22.6 ± 4.3 |
| ssb3+, recO::cat | 1.0 ± 1.1 | 0.4 ± 0.7 | ND | 0.3 ± 0.6 | ND |
| ssbΔ35, recO::cat | 8.8 ± 2.3 | 9.7 ± 3.4 | ND | 9.8 ± 2.9 | ND |
| ssb3+, recR::cat | 0.3 ± 0.6 | 2.8 ± 1.8 | ND | 2.9 ± 1.7 | ND |
| ssbΔ35, recR::cat | 11.0 ± 3.2 | 11.0 ± 3.1 | ND | 13.1 ± 2.8 | ND |
ND, not determined. Error bars represent the 95% confidence interval with n > 282 cells scored for each strain and condition.
In the ssbΔ35 background, we observed similar percentages of cells with RecA-GFP repair centers left untreated (∼20%) and following challenge with MMC, phleomycin, HPUra, or UV, where the percentage of cells ranged from ∼21% to ∼26% (Table 1). These results indicate a failure in the organization of RecA into repair centers following damage or fork arrest in the ssbΔ35 strain (Table 1). We combined the recO- or recR-deficient alleles with the ssb3+ or ssbΔ35 cells. We found that loss of recO or recR blocked RecA repair center formation in cells left untreated or challenged with MMC and HPUra in the ssb3+ wild-type control (Table 1). In ssbΔ35 cells, we found that loss of recO and recR function reduced RecA-GFP localization for less than 13% of cells (Table 1). Therefore, in the ssbΔ35 background, deficiencies in recO and recR did not block RecA localization as we observed in the ssb3+ or wild-type background; however, induced focus formation is blocked. Prior work showed that ssbΔ35 causes chronic SOS induction, possibly explaining the elevated RecA-GFP levels (56). We performed immunoblot analyses for RecA-GFP and indeed found that RecA levels increased in ssbΔ35 cells (Fig. 5A). We also found that DnaN levels were increased, suggesting that dnaN is damage inducible. We suggest that the basal recOR-independent RecA localization we observe in ssbΔ35 cells is caused by elevated RecA levels in vivo, which may be caused or exacerbated by a decrease in the ability of ssbΔ35 to bind ssDNA (62).
To place ssbΔ35 in the recOR pathway, we performed survival assays following exposure to MMC (Fig. 5B). We found that the wild-type control ssb3+ cells experienced virtually no killing at the concentrations tested, whereas the isogenic ssbΔ35 phenocopied a recA-deficient strain. Furthermore, we found that ssbΔ35 cells in combination with recO or recR deficiencies showed the same killing as either single mutant (Fig. 5B). These results provide evidence that ssb is in the same pathway as recO and recR for survival in response to MMC damage. We conclude that SSB is an important component of the RecOR-dependent loading system for RecA in response to DNA damage and damage-independent fork arrest.
DISCUSSION
RecO and RecR are necessary for RecA focus assembly.
In B. subtilis, the proteins necessary to elicit formation of spontaneous and DNA damage-induced RecA-GFP repair centers (foci) have been unknown. We showed that RecOR is necessary for the assembly of RecA-mediated DNA repair centers in live B. subtilis cells. Importantly, RecO and RecR are required for the RecA response to both endogenous and exogenous sources of DNA damage, as well as in response to HPUra, a drug that causes DNA damage-independent replication fork arrest (54). In E. coli, DSBs are initially processed by RecBCD to provide the 3′ ssDNA segment, followed by RecA loading, making most spontaneous RecA-GFP foci in rich medium recB dependent (8, 18). In B. subtilis, AddAB provides a 3′ extension (25); however, it is unclear whether AddAB is able to physically load RecA. Our results with phleomycin and an I-SceI site-specific DSB demonstrated that following AddAB end processing, RecOR is still necessary for the assembly of RecA repair centers, making it unlikely that AddAB can physically load RecA on processed DNA ends. We suggest that SSB binds the 3′ extension to protect the DNA end, followed by recruitment of RecOR through interaction with the SSB C terminus (Fig. 6).
FIG 6.
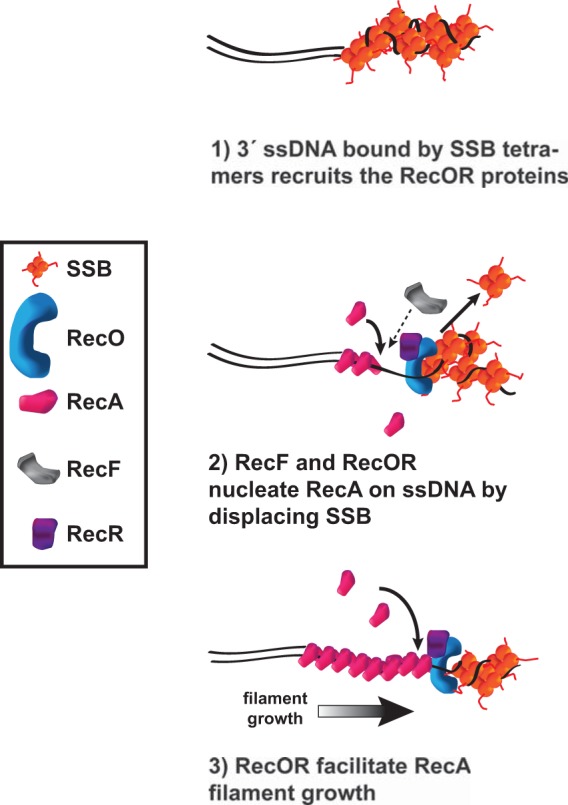
Model for RecOR-dependent RecA loading and establishment of repair centers in B. subtilis at a DSB. We propose that after AddAB catalyzes end resection to generate a 3′ ssDNA extension, RecOR is recruited to the large stretches of ssDNA by SSB and then helps to displace SSB and facilitate RecA loading and nucleation of RecA filament formation for homology-directed repair.
Prior work showed that GFP-RecA is mostly unaffected with respect to focus formation in recO- or recR-deficient cells (5). In that work, cells deficient for RecO still showed GFP-RecA foci in ∼25% of cells challenged with 50 ng/ml MMC (5). We suggest that ectopic expression of GFP-RecA may have increased the levels of RecA to a point where RecA was able to bypass the requirement for recO and recR. In support of our argument, ssbΔ35 cells have elevated RecA levels compared with the wild-type control and show recOR-independent RecA foci in untreated cells. B. subtilis RecA can self-assemble on SSB-coated ssDNA in vitro although the reaction is accelerated with RecO (28). Taking this prior work into consideration in combination with our study, we suggest that the recOR genes are necessary for RecA to load in vivo when RecA is expressed at its native levels. The RecA dependence on RecOR also provides a convenient regulatory mechanism to prevent aberrant RecA repair centers from assembling in replicating cells. Another important finding of our work is that although B. subtilis RecO can aid in nucleation of RecA onto SSB-coated ssDNA in vitro in the absence of RecR (28), we did not observe RecA-GFP foci in the recR::cat strain (Fig. 3 and 4). Therefore, we showed that RecR is also necessary for RecA-GFP focus formation in vivo. Together, our results showed that RecOR represents the major RecA loading pathway in B. subtilis in response to several types of DNA damage and damage-independent replication fork arrest.
In further support of our findings, it was recently shown that RecO was required for site-specific DSB repair in Mycobacterium smegmatis, which uses AdnAB for end resectioning (63). Furthermore, in M. smegmatis, a recO deletion phenocopies a recA deletion, further highlighting the reliance of M. smegmatis on RecO for RecA-dependent repair (63). Therefore, considering the M. smegmatis studies and ours, we suggest that RecOR represents the major RecA loading pathway for homology-directed repair in bacteria containing the AddAB or AdnAB pathways. Furthermore, many more bacteria use AddAB or AdnAB for end processing of DSBs than RecBCD (30). We suggest that these bacteria may also rely on RecOR for RecA loading during repair and stabilization of stalled replication forks.
RecF and SSB increase the efficiency of RecA loading in vivo.
RecF is in the recOR pathway (27). We therefore tested the effect of a recF-deficient allele on the ability of RecA to assemble into repair centers. We found that RecF was not necessary to elicit RecA-GFP repair center formation; however, our results suggest that it plays a necessary role in facilitating efficient RecA filament nucleation or elongation. These results support in vitro studies directly imaging E. coli RecA loading where the presence of RecF was not necessary for filament elongation and yet increased the kinetics of RecA nucleation and loading (55).
Because we found a complete dependency on RecO for assembly of RecA repair centers and RecO is an SSB binding partner (56), we asked if SSB contributes to RecA filament formation in B. subtilis. SSB contains a C-terminal tail which binds to several proteins, recruiting them to sites of excess ssDNA (56, 57, 64, 65). In B. subtilis, RecO binds SSB, and ectopic expression of GFP-RecO relies on the SSB C terminus for focus formation (56). The data we present here showed that the SSB C-terminal 35 amino acids are important for the DNA damage-dependent and damage-independent localization of RecA and that a strain with the ssbΔ35 allele is as sensitive to DNA damage as a strain deficient for recO or recR. In E. coli, deletion of the SSB C-terminal PF is lethal and the lethality is attributed to these residues mediating protein-protein interactions important for essential replication functions (58, 59). Therefore, how can B. subtilis tolerate truncation of the C-terminal 35 amino acids, including the ultimate PF motif? In contrast to E. coli, B. subtilis SSB contains three PF repeats in the C-terminal 38 amino acids and the Δ35 truncation leaves the sequence PFG on the C terminus (see Fig. S18 in the supplemental material). We suggest that the remaining PF residues in the Δ35 truncation allow viability and recruitment of a limited number of SSB binding partners. Our results showed that the ssbΔ35 allele supported RecA-GFP assembly during normal growth but prevented a DNA damage-induced increase. Our work and another study also showed that the ssbΔ35 strains are sensitive to DNA damage (Fig. 5 and reference 56). Based on our results and those of Costes et al. (56), we propose that the three PF repeats in the B. subtilis protein function to accommodate several binding partners and are crucial for activating the DNA damage response in B. subtilis.
HPUra-dependent inhibition of DNA replication.
6-(p-Hydroxyphenylazo)-uracil (HPUra) is a PolC-specific inhibitor that has been widely used in B. subtilis and related Gram-positive bacteria (see, e.g., references 10, 37, 47, 48, 49, 50, and 51). HPUra has been a very important tool for investigating the involvement of DNA replication in a number of different DNA transactions in vivo (see, e.g., references 10, 48, 49, and 54). Prior work suggested that the hydrazine congener (H2-HPUra) is the active form that inhibits DNA synthesis (37, 47, 50, 51). We were unable to synthesize H2-HPUra and could obtain only the oxidized form (HPUra). We cannot exclude the possibility that HPUra is reduced in vivo to form H2-HPUra; however, an HPUra tautomer (compound 3a) could form the proper hydrogen bonds to pair with dCMP in vivo if it is not reduced to H2-HPUra. With these results, we suggest that the penultimate compound (HPUra), which is readily synthesized as described (see reference 50 and the supplemental material), is the appropriate compound for rapidly inhibiting DNA synthesis in B. subtilis and other Gram-positive bacteria.
We also showed that HPUra is indeed both a potent inhibitor of DNA synthesis and triggers replication stress in live cells (Fig. 1 and 2). We speculate that HPUra-induced replication stress causes the replicative polymerase PolC to uncouple from the replicative helicase DnaC. We suggest that a blockage to PolC movement and continued unwinding of the DNA by the replicative helicase DnaC ahead of the blocked fork would form excess ssDNA, providing a substrate for RecA binding. Interestingly, it was shown that activation of the stringent response with arginine hydroxamate, which stops replication elongation via inhibition of primase activity, fails to result in the formation of RecA-GFP filaments (54). We suggest that by directly inhibiting primase, the primase-DnaC complex is maintained, causing DnaC to remain at the blocked fork and failing to cause excess ssDNA and RecA-GFP recruitment.
Supplementary Material
ACKNOWLEDGMENTS
We thank members of the Simmons laboratory for their critical reading of the manuscript, and we thank two anonymous referees for their thoughtful suggestions. We thank C. Lee and Alan D. Grossman (Massachusetts Institute of Technology) for the gift of a recA overexpression plasmid.
This work was supported by a grant (MCB1050948) from the National Science Foundation to L.A.S. In addition, E.R.B. was supported in part by an undergraduate summer fellowship from the Honors College at the University of Michigan. J.S.L. was supported by a predoctoral fellowship from the Rackham Graduate School at the University of Michigan. J.W.S. was supported in part by an NIH Genetics Training grant (T32GM007544). H.D.S. and R.J.S. acknowledge generous support by the University of Michigan College of Pharmacy Ella and Hans Vahlteich Research Fund.
Footnotes
Published ahead of print 2 June 2014
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JB.01494-14.
REFERENCES
- 1.Friedberg EC, Walker GC, Siede W, Wood RD, Schultz RA, Ellenberger T. 2006. DNA repair and mutagenesis, 2nd ed. American Society for Microbiology, Washington, DC [Google Scholar]
- 2.Simmons LA, Foti JJ, Cohen SE, Walker GC. 2008. The SOS regulatory network. EcoSal Plus. 10.1128/ecosalplus.5.4.3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cox MM. 2007. Regulation of bacterial RecA protein function. Crit. Rev. Biochem. Mol. Biol. 42:41–63. 10.1080/10409230701260258 [DOI] [PubMed] [Google Scholar]
- 4.Little JW, Mount DW. 1982. The SOS regulatory system of Escherichia coli. Cell 29:11–22. 10.1016/0092-8674(82)90085-X [DOI] [PubMed] [Google Scholar]
- 5.Kidane D, Graumann PL. 2005. Dynamic formation of RecA filaments at DNA double strand break repair centers in live cells. J. Cell Biol. 170:357–366. 10.1083/jcb.200412090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Simmons LA, Grossman AD, Walker GC. 2007. Replication is required for the RecA localization response to DNA damage in Bacillus subtilis. Proc. Natl. Acad. Sci. U. S. A. 104:1360–1365. 10.1073/pnas.0607123104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Centore RC, Lestini R, Sandler SJ. 2008. XthA (exonuclease III) regulates loading of RecA onto DNA substrates in log phase Escherichia coli cells. Mol. Microbiol. 67:88–101. 10.1111/j.1365-2958.2007.06026.x [DOI] [PubMed] [Google Scholar]
- 8.Renzette N, Gumlaw N, Nordman JT, Krieger M, Yeh SP, Long E, Centore R, Boonsombat R, Sandler SJ. 2005. Localization of RecA in Escherichia coli K-12 using RecA-GFP. Mol. Microbiol. 57:1074–1085. 10.1111/j.1365-2958.2005.04755.x [DOI] [PubMed] [Google Scholar]
- 9.Renzette N, Gumlaw N, Sandler SJ. 2007. DinI and RecX modulate RecA-DNA structures in Escherichia coli K-12. Mol. Microbiol. 63:103–115. 10.1111/j.1365-2958.2006.05496.x [DOI] [PubMed] [Google Scholar]
- 10.Bernard R, Marquis KA, Rudner DZ. 2010. Nucleoid occlusion prevents cell division during replication fork arrest in Bacillus subtilis. Mol. Microbiol. 78:866–882. 10.1111/j.1365-2958.2010.07369.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Renzette N, Sandler SJ. 2008. Requirements for ATP binding and hydrolysis in RecA function in Escherichia coli. Mol. Microbiol. 67:1347–1359. 10.1111/j.1365-2958.2008.06130.x [DOI] [PubMed] [Google Scholar]
- 12.Meile JC, Wu LJ, Ehrlich SD, Errington J, Noirot P. 2006. Systematic localisation of proteins fused to the green fluorescent protein in Bacillus subtilis: identification of new proteins at the DNA replication factory. Proteomics 6:2135–2146. 10.1002/pmic.200500512 [DOI] [PubMed] [Google Scholar]
- 13.Clark AJ, Sandler SJ. 1994. Homologous genetic recombination: the pieces begin to fall into place. Crit. Rev. Microbiol. 20:125–142. 10.3109/10408419409113552 [DOI] [PubMed] [Google Scholar]
- 14.Kowalczykowski SC, Dixon DA, Eggleston AK, Lauder SD, Rehrauer WM. 1994. Biochemistry of homologous recombination in Escherichia coli. Microbiol. Rev. 58:401–465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lenhart JS, Schroeder JW, Walsh BW, Simmons LA. 2012. DNA repair and genome maintenance in Bacillus subtilis. Microbiol. Mol. Biol. Rev. 76:530–564. 10.1128/MMBR.05020-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dillingham MS, Kowalczykowski SC. 2008. RecBCD enzyme and the repair of double-stranded DNA breaks. Microbiol. Mol. Biol. Rev. 72:642–671. 10.1128/MMBR.00020-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Spies M, Amitani I, Baskin RJ, Kowalczykowski SC. 2007. RecBCD enzyme switches lead motor subunits in response to chi recognition. Cell 131:694–705. 10.1016/j.cell.2007.09.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Arnold DA, Kowalczykowski SC. 2000. Facilitated loading of RecA protein is essential to recombination by RecBCD enzyme. J. Biol. Chem. 275:12261–12265. 10.1074/jbc.275.16.12261 [DOI] [PubMed] [Google Scholar]
- 19.Spies M, Kowalczykowski SC. 2006. The RecA binding locus of RecBCD is a general domain for recruitment of DNA strand exchange proteins. Mol. Cell 21:573–580. 10.1016/j.molcel.2006.01.007 [DOI] [PubMed] [Google Scholar]
- 20.Umezu K, Kolodner RD. 1994. Protein interactions in genetic recombination in Escherichia coli. Interactions involving RecO and RecR overcome the inhibition of RecA by single-stranded DNA-binding protein. J. Biol. Chem. 269:30005–30013 [PubMed] [Google Scholar]
- 21.Morimatsu K, Wu Y, Kowalczykowski SC. 2012. RecFOR proteins target RecA protein to a DNA gap with either DNA or RNA at the 5′ terminus: implication for repair of stalled replication forks. J. Biol. Chem. 287:35621–35630. 10.1074/jbc.M112.397034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ryzhikov M, Koroleva O, Postnov D, Tran A, Korolev S. 2011. Mechanism of RecO recruitment to DNA by single-stranded DNA binding protein. Nucleic Acids Res. 39:6305–6314. 10.1093/nar/gkr199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Umezu K, Chi N-W, Kolodner RD. 1993. Biochemical interactions of the Escherichia coli RecF, RecO, and RecR proteins with RecA protein and single-stranded binding protein. Proc. Natl. Acad. Sci. U. S. A. 90:3875–3879. 10.1073/pnas.90.9.3875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lia G, Rigato A, Long E, Chagneau C, Le Masson M, Allemand JF, Michel B. 2013. RecA-promoted, RecFOR-independent progressive disassembly of replisomes stalled by helicase inactivation. Mol. Cell 49:547–557. 10.1016/j.molcel.2012.11.018 [DOI] [PubMed] [Google Scholar]
- 25.Chédin F, Ehrlich SD, Kowalczykowski SC. 2000. The Bacillus subtilis AddAB helicase/nuclease is regulated by its cognate Chi sequence in vitro. J. Mol. Biol. 298:7–20. 10.1006/jmbi.2000.3556 [DOI] [PubMed] [Google Scholar]
- 26.Saikrishnan K, Yeeles JT, Gilhooly NS, Krajewski WW, Dillingham MS, Wigley DB. 2012. Insights into Chi recognition from the structure of an AddAB-type helicase-nuclease complex. EMBO J. 31:1568–1578. 10.1038/emboj.2012.9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fernández S, Kobayashi Y, Ogasawara N, Alonso JC. 1999. Analysis of the Bacillus subtilis recO gene: RecO forms part of the RecFLOR function. Mol. Gen. Genet. 261:567–573. 10.1007/s004380051002 [DOI] [PubMed] [Google Scholar]
- 28.Manfredi C, Carrasco B, Ayora S, Alonso JC. 2008. Bacillus subtilis RecO nucleates RecA onto SsbA-coated single-stranded DNA. J. Biol. Chem. 283:24837–24847. 10.1074/jbc.M802002200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sanchez H, Kidane D, Castillo Cozar M, Graumann PL, Alonso JC. 2006. Recruitment of Bacillus subtilis RecN to DNA double-strand breaks in the absence of DNA end processing. J. Bacteriol. 188:353–360. 10.1128/JB.188.2.353-360.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cromie GA. 2009. Phylogenetic ubiquity and shuffling of the bacterial RecBCD and AddAB recombination complexes. J. Bacteriol. 191:5076–5084. 10.1128/JB.00254-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hardwood CR, Cutting SM. 1990. Molecular biological methods for Bacillus. John Wiley & Sons, Chichester, United Kingdom [Google Scholar]
- 32.Smith BT, Grossman AD, Walker GC. 2001. Visualization of mismatch repair in bacterial cells. Mol. Cell 8:1197–1206. 10.1016/S1097-2765(01)00402-6 [DOI] [PubMed] [Google Scholar]
- 33.Lemon KP, Grossman AD. 2000. Movement of replicating DNA through a stationary replisome. Mol. Cell 6:1321–1330. 10.1016/S1097-2765(00)00130-1 [DOI] [PubMed] [Google Scholar]
- 34.Dupes NM, Walsh BW, Klocko AD, Lenhart JS, Peterson HL, Gessert DA, Pavlick CE, Simmons LA. 2010. Mutations in the Bacillus subtilis beta clamp that separate its roles in DNA replication from mismatch repair. J. Bacteriol. 192:3452–3463. 10.1128/JB.01435-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Klocko AD, Schroeder JW, Walsh BW, Lenhart JS, Evans ML, Simmons LA. 2011. Mismatch repair causes the dynamic release of an essential DNA polymerase from the replication fork. Mol. Microbiol. 82:648–663. 10.1111/j.1365-2958.2011.07841.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Klocko AD, Crafton KM, Walsh BW, Lenhart JS, Simmons LA. 2010. Imaging mismatch repair and cellular responses to DNA damage in Bacillus subtilis. J. Vis. Exp. 2010:1736. 10.3791/1736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wright GE, Brown NC. 1974. Synthesis of 6-(phenylhydrazino)uracils and their inhibition of a replication-specific deoxyribonucleic acid polymerase. J. Med. Chem. 17:1277–1282. 10.1021/jm00258a009 [DOI] [PubMed] [Google Scholar]
- 38.Simmons LA, Goranov AI, Kobayashi H, Davies BW, Yuan DS, Grossman AD, Walker GC. 2009. Comparison of responses to double-strand breaks between Escherichia coli and Bacillus subtilis reveals different requirements for SOS induction. J. Bacteriol. 191:1152–1161. 10.1128/JB.01292-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Davies BW, Bogard RW, Dupes NM, Gerstenfeld TA, Simmons LA, Mekalanos JJ. 2011. DNA damage and reactive nitrogen species are barriers to Vibrio cholerae colonization of the infant mouse intestine. PLoS Pathog. 7:e1001295. 10.1371/journal.ppat.1001295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kobayashi H, Simmons LA, Yuan DS, Broughton WJ, Walker GC. 2008. Multiple Ku orthologues mediate DNA non-homologous end-joining in the free-living form and during chronic infection of Sinorhizobium meliloti. Mol. Microbiol. 67:350–363. 10.1111/j.1365-2958.2007.06036.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Schroeder JW, Simmons LA. 2013. Complete genome sequence of Bacillus subtilis strain PY79. Genome Announc. 1:e01085–13. 10.1128/genomeA.01085-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Li H, Durbin R. 2009. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25:1754–1760. 10.1093/bioinformatics/btp324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lesterlin C, Ball G, Schermelleh L, Sherratt DJ. 22 December 2013. RecA bundles mediate homology pairing between distant sisters during DNA break repair. Nature. 10.1038/nature12868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kidane D, Sanchez H, Alonso JC, Graumann PL. 2004. Visualization of DNA double-strand break repair in live bacteria reveals dynamic recruitment of Bacillus subtilis RecF, RecO and RecN proteins to distinct sites on the nucleoids. Mol. Microbiol. 52:1627–1639. 10.1111/j.1365-2958.2004.04102.x [DOI] [PubMed] [Google Scholar]
- 45.Dronkert ML, Kanaar R. 2001. Repair of DNA interstrand cross-links. Mutat. Res. 486:217–247. 10.1016/S0921-8777(01)00092-1 [DOI] [PubMed] [Google Scholar]
- 46.Sleigh MJ. 1976. The mechanism of DNA breakage by phleomycin in vitro. Nucleic Acids Res. 3:891–901. 10.1093/nar/3.4.891 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gass KB, Low RL, Cozzarelli NR. 1973. Inhibition of a DNA polymerase from Bacillus subtilis by hydroxyphenylazopyrimidines. Proc. Natl. Acad. Sci. U. S. A. 70:103–107. 10.1073/pnas.70.1.103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Goranov AI, Katz L, Breier AM, Burge CB, Grossman AD. 2005. A transcriptional response to replication status mediated by the conserved bacterial replication protein DnaA. Proc. Natl. Acad. Sci. U. S. A. 102:12932–12937. 10.1073/pnas.0506174102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Goranov AI, Kuester-Schoeck E, Wang JD, Grossman AD. 2006. Characterization of the global transcriptional responses to different types of DNA damage and disruption of replication in Bacillus subtilis. J. Bacteriol. 188:5595–5605. 10.1128/JB.00342-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mackenzie JM, Neville MM, Wright GE, Brown NC. 1973. Hydroxyphenylazopyrimidines: characterization of the active forms and their inhibitory action on a DNA polymerase from Bacillus subtilis. Proc. Natl. Acad. Sci. U. S. A. 70:512–516. 10.1073/pnas.70.2.512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Neville MM, Brown NC. 1972. Inhibition of a discrete bacterial DNA polymerase by 6-(p-hydroxyphenylazo)-uracil and 6-(p-hydroxyphenylazo-)-isocytosine. Nat. New Biol. 240:80–82 [DOI] [PubMed] [Google Scholar]
- 52.Brown NC. 1970. 6-(p-Hydroxyphenylazo)-uracil: a selective inhibitor of host DNA replication in phage-infected Bacillus subtilis. Proc. Natl. Acad. Sci. U. S. A. 67:1454–1461. 10.1073/pnas.67.3.1454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Walsh BW, Bolz SA, Wessel SR, Schroeder JW, Keck JL, Simmons LA. 2014. RecD2 helicase limits replication fork stress in Bacillus subtilis. J. Bacteriol. 196:1359–1368. 10.1128/JB.01475-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wang JD, Sanders GM, Grossman AD. 2007. Nutritional control of elongation of DNA replication by (p)ppGpp. Cell 128:865–875. 10.1016/j.cell.2006.12.043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bell JC, Plank JL, Dombrowski CC, Kowalczykowski SC. 2012. Direct imaging of RecA nucleation and growth on single molecules of SSB-coated ssDNA. Nature 491:274–278. 10.1038/nature11598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Costes A, Lecointe F, McGovern S, Quevillon-Cheruel S, Polard P. 2010. The C-terminal domain of the bacterial SSB protein acts as a DNA maintenance hub at active chromosome replication forks. PLoS Genet. 6:e1001238. 10.1371/journal.pgen.1001238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Shereda RD, Kozlov AG, Lohman TM, Cox MM, Keck JL. 2008. SSB as an organizer/mobilizer of genome maintenance complexes. Crit. Rev. Biochem. Mol. Biol. 43:289–318. 10.1080/10409230802341296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Chase JW, L'Italien JJ, Murphy JB, Spicer EK, Williams KR. 1984. Characterization of the Escherichia coli SSB-113 mutant single-stranded DNA-binding protein. Cloning of the gene, DNA and protein sequence analysis, high pressure liquid chromatography peptide mapping, and DNA-binding studies. J. Biol. Chem. 259:805–814 [PubMed] [Google Scholar]
- 59.Curth U, Genschel J, Urbanke C, Greipel J. 1996. In vitro and in vivo function of the C-terminus of Escherichia coli single-stranded DNA binding protein. Nucleic Acids Res. 24:2706–2711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Johnson BF. 1977. Genetic mapping of the lexC-113 mutation. Mol. Gen. Genet. 157:91–97 [DOI] [PubMed] [Google Scholar]
- 61.Lecointe F, Serena C, Velten M, Costes A, McGovern S, Meile JC, Errington J, Ehrlich SD, Noirot P, Polard P. 2007. Anticipating chromosomal replication fork arrest: SSB targets repair DNA helicases to active forks. EMBO J. 26:4239–4251. 10.1038/sj.emboj.7601848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Antony E, Weiland E, Yuan Q, Manhart CM, Nguyen B, Kozlov AG, McHenry CS, Lohman TM. 2013. Multiple C-terminal tails within a single E. coli SSB homotetramer coordinate DNA replication and repair. J. Mol. Biol. 425:4802–4819. 10.1016/j.jmb.2013.08.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Gupta R, Ryzhikov M, Koroleva O, Unciuleac M, Shuman S, Korolev S, Glickman MS. 2013. A dual role for mycobacterial RecO in RecA-dependent homologous recombination and RecA-independent single-strand annealing. Nucleic Acids Res. 41:2284–2295. 10.1093/nar/gks1298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Lu D, Keck JL. 2008. Structural basis of Escherichia coli single-stranded DNA-binding protein stimulation of exonuclease I. Proc. Natl. Acad. Sci. U. S. A. 105:9169–9174. 10.1073/pnas.0800741105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Shereda RD, Bernstein DA, Keck JL. 2007. A central role for SSB in Escherichia coli RecQ DNA helicase function. J. Biol. Chem. 282:19247–19258. 10.1074/jbc.M608011200 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.