

Abstract

The Src homology 2 domain containing protein tyrosine phosphatase-2 (SHP2) is an oncogenic phosphatase associated with various kinds of leukemia and solid tumors. Thus, there is substantial interest in developing SHP2 inhibitors as potential anticancer and antileukemia agents. Using a structure-guided and fragment-based library approach, we identified a novel hydroxyindole carboxylic acid-based SHP2 inhibitor 11a-1, with an IC50 value of 200 nM and greater than 5-fold selectivity against 20 mammalian PTPs. Structural and modeling studies reveal that the hydroxyindole carboxylic acid anchors the inhibitor to the SHP2 active site, while interactions of the oxalamide linker and the phenylthiophene tail with residues in the β5–β6 loop contribute to 11a-1’s binding potency and selectivity. Evidence suggests that 11a-1 specifically attenuates the SHP2-dependent signaling inside the cell. Moreover, 11a-1 blocks growth factor mediated Erk1/2 and Akt activation and exhibits excellent antiproliferative activity in lung cancer and breast cancer as well as leukemia cell lines.
Introduction
Proper levels of tyrosine phosphorylation, maintained by the reversible and coordinated actions of protein tyrosine kinases (PTKs) and protein tyrosine phosphatases (PTPs), are critical for a wide range of cellular processes including growth, differentiation, metabolism, migration, and survival.1,2 Aberrant tyrosine phosphorylation, as a result of perturbed balance between the activities of PTKs and PTPs, is linked to the pathogenesis of numerous human diseases, including cancer.1,3,4 Consequently, signaling events driven by tyrosine phosphorylation offer a rich source of molecular targets for therapeutic interventions.5,6 The success of such targeted approaches has been well demonstrated by the more than two dozen PTK inhibitors that are already approved for clinical uses.7 However, the therapeutic potential of modulating the PTPs is still underexplored despite the fact that several PTPs have also been identified as high-value targets.3,4
Excessive tyrosine phosphorylation is a hallmark of cancer, usually caused by abnormal expression and/or activation of receptor PTKs, such as epidermal growth factor receptor, and nonreceptor PTKs such as Src and Abl.5 By catalyzing the dephosphorylation of phosphotyrosine residues, PTPs are usually viewed as negative regulators of signal transduction and therefore perceived as products of tumor suppressor genes. Indeed, several PTPs, including PTEN, have been identified as tumor suppressors.8,9 However, recent genetic analyses of human tumors reveal that at least 22 PTPs are candidate oncoproteins.4 To this end, the Src homology 2 (SH2) domain containing protein tyrosine phosphatase-2 (SHP2), encoded by the Ptpn11 gene, positively potentiates cellular signaling.10−12 Biochemical and genetic studies establish that SHP2 promotes signaling downstream from almost all growth factor and cytokine receptors and upstream of Ras and is required for growth factor/cytokine-induced cell proliferation, migration, and survival. In addition, considerable evidence indicates that SHP2 is a bona fide oncoprotein. Germline gain-of-function SHP2 mutations cause 50% of Noonan syndrome, an autosomal dominant developmental disorder with increased risk of malignancy.13,14 Moreover, somatic activating mutations in SHP2 are associated with juvenile myelomonocytic leukemia, acute myeloid leukemia, myelodysplastic syndrome, acute lymphoid leukemia, and several types of solid tumors including lung adenocarcinoma, colon cancer, neuroblastoma, glioblastoma, melanoma, hepatocellular carcinoma, and prostate cancer.15−24 Furthermore, SHP2 has also been implicated as a therapeutic target for triple-negative and HER2+ breast cancer.25 Finally, given the obligatory requirement of SHP2 in multiple growth factor-mediated pathways, thwarting SHP2 activity may also prove effective for cancers caused by abnormal activation of receptor PTKs, some of which respond poorly to kinase inhibitor monotherapy.26 This strong validation of SHP2 as an oncology target has generated considerable interest in the development of small molecule SHP2 inhibitors as potential therapeutic agents for hematologic malignancies and solid tumors.27−41
Unfortunately, PTPs, including SHP2, have proven to be an extremely difficult target for drug discovery, due primarily to the highly conserved and positively charged nature of its PTP active site. Indeed, the majority of reported SHP2 inhibitors lack either appropriate selectivity or membrane permeability, limiting their utility in modulating the activity of the intracellular SHP2 phosphatase. We describe here a structure-guided and fragment-based focused library approach, which led to the development of a novel SHP2 inhibitor from the hydroxyindole carboxylic acid scaffold. This inhibitor has an IC50 of 200 nM for SHP2 that is over 5-fold more selective for SHP2 than any of the over 20 tested PTPs. In addition, this inhibitor efficaciously blocks growth factor stimulated Erk1/2 activation and proliferation in a number of cancer cell lines, validating the concept that small molecule SHP2 inhibitors may be developed that effectively treat solid cancers and leukemias.
Results and Discussion
Structure-Guided and Fragment-Based Library Approach for Acquisition of SHP2 Inhibitors
The goal for this study was to combine structure-guided design and fragment-based focused library approach to obtain potent and selective SHP2 inhibitors for preclinical evaluation and pharmacological validation of SHP2 as an oncology drug target. However, targeting PTPs for therapeutic development has historically been shrouded with two major challenges. First, the similarities between their active sites (i.e., the pTyr-binding pockets) make it nearly impossible to develop small molecules capable of inhibiting just one of the 100 plus PTPs encoded by the human genome without inhibiting other closely related family members. Second, finding compounds with high affinity for the positively charged PTP active site while at the same time possessing favorable cell permeability also seems an insurmountable mountain to climb. To enhance both inhibitor potency and selectivity, we have advanced a novel paradigm to acquire compounds that interact not only with the pTyr-binding pocket but also nearby peripheral sites that are unique to particular PTPs.42,43 Application of this strategy has enabled the identification of a number of potent and selective PTP inhibitors.44 To address the bioavailability issue, we sought to identify nonhydrolyzable pTyr mimetics that possess sufficient polarity to bind the PTP active site yet still capable of penetrating cell membranes. We discovered that bicyclic salicylic acids could serve as novel nonphosphorus-containing pTyr mimetics and PTP inhibitors carrying a bicyclic salicylic acid scaffold have excellent cellular efficacies.35,45−49
To target both the active site and adjacent secondary binding cavities in SHP2, Click reaction was initially utilized to tether an alkyne-containing hydroxyindole carboxylic acid with 53 azide-containing amines and hydrazines.35 This led to the identification of II-B08 (Scheme 1), which exhibits an IC50 of 5.5 μM for SHP2 and several-fold selectivity against a panel of mammalian PTPs. However, the potency and selectivity displayed by II-B08 are still modest and insufficient for further pharmacological investigation. Given its promising in vitro and in vivo activities,35,50,51 we envisioned that II-B08 could serve as an excellent starting point upon which more potent and selective SHP2 inhibitors can be developed. To guide the design of next-generation SHP2 inhibitors, we solved the X-ray crystal structure of SHP2 in complex with II-B08.35 The co-crystal structure reveals that in addition to the hydroxyindole carboxylic acid-dependent interactions with the SHP2 active site, the distal phenyl ring in the biphenyl moiety is sandwiched between the side chains of R362 and K364 in the β5–β6 loop (residues 362–365). Interestingly, the β5–β6 loop is variable among the PTPs, and interactions between the terminal biphenyl group and residues R362 and K364 likely contribute to the observed potency and selectivity of II-B08.35 Little interaction was observed between SHP2 and the 2-phenyl group (the α-ring) directly attached to the hydroxyindole carboxylic acid core. In addition, there was no significant contact between the triazol linker and SHP2. Molecular modeling indicated that appropriately functionalized substituents placed at the meta-position of the α-ring could increase inhibitor binding to SHP2 active site and the β5–β6 loop. To this end, we devised a fragment-based focused library strategy (Scheme 1) to transform the hydroxyindole carboxylic acid scaffold into highly potent and selective SHP2 inhibitors. The libraries contain (1) the hydroxyindole carboxylic acid core to target SHP2 active site, (2) proper linkers to connect the hydroxyindole carboxylic acid to (3) a structurally diverse collection of amines, intended to capture additional interactions with the adjacent β5–β6 loop. In the interest of keeping the libraries to a manageable size, we chose 192 commercially available amines that differ in size, charge, hydrophobicity, polarity, solubility, and drug-like properties, which should provide a reasonable (albeit limited) structural diversity to increase the number and strength of noncovalent interactions between SHP2 and the inhibitor. To ensure that library components can optimally bridge both the active site cavity and the adjacent peripheral site in SHP2, a spacer of 0–3 methylene units was introduced between the two carbonyls in the amino-oxo acylamido phenyl linker (Scheme 1).
Scheme 1. Design and Synthesis of Hydroxyindole Carboxylic Acid Based Libraries 11a–d.

Conditions: (a) FmocOSu, THF, reflux, 20 h, 81.4%; (b) Pd(PPh3)2Cl2, CuI, Na2CO3, DMF, 44%; (c) I2, NaHCO3, CH2Cl2 or AcCN, rt, 86%; (d) 50% diethylamine in DCM, 3 h, 85%; (e) 5% LiOH/THF = 1:2, 80 °C, 2 h, 97.2%; (f) corresponding acyl chloride, Et3N, DMF, 0 °C, 80–90%; (g) 5% LiOH/THF = 1:2, rt, 2 h, 80–90%; (h) 192 amines, HOBT, HBTU, DIPEA, DMF, rt, overnigh,. 60–80%.
To construct the bidentate libraries (Scheme 1), amine 1 was protected with FmocOSu to produce alkyne 2. Alkyne 2 was coupled with iodide 3 by Sonogashira reaction to afford compound 4 with 44% yield.47 Electrophilic cyclization of 4 by I2 furnished compound 5 in 86% yield. Deprotection of 5 under 50% diethylamine in DCM afforded amine 6. Compound 7 was obtained by hydrolysis of 6 in 5% LiOH under 80 °C for 2 h. Compound 7 upon treatment with acetyl chlorides 8a–d yielded compounds 9a–d, which were hydrolyzed in 5% LiOH at room temperature for 2 h to produce compounds 10a–d. To assemble libraries 11a–d, 192 structurally diverse amines were introduced, in equal quantities, into individual wells of two 96-well plates in the presence of HOBT, HBTU, and DIPEA in DMF to condense with the carboxylic acid in compounds 10a–d overnight. The quality of the reactions in the wells was randomly monitored by LC-MS, indicating that 60–80% of compounds 10a–d was converted to the desired products.
Libraries 11a–d were screened at ∼5 μM for SHP2 inhibitors without further purification. The ability of the library components to inhibit the SHP2-catalyzed hydrolysis of p-nitrophenyl phosphate (pNPP) was evaluated at pH 7 and 25 °C. It became immediately evident from the initial screening results that the linker length is of critical importance. In fact, all of the top hits were from library 11a, which had the shortest oxalic linker. The top 20 hits from library 11a were resynthesized, purified by HPLC, and their IC50 values determined. As can be seen from Table 1, the IC50 values matched well with the percent inhibition data measured at ∼5 μM compound concentration. The best hit compound 11a-1 identified from the screen also exhibited the lowest IC50 value of 0.20 ± 0.02 μM against SHP2. Similar to 11a-1, other compounds bearing a biaryl substituent (e.g., 11a-2 and 11a-5) also strongly inhibited SHP2. Interestingly, compounds with the benzyloxyphenyl amino scaffold (e.g., 11a-3 and 11a-6) also inhibited SHP2 at submicromolar concentration. To further establish biaryl substituents as privileged structures for SHP2 binding, we synthesized six additional biaryl substituted derivatives of 11a-1 (Table 2). As expected, the IC50 values of compounds 11a-21 to 11a-26 are comparable to that of 11a-1. To confirm the effectiveness of oxalic linker, we resynthesized and characterized 11 top hits from library 11c (with a succinic linker). In agreement with the results from screening, the hits selected from library 11c were less potent than their library 11a counterparts (Table 1 and Table 3). For example, 11c-1, the top hit from library 11c, had an IC50 of 2.3 μM, more than 10 times higher than that of 11a-1 (IC50 = 0.20 μM). Collectively, the structure and activity data indicate that the aromatic oxalic linker and biaryl substituents are preferred for enhanced inhibitor binding toward SHP2. Selectivity profiling revealed that 11a-1 exhibits over 5-fold selectivity for SHP2 over a large panel of 20 mammalian PTPs, including receptor-like PTPs, PTPμ, PTPε, PTPα, PTPσ, and PTPγ, cytosolic PTPs, PTP1B, Lyp, SHP1, PTPH1, HePTP, STEP, and PEZ, the dual specificity phosphatase VHR, VHZ, MKP5, CDC14A, UBLCP1, and Laforin, and the low-molecular-weight PTP and SSu72 (Table 4). Of particular note, compound 11a-1 displayed a 7- and 11-fold preference for SHP2 over its closely related homologue SHP1 and PTP1B. To the best of our knowledge, 11a-1 is the most potent SHP2 inhibitor reported to date.
Table 1. IC50 Values (μM) of Top Hits from Library 11a for SHP2.
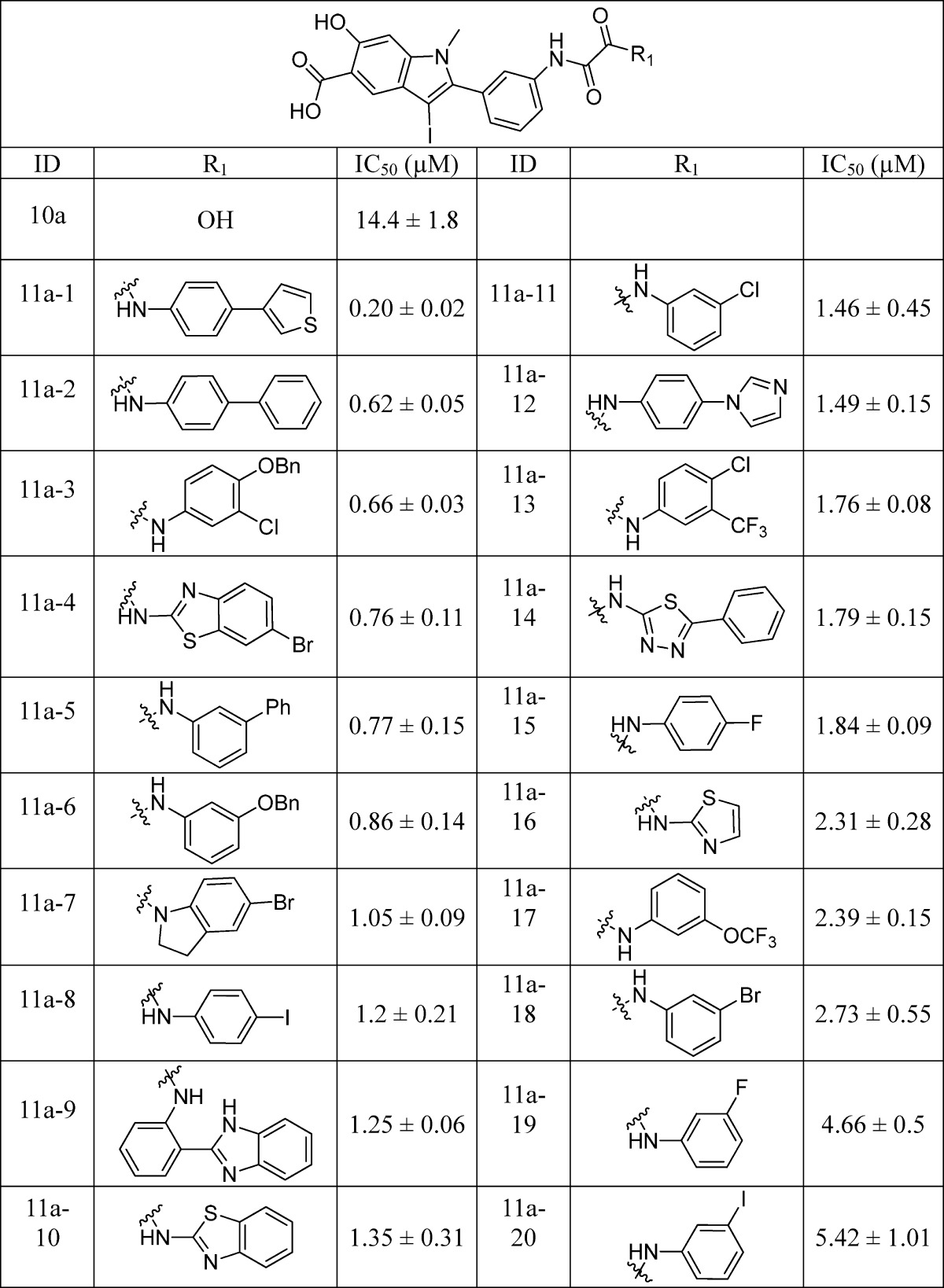
Table 2. IC50 Values (μM) of 11a-21 to 11a-26 for SHP2.
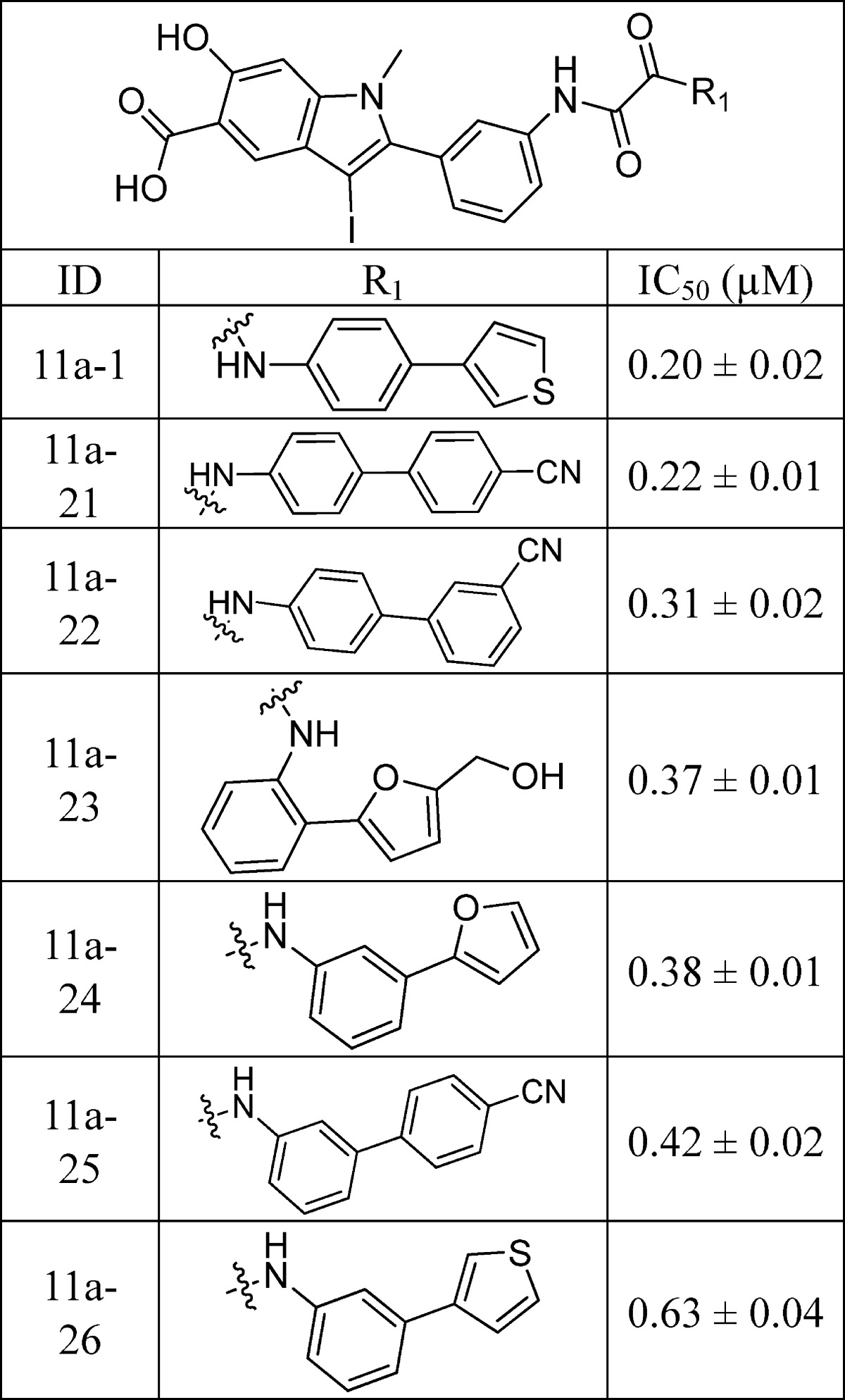
Table 3. IC50 Values (μM) of Top Hits from Library 11c for SHP2.
Table 4. Selectivity of 11a-1 against a Panel of PTPs.
| PTP | IC50 (μM) | PTP | IC50 (μM) |
|---|---|---|---|
| SHP2 | 0.20 ± 0.02 | VHR | 3.2 ± 0.1 |
| LYP | 1.05 ± 0.02 | PTPμ | 3.3 ± 0.2 |
| HePTP | 1.03 ± 0.08 | STEP | 4.0 ± 0.2 |
| PTPH1 | 1.07 ± 0.07 | PEZ | 5.3 ± 0.2 |
| SHP1 | 1.44 ± 0.04 | PTPσ | 8.6 ± 0.4 |
| Ssu72 | 1.3 ± 0.2 | UBLCP1 | 9.7 ± 0.5 |
| PTP1B | 2.29 ± 0.03 | laforin | 12.2 ± 2 |
| LMWPTP | 2.34 ± 0.02 | CDC14A | 16 ± 4 |
| VHZ | 2.3 ± 0.2 | PTPε | >20 |
| PTPγ | 2.4 ± 0.4 | PTPα | >100 |
| MKP5 | 2.9 ± 0.1 |
Structural Basis of SHP2 Inhibition by Compound 11a-1 and 11c-9
To elucidate the structural basis of SHP2 inhibition by 11a-1, we sought to determine the crystal structure of the SHP2 PTP domain (residues 240–528) in complex with 11a-1. Unfortunately, no diffraction quality crystals were obtained for the complex. Instead, we were able to cocrystallize SHP2 with 11c-9 and solved the crystal structure of SHP2 in complex with 11c-9 (Table 3, IC50 = 14 μM). The SHP2·11c-9 complex crystallized in space group P1 with one molecule in the asymmetric unit. The crystals of the SHP2·11c-9 complex diffracted to 2.1 Å resolution, and the structure was solved by molecular replacement, using the apo-form of SHP2 catalytic domain (PDB: 3B7O)52 as the search model. The three-dimensional structure of SHP2·11c-9 was refined to a crystallographic R-factor of 18.7% (Rfree = 22.1%), and the statistics for data collection and refinement are shown in Table 5. The atomic model includes SHP2 residues 246–314 and 323–526, and all atoms of the compound 11c-9 (Figure 1).
Table 5. Data Collection and Refinement Statistics.
| SHP2·11c-9 | |
|---|---|
| crystal parameters | |
| space group | P1 |
| cell dimensions | |
| a (Å) | 40.0 |
| b (Å) | 40.9 |
| c (Å) | 48.9 |
| α (deg) | 94.6 |
| β (deg) | 109.2 |
| γ (deg) | 110.0 |
| data collection | |
| resolution range (Å) | 50.0–2.1 |
| no. of unique reflections | 12808 |
| completeness (%) | 82.3 |
| redundancy | 2.5 |
| Rmergea | 0.071 |
| refinement | |
| resolution range (Å) | 50.0–2.5 |
| no. of reflections used (F ≥ 2.0δ (F)) | 8843 |
| completeness (%) | 94.9 |
| no. of protein atoms | 2219 |
| no. of inhibitors | 1 |
| Rworkb/Rfreec | 18.7/22.1 |
| rms deviations from ideal geometry | |
| bond length (Å) | 0.0079 |
| bond angle (deg) | 1.33 |
Rmerge = ∑h ∑i|I(h)i – ⟨I(h)⟩|/∑h∑iI(h)i.
Rwork = ∑h|F(h)calcd – F(h)obsd|/∑hF(h)obsd, where F(h)calcd and F(h)obsd were the refined calculated and observed structure factors, respectively.
Rfree was calculated for a randomly selected 3.9% of the reflections that were omitted from refinement.
Figure 1.
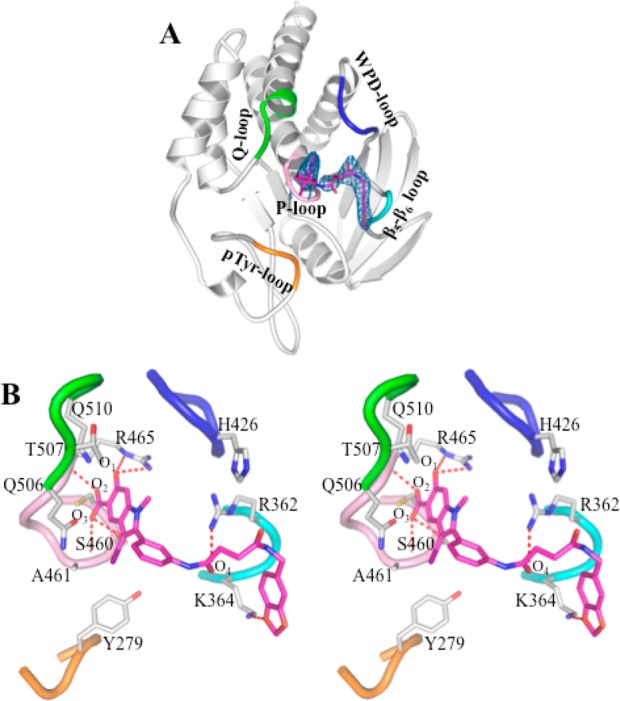
Crystal structure of SHP2 in complex with compound 11c-9. (A) Overall structure of SHP2 in complex with 11c-9. The P-loop is shown in pink, the WPD-loop in blue, the Q-loop in green, the pY-loop in orange, and the β5–β6 loop in cyan. Compound 11c-9 is shown in stick model with unbiased Fo – Fc map contoured at 2.5σ calculated before the ligand and water molecules were added to the model. (B) Detailed interactions between compound 11c-9 and SHP2. Polar interactions or H-bonds are shown by red dashed lines.
The overall structure of SHP2·11c-9 is similar to the ligand free SHP2 structure used for molecular replacement, with the root-mean-square-deviation (rmsd) for all α-carbon positions between the two being 0.53 Å. A major difference between the two structures are electron density in the SHP2 active site corresponding to 11c-9, which was confirmed by the |Fo| – |Fc| difference map contoured at 2.5σ (Figure 1A). As expected, the hydroxyindole carboxylic acid moiety in 11c-9 occupies the SHP2 active-site pocket and forms extensive interactions with residues in the P-loop (residues 458–465), the pY recognition loop (residues 277–284), and the Q loop (residues 501–507) (Figure 1B). Specifically, the phenolic oxygen O1 within the hydroxyindole carboxylic acid core makes two hydrogen bonds with the side chain of R465 in the P-loop; the carboxylate O2 is hydrogen bonded to the main chain amide of R465, and O3 contributes one H-bond with the main chain amide of A461 and one polar interaction with the side chain of S460. The indole ring interacts favorably with the side chains of S460, A461, and R465 in the P-loop as well as the side chains of Q506, T507, and Q510 in the Q-loop, and the iodine atom at the 3-position of the indole ring has van der Waals contacts with the side chain of Y279. Interestingly, the distal benzodioxol ring in 11c-9 is also involved in polar and nonpolar interactions with the side chain of K364 in the β5–β6 loop (residues 362–365). Finally, the succinic linker makes van der Waals contacts with both the side chain of R362 in the β5–β6 loop and the side chain of H426 in the WPD loop (residues 421–431), whereas the carbonyl oxygen O4 in the linker forms a polar interaction with the side chain of R362.
Guided by our previous SHP2·II-B08 structure35 and the current SHP2·11c-9 structure, we sought to understand the molecular basis of compound 11a-1 mediated SHP2 inhibition by carrying out docking studies within the area that covers both II-B08 and 11c-9 around the SHP2 active site. As shown in Figure 2A, the overall SHP2 binding mode of 11a-1 (green carbon) is similar to those of II-B08 (cyan carbon) and 11c-9 (purple carbon). In all three cases, the hydroxyindole carboxylic acid motif penetrates into the active site, and the distal heterocycle tail interacts with a groove formed by residues in the β5–β6 loop. However, unlike II-B08 or 11c-9, which extend their heterocycle tail to the groove via their flexible linkers primarily along the Q-loop and WPD-loop, compound 11a-1 spreads its tail through the rigid oxalic linker along the pY recognition loop. Consequently, the hydroxyindole carboxylic acid in 11a-1 binds more deeply into the active site along the pY recognition cleft, occupies most of the active site pocket (Figure 2B), and the adjacent α-phenyl ring forms strong π–π stacking interaction with Y279 in the pY recognition loop (Figure 2C), which is not observed in SHP2·II-B08 and SHP2·11c-9 structures. In addition, the oxalamide linker properly places the phenylthiophene tail to be well sandwiched by R362 and K364 in the β5–β6 loop (Figure 2D). In more detail (Figure 2E), the hydroxyindole carboxylic acid forms four H-bonds with the backbone amides of S460, I463, G464, and R465 in the P-loop, which anchors the indole core in the active site to be in van der Waals contacts with several P-loop residues, the 1-methyl group makes hydrophobic interactions with A461, Y279, and I282 and the α-phenyl ring π–π stacks with Y279, and one of the carbonyls within the oxalamide linker forms H-bond with the side chain of K364, which thus orients the phenylthiophene tail between R362 and K364 in the β5–β6 loop. Collectively, the increased and more favorable interactions between 11a-1 and SHP2 likely contribute to 11a-1’s enhanced inhibition potency and selectivity toward SHP2. To corroborate the proposed 11a-1 binding mode, we carried out site-directed mutagenesis to convert the β5–β6 loop residues R362 and K364 to Ala and Ser, respectively. The R362A and K364S mutants displayed similar kcat and Km to those of the wild-type SHP2. However, the IC50 values of 11a-1 for R363A and K364S were 2.2- and 1.5-fold higher than that of wild-type SHP2. These results are in agreement with the binding mode observed from the docking analysis, supporting the conclusion that interactions between 11a-1 and R362/K364 contribute to the enhanced potency of 11a-1 for SHP2.
Figure 2.
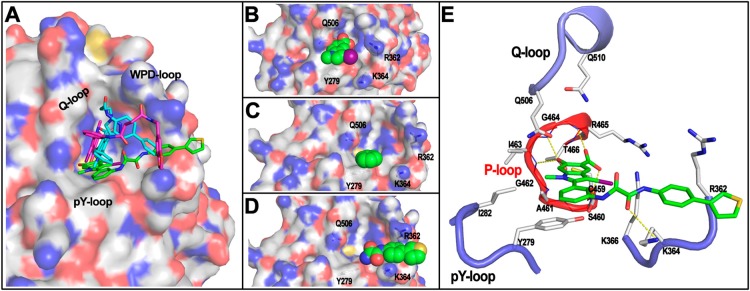
SHP2 binding mode by 11a-1 revealed by molecular docking. (A) The overall binding mode of 11a-1 (green carbon stick) with SHP2 (represented by transparent surface). The binding modes of II-B08 (cyan carbon stick) and 11c-9 (purple carbon stick) from the complex structures are shown for comparison. (B) The hydroxyindole carboxylic acid motif (spheres) penetrating deeply into the SHP2 active site along the pY recognition cleft. (C) The α-phenyl ring (spheres) forms strong π–π stack interaction with Y279 (gray carbon stick with dotted surface). (D) The rigid oxalamide linker orients the phenylthiophene (spheres) to be well sandwiched by R362 and K364 (gray carbon stick). (E) The interaction details between 11a-1 (green carbon stick) with SHP2. Residues within 5 Å distance to 11a-1 are shown in stick. P-loop is highlighted in red, and the other three loops are in blue.
SHP2 Inhibitor 11a-1 Displays Activity in Several Cancerous Cell Lines
Our goal is to develop potent and specific SHP2 inhibitors as novel anticancer and antileukemia agents. Previous studies have established that SHP2 promotes growth factor-mediated Ras-Erk1/2 activation and cell proliferation.10−12 Given 11a-1’s excellent potency and selectivity toward SHP2, we evaluated its ability to inhibit SHP2-dependent signaling and proliferation in a number of cancer cell lines. We previously demonstrated that SHP2 is required for growth of H1975,51 a nonsmall cell lung cancer (NSCLC) patient-derived cell line with secondary gatekeeper mutations in EGF receptor and showing resistance to EGF receptor inhibitors gefitinib and erlotinib.53,54 To determine the effect of SHP2 inhibition by 11a-1, H1975 cells were treated with various concentrations of 11a-1 and the cell number was measured by the MTT assay. As shown in Figure 3A, 11a-1 inhibited H1975 cell proliferation in a dose-dependent manner with an IC50 of 0.17 ± 0.02 μM.
Figure 3.

SHP2 inhibitor 11a-1 reduced lung cancer cell proliferation and specifically blocked SHP2-dependent signaling. (A) 11a-1 dose dependently inhibited H1975 proliferation with an IC50 of 0.17 ± 0.02 μM. (B) 11a-1 decreased EGF induced Erk1/2 phosphorylation and increased EGF induced paxillin (Y118) phosphorylation in a dose-dependent manner. (C) The structurally related negative control 10a failed to block SHP2-dependent signaling at 2 μM. (D) the ability of 11a-1 to inhibit Erk1/2 activation was blunted in SHP2 knocked down cells, and (E) 11a-1 had no effect on PMA-stimulated Erk1/2 phosphorylation.
Because SHP2 phosphatase activity is required for the full activation of the Ras-Erk1/2 pathway,10,55 we assessed the cellular effect of 11a-1 on EGF-induced Erk1/2 activation in H1975 cells. As expected, 11a-1 effectively reduced the EGF-induced Erk1/2 phosphorylation in a dose-dependent manner (Figure 3B). To provide direct evidence that 11a-1 can block the phosphatase activity of SHP2 inside the cell, we measured the phosphorylation level of Paxillin (pY118), a physiological substrate of SHP2 whose dephosphorylation by SHP2 enhances EGF-stimulated Erk1/2 activation.56 As shown in Figure 3B, treatment of H1975 cells with 11a-1 dose dependently increased Paxillin phosphorylation on Y118, which is consistent with the decreased EGF-induced Erk1/2 activation in the presence of 11a-1.
To ensure that the cellular activity of 11a-1 is manifested through SHP2 inhibition and not due to nonspecific effects, a structurally related but significantly less inhibitory compound 10a (IC50 of 14.4 μM for SHP2, Table 1) was also evaluated. As expected, at 2 μM compound 10a exerted no effect on Paxillin and Erk1/2 phosphorylation while at the same concentration compound 11a-1 reduced Erk1/2 phosphorylation by 40% and augmented Paxillin phosphorylation by 30% (Figure 3C). Moreover, the ability of 11a-1 to attenuate EGF-mediated Erk1/2 activation was blunted when the level of SHP2 was knocked down with siRNA. As shown in Figure 3D, at 2 μM concentration 11a-1 reduced Erk1/2 phosphorylation by 2.2-fold in cells treated with scrambled siRNA but only 1.5-fold in cells treated with SHP2 siRNA. To provide further evidence that the observed cellular effect of 11a-1 is SHP2 dependent, we examined the effect of 11a-1 on PMA (phorbol 12-myristate 13-acetate)-induced Erk1/2 activation, which is SHP2 independent57 and instead depends on the activation of protein kinase C and Raf58 in a Ras-independent manner.59 Thus, SHP2 inhibitors would not be expected to affect PMA-induced Erk1/2 phosphorylation. Indeed, compound 11a-1 had no effect on PMA-induced Erk1/2 phosphorylation (Figure 3E). Taken together, the results indicate that 11a-1 specifically inhibits SHP2-mediated cellular signaling events.
The promising activity of 11a-1 in inhibiting H1975 lung cancer cell proliferation spurred an investigation into its effectiveness on inhibiting the growth of the ErbB2 positive SKBR3 breast cancer cell line. These cells through upregulation of ErbB2 strongly activate Ras, which then promotes either Akt signaling when grown on plastic or Erk1/2 signaling when grown in Matrigel.60 Given the multitude of studies that show that cancer cells grow in Matrigel as a mass that much more closely models the formation and signaling properties of real human tumors,61 the effectiveness of 11a-1 in blocking the growth of these cells in Matrigel was determined. Consistently, SKBR3 cells formed tumor like growths in Matrigel treated with vehicle but their growth within 24 h was potently inhibited by 3 μM of 11a-1 (Figure 4A). Further, while both control and 11a-1 treated cells showed equivalent levels of total Erk1/2 and Akt over 4 days, their levels of phosphorylation were concordantly inhibited by increasing concentrations 11a-1 from 1 to 10 μM (Figure 4B). Taken together, 11a-1 potently inhibits the growth of the ErbB2 positive SKBR3 cell line, most likely through its ability to block SHP2-dependent Erk1/2 and Akt activation.
Figure 4.

Compound 11a-1 inhibits Erk1/2 and Akt activity and ErbB2+ breast cancer cell growth in a 3D Matrigel environment. (A) SKBR3 cells were seeded into Matrigel, and their growth was then monitored over 4 days in the presence of vehicle or the indicated concentrations of 11a-1. (B) Cells after 4 days of growth in Matrigel were recovered and the levels of the total and phospho forms of Erk1/2 and Akt were measured by immunoblot.
Gain-of-function mutations in KIT receptor (KITD814V) in humans are associated with gastrointestinal stromal tumors, systemic mastocytosis, and acute myelogenous leukemia.62,63 We have previously shown that our first-generation SHP2 inhibitor II-B08 suppresses the growth of oncogenic KITD814V-induced ligand-independent growth.50 To further determine the efficacy of the new SHP2 inhibitor 11a-1, we compared the growth of oncogenic KITD814V-bearing 32D myeloid cells in the presence of II-B08 and 11a-1. As seen in Figure 5A, we did not see any significant suppression in the growth of wild-type KIT-bearing cells in the presence of II-B08 or 11a-1 at the tested concentrations. This was to be expected because SHP2 exists in an autoinhibited conformation in the absence of ligand stimulated KIT activation.50 In contrast, treatment with II-B08 at 5 μM showed significant suppression in ligand-independent growth of KITD814V-bearing cells but not at 1 μM concentration. Furthermore, treatment with 11a-1 showed significant inhibition of ligand-independent growth of KITD814V-bearing cells at both 1 and 5 μM concentrations. To further determine the effect of II-B08 and 11a-1 on primary bone marrow cells bearing oncogenic KITD814V, we transduced primary low-density bone marrow cells from C57BL/6 mice with KITD814V and sorted cells were subjected to proliferation assay in the presence or absence of SHP2 inhibitors II-B08 and 11a-1. Similar to 32D myeloid cells, primary bone marrow cells bearing KITD814V showed significant reduced growth in the presence of 11a-1 compared to II-B08 at both 1 and 5 μM concentrations (Figure 5B). These results suggest that 11a-1 is more potent in inhibiting oncogenic KITD814V-induced constitutive growth compared to II-B08.
Figure 5.

Effects of compound 11a-1 on 32D cells. (A) 32D myeloid cells bearing wild-type (WT) KIT or KITD814V were starved of serum and growth factors for 6 h and subjected to proliferation assay in the presence or absence of indicated concentration of II-B08 or 11a-1. Assay was performed in the presence of IL-3 (10 ng/mL) for cells bearing WT KIT and in the absence of growth factors for cells bearing oncogenic KITD814V. Bars denote the mean thymidine incorporation (CPM ± SEM) consolidated from four independent experiments performed in triplicate. *p < 0.05. (B) WT hematopoietic stem and progenitor cells bearing KITD814V were starved of serum and growth factors for 6 h and subjected to proliferation assay in the presence or absence of indicated concentration of II-B08 or 11a-1. Bars denote the mean thymidine incorporation (CPM ± SD) performed in triplicate from one experiment. *p < 0.05. (C) 32D myeloid cells bearing KITD814V were starved of serum and growth factors for 6 h and incubated with the indicated concentration of 11a-1 for 2 h. After treatment, cells were lysed and equal amount of protein lysates were subjected to Western blot analysis using indicated antibodies. Similar results were observed in two independent experiments.
Previous studies have shown that SHP2 controls the cell growth and survival through regulation of the activation of Akt and Erk1/2 signaling. We also found that SHP2 inhibitor II-B08 suppresses the activation of Akt in oncogenic KITD814V-bearing cells. To determine whether SHP2 inhibitor 11a-1 also suppress the ligand-independent activation of Akt and Erk1/2 signaling, we starved the 32D myeloid cells bearing wild-type KIT or KITD814V and treated them with 11a-1. Consistent with our previous results, cells bearing wild-type KIT did not show any constitutive activation of Akt or Erk1/2 in the presence or absence of 11a-1 (data not shown). Likewise, constitutive phosphorylation of Akt and Erk1/2 was observed in oncogenic KITD814V-bearing cells (Figure 5C, lane 1). There was dose-dependent inhibition of phosphorylation of Akt and Erk1/2 in the presence of 11a-1 (Figure 5C, lanes 2–4). These results suggest that 11a-1 inhibits the activation of Akt and Erk1/2 signaling, leading to reduced growth of oncogenic KITD814V-bearing cells. Collectively, the results described above indicate that 11a-1 is highly efficacious in blocking SHP2 activity and cell proliferation in H1975 lung cancer cells, ErbB2 positive SKBR3 breast cancer cells, and oncogenic KITD814V-bearing 32D myeloid cells and primary low-density bone marrow cells.
Conclusion
In summary, we report a novel hydroxyindole carboxylic acid-based inhibitor for SHP2, an oncogenic phosphatase required for multiple growth factor receptor signaling and implicated in a broad-spectrum of cancer and leukemia. We took a structure-guided approach that targets the SHP2 active site with hydroxyindole carboxylic acid and relies on a fragment-based library strategy to introduce additional functionalities intended to interact with peripheral sites proximal to the catalytic pocket to increase binding affinity and selectivity for SHP2. This led to the transformation of a modest 5.5 μM hit II-B08 into a 200 nM SHP2 inhibitor 11a-1 with greater than 5-fold selectivity against a large panel of mammalian PTPs. Structural analysis and molecular modeling reveal that the hydroxyindole carboxylic acid anchors the inhibitor to the SHP2 active site while interactions of the oxalamide linker and phenylthiophene tail with residues in the β5–β6 loop contribute significantly to 11a-1’s binding potency and selectivity toward SHP2. We provide evidence that 11a-1 displays highly efficacious cellular activity and can specifically block the SHP2-dependent signaling inside the cell. Moreover, 11a-1 attenuates growth factor mediated Erk1/2 and Akt activation and exhibits excellent antiproliferative activity in nonsmall cell lung cancer cell line H1975, ErbB2 positive breast cancer cell line SKBR3, and oncogenic KITD814V expressing 32D myeloid cells and primary low-density bone marrow cells. Thus, compound 11a-1 not only serves as a promising candidate for the development of novel agents for a wide range of neoplastic disorders but also a useful tool to interrogate the function of SHP2 in normal physiology and to elucidate the events underlying SHP2-evoked transformation. Obtaining this knowledge is vital for understanding SHP2-mediated oncogenic mechanisms and for the development of novel anticancer and antileukemia therapies targeted to SHP2.
Experimental Section
Materials
para-Nitrophenyl phosphate (pNPP) was purchased from Fisher Scientific. For organic synthesis, reagents were used as purchased (Aldrich, Acros, Alfa Aesar, TCI) except where noted. Rabbit antiphospho-Akt, antitotal Akt, antiphospho-Erk1/2, antitotal Erk1/2, and anti-phospho-Paxillin (Tyr118) antibodies were purchased from Cell Signaling Technology (Beverly, MA). Anti-Paxilin antibodies were from BD Transduction Laboratories. Recombinant murine interleukin-3 (IL-3), murine stem cell factor (SCF), murine thrombopoietin (TPO), and murine FLT3 ligand (FLT3-L) were purchased from Peprotech (Rocky Hill, NJ). Iscove’s Modified Dulbecco’s Medium (IMDM) was purchased from Invitrogen (Carlsbad, CA). [3H] Thymidine was purchased from PerkinElmer (Boston, MA).
General Procedures
1H and 13C NMR spectra were obtained on Bruker 500 spectrometers with TMS or residual solvent as standard. All column chromatography was performed using Dynamic Adsorbents 230–400 mesh silica gel (SiO2) with the indicated solvent system unless otherwise noted. TLC analysis was performed using 254 nm glass-backed plates and visualized using UV light (254 nm), and low-resolution mass spectra and purity data were obtained using an Agilent Technologies 6130 Quadrupole LC/MS. The purity of the compounds were assessed by analytical HPLC, with the solvent gradient started at 0% methanol in water and ended at 100% methanol after 8 min with 0.1% of TFA. The purity of all final tested compounds was determined to be >95% (UV, λ = 254 nm). High-resolution mass spectrum data were collected on an Agilent 6520 Accurate-Mass Q-TOF LC/MS. HPLC purification was carried out on a Waters Delta 600 equipped with a Sunfire Prep C18 OBD column (30 mm × 150 mm, 5 μm) with methanol–water (both containing 0.1% TFA) as mobile phase. The preparative HPLC gradient started at 50% methanol in water and ended at 100% methanol after 40 min with 0.1% of TFA.
(9H-Fluoren-9-yl)methyl (3-Ethynylphenyl)carbamate (2)
Mixtures of compound 1 (0.75 mL, 7.18 mmol) and Fmoc-OSu (2.664 g, 7.89 mmol) in 200 mL of THF were refluxed overnight. The solution was partitioned between EtOAc and water. The residue was purified by flash silica chromatography (Hex/EtOAc = 8:1) to afford titled compound as white solid (1.98 g, 81.4%); mp 137–138 °C. 1H NMR (500 MHz, CDCl3): δ 7.83 (m, 2H), 7.61 (m, 2H), 7.54 (brs, 1H), 7.36 (m, 3H), 7.28 (m, 2H), 7.23 (m, 1H), 7.21 (m, 1H), 6.66 (brs, 1H), 4.57 (d, J = 6.6 Hz, 2H), 4.30 (t, J = 6.6 Hz, 1H), 3.08 (s, 1H). 13C NMR (125 MHz, DMSO): δ 153.8, 144.1, 141.2, 139.8, 129.6, 128.1, 127.5, 126.2, 125.5, 122.5, 121.5, 120.6, 119.3, 83.5, 80.9, 66.1, 47.0. HRMS (ESI): (M + H)+ calcd for C23H18NO2, 340.1332; found, 340.1341. LC-MS (ESI): 362.0 (M + Na)+. Purity: >95% (UV, λ = 254 nm).
Methyl 5-((3-((((9H-Fluoren-9-yl)methoxy)carbonyl)amino)phenyl)ethynyl)-4-(dimethylamino)-2-hydroxybenzoate (4)
A mixture of methyl 4-(dimethylamino)-2-hydroxy-5-iodobenzoate 3 (5.13 g, 16 mmol), compound 2 (6.5 g, 19 mmol), Na2CO3 (2.03 g, 19.2 mmol), bis(triphenylphosphine)palladium(II) chloride (0.576 g, 0.8 mmol), and CuI (304 mg, 1.6 mmol) were loaded in a flask, which was degassed and backfilled with nitrogen. then 15 mL of DMF were added. The resulting mixture was stirred under a nitrogen atmosphere at room temperature for 4 h. The reaction was monitored by TLC to establish completion. The solution was partitioned between EtOAc (200 mL) and brine (200 mL). The organic layers were washed with brine (3 × 200 mL), dried over sodium sulfate, and concentrated in vacuum. The residue was purified by flash silica chromatography (Hex/EtOAc = 4:1) to afford titled compound as viscous oil (3.74 g, 44%). 1H NMR (500 MHz, CDCl3): δ 10.96 (s, 1H), 7.97 (s, 1H), 7.80 (m, 2H), 7.64 (m, 2H), 7.58 (brs, 1H), 7.45 (m, 2H), 7.20–7.31 (m, 6H), 6.72 (brs, 1H), 6.33 (s, 1H), 4.57 (d, J = 6.6 Hz, 2H), 4.30 (t, J = 6.6 Hz, 1H), 3.93 (s, 3H), 3.15 (s, 6H). 13C NMR (125 MHz, CDCl3): δ 169.8, 162.6, 159.2, 153.2, 153.2, 143.6, 141.3, 137.8, 137.4, 129.0, 127.8, 127.6, 127.1, 126.1, 124.9, 124.7, 120.7, 120.0, 104.1, 103.9, 102.8, 92.1, 89.0, 66.9, 51.9, 47.1, 42.5. HRMS (ESI): (M + H)+ calcd for C33H29N2O5, 533.2071; found, 533.2072. LC-MS (ESI): 533.2 (M + H)+, 530.8 (M −H)−. Purity: >95% (UV, λ = 254 nm).
Methyl 2-(3-((((9H-Fluoren-9-yl)methoxy)carbonyl)amino)phenyl)-6-hydroxy-3-iodo-1-methyl-1H-indole-5-carboxylate (5)
To a solution of 4 (1.72 g, 3.23 mmol) and NaHCO3 (0.408 g, 4.85 mmol) in CH2Cl2 (500 mL) was added iodine (1.23 g, 4.85 mmol). The resulting mixture was stirred at room temperature for 4 h, then 100 mL of CH2Cl2 was added, then washed with saturated aqueous Na2SO3 solution (2 × 500 mL) and brine (500 mL), dried over Na2SO4, and concentrated in vacuum. The residue was purified by flash chromatography (hexane/THF = 1:1) to afford the title compound as colorless solid (1.79 g, 86%); mp 131–132 °C. 1H NMR (500 MHz, CDCl3): δ 10.92 (s, 1H), 8.03 (s, 1H), 7.79 (m, 2H), 7.65 (m, 2H), 7.48 (m, 5H), 7.36 (m, 2H), 7.18 (m, 1H), 6.85 (s, 1H), 6.80 (s, 1H), 4.61 (m, 2H), 4.30 (m, 1H), 4.03 (s, 3H), 3.60 (s, 3H). 13C NMR (125 MHz, CDCl3): δ 171.2, 158.2, 153.4, 143.6, 142.5, 142.2, 141.3, 137.9, 131.9, 129.2, 127.8, 127.1, 127.0, 125.8, 124.8, 124.3, 124.1, 120.0, 107.6, 96.2, 66.9, 60.0, 52.3, 47.1, 32.1. HRMS (ESI): (M + H)+ calcd for C32H26IN2O5, 645.0881; found, 645.0851. LC-MS (ESI): 667.0 (M + Na)+. Purity: >95% (UV, λ = 254 nm).
Methyl 2-(3-Aminophenyl)-6-hydroxy-3-iodo-1-methyl-1H-indole-5-carboxylate (6)
Compound 5 (1.79 g, 2.77 mmol) was dissolved in 10 mL of CH2Cl2, and then 10 mL of diethylamine was added to the solution under room temperature for 3 h. The solution was concentrated in vacuum. The residue was partitioned between CH2Cl2 (200 mL) and brine (200 mL). The organic layer was dried over sodium sulfate and concentrated in vacuum. The residue was purified by flash silica chromatography (CH2Cl2 as elution) to afford the title compound as white solid (1 g, 85.5%); mp 83–85 °C. 1H NMR (500 MHz, CDCl3): δ 10.90 (s, 1H), 8.02 (s, 1H), 7.31 (m, 1H), 6.84 (s, 1H), 6.80 (m, 2H), 6.76 (m, 1H), 4.03 (s, 3H), 3.83 (brs, 2H), 3.60 (s, 3H). 13C NMR (125 MHz, CDCl3): δ 171.2, 158.1, 146.4, 143.1, 142.4, 132.1, 129.4, 124.2, 124.1, 120.8, 117.1, 115.6, 107.4, 96.1, 59.4, 52.1, 32.1. HRMS (ESI): (M + H)+ calcd for C17H16IN2O3, 423.0200; found, 423.0190. LC-MS (ESI): 423.0 (M + H)+, 420.8 (M – H)−. Purity: >95% (UV, λ = 254 nm).
2-(3-Aminophenyl)-6-hydroxy-3-iodo-1-methyl-1H-indole-5-carboxylic Acid (7)
Compound 6 (1 g, 2.37 mmol) was dissolved in 8 mL of THF. Then 5% LiOH (4 mL) solution was added. The mixture was heated to 80 °C for 2 h, cooled to room temperature, diluted by brine (200 mL), acidified by 2 N HCl to pH 5, and extracted with EtOAc (2 × 200 mL). The organic layers were combined, washed with brine, dried over sodium sulfate, and concentrated in vacuum to give the title compound as pale solid (940 mg, 97.2%); decomposed at 148 °C. 1H NMR (500 MHz, DMSO): δ 7.85 (s, 1H), 7.46 (m, 1H), 7.11 (m, 3H), 7.04 (s, 1H), 4.11 (m, 2H), 3.59 (s, 3H). 13C NMR (125 MHz, DMSO): δ 173.0, 158.3, 142.6, 142.5, 140.9, 132.1, 130.0, 124.5, 124.0, 123.8, 120.9, 119.5, 108.1, 97.0, 60.6, 32.5. HRMS (ESI): (M – H)− calcd for C16H12IN2O3, 406.9898; found, 406.9894. LC-MS (ESI): 409.0 (M + H)+, 407.0 (M – H)−. Purity: >95% (UV, λ = 254 nm).
General Method for the Synthesis of 9a–d
Compound 7 (30 mg, 0.074 mmol) was dissolved in 1 mL of DMF under 0 °C, and then 1.5 equiv of corresponding acyl chloride 8a–d was added to the solution for overnight. The solution was partitioned between EtOAc (200 mL) and brine (200 mL). The organic layers were washed with brine (3 × 200 mL), dried over sodium sulfate, and concentrated in vacuum to afford the title compounds as pale solids without purification for further use.
General Method for the Synthesis of 10a–d
Compound 9a–d (0.074 mmol) was dissolved in 2 mL of THF. Then 5% LiOH (2 mL) solution was added. The mixture was stirred under room temperature for overnight, acidified by 2 N HCl to pH 5, and extracted with EtOAc (2 × 200 mL). The organic layers were combined, washed with brine, dried over sodium sulfate, and concentrated in vacuum. This crude product was purified by Prep-HPLC to give the title compounds.
2-(3-(Carboxyformamido)phenyl)-6-hydroxy-3-iodo-1-methyl-1H-indole-5-carboxylic Acid (10a)
Pale solid (26 mg, 71%, two steps). 1H NMR (500 MHz, DMSO): δ 11.38 (brs, 1H), 10.95 (s, 1H), 7.94 (m, 2H), 7.86 (s, 1H), 7.54 (m, 1H), 7.30 (m, 1H), 7.05 (s, 1H), 3.61 (s, 3H). 13C NMR (125 MHz, DMSO): δ 173.0, 162.4, 158.3, 157.4, 142.5, 142.4, 138.3, 131.5, 129.4, 127.1, 124.0, 123.8, 122.6, 121.1, 108.1, 97.0, 60.8, 32.5. HRMS (ESI): (M – H)− calcd for C18H12IN2O6, 478.9746; found, 478.9762. LC-MS (ESI): 481.0 (M + H)+, 478.8 (M – H)−. Purity: >95% (UV, λ = 254 nm).
2-(3-(2-Carboxyacetamido)phenyl)-6-hydroxy-3-iodo-1-methyl-1H-indole-5-carboxylic Acid (10b)
Pale solid (30 mg, 82%, two steps). 1H NMR (500 MHz, DMSO): δ 10.37 (s, 1H), 7.86 (s, 1H), 7.74 (m, 2H), 7.50 (m, 1H), 7.21 (m, 1H), 7.04 (s, 1H), 3.61 (s, 3H), 3.40 (s, 2H). HRMS (ESI): (M – H)− calcd for C19H14IN2O6, 492.9902; found, 492.9933. LC-MS (ESI): 495.0 (M + H)+, 492.8 (M – H)−. Purity: >95% (UV, λ = 254 nm).
2-(3-(3-Carboxypropanamido)phenyl)-6-hydroxy-3-iodo-1-methyl-1H-indole-5-carboxylic Acid (10c)
Pale solid (31 mg, 82%, two steps). 1H NMR (500 MHz, DMSO): δ 12.16 (brs, 1H), 11.36 (brs, 1H), 10.19 (s, 1H), 7.84 (s, 1H), 7.42 (m, 2H), 7.47 (s, 1H), 7.16 (m, 1H), 7.04 (s, 1H), 3.58 (s, 3H), 2.60 (m, 4H). 13C NMR (125 MHz, DMSO): δ 174.2, 173.0, 170.8, 158.3, 142.8, 142.5, 139.9, 131.5, 129.4, 125.4, 123.9, 123.8, 121.2, 119.7, 108.0, 97.1, 60.6, 32.5, 31.5, 29.1. HRMS (ESI): (M – H)− calcd for C20H16IN2O6, 507.0059; found, 507.0080. LC-MS (ESI): 509.0 (M + H)+, 507.0 (M – H)−. Purity: >95% (UV, λ = 254 nm).
2-(3-(4-Carboxybutanamido)phenyl)-6-hydroxy-3-iodo-1-methyl-1H-indole-5-carboxylic Acid (10d)
Pale solid (21 mg, 54%, two steps). 1H NMR (500 MHz, DMSO): δ 11.38 (brs, 1H), 10.12 (s, 1H), 7.85 (s, 1H), 7.74 (m, 2H), 7.48 (m, 1H), 7.17 (m, 1H), 7.04 (s, 1H), 3.59 (s, 3H), 2.40 (m, 2H), 2.29 (m, 2H), 1.83 (m, 2H). HRMS (ESI): (M – H)− calcd for C21H18IN2O6, 521.0215; found, 520.9139. LC-MS (ESI): 523.0 (M + H)+, 520.8 (M – H)−. Purity: >95% (UV, λ = 254 nm).
Procedure for the Assembling of Library 11a–d
Compound 10a–d (65 mM, 3 μL) in DMF reacted with 192 amines (200 mM, 3 μL) in DMF respectively in the presence of HOBT (14 mM), HBTU (17 mM), and DIPEA (28 mM) in 14 μL of DMF overnight to assemble the combinatorial amide library 11a–d in 96-well plates. Ten of the reactions were picked up randomly and monitored by LC-MS, which showed an average of 70% yield desired products.
General Method for the Synthesis of (11a-1 to 11a-26 and 11c-1 to 11c-11)
Compound 11a or 11c (0.02 mmol) dissolved in 0.5 mL of DMF was added to a solution of corresponding amines (0.04 mmol), HOBT (3.06 mg, 0.02 mmol), HBTU (7.58 mg, 0.02 mmol), and DIPEA (5.16 μL, 0.04 mmol) in 1 mL of DMF. The mixture was stirred under room temperature for 4 h. This crude product was KJMM purified by Prep-HPLC to give titled compounds.
6-Hydroxy-3-iodo-1-methyl-2-(3-(2-oxo-2-((4-(thiophen-3-yl)phenyl)amino)acetamido)phenyl)-1H-indole-5-carboxylic Acid (11a-1)
Pale solid (3.8 mg, 29%); decomposed at 185 °C. 1H NMR (500 MHz, DMSO): δ 11.08 (s, 1H), 10.95 (s, 1H), 8.06 (s, 1H), 8.01 (m, 1H), 7.92 (m, 2H), 7.85 (m, 2H), 7.75 (m, 2H), 7.63 (m, 1H), 7.57 (m, 2H), 7.33 (m, 1H), 7.06 (s, 1H), 3.63 (s, 3H). 13C NMR (125 MHz, DMSO): δ 173.0, 159.2, 158.8, 158.3, 142.6, 142.5, 141.4, 138.3, 137.1, 132.0, 131.5, 129.5, 127.5, 127.2, 126.8, 126.5, 124.1, 123.9, 122.8, 121.3, 120.9, 108.1, 97.1, 60.9, 32.6. HRMS (ESI): (M – H)− calcd for C28H19IN3O5S, 636.0096; found, 636.0098. LC-MS (ESI): 659.8 (M + Na)+, 635.8 (M – H)−. Purity: >95% (UV, λ = 254 nm).
2-(3-(2-([1,1′-Biphenyl]-4-ylamino)-2-oxoacetamido)phenyl)-6-hydroxy-3-iodo-1-methyl-1H-indole-5-carboxylic Acid (11a-2)
Pale solid (3.5 mg, 27%). 1H NMR (500 MHz, DMSO): δ 11.11 (s, 1H), 11.02 (s, 1H), 8.08 (s, 1H), 8.03 (m, 1H), 7.99 (m, 2H), 7.87 (s, 1H), 7.70 (m, 4H), 7.59 (m, 1H), 7.37 (m, 2H), 7.33 (m, 2H), 7.06 (s, 1H), 3.64 (s, 3H). HRMS (ESI): (M – H)− calcd for C30H21IN3O5, 630.0531; found, 630.0546. LC-MS (ESI): 653.8 (M + Na)+, 629.8 (M – H)−. Purity: >95% (UV, λ = 254 nm).
2-(3-(2-((4-(Benzyloxy)-3-chlorophenyl)amino)-2-oxoacetamido)phenyl)-6-hydroxy-3-iodo-1-methyl-1H-indole-5-carboxylic Acid (11a-3)
Pale solid (4.2 mg, 30%). 1H NMR (500 MHz, DMSO): δ 11.37 (s, 1H), 11.07 (s, 1H), 10.97 (s, 1H), 8.04 (m, 2H), 7.93 (m, 1H), 7.86 (s, 1H), 7.80 (m, 1H), 7.59–7.27 (m, 8H), 7.07 (s, 1H), 6.82 (m, 1H), 5.21 (s, 2H), 3.63 (s, 3H). HRMS (ESI): (M – H)− calcd for C31H22ClIN3O6, 694.0247; found, 694.0233. LC-MS (ESI): 718.0 (M + Na)+, 693.8 (M – H)−. Purity: >95% (UV, λ = 254 nm).
2-(3-(2-((6-Bromobenzo[d]thiazol-2-yl)amino)-2-oxoacetamido)phenyl)-6-hydroxy-3-iodo-1-methyl-1H-indole-5-carboxylic Acid (11a-4)
Pale solid (3.4 mg, 24%). 1H NMR (500 MHz, DMSO): δ 13.16 (s, 1H), 11.37 (s, 1H), 11.27 (s, 1H), 8.37 (s, 1H), 8.02 (m, 2H), 7.87 (s, 1H), 7.78 (m, 1H), 7.63 (m, 2H), 7.34 (m, 1H), 7.07 (s, 1H), 3.63 (s, 3H). HRMS (ESI): (M – H)− calcd for C25H15BrIN4O5S, 688.8997; found, 688.9008. LC-MS (ESI): 690.6 (M – H)−. Purity: >95% (UV, λ = 254 nm).
2-(3-(2-([1,1′-Biphenyl]-3-ylamino)-2-oxoacetamido)phenyl)-6-hydroxy-3-iodo-1-methyl-1H-indole-5-carboxylic Acid (11a-5)
Pale solid (4.5 mg, 35%). 1H NMR (500 MHz, DMSO): δ 11.37 (s, 1H), 11.12 (s, 1H), 10.99 (s, 1H), 8.23 (s, 1H), 8.05 (m, 2H), 7.90 (m, 1H), 7.87 (s, 1H), 7.80 (m, 1H), 7.65 (m, 3H), 7.52 (m, 6H), 7.08 (s, 1H), 3.63 (s, 3H). HRMS (ESI): (M – H)− calcd for C30H21IN3O5, 630.0531; found, 630.0533. LC-MS (ESI): 629.8 (M – H)−. Purity: >95% (UV, λ = 254 nm).
2-(3-(2-((3-(Benzyloxy)phenyl)amino)-2-oxoacetamido)phenyl)-6-hydroxy-3-iodo-1-methyl-1H-indole-5-carboxylic Acid (11a-6)
Pale solid (3.6 mg, 27%). 1H NMR (500 MHz, DMSO): δ 11.38 (s, 1H), 11.09 (s, 1H), 10.86 (s, 1H), 8.06 (s, 1H), 8.00 (m, 1H), 7.86 (s, 1H), 7.64 (s, 1H), 7.58–7.27 (m, 9H), 7.07 (s, 1H), 6.82 (m, 1H), 5.10 (s, 2H), 3.63 (s, 3H). HRMS (ESI): (M – H)− calcd for C31H23IN3O6, 660.0637; found, 660.0627. LC-MS (ESI): 684.0 (M + Na)+, 659.8 (M – H)−. Purity: >95% (UV, λ = 254 nm).
2-(3-(2-(5-Bromoindolin-1-yl)-2-oxoacetamido)phenyl)-6-hydroxy-3-iodo-1-methyl-1H-indole-5-carboxylic Acid (11a-7)
Pale solid (4.6 mg, 34%). 1H NMR (500 MHz, DMSO): δ 11.36 (s, 1H), 11.09 (s, 1H), 8.06 (m, 1H), 7.92 (m, 1H), 7.89 (s, 1H), 7.86 (s, 1H), 7.57 (m, 2H), 7.46 (m, 1H), 7.31 (m, 1H), 7.07 (s, 1H), 4.39 (m, 2H), 3.64 (s, 3H), 3.18 (m, 2H). HRMS (ESI): (M – H)− calcd for C26H18BrIN3O5, 657.9480; found, 657.9501.; LC-MS (ESI): 661.8 (M + H)+, 659.8 (M – H)−. Purity: >95% (UV, λ = 254 nm).
6-Hydroxy-3-iodo-2-(3-(2-((4-iodophenyl)amino)-2-oxoacetamido)phenyl)-1-methyl-1H-indole-5-carboxylic Acid (11a-8)
Pale solid (3.5 mg, 25%). 1H NMR (500 MHz, DMSO): δ 11.36 (s, 1H), 11.08 (s, 1H), 11.01 (s, 1H), 8.05 (s, 1H), 8.01 (m, 1H), 7.86 (s, 1H), 8.01 (m, 1H), 7.73 (m, 4H), 7.58 (m, 1H), 7.33 (m, 1H), 7.07 (s, 1H), 3.63 (s, 3H). HRMS (ESI): (M – H)− calcd for C24H16I2N3O5, 679.9185; found, 679.9183. LC-MS (ESI): 703.8 (M + Na)+, 679.8 (M – H)−. Purity: >95% (UV, λ = 254 nm).
2-(3-(2-((2-(1H-Benzo[d]imidazol-2-yl)phenyl)amino)-2-oxoacetamido)phenyl)-6-hydroxy-3-iodo-1-methyl-1H-indole-5-carboxylic Acid (11a-9)
Pale solid (4.7 mg, 35%). 1H NMR (500 MHz, DMSO): δ 11.38 (s, 1H), 11.13 (s, 1H), 8.81 (m, 1H), 8.17 (m, 1H), 8.12 (s, 2H), 7.78 (s, 1H), 7.64 (m, 4H), 7.39 (m, 5H), 7.07 (s, 1H), 3.64 (s, 3H). HRMS (ESI): (M – H)− calcd for C31H21IN5O5, 670.0593; found, 670.0594. LC-MS (ESI): 672.0 (M + H)+, 669.8 (M – H)−. Purity: >95% (UV, λ = 254 nm).
2-(3-(2-(Benzo[d]thiazol-2-ylamino)-2-oxoacetamido)phenyl)-6-hydroxy-3-iodo-1-methyl-1H-indole-5-carboxylic Acid (11a-10)
Pale solid (3.5 mg, 28%). 1H NMR (500 MHz, DMSO): δ 13.02 (s, 1H), 11.37 (s, 1H), 11.28 (s, 1H), 8.12 (m, 1H), 8.06 (s, 2H), 7.85 (m, 2H), 7.60 (m, 1H), 7.52 (m, 1H), 7.38 (m, 2H), 7.06 (s, 1H), 3.63 (s, 3H). HRMS (ESI): (M – H)− calcd for C25H16IN4O5S, 610.9892; found, 610.9884. LC-MS (ESI): 613.0 (M + H)+, 610.8 (M – H)−. Purity: >95% (UV, λ = 254 nm).
2-(3-(2-((3-Chlorophenyl)amino)-2-oxoacetamido)phenyl)-6-hydroxy-3-iodo-1-methyl-1H-indole-5-carboxylic Acid (11a-11)
Pale solid (4.1 mg, 34%). 1H NMR (500 MHz, DMSO): δ 11.36 (s, 1H), 11.11 (s, 1H), 11.10 (s, 1H), 8.03 (m, 3H), 7.85 (m, 2H), 7.57 (m, 1H), 7.42 (m, 1H), 7.34 (m, 1H), 7.24 (m, 1H), 7.07 (s, 1H), 3.63 (s, 3H). HRMS (ESI): (M – H)− calcd for C24H16ClIN3O5, 587.9829; found, 587.9831. LC-MS (ESI): 587.8 (M – H)−. Purity: >95% (UV, λ = 254 nm).
2-(3-(2-((4-(1H-Imidazol-1-yl)phenyl)amino)-2-oxoacetamido)phenyl)-6-hydroxy-3-iodo-1-methyl-1H-indole-5-carboxylic Acid (11a-12)
Pale solid (4.8 mg, 38%). 1H NMR (500 MHz, DMSO): δ 11.24 (s, 1H), 11.15 (s, 1H), 9.53 (s, 1H), 8.23 (s, 1H), 8.13 (m, 2H), 8.08 (s, 1H), 8.01 (m, 1H), 7.87 (s, 1H), 7.82 (m, 3H), 7.59 (m, 1H), 7.35 (m, 1H), 7.08 (s, 1H), 3.63 (s, 3H). HRMS (ESI): (M – H)− calcd for C27H19IN5O5, 620.0436; found, 620.0442. LC-MS (ESI): 622.0 (M + H)+, 619.8 (M – H)−. Purity: >95% (UV, λ = 254 nm).
2-(3-(2-((4-Chloro-3-(trifluoromethyl)phenyl)amino)-2-oxoacetamido)phenyl)-6-hydroxy-3-iodo-1-methyl-1H-indole-5-carboxylic Acid (11a-13)
Pale solid (3.6 mg, 27%). 1H NMR (500 MHz, DMSO): δ 11.38 (s, 2H), 11.14 (s, 1H), 10.99 (s, 1H), 8.52 (s, 1H), 8.19 (m, 1H), 8.06 (s, 1H), 8.01 (m, 1H), 7.86 (s, 1H), 7.77 (m, 1H), 7.59 (m, 1H), 7.34 (m, 1H), 7.07 (s, 1H), 3.63 (s, 3H). HRMS (ESI): (M – H)− calcd for C25H15ClF3IN3O5, 655.9702; found, 655.9720. LC-MS (ESI): 679.8 (M + Na)+, 655.8 (M – H)−. Purity: >95% (UV, λ = 254 nm).
6-Hydroxy-3-iodo-1-methyl-2-(3-(2-oxo-2-((5-phenyl-1,3,4-thiadiazol-2-yl)amino)acetamido)phenyl)-1H-indole-5-carboxylic Acid (11a-14)
Pale solid (4.5 mg, 35%). 1H NMR (500 MHz, DMSO): δ 13.61 (s, 1H), 11.38 (s, 1H), 11.29 (s, 1H), 8.01 (m, 4H), 7.86 (s, 1H), 7.58 (m, 4H), 7.34 (m, 4H), 7.07 (s, 1H), 3.62 (s, 3H). HRMS (ESI): (M – H)− calcd for C26H17ClIN5O5S, 638.0001; found, 638.0015. LC-MS (ESI): 637.8 (M – H)−. Purity: >95% (UV, λ = 254 nm).
2-(3-(2-((4-Fluorophenyl)amino)-2-oxoacetamido)phenyl)-6-hydroxy-3-iodo-1-methyl-1H-indole-5-carboxylic Acid (11a-15)
Pale solid (4.7 mg, 41%). 1H NMR (500 MHz, DMSO): δ 11.39 (s, 1H), 11.11 (s, 1H), 11.02 (s, 1H), 8.06 (s, 1H), 8.01 (m, 1H), 7.92 (m, 2H), 7.88 (s, 1H), 7.58 (m, 1H), 7.34 (m, 1H), 7.23 (m, 2H), 7.06 (s, 1H), 3.63 (s, 3H). HRMS (ESI): (M – H)− calcd for C24H16FIN3O5, 572.0124; found, 572.0148. LC-MS (ESI): 571.8 (M – H)−. Purity: >95% (UV, λ = 254 nm).
6-Hydroxy-3-iodo-1-methyl-2-(3-(2-oxo-2-(thiazol-2-ylamino)acetamido)phenyl)-1H-indole-5-carboxylic Acid (11a-16)
Pale solid (3.9 mg, 34%). 1H NMR (500 MHz, DMSO): δ 12.73 (s, 1H), 11.36 (s, 1H), 11.20 (s, 1H), 8.01 (s, 1H), 7.99 (s, 1H), 7.86 (s, 1H), 7.60 (m, 2H), 7.42 (m, 1H), 7.32 (m, 1H), 7.07 (s, 1H), 3.62 (s, 3H). HRMS (ESI): (M – H)− calcd for C21H14IN4O5S, 560.9735; found, 560.9744. LC-MS (ESI): 563.0 (M + H)+, 560.8 (M – H)−. Purity: >95% (UV, λ = 254 nm).
6-Hydroxy-3-iodo-1-methyl-2-(3-(2-oxo-2-((3-(trifluoromethoxy)phenyl)amino)acetamido)phenyl)-1H-indole-5-carboxylic Acid (11a-17)
Pale solid (3.3 mg, 25%). 1H NMR (500 MHz, DMSO): δ 11.37 (s, 1H), 11.21 (s, 1H), 11.13 (s, 1H), 8.05 (m, 3H), 7.90 (m, 1H), 7.87 (s, 1H), 7.76–7.51 (m, 2H), 7.33 (m, 1H), 7.17 (m, 1H), 7.07 (s, 1H), 3.63 (s, 3H). HRMS (ESI): (M – H)− calcd for C25H16F3IN3O6, 638.0041; found, 638.0039. LC-MS (ESI): 661.8 (M + Na)+, 637.8 (M – H)−. Purity: >95% (UV, λ = 254 nm).
2-(3-(2-((3-Bromophenyl)amino)-2-oxoacetamido)phenyl)-6-hydroxy-3-iodo-1-methyl-1H-indole-5-carboxylic Acid (11a-18)
Pale solid (3.8 mg, 30%). 1H NMR (500 MHz, DMSO): δ 11.38 (s, 1H), 11.10 (s, 1H), 11.08 (s, 1H), 8.17 (s, 1H), 8.06 (s, 1H), 8.01 (m, 1H), 7.89 (m, 1H), 7.86 (s, 1H), 7.58 (m, 1H), 7.36 (m, 3H), 7.07 (s, 1H), 3.63 (s, 3H). HRMS (ESI): (M – H)− calcd for C24H16BrIN3O5, 631.9323; found, 631.9326. LC-MS (ESI): 655.8 (M + Na)+, 631.8 (M – H)−. Purity: >95% (UV, λ = 254 nm).
2-(3-(2-((3-Fluorophenyl)amino)-2-oxoacetamido)phenyl)-6-hydroxy-3-iodo-1-methyl-1H-indole-5-carboxylic Acid (11a-19)
Pale solid (4.7 mg, 41%). 1H NMR (500 MHz, DMSO): δ 11.37 (s, 1H), 11.13 (s, 2H), 8.08 (s, 1H), 8.03 (m, 1H), 7.99 (m, 2H), 7.87 (s, 1H), 7.82 (m, 1H), 7.76 (m, 1H), 7.58 (m, 1H), 7.43 (m, 1H), 7.32 (m, 1H), 7.06 (s, 1H), 7.01 (m, 1H), 3.64 (s, 3H). HRMS (ESI): (M – H)− calcd for C24H16FIN3O5, 572.0124; found, 572.0136. LC-MS (ESI): 573.8 (M + H)+, 571.8 (M – H)−. Purity: >95% (UV, λ = 254 nm).
6-Hydroxy-3-iodo-2-(3-(2-((3-iodophenyl)amino)-2-oxoacetamido)phenyl)-1-methyl-1H-indole-5-carboxylic Acid (11a-20)
Pale solid (4.5 mg, 33%). 1H NMR (500 MHz, DMSO): δ 11.36 (s, 1H), 11.08 (s, 1H), 11.01 (s, 1H), 8.34 (s, 1H), 8.05 (s, 1H), 8.01 (m, 1H), 8.01 (m, 1H), 7.90 (m, 1H), 7.87 (s, 1H), 7.57 (m, 2H), 7.33 (m, 1H), 7.19 (m, 1H), 7.07 (s, 1H), 3.63 (s, 3H). HRMS (ESI): (M – H)− calcd for C24H16I2N3O5, 679.9185; found, 679.9184. LC-MS (ESI): 679.8 (M – H)−. Purity: >95% (UV, λ = 254 nm).
2-(3-(2-((4′-Cyano-[1,1′-biphenyl]-4-yl)amino)-2-oxoacetamido)phenyl)-6-hydroxy-3-iodo-1-methyl-1H-indole-5-carboxylic Acid (11a-21)
Pale solid (2.3 mg, 17%). 1H NMR (500 MHz, DMSO): δ 11.37 (s, 1H), 11.12 (s, 1H), 11.10 (s, 1H), 8.14–8.04 (m, 4H), 7.93 (m, 4H), 7.87–7.81 (m, 3H), 7.59 (m, 1H), 7.35 (m, 1H), 7.07 (s, 1H), 3.64 (s, 3H). HRMS (ESI): (M – H)− calcd for C31H20IN4O5, 655.0484; found, 655.0509. LC-MS (ESI): 654.8 (M – H)−. Purity: >95% (UV, λ = 254 nm).
2-(3-(2-((3′-Cyano-[1,1′-biphenyl]-4-yl)amino)-2-oxoacetamido)phenyl)-6-hydroxy-3-iodo-1-methyl-1H-indole-5-carboxylic Acid (11a-22)
Pale solid (2.9 mg, 21%). 1H NMR (500 MHz, DMSO): δ 11.37 (s, 1H), 11.11 (s, 1H), 11.07 (s, 1H), 8.18 (s, 1H), 8.03 (m, 5H), 7.87 (s, 1H), 7.82 (m, 3H), 7.67 (m, 1H), 7.59 (m, 1H), 7.34 (m, 1H), 7.08 (s, 1H), 3.64 (s, 3H). HRMS (ESI): (M – H)− calcd for C31H20IN4O5, 655.0484; found, 655.0499. LC-MS (ESI): 654.8 (M – H)−. Purity: >95% (UV, λ = 254 nm).
6-Hydroxy-2-(3-(2-((2-(5-(hydroxymethyl)furan-2-yl)phenyl)amino)-2-oxoacetamido)phenyl)-3-iodo-1-methyl-1H-indole-5-carboxylic Acid (11a-23)
Pale solid (2.4 mg, 18%). 1H NMR (500 MHz, DMSO): δ 11.15 (s, 1H), 10.76 (s, 1H), 8.05 (m, 3H), 7.87 (s, 1H), 7.82 (s, 1H), 7.76 (m, 1H), 7.58 (m, 2H), 7.41 (m, 3H), 7.08 (s, 1H), 4.51 (s, 2H), 3.65 (s, 1H), 3.64 (s, 3H). HRMS (ESI): (M – H)− calcd for C29H21IN3O7, 650.0430; found, 650.0422. LC-MS (ESI): 649.8 (M – H)−. Purity: >95% (UV, λ = 254 nm).
2-(3-(2-((3-(Furan-2-yl)phenyl)amino)-2-oxoacetamido)phenyl)-6-hydroxy-3-iodo-1-methyl-1H-indole-5-carboxylic Acid (11a-24)
Pale solid (4.6 mg, 37%). 1H NMR (500 MHz, DMSO): δ 11.36 (s, 1H), 11.10 (s, 1H), 10.98 (s, 1H), 8.29 (s, 1H), 8.07 (s, 1H), 8.03 (m, 1H), 7.94 (m, 1H), 7.87 (s, 1H), 7.77 (m, 2H), 7.50 (m, 4H), 7.34 (m, 1H), 7.07 (s, 1H), 6.90 (m, 1H), 3.64 (s, 3H). HRMS (ESI): (M – H)− calcd for C28H19IN3O6, 620.0324; found, 620.0327. LC-MS (ESI): 619.8 (M – H)−. Purity: >95% (UV, λ = 254 nm).
2-(3-(2-((4′-Cyano-[1,1′-biphenyl]-3-yl)amino)-2-oxoacetamido)phenyl)-6-hydroxy-3-iodo-1-methyl-1H-indole-5-carboxylic Acid (11a-25)
Pale solid (3.4 mg, 26%). 1H NMR (500 MHz, DMSO): δ 11.12 (s, 1H), 11.05 (s, 1H), 8.29 (s, 1H), 8.08 (s, 1H), 8.03 (m, 4H), 7.87 (s, 1H), 7.86 (s, 1H), 7.56 (m, 3H), 7.33 (m, 1H), 7.09 (s, 1H), 3.64 (s, 3H). HRMS (ESI): (M – H)− calcd for C31H20IN4O5, 655.0484; found, 655.0485. LC-MS (ESI): 654.8 (M – H)−. Purity: >95% (UV, λ = 254 nm).
6-Hydroxy-3-iodo-1-methyl-2-(3-(2-oxo-2-((3-(thiophen-3-yl)phenyl)amino)acetamido)phenyl)-1H-indole-5-carboxylic Acid (11a-26)
Pale solid (3.1 mg, 24%). 1H NMR (500 MHz, DMSO): δ 11.12 (s, 1H), 10.92 (s, 1H), 8.22 (s, 1H), 8.08 (s, 1H), 8.02 (m, 4H), 7.87 (s, 1H), 7.84 (m, 2H), 7.68 (m, 1H), 7.59 (m, 1H), 7.52 (m, 2H), 7.43 (m, 1H), 7.34 (m, 1H), 7.08 (s, 1H), 3.64 (s, 3H). HRMS (ESI): (M – H)− calcd for C28H19IN3O5S, 636.0096; found, 636.0102. LC-MS (ESI): 635.8 (M – H)−. Purity: >95% (UV, λ = 254 nm).
6-Hydroxy-3-iodo-1-methyl-2-(3-(4-oxo-4-((4-(thiophen-3-yl)phenyl)amino)butanamido)phenyl)-1H-indole-5-carboxylic Acid (11c-1)
Pale solid (3.8 mg, 28%). 1H NMR (500 MHz, DMSO): δ 11.35 (brs, 1H), 10.24 (s, 1H), 10.08 (s, 1H), 7.86 (s, 1H), 7.73 (m, 4H), 7.62 (m, 4H), 7.52 (m, 3H), 7.17 (m, 1H), 7.04 (s, 1H), 3.56 (s, 3H), 2.69 (m, 4H). HRMS (ESI): (M – H)− calcd for C30H23IN3O5S, 664.0409; found, 664.0414. LC-MS (ESI): 663.8 (M – H)−. Purity: >95% (UV, λ = 254 nm).
6-Hydroxy-3-iodo-1-methyl-2-(3-(4-oxo-4-((4-phenoxyphenyl)amino)butanamido)phenyl)-1H-indole-5-carboxylic Acid (11c-2)
Pale solid (1.6 mg, 11%). 1H NMR (500 MHz, DMSO): δ 10.22 (s, 1H), 10.04 (s, 1H), 7.83 (s, 1H), 7.76 (m, 2H), 7.60 (m, 2H), 7.47 (m, 1H), 7.35 (m, 2H), 7.09 (m, 3H), 6.96 (m, 4H), 3.58 (s, 3H), 2.67 (m, 4H). HRMS (ESI): (M – H)− calcd for C32H25IN3O6, 674.0794; found, 664.0796. LC-MS (ESI): 673.8 (M – H)−. Purity: >95% (UV, λ = 254 nm).
2-(3-(4-((4-(Benzyloxy)-3-chlorophenyl)amino)-4-oxobutanamido)phenyl)-6-hydroxy-3-iodo-1-methyl-1H-indole-5-carboxylic Acid (11c-3)
Pale solid (5.7 mg, 39%). 1H NMR (500 MHz, DMSO): δ 10.23 (s, 1H), 10.03 (s, 1H), 7.84 (s, 1H), 7.80 (m, 2H), 7.49–7.33 (m, 8H), 7.17 (m, 2H), 7.03 (s, 1H), 5.15 (s, 2H), 3.58 (s, 3H), 2.67 (m, 4H). HRMS (ESI): (M – H)− calcd for C33H26ClIN3O6, 722.0560; found, 722.0563. LC-MS (ESI): 746.0 (M + Na)+, 721.8 (M – H)−. Purity: >95% (UV, λ = 254 nm).
2-(3-(4-((3-(Benzyloxy)phenyl)amino)-4-oxobutanamido)phenyl)-6-hydroxy-3-iodo-1-methyl-1H-indole-5-carboxylic Acid (11c-4)
Pale solid (2.2 mg, 15%). 1H NMR (500 MHz, DMSO): δ 11.35 (brs, 1H), 10.22 (s, 1H), 9.99 (s, 1H), 7.84 (s, 1H), 7.73 (m, 2H), 7.50–7.32 (m, 7H), 7.15 (m, 3H), 7.04 (s, 1H), 6.68 (m, 1H), 5.05 (s, 2H), 3.58 (s, 3H), 2.67 (m, 4H). HRMS (ESI): (M – H)− calcd for C33H27IN3O6, 688.0950; found, 688.0950. LC-MS (ESI): 687.8 (M – H)−. Purity: >95% (UV, λ = 254 nm).
2-(3-(4-([1,1′-Biphenyl]-3-ylamino)-4-oxobutanamido)phenyl)-6-hydroxy-3-iodo-1-methyl-1H-indole-5-carboxylic Acid (11c-5)
Pale solid (4.8 mg, 36%). 1H NMR (500 MHz, DMSO): δ 11.35 (brs, 1H), 10.24 (s, 1H), 10.12 (s, 1H), 7.93 (m, 2H), 7.84 (s, 1H), 7.76 (m, 2H), 7.60–7.40 (m, 8H), 7.16 (m, 1H), 7.03 (s, 1H), 3.58 (s, 3H), 2.64 (m, 4H). HRMS (ESI): (M – H)− calcd for C32H25IN3O5, 658.0844; found, 658.0861. LC-MS (ESI): 683.4 (M + H)+, 657.8 (M – H)−. Purity: >95% (UV, λ = 254 nm).
2-(3-(4-(5-Bromoindolin-1-yl)-4-oxobutanamido)phenyl)-6-hydroxy-3-iodo-1-methyl-1H-indole-5-carboxylic Acid (11c-6)
Pale solid (3.2 mg, 23%). 1H NMR (500 MHz, DMSO): δ 11.35 (brs, 1H), 10.25 (s, 1H), 7.97 (m, 1H), 7.84 (s, 1H), 7.76 (m, 2H), 7.47 (m, 1H), 7.42 (s, 1H), 7.29 (m, 1H), 7.17 (m, 1H), 7.04 (s, 1H), 4.16 (m, 2H), 3.56 (s, 3H), 3.17 (m, 2H), 2.77 (m, 2H), 2.71 (m, 2H). HRMS (ESI): (M – H)− calcd for C28H22BrIN3O5, 685.9793; found, 685.9798. LC-MS (ESI): 687.8 (M – H)−. Purity: >95% (UV, λ = 254 nm).
6-Hydroxy-3-iodo-1-methyl-2-(3-(4-oxo-4-((4-(trifluoromethoxy)phenyl)amino)butanamido)phenyl)-1H-indole-5-carboxylic Acid (11c-7)
Pale solid (6.1 mg, 45%). 1H NMR (500 MHz, DMSO): δ 11.35 (brs, 1H), 10.23 (s, 1H), 10.22 (s, 1H), 7.84 (s, 1H), 7.71 (m, 5H), 7.48 (m, 1H), 7.30 (m, 2H), 7.16 (m, 1H), 7.03 (s, 1H), 3.58 (s, 3H), 2.70 (m, 4H). HRMS (ESI): (M – H)− calcd for C27H20F3IN3O6, 666.0354; found, 666.0329. LC-MS (ESI): 665.8 (M – H)−. Purity: >95% (UV, λ = 254 nm).
6-Hydroxy-3-iodo-2-(3-(4-((4-isopropylphenyl)amino)-4-oxobutanamido)phenyl)-1-methyl-1H-indole-5-carboxylic Acid (11c-8)
Pale solid (2.1 mg, 16%). 1H NMR (500 MHz, DMSO): δ 9.91 (s, 1H), 7.70 (s, 1H), 7.50–7.45 (m, 5H), 7.15 (m, 5H), 6.66 (s, 1H), 3.58 (s, 3H), 2.82 (m, 1H), 2.64 (m, 4H), 1.17 (d, J = 6.9 Hz, 1H). HRMS (ESI): (M – H)− calcd for C29H27IN3O5, 624.1001; found, 624.1037. LC-MS (ESI): 648.0 (M + Na)+, 624.0 (M – H)−. Purity: >95% (UV, λ = 254 nm).
2-(3-(4-((Benzo[d][1,3]dioxol-5-ylmethyl)amino)-4-oxobutanamido)phenyl)-6-hydroxy-3-iodo-1-methyl-1H-indole-5-carboxylic Acid (11c-9)
Pale solid (8.1 mg, 63%). 1H NMR (500 MHz, DMSO): δ 11.35 (brs, 1H), 10.19 (s, 1H), 8.37 (m, 1H), 7.85 (s, 1H), 7.61 (s, 1H), 7.72 (m, 1H), 7.47 (m, 1H), 7.16 (m, 1H), 7.03 (s, 1H), 6.80 (m, 2H), 6.70 (m, 1H), 5.93 (s, 2H), 4.17 (m, 2H), 3.58 (s, 3H), 2.64 (m, 4H). 13C NMR (125 MHz, DMSO): δ 172.8, 171.8, 171.3, 158.2, 147.6, 146.3, 142.9, 142.5, 139.8, 133.9, 131.5, 129.4, 125.5, 123.9, 123.8, 121.2, 120.7, 119.8, 108.3, 108.2, 108.0, 101.1, 97.0, 60.5, 42.2, 32.5, 32.0, 30.6. HRMS (ESI): (M – H)− calcd for C28H23IN3O7, 640.0586; found, 640.0569. LC-MS (ESI): 642.0 (M + H)+, 639.6 (M – H)−. Purity: >95% (UV, λ = 254 nm).
2-(3-(4-((Cyclopropylmethyl)(propyl)amino)-4-oxobutanamido)phenyl)-6-hydroxy-3-iodo-1-methyl-1H-indole-5-carboxylic Acid (11c-10)
Pale solid (10.6 mg, 87%). 1H NMR (500 MHz, DMSO): δ 11.35 (brs, 1H), 10.16 (s, 1H), 7.85 (s, 1H), 7.73 (m, 2H), 7.48 (m, 1H), 7.16 (m, 1H), 7.04 (s, 1H), 3.58 (s, 3H), 3.33–3.14 (m, 4H), 2.64 (m, 2H), 1.59–1.46 (m, 2H), 0.88 (m, 4H), 0.50–0.10 (m, 4H). HRMS (ESI): (M – H)− calcd for C27H29IN3O5, 602.1157; found, 602.1171. LC-MS (ESI): 604.0 (M + H)+, 602.0 (M – H)−. Purity: >95% (UV, λ = 254 nm).
6-Hydroxy-3-iodo-1-methyl-2-(3-(4-oxo-4-(thiazol-2-ylamino)butanamido)phenyl)-1H-indole-5-carboxylic Acid (11c-11)
Pale solid (0.9 mg, 7%). 1H NMR (500 MHz, DMSO): δ 12.15 (s, 1H), 10.25 (s, 1H), 7.84 (s, 1H), 7.74 (m, 2H), 7.46 (m, 3H), 7.17 (m, 3H), 6.88 (s, 1H), 3.58 (s, 3H), 2.64 (m, 4H). HRMS (ESI): (M – H)− calcd for C23H18IN4O5S, 589.0048; found, 589.0056. LC-MS (ESI): 591.0 (M + H)+. Purity: >95% (UV, λ = 254 nm).
SHP2 Crystallization and X-ray Data Collection
Recombinant SHP2 expression and purification were described previously.35 All crystallization experiments were performed at room temperature using the sitting drop vapor diffusion method. For co-crystallization, 100 μL of SHP2 stock (7.0 mg/mL) in 20 mM MES (pH 6.0), 50 mM NaCl, 0.1 mM EDTA, and 4 mM DTT was mixed with 1 μL of compound 11a-1 or 11c-9 stock solution (50 mM in DMSO). Crystals of SHP2·11c-9 complex were obtained at room temperature by vapor diffusion in sitting drops. Protein drops were equilibrated against a reservoir solution containing 20% w/v polyethylene glycol 3350, 200 mM magnesium acetate tetrahydrate, and 100 mM HEPES buffer (pH 7.7). For X-ray data collection, the crystals were transferred into 4 μL of cryoprotectant buffer containing 30% w/v polyethylene glycol 3350, 100 mM NaCl, 1 mM 11c-9, and 100 mM HEPES (pH 7.7) and were allowed to soak for 30 min. The crystals were then flash-cooled by liquid nitrogen. X-ray data were collected at 19 μB beamline at APS (Argonne, IL). Data were processed using the program HKL3000.64 The space group of the crystals is P1. The statistics are provided in Table 5.
Structural Determination and Refinement
The structure of SHP2·11c-9 was solved by molecular replacement using the program MolRep.65 The structure of SHP2 (PDB: 3B7O),52 without the solvent molecules, was used as a search model. The resulting difference Fourier map indicated the first 16 N-terminal residues has different conformation. The map also revealed the density for the compound 11c-9 in the active site of SHP2. The N-terminal peptide 246–261 was first rebuilt according to the Fo – Fc density map, then the structure was refined to 2.4 Å resolution with the program CNS1.1,66 using simulated annealing at 2500 K and then alternating positional and individual temperature factor refinement cycles. The density in the active site was interpreted to be 11c-9. Then alternating positional and individual temperature factor refinement cycles were applied to the structure. The progress of the refinement was evaluated by improvement in the quality of the electron density maps, and the reduced values of the conventional R factor (R = ∑h||Fo| – |Fc||/∑h|Fo|) and the free R factor (3.7% of the reflections omitted from the refinement).67 Electron density maps were inspected, and the model was modified on Coot.68 Finally, water molecules were added gradually as the refinement progressed. They were assigned in the |Fo| – |Fc| difference Fourier maps with a 2.5σ cutoff level for inclusion in the model.
Docking Studies
The 3D structure of 11a-1 was built and energy-minimized using Chem3D program, and three SHP2 catalytic domain structures, 3B7O.pdb52 (apo form SHP2 catalytic domain), 3O5X.pdb35 (SHP2·II-B08 complex structure) and 4PVG.pdb (SHP2·11c-9 complex structure), were used for ensemble docking in the AutoDock4.2.5 software suite.69 The ligand and receptor were predocking processed as following in AutoDockTools1.4.6 program:70 merge nonpolar hydrogens, add Gasteiger charges, set rotatable bond for ligand, add solvation parameter for receptor, and so on. To define the common docking area, the above two SHP2 complex structures were superimposed onto the apo form of SHP2, a rectangle docking area was visually set to adequately cover both II-B08 and 11c-9 around the active site, the energy grid size was set to 54 × 54 × 36 points with 0.375 Å spacing on each axis, and the energy grid maps for each atom type (i.e., A, C, I, OA, N, SA, and HD), as well as the electrostatics and desolvation maps were calculated using the AutoGrid4. The molecular docking were carried out using AutoDock4.2.5, the optimal binding conformation was determined by LGALS (Lamarckian Genetic Algorithm with Local Search) algorithm with the following parameters during each docking run: energy evaluations of 2500000, population size of 100, mutation rate of 0.02, crossover rate of 0.8, Solis and Wets local search iterations of 300 with probability of 0.06. For each SHP2 structure, 200 docking runs were performed and the resulted binding modes were conformation-clustered and energy-ranked. The final binding mode was determined by visual inspections, cluster analyses, and energy comparisons.
Inhibition Studies
The phosphatase activity of SHP2 was assayed using p-nitrophenyl phosphate (pNPP) as a substrate at 25 °C in 50 mM 3,3-dimethylglutarate buffer, pH 7.0, containing 1 mM EDTA with an ionic strength of 0.15 M adjusted by NaCl. The hydroxyindole carboxylic acid-based libraries were screened in a 384-well format at 5 μM compound concentration. The reaction was initiated by the addition of 20 μL of the enzyme to 30 μL of reaction mixture containing 5 μM library compound and 2.9 mM (the Km value) pNPP and quenched after 10 min by the addition of 20 μL of 5N NaOH. The nonenzymatic hydrolysis of pNPP was corrected by measuring the control without the addition of enzyme. The amount of product p-nitrophenol was determined from the absorbance at 405 nm detected by a Spectra MAX340 microplate spectrophotometer (Molecular Devices) using a molar extinction coefficient of 18000 M–1cm–1. Inhibitor concentrations used for IC50 measurements cover the range from 0.2 to 5× of the IC50 value. IC50 values for selected resynthesized and purified hits were calculated by fitting the absorbance at 405 nm versus inhibitor concentration to the following equation:
where AI is the absorbance at 405 nm of the sample in the presence of inhibitor, A0 is the absorbance at 405 nm in the absence of inhibitor, and [I] is the concentration of the inhibitor.
For selectivity studies, the PTPs, including LYP, HePTP, PTPH1, SHP1, Ssu72, PTP1B, LMWPTP, VHZ, PTPγ, MKP5, VHR, PTPμ, STEP, PEZ, PTPσ, UBLCP1, Laforin, CDC14A, PTPε, and PTPα, were expressed and purified from Escherichia coli. The IC50 determination for these PTPs was performed under the same conditions as for SHP2, except the pNPP concentrations used corresponded to the Km values of the PTPs studied.
Effects of SHP2 Inhibitor 11a-1 on Lung Cancer Cell Line H1975
Human nonsmall cell lung carcinoma cell line H1975 was obtained from the American Tissue Culture Collection and cultured at 37 °C and 5% CO2 in RPMI-1640 (Corning) supplemented with 10% fetal bovine serum (HyClone). 3 ×103 cells were seeded in each well of 96-well plates. After treatment with 11a-1 for 2 days in RPMI-1640 supplement with 0.5% FBS, cells were incubated with 50 μg/mL MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) (Sigma) for 3–4 h. Then the culture medium was removed, and DMSO was added to dissolve the formazan crystals. Wells containing only media were used for background correction. The optical density was measured spectrophotometrically at 540 nm. For biochemical studies, H1975 cells were serum-starved overnight followed by treatment with vehicle or 11a-1 for 3 h, and then either left unstimulated or stimulated with 5 ng/mL EGF (Sigma) for 60 min. Lysates were then resolved by SDS-PAGE and the protein phosphorylation levels were detected by immunoblot analysis.
Effect of SHP2 Inhibitor 11a-1 on Breast Cancer Cells
Approximately 300000 SKBR3 cells were seeded into 150 μL of growth factor reduced Matrigel (Corning) in 35 mm dishes. To three dishes for each condition, 2 mL of media containing either 10 μL of vehicle (DMSO) or 11a-1 at the indicated concentration was then added. Every 24 h, the cells were imaged with a NIKON SMZ1500 stereo microscope. The cross sectional area of individual colonies from each image were measured using Adobe Photoshop software. After 4 days, the cells were recovered from the Matrigel and lysed in PLC buffer containing protease inhibitor cocktail. Lysates were then resolved by SDS-PAGE, and relative levels of the indicated proteins were detected by immunoblot analysis.
Effect of SHP2 Inhibitor II-B08 and 11a-1 on D32 Myeloid Cells and Primary Hematopoietic Stem and Progenitor Cells
The WT KIT and KITD814V were inserted into the bicistronic retroviral vector, MIEG3, upstream of the internal ribosome entry site (IRES) and the enhanced green fluorescent protein (EGFP) gene as previously described.50 Retroviral supernatants for transduction of 32D myeloid cells and primary hematopoietic stem and progenitor cells were generated using Phoenix ecotropic packaging cell line transfected with retroviral vector plasmids using a calcium phosphate transfection kit (Invitrogen, Carlsbad, CA). Supernatants were collected 48 h after transfection and filtered through 0.45 μm membranes. 32D myeloid cells were infected twice at 24 h intervals with 2 mL of high-titer virus supernatant in the presence of 10 ng/mL IL-3 and 8 μg/mL Polybrene. Forty-eight hours after infection, 32D cells expressing WT KIT or KITD814V receptors were sorted to homogeneity based on EGFP expression and utilized to perform all the experiments.
To express WT KIT and KITD814V receptors in bone marrow cells, low-density bone marrow cells were collected from WT mice, and prestimulated in IMDM supplemented with 20% FBS, 2% penicillin/streptomycin, and cytokines (100 ng/mL SCF, 100 ng/mL TPO, 50 ng/mL FLT3-L, and 4 ng/mL IL-6) for 48 h prior to retroviral infection on fibronectin fragments (Retronectin) in nontissue culture plates. On the third day, cells were infected with 2 mL of high-titer retroviral supernatants for WT KIT or KITD814V prepared as described above. A second shot of viral infection was given 20 4 h later. Forty-eight hours after the second infection, cells expressing EGFP were sorted and utilized to perform all experiments.
Proliferation was assessed by conducting a thymidine incorporation assay as previously described (Mali et al., 201250). Cells were washed and starved in IMDM containing 0.2% BSA without serum or any growth factors for 6–8 h. Cells (5 × 104) were plated in replicates of three in a 96-well plate in 200 μL of complete medium (IMDM, 10% FBS, 2% penicillin–streptomycin) either in the presence or absence of indicated growth factors with or without SHP2 inhibitors. Cells were cultured for 48 h and subsequently pulsed with 1.0 μCi (0.037 MBq) [3H] thymidine (PerkinElmer, Shelton, CT) for 6 h. Cells were harvested using an automated 96-well cell harvester (Brandel, Gaithersburg, MD), and thymidine incorporation was determined as counts per minute (CPM).
Immunoblot Analysis
Cells were lysed in lysis buffer (1.0% Nonidet P-40, 50 mM Tris-HCl (pH 7.4), 150 mM NaCl, 5 mM EDTA, 2 mM NaVO3, 10 mM NaF) plus a protease inhibitor mixture (Roche) and centrifuged at 10000 rpm for 5 min at 4 °C. Supernatants were collected and protein concentration was determined using the BCA protein assay (Thermo Fisher Scientific, Rockford, IL). Equal amount of protein extracts were mixed with gel loading buffer and separated on SDS-polyacrylamide gels. After electrophoresis, the proteins were transferred onto nitrocellulose membranes and nonspecific binding was blocked with 5% nonfat dry milk in Tris-buffered saline containing 0.1% Tween-20 (TBS-T). Membranes were then probed with various antibodies overnight at 4 °C on a rocker. After incubation, membranes were washed with TBS-T and incubated with appropriate horseradish peroxidase (HRP)-conjugated secondary antibodies for 1 h at room temperature. Finally, the proteins on the membranes were detected using SuperSignal West Dura Luminol/Enhancer solution (Thermo Fisher Scientific, Rockford, IL) and membranes were analyzed using Bio-Rad ChemiDoc XRS Imaging System.
RNA Interference Studies
Small interfering RNA (siRNA) specific for SHP2 (5′-PCACGCAUGACGCCAUAUUCTT-3′) and scrambled SHP2 siRNA (5′-PGCACGACCGCCUUAUAACUTT-3′) were synthesized by Dharmacon Research. siRNAs were transfected into H1975 cells by using Lipofectamine 2000 reagent (Invitrogen Life Technologies) for 6 h and then recovered in RPMI1640 plus 10% FBS.
Acknowledgments
This work was supported by National Institutes of Health Grants RO1CA152194, R01CA151765, R01HL077177, R01HL081111, R01CA173852, and R01CA134777.
Glossary
Abbreviations Used
- SHP2
Src homology-2 domain containing protein tyrosine phosphatase-2
- PTP
protein tyrosine phosphatase
- PTK
protein tyrosine kinase
- Erk
extracellular signal-regulated protein kinase
- EGF
epidermal growth factor
Supporting Information Available
1H and 13C NMR spectra for all of the intermediates (compounds 2, 4, 5, 6, and 7) and final compounds (10a–d, 11a-1 to 11a-26 and 11c-1 to 11c-11) tested for biological activity. This material is available free of charge via the Internet at http://pubs.acs.org.
Accession Codes
The coordinates for the structure of the SHP2·11c-9 complex (PDB: 4PVG) have been deposited in the Protein Data Bank.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Hunter T. Tyrosine phosphorylation: thirty years and counting. Curr. Opin. Cell Biol. 2009, 21, 140–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tonks N. K. Protein tyrosine phosphatases: from genes, to function, to disease. Nature Rev. Mol. Cell Biol. 2006, 7, 833–846. [DOI] [PubMed] [Google Scholar]
- Zhang Z.-Y. Protein tyrosine phosphatases: prospects for therapeutics. Curr. Opin. Chem. Biol. 2001, 5, 416–423. [DOI] [PubMed] [Google Scholar]
- Julien S. G.; Dubé N.; Hardy S.; Tremblay M. L. Inside the human cancer tyrosine phosphatome. Nature Rev. Cancer 2011, 11, 35–49. [DOI] [PubMed] [Google Scholar]
- Krause D. S.; Van Etten R. A. Tyrosine kinases as targets for cancer therapy. N. Engl. J. Med. 2005, 353, 172–187. [DOI] [PubMed] [Google Scholar]
- Ventura J. J.; Nebreda A. R. Protein kinases and phosphatases as therapeutic targets in cancer. Clin. Transl. Oncol. 2006, 8, 153–160. [DOI] [PubMed] [Google Scholar]
- Cohen P.; Alessi D. R. Kinase Drug Discovery - What’s Next in the Field?. ACS Chem. Biol. 2013, 8, 96–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J.; Yen C.; Liaw D.; Podsypanina K.; Bose S.; Wang S. I.; Puc J.; Miliaresis C.; Rodgers L.; McCombie R.; Bigner S. H.; Giovanella B. C.; Ittmann M.; Tycko B.; Hibshoosh H.; Wigler M. H.; Parsons R. PTEN, a putative protein tyrosine phosphatase gene mutated in human brain, breast, and prostate cancer. Science 1997, 275, 1943–1947. [DOI] [PubMed] [Google Scholar]
- Wang Z.; Shen D.; Parsons D. W.; Bardelli A.; Sager J.; Szabo S.; Ptak J.; Silliman N.; Peters B. A.; van der Heijden M. S.; Parmigiani G.; Yan H.; Wang T. L.; Riggins G.; Powell S. M.; Willson J. K.; Markowitz S.; Kinzler K. W.; Vogelstein B.; Velculescu V. E. Mutational analysis of the tyrosine phosphatome in colorectal cancers. Science 2004, 304, 1164–1166. [DOI] [PubMed] [Google Scholar]
- Neel B. G.; Gu H. H.; Pao L. The ‘Shp’ing news: SH2 domain-containing tyrosine phosphatases in cell signaling. Trends Biochem. Sci. 2003, 28, 284–293. [DOI] [PubMed] [Google Scholar]
- Chan R. J.; Feng G. S. PTPN11 is the first identified proto-oncogene that encodes a tyrosine phosphatase. Blood 2007, 109, 862–867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grossmann K. S.; Rosário M.; Birchmeier C.; Birchmeier W. The tyrosine phosphatase Shp2 in development and cancer. Adv. Cancer Res. 2010, 106, 53–89. [DOI] [PubMed] [Google Scholar]
- Tartaglia M.; Mehler E. L.; Goldberg R.; Zampino G.; Brunner H. G.; Kremer H.; van der Burgt I.; Crosby A. H.; Ion A.; Jeffery S.; Kalidas K.; Patton M. A.; Kucherlapati R. S.; Gelb B. D. Mutations in PTPN11, encoding the protein tyrosine phosphatase SHP-2, cause Noonan syndrome. Nature Genet. 2001, 29, 465–468. [DOI] [PubMed] [Google Scholar]
- Tartaglia M.; Gelb B. D. Noonan syndrome and related disorders: genetics and pathogenesis. Annu. Rev. Genomics Hum. Genet. 2005, 6, 45–68. [DOI] [PubMed] [Google Scholar]
- Tartaglia M.; Niemeyer C. M.; Fragale A.; Song X.; Buechner J.; Jung A.; Hahlen K.; Hasle H.; Licht J. D.; Gelb B. D. Somatic mutations in PTPN11 in juvenile myelomonocytic leukemia, myelodysplastic syndromes and acute myeloid leukemia. Nature Genet. 2003, 34, 148–150. [DOI] [PubMed] [Google Scholar]
- Tartaglia M.; Martinelli S.; Cazzaniga G.; Cordeddu V.; Iavarone I.; Spinelli M.; Palmi C.; Carta C.; Pession A.; Arico M.; Masera G.; Basso G.; Sorcini M.; Gelb B. D.; Biondi A. Genetic evidence for lineage-related and differentiation stage-related contribution of somatic PTPN11 mutations to leukemogenesis in childhood acute leukemia. Blood 2004, 104, 307–313. [DOI] [PubMed] [Google Scholar]
- Loh M. L.; Vattikuti S.; Schubbert S.; Reynolds M. G.; Carlson E.; Lieuw K. H.; Cheng J. W.; Lee C. M.; Stokoe D.; Bonifas J. M.; Curtiss N. P.; Gotlib J.; Meshinchi S.; Le Beau M. M.; Emanuel P. D.; Shannon K. M. Mutations in PTPN11 implicate the SHP-2 phosphatase in leukemogenesis. Blood 2004, 103, 2325–2331. [DOI] [PubMed] [Google Scholar]
- Loh M. L.; Reynolds M. G.; Vattikuti S.; Gerbing R. B.; Alonzo T. A.; Carlson E.; Cheng J. W.; Lee C. M.; Lange B. J.; Meshinchi S.; PTPN11 mutations in pediatric patients with acute myeloid leukemia: results from the Children’s Cancer Group. Leukemia 2004, 18, 1831–1834. [DOI] [PubMed] [Google Scholar]
- Kratz C. P.; Niemeyer C. M.; Castleberry R. P.; Cetin M.; Bergsträsser E.; Emanuel P. D.; Hasle H.; Kardos G.; Klein C.; Kojima S.; Stary J.; Trebo M.; Zecca M.; Gelb B. D.; Tartaglia M.; Loh M. L. The mutational spectrum of PTPN11 in juvenile myelomonocytic leukemia and Noonan syndrome/myeloproliferative disease. Blood 2005, 106, 2183–2185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bentires-Alj M.; Paez J. G.; David F. S.; Keilhack H.; Halmos B.; Naoki K.; Maris J. M.; Richardson A.; Bardelli A.; Sugarbaker D. J.; Richards W. G.; Du J.; Girard L.; Minna J. D.; Loh M. L.; Fisher D. E.; Velculescu V. E.; Vogelstein B.; Meyerson M.; Sellers W. R.; Neel B. G. Activating mutations of the noonan syndrome-associated SHP2/PTPN11 gene in human solid tumors and adult acute myelogenous leukemia. Cancer Res. 2004, 64, 8816–8820. [DOI] [PubMed] [Google Scholar]
- Miyamoto D.; Miyamoto M.; Takahashi A.; Yomogita Y.; Higashi H.; Kondo S.; Hatakeyama M. Isolation of a distinct class of gain-of-function SHP-2 mutants with oncogenic RAS-like transforming activity from solid tumors. Oncogene 2008, 27, 3508–3515. [DOI] [PubMed] [Google Scholar]
- Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature 2008, 455, 1061–1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor B. S.; Schultz N.; Hieronymus H.; Gopalan A.; Xiao Y.; Carver B. S.; Arora V. K.; Kaushik P.; Cerami E.; Reva B.; Antipin Y.; Mitsiades N.; Landers T.; Dolgalev I.; Major J. E.; Wilson M.; Socci N. D.; Lash A. E.; Heguy A.; Eastham J. A.; Scher H. I.; Reuter V. E.; Scardino P. T.; Sander C.; Sawyers C. L.; Gerald W. L. Integrative genomic profiling of human prostate cancer. Cancer Cell 2010, 18, 11–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brennan C. W.; Verhaak R. G.; McKenna A.; Campos B.; Noushmehr H.; Salama S. R.; Zheng S.; Chakravarty D.; Sanborn J. Z.; Berman S. H.; Beroukhim R.; Bernard B.; Wu C. J.; Genovese G.; Shmulevich I.; Barnholtz-Sloan J.; Zou L.; Vegesna R.; Shukla S. A.; Ciriello G.; Yung W. K.; Zhang W.; Sougnez C.; Mikkelsen T.; Aldape K.; Bigner D. D.; Van Meir E. G.; Prados M.; Sloan A.; Black K. L.; Eschbacher J.; Finocchiaro G.; Friedman W.; Andrews D. W.; Guha A.; Iacocca M.; O’Neill B. P.; Foltz G.; Myers J.; Weisenberger D. J.; Penny R.; Kucherlapati R.; Perou C. M.; Hayes D. N.; Gibbs R.; Marra M.; Mills G. B.; Lander E.; Spellman P.; Wilson R.; Sander C.; Weinstein J.; Meyerson M.; Gabriel S.; Laird P. W.; Haussler D.; Getz G.; Chin L.; The somatic genomic landscape of glioblastoma. Cell 2013, 155, 462–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aceto N.; Sausgruber N.; Brinkhaus H.; Gaidatzis D.; Martiny-Baron G.; Mazzarol G.; Confalonieri S.; Quarto M.; Hu G.; Balwierz P. J.; Pachkov M.; Elledge S. J.; van Nimwegen E.; Stadler M. B.; Bentires-Alj M. Tyrosine phosphatase SHP2 promotes breast cancer progression and maintains tumor-initiating cells via activation of key transcription factors and a positive feedback signaling loop. Nature Med. 2012, 18, 529–537. [DOI] [PubMed] [Google Scholar]
- Stommel J. M.; Kimmelman A. C.; Ying H.; Nabioullin R.; Ponugoti A. H.; Wiedemeyer R.; Stegh A. H.; Bradner J. E.; Ligon K. L.; Brennan C.; Chin L.; DePinho R. A. Coactivation of receptor tyrosine kinases affects the response of tumor cells to targeted therapies. Science 2007, 318, 287–290. [DOI] [PubMed] [Google Scholar]
- Noren-Muller A.; Reis-Correa I.; Prinz H.; Rosenbaum C.; Saxena K.; Schwalbe H. J.; Vestweber D.; Cagna G.; Schunk S.; Schwarz O.; Schiewe H.; Waldmann H. Discovery of protein phosphatase inhibitor classes by biology-oriented synthesis. Proc. Natl. Acad. Sci. U. S. A. 2006, 103, 10606–10611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L. W.; Sung S. S.; Yip M. L. R.; Lawrence H. R.; Ren Y.; Guida W. C.; Sebti S. M.; Lawrence N. J.; Wu J. Discovery of a novel shp2 protein tyrosine phosphatase inhibitor. Mol. Pharmacol. 2006, 70, 562–570. [DOI] [PubMed] [Google Scholar]
- Dawson M. I.; Xia Z.; Jiang T.; Ye M.; Fontana J. A.; Farhana L.; Patel B.; Xue L. P.; Bhuiyan M.; Pellicciari R.; Macchiarulo A.; Nuti R.; Zhang X. K.; Han Y. H.; Tautz L.; Hobbs P. D.; Jong L.; Waleh N.; Chao W. R.; Feng G. S.; Pang Y.; Su Y. Adamantyl-substituted retinoid-derived molecules that interact with the orphan nuclear receptor small heterodimer partner: effects of replacing the 1-adamantyl or hydroxyl group on inhibition of cancer cell growth, induction of cancer cell apoptosis, and inhibition of SRC homology 2 domain-containing protein tyrosine phosphatase-2 activity. J. Med. Chem. 2008, 51, 5650–5662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geronikaki A.; Eleftheriou P.; Vicini P.; Alam I.; Dixit A.; Saxena A. K. 2-Thiazolylimino/heteroarylimino-5-arylidene-4-thiazolidinones as new agents with SHP-2 inhibitory action. J. Med. Chem. 2008, 51, 5221–5228. [DOI] [PubMed] [Google Scholar]
- Lawrence H. R.; Pireddu R.; Chen L. W.; Luo Y. T.; Sung S. S.; Szymanski A. M.; Yip M. L. R.; Guida W. C.; Sebti S. M.; Wu J.; Lawrence N. J. Inhibitors of Src homology-2 domain containing protein tyrosine phosphatase-2 (Shp2) based on oxindole scaffolds. J. Med. Chem. 2008, 51, 4948–4956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hellmuth K.; Grosskopf S.; Lum C. T.; Wurtele M.; Roder N.; Kries J. P. V.; Rosario M.; Rademann J.; Birchmeier W. Specific inhibitors of the protein tyrosine phosphatase Shp2 identified by high-throughput docking. Proc. Natl. Acad. Sci. U. S. A. 2008, 105, 7275–7280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu W. M.; Guvench O.; MacKerell A. D.; Qu C. K. Identification of small molecular weight inhibitors of Src homology 2 domain-containing tyrosine phosphatase 2 (SHP-2) via in silico database screening combined with experimental assay. J. Med. Chem. 2008, 51, 7396–7404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu D.; Pang Y.; Ke Y.; Yu J.; He Z.; Tautz L.; Mustelin T.; Ding S.; Huang Z.; Feng G.-S. A conserved mechanism for control of human and mouse embryonic stem cell pluripotency and differentiation by shp2 tyrosine phosphatase. PLoS One 2009, 4, e4914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X.; He Y.; Liu S.; Yu Z.; Jiang Z.-X.; Yang Z.; Dong Y.; Nabinger S. C.; Wu L.; Gunawan A. M.; Wang L.; Chan R. J.; Zhang Z.-Y. Salicylic acid-based small molecule inhibitor for the oncogenic Src homology-2 domain containing protein tyrosine phosphatase-2 (SHP2). J. Med. Chem. 2010, 53, 2482–2493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu D.; Kong G.; Chen Q. C.; Wang G.; Li J.; Xu Y.; Lin T.; Tian Y.; Zhang X.; Yao X.; Feng G.; Lu Z.; Chen H. Fatty acids as natural specific inhibitors of the proto-oncogenic protein Shp2. Bioorg. Med. Chem. Lett. 2011, 21, 6833–6837. [DOI] [PubMed] [Google Scholar]
- Scott L. M.; Chen L.; Daniel K. G.; Brooks W. H.; Guida W. C.; Lawrence H. R.; Sebti S. M.; Lawrence N. J.; Wu J. Shp2 protein tyrosine phosphatase inhibitor activity of estramustine phosphate and its triterpenoid analogs. Bioorg. Bioorg. Med. Chem. Lett. 2011, 21, 730–733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu Z.-H.; Chen L.; Wu L.; Liu S.; Wang L.; Zhang Z.-Y. Small molecule inhibitors of SHP2 tyrosine phosphatase discovered by virtual screening. Bioorg. Med. Chem. Lett. 2011, 21, 4238–4242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu S.; Yu Z.; Yu X.; Huang S.-X.; Luo Y.; Wu L.; Shen W.; Yang Z.; Wang L.; Gunawan A. M.; Chan R. J.; Shen B.; Zhang Z.-Y. SHP2 is a target of the immunosuppressant tautomycetin. Chem. Biol. 2011, 18, 101–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu W.; Yu B.; Xu G.; Xu W. R.; Loh M. L.; Tang L. D.; Qu C. K. Identification of cryptotanshinone as an inhibitor of oncogenic protein tyrosine phosphatase SHP2 (PTPN11). J. Med. Chem. 2013, 56, 7212–7221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu B.; Liu W.; Yu W. M.; Loh M. L.; Alter S.; Guvench O.; Mackerell A. D. Jr.; Tang L. D.; Qu C. K. Targeting protein tyrosine phosphatase SHP2 for the treatment of PTPN11-associated malignancies. Mol. Cancer Ther. 2013, 12, 1738–1748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puius Y. A.; Zhao Y.; Sullivan M.; Lawrence D. S.; Almo S. C.; Zhang Z.-Y. Identification of a second aryl phosphate-binding site in protein-tyrosine phosphatase 1B: A paradigm for inhibitor design. Proc. Natl. Acad. Sci. U. S. A. 1997, 94, 13420–13425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z.-Y. Protein tyrosine phosphatases: structure and function, substrate specificity, and inhibitor development. Annu. Rev. Pharmacol. Toxicol. 2002, 42, 209–234. [DOI] [PubMed] [Google Scholar]
- He R.; Zeng L.-F.; He Y.; Zhang S.; Zhang Z.-Y. Small molecule tools for functional interrogation of protein tyrosine phosphatases. FEBS J. 2013, 280, 731–750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu X.; Sun J.-P.; He Y.; Guo X.-L.; Liu S.; Zhou B.; Hudmon A.; Zhang Z.-Y. Structure, inhibitor, and regulatory mechanism of Lyp, a lymphoid-specific tyrosine phosphatase implicated in autoimmune diseases. Proc. Natl. Acad. Sci. U. S. A. 2007, 104, 19767–19772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou B.; He Y.; Zhang X.; Xu J.; Luo Y.; Wang Y.; Franzblau S. G.; Yang Z.; Chan R. J.; Liu Y.; Zheng J.; Zhang Z.-Y. Targeting Mycobacterium Protein Tyrosine Phosphatase B for Anti-Tuberculosis Agents. Proc. Natl. Acad. Sci. U. S. A. 2010, 107, 4573–4578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng L.-F.; Xu J.; He Y.; He R.; Wu L.; Gunawan A. M.; Zhang Z.-Y. A Facile Hydroxyindole Carboxylic Acid-Based Focused Library Approach for Potent and Selective Inhibitors of Mycobacterium Protein Tyrosine Phosphatase B. ChemMedChem 2013, 8, 904–908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He Y.; Xu J.; Yu Z.-H.; Gunawan A. M.; Wu L.; Wang L.; Zhang Z.-Y. Discovery and Evaluation of Novel Inhibitors of Mycobacterium Protein Tyrosine Phosphatase B from the 6-Hydroxy-Benzofuran-5-Carboxylic Acid Scaffold. J. Med. Chem. 2013, 56, 832–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He Y.; Liu S.; Menon A.; Stanford S.; Oppong E.; Gunawan A. M.; Wu L.; Wu D. J.; Barrios A. M.; Bottini N.; Cato A. C.; Zhang Z.-Y. A potent and selective small molecular inhibitor for the lymphoid-specific tyrosine phosphatase (LYP), a target associated with autoimmune diseases. J. Med. Chem. 2013, 56, 4990–5008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mali R. S.; Ma P.; Zeng L.-F.; Martin H.; Ramdas B.; He Y.; Sims E.; Gosh J.; Nabinger S.; Li S.; Munugalavadla V.; Craig A. W.; Bunting K. D.; Feng G.-S.; Chan R. J.; Zhang Z.-Y.; Kapur R. Role of SHP2 phosphatase in KIT-induced transformation: identification of SHP2 as a druggable target in diseases involving oncogenic KIT. Blood 2012, 120, 2669–2678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J.; Zeng L.-F.; Shen W.; Turchi J. J.; Zhang Z.-Y. Targeting SHP2 for EGFR inhibitor resistant non small cell lung carcinoma. Biochem. Biophys. Res. Commun. 2013, 439, 586–590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barr A. J.; Ugochukwu E.; Lee W. H.; King O. N.; Filippakopoulos P.; Alfano I.; Savitsky P.; Burgess-Brown N. A.; Muller S.; Knapp S. Large-scale structural analysis of the classical human protein tyrosine phosphatome. Cell 2009, 136, 352–363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sordella R.; Bell D. W.; Haber D. A.; Settleman J. Gefitinib-sensitizing EGFR mutations in lung cancer activate anti-apoptotic pathways. Science 2004, 305, 1163–1167. [DOI] [PubMed] [Google Scholar]
- Pao W.; Miller V. A.; Politi K. A.; Riely G. J.; Somwar R.; Zakowski M. F.; Kris M. G.; Varmus H. Acquired resistance of lung adenocarcinomas to gefitinib or erlotinib is associated with a second mutation in the EGFR kinase domain. PLoS Med. 2005, 2, e73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dance M.; Montagner A.; Salles J. P.; Yart A.; Raynal P. The molecular functions of Shp2 in the Ras/Mitogen-activated protein kinase (ERK1/2) pathway. Cell Signalling 2008, 20, 453–459. [DOI] [PubMed] [Google Scholar]
- Ren Y.; Meng S.; Mei L.; Zhao Z. J.; Jove R.; Wu J. (2004) Roles of Gab1 and SHP2 in paxillin tyrosine dephosphorylation and Src activation in response to epidermal growth factor. J. Biol. Chem. 2004, 279, 8497–8505. [DOI] [PubMed] [Google Scholar]
- Yamauchi K.; Milarski K. L.; Saltiel A. R.; Pessin J. E. Protein-tyrosine-phosphatase SHPTP2 is a required positive effector for insulin downstream signaling. Proc. Natl. Acad. Sci. U. S. A. 1995, 92, 664–668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marquardt B.; Frith D.; Stabel S. Signalling from TPA to MAP kinase requires protein kinase C, raf and MEK: reconstitution of the signalling pathway in vitro. Oncogene 1994, 9, 3213–3218. [PubMed] [Google Scholar]
- Ueda Y.; Hirai S.; Osada S.; Suzuki A.; Mizuno K.; Ohno S. Protein kinase C activates the MEK-ERK pathway in a manner independent of Ras and dependent of Raf. J. Biol. Chem. 1996, 271, 23512–23519. [DOI] [PubMed] [Google Scholar]
- Weigelt B.; Lo A. T.; Park C. C.; Gray J. W.; Bissell M. J. HER2 signaling pathway activation and response of breast cancer cells to HER2-targeting agents is dependent strongly on the 3D microenvironment. Breast Cancer Res. Treat. 2010, 122, 35–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muthuswamy S. K.; Xue B. Cell polarity as a regulator of cancer cell behavior plasticity. Annu. Rev. Cell Dev. Biol. 2012, 28, 599–625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirota S.; Isozaki K.; Moriyama Y.; Hashimoto K.; Nishida T.; Ishiguro S.; Kawano K.; Hanada M.; Kurata A.; Takeda M.; Muhammad Tunio G.; Matsuzawa Y.; Kanakura Y.; Shinomura Y.; Kitamura Y. Gain-of-function mutations of c-kit in human gastrointestinal stromal tumors. Science 1998, 279, 577–580. [DOI] [PubMed] [Google Scholar]
- Longley B. J.; Reguera M. J.; Ma Y. Classes of c-KIT activating mutations: proposed mechanisms of action and implications for disease classification and therapy. Leuk. Res. 2001, 25, 571–576. [DOI] [PubMed] [Google Scholar]
- Minor W.; Cymborowski M.; Otwinowski Z.; Chruszcz M. HKL-3000: the integration of data reduction and structure solution–from diffraction images to an initial model in minutes. Acta. Crystallogr., Sect. D: Biol. Crystallogr. 2006, 62, 859–866. [DOI] [PubMed] [Google Scholar]
- Vagin A.; Teplyakov A. MOLREP: an automated program for molecular replacement. J. Appl. Crystallogr. 1997, 30, 1022–1025. [Google Scholar]
- Brünger A. T.; Adams P. D.; Clore G. M.; DeLano W. L.; Gros P.; Grosse-Kunstleve R. W.; Jiang J. S.; Kuszewski J.; Nilges M.; Pannu N. S.; Read R. J.; Rice L. M.; Simonson T.; Warren G. L. Crystallography and NMR system: a new software suite for macro-molecular structure determination. Acta Crystallogr., Sect. D: Biol. Crystallogr. 1998, 54, 905–921. [DOI] [PubMed] [Google Scholar]
- Brünger A. T. The free R value: a novel statistical quantity for assessing the accuracy of crystal structures. Nature 1992, 355, 472–475. [DOI] [PubMed] [Google Scholar]
- Emsley P.; Lohkamp B.; Scott W.; Cowtan K. Features and Development of Coot. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2010, 66, 486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris G. M.; Huey R.; Lindstrom W.; Sanner M. F.; Belew R. K.; Goodsell D. S.; Olson A. J. AutoDock4 and AutoDockTools4: Automated Docking with Selective Receptor Flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanner M. F. Python: A programming language for software integration and development. Journal Mol. Graphics Modell. 1999, 17, 57–61. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.