

ABSTRACT
Herpes simplex virus 1 (HSV-1) establishes lifelong latent infections in the sensory neurons of the trigeminal ganglia (TG), wherein it retains the capacity to reactivate. The interferon (IFN)-driven antiviral response is critical for the control of HSV-1 acute replication. We therefore sought to further investigate this response in TG neurons cultured from adult mice deficient in a variety of IFN signaling components. Parallel experiments were also performed in fibroblasts isolated concurrently. We showed that HSV-1 replication was comparable in wild-type (WT) and IFN signaling-deficient neurons and fibroblasts. Unexpectedly, a similar pattern was observed for the IFN-sensitive vesicular stomatitis virus (VSV). Despite these findings, TG neurons responded to IFN-β pretreatment with STAT1 nuclear localization and restricted replication of both VSV and an HSV-1 strain deficient in γ34.5, while wild-type HSV-1 replication was unaffected. This was in contrast to fibroblasts in which all viruses were restricted by the addition of IFN-β. Taken together, these data show that adult TG neurons can mount an effective antiviral response only if provided with an exogenous source of IFN-β, and HSV-1 combats this response through γ34.5. These results further our understanding of the antiviral response of neurons and highlight the importance of paracrine IFN-β signaling in establishing an antiviral state.
IMPORTANCE Herpes simplex virus 1 (HSV-1) is a ubiquitous virus that establishes a lifelong latent infection in neurons. Reactivation from latency can cause cold sores, blindness, and death from encephalitis. Humans with deficiencies in innate immunity have significant problems controlling HSV infections. In this study, we therefore sought to elucidate the role of neuronal innate immunity in the control of viral infection. Using neurons isolated from mice, we found that the intrinsic capacity of neurons to restrict virus replication was unaffected by the presence or absence of innate immunity. In contrast, neurons were able to mount a robust antiviral response when provided with beta interferon, a molecule that strongly stimulates innate immunity, and that HSV-1 can combat this response through the γ34.5 viral gene. Our results have important implications for understanding how the nervous system defends itself against virus infections.
INTRODUCTION
Herpes simplex virus 1 (HSV-1) is a prevalent neurotropic virus that persists for the lifetime of the host (1). It infects primarily via the orofacial route, where it undergoes rounds of lytic replication in the mucosal epithelium of the eyes, nose, and mouth. After entering the axonal terminals of innervating sensory neurons, the virus then travels in a retrograde direction to the cell body, which resides in the trigeminal ganglia (TG), where it establishes latency, facilitating persistence in the host. Spontaneous reactivation from latency can occur, and the virus travels in the anterograde direction to undergo subsequent rounds of lytic replication in the mucosal epithelium, resulting in viral shedding and enabling host-to-host transmission (2, 3). It is notable that the successful completion of the HSV-1 life cycle in vivo requires that the virus can infect and replicate in multiple cell types.
HSV-1 replication results in the production of type I interferon (IFN), and IFN responses are essential to controlling infection (4–10). Classically, infected cells can detect the presence of virus through pathogen recognition receptors (PRRs), which leads to the activation of key transcription factors, including interferon regulatory factor 3 (IRF-3) and nuclear factor κB (NF-κB) (11). This induces the production and secretion of type I IFN, which acts in an autocrine or paracrine manner to engage the IFN receptor (IFNR) and activate the Janus-activated kinase (JAK) and signal transducer and activator of transcription (STAT) pathway through the transcription factor STAT1. Once activated, STAT1 localizes to the nucleus and subsequently induces production of interferon-stimulated genes (ISGs) (12). This pathway self-amplifies via successive secretion of IFN-α/β and stimulation of IFN receptors. ISGs establish an antiviral state in the cell through inhibition of transcription and translation, stimulation of cytokine production, and promotion of apoptosis. Uninfected cells can also respond to secreted exogenous type I IFN, establishing an antiviral state to further halt viral spread (13).
The importance of the IFN-driven antiviral response in combating HSV-1 has been demonstrated in humans with genetic impairments in Toll-like receptor 3 (TLR3)- and STAT1-dependent pathways manifesting as increased frequency of recurrent herpes simplex encephalitis (HSE) (4–7). This is recapitulated in mouse models of acute HSV-1 infection, where mice lacking components of antiviral signaling succumb rapidly to disease (8–10). The importance of this pathway to both host and pathogen is further underscored by the presence of many HSV genes that counter this antiviral response (14, 15). Of note is the γ34.5 viral protein, which reverses the double-stranded RNA (dsRNA)-dependent protein kinase R (PKR)-mediated phosphorylation of eIF2α, thereby relieving translational arrest, and prevents TANK-binding kinase (TBK) phosphorylation of IRF3 (16–18). γ34.5 also serves to counter the host autophagy response, thereby suppressing xenophagy and antigen presentation (19, 20). HSV-1 strains lacking γ34.5 are attenuated in humans (21) and in mouse models of intracerebral and corneal infection (22, 23). The virulence of Δγ34.5 mutant viruses is fully and specifically restored in IFN-αβγR−/− and PKR−/− mice, demonstrating the role of γ34.5 in counteracting the IFN response (22). Additionally, the demonstrated safety of γ34.5-deficient oncolytic HSV-1 vectors upon intracerebral infection in clinical trials highlights the importance of γ34.5 as a neurovirulence factor in humans (21).
Recent work has shown that autophagy rather than IFN signaling is the dominant pathway for the neuronal antiviral response to HSV-1 (24). Autophagy is a catabolic process of cytoplasmic protein and organelle degradation, which can also be triggered by invading pathogens in a process known as xenophagy. This occurs through engulfment of cytoplasmic contents by autophagosomes and subsequent fusion with the lysosome (25, 26). Other studies, however, have shown an important role for type I IFN signaling in combating HSV-1 infection in neurons (27). For example, HSV-1 replication was reduced in IFN-pretreated sensory neurons infected via axon terminals (28), and this promoted a quiescent state that resembled bona fide latency (29, 30). Additionally, neurons derived from stem cells from patients with genetic defects in IFN-driven antiviral signaling showed increased permissivity to HSV-1 (31). Given these findings, a clearer understanding of the nature of the neuronal antiviral response to HSV-1 is needed to elucidate the factors that regulate establishment, maintenance, and reactivation from latency.
Many previous in vitro studies of neuronal HSV infection have utilized sensory or sympathetic neurons derived from embryonic or early postnatal animals (30, 32–34). While these models have been informative, the innate responses in these cells may not accurately reflect those of adult sensory neurons, since subtype and differentiation state significantly impact the magnitude and quality of antiviral responses (35, 36). We therefore cultured TG neurons from adult mice in an attempt to more accurately reflect the neuronal subpopulations and differentiation state of the neurons infected by HSV-1 in vivo (37). These cultures have been characterized previously and represent the neuronal heterogeneity of the adult murine trigeminal ganglia seen in vivo (37–40).
To better dissect the antiviral response of neurons and gain perspective on responses to RNA virus infection, we also used the prototypic interferon-sensitive virus, vesicular stomatitis virus (VSV) (41, 42). IFN has been show to restrict VSV replication in other neuronal cultures (43, 44), and mouse models of intranasal VSV infection have demonstrated a critical role for IFN in protection against neuropathogenesis (45, 46). Using VSV and HSV-1 mutants in combination with genetic knockouts of IFN signaling, we sought to further our understanding of the antiviral response of sensory neurons. For comparison purposes, and to gain a wider perspective on the potential impact of these cells on pathogenesis, we also performed parallel experiments using bone marrow-derived dendritic cells (BMDCs) and adult fibroblast cultures. Dendritic cells are able to mount robust antiviral responses and were used as a positive control (47, 48). Murine embryonic fibroblasts (MEFs) have been used in many studies as an IFN-responsive nonimmune cell capable of responding to viral infection, including HSV-1 and VSV, through upregulation of IFN-β transcript and protein (49–52). Rather than using MEFs, in this study we chose to utilize fibroblasts derived from adult mice to provide a more appropriate comparison with neurons derived from mice of the same developmental age.
Our results showed that the intrinsic innate antiviral response of adult TG neurons is insufficient to counter viral replication. TG neurons did, however, respond robustly to exogenous IFN-β to effectively restrict VSV replication, demonstrating the presence of a functional antiviral response through paracrine IFN signaling. This response was insufficient, however, to restrict HSV-1 replication. This is in contrast to adult fibroblasts and BMDCs, in which HSV-1 replication was significantly reduced by exogenous IFN-β. We further showed that resistance of HSV-1 to IFN-β signaling in neurons is mediated by γ34.5 and is only partly dependent on the ability of γ34.5 to bind the essential autophagy protein Beclin 1. Importantly, these data demonstrate a critical role for exogenous IFN-β in establishing a functional antiviral state in TG neurons and have significant implications for our understanding of acute and latent HSV infections.
MATERIALS AND METHODS
Neuron isolation and culture.
Coverslips (12 mm) were coated with poly-d-lysine (BD Biosciences) at 20 μg/ml in Hanks balanced salt solution minus calcium/magnesium (HBSS; Cellgro) for a minimum of 3 h. Coverslips were then washed three times with sterile distilled water and coated with natural mouse laminin (Invitrogen) at a concentration of 18 μg/ml in HBSS overnight. Trigeminal ganglia (TG) neurons were isolated as described previously with a few modifications (37). Mice 6 to 10 weeks old were euthanized by CO2 and transcardially perfused with phosphate-buffered saline (PBS; HyClone). TGs were harvested and enzymatically digested in papain solution consisting of 40 units/ml papain (Worthington) in HBSS with 1 mg l-cysteine (Sigma) and 3 μl saturated sodium bicarbonate (Sigma) for 20 min at 37°C on a rotator. This was followed by a subsequent 20-min incubation at 37°C on a rotator in a solution of 5 mg/ml collagenase type II (Invitrogen) and 5.5 mg/ml neutral protease (Worthington) dissolved in HBSS. TGs were then triturated in Neurobasal-A (NB-A) working medium consisting of Neurobasal-A (Invitrogen), 2% SM1 (StemCell), and 1% penicillin-streptomycin (penn/strep; HyClone). The resulting homogenate was spun over a four-layer density gradient made with Optiprep and NB-A working medium. Optiprep was first diluted to 50.5% with 0.8% sodium chloride and then combined with NB-A working medium to obtain the following gradient layers (Optiprep:NB-A working medium); 450 μl:550 μl, 350 μl:650 μl, 250 μl:750 μl, and 150 μl:850 μl. After a 20-min spin at 800 × g, two bands of lower density were collected and washed three times. Neurons were counted and seeded at a density of 3,600 neurons/12-mm coated coverslip in a volume of 60 μl. After 1 to 2 h, coverslips were transferred to 24-well plates and neurons were cultured in NB-A complete medium with the antimitotic 5′-fluro-2′deoxyuridine (FUDR; Sigma) for a minimum of 3 days prior to use. NB-A complete medium consisted of Neurobasal-A, 2% SM1, 1% GlutaMAX (Invitrogen), 1% penn/strep, 50 ng/ml Neurturin (R&D Systems), 50 ng/ml neuronal growth factor (NGF; Invitrogen), 50 ng/ml glial-derived neurotrophic factor (GDNF; R&D Systems), and 60 μM FUDR.
BMDC isolation and culture.
Bone marrow-derived dendritic cells (BMDCs) were isolated and cultured as described previously (8). Briefly, femurs were removed from mice that had been lightly perfused for TG neuronal isolation. Bone marrow was flushed and filtered through a 100-μM-pore-size mesh. Cells were seeded in 6-well plates at a density of 3 million cells per well. Cells were differentiated through culture with RPMI 1640 (HyClone), 1% sodium pyruvate (HyClone), 10% fetal bovine serum (FBS; Atlanta Biologicals), 0.5% penn/strep, 1% l-glutamine (HyClone), and 15% granulocyte-macrophage colony-stimulating factor (GM-CSF). Isolation of BMDCs and resulting experiments were done separately and independently for each knockout and respective wild-type strain.
Fibroblast isolation and culture.
Fibroblasts from adult mice were obtained through ear clippings and subsequently minced and digested in 1,000 U/ml collagenase type II (Invitrogen) followed by 0.05% trypsin (Cellgro). Resulting cell lysate was triturated and plated in 6-well plates in Dulbecco's modified Eagle's medium (DMEM) (HyClone) with 10% FBS, 1% nonessential amino acids, 1% GlutaMAX (Invitrogen), and 1% penn/strep.
Virus infection and interferon treatment.
Neurons cultured on coverslips were infected in NB-A complete medium (minus FUDR) in 60 μl for 1 h. They were then washed in NB-A working medium and replaced with NB-A complete. For viral growth curves, samples underwent a freeze-thaw prior to determining the titer of supernatant and cell lysate combined. A multiplicity of infection (MOI) of 20 (neurons) and 0.5 (fibroblasts) was used for all growth curves unless otherwise noted, which corresponds to an approximate effective MOI of 0.01, as determined by infection and staining 6 h postinfection (hpi) with a polyclonal HSV-1 antibody (B0114; Dako). BMDCs were harvested and resuspended in medium without GM-CSF in a 15-ml conical tube in 500 μl. Cells were incubated with virus for 1 h at 37°C, agitating every 15 min. Cells were spun down and washed with PBS and then resuspended and seeded in a 12-well dish. An MOI of 0.1 was used for all BMDC growth curves. Viral titers were assessed via plaque assay on Vero cells as described previously (53). When noted, cells were treated with IFN-β (PBL Interferon Source) at 12.5 units/ml 18 h prior to infection.
Immunofluorescence.
Cells were fixed in 3% paraformaldehyde and then permeabilized with 0.1% Triton X-100 (Sigma) in 2% normal goat serum (NGS; Vector Laboratories) in PBS. Primary and secondary antibody incubations were done in 2% NGS for 30 min at 37°C and room temperature, respectively. Primary antibodies used were rabbit anti-STAT1α91 (M-23; Santa Cruz Biotechnology), rabbit anti-phosphoSTAT1 (A-2; Santa Cruz Biotechnology), rabbit anti-beta III tubulin (Abcam), mouse anti-NeuN (A60; Millipore), mouse anti-ICP0 (Virusys), mouse anti-ICP8 (39S; kindly provided by David Knipe) (54), and rabbit anti-HSV-1 (B0114, Dako). Secondary antibodies used were goat anti-mouse Alexa 555 and goat anti mouse/rabbit Alexa 488 (Invitrogen). STAT1 staining was quantified and scored as the percentage of cells exhibiting predominantly nuclear localization. This was done for a minimum of 100 cells per group per sample.
Viruses and mice.
Strains KOS and 17 syn+ and the γ34.5-null mutant (Δγ34.5 mutant) and Beclin-binding domain mutant (ΔBBD mutant) were made as previously described (55–58). ICP0 virus (dl1403) (59) was kindly provided by David Bloom. STING−/− mice were generously provided by Glen Barber and described previously (60). STAT1−/− mice were generously provided by Joan Durbin and previously described (61). IFN-αβγR−/− (AG129) mice were described previously (62). IRF3/7 double knockout mice were generated by crossing IRF3−/− and IRF7−/− mice (50) and have been described previously (63). This study was carried out in strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. The protocol was approved by the Dartmouth IACUC Committee (5 June 2012, permit number leib.da.1). No surgery was performed, and all efforts were made to minimize suffering.
IFN-β enzyme-linked immunosorbent assay (ELISA).
Neurons, fibroblasts, and BMDCs were infected with virus as described above, washed, and then cultured for 24 h in minimal medium. Approximately 5,000 neurons, 5,000 fibroblasts, and 20,000 BMDCs were infected per sample, and results were normalized to cell number. Supernatants were harvested and processed on an IFN-β ELISA-HS kit as per the manufacture's protocol (PBL Interferon Source).
RESULTS
The intrinsic antiviral response of TG neurons is insufficient to control HSV-1 replication.
To assess the impact of intrinsic antiviral signaling of TG neurons on HSV-1 replication, we isolated neurons from wild-type (WT) mice and mice deficient in successive components of the antiviral signaling cascade, namely, the DNA sensor/adaptor molecule stimulator of interferon genes (STING), the transcription factors IRF3 and IRF7, type I and II interferon receptors (IFN-αβγR), and STAT1. Neuronal cultures were infected with HSV-1 strain KOS, and viral replication was measured over time in parallel with concurrently isolated bone marrow-derived dendritic cells (BMDCs). BMDCs are well characterized in their ability to mount a robust and effective antiviral response and were therefore used as a positive control (47, 48). As expected (10), WT BMDCs controlled HSV-1 replication, while BMDCs deficient in antiviral signaling supported significantly higher viral titers (Fig. 1A). Contrary to this, however, WT and antiviral signaling-deficient neuronal cultures yielded comparable titers of HSV-1 with no significant differences (Fig. 1B). Since KOS is a relatively avirulent HSV-1 strain in vivo (64), we wished to rule out the possibility that it replicates inherently poorly in neurons, even in the absence of IFN signaling. We therefore performed analogous experiments with HSV-1 strain 17 in WT and STAT1−/− neurons and BMDCs (Fig. 1C). Strain 17 showed the identical pattern to KOS whereby there was no difference in replication between WT and STAT1−/− neurons, while STAT1−/− BMDCs yielded significantly increased titers compared to those of the WT. This pattern of results was also observed at lower MOIs (data not shown).
FIG 1.

The intrinsic antiviral response of TG neurons is insufficient to effectively control HSV-1 replication. (A) Replication of HSV-1 KOS in BMDCs derived from mice deficient in antiviral signaling (STING−/−, STAT1−/−, IRF3/7−/−, and IFN-αβγR−/−) or respective wild-type background strains. Cells were infected at an MOI of 0.1. For all experiments, viral yield was determined by plaque assay on Vero cells at 12 and 48 h postinfection (hpi). (B) HSV-1 KOS replication in TG neurons derived from the mice used for panel A, infected at an MOI of 20. (C) HSV-1 strain 17 replication in WT (129SVEV) and STAT1−/− BMDCs, TG neurons, and fibroblasts infected at an MOI of 0.1, 20, and 0.5, respectively. In all experiments, time zero represents input inoculum. Data points represent a total from at least three (for neurons) or two (for BMDCs and fibroblasts) independent experiments each performed in triplicate. Error bars represent standard errors of the means (SEM). Significance was evaluated by two-way analysis of variance (ANOVA) with Bonferroni posttests. ***, P < 0.001.
Dendritic cells (DCs) are uniquely equipped to mount a robust antiviral response to HSV and other viruses and are a significant source of type I IFN (48, 65, 66). To gain further insight into the cell specificity of the antiviral response, we measured HSV replication in fibroblasts, a cell type that is important to the HSV life cycle and has been used to examine antiviral responses in other studies (49–52). WT (129SVEV) or STAT1−/− fibroblasts were seeded and infected at a density comparable to our experiments with TG neurons. As judged by antibody staining 6 hpi, an MOI of 0.5 for fibroblasts and an MOI of 20 for neurons achieved infection of approximately 1% of cells and was used for all experiments unless noted otherwise. The relatively high MOI needed to infect 1% of cultured neurons was consistent with previous findings (37). A comparison of strain 17 replication in WT and STAT1−/− fibroblasts revealed no difference in replication (Fig. 1C), demonstrating that these cells, like TG neurons, may be impaired in mounting an intrinsic antiviral response to HSV.
Exogenous IFN-β but not viral infection causes STAT1 relocalization to the nucleus of TG neurons.
Given the above-described data, we wished to test the hypothesis that TG neurons lack the capacity to mount an effective IFN-driven antiviral response. We exposed neurons, fibroblasts, and BMDCs to IFN-β and examined cells for STAT1 nuclear localization by immunofluorescence as a readout for IFN responsiveness (Fig. 2). One hour post-IFN-β exposure, we observed STAT1 nuclear localization in ∼99% of BMDCs (Fig. 2A and B), 90% of fibroblasts (Fig. 2C and D), and 90% of neurons (Fig. 2E and F). Having shown that these cell types could all respond robustly to IFN-β, we next measured STAT1 relocalization upon HSV infection. Cells were infected with wild-type HSV-1 strain 17 and stained for both STAT1 and the abundant HSV-1 DNA replication protein ICP8 as a marker for infected cells (67). In these infected cultures, BMDCs exhibiting both ICP8-positive and ICP8-negative staining showed significantly increased nuclear STAT1 staining 5 h postinfection (Fig. 2A and B). This indicates that HSV infection of BMDCs was sufficient to initiate autocrine IFN signaling in infected cells and a paracrine response in naive cells (Fig. 2A and B). In contrast, in infected TG neuron and fibroblast cultures, there was minimal STAT1 nuclear localization in ICP8-positive or ICP8-negative cells 5 h postinfection (Fig. 2C to F), with similar results observed at 3 and 12 hpi (data not shown). The relatively faint STAT1 staining in mock and infected fibroblasts (Fig. 2C) is likely due to the large cell size and diffuse cytoplasmic STAT1 distribution.
FIG 2.

Exogenous IFN-β but not viral infection causes STAT1 relocalization to the nucleus of TG neurons. (A) Immunofluorescence staining for STAT1 (red) or HSV-1 ICP8 or VSV-G (green) in BMDCs that were mock infected, treated with 12.5 units/ml IFN-β for 1 h, or infected with HSV-1 or VSV for 5 h. Images were merged with nuclear stain (blue; Hoechst). (B) Quantification of nuclear STAT1 staining shown in panel A. Separate quantification is shown for both infected (ICP8+ or VSVG+) and uninfected (ICP8− or VSVG−) cells of the same culture. (C) Immunofluorescence staining for STAT1 (red) or HSV-1 ICP8 or VSV-G (green) in fibroblasts. Cells were either mock infected, exposed to 12.5 units/ml IFN-β for 1 h, infected with HSV-1 or VSV for 5 h, or infected with HSV-1 for 4 h and then exposed to IFN-β for 1 h. Images were merged with nuclear stain (blue; Hoechst). (D) Quantification of nuclear STAT1 staining shown in panel C. Separate quantification is shown for both infected and uninfected cells in the same culture. (E) Analogous infection/treatment and staining as for panel C done in TG neuron cultures. Images were merged with nuclear stain (blue; Hoechst), and white arrowheads mark neurons as determined by phase contrast. (F) Quantification of nuclear STAT1 staining in panel E. Scale bar = 10 μm. Quantification was done in ≥3 replicates with ≥100 cells quantified for each group. Error bars represent SEM. Significance was evaluated in each group compared to mock (unless noted by brackets) by one-way ANOVA with Bonferroni posttests. ***, P < 0.001.
HSV-1 encodes proteins that interfere with STAT1 phosphorylation (14, 15). Given the observed lack of STAT1 nuclear localization in infected neurons and fibroblasts, we wished to distinguish whether this was due to a suppressive activity mediated by HSV or a limited cellular response to infection. To test this, we infected TG neurons, fibroblasts, or BMDCs with vesicular stomatitis virus (VSV), a prototypic IFN-sensitive virus (41). Cells were examined by immunofluorescence for STAT1 (neurons and fibroblasts) or phosphorylated STAT1 (pSTAT1; BMDCs) localization and presence of the VSV spike glycoprotein VSV-G (Fig. 2A, C, and E, right panels). In VSV-infected BMDCs, the majority of both VSV-G-positive and -negative cells showed STAT1 staining in the nucleus (Fig. 2A and B) consistent with autocrine and paracrine responses to IFN. In contrast, in TG neuron and fibroblast cultures, we observed minimal STAT1 nuclear staining in VSV-G-positive or -negative cells (Fig. 2C to F), suggesting these cells may have inefficient responses to infection. We subsequently tested whether exogenous IFN-β exposure could induce STAT1 nuclear localization in HSV-1-infected neurons and fibroblasts. In infected fibroblast cultures, nearly 100% of ICP8-positive and ICP8-negative cells had STAT1 nuclear staining, demonstrating that the addition of exogenous IFN-β is sufficient to induce STAT1 nuclear localization in infected fibroblasts (Fig. 2C and D). In TG neurons, we observed STAT1 nuclear localization in 41% of ICP8-positive and 83% of ICP8-negative neurons (Fig. 2E and F). These results are consistent with the ability of HSV to inhibit STAT1 translocation (15, 68). These data also suggest that fibroblasts may be more sensitive to exogenous IFN or HSV may be more efficient at blocking STAT1 nuclear localization in neurons.
Given the efficient translocation of STAT1 in IFN-β-treated neurons, the observed lack of STAT1 nuclear staining in HSV- and VSV-infected neurons led to the hypothesis that infected cells may make only a limited amount of IFN. We therefore measured IFN-β secretion by HSV- or VSV-infected TG neuronal cultures, fibroblasts, and BMDCs (Fig. 3). Consistent with our hypothesis and previous studies (24), we observed minimal levels of IFN-β production from neuron cultures in response to VSV or HSV-1 infection and almost undetectable IFN-β production by infected fibroblasts. In contrast, and as expected, we observed robust IFN-β production from infected BMDCs. Together, these data show that adult TG neurons and fibroblasts are limited in their ability to respond directly to viral infection but are capable of responding to exogenous IFN-β through STAT1 nuclear localization.
FIG 3.
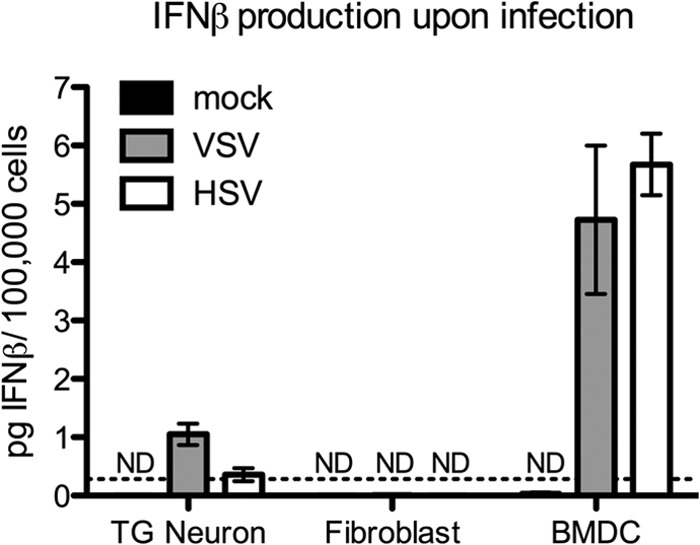
Limited IFN-β production by infected TG neuron cultures. TG neurons, fibroblasts, or BMDCs were mock treated or infected with an MOI of 20, 0.5, and 0.1, respectively, to achieve comparable numbers of infected cells. Supernatants were collected at 24 hpi, and IFN-β production measured by ELISA. Data points represent the averages of the means from at least two independent experiments. Error bars represent SEM. ND, not detectable. Dotted line represents the limit of detection.
Exogenous IFN-β effectively restricts VSV but not HSV-1 replication in TG neurons.
To test whether exogenous IFN-β and STAT1 translocation is sufficient to establish a functional antiviral response in TG neurons, we measured replication of VSV in WT and STAT1−/− neurons (Fig. 4A). VSV replicated to high titers, showing a modest but statistically significant 5-fold increase in replication in STAT1−/− neurons relative to WT. In fibroblasts, we observed no difference in VSV replication between WT and STAT1−/− cells. Upon exogenous IFN-β exposure, however, VSV replication in neurons and fibroblasts was reduced by 1,000 to 100,000-fold, compared to untreated WT or STAT1−/− cells (Fig. 4A). Interestingly, we observed a significant decrease in VSV replication in STAT1−/− fibroblasts treated with IFN-β compared to that of untreated cells, suggesting that fibroblasts may also utilize STAT1-independent IFN signaling pathways. This is in contrast to VSV replication in BMDCs where we observed a 1,000-fold decrease in titers in WT compared to in STAT1−/− cells even without the addition of exogenous IFN-β (Fig. 4A).
FIG 4.

Exogenous IFN-β effectively restricts VSV but not HSV-1 replication in TG neurons. (A) VSV replication in cells isolated from WT (129SVEV) and STAT1−/− mice. TG neurons, fibroblasts, and BMDCs were untreated or pretreated with 12.5 units/ml IFN-β for 18 h and were then infected with VSV at an MOI of 20, 0.5, and 0.1, respectively. (B) Replication of HSV-1 in 129SVEV and STAT1−/− TG neurons, fibroblasts, and BMDCs. Cells were untreated or treated with IFN-β and infected as described for panel A. Data points represent the averages of the means from at least two independent experiments. Error bars represent SEM. Significance was evaluated by two-way ANOVA with Bonferroni posttests. On the graphs, one symbol represents P values of <0.05; two symbols represent P values of <0.01; and three symbols represent P values of <0.001. * indicates significant differences between 129SVEV and 129SVEV + IFN-β; #, between 129SVEV and STAT1−/−; and †, between STAT1−/− and STAT1−/− + IFN-β.
Having shown that exogenous IFN-β is sufficient to establish an antiviral response to VSV in neurons, we next assessed the ability of IFN-pretreated TG cultures to control HSV-1 infection (Fig. 4B). In contrast to VSV, we observed no significant difference in HSV-1 replication in untreated or IFN-β-treated TG neurons, regardless of the presence of an intact antiviral response. Additionally, we observed statistically significant restricted HSV-1 replication in WT BMDCs and fibroblasts with the addition of exogenous IFN-β in a STAT1-dependent manner (Fig. 4B). Comparable results showing no impact of IFN on HSV-1 titers were also obtained from experiments performed at an MOI of 2 in neurons (data not shown).
Virus lacking γ34.5 is restricted in replication in IFN-β-treated neurons by a STAT1-dependent pathway.
The data given above strongly suggested that neurons are inefficient at establishing an IFN-dependent antiviral state upon HSV infection. We therefore wished to address the hypothesis that HSV controls the IFN-induced antiviral response of TG neurons (14, 15). To test this, we utilized an HSV-1 strain lacking γ34.5 (Δγ34.5 mutant), a neurovirulence factor capable of reversing establishment of the antiviral state and meditating resistance to IFN (16, 17). Consistent with the idea that neurons are inefficient at establishing an IFN-dependent antiviral state, there was no significant difference in replication between WT virus (strain 17) and the Δγ34.5 mutant in TG neurons, regardless of the presence of an intact IFN signaling pathway (Fig. 5A and B). These results were comparable at 24 h and at lower MOIs (data not shown). This is in contrast to a significant STAT1-dependent attenuation of Δγ34.5 mutant replication in fibroblasts and BMDCs relative to strain 17 (Fig. 5A and B). Notably, at 48 h postinfection, the fibroblast cultures were largely destroyed by virus infection. This likely explains the similar titers of strain 17 and the Δγ34.5 mutant at this time point. Upon IFN-β pretreatment, however, we observed a 100-fold decrease in the yield of the Δγ34.5 mutant from WT neurons, and this decrease was dependent upon the presence of STAT1 (Fig. 5A and B). These results were further validated by costaining WT cultures for the neuronal marker βIII-tubulin and the HSV immediate early protein, ICP0 (Fig. 5C and D). At 48 hpi, ICP0 expression was observed in approximately 60% of neurons in strain 17-infected cultures with or without IFN-β and in 60% of neurons in untreated Δγ34.5 mutant-infected cultures. In contrast, only 15% of neurons were ICP0 positive in IFN-β-treated Δγ34.5 mutant-infected cultures, consistent with decreases in viral titers described above (Fig. 5A). From these results, we conclude that γ34.5 neutralizes the antiviral impact of exogenous IFN-β on HSV-1 replication in TG neuronal cultures.
FIG 5.

Virus lacking γ34.5 is restricted in replication in IFN-β-treated neurons in a STAT1-dependent manner. Replication of HSV-1 in 129SVEV (A) or STAT1−/− (B) cells. TG neurons, fibroblasts, or BMDCs were untreated or treated with 12.5 units/ml IFN-β for 18 h and then infected at an MOI of 20, 0.5, and 0.1, respectively, with strain 17 or the Δγ34.5 mutant. Data points represent the averages of the means from at least two independent experiments. (C) 129SVEV TG neurons were infected with st17 and the Δγ34.5 mutant with or without pretreatment with 12.5 units/ml IFN-β and then fixed at 48 hpi. Immunofluorescent staining was done for neuronal marker βIII tubulin (red), HSV protein ICP0 (green), and nuclear stain (blue; DAPI). Scale bar = 100 μm. (D) Quantification of ICP0-positive neurons in panel C expressed as a percentage of total neurons, with at least 100 cells counted per sample, and performed in triplicate. Error bars represent SEM. Significance was evaluated by two-way ANOVA with Bonferroni posttests. One symbol represents P values of <0.05; two symbols represent P values of <0.01; and three symbols represent P values of <0.001. * indicates significant difference between st17 and st17 + IFN-β; #, between st17 and Δγ34.5 mutant; and †, between Δγ34.5 mutant and Δγ34.5 mutant + IFN-β.
The role of γ34.5 in promoting replication in IFN-β-treated neurons is only partly dependent on the control of autophagy.
γ34.5 also modulates the autophagic pathway through binding to the host protein Beclin 1 (19). Recently, autophagy was implicated in a dominant role in neuronal defense against HSV-1 (24). To address the contribution of autophagy modulation by γ34.5 in promoting HSV-1 replication in TG neurons, we measured replication of an HSV-1 strain that lacks the γ34.5 Beclin-binding domain (ΔBBD mutant). This in-frame deletion mutant virus has been characterized previously and retains the ability to dephosphorylate eIF2α and is thereby capable of relieving translational arrest but is incapable of binding Beclin 1 and therefore inefficiently interferes with autophagy (19). In WT neurons, there were no differences in viral yields between strain 17, the Δγ34.5 mutant, or the ΔBBD mutant at 48 hpi (Fig. 6). Upon IFN-β treatment, however, we observed a 10-fold reduction in the yield of the ΔBBD mutant and a 100-fold reduction of the Δγ34.5 mutant (Fig. 6). All 3 viruses replicated equivalently with or without IFN-β treatment in STAT1−/− neurons. This suggests that the large decrease in Δγ34.5 mutant replication in IFN-β-treated neurons is a combination of a failure to counter autophagy as well as an inability to counter the IFN-driven antiviral state. That said, the countering of the antiviral state through dephosphorylation of eIF2α likely represents the dominant function of γ34.5 in promoting viral replication in the presence of exogenous IFN-β.
FIG 6.
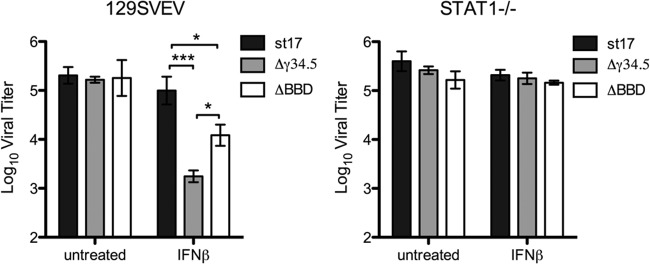
HSV resistance to IFN-β in neurons is only partly dependent on control of autophagy by the Δγ34.5 mutant. Replication of HSV strain 17, Δγ34.5 mutant, or ΔBBD mutant in 129SVEV or STAT1−/− neurons. Cells were untreated or treated with 12.5 units/ml IFN-β for 18 h and then infected at an MOI of 20, and sample titers were determined at 48 hpi. Error bars represent SEM. Experiments were performed twice in triplicate, and significance was evaluated by two-way ANOVA with Bonferroni posttests. *, P < 0.05; ***, P < 0.001.
DISCUSSION
Neuronal populations vary greatly, and this heterogeneity is reflected by a corresponding diversity in the nature and magnitude of innate antiviral responses (35, 36). This diversity, coupled with discordant paradigms of IFN-driven responses established in other cell types, significantly complicates interpretation of data derived from neurons infected in vitro and in vivo. Using cultured TG neurons from adult mice, we have demonstrated that the infection-driven antiviral response of TG neurons is insufficient to alter HSV-1 replication and only modestly reduces VSV replication. This relatively weak antiviral response correlates with the modest nuclear relocalization of STAT1 and low-level IFN-β production upon virus infection. Limited production of IFN-β by adult sensory neurons is consistent with previously published results (24). Importantly, we show that TG neurons are capable of responding to exogenous IFN-β through STAT1 nuclear localization and effective restriction of VSV and the Δγ34.5 mutant. The ability of exogenous IFN-β to restrict VSV replication in TG neurons in a STAT1-dependent manner is consistent with previous findings (43, 69).
MEFs are capable of responding to virus infection (49–52) with production of an IFN-driven response, but many viruses, including HSV and VSV, are capable of at least partially counteracting this response (70). Consistent with this idea, adult fibroblasts showed limited STAT1 nuclear localization and IFN-β production upon infection. It is likely, therefore, that adult fibroblasts are fully capable of detecting infection and mounting a response, but this pathway is counteracted by HSV and VSV. Consistent with this hypothesis, Δγ34.5 mutant replication was attenuated in a STAT1-dependent manner, suggesting that the intrinsic antiviral response of fibroblasts is suppressed by γ34.5. Additionally, there was no difference in VSV or HSV-1 replication between untreated WT and STAT1−/− adult fibroblasts, in accordance with a recent study in MEFs (71). Notably, this mirrors the pattern of virus replication observed in neurons.
Neurons and fibroblasts differ significantly in their abilities to control HSV-1 replication following IFN treatment. IFN-treated neurons were incapable of inhibiting HSV-1 replication, and these dampened IFN responses of primary neurons are consistent with previous work (24). In contrast to our findings, other studies have shown that when provided with high concentrations of IFN (100 to 1,000 U/ml), adult sensory neurons can modestly restrict HSV-1 replication (24) and viral gene expression (72). We employed a low concentration (12.5 U/ml) of IFN-β in this study to more accurately reflect the limiting concentrations found during acute HSV infection of the murine nervous system (70) (Z. M. Parker and D. A. Leib, unpublished data). The difference in IFN concentration likely explains the disparity between our data and those previously published.
These differences notwithstanding, we have demonstrated a critical and specific role for γ34.5 in promoting HSV replication in neurons by combating the antiviral state established in neurons upon IFN-β exposure. Indeed, in the absence of γ34.5, HSV-1 is as sensitive to IFN as VSV. Additionally, a virus lacking ICP0, an HSV gene also shown to mediate resistance to IFN responses (73, 74), showed no difference in replication in STAT1 and WT neurons with or without IFN (data not shown). Together these data suggest that the resistance of HSV to the neuronal antiviral response is specific to γ34.5.
Previous work has shown that HSV strains lacking γ34.5 are significantly attenuated in the nervous systems of mice and humans and replicate poorly in neurons in vitro (16, 21–23, 75). It was unexpected, therefore, to find comparable replication of WT and Δγ34.5 mutant viruses in untreated TG cultures. This contrasts viral replication in untreated fibroblasts and BMDCs where we observed the predictable STAT1-dependent attenuation of the Δγ34.5 mutant, further highlighting the lack of an effective intrinsic antiviral response in TG neurons. The significant attenuation of Δγ34.5 mutant replication upon IFN-β pretreatment of neurons suggests that the neuroattenuation of the Δγ34.5 mutant seen in other studies (16, 21, 23, 75) is at least partially dependent on paracrine IFN-β signaling. During HSV-1 infection in vivo, IFN-β is present both in the peripheral nervous system (76) and central nervous system (77), which likely restricts further Δγ34.5 mutant replication, spread, and neurovirulence. It is also probable that in previous in vitro studies, nonneuronal cells were a significant source of IFN that primed infected and uninfected neurons leading to reduced yields of the Δγ34.5 mutant (78, 79). To our knowledge, this is the first study to examine replication of the Δγ34.5 mutant in adult murine TG neurons and in neurons of such high purity (32, 34). These factors are all likely important determinants of the ability of neurons to exert innate control of HSV replication in the absence of γ34.5. It is possible that a limiting level of γ34.5 expression during natural infections in vivo may contribute to the susceptibility of HSV-1 to innate defense, thereby promoting the establishment of latency and a successful viral life cycle (80, 81).
In addition to modulating establishment of the antiviral state, γ34.5 also modulates autophagy through its Beclin-binding domain (BBD) (19), and this activity promotes HSV replication in the nervous system (24). This previous study furthered the idea that autophagy can act as a nondestructive antiviral defense in neurons (82). In the present study, however, ΔBBD mutant replication was comparable to that of strain 17 in untreated neurons but showed a significant STAT1-dependent decrease following IFN-β treatment. While IFN-induced autophagy has been directly studied only in other cell types (83–85), these results are consistent with a role for IFN in the upregulation of antiviral autophagy (xenophagy) in neurons. The 10-fold decrease in ΔBBD mutant replication following IFN pretreatment suggests that autophagy cannot completely account for the 100-fold reduction of Δγ34.5 mutant replication upon IFN-β treatment. It seems more likely, therefore, that the neuronal antiviral response is substantially responsible for this attenuation and that the autophagy and antiviral countering functions of γ34.5 synergize to combat the IFN-driven neuronal response to infection.
This study demonstrates a requirement for exogenously supplied IFN for establishment of the antiviral state in neurons. As previously suggested, the requirement of exogenous IFN-β signaling to upregulate antiviral responses may help ensure neuronal survival (24, 72, 86–89) while promoting apoptosis in other cell types (24, 90). Indeed, it has been shown that IFN can promote survival of HSV-1-infected sensory neurons (72). Tight regulation of antiviral signaling in neurons may be a potential safety mechanism to preclude a vigorous antiviral response that may risk death or damage of an irreplaceable cell population (24, 82). The lack of an effective autocrine response in infected neurons and the apparent requirement for paracrine IFN signaling may help ensure that the antiviral response acts to balance viral clearance and cell survival. Additionally, these data highlight a more important role for paracrine IFN responses in nonimmune cells in establishing an effective antiviral response. Our data further our understanding of the neuronal antiviral response and suggest an important role for a measured neuronal IFN response in order to promote latent HSV infections and to balance control of virus replication and neuronal survival.
ACKNOWLEDGMENTS
This study was supported by National Institutes of Health grants to D.A.L. (RO1 EY09083 and P01 AI098681). The project was also supported by P20RR016437 from the National Center for Research Resources to Dartmouth and a Geisel School of Medicine Microbiology and Molecular Pathogenesis Program Training Grant (5T32AI007519) to P.C.R.
We also acknowledge Sarah Katzenell, David Bloom, Don Coen, and David Knipe for reagents and helpful discussions. We especially thank Todd Margolis and Andrea Bertke for thoughtful discussions and generous help with the TG neuronal culture system and Bill Halford for helpful comments on a previous version of the manuscript.
Footnotes
Published ahead of print 18 June 2014
REFERENCES
- 1.Smith JS, Robinson NJ. 2002. Age-specific prevalence of infection with herpes simplex virus types 2 and 1: a global review. J. Infect. Dis. 186:S3–S28. 10.1086/343739 [DOI] [PubMed] [Google Scholar]
- 2.Gilden DH, Mahalingam R, Cohrs RJ, Tyler KL. 2007. Herpesvirus infections of the nervous system. Nat. Clin. Pract. Neurol. 3:82–94. 10.1038/ncpneuro0401 [DOI] [PubMed] [Google Scholar]
- 3.Smith G. 2012. Herpesvirus transport to the nervous system and back again. Annu. Rev. Microbiol. 66:153–176. 10.1146/annurev-micro-092611-150051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dupuis S, Jouanguy E, Al-Hajjar S, Fieschi C, Al-Mohsen IZ, Al-Jumaah S, Yang K, Chapgier A, Eidenschenk C, Eid P, Ghonaium AA, Tufenkeji H, Frayha H, Al-Gazlan S, Al-Rayes H, Schreiber RD, Gresser I, Casanova J-L. 2003. Impaired response to interferon-[alpha]/[beta] and lethal viral disease in human STAT1 deficiency. Nat. Genet. 33:388–391. 10.1038/ng1097 [DOI] [PubMed] [Google Scholar]
- 5.Casrouge A, Zhang S-Y, Eidenschenk C, Jouanguy E, Puel A, Yang K, Alcais A, Picard C, Mahfoufi N, Nicolas N, Lorenzo L, Plancoulaine S, Sénéchal B, Geissmann F, Tabeta K, Hoebe K, Du X, Miller RL, Héron B, Mignot C, de Villemeur TB, Lebon P, Dulac O, Rozenberg F, Beutler B, Tardieu M, Abel L, Casanova J-L 2006. Herpes simplex virus encephalitis in human UNC-93B deficiency. Science 314:308–312. 10.1126/science.1128346 [DOI] [PubMed] [Google Scholar]
- 6.Zhang S-Y, Jouanguy E, Ugolini S, Smahi A, Elain G, Romero P, Segal D, Sancho-Shimizu V, Lorenzo L, Puel A, Picard C, Chapgier A, Plancoulaine S, Titeux M, Cognet C, von Bernuth H, Ku C-L, Casrouge A, Zhang X-X, Barreiro L, Leonard J, Hamilton C, Lebon P, Héron B, Vallée L, Quintana-Murci L, Hovnanian A, Rozenberg F, Vivier E, Geissmann F, Tardieu M, Abel L, Casanova J-L. 2007. TLR3 deficiency in patients with herpes simplex encephalitis. Science 317:1522–1527. 10.1126/science.1139522 [DOI] [PubMed] [Google Scholar]
- 7.Pérez de Diego R, Sancho-Shimizu V, Lorenzo L, Puel A, Plancoulaine S, Picard C, Herman M, Cardon A, Durandy A, Bustamante J, Vallabhapurapu S, Bravo J, Warnatz K, Chaix Y, Cascarrigny F, Lebon P, Rozenberg F, Karin M, Tardieu M, Al-Muhsen S, Jouanguy E, Zhang S-Y, Abel L, Casanova J-L. 2010. Human TRAF3 adaptor molecule deficiency leads to impaired Toll-like receptor 3 response and susceptibility to herpes simplex encephalitis. Immunity 33:400–411. 10.1016/j.immuni.2010.08.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Menachery VD, Pasieka TJ, Leib DA. 2010. Interferon regulatory factor 3-dependent pathways are critical for control of herpes simplex virus type 1 central nervous system infection. J. Virol. 84:9685–9694. 10.1128/JVI.00706-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Leib DA, Harrison TE, Laslo KM, Machalek MA, Moorman NJ, Virgin HW. 1999. Interferons regulate the phenotype of wild-type and mutant herpes simplex viruses in vivo. J. Exp. Med. 189:663–672. 10.1084/jem.189.4.663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Luker GD, Prior JL, Song J, Pica CM, Leib DA. 2003. Bioluminescence imaging reveals systemic dissemination of herpes simplex virus type 1 in the absence of interferon receptors. J. Virol. 77:11082–11093. 10.1128/JVI.77.20.11082-11093.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Paludan SR, Bowie AG, Horan KA, Fitzgerald KA. 2011. Recognition of herpesviruses by the innate immune system. Nat. Rev. Immunol. 11:143–154. 10.1038/nri2937 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Schindler C, Levy DE, Decker T. 2007. JAK-STAT signaling: from interferons to cytokines. J. Biol. Chem. 282:20059–20063. 10.1074/jbc.R700016200 [DOI] [PubMed] [Google Scholar]
- 13.Levy DE, Marié IJ, Durbin JE. 2011. Induction and function of type I and III interferon in response to viral infection. Curr. Opin. Virol. 1:476–486. 10.1016/j.coviro.2011.11.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Leib DA. 2002. Counteraction of interferon-induced antiviral responses by herpes simplex viruses. Curr. Top. Microbiol. Immunol. 269:171–185 [DOI] [PubMed] [Google Scholar]
- 15.Paladino P, Mossman KL. 2009. Mechanisms employed by herpes simplex virus 1 to inhibit the interferon response. J. Interferon Cytokine Res. 29:599–607. 10.1089/jir.2009.0074 [DOI] [PubMed] [Google Scholar]
- 16.Chou J, Kern ER, Whitley RJ, Roizman B. 1990. Mapping of herpes simplex virus-1 neurovirulence to gamma 134.5, a gene nonessential for growth in culture. Science 250:1262–1266. 10.1126/science.2173860 [DOI] [PubMed] [Google Scholar]
- 17.Li Y, Zhang C, Chen X, Yu J, Wang Y, Yang Y, Du M, Jin H, Ma Y, He B, Cao Y. 2011. ICP34.5 protein of herpes simplex virus facilitates the initiation of protein translation by bridging eukaryotic initiation factor 2alpha (eIF2alpha) and protein phosphatase 1. J. Biol. Chem. 286:24785–24792. 10.1074/jbc.M111.232439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Verpooten D, Ma Y, Hou S, Yan Z, He B. 2009. Control of TANK-binding kinase 1-mediated signaling by the gamma(1)34.5 protein of herpes simplex virus 1. J. Biol. Chem. 284:1097–1105. 10.1074/jbc.M805905200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Orvedahl A, Alexander D, Tallóczy Z, Sun Q, Wei Y, Zhang W, Burns D, Leib DA, Levine B. 2007. HSV-1 ICP34.5 confers neurovirulence by targeting the Beclin 1 autophagy protein. Cell Host Microbe 1:23–35. 10.1016/j.chom.2006.12.001 [DOI] [PubMed] [Google Scholar]
- 20.Gobeil PAM, Leib DA. 2012. Herpes simplex virus? 34.5 interferes with autophagosome maturation and antigen presentation in dendritic cells. mBio 3(5):e00267–12. 10.1128/mBio.00267-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rampling R, Cruickshank G, Papanastassiou V, Nicoll J, Hadley D, Brennan D, Petty R, MacLean A, Harland J, McKie E, Mabbs R, Brown M. 2000. Toxicity evaluation of replication-competent herpes simplex virus (ICP 34.5 null mutant 1716) in patients with recurrent malignant glioma. Gene Ther. 7:859–866. 10.1038/sj.gt.3301184 [DOI] [PubMed] [Google Scholar]
- 22.Leib DA, Machalek MA, Williams BR, Silverman RH, Virgin HW. 2000. Specific phenotypic restoration of an attenuated virus by knockout of a host resistance gene. Proc. Natl. Acad. Sci. U. S. A. 97:6097–6101. 10.1073/pnas.100415697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Thompson RL, Stevens JG. 1983. Biological characterization of a herpes simplex virus intertypic recombinant which is completely and specifically non-neurovirulent. Virology 131:171–179. 10.1016/0042-6822(83)90543-3 [DOI] [PubMed] [Google Scholar]
- 24.Yordy B, Iijima N, Huttner A, Leib D, Iwasaki A. 2012. A neuron-specific role for autophagy in antiviral defense against herpes simplex virus. Cell Host Microbe 12:334–345. 10.1016/j.chom.2012.07.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.He C, Klionsky DJ. 2009. Regulation mechanisms and signaling pathways of autophagy. Annu. Rev. Genet. 43:67–93. 10.1146/annurev-genet-102808-114910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kroemer G, Mariño G, Levine B. 2010. Autophagy and the integrated stress response. Mol. Cell 40:280–293. 10.1016/j.molcel.2010.09.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Carr DJJ, Al-khatib Khaldun James CM, Silverman R. 2003. Interferon-β suppresses herpes simplex virus type 1 replication in trigeminal ganglion cells through an RNase L-dependent pathway. J. Neuroimmunol. 141:40–46. 10.1016/S0165-5728(03)00216-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Svennerholm B, Ziegler R, Lycke E. 1989. Herpes simplex virus infection of the rat sensory neuron effects of interferon on cultured cells. Arch. Virol. 104:153–156. 10.1007/BF01313816 [DOI] [PubMed] [Google Scholar]
- 29.De Regge N, Van Opdenbosch N, Nauwynck HJ, Efstathiou S, Favoreel HW. 2010. Interferon alpha induces establishment of alphaherpesvirus latency in sensory neurons in vitro. PLoS One 5:e13076. 10.1371/journal.pone.0013076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wigdahl BL, Ziegler RJ, Sneve M, Rapp F. 1983. Herpes simplex virus latency and reactivation in isolated rat sensory neurons. Virology 127:159–167. 10.1016/0042-6822(83)90380-X [DOI] [PubMed] [Google Scholar]
- 31.Lafaille FG, Pessach IM, Zhang S-Y, Ciancanelli MJ, Herman M, Abhyankar A, Ying S-W, Keros S, Goldstein PA, Mostoslavsky G, Ordovas-Montanes J, Jouanguy E, Plancoulaine S, Tu E, Elkabetz Y, Al-Muhsen S, Tardieu M, Schlaeger TM, Daley GQ, Abel L, Casanova J-L, Studer L, Notarangelo LD. 2012. Impaired intrinsic immunity to HSV-1 in human iPSC-derived TLR3-deficient CNS cells. Nature 491:769–773. 10.1038/nature11583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wilson AC, Mohr I. 2012. A cultured affair: HSV latency and reactivation in neurons. Trends Microbiol. 20:604–611. 10.1016/j.tim.2012.08.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hafezi W, Lorentzen EU, Eing BR, Müller M, King NJC, Klupp B, Mettenleiter TC, Kühn JE. 2012. Entry of herpes simplex virus type 1 (HSV-1) into the distal axons of trigeminal neurons favors the onset of nonproductive, silent infection. PLoS Pathog. 8:e1002679. 10.1371/journal.ppat.1002679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kobayashi M, Kim J-Y, Camarena V, Roehm PC, Chao MV, Wilson AC, Mohr I. 2012. A primary neuron culture system for the study of herpes simplex virus latency and reactivation. J. Vis. Exp. 10.3791/3823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Farmer JR, Altschaefl KM, O'Shea KS, Miller DJ. 2013. Activation of the type I interferon pathway is enhanced in response to human neuronal differentiation. PLoS One 8:e58813. 10.1371/journal.pone.0058813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cho H, Proll SC, Szretter KJ, Katze MG, Gale M, Jr, Diamond MS. 2013. Differential innate immune response programs in neuronal subtypes determine susceptibility to infection in the brain by positive-stranded RNA viruses. Nat. Med. 19:458–464. 10.1038/nm.3108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bertke AS, Swanson SM, Chen J, Imai Y, Kinchington PR, Margolis TP. 2011. A5-positive primary sensory neurons are nonpermissive for productive infection with herpes simplex virus 1 in vitro. J. Virol. 85:6669–6677. 10.1128/JVI.00204-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Harkness JM, Kader M, DeLuca NA. 2014. Transcription of the herpes simplex virus 1 genome during productive and quiescent infection of neuronal and nonneuronal cells. J. Virol. 88:6847–6861. 10.1128/JVI.00516-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bertke AS, Ma A, Margolis MS, Margolis TP. 2013. Different mechanisms regulate productive herpes simplex virus 1 (HSV-1) and HSV-2 infections in adult trigeminal neurons. J. Virol. 87:6512–6516. 10.1128/JVI.00383-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bertke AS, Apakupakul K, Ma A, Imai Y, Gussow AM, Wang K, Cohen JI, Bloom DC, Margolis TP. 2012. LAT region factors mediating differential neuronal tropism of HSV-1 and HSV-2 do not act in trans. PLoS One 7:e53281. 10.1371/journal.pone.0053281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Stewart WE, II, Scott WD, Sulkin SE. 1969. Relative sensitivities of viruses to different species of interferon. J. Virol. 4:147–153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rubinstein S, Familletti PC, Pestka S. 1981. Convenient assay for interferons. J. Virol. 37:755–758 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tsukamoto LF, Price RW. 1982. Interferon protects neurons in culture infected with vesicular stomatitis and herpes simplex viruses. J. Neurol. Sci. 56:115–128. 10.1016/0022-510X(82)90066-1 [DOI] [PubMed] [Google Scholar]
- 44.Trottier MD, Jr, Palian BM, Reiss CS. 2005. VSV replication in neurons is inhibited by type I IFN at multiple stages of infection. Virology 333:215–225. 10.1016/j.virol.2005.01.009 [DOI] [PubMed] [Google Scholar]
- 45.Detje CN, Meyer T, Schmidt H, Kreuz D, Rose JK, Bechmann I, Prinz M, Kalinke U. 2009. Local type I IFN receptor signaling protects against virus spread within the central nervous system. J. Immunol. 182:2297–2304 [DOI] [PubMed] [Google Scholar]
- 46.Fensterl V, Wetzel JL, Ramachandran S, Ogino T, Stohlman SA, Bergmann CC, Diamond MS, Virgin HW, Sen GC. 2012. Interferon-induced Ifit2/ISG54 protects mice from lethal VSV neuropathogenesis. PLoS Pathog. 8:e1002712. 10.1371/journal.ppat.1002712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Liu Y-J. 2005. IPC: professional type 1 interferon-producing cells and plasmacytoid dendritic cell precursors. Annu. Rev. Immunol. 23:275–306. 10.1146/annurev.immunol.23.021704.115633 [DOI] [PubMed] [Google Scholar]
- 48.Colonna M, Trinchieri G, Liu Y-J. 2004. Plasmacytoid dendritic cells in immunity. Nat. Immunol. 5:1219–1226. 10.1038/ni1141 [DOI] [PubMed] [Google Scholar]
- 49.Rasmussen SB, Sørensen LN, Malmgaard L, Ank N, Baines JD, Chen ZJ, Paludan SR. 2007. Type I interferon production during herpes simplex virus infection is controlled by cell-type-specific viral recognition through Toll-like receptor 9, the mitochondrial antiviral signaling protein pathway, and novel recognition systems. J. Virol. 81:13315–13324. 10.1128/JVI.01167-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sato M, Suemori H, Hata N, Asagiri M, Ogasawara K, Nakao K, Nakaya T, Katsuki M, Noguchi S, Tanaka N, Taniguchi T. 2000. Distinct and essential roles of transcription factors IRF-3 and IRF-7 in response to viruses for IFN-alpha/beta gene induction. Immunity 13:539–548. 10.1016/S1074-7613(00)00053-4 [DOI] [PubMed] [Google Scholar]
- 51.Choi MK, Wang Z, Ban T, Yanai H, Lu Y, Koshiba R, Nakaima Y, Hangai S, Savitsky D, Nakasato M, Negishi H, Takeuchi O, Honda K, Akira S, Tamura T, Taniguchi T. 2009. A selective contribution of the RIG-I-like receptor pathway to type I interferon responses activated by cytosolic DNA. Proc. Natl. Acad. Sci. 10.1073/pnas.0909545106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Randall RE, Goodbourn S. 2008. Interferons and viruses: an interplay between induction, signalling, antiviral responses and virus countermeasures. J. Gen. Virol. 89:1–47. 10.1099/vir.0.83391-0 [DOI] [PubMed] [Google Scholar]
- 53.Rader KA, Ackland-Berglund CE, Miller JK, Pepose JS, Leib DA. 1993. In vivo characterization of site-directed mutations in the promoter of the herpes simplex virus type 1 latency-associated transcripts. J. Gen. Virol. 74(Part 9):1859–1869 [DOI] [PubMed] [Google Scholar]
- 54.Showalter SD, Zweig M, Hampar B. 1981. Monoclonal antibodies to herpes simplex virus type 1 proteins, including the immediate-early protein ICP 4. Infect. Immun. 34:684–692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Smith KO. 1964. Relationship between the envelope and the infectivity of herpes simplex virus. Proc. Soc. Exp. Biol. Med. 115:814–816. 10.3181/00379727-115-29045 [DOI] [PubMed] [Google Scholar]
- 56.Alexander DE, Ward SL, Mizushima N, Levine B, Leib DA. 2007. Analysis of the role of autophagy in replication of herpes simplex virus in cell culture. J. Virol. 81:12128–12134. 10.1128/JVI.01356-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.English L, Chemali M, Duron J, Rondeau C, Laplante A, Gingras D, Alexander D, Leib D, Norbury C, Lippé R, Desjardins M. 2009. Autophagy enhances the presentation of endogenous viral antigens on MHC class I molecules during HSV-1 infection. Nat. Immunol. 10:480–487. 10.1038/ni.1720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Brown SM, Ritchie DA, Subak-Sharpe JH. 1973. Genetic studies with herpes simplex virus type 1. The isolation of temperature-sensitive mutants, their arrangement into complementation groups and recombination analysis leading to a linkage map. J. Gen. Virol. 18:329–346 [DOI] [PubMed] [Google Scholar]
- 59.Stow ND, Stow EC. 1986. Isolation and characterization of a herpes simplex virus type 1 mutant containing a deletion within the gene encoding the immediate early polypeptide Vmw110. J. Gen. Virol. 67(Part 12):2571–2585 [DOI] [PubMed] [Google Scholar]
- 60.Ishikawa H, Barber GN. 2008. STING is an endoplasmic reticulum adaptor that facilitates innate immune signalling. Nature 455:674–678. 10.1038/nature07317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Durbin JE, Hackenmiller R, Simon MC, Levy DE. 1996. Targeted disruption of the mouse Stat1 gene results in compromised innate immunity to viral disease. Cell 84:443–450. 10.1016/S0092-8674(00)81289-1 [DOI] [PubMed] [Google Scholar]
- 62.Van den Broek MF, Muller U, Huang S, Aguet M, Zinkernagel RM. 1995. Antiviral defense in mice lacking both alpha/beta and gamma interferon receptors. J. Virol. 69:4792–4796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Murphy AA, Rosato PC, Parker ZM, Khalenkov A, Leib DA. 2013. Synergistic control of herpes simplex virus pathogenesis by IRF-3, and IRF-7 revealed through non-invasive bioluminescence imaging. Virology 444:71–79. 10.1016/j.virol.2013.05.034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Dix RD, McKendall RR, Baringer JR. 1983. Comparative neurovirulence of herpes simplex virus type 1 strains after peripheral or intracerebral inoculation of BALB/c mice. Infect. Immun. 40:103–112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Krug A, Luker GD, Barchet W, Leib DA, Akira S, Colonna M. 2004. Herpes simplex virus type 1 activates murine natural interferon-producing cells through Toll-like receptor 9. Blood 103:1433–1437. 10.1182/blood-2003-08-2674 [DOI] [PubMed] [Google Scholar]
- 66.Swiecki M, Wang Y, Gilfillan S, Colonna M. 2013. Plasmacytoid dendritic cells contribute to systemic but not local antiviral responses to HSV infections. PLoS Pathog. 9:e1003728. 10.1371/journal.ppat.1003728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Conley AJ, Knipe DM, Jones PC, Roizman B. 1981. Molecular genetics of herpes simplex virus. VII. Characterization of a temperature-sensitive mutant produced by in vitro mutagenesis and defective in DNA synthesis and accumulation of gamma polypeptides. J. Virol. 37:191–206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Reich NC, Liu L. 2006. Tracking STAT nuclear traffic. Nat. Rev. Immunol. 6:602–612. 10.1038/nri1885 [DOI] [PubMed] [Google Scholar]
- 69.Chesler DA, Dodard C, Lee GY, Levy DE, Reiss CS. 2004. Interferon-gamma-induced inhibition of neuronal vesicular stomatitis virus infection is STAT1 dependent. J. Neurovirol. 10:57–63. 10.1080/13550280490261707 [DOI] [PubMed] [Google Scholar]
- 70.Kato H, Takeuchi O, Sato S, Yoneyama M, Yamamoto M, Matsui K, Uematsu S, Jung A, Kawai T, Ishii KJ, Yamaguchi O, Otsu K, Tsujimura T, Koh C-S, Reis e Sousa C, Matsuura Y, Fujita T, Akira S. 2006. Differential roles of MDA5 and RIG-I helicases in the recognition of RNA viruses. Nature 441:101–105. 10.1038/nature04734 [DOI] [PubMed] [Google Scholar]
- 71.Katzenell S, Chen Y, Parker ZM, Leib DA. 2014. The differential interferon responses of two strains of Stat1-deficient mice do not alter susceptibility to HSV-1 and VSV in vivo. Virology 451:350–354. 10.1016/j.virol.2013.12.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Low-Calle AM, Prada-Arismendy J, Castellanos JE. 2013. Study of interferon-β antiviral activity against herpes simplex virus type 1 in neuron-enriched trigeminal ganglia cultures. Virus Res. 180:49–58. 10.1016/j.virusres.2013.12.022 [DOI] [PubMed] [Google Scholar]
- 73.Mossman KL, Saffran HA, Smiley JR. 2000. Herpes simplex virus ICP0 mutants are hypersensitive to interferon. J. Virol. 74:2052–2056. 10.1128/JVI.74.4.2052-2056.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Härle P, Sainz B, Jr, Carr DJJ, Halford WP. 2002. The immediate-early protein, ICP0, is essential for the resistance of herpes simplex virus to interferon-alpha/beta. Virology 293:295–304. 10.1006/viro.2001.1280 [DOI] [PubMed] [Google Scholar]
- 75.Chou J, Roizman B. 1992. The gamma 1(34.5) gene of herpes simplex virus 1 precludes neuroblastoma cells from triggering total shutoff of protein synthesis characteristic of programed cell death in neuronal cells. Proc. Natl. Acad. Sci. U. S. A. 89:3266–3270. 10.1073/pnas.89.8.3266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Conrady CD, Jones H, Zheng M, Carr DJJ. 2011. A functional type I interferon pathway drives resistance to cornea herpes simplex virus type 1 infection by recruitment of leukocytes. J. Biomed. Res. 25:111–119. 10.1016/S1674-8301(11)60014-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Wang JP, Bowen GN, Zhou S, Cerny A, Zacharia A, Knipe DM, Finberg RW, Kurt-Jones EA. 2012. Role of specific innate immune responses in herpes simplex virus infection of the central nervous system. J. Virol. 86:2273–2281. 10.1128/JVI.06010-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Kallfass C, Ackerman A, Lienenklaus S, Weiss S, Heimrich B, Staeheli P. 2012. Visualizing production of beta interferon by astrocytes and microglia in brain of La Crosse virus-infected mice. J. Virol. 86:11223–11230. 10.1128/JVI.01093-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Roth-Cross JK, Bender SJ, Weiss SR. 2008. Murine coronavirus mouse hepatitis virus is recognized by MDA5 and induces type I interferon in brain macrophages/microglia. J. Virol. 82:9829–9838. 10.1128/JVI.01199-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Nichol PF, Chang JY, Johnson EM, Jr, Olivo PD. 1996. Herpes simplex virus gene expression in neurons: viral DNA synthesis is a critical regulatory event in the branch point between the lytic and latent pathways. J. Virol. 70:5476–5486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Kemp LM, Dent CL, Latchman DS. 1990. Octamer motif mediates transcriptional repression of HSV immediate-early genes and octamer-containing cellular promoters in neuronal cells. Neuron 4:215–222. 10.1016/0896-6273(90)90096-X [DOI] [PubMed] [Google Scholar]
- 82.Alexander DE, Leib DA. 2008. Xenophagy in herpes simplex virus replication and pathogenesis. Autophagy 4:101–103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Schmeisser H, Bekisz J, Zoon KC. 2014. New function of type I IFN-: induction of autophagy. J. Interferon Cytokine Res. 34:71–78. 10.1089/jir.2013.0128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Schmeisser H, Fey SB, Horowitz J, Fischer ER, Balinsky CA, Miyake K, Bekisz J, Snow AL, Zoon KC. 2013. Type I interferons induce autophagy in certain human cancer cell lines. Autophagy 9:683–696. 10.4161/auto.23921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Matsuzawa T, Kim B-H, Shenoy AR, Kamitani S, Miyake M, Macmicking JD. 2012. IFN-γ elicits macrophage autophagy via the p38 MAPK signaling pathway. J. Immunol. 189:813–818. 10.4049/jimmunol.1102041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Chang JY, Martin DP, Johnson EM. 1990. Interferon suppresses sympathetic neuronal cell death caused by nerve growth factor deprivation. J. Neurochem. 55:436–445. 10.1111/j.1471-4159.1990.tb04155.x [DOI] [PubMed] [Google Scholar]
- 87.Yang CH, Murti A, Pfeffer SR, Basu L, Kim JG, Pfeffer LM. 2000. IFN-α/β promotes cell survival by activating NF-κB. Proc. Natl. Acad. Sci. U. S. A. 97:13631–13636. 10.1073/pnas.250477397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Samuel MA, Diamond MS. 2005. Alpha/beta interferon protects against lethal West Nile virus infection by restricting cellular tropism and enhancing neuronal survival. J. Virol. 79:13350–13361. 10.1128/JVI.79.21.13350-13361.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Jin S, Kawanokuchi J, Mizuno T, Wang J, Sonobe Y, Takeuchi H, Suzumura A. 2007. Interferon-beta is neuroprotective against the toxicity induced by activated microglia. Brain Res. 1179:140–146. 10.1016/j.brainres.2007.08.055 [DOI] [PubMed] [Google Scholar]
- 90.Balachandran S, Roberts PC, Kipperman T, Bhalla KN, Compans RW, Archer DR, Barber GN. 2000. Alpha/beta interferons potentiate virus-induced apoptosis through activation of the FADD/caspase-8 death signaling pathway. J. Virol. 74:1513–1523. 10.1128/JVI.74.3.1513-1523.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]