

Abstract
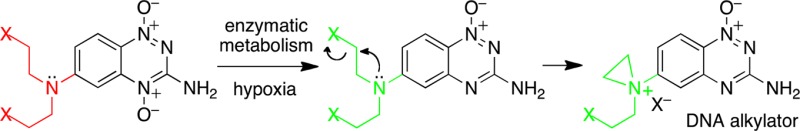
Tirapazamine (3-amino-1,2,4-benzotriazine 1,4-dioxide) is a heterocyclic di-N-oxide that undergoes enzymatic deoxygenation selectively in the oxygen-poor (hypoxic) cells found in solid tumors to generate a mono-N-oxide metabolite. This work explored the idea that the electronic changes resulting from the metabolic deoxygenation of tirapazamine analogues might be exploited to activate a DNA-alkylating species selectively in hypoxic tissue. Toward this end, tirapazamine analogues bearing nitrogen mustard units were prepared. In the case of the tirapazamine analogue 18a bearing a nitrogen mustard unit at the 6-position, it was found that removal of the 4-oxide from the parent di-N-oxide to generate the mono-N-oxide analogue 17a did indeed cause a substantial increase in reactivity of the mustard unit, as measured by hydrolysis rates and DNA-alkylation yields. Hammett sigma values were measured to quantitatively assess the magnitude of the electronic changes induced by metabolic deoxygenation of the 3-amino-1,2,4-benzotriazine 1,4-dioxide heterocycle. The results provide evidence that the 1,2,4-benzotiazine 1,4-dioxide unit can serve as an oxygen-sensing prodrug platform for the selective unmasking of bioactive agents in hypoxic cells.
Introduction
The nitrogen mustard
mechlorethamine (1), developed
in the 1940s, was the first cancer chemotherapeutic agent.1,2 Analogues such as chlorambucil (2), melphalan (3), bendamustine (4), estramustine (5), uramustine (6), cyclophosphamide (7),
and ifosfamide (8) see widespread clinical use today.3,4 Nitrogen mustards generate aziridinium ions that alkylate DNA at
a variety of positions including N7-guanosine, N3-adenosine, N1-adenosine,
and N3-cytosine.3,5−12 The predominant site of DNA alkylation by nitrogen mustards is the
N7-atom of guanine residues.6,8,9
Nitrogen mustards cause serious side effects that arise from the alkylation of DNA and other biomolecules in nonmalignant tissue.4 Consequently, there have been many efforts to develop “targeted” nitrogen mustards with improved potency and selectivity against cancer cells.13−21 One promising approach for the design of cancer-cell-selective mustards exploits selective enzymatic reduction of nitroaryl compounds in the oxygen-starved (hypoxic) cells found in solid tumors.22−32 One-electron reductases such as NADPH:cytochrome P450 reductase, cytochrome b5 reductase, xanthine oxidase, and aldehyde oxidase can convert nitroaryl compounds to nitroso, hydroxylamino, and amino metabolites selectively under hypoxic conditions (Scheme 1).22−32 In normal tissue, O2 blocks production of reduced metabolites via oxidation of the radical anion intermediates involved in this process (reverse reactions, Scheme 1).28,33,34 The initial nitro-to-nitroso conversion typically is a key oxygen-sensitive step in the bioreduction of nitroaryl compounds,26−28 but there is also evidence that the hydroxylamino-to-aniline step can be inhibited by O2.22
Scheme 1.

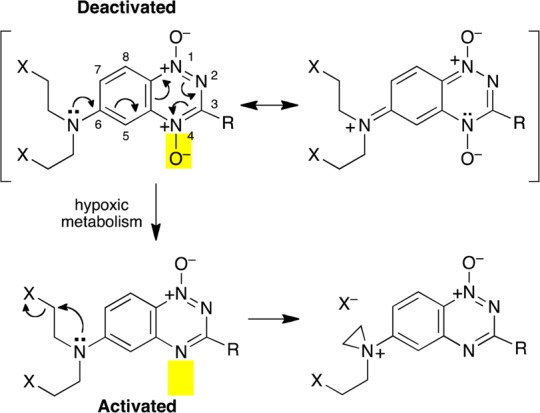
It is well established that aziridinum ion formation by N-aryl nitrogen mustards is suppressed by electron-withdrawing substituents and favored by electron-releasing substituents on the aromatic ring.35,36 Thus, hypoxia-selective conversion of the electron-withdrawing nitro substituent (Hammett σ = 0.78) to the electron-donating hydroxylamino (σ = −0.34) or amino substituents (σ = −0.66) constitutes an “electronic switch” that can transform a deactivated N-aryl nitrogen mustard into an activated nitrogen mustard selectively in tumor tissue (Scheme 2).28,37 Two anticancer drug candidates, TH-302 and PR-104, that employ this design principle are currently undergoing phase I and II clinical trials.38,39
Scheme 2.

In the pursuit of clinically useful hypoxia-selective DNA cross-linking agents, it may be important to explore the utility of oxygen-sensing units other than the nitroaryl motif. In this regard, the 1,2,4-benzotriazine 1,4-dioxide scaffold deserves consideration. In terms of both basic and clinical research, the compound 3-amino-1,2,4-benzotriazine 1,4-dioxide (tirapazamine, 9) may be the best-characterized hypoxia-selective antitumor agent.40−43 On the basis of its potent hypoxia-selective cytotoxicity in preclinical testing,44−46 this compound was examined in a wide variety of phase I, II, and III clinical trials.40−43 In early clinical trials, the drug showed promise47,48 that was not realized in subsequent studies.43 The disappointing clinical performance of 9 may stem, in part, from failure to stratify patients on the basis of the hypoxic character of their tumors.49 In addition, pharmacokinetic models suggested that 9 may be metabolized in a small zone of hypoxic tumor cells, leaving a significant fraction of neighboring cancer cells untouched by the drug.49 Second-generation analogues of 9 are in development.49
Rational use of the 1,2,4-benzotriazine 1,4-dioxides as components in the design of hypoxia-selective DNA-alkylating agents rests upon an existing understanding of the mechanisms by which these agents selectively kill hypoxic cells. Intracellular one-electron reductases convert 9 to an oxygen-sensitive radical intermediate 10 (Scheme 3).50,51 We have presented evidence that, under hypoxic conditions, the protonated drug radical 11 fragments to release the DNA-damaging agent, hydroxyl radical,52−57 though other mechanisms also have been considered.58−62 The deoxygenation product, 1,2,4-benzotriazine 1-oxide 12, is the major metabolite generated by hypoxic metabolism of 9,63−65 and other 1,2,4-benzotriazine 1,4-dioxides similarly are converted to the 1-oxide products.53,66 Compound 9 is not extensively metabolized to the mono-N-oxide 12 by oxygen-insensitive pathways involving two-electron reductases such as DT-diaphorase.67,68 The mono-N-oxide metabolite of tirapazamine is not cytotoxic on its own,45,69 although it does display useful oxygen mimetic (radiosensitizing) properties that potentiate the DNA strand-cleaving properties of hydroxyl radical.70−73
Scheme 3.

We envisioned that electronic changes resulting from the deoxygenation of tirapazamine analogues might be exploited to activate DNA-alkylating functional groups selectively in hypoxic tissue. Indeed, the charge distribution in the mono-N-oxide metabolite 12 is quite different than that of the parent di-N-oxide 9.57 We describe the design, synthesis, and characterization of hypoxia-selective DNA-alkylating agents constructed by grafting nitrogen mustard units onto the 1,2,4-benzotriazine 1,4-dioxide scaffold (Scheme 4).
Scheme 4.
Results and Discussion
Synthesis of 1,2,4-Benzotriazine 1,4-Dioxide Mustards
Our synthetic approaches to the desired tirapazamine-mustard
derivatives
were informed by previous reports describing the synthesis of tirapazamine
and its analogues.74,75 The compound 6-fluoro-1,2,4-benzotriazine
1-oxide 13 was prepared via condensation of 5-fluoro-2-nitroaniline
with cyanamide.74,75 Oxidation of 13 with
trifluoroacetic acid/80% H2O2 gave the di-N-oxide 14 in 43% yield (Scheme 5). Treatment with diethanolamine in acetonitrile afforded
a 73% yield of 15. Attempts to convert 15 to the tosyl mustard 17b via treatment with tosyl chloride
gave a complex mixture of products, from which we isolated 19 as a major component. The structure of 19 was confirmed
by single-crystal X-ray crystallographic analysis (Supplementary Figure S1). This product may arise via the initial
attack of the 4-oxide unit of 15 on tosyl chloride, followed
by a nucleophilic deoxygenative rearrangement.76,77 As an alternate route to the target mustards, 13 was
treated with diethanolamine in N-methylpyrrolidone
to give the mono-N-oxide 16 (94%). This
product was converted in good yields to the ditosylate 17b by treatment with tosyl chloride or to the dimesylate 17a by treatment with mesyl chloride. Oxidation to the di-N-oxides 18 was effected by oxone or m-CPBA in modest yields (15–37%). The chlorinated mustard 17c was synthesized in 94% yield via treatment of 17b with lithium chloride in DMF, and 18c was prepared
in 30% yield by oxidation with oxone.
Scheme 5. Synthesis of 3-Amino-1,2,4-benzotriazine Mustards.

Reagents and conditions: (a) CF3COOH, 70% H2O2, 50 °C, 43%; (b) HN(CH2CH2OH)2, CH3CN, 73%; (c) HN(CH2CH2OH)2, NMP, 100 °C, 94%; (d) mesyl chloride, TEA, DMF, 0 °C, 72%; (e) tosyl chloride, NaOH, THF/H2O, 0 °C, 74%; (f) NaCl, DMF, 110 °C, 94%; (g) oxone, NaHCO3, MeOH/H2O, 50 °C, 15-30%; (h) m-CPBA, THF, 37%.
To investigate how the 3-amino substituent and the location of the mustard unit on the benzotriazine ring system affect reactivity of an appended nitrogen mustard unit, we sought to prepare the 3-desamino tirapazamine analogues 21, 22, 25, and 26. Toward this end, 13 was deaminated by treatment with tert-butylnitrite in dimethylformamide to give 20 (50% yield, Scheme 6).66,78 Nucleophilic aromatic substitution with diethanolamine, followed by treatment with tosyl chloride, gave the mono-N-oxide mustard 21 in 67% yield for the two steps. Oxidation of 21 with oxone gave the di-N-oxide mustard 22 (10%). Synthesis of compounds with the mustard located on the 7-position of the benzotriazine ring system started with preparation of 7-fluoro-1,2,4-benzotriazine 1-oxide 23 via condensation of 6-fluoro-2-nitroaniline with cyanamide (Scheme 7).74,75 Treatment with tert-butylnitrite in dimethylformamide gave the deaminated analogue 24 in 50% yield. Subsequent reaction of this compound with diethanolamine, followed by tosyl chloride, gave a 57% yield of 25. Finally, oxidation with either trifluoroacetic anhydride/70% H2O2 in CH2Cl2 or oxone gave the target di-N-oxide 26 in low (1%) yield. Formation of the desired compound 26, in this case, was accompanied by extensive degradation during the reaction and workup.
Scheme 6. Synthesis of 1,2,4-Benzotriazine 6-Mustards.

Reagents and conditions: (a) t-BuNO2, DMF, 60 °C, 50%; (b) HN(CH2CH2OH)2, CH3CN; (c) TsCl, NaOH, THF 0 °C, 61% from 2 steps; (d) Oxone, NaHCO3, MeOH, H2O, 60 °C, 10%.
Scheme 7. Synthesis of 1,2,4-Benzotriazine 7-Mustards.

Reagents and conditions: (a) t-BuNO2, DMF, 60 °C, 50%; (b) HN(CH2CH2OH)2, CH3CN; (c) TsCl, NaOH, THF 0 °C, 57% from 2 steps; (d) TFA, 70% H2O2, CH2Cl2, 0 °C, 1%.
Reactivity of the 1,2,4-Benzotriazine Oxide Mustards
This work explores the idea that mustard units in the parent di-N-oxides will be relatively unreactive, while the mustard units in the mono-N-oxide deoxygenation products will be active alkylating agents. To learn whether the mustard units in the mono-N-oxides 17, 21, and 25 are in fact more reactive than those in the corresponding di-N-oxides (18, 22, 26) as illustrated in Scheme 4, we first examined the hydrolytic stability of these agents in an organic/aqueous solvent mixture. Hydrolysis of aromatic nitrogen mustards typically proceeds via a rate-determining formation of the aziridinium ion intermediate.5,36 We used 1H NMR to monitor the hydrolysis of these mustard derivatives (∼1 mM) in 50:50 CD3CN/D2O at 50 °C. This mixed organic/aqueous solvent system ensured solubility of the compounds at the concentrations required for the NMR experiments and also served to slow aziridinium ion formation and hydrolysis to easily measurable rates.79 The disappearance of the compounds followed first-order kinetics (Figure 1). The mesylate mustard analogue of tirapazamine 18a disappeared with a half-life of 15.4 ± 0.6 d (Table 1), while the corresponding mono-N-oxide mustard 17a disappeared approximately 5-fold faster, with a half-life of 2.7 ± 0.4 d. The tosylate mustards displayed a similar trend, in which the di-N-oxide 18b was more stable (t1/2 = 15.4 ± 0.6 d) than the mono-N-oxide 17b (t1/2 = 5.5 ± 0.6 d). The similar reactivities of the tosyl (17b/18b) and mesyl (17a/18a) systems is consistent with previous analyses showing that tosylate and mesylate are comparable leaving groups in aqueous/organic solvent mixtures.80 The chlorinated mustard analogues 17c and 18c were quite stable, with 17c displaying an estimated half-life of more than 51 d under the conditions of our NMR experiment (85% of the starting material remained after 10 d). Compound 18c showed no detectable reaction over the course of 10 d. Due to the sluggishness of these reactions we did not examine the hydrolysis of 17c and 18c further.
Figure 1.
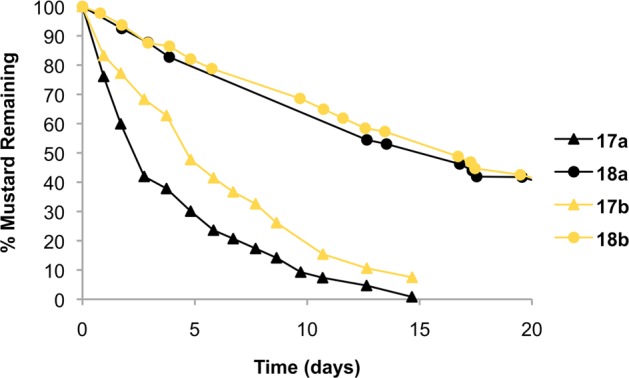
Rate of decay for compounds 17a, 17b, 18a, and 18b in 1:1 acetonitrile-d6/D2O measured by 1H NMR at 50 °C.
Table 1. Decay Rates of 1,2,4-Benzotriazine Mustards in Acetonitrile-d6/D2O (1:1) at 50 °C Measured by 1H NMR.
| compd | R1 | R2 | R3 | oxidation | k (d–1) |
|---|---|---|---|---|---|
| 17a | H2 | N(CH2CH2OMs)2 | H | 1-oxide | 0.259 ± 0.060 |
| 18a | NH2 | N(CH2CH2OMs)2 | H | 1,4-dioxide | 0.045 ± 0.002 |
| 17b | NH2 | N(CH2CH2OTs)2 | H | 1-oxide | 0.127 ± 0.014 |
| 18b | NH2 | N(CH2CH2OTs)2 | H | 1,4-dioxide | 0.045 ± 0.002 |
| 21 | H | N(CH2CH2OTs)2 | H | 1-oxide | 0.050 ± 0.004 |
| 22 | H | N(CH2CH2OTs)2 | H | 1,4-dioxide | 0.055 ± 0.006 |
| 25 | H | H | N(CH2CH2OTs)2 | 1-oxide | 0.107 ± 0.013 |
| 26 | H | H | N(CH2CH2OTs)2 | 1,4-dioxide | 0.106 ± 0.001 |
The products resulting from decomposition of the nitrogen mustards in these NMR experiments were identified as the expected diol (16) and the morpholino compound (27), presumably derived from intramolecular attack of the hydroxyethyl arm of the half-mustard intermediates (28) on the adjacent mustard group. Analogous metabolites have been observed for the clinically used mustard 2.81

The desamino analogues 21 and 22, bearing a tosyl mustard at the 6-position, disappeared with half-lives that were the same within experimental error (Table 1). The half-lives of these compounds (t1/2 ≈ 13 d) were very similar to that of the deactivated dioxide tosyl mustard 18b in the 3-amino series. The desamino analogues 25 and 26, bearing the tosyl mustard unit on the 7-position, also disappeared at the same rate within experimental error (Table 1). The decomposition rates of the 7-mustards 25 and 26 (t1/2 ≈ 6.5 d) was approximately two times faster than the decomposition of the desamino 6-mustards 21 and 22 (t1/2 ≈ 13 d). In the Conclusion, we consider possible reasons why there is no significant difference between the reactivities of the desamino mono-N-oxide and di-N-oxide mustards.
We also measured decomposition of the mesyl mustards 17a and 18a (250 μM) at 50 °C in a predominantly aqueous solvent mixture composed of sodium phosphate buffer (25 mM, pH 7) containing DMF (2.5% v/v). We used HPLC analysis to monitor the disappearance of the starting mustards (17a and 18a). As expected, the hydrolysis rates of 17a and 18a were substantially faster in this solvent as compared to the 50:50 CD3CN/D2O mixture employed in the NMR experiments.79 Compounds 17a and 18a disappeared with half-lives of 12 ± 0.5 h and 96 ± 21 h, respectively, in phosphate-buffered water. Under these conditions, the “activated” mono-N-oxide metabolite 17a reacted approximately seven times faster than the parent dioxide 18a. Comparison with authentic synthetic standards revealed that the major products generated in the decomposition of 17a were the diol 16 and the morpholino 27, both presumably formed via the half-mustard 28 (Supplementary Figure S2).
Measurement of the Electron-Withdrawing Power of the Triazine Ring Systems in 17 and 18
We felt it would be useful to quantitatively measure the electron-withdrawing power of the triazine ring systems in 17 and 18. Electronic effects exerted by various substituents on an aromatic ring typically are assessed using Hammett sigma (σ) constants.82 Hammett σ values are determined by measuring the affect that a substituent exerts on the acidity of benzoic acid, where σsubstituent = log(Ka(substituted benzoic acid)/Ka(benzoic acid)). Therefore, we prepared the carboxylic acid derivatives 29 and 30 via reaction of guanidine with 3-fluoro-4-nitrobenzoic acid under basic conditions, as shown in Scheme 8.83
Scheme 8. Synthesis of 6-Carboxy-1,2,4-benzotriazines.

Reagents and conditions: (a) guanidine, THF, EtOH 90 °C; (b) KOtBu, THF, 90 °C, 95% after 2 steps; (c) TFA, 70% H2O2, 50 °C, 36%.
We measured the acidity of 29 by monitoring the changes in its UV–vis spectrum as a function of pH (Supplementary Figure S3). From the measured pKa of 2.9, we were able to calculate a σ value of 1.3 for the mono-N-oxide 29. The establishes the 1,2,4-triazine-1-oxide ring as a strongly electron-withdrawing substituent comparable to a p-sulfonyl cyanide group (-SO2CN).82 Unfortunately, we were not able to measure the pKa for the di-N-oxide 30 because the UV–vis spectral changes associated with protonation of the carboxylic acid group were obscured by another process, perhaps involving protonation of the oxygen in the 4-oxide group.
Electronic properties of substituents can also be measured using a σ– parameter that is obtained by determining the effect of substituents on the acidity of phenol.82,84 While the σ– parameter is probably less relevant than σ to the aziridinium ion-forming reactions that are the subject of this work,36 we felt this measurement would nonetheless provide a useful evaluation of the electronic properties of the triazine mono-oxide and dioxide ring systems. Therefore, we synthesized the phenol derivatives 31 and 32 by reaction of the corresponding fluoro compounds with basic hydrogen peroxide, followed by workup with sodium thiosulfate (Scheme 9).85 We then measured the acidity of these compounds by monitoring the changes in their UV–vis spectra as a function of pH (Figure 2). The pKa of 31 was found to be 6.3, and that of 32 was 5.3 (Supplementary Figure S4). The pKa measurements allowed us to calculate σ– values of 1.81 for the 1,2,4-triazine 1-oxide “substituent” in 31 and 2.31 for the 1,2,4-triazine 1,4-dioxide “substituent” in 32. These values indicate that both the triazine mono-N-oxide and the di-N-oxide rings are strongly electron-withdrawing. By way of comparison, the sulfonyl perfluoropropane group (-SO2(CF2)2CF3) has a σ– value of 1.75, and the diazonium group (-N2+) has a σ– value of 3.43.82 Importantly, there is a substantial difference (0.5) between the σ– values of the 1,2,4-triazine 1-oxide and 1,2,4-triazine 1,4-dioxide substituents in 31 and 32. This is similar to the difference between σ– values of the nitro group (1.27) and the acetyl group (0.84).82 Overall, the results validate the idea that transformation of the 1,4-di-N-oxide (9) to the 1-N-oxide (12) is accompanied by a substantial change in electron density at the 6-position of the benzo ring.57
Scheme 9. Synthesis of 6-Hydroxy-1,2,4-benzotriazines.

Reagents and conditions: (a) NaOH, 30% H2O2, H2O/NMP 60 °C, 50%; (b) NaOH, 30% H2O2, H2O/NMP 50 °C, 51%.
Figure 2.
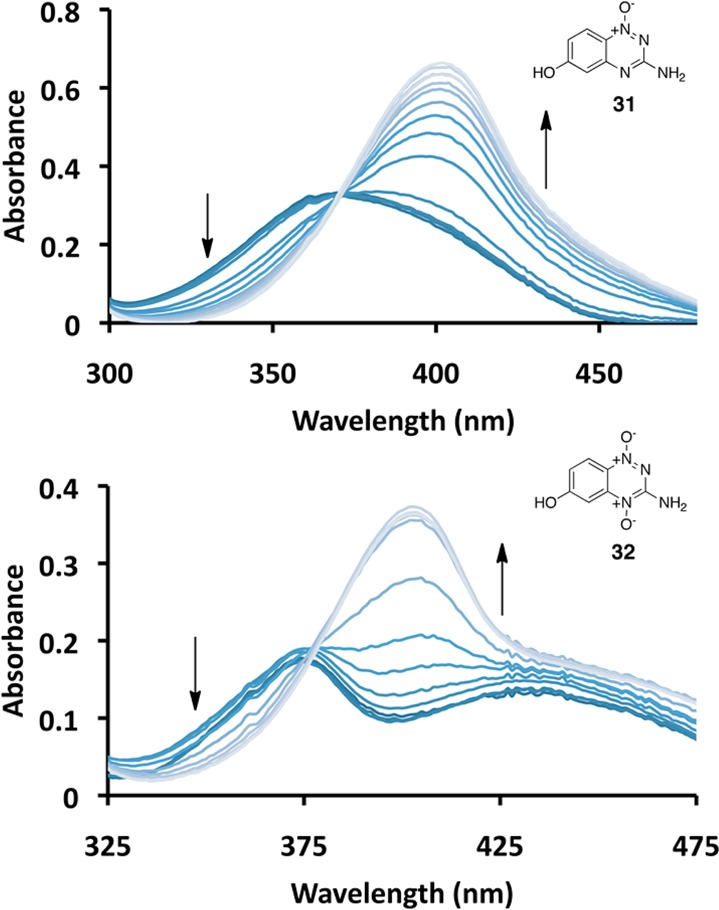
UV–vis spectra of compounds 31 (top) and 32 (bottom) from pH 3.4 to 9.0 (31) and pH 2.4 to 9.0 (32).
DNA Alkylation by 17a
We next examined the abilities of 17a and 18a to alkylate DNA. Nitrogen mustards alkylate DNA primarily at the N7-position of guanine residues, with smaller amounts of reaction also occurring at the N3-position of adenine residues.6,8,9,86 The resulting lesions can be converted to strand breaks by treatment of the DNA with warm piperidine (Maxam–Gilbert workup).87 We incubated mustards 17a and 18a with the 5′-32P-labeled DNA duplex 33 in HEPES buffer (50 mM, pH 7) containing NaCl (100 mM) and DMF (10% v/v), followed by piperidine workup (Figure 3). The resulting labeled DNA fragments were resolved on a 20% denaturing polyacrylamide gel and visualized by phosphorimager analysis. We found that treatment of DNA with the dioxide 18a, followed by piperidine workup, generated little strand cleavage at the guanine residues in the labeled strand of duplex 33. The mono-N-oxide 17a generated significantly higher yields of alkali-labile lesions at both guanine residues in duplex 33. Specifically, compound 17a generated a 30-fold higher yield of alkylation at G2 than 18a (lanes 3 and 4, Figure 3B). The control diol 16 did not induce strand cleavage that was significantly above background (lane 2, Figure 3B). We compared DNA alkylation by 17a to that by the clinically used nitrogen mustard chlorambucil (2, lanes 4 and 5, Figure 3B). Chlorambucil generated piperidine-labile lesions predominantly at the guanine residues in duplex 31, alongside weaker cleavage that may arise from alkylation at adenine residues in the DNA.88
Figure 3.
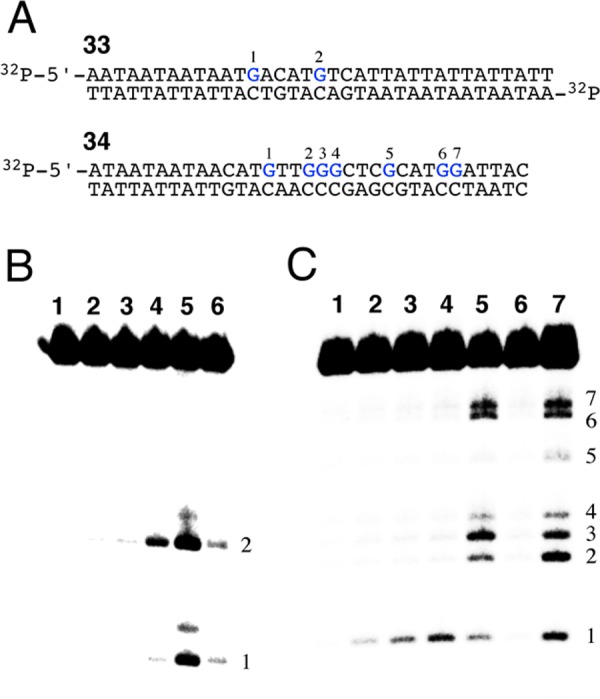
DNA Alkylation by 17a. Conditions: HEPES buffer (50 mM, pH 7), NaCl (100 mM), mustard (1 mM unless otherwise specified), 10% DMF, 37 °C for 3 d followed by piperidine workup (1 M, 95 °C 30 min). Following incubation labeled DNA fragments were resolved on a 20% denaturing polyacrylamide gel. Labeled DNA was visualized by phosphorimager analysis. (A) 32P-labeled duplexes 33 and 34 used to examine DNA-alkylating properties of mustards. (B) Lane 1: duplex 33 (no mustard), Lane 2: duplex 33 with 16, Lane 3: duplex 33 with 18a, Lane 4: duplex 33 with 17a, Lane 5: duplex 33 with 2, Lane 6: G-sequencing of duplex 33. (C) Lane 1: duplex 34 (no mustard), Lanes 2–4: duplex 34 with 17a at 0.25, 0.5, and 1 mM concentrations, Lane 5: duplex 34 with 1 (0.25 mM), Lane 6: duplex 34 with 2 (0.25 mM), and Lane 7: G-sequencing of duplex 34.
Compound 17a showed a marked selectivity for alkylation at G2 of duplex 33 that is distinct from the sequence specificity of 2 (Figure 3B). This observation inspired us to examine the alkylation of duplex 34 containing a larger number of guanine residues embedded in different sequence contexts. Again, compound 17a showed a strong sequence preference for alkylation of a guanine residue residing in a 5′-GT sequence (G1 in duplex 34, Figure 3C). Mechlorethamine (1) displayed relatively low sequence specificity compared to 17a, with 5′-GGG as a favored sequence and 5′-GC as a disfavored sequence, consistent with literature reports (lane 5, Figure 3C).86 Possible origins of the sequence specificity displayed by 17a are discussed in the Conclusions below. These experiments provided evidence that the anticipated hypoxic metabolite 17a is a significantly better DNA-alkylating agent than the parent di-N-oxide 18a.
Hypoxia-Selective, In Vitro Metabolic Conversion of 15 to 16
Successful deployment of the activated mustard 17a requires hypoxia-selective enzymatic reduction of the dioxide 18a. Thus, it was important to examine whether the dialkylamine substituent at the 6-position of the tirapazamine analogue perturbs bioreductive, deoxygenative metabolism of the 1,2,4-benzotriazine 1,4-dioxide “core”. To simplify product analysis in these experiments, we examined the in vitro hypoxic metabolism of an analogue, 15, bearing the chemically stable bis(2-hydroxyethyl)amino substituent. We employed recombinant human NADPH:cytochrome P450 reductase as the bioreductive enzyme because this, or a closely related enzyme, is thought to be responsible for bioactivation of 9 and related compounds in mammalian cells.68,89−91
Compound 15 was incubated with NADPH:cytochrome P450 reductase (1 U/mL) and NADPH (500 μM) in sodium phosphate buffer (25 mM, pH 7) under hypoxic conditions. HPLC analysis of the resulting metabolites revealed conversion of the di-N-oxide 15 to the corresponding mono-N-oxide 16 in approximately 16% yield (Figure 4). A parallel experiment carried out under aerobic conditions produced no detectable yields of 16 (Supplementary Figure S6). A separate control experiment showed that tirapazamine is a better substrate for NADPH:cytochrome P450 reductase than is 15 under our reaction conditions (47% conversion of 9 to the mono-N-oxide versus 16% for 15). Likely this is because the electron-donating bis(2-hydroxyethyl)amino group in 15 decreases the electron-affinity of the 1,2,4-benzotriazine 1,4-dioxide core. Nonetheless, the results provide evidence that 15 can undergo hypoxia-selective metabolism in a manner similar to that of the parent compound tirapazamine (Figure S7).
Figure 4.
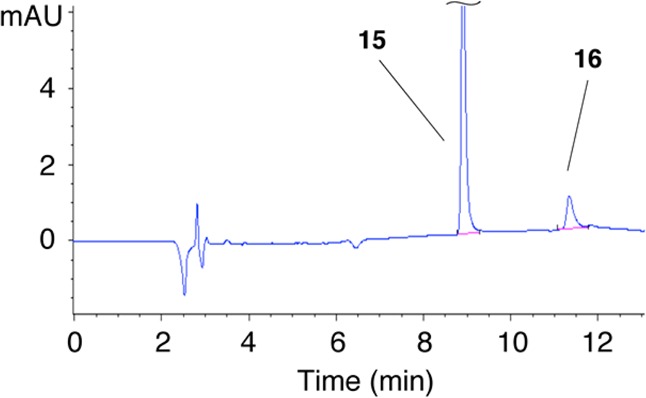
HPLC chromatogram showing the in vitro metabolic conversion of 15 to 16 by NADPH:cytochrome P450 reductase under anaerobic conditions.
Conclusions
In the work described here, we explored the idea that the hypoxia-selective enzymatic reduction of 1,2,4-benzotriazine 1,4-dioxides to the corresponding 1,2,4-benzotriazine 1-oxide metabolites can be exploited for the selective generation of DNA-alkylating species in the oxygen-poor cells found in tumor tissue. In the case of the tirapazamine analogue 18a bearing a nitrogen mustard unit at the 6-position, it was found that removal of the 4-oxide from the parent di-N-oxide to generate the mono-N-oxide analogue 17a does indeed cause a substantial increase in reactivity of the mustard unit, as measured by the rates of mustard hydrolysis. Hammett σ and σ– values measured for the 3-amino-1,2,4-triazine 1-oxide and 3-amino-1,2,4-triazine 1,4-dioxide “substituents” in 17a and 18a, respectively, confirmed that removal of the 4-oxide causes a significant decrease in the electron-withdrawing effects exerted by the triazine ring system on substituents at the 6-position of the benzo ring.
In contrast to the tirapazamine analogues 17 and 18, the reactivities of the mustard units in the desamino series 21, 22, 25, and 26 were unaffected by the presence (or absence) of the 4-oxide group. It is interesting to consider why the reactivities of the mustard groups in the 3-desaminotirapazamine analogues (21, 22, 25, and 26) are not “switched” by deoxygenation. First, it may be noteworthy that the N=N(O) group in 17 and 18 is cross-conjugated with the mustard nitrogen at the 6-position and the 3-amino group. Thus, in these tirapazamine analogues, the 3-amino group may serve to mitigate the electron-withdrawing properties of the N=N(O) group. On the other hand, in analogues 21 and 22 lacking the 3-amino group, the N=N(O) group may exert a strong electron-withdrawing (deactivating) effect on the mustard unit in the 6-position that altogether prevents participation of the nitrogen lone pair in aziridinium ion formation. The hydrolysis rates of 25 and 26 are higher than those of 21 and 22 but still are unaffected by the presence or absence of the 4-oxide unit. Evidently, the N=N(O) unit in the meta position relative to the mustard unit is less electron-withdrawing than when it resides in the para position. In this regard, the N=N(O) group behaves similarly to the nitro group. The observation that the reactivity of the mustard unit in the 7-position is not affected by the presence or absence of the 4-oxide unit may be rationalized by the fact that the nitrogen lone pair of the mustard substituent in this location is not “through conjugated” with the 4-oxide unit.
The DNA-alkylating properties of the nitrogen mustard unit in the mono-N-oxide 17a are “switched on” compared to those of the parent di-N-oxide 18a. Under our assay conditions, the activated analogue 17a generates approximately 30-fold greater yields of guanine alkylation than does 18a. The mono-N-oxide 17a displays a striking and unexpected preference for reaction at guanine residues located in 5′-GT sequences. In contrast, typical mustards such as mechlorethamine (1) alkylate guanine residues with modest sequence selectivity (lane 5 of Figure 3C).86 We speculate that the sequence specificity of 17a arises via formation of Hoogsteen-type hydrogen bonds between the 3-amino-1,2,4-benzotriazine ring system and the adenine residue in the target 5′-GT/5′-AC sequence. Such an interaction could deliver the mustard unit of 17a to the N7-atom of the guanine residue in the major groove of the duplex (Figure 5). A similar scenario has been proposed to explain the unusual 5′-GC sequence specificity for uramustine 6.86 Finally, we provided evidence that the dialkylamino substituent on the 6-position of the 3-amino-1,2,4-benzotriazine 1,4-dioxide ring in analogues 18 is compatible with the hypoxia-selective enzymatic deoxygenative metabolism required to deploy the activated mustards such as 17a.
Figure 5.
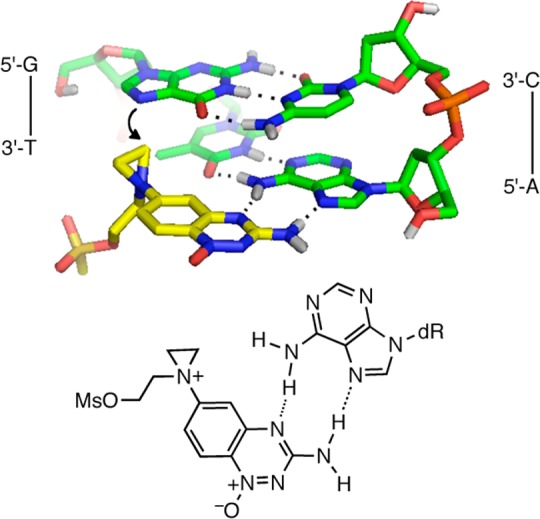
Molecular model depicting Hoogsteen base pairing by 17a with the adenine residue in a 5′-GT/5′-AC sequence.
The compounds described here result from the combination of two well-studied anticancer drugs. The drug tirapazamine has oxygen sensing properties and generates oxidative DNA damage selectively under hypoxic conditions,44−46,52−57 while the nitrogen mustards are clinically used DNA-alkylating agents.3 Our work provides evidence that a judicious union of these two anticancer drug motifs can yield new agents with the potential to deliver both DNA-alkylating and DNA-oxidizing power selectively to oxygen-poor tumor tissue. A potential disadvantage of tirapazamine is that the agent kills only the small subset of tumor cells in which bioreductive metabolism occurs. Addition of a DNA-alkylating unit to the tirapazamine scaffold may yield agents with an ability to diffuse into–and kill–neighboring cells that exist under conditions of both more modest and more severe hypoxia. More generally, our results provide evidence that the 1,2,4-benzotriazine 1,4-dioxide unit can serve as an oxygen-sensing prodrug platform for the selective release or activation of various bioactive agents in hypoxic tissue.
Experimental Section
Materials and Methods
All chemicals were used as purchased. The compound 5-fluoro-2-nitroaniline was purchased from Ak Scientific. The compound 3-fluoro-4-nitrobenzoic acid was purchased from Oakwood Chemical. Human cytochrome P450 reductase was purchased from Sigma-Aldrich. For the kinetic measurements in the NMR, S500 select series NMR tubes from Norell were used. NMR spectra were recorded at 500 MHz for 1H NMR and 125 MHz for 13C NMR. Oligonucleotides were obtained from Integrated DNA Technologies. T4 polynucleotide kinase was purchased from New England Biolabs. [γ-32P]-ATP (6000 Ci/mmol) was purchased from PerkinElmer. Compounds 9, 12, 13, 14, and 23 were prepared according to literature methods.65,74,75,78,92 Mass spectra were recorded using ESI-QTOF MS.
Synthesis of 3-Amino-6-(bis(2-hydroxyethyl)amino)-1,2,4-benzotriazine 1,4-Oxide (15)
Compound 14 (240 mg, 1.3 mmol) was suspended in a mixture of acetonitrile (35 mL), water (1 mL), and diethanolamine (1.4 g, 10 equiv). The suspension was stirred at room temperature for 2 d protected from light. The solvent was removed by rotary evaporation, the orange residue resuspended in cold ethanol, and the solid collected by vacuum filtration. The solid was washed with cold ethanol followed by diethyl ether and dried in an oven overnight at 70 °C to give 15 as an orange powder (270 mg, 73%): mp 210–212 °C dec; 1H NMR (500 MHz, DMSO-d6) δ 7.94 (d, J = 9.8 Hz, 1H), 7.71 (bs, 2H), 7.20 (dd, J = 9.9, 2.7 Hz, 1H), 6.96 (d, J = 2.7, 1H), 4.93 (t, J = 5.1 Hz, 2H), 3.64 (bs, 8H); 13C NMR (125 MHz, DMSO-d6) δ 153.7, 151.8, 140.1, 123.0, 122.8, 116.5, 91.2, 58.2, 53.7; HRMS (ESI) m/z calculated for C11H16N5O4 (M + H+) 282.1197, found 282.1201.
Synthesis of 3-Amino-6-(bis(2-hydroxyethyl)amino)-1,2,4-benzotriazine 1-Oxide (16)
Compound 13 (1.97 g, 11 mmol) and diethanolamine (5.75 g, 5 equiv) were suspended in 1-methyl-2-pyrrolidinone (18 mL) and heated at 100 °C for 18 h. The resulting orange suspension was cooled to room temperature, triturated with cold ethanol (40 mL), and collected by vacuum filtration. The solid was washed with dichloromethane and dried in an oven overnight at 70 °C to give 16 as an orange powder (2.73 g, 94%). (For further purification the solid was suspended in ethanol, briefly heated with stirring, cooled, and collected by vacuum filtration): mp 205–210 °C dec; 1H NMR (500 MHz, DMSO-d6) δ 7.88 (d, J = 9.7 Hz, 1H), 6.99 (dd, J = 9.7, 2.4 Hz, 2H), 6.83 (bs, 2H), 6.41 (d, J = 2.3, 1H), 4.85 (t, J = 5.2 Hz, 2H), 3.60–3.56 (m, 8H); 13C NMR (125 MHz, DMSO-d6) δ 160.6, 153.3, 150.8, 122.0, 120.9, 114.4, 99.7, 58.0, 53.2; HRMS (ESI) m/z calculated for C11H16N5O3 (M + H+) 266.1248, found 266.1252.
Preparation of 3-Amino-6-(bis(2-((methylsulfonyl)oxy)ethyl)amino)-1,2,4-benzotriazine 1-Oxide (17a)
Compound 16 (500 mg, 1.9 mmol) was dissolved in dimethylformamide (4.5 mL), followed by the addition of triethylamine (800 μL, 3 equiv) and cooling in an ice bath. Methanesulfonyl chloride (370 μL, 2.5 equiv) was added dropwise over 15 min as an ice-cold solution in dimethylformamide (0.5 mL), and the resulting mixture was stirred at room temperature for 2.5 h. The solution was poured into 30 mL of cold water with stirring and refrigerated overnight at 4 °C. The resulting orange precipitate was collected by vacuum filtration. The solid was washed with water and diethyl ether and dried in a desiccator to give 17a (570 mg, 72%): 1H NMR (500 MHz, DMSO-d6) δ 7.94 (d, J = 9.6 Hz, 1H), 7.06 (dd, J = 9.7, 1.8 Hz, 2H), 6.98 (bs, 2H), 6.56 (s, 1H), 4.38 (t, J = 5.3 Hz, 4H), 3.90 (t, J = 5.2 Hz, 4H), 3.17 (s, 6H); 13C NMR (125 MHz, DMSO-d6) δ 161.0, 152.8, 151.1, 123.1, 121.6, 114.4, 101.6, 67.4, 49.7, 37.1; HRMS (ESI) m/z calculated for C13H20N5O7S2 (M + H+) 422.0799, found 422.0800.
Preparation of 3-Amino-6-(bis(2-(tosyloxy)ethyl)amino)-1,2,4-benzotriazine 1-Oxide (17b)
Compound 16 (126 mg, 0.5 mmol) was suspended in a biphasic mixture of tetrahydrofuran (2 mL) and NaOH (1.5 mL of a 4 M solution in water) and cooled in an ice bath. To this orange biphasic mixture was added dropwise an ice-cold solution of p-toluenesulfonyl chloride (200 mg, 2.2 equiv) in tetrahydrofuran (3 mL). The resulting suspension was stirred vigorously with cooling in an ice bath for 5 h. During this time the mixture turned yellow. The mixture was then poured into an ice–water slurry (10 mL) and stirred for 20 min to give a bright yellow powder. This solid was collected by vacuum filtration, washed with water and then diethyl ether, and then dried in a desiccator in to give 17b (202 mg, 74%): 1H NMR (500 MHz, DMSO-d6) δ 7.73 (d, J = 9.7 Hz, 1H), 7.60 (d, J = 8.2 Hz, 4H), 7.24 (d, J = 8.1 Hz, 4H), 6.97 (bs, 2H), 6.63 (dd, J = 9.7, 2.5 Hz, 1H), 6.15 (d, J = 7.5 Hz, 1H), 4.13 (t, J = 5.1 Hz, 4H), 3.64 (t, J = 5.1 Hz, 4H), 2.25 (s, 6H); 13C NMR (125 MHz, DMSO) δ 160.6, 151.4, 150,4, 144.9, 131.6, 129.9, 127.4, 122.5, 120.6, 113.7, 101.1, 67.29, 48.9, 20.9. HRMS (ESI) m/z calculated for C25H28N5O7S2 (M + H+) 574.1425, found 574.1433.
Synthesis of 3-Amino-6-(bis(2-chloroethyl)amino)-1,2,4-benzotriazine 1-Oxide (17c)
Compound 17b (200 mg, 0.35 mmol) and LiCl (148 mg, 10 equiv) were dissolved in dimethylformamide (1.5 mL) and heated at 110 °C for 2 h under a N2 atmosphere before being cooled on ice and mixed with ice-cold water (20 mL). The resulting yellow precipitate was collected by vacuum filtration, washed with water and diethyl ether, and then dried in a desiccator to give 17c (104 mg, 94% yield): 1H NMR (500 MHz, DMSO-d6) δ 7.95 (d, J = 9.6 Hz, 1H), 7.04 (dd, 9.7, 2.6 Hz, 1H), 6.98 (bs, 2H), 6.48 (d, J = 2.5 Hz, 1H), 3.89 (t, J = 6.6 Hz, 4H), 3.80 (t, J = 6.6 Hz, 4H); 13C NMR (125 MHz, DMSO-d6) δ 160.7 (d, J = 253.90 Hz), 152.1, 150.8, 122.8, 121.4, 113.9, 101.1, 51.8, 40.9; HRMS (ESI) m/z calculated for C11H14Cl2N5O (M + H+) 302.0570, found 302.0576.
Preparation of 3-Amino-6-(bis(2-((methylsulfonyl)oxy)ethyl)amino)-1,2,4-benzotriazine 1,4-Dioxide (18a)
Compound 17a (70 mg, 0.17 mmol) and NaHCO3 (86 mg, 6 equiv) were suspended in methanol (25 mL). Oxone (245 mg, 1.2 equiv) was added, followed by water (10 mL). The mixture was stirred at 50 °C under a N2 atmosphere for 24 h. Thin layer chromatographic analysis indicated that most of the reaction progress occurred within the first 5 h. The reaction was cooled to room temperature, and the white solid was removed by vacuum filtration and washed with CH2Cl2 (10 mL). The filtrate was extracted with CH2Cl2 (2 × 125 mL), dried with anhydrous sodium sulfate, and evaporated under reduced pressure, and the resulting residue was subjected to column chromatography on silica gel eluted with a gradient of 1–5% methanol in CH2Cl2 to give 18a as an orange solid (11 mg, 15%): 1H NMR (500 MHz, DMSO-d6) δ 8.01 (d, J = 9.8 Hz, 1H), 7.82 (bs, 2H), 7.28 (dd, J = 9.9, 2.7 Hz, 2H), 7.09 (d, J = 2.7, 1H), 4.43 (t, J = 5.3 Hz, 4H), 3.97 (t, J = 5.3 Hz, 4H), 3.19 (s, 6H); 13C NMR (125 MHz, DMSO-d6) δ 153.0, 151.9, 140.1, 123.7, 123.2, 116.2, 92.8, 67.2, 49.7, 37.1; HRMS (ESI) m/z calculated for C13H20N5O8S2 (M + H+) 438.0748, found 438.0746.
Preparation of 3-Amino-6-(bis(2-(tosyloxy)ethyl)amino)-1,2,4-benzotriazine 1,4-Dioxide (18b)
Compound 17b (501 mg, 0.9 mmol) was dissolved in tetrahydrofuran (100 mL) and cooled with stirring in an ice bath. To this mixture was added m-CPBA (300 mg, ∼1.5 equiv of a 77% maximum purity material), and the reaction was stirred overnight in an ice bath. An additional portion of m-CPBA was added, and the reaction was stirred at room temperature for an additional 3 d. The solution was concentrated under vacuum and poured into cold water, and the solid was collected by vacuum filtration. The material was purified by column chromatography on silica gel eluted with a gradient of 1–3% methanol in CH2Cl2 to give 18b as a yellow solid (194 mg, 37%): 1H NMR (500 MHz, DMSO-d6) 7.84 (bs, 2H), 7.80 (d, J = 9.8 Hz, 1H), 7.60 (d, J = 8.2 Hz, 4H), 7.22 (d, J = 8.1 Hz, 4H), 6.86 (dd, J = 9.8, 2.7 Hz, 1H), 6.65 (d, J = 2.7 Hz, 1H), 4.19 (t, J = 5.0 Hz, 4H), 3.67 (t, J = 4.9 Hz, 4H), 2.24 (s, 6H); 13C NMR (125 MHz, DMSO-d6) δ 151.6, 151.5, 144.9, 139.3, 131.6, 129.9, 127.4, 123.0, 122.1, 115.6, 92.2, 67.1, 48.9, 20.8. HRMS (ESI) m/z calculated for C25H28N5O8S2 (M + H+) 590.1374, found 590.1371.
Preparation of 3-Amino-6-(bis(2-chloroethyl)amino)-1,2,4-benzotriazine 1,4-Dioxide 18c
Compound 17c (50 mg, 0.17 mmol) and NaHCO3 (42 mg, 3 equiv) were suspended in methanol (25 mL). Oxone (122 mg, 1.2 equiv) was added, followed by water (10 mL). The mixture was stirred at 50 °C under a N2 atmosphere. The reaction mixture was filtered while still warm, and the solids were washed with methanol (5 mL). The filtrate was cooled to room temperature, diluted with water (50 mL), and extracted methylene chloride (3 × 100 mL). Column chromatography on silica gel eluted with a gradient of 2–6% methanol in CH2Cl2 gave 18c as an orange solid (16 mg, 30%): 1H NMR (500 MHz, DMSO-d6) δ 8.01 (d, J = 9.8, Hz, 1H), 7.81 (bs 2H), 7.27 (dd, J = 9.8, 2.7 Hz, 1H), 7.05 (d, J = 2.7 Hz, 1H), 3.96 (t, J = 6.7 Hz, 4H), 3.85 (t J = 6.7 Hz, 4H); 13C NMR (125 MHz, DMSO-d6) δ 152.3, 151.5, 139.7, 123.4, 123.0, 115.8, 92.2; HRMS (ESI) m/z calculated for C11H14Cl2N5O2 (M + H+) 318.0519, found 318.0519.
Synthesis of 3-Amino-5-tosyl-6-(bis(2-hydroxyethyl)amino)-1,2,4-benzotriazine 1-Oxide (19)
Compound 15 (52 mg, 0.18 mmol) was suspended in a stirred solution of pyridine in an ice bath, and tosyl chloride (172 mg, 5 equiv) was added. The mixture was stirred in an ice bath for 12 h, poured into ice-cold water (20 mL), and extracted with methylene chloride (5 × 25 mL). The combined organic fractions were washed with brine and dried over Na2SO4, and solvent was removed by rotary evaporation. Column chromatography on silica gel eluted with 1% methanol in methylene chloride 19 as an orange solid (6 mg, 10%): 1H NMR (500 MHz, DMSO-d6) δ 7.85 (d, J = 9.8 Hz, 1H), 7.75 (d, J = 8.2 Hz, 2H), 7.739 (d, J = 8.2 Hz, 2H), 7.19 (d, J = 9.8 Hz, 1H), 6.94 (bs 2H), 4.65 (t, J = 5.1 Hz, 2H), 3.47–3.40 (m, 8H); 13C NMR (125 MHz, DMSO-d6) δ 160.1, 148.2, 145.9, 145.5, 132.6, 129.5, 128.8, 126.6, 123.6, 118.8, 118.7, 58.4, 54.3, 21.3; HRMS (ESI) m/z calculated for C18H22N5O6S (M + H+) 436.1285, found 436.1293. Crystals suitable for X-ray crystallography were prepared by vapor diffusion with ethyl acetate and hexane. The crystal structure and crystallographic data for 19 are shown in Supplementary Figure S1 and Table S1, respectively.
Synthesis of Desamino-tirapazamine Derivatives 6-Fluoro-1,2,4-benzotriazine 1-Oxide (20) and 7-Fluoro-1,2,4-benzotriazine 1-Oxide (24)
Following the general procedure of Boyd et al.9323 (930 mg, 5 mmol) was dissolved in anhydrous dimethylformamide (50 mL), and the mixture was degassed by bubbling argon through the solution for 30 min. To this mixture was added tert-butyl nitrite (3.5 mL, 5 equiv, 90%) by syringe, and the mixture was heated in a 60 °C oil bath for 2 h under an atmosphere of argon gas. The reaction was cooled, and the solvent was removed under vacuum. The resulting dark residue was taken up in ethyl acetate (300 mL), mixed with brine (150 mL), and stirred vigorously. The phases were allowed to separate, the organic layer was washed with brine (2 × 150 mL) and dried over anhydrous sodium sulfate, and the solvent was removed by rotary evaporation. Column chromatography on silica gel eluted with a gradient of 0–10% ethyl acetate in CH2Cl2 gave 24 as a pale white solid (438 mg, 50%): mp 127–129 °C; 1H NMR (500 MHz, DMSO-d6) δ 9.20 (s, 1H), 8.24 (dd, J = 9.3, 5.3 Hz, 1H), 8.20 (dd, J = 8.4, 2.8 Hz, 1H), 8.08 (m, 1H); 13C NMR (125 MHz, DMSO-d6) δ 162.6 (d, J = 253.9), 154.0 (d, J = 2.6 Hz), 144.9, 136.0, 132.5 (d, J = 9.4 Hz), 126.7 (d, J = 25.9 Hz), 105.2 (d, J = 29.3 Hz); HRMS (ESI) m/z calculated for C7H5FN3O (M + H+) 166.0411, found 166.0419. Compound 20 was prepared from 13 (980 mg) by the same method (447 mg, 50%): mp 160–162 °C; 1H NMR (500 MHz, DMSO-d6) δ 9.19 (s, 1H), 8.50 (dd, J = 9.6, 5.5 Hz, 1H), 7.99 (dd, J = 9.1, 2.7 Hz, 1H), 7.82 (m, 1H); 13C NMR (125 MHz, DMSO-d6) δ 166.1 (d, J = 256.7), 155.5, 149.3 (d, J = 15.1 Hz), 133.1, 123.8 (d, J = 11.2 Hz), 121.8 (d, J = 26.6 Hz), 113.3 (d, J = 22.7 Hz); HRMS (ESI) m/z calculated for C7H5FN3O (M + H+) 166.0411, found 166.0417.
Synthesis of 6-(Bis(2-(tosyloxy)ethyl)amino))-1,2,4-benzotriazine 1-Oxide (21) and 7-(Bis(2-(tosyloxy)ethyl)amino)benzo[e][1,2,4]triazine 1-Oxide (25)
Compound 20 (361 mg, 2.2 mmol) and diethanolamine (500 mg, 2.5 equiv) were dissolved in acetonitrile (7 mL), and the mixture was heated in an 80 °C oil bath overnight. The reaction mixture was cooled, and solvent was removed under reduced pressure by rotary evaporation. The resulting solid was suspended in ethanol (10 mL), briefly heated to 80 °C with stirring, and then cooled to 0 °C, and the resulting precipitate was collected by vacuum filtration. The orange solid was washed with cold ethanol and diethyl ether and dried in an oven overnight at 70 °C. The resulting solid (200 mg) was dissolved in a biphasic mixture of tetrahydrofuran (9 mL) and NaOH (2.4 mL of a 4 M solution in water) and cooled in an ice bath. To this orange biphasic mixture was added dropwise an ice-cold solution of p-toluenesulfonyl chloride (455 mg, 3 equiv) in tetrahydrofuran (1.5 mL). The resulting mixture was stirred vigorously in an ice bath for 1.5 h. During this time the mixture turned yellow. The mixture was then poured into an ice–water slurry (100 mL) and stirred for 30 min to give a yellow precipitate. The solid was collected by vacuum filtration, then washed with water and diethyl ether, and dried in a desiccator to give 21 (356 mg, 61% overall yield over two steps): 1H NMR (500 MHz, DMSO-d6) δ 8.87 (s, 1H), 7.96 (d, J = 9.2 Hz, 1H), 7.57 (d, J = 8.3 Hz, 4H), 7.19 (d, J = 8.0, 4H), 7.14 (dd, J = 9.8, 2.6 Hz, 1H), 6.64 (d, J = 2.7 Hz, 1H), 4.18 (t, J = 5.1 Hz, 4H), 3.71 (t, J = 5.1 Hz, 4H), 2.20 (s, 6H); 13C NMR (500 MHz, DMSO-d6) δ 154.8, 152.1, 149.2, 145.2, 131.9, 130.2, 127.8, 127.4, 120.7, 120.3, 103.7, 67.6, 49.3, 21.2. HRMS (ESI) m/z calculated for C25H27N4O7S2 (M + H+) 559.1316, found 559.1320. Compound 25 was prepared in from 24 by heating for 4 days using the same conditions (390 mg, 57% overall yield over two steps): 1H NMR (500 MHz, DMSO-d6) δ 8.89 (s, 1H), 7.73 (d, J = 9.4 Hz, 1H), 7.56 (d, J = 8.2 Hz, 4H), 7.44 (dd, J = 9.5, 2.8, 1H), 7.16 (d, J = 8.1 Hz, 4H), 6.86 (d, J = 2.8 Hz, 1H), 4.19 (t, J = 5.0 Hz, 4H), 3.69 (t, J = 5.0 Hz, 4H), 2.17 (s, 6H); 13C NMR (125 MHz, DMSO-d6) δ 150.2, 148.6, 145.2, 140.7, 135.8, 132.0, 130.1, 129.5, 127.7, 125.0, 94.8, 67.5, 49.1, 21.2. HRMS (ESI) m/z calculated for C25H27N4O7S2 (M + H+) 559.1316, found 559.1317.
Preparation of 6-(Bis(2-(tosyloxy)ethyl)amino))-1,2,4-benzotriazine 1,4-Dioxide (22)
Compound 21 (20 mg, 0.04 mmol) and NaHCO3 (30 mg, 10 equiv) were suspended in methanol (6.25 mL). Oxone (110 mg, 5 equiv) was added, followed by water (2.5 mL), and the mixture was stirred at 50 °C under an atmosphere of nitrogen gas for 16 h. The reaction was cooled to room temperature and extracted with methylene chloride (5 × 10 mL). The organic layers were combined, washed with brine, and dried over anhydrous sodium sulfate. Column chromatography on silica gel eluted with 1% MeOH in CH2Cl2 gave compound 22 as an red-orange solid (2 mg, 10%) with 50% recovery of starting materials: 1H NMR (500 MHz, DMSO-d6) δ 9.15 (s, 1H), 7.96 (d, J = 9.8 Hz, 1H), 7.57 (d, J = 8.2 Hz, 4H), 7.25–7.18 (m, 5H), 6.84 (d, J = 2.8 Hz, 1H), 4.21 (t, J = 5.0 Hz, 4H), 3.70 (t, J = 4.9 Hz, 4H), 2.25 (s, 6H); 13C NMR (125 MHz, DMSO-d6) δ 152.0, 145.3, 142.5, 140.7, 132.0, 130.2, 127.8, 127.3, 122.4, 120.9, 84.6, 67.4, 49.1, 21.2. HRMS (ESI) m/z calculated for C25H27N4O8S2 (M + H+) 575.1265, found 575.1263.
Synthesis of 7-(Bis(2-(tosyloxy)ethyl)amino)-1,2,4-benzotriazine 1,4-Dioxide (26)
Using a procedure adapted from Pchalek and Hay,94 trifluoroacetic anhydride (300 μL) and methylene chloride (1.5 mL) were mixed with stirring in an ice bath, and 70% H2O2 (105 μL) was added dropwise. The mixture was stirred for 10 min and then allowed to warm to room temperature. This solution was cooled in an ice bath and slowly added to an ice-cold solution of 25 (110 mg, 0.2 mmol) in methylene chloride (10 mL). The reaction was stirred in an ice bath for 30 min before being diluted with methylene chloride (100 mL) and washed with cold water, cold NaHCO3 (saturated), and then brine. The organic layer was dried over anhydrous sodium sulfate, and column chromatography on silica gel eluted with 0.5% MeOH in CH2Cl2 gave 26 as a red solid (1 mg, 1%): 1H NMR (500 MHz, DMSO-d6) δ 9.06 (s, 1H), 7.96 (d, J = 9.7 Hz, 1H), 7.57 (d, J = 8.3 Hz, 4H), 7.41 (dd, J = 9.7, 2.7, 1H), 7.22 (d, J = 8.0 Hz, 4H), 6.85 (d, J = 2.6 Hz, 1H), 4.19 (t, J = 5.0 Hz, 4H), 3.67 (t, J = 4.9 Hz, 4H), 2.25 (s, 6H); 13C NMR (125 MHz, DMSO-d6) δ 149.3, 144.9, 138.8, 135.6, 131.8, 131.7, 129.9, 127.4, 123.2, 119.5, 96.1, 67.1, 48.6, 20.9. HRMS (ESI) m/z calculated for C25H27N4O8S2 (M + H+) 575.1265, found 575.1275.
Synthesis of 3-Amino-6-N-morpholino-1,2,4-benzotriazine 1-Oxide (27)
Compound 13 (503 mg, 2.8 mmol) and morpholine (0.72 mL, 3 equiv) were suspended in 1-methyl-2-pyrrolidinone (4 mL) and heated to 100 °C overnight. The resulting orange suspension was cooled to room temperature, diluted with water (40 mL), and filtered. The precipitate was washed with water and diethyl ether and then dried in an oven overnight at 70 °C. The orange-yellow powder was collected by vacuum filtration to give 27 (650 mg, 94%): mp 245–250 °C dec; 1H NMR (500 MHz, DMSO-d6) δ 7.94 (d, J = 9.6 Hz, 1H), 7.16 (dd, J = 9.7, 2.5 Hz, 2H), 7.00 (bs, 2H), 6.60 (d, J = 2.5, 1H), 3.73 (t, J = 4.7 Hz, 4H), 3.38 (t, J = 4.8 Hz, 4H); 13C NMR (125 MHz, DMSO-d6) δ 160.7, 155.3, 150.8, 123.3, 120.9, 115.2, 103.0, 65.8, 46.8; HRMS (ESI) m/z calculated for C11H14N5O2 (M + H+) 248.1142, found 248.1142
Synthesis of 3-Amino-6-carboxy-1,2,4-benzotriazine 1-Oxide (29)
Using a procedure adapted from the method of Ligthart et al.,95 NaOH (528 mg, 10 equiv) was dissolved in hot ethanol (30 mL), guanidine hydrochloride (1.2 g, 10 equiv) was added, and the resulting mixture was stirred for 20 min. The reaction was cooled to room temperature and filtered to remove the white precipitate. To the filtrate were added tetrahydrofuran (20 mL) and 3-fluoro-4-nitrobenzoic acid (230 mg, 1.2 mmol), and the resulting heterogeneous mixture was refluxed for 4 h. Potassium tert-butoxide (150 mg, 10 equiv) was added, and the mixture was stirred at reflux for an additional 2 h. After the reaction was complete, the tetrahydrofuran was decanted of,f and water (60 mL) was added with vigorous stirring. Acidification with HCl (1 M) resulted in precipitation of a yellow solid that was collected by vacuum filtration and washed with dilute HCl, followed by minimal amounts of water and diethyl ether. The solid was then dried in an oven overnight at 70 °C to give 29 (244 mg, 95% yield): mp >280 °C; 1H NMR (500 MHz, DMSO-d6) δ 13.63 (bs, 1H), 8.20 (d, J = 8.9 Hz, 1H), 7.98 (d, J = 1.5 Hz, 1H), 7.74 (dd, J = 2 Hz, 1.6 Hz, 1H), and 7.52 (s, 2H); 13C NMR (125 MHz, DMSO-d6) δ 166.1, 160.6, 148.6, 137.1, 131.7, 127.4, 123.6, 120.7; HRMS (ESI) m/z calculated for C8H7N4O3 (M + H+) 207.0513, found 207.0519.
Synthesis of 3-Amino-6-carboxy-1,2,4-benzotriazine 1,4-Dioxide (30)
Compound 29 (40 mg, 0.2 mmol) was suspended in trifluoroacetic acid (1 mL) and H2O2 (70%, 0.8 mL), and the mixture was stirred at 50 °C for 48 h. After the reaction was complete (as judged by thin layer chromatography), the solvent was removed by rotary evaporation under reduced pressure, and the resulting residue was triturated with ethanol (5 mL). The resulting suspension was chilled, and the red precipitate was collected by vacuum filtration. The solid was washed with cold ethanol and dried under vacuum to provide compound 30 (16 mg, 36%): mp 260 °C dec; 1H NMR (500 MHz, DMSO-d6) δ 13.84 (bs, 1H), 8.61 (s, 1H), 8.28 (d, J = 8.8 Hz, 1H), 8.19 (bs, 2H), and 7.94 (d, J = 8.7 Hz, 1H); 13C NMR (125 MHz, DMSO-d6) δ 165.6, 151.8, 138.3, 136.3, 132.1, 125.8, 122.2, 118.8; HRMS (ESI) m/z calculated for C8H7N4O4 (M + H+) 223.0462, found 223.0467.
Synthesis of 3-Amino-6-hydroxy-1,2,4-benzotriazine 1-Oxide (31)
Using a procedure adapted from Cantrell et al.96 compound 13 (504 mg, 2.8 mmol) was suspended in 1-methyl-2-pyrrolidinone (4 mL), and an aqueous solution of NaOH (400 mg in 20 mL, 4 equiv) was added. To this mixture, a solution of H2O2 (30%, 480 μL, 2 equiv) was added, and the mixture was stirred for 4 h at 60 °C. The reaction was cooled to room temperature and filtered to remove undissolved starting material. Na2S2O3 (700 mg) was added to decompose peroxides. The resulting solution was acidified with HCl (1 M), and the resulting orange-yellow precipitate was collected by vacuum filtration. The solid was then washed with water and diethyl ether and dried in an oven overnight at 70 °C to give 31 (360 mg, 50%): mp 210 °C dec; 1H NMR (500 MHz, DMSO-d6) δ 10.94 (bs, 1H), 8.00 (d, J = 9.3 Hz, 1H), 7.13 (bs, 2H), 6.84 (dd, J = 9.3 Hz, 1.9 Hz, 1H), and 6.68 (d, J = 1.9 Hz, 1H); 13C NMR (125 MHz, DMSO-d6) δ 163.8, 160.6, 151.1, 124.4, 122.0, 117.2, 106.1; HRMS (ESI) m/z calculated for C7H7N4O2 (M + H+) 179.0564, found 179.0566.
Synthesis of 3-Amino-6-hydroxy-1,2,4-benzotriazine 1,4-Dioxide (32)
Compound 14 (160 mg, 0.83 mmol) was suspended in 1-methyl-2-pyrrolidinone (1.2 mL), and NaOH (120 mg, 4 equiv, in 6 mL of water) was added. To this mixture was added H2O2 (30%, 144 μL, 2 equiv), followed by stirring for 1.5 h at 50 °C. The reaction was cooled to room temperature and acidified with HCl (1 M). The resulting precipitate was collected by vacuum filtration and washed with water and diethyl ether. Drying under vacuum gave 32 as a dark red solid (81 mg, 51%): mp 202 °C dec; 1H NMR (500 MHz, DMSO-d6) δ 11.51 (bs, 1H), 8.08 (d, J = 9.5 Hz, 1H), 7.92 (bs, 2H), 7.31 (d, J = 2.5 Hz, 1H), and 7.04 (dd, J = 9.5 Hz, 2.5 Hz, 1H); 13C NMR (125 MHz, DMSO-d6) δ 164.1, 151.5, 140.1, 125.1, 123.7, 119.2, 98.0; HRMS (ESI) m/z calculated for C7H7N4O3 (M + H+) 195.0513, found 195.0516.
Measurement of Mustard Hydrolysis Rates Using 1H NMR
Solutions containing the 1,2,4-benzotriazine nitrogen mustards (1 mM final concentration) were prepared in CD3CN/D2O (1:1) containing disodium maleate (5 mM) as an internal standard. Solutions were placed in a capped S500 Norell NMR tube and warmed in a 50 °C water bath. At various time points the NMR tubes were removed from the heat bath and cooled in a room temperature water bath, and 1H NMR spectra were obtained using a 500 MHz NMR equipped with a 5 mm HCN cryo-probe. The probe temperature was 298 K during the experiment. Total number of scans acquired was 32 with repetition delay of 4.1719923 s to ensure that the integration of resonances in various compounds in the mixture was quantitatively accurate. The amount of unreacted mustard at each time point was assessed by measuring the integration of the CH2 resonances of the starting material in comparison to the CH2 peak of the internal standard at each time point. The pseudo-first-order rates for the hydrolysis reactions were obtained by a least-squares fit to the equation ln A/A0 = −kt.
Measurement of Mustard Hydrolysis Rates and Decomposition Products Using HPLC
Solutions of the mono- or di-N-oxide mustards (250 μM) were prepared in sodium phosphate buffer (25 mM, pH 7) containing 2.5% DMF (v/v). The samples were incubated at 50 °C for 24 h. At various time-points aliquots were removed and frozen at −20 °C for later analysis. The samples were then analyzed by HPLC using a Varian Microsorb-MV C-18 column (100 Å sphere size, 5 μm pore size, 250 mm length, and 2.6 mm i.d. eluted with a gradient mobile phase composed of solvent A (0.5% AcOH in H2O) and solvent B (methanol). For compound 17a a gradient of 25–50% B over 5 min, followed by 50–100% B over 5 min, followed by 100% B for 5 min was used. The mobile phase was returned to 25% B over 5 min and held at 25% B for 5 min postrun. For compound 18a the mobile phase was held at 25% B for 5 min following injection, before increasing to 50% B over 5 min and holding at 50% B for 4 min. The mobile phase was then returned to 25% B over 1 min and held at 25% B for 5 min postrun. The products were detected by monitoring absorbance at 280 nm. The identity of the major hydrolysis products was confirmed by comparison to authentic synthetic standards.
Determination of pKa’s for 29, 31, and 32
UV–vis spectra were taken for compounds 29, 30, 31, and 32 (50 μM) in solutions with pH values from 0 to 9 using HCl solutions and universal buffers prepared as in Britton and Robinson except containing 0.5% DMF (v/v).97 Buffer pH was determined with via pH meter. Absorbance values were measured at 256 and 282 nm for 29, 260 and 270 nm for 30, 345 and 400 nm for 31, and 360 and 400 nm for compound 32. Changes in absorbance were plotted against pH and the pKa values were calculated by fitting the data to the equation: pKa = pH + log(dM – d)/(d – dI), where dM is the absorbance for the un-ionized species (starting absorbance), dI is the absorbance of the ionized species (final absorbance), and d is the absorbance at any point in the pH titration.98 Taking the midpoint of the titration data gave a very similar result. To confirm that pKa values measured reflected protonation of the 6-COOH or 6-OH substituents in compounds 29, 31, and 32, pH-absorbance measurements were performed with 9 and 12 to show that these control compounds displayed no significant changes in absorbance in the pH regions of interest.
Examination of the DNA-Alkylating Properties of 17a and 18a
The 2′-deoxyoligonucleotides (33 and 34, Figure 3) were labeled on the 5′-end with 32P and annealed to their complements using standard methods.99 DNA duplexes were mixed with the indicated compounds (1 mM final concentrations) and incubated at 37 °C for 3 d in HEPES buffer (50 mM, pH 7) containing NaCl (100 mM) and DMF (10% v/v). The DNA was then ethanol precipitated, washed with 80% ethanol twice, and briefly dried in a SpeedVac concentrator (5 min at room temperature). The DNA was resuspended in piperidine (1 M in water) and heated to 95 °C for 30 min. The piperidine solution was removed under vacuum in a SpeedVac concentrator, and the resulting residue was resuspended in formamide loading buffer,99 warmed briefly, and loaded onto a 20% denaturing polyacrylamide gel. The gel was electrophoresed for 3 h at 1500 V to resolve the labeled DNA fragments. The labeled DNA fragments in the gel were visualized using phosphorimaging.
Hypoxia-Selective Enzymatic Reduction of N-Mustard Containing 1,2,4-Benzotriazine 1,4-Dioxides with Cytochrome P450
For experiments involving in vitro hypoxic metabolism, sodium phosphate buffer and HPLC-grade water were degassed by bubbling argon through the solutions for at least 30 min inside a glovebag filled with argon gas. Stock solutions of compounds 15 or 9 in DMF were degassed by three cycles of freeze–pump–thaw inside pyrex tubes. The tubes were then torch-sealed, scored, and transferred to the argon-purged glovebag. The enzyme NADPH:cytochrome P450 reductase was used without degassing. The enzyme substrate NADPH was dissolved in degassed water inside the glovebag. In the glovebag, solutions containing sodium phosphate buffer (25 mM, pH 7), the di-N-oxide 15 (100 μM), NADPH (500 μM), and NADPH:cytochrome P450 reductase (1 unit/mL) and DMF (1% v/v) were incubated in a sealed microcentrifuge tube at room temperature protected from light for 24 h. A similar reaction was prepared using nondegassed solutions and incubated open to air as a aerobic control. After 24 h, the solutions were passed through Millipore (YM-3) centrifuge filters (30 min, 7500 × g). The filtrate was analyzed by HPLC using a Varian Microsorb-MV C-18 column (100 Å sphere size, 5 μm pore size, 250 mm length, and 2.6 mm i.d.). The column was eluted with a mobile phase composed of solvent A (0.5% AcOH in H2O) and solvent B (methanol). The column was eluted at a flow rate of 0.9 mL/min for 2 min at 10% B, a gradient of 10–50% B over 8 min, 50% B for 5 min, then returned to 10% B over 5 min and held at 10% B for 5 min postrun. Compounds were detected by their absorbance at 420 nm, and the identity of products 15 and 16 was confirmed by comparison to authentic synthetic standards.
Acknowledgments
We are grateful to the National Institutes of Health for partial support of this work (ES021007) and the National Science Foundation and the National Institutes of Health for support of the MU NMR facility (NSF CHE-89-08304, NIH/NCRR S10 RR022341-01).
Supporting Information Available
Crystallographic information for compound 19. HPLC analysis. UV–vis spectra associated with pKa determinations. Spectroscopic data for all compounds. This materials is available free of charge via the Internet at http://pubs.acs.org
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- de Vita V. T. J.; Chu E. Cancer Res. 2008, 68, 8643–8653. [DOI] [PubMed] [Google Scholar]
- Goodman L. S.; Wintrobe M. M.; Dameshek W.; Goodman M. J.; Gilman A.; McClennan M. T. J. Am. Med. Assoc. 1946, 132, 126. [DOI] [PubMed] [Google Scholar]
- Povirk L. F.; Shuker D. E. Mutat. Res. 1994, 318, 205–226. [DOI] [PubMed] [Google Scholar]
- Physicians’ Desk Reference, 65th ed.; Thomsom PDR: Montvale, NJ, 2011. [Google Scholar]
- Cullis P. M.; Green R. E.; Malone M. E. J. Chem. Soc., Perkin Trans. 2 1995, 1503–1511. [Google Scholar]
- Mohamed D.; Mowaka S.; Thomale J.; Linscheid M. W. Chem. Res. Toxicol. 2009, 22, 1435–1446. [DOI] [PubMed] [Google Scholar]
- Gates K. S. Chem. Res. Toxicol. 2009, 22, 1747–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gates K. S.; Nooner T.; Dutta S. Chem. Res. Toxicol. 2004, 17, 839–856. [DOI] [PubMed] [Google Scholar]
- Haapala E.; Hakala K.; Jokipelto E.; Vilpo J.; Hovinen J. Chem. Res. Toxicol. 2001, 14, 988–995. [DOI] [PubMed] [Google Scholar]
- Balcome S.; Park S.; Quirk Dorr D. R.; Hafner L.; Phillips L.; Tretyakova N. Chem. Res. Toxicol. 2004, 17, 950–962. [DOI] [PubMed] [Google Scholar]
- Dutta S.; Abe H.; Aoyagi S.; Kibayashi C.; Gates K. S. J. Am. Chem. Soc. 2005, 127, 15004–15005. [DOI] [PubMed] [Google Scholar]
- Millard J. T.; Raucher S.; Hopkins P. B. J. Am. Chem. Soc. 1990, 112, 2459–2460. [Google Scholar]
- Sunavala-Dossabhoy G.; Van Dyke M. W. Biochemistry 2005, 44, 2510–2522. [DOI] [PubMed] [Google Scholar]
- Turner P. R.; Denny W. A.; Ferguson L. R. Anti-Cancer Drug Des. 2000, 15, 245–253. [PubMed] [Google Scholar]
- Dickinson L. A.; Burnett R.; Melander C.; Edelson B. S.; Arora P. S.; Dervan P. B.; Gottesfeld J. M. Chem. Biol. 2004, 11, 1583–1594. [DOI] [PubMed] [Google Scholar]
- Marquis J. C.; Hillier S. M.; Dinaut A. N.; Rodrigues D.; Mitra K.; Essigmann J. M.; Croy R. G. Chem. Biol. 2005, 12, 779–787. [DOI] [PubMed] [Google Scholar]
- Cullis P. M.; Merson-Davies L.; Weaver R. J. Am. Chem. Soc. 1995, 117, 8033–8034. [Google Scholar]
- Rink S. M.; Yarema K. J.; Solomon M. S.; Paige L. A.; Tadayoni-Rebek B. M.; Essigmann J. M.; Croy R. G. Proc. Natl. Acad. Sci. U.S.A. 1996, 93, 15063–15068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan J. Y.; Ohms S. J.; Boyd M.; Denny W. A. Chem. Res. Toxicol. 1999, 12, 1166–1172. [DOI] [PubMed] [Google Scholar]
- Fonseca S. B.; Pereira M. P.; Mourtada R.; Gronda M.; Horton K. L.; Hurren R.; Minden M. D.; Schimmer A. D.; Kelley S. O. Chem. Biol. 2011, 18, 445–453. [DOI] [PubMed] [Google Scholar]
- Kuang Y.; Balakrishnan K.; Gandhi V.; Peng X. J. Am. Chem. Soc. 2011, 133, 19278–19231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajapakse A.; Linder C.; Morrison R. D.; Sarkar U.; Leigh N.; Barnes C. L.; Daniels J. S.; Gates K. S. Chem. Res. Toxicol. 2013, 26, 555–563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wardman P.; Dennis M. F.; Everett S. A.; Patel K. B.; Stratford M. R. L.; Tracy M. Biochem. Soc. Trans. 1995, 61, 171–194. [DOI] [PubMed] [Google Scholar]
- Fitzsimmons S. A.; Workman P. A.; Grever M.; Paull K.; Camalier R.; Lewis A. D. J. Natl. Cancer Inst. 1996, 88, 259–269. [DOI] [PubMed] [Google Scholar]
- Rooseboom M.; Commandeur J. N. M.; Vermeulen N. P. E. Pharm. Rev. 2004, 56, 53–102. [DOI] [PubMed] [Google Scholar]
- Chen Y.; Hu L. Med. Res. Rev. 2009, 29, 29–64. [DOI] [PubMed] [Google Scholar]
- Wilson W. R.; Anderson R. F.; Denny W. A. J. Med. Chem. 1989, 32, 23–30. [DOI] [PubMed] [Google Scholar]
- Denny W. A.; Wilson W. R. J. Med. Chem. 1986, 29, 879–887. [DOI] [PubMed] [Google Scholar]
- Walton M. I.; Wolf C. R.; Workman P. Int. J. Radiat. Oncol. Biol. Phys. 1989, 16, 983–986. [DOI] [PubMed] [Google Scholar]
- Wen B.; Coe K. J.; Rademacher P.; Fitch W. L.; Monshouwer M.; Nelson S. D. Chem. Res. Toxicol. 2008, 21, 2393–2406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- James A. L.; Perry J. D.; Jay C.; Monget D.; Rasburn J. W.; Gould F. K. Lett. Appl. Microbiol. 2001, 33, 403–408. [DOI] [PubMed] [Google Scholar]
- Helsby N. A.; Goldthorpe M. A.; Tang M. H. Y.; Atwell G. J.; Smith E. M.; Wilson W. R.; Tingle M. D. Drug Metab. Dispos. 2008, 36, 353–360. [DOI] [PubMed] [Google Scholar]
- Mason R. P.; Holtzman J. L. Biochemistry 1975, 14, 1626–1632. [DOI] [PubMed] [Google Scholar]
- Mason R. P.; Holtzman J. L. Biochem. Biophys. Res. Commun. 1975, 67, 1267–1274. [DOI] [PubMed] [Google Scholar]
- Wilman D. E. V.; Palmer B. D.; Denny W. A. J. Med. Chem. 1995, 38, 2256–2258. [DOI] [PubMed] [Google Scholar]
- O’Connor C. J.; Denny W. A.; Fan J.-Y.; Gravatt G. L.; Grigor B. A.; McLennan D. J. J. Chem. Soc., Perkin Trans. 2 1991, 1933–1939. [Google Scholar]
- Palmer B. D.; Wilson W. R.; Pullen S. M.; Denny W. A. J. Med. Chem. 1990, 33, 112–121. [DOI] [PubMed] [Google Scholar]
- Duan J.-X.; Jiao H.; Kaizerman J.; Stanton T.; Evans J. W.; Lan L.; Lorente G.; Banica M.; Jung D.; Wang J.; Ma H.; Li X.; Yang Z.; Hoffman R. M.; Ammons W. S.; Hart C. P.; Matteucci M. J. Med. Chem. 2008, 51, 2412–2420. [DOI] [PubMed] [Google Scholar]
- Patterson A. V.; Ferry D. M.; Edmunds S. J.; Gu Y.; Singleton R. S.; Patel K. B.; Pullen S. M.; Hicks K. O.; Syddall S. P.; Atwell G. J.; Yang S.; Denny W. A.; Wilson W. R. Clin. Cancer Res. 2007, 13, 3922–3932. [DOI] [PubMed] [Google Scholar]
- Brown J. M. Cancer Res. 1999, 59, 5863–5870. [PubMed] [Google Scholar]
- Brown J. M.; Wilson W. R. Nat. Rev. Cancer 2004, 4, 437–447. [DOI] [PubMed] [Google Scholar]
- Wilson W. R.; Hay M. P. Nat. Rev. Cancer 2011, 11, 393–409. [DOI] [PubMed] [Google Scholar]
- Marcu L.; Olver I. Curr. Clin. Oncol. 2006, 1, 71–79. [DOI] [PubMed] [Google Scholar]
- Zeman E. M.; Baker M. A.; Lemmon M. J.; Pearson B. A.; Adams J. A.; Brown J. M.; Lee W. W.; Tracy M. Int. J. Radiat. Oncol. Biol. Phys. 1989, 16, 977–981. [DOI] [PubMed] [Google Scholar]
- Zeman E. M.; Brown J. M.; Lemmon M. J.; Hirst V. K.; Lee W. W. Int. J. Radiat. Oncol. Biol. Phys. 1986, 12, 1239–1242. [DOI] [PubMed] [Google Scholar]
- Brown J. M. Br. J. Cancer 1993, 67, 1163–1170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Pawel J.; von Roemeling R.; Gatzmeier U.; et al. J. Clin. Oncol. 2000, 18, 1351–1359. [DOI] [PubMed] [Google Scholar]
- Rischin D.; Peters L.; Fisher R.; et al. J. Clin. Oncol. 2005, 23, 79–87. [DOI] [PubMed] [Google Scholar]
- Hicks K. O.; Siim B. G.; Jaiswal J. K.; Pruijin F. B.; Fraser A. M.; Patel R.; Hogg A.; Liyanage H. D. S.; Dorie M. J.; Brown J. M.; Denny W. A.; Hay M. P.; Wilson W. R. Clin. Cancer Res. 2010, 16, 4946–4957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laderoute K. L.; Wardman P.; Rauth M. Biochem. Pharmacol. 1988, 37, 1487–1495. [DOI] [PubMed] [Google Scholar]
- Lloyd R. V.; Duling D. R.; Rumyantseva G. V.; Mason R. P.; Bridson P. K. Mol. Pharmacol. 1991, 40, 440–445. [PubMed] [Google Scholar]
- Daniels J. S.; Gates K. S. J. Am. Chem. Soc. 1996, 118, 3380–3385. [Google Scholar]
- Junnotula V.; Sarkar U.; Sinha S.; Gates K. S. J. Am. Chem. Soc. 2009, 131, 1015–1024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birincioglu M.; Jaruga P.; Chowdhury G.; Rodriguez H.; Dizdaroglu M.; Gates K. S. J. Am. Chem. Soc. 2003, 125, 11607–11615. [DOI] [PubMed] [Google Scholar]
- Chowdhury G.; Junnutula V.; Daniels J. S.; Greenberg M. M.; Gates K. S. J. Am. Chem. Soc. 2007, 129, 12870–12877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin J.; Glaser R.; Gates K. S. Chem. Res. Toxicol. 2012, 25, 634–645. [DOI] [PubMed] [Google Scholar]
- Yin J.; Glaser R.; Gates K. S. Chem. Res. Toxicol. 2012, 25, 620–633. [DOI] [PubMed] [Google Scholar]
- Shinde S. S.; Anderson R. F.; Hay M. P.; Gamage S. A.; Denny W. A. J. Am. Chem. Soc. 2004, 126, 7865–7874. [DOI] [PubMed] [Google Scholar]
- Shinde S. S.; Hay M. P.; Patterson A. V.; Denny W. A.; Anderson R. F. J. Am. Chem. Soc. 2009, 131, 14220–14221. [DOI] [PubMed] [Google Scholar]
- Shinde S. S.; Maroz A.; Hay M. P.; Patterson A. V.; Denny W. A.; Anderson R. F. J. Am. Chem. Soc. 2010, 132, 2591–2599. [DOI] [PubMed] [Google Scholar]
- Anderson R. F.; Shinde S. S.; Hay M. P.; Gamage S. A.; Denny W. A. J. Am. Chem. Soc. 2003, 125, 748–756. [DOI] [PubMed] [Google Scholar]
- Anderson R. F.; Shinde S. S.; Hay M. P.; Gamage S. A.; Denny W. A. Org. Biomol. Chem. 2005, 3, 2167–2174. [DOI] [PubMed] [Google Scholar]
- Laderoute K.; Rauth A. M. Biochem. Pharmacol. 1986, 35, 3417–3420. [DOI] [PubMed] [Google Scholar]
- Walton M. I.; Workman P. J. Pharmacol. Exp. Ther. 1993, 265, 938–947. [PubMed] [Google Scholar]
- Fuchs T.; Chowdhary G.; Barnes C. L.; Gates K. S. J. Org. Chem. 2001, 66, 107–114. [DOI] [PubMed] [Google Scholar]
- Shen X.; Rajapakse A.; Galazzi F.; Junnotula V.; Fuchs-Knotts T.; Glaser R.; Gates K. S. Chem. Res. Toxicol. 2014, 27, 1015–1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cahill A.; Jenkins T. C.; White I. N. H. Biochem. Pharmacol. 1993, 45, 321–329. [DOI] [PubMed] [Google Scholar]
- Patterson A. V.; Saunders M. P.; Chinje E. C.; Patterson L. H.; Stratford I. J. Anti-Cancer Drug Des. 1998, 13, 541–573. [PubMed] [Google Scholar]
- Baker M. A.; Zeman E. M.; Hirst V. K.; Brown J. M. Cancer Res. 1988, 48, 5947–5952. [PubMed] [Google Scholar]
- Jones G. D. D.; Weinfeld M. Cancer Res. 1996, 56, 1584–1590. [PubMed] [Google Scholar]
- Daniels J. S.; Gates K. S.; Tronche C.; Greenberg M. M. Chem. Res. Toxicol. 1998, 11, 1254–1257. [DOI] [PubMed] [Google Scholar]
- Hwang J.-T.; Greenberg M. M.; Fuchs T.; Gates K. S. Biochemistry 1999, 38, 14248–14255. [DOI] [PubMed] [Google Scholar]
- Siim B. G.; Pruijn F. B.; Sturman J. R.; Hogg A.; Hay M. P.; Brown J. M.; Wilson W. R. Cancer Res. 2004, 64, 736–742. [DOI] [PubMed] [Google Scholar]
- Mason J. C.; Tennant G. J. Chem. Soc. B 1970, 911–916. [Google Scholar]
- Hay M. P.; Gamage S. A.; Kovacs M. S.; Pruijn F. B.; Anderson R. F.; Patterson A. V.; Wilson W. R.; Brown J. M.; Denny W. A. J. Med. Chem. 2003, 46, 169–182. [DOI] [PubMed] [Google Scholar]
- Deady L. W.; Stanborough M. S. Aust. J. Chem. 1982, 35, 1841–1849. [Google Scholar]
- Cohen T.; Deets G. L. J. Am. Chem. Soc. 1967, 89, 3939–3940. [Google Scholar]
- Boyd M.; Hay M. P.; Boyd P. D. W. Magn. Reson. Chem. 2006, 44, 948–954. [DOI] [PubMed] [Google Scholar]
- The formation of charged alkylating species such as episulfonium ions is solvent dependent, see:Bae S. Y.; Winemiller M. D. J. Org. Chem. 2013, 6457–6470. [DOI] [PubMed] [Google Scholar]
- Streidl N.; Denegri B.; Kronja O.; Mayr H. Acc. Chem. Res. 2010, 43, 1537–1549. [DOI] [PubMed] [Google Scholar]
- Mitoma C.; Onodera T.; Takegoshi T.; Thomas D. W. Xenobiotica 1977, 7, 205–220. [DOI] [PubMed] [Google Scholar]
- Hansch C.; Leo A.; Taft R. W. Chem. Rev. 1991, 91, 165–195. [Google Scholar]
- Suzuki H.; Kawakami T. Synthesis 1997, 855. [Google Scholar]
- Hammett L. P.Physical Organic Chemistry; McGraw-Hill: New York, 1940. [Google Scholar]
- Cantrell W. R. J.; Bauta W. E.; Engles T. Tetrahedron Lett. 2006, 47, 4249–4251. [Google Scholar]
- Kohn K. W.; Hartley J. A.; Mattes W. B. Nucleic Acids Res. 1987, 15, 10531–10549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maxam A. M.; Gilbert W. Methods Enzymol. 1980, 65, 499–560. [DOI] [PubMed] [Google Scholar]
- Mattes W. B.; Lee C.-S.; Laval J.; O’Connor T. R. Carcinogenesis 1996, 17, 643–648. [DOI] [PubMed] [Google Scholar]
- Delahoussaye Y. M.; Evans J. M.; Brown J. M. Biochem. Pharmacol. 2001, 62, 1201–1209. [DOI] [PubMed] [Google Scholar]
- Walton M. I.; Wolf C. R.; Workman P. Biochem. Pharmacol. 1992, 44, 251–259. [DOI] [PubMed] [Google Scholar]
- Fitzsimmons S. A.; Lewis A. D.; Riley R. J.; Workman P. Carcinogenesis 1994, 15, 1503–1510. [DOI] [PubMed] [Google Scholar]
- Hay M. P.; Denny W. A. Tetrahedron Lett. 2002, 43, 9569–9571. [Google Scholar]
- Boyd M.; Hay M. P.; Boyd P. D. W. Magn. Reson. Chem. 2006, 44, 948–954. [DOI] [PubMed] [Google Scholar]
- Pchalek K.; Hay M. P. J. Org. Chem. 2006, 71, 6530–6535. [DOI] [PubMed] [Google Scholar]
- Ligthart G. B. W. L.; Guo D.; Spek A. L.; Kooijman H.; Zuilhof H.; Sijbesma R. P. J. Org. Chem. 2007, 73, 111–117. [DOI] [PubMed] [Google Scholar]
- Cantrell W. R. Jr; Bauta W. E.; Engles T. Tetrahedron Lett. 2006, 47, 4249–4251. [Google Scholar]
- Britton H. T. S.; Robinson R. A. J. Chem. Soc. 1931, 1456–1462. [Google Scholar]
- Pandey M. M.; Jaipal A.; Kumar R. M.; Charde S. Y. Spectrochim. Acta, Part A 2013, 115, 887–890. [DOI] [PubMed] [Google Scholar]
- Sambrook J.; Fritsch E. F.; Maniatis T.. Molecular Cloning: A Lab Manual; Cold Spring Harbor Press: Cold Spring Harbor, NY, 1989. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.