

Background: Procollagen biosynthesis requires a large number of foldases, chaperones, and modifying enzymes.
Results: FKBP22 is a foldase and chaperone that interacts with a subset of collagens.
Conclusion: Collagen type-specific chaperones and foldases exist in the rER.
Significance: The lack of collagen type-specific rER proteins can lead to broader or overlapping phenotypes of connective tissue disorders.
Keywords: Biosynthesis, Collagen, Endoplasmic Reticulum (ER), Molecular Chaperone, Protein Folding, FK506-binding Protein, Peptidyl-Prolyl Cis-Trans Isomerase
Abstract
The biosynthesis of collagens occurs in the rough endoplasmic reticulum and requires a large numbers of molecular chaperones, foldases, and post-translational modification enzymes. Collagens contain a large number of proline residues that are post-translationally modified to 3-hydroxyproline or 4-hydroxyproline, and the rate-limiting step in formation of the triple helix is the cis-trans isomerization of peptidyl-proline bonds. This step is catalyzed by peptidyl-prolyl cis-trans isomerases. There are seven peptidyl-prolyl cis-trans isomerases in the rER, and so far, two of these enzymes, cyclophilin B and FKBP65, have been shown to be involved in collagen biosynthesis. The absence of either cyclophilin B or FKBP65 leads to a recessive form of osteogenesis imperfecta. The absence of FKBP22 leads to a kyphoscoliotic type of Ehlers-Danlos syndrome (EDS), and this type of EDS is classified as EDS type VI, which can also be caused by a deficiency in lysyl-hydroxylase 1. However, the lack of FKBP22 shows a wider spectrum of clinical phenotypes than the absence of lysyl-hydroxylase 1 and additionally includes myopathy, hearing loss, and aortic rupture. Here we show that FKBP22 catalyzes the folding of type III collagen and interacts with type III collagen, type VI collagen, and type X collagen, but not with type I collagen, type II collagen, or type V collagen. These restrictive interactions might help explain the broader phenotype observed in patients that lack FKBP22.
Introduction
Collagen is the most abundant protein in the body, and 29 different types have been reported in humans. Collagens are the major components of structural frameworks, such as bone, tendon, cartilage, and skin. Each type of collagen displays different functions and distributions in tissues (1, 2). Among these, type I collagen is the most ubiquitous and best characterized. Folding, secretion, and quality control of type I collagen requires multiple steps that may be shared with other types of collagen (3, 4). Biosynthesis of procollagen, the biosynthetic precursor molecule of collagen, takes place in the rough endoplasmic reticulum (rER)2 and is secreted to the extracellular matrix through the Golgi. Many molecular chaperones, protein foldases, and posttranslational modifying enzymes are involved in this process. This folding machinery consists of a molecular ensemble of proteins (4). Defects in molecules of this ensemble result in connective tissue disorders, such as osteogenesis imperfecta (OI) and Ehlers-Danlos syndrome (5). Proline is one of the common amino acids in the collagen sequences, and proline isomerization is the rate-limiting step during triple helix formation. This isomerization is catalyzed by peptidyl-prolyl cis-trans isomerases (PPIases) (6–9). PPIases are found ubiquitously in all cellular compartments (10, 11). Seven PPIases reside in the rER, and three of them, cyclophilin B (CypB) and FK506-binding protein (FKBP) 65 and 22, were shown to be involved in procollagen biosynthesis (4).
Both CypB and FKBP65 are involved in type I procollagen biosynthesis (7, 12–13). Mutations in either PPIB (coding for CypB) or FKBP10 (coding for FKBP65) lead to a recessive form of OI, and mutations in FKBP10 are also found in Bruck syndrome and in Kuskokwim syndrome (14–26). CypB is an ∼18-kDa PPIase (27) and forms protein complexes with other molecular chaperones and with modifying enzymes in the rER (28–31). Both the enzyme activity and complex formation are important during procollagen biosynthesis. Hyperelastosis cutis, which is known as hereditary equine regional dermal asthenia, is an autosomal recessive skin fragility syndrome observed in Quarter Horses, and this disease is caused by a mutation in the protein-protein interaction site of CypB (13). This mutation does not directly affect the PPIase activity but does affect protein-protein interactions (32). The lack of binding to other proteins altered the post-translational modifications, rate of folding, and secretion of type I procollagen (13). The absence of CypB due to mutations or in knock-out mice results in OI with an absence of 3-hydroxylation at Pro-986 of the α1 chain of type I collagen (16, 19, 25, 33). It is still unclear which function of CypB is crucial during procollagen biosynthesis. FKBP65 was initially proposed to be a tropoelastin chaperone (34) and later was identified as a procollagen molecular chaperone during biosynthesis and maturation (12, 35). This protein consists of four FKBP domains, two EF-hand motifs, and an rER retention signal. The FKBP domains are all probably catalytic sites and are homologous to FKBP13. The EF-hand motifs have a helix-loop-helix structure and bind calcium (36–38). FKBP65 has PPIase activity against peptide model substrates but only weak activity for triple helix formation. FKBP65 was also only marginally inhibited by FK506, a potent inhibitor for other FKBPs (35). On the other hand, this protein prevents fibril formation of type I collagen and provides an increase in the thermal stability of type I and III collagen (12). These results indicate that FKBP65 interacts with triple helices and prevents premature association between procollagen molecules. In vivo studies also indicate that FKBP65 is a procollagen molecular chaperone because mutations in FKBP10 (coding for FKBP65) lead to OI and Bruck syndrome. A recent publication proposes that FKBP65 might also be involved in the activity of lysyl-hydroxylase 2, which hydroxylates lysines in the telopeptide region of type I procollagen (20).
FKBP22 is composed of a single FKBP domain, two EF-hand motifs, and a rER retention signal. This is similar to the carboxyl-terminal portion of FKBP65 (10). The function of mammalian FKBP22 is not well understood and differs from the well characterized bacterial system in key aspects (39–41). In bacterial FKBP22, the FKBP domain is located at the carboxyl-terminal end instead of the amino-terminal region as in mammals. The two EF-hand motifs are missing, but it has the dimerization domain in the amino-terminal region (42). The lack of FKBP22 in Drosophila leads to embryonic lethality and may be involved in the Notch signaling pathway (43). Recently, FKBP22 was reported to be possibly involved in collagen biosynthesis in mammals. Human mutations in FKBP14 (coding for FKBP22) cause a kyphoscoliotic type of Ehlers-Danlos syndrome (44). This type of Ehlers-Danlos syndrome is classified as EDS type VI, which is also caused by a deficiency of lysyl-hydroxylase 1 (45, 46). The clinical diagnosis of EDS type VI is characterized by severe muscle hypotonia at birth, progressive kyphoscoliosis, marked skin hyperelasticity with widened atrophic scars, and joint hypermobility (47, 48). Interestingly, FKBP14 patients also displayed a wide spectrum of clinical features, such as myopathy, hearing loss, and aortic rupture (44). The typical Ehlers-Danlos syndrome is the result of gene defects of type III and V procollagen, whereas OI is caused by defects in type I procollagen (5, 49). Immunofluorescence studies showed that networks of extracellular matrix proteins, including type I, III, and VI collagen, were disrupted in patient cells lacking FKBP22. Additionally, abnormally enlarged rER was observed in these cells. This suggests that FKBP22 may be a part of the molecular ensemble for procollagen maturation. To address this question, we examined the interaction of recombinant human FKBP22 and various collagens in vitro. Here we show that FKBP22 preferentially recognizes type III, VI, and X collagen in vitro.
EXPERIMENTAL PROCEDURES
Expression and Purification of Human Recombinant FKBP22
DNA encoding human FKBP22, without the signal peptide sequence, was isolated from the MGC full-length clone ID 4042173 (Invitrogen) by PCR using primers containing an EcoRI site at the 5′ end and a XhoI site after the stop codon at the 3′ end. That DNA was inserted between the EcoRI and XhoI restriction sites of a pET30a(+) expression vector (Invitrogen). The expression vectors were transformed into Escherichia coli BL21(DE3) and grown at 37 °C to an optical density of 0.6 at 600 nm, and expression was induced with 1 mm isopropyl 1-thio-β-d-galactopyranoside. After incubation at 20 °C overnight, the cells were harvested by centrifugation and resuspended in Tris base B-PER (Thermo Scientific) containing 1 mm CaCl2. Insoluble material was removed by centrifugation, and proteins in the soluble fraction were precipitated with ammonium sulfate at a final concentration of 30% (w/v). After overnight incubation at 4 °C, the sample was centrifuged, and the precipitated materials were dissolved in HEPES buffer (20 mm HEPES buffer, pH 7.5, containing 1.0 m NaCl, 20 mm imidazole, and 1 mm CaCl2). The protein solution was passed through a 0.22-μm filter and loaded onto a Co2+-chelating column. After washing with HEPES buffer (minimum 5 column volumes), FKBP22 was eluted with elution buffer (20 mm HEPES buffer, pH 7.5, containing 1.0 m NaCl, 500 mm imidazole, and 1 mm CaCl2). The fractions containing FKBP22 were dialyzed into enterokinase cleavage buffer (50 mm Tris/HCl buffer, pH 8.0, containing 1 mm CaCl2 and 0.1% Tween 20). Enterokinase (1 unit/ml reaction volume) (Invitrogen) was used to cleave the His tag at 4 °C overnight, and the sample was dialyzed into the HEPES buffer of the chelating column. The protein solution, including FKBP22, was treated with 1 μl/ml diisopropyl fluorophosphate to inactivate proteases derived from E. coli and enterokinase and gently stirred for 4 h on ice. This solution was applied onto a Co2+-chelating column to remove the cleaved His tag fragment. FKBP22 passed through the Co2+-chelating column, and the flow-through fraction was dialyzed against 20 mm triethanolamine/HCl buffer, pH 7.5, containing 20 mm NaCl and 0.5 m urea, loaded onto a HiTrapQ XL column (GE Healthcare), and washed with the same buffer (minimum 5 column volumes). The contaminating proteins and FKBP22 were eluted with 20 and 30% 20 mm triethanolamine/HCl buffer, pH 7.5, containing 500 mm NaCl and 0.5 m urea, respectively. The purified FKBP22 was then dialyzed against different reaction buffers to remove urea and used for further experiments.
Circular Dichroism Measurements
Circular dichroism spectra were recorded on an Aviv 202 spectropolarimeter (Aviv, Lakewood, NJ) using a Peltier thermostated cell holder and a 1-mm path length cell (Starna Cells, Atascadero, CA). Protein concentrations were determined by amino acid analysis. The spectra represent the average of at least 10 scans recorded at a wavelength resolution of 0.1 nm. The proteins were measured in 1 mm Tris buffer containing 0.05 mm calcium chloride, pH 7.5, at 4 °C. The spectra were analyzed using the CD Spectra deconvolution software (CDnn) (50).
Collagen Purification
Type I, III, and V collagens were purified from fetal bovine calf skin by methods described previously (51, 52). Human type X collagen was recombinantly expressed and purified as described previously (53). Human type VI collagen was provided by Dr. Takako Sasaki. Type II collagen was purified from bovine cartilage as follows. All procedures were performed at 4 °C. Pieces of cartilage were dissolved into 0.1 m acetic acid and stirred in 1 mg/ml pepsin at 4 °C overnight. After spinning down the pieces of cartilage, the supernatant was adjusted to 0.7 m NaCl (final concentration). Type II collagen containing pellets were collected by centrifugation for 1 h at 13,000 rpm and redissolved in 0.1 m acetic acid. This solution was dialyzed into 0.1 m Tris/HCl buffer, pH 7.0, and then NaCl was added to a final concentration of 2.5 m to precipitate contaminating type I and III collagens. These collagens were removed by centrifugation for 1 h at 13,000 rpm. Type II collagen was precipitated by adding additional NaCl to 4.0 m and then redissolved in 0.1 m acetic acid. These collagens were identified by mass spectrometry. Preparations of carboxyl-terminal quarter fragments of type III collagen with and without prolyl 4-hydroxylation were prepared as described previously (54, 55)
Type I and Type III Collagen Fibril Formation Assay
Stock solutions of type I and type III collagen in 50 mm acetic acid were diluted in 0.1 m sodium bicarbonate buffer, pH 7.8, containing 0.15 m NaCl and 1 mm CaCl2 to a final concentration of 0.1 μm and 0.2 μm, respectively. Measurements were performed at 34 °C, and the absorbance (light scattering) was monitored at 313 nm as a function of time. All curves are the average of at least three independent measurements.
Thermal Stability of Collagens in the Presence of FKBP22
The thermal stability of type I and type III collagen was monitored at 220 nm using circular dichroism measurements. The temperature was increased from 25 to 50 °C at a rate of 30 °C/h. Stock solutions of collagens in 50 mm acetic acid were diluted into 50 mm Tris/HCl buffer, pH 7.5, containing 0.4 m NaCl and 1 mm CaCl2. The final protein concentrations were 0.2 and 0.6 μm for collagens and FKBP22, respectively. The curve in presence of FKBP22 was subtracted by the curve obtained for FKBP22 alone. Experiments were performed at least three times independently.
Refolding of Full-length Type III Collagen Measured by Optical Rotary Dispersion
Refolding of type III collagen was monitored at 365 nm using a model 341 polarimeter (PerkinElmer Life Sciences) with a 10-cm path length thermostated cell. The temperature was controlled by a circulating water bath and programmable temperature controller (RCS, Lauda Division, Brinkmann Instruments, Inc., Westbury, NY). The optical rotatory dispersion signals were recorded and digitized on an HP9070A measurement and plotting system (Hewlett-Packard, Palo Alto, CA). Stock solutions of type III collagen (final concentration 0.075 μm) in 0.05 m acetic acid were diluted into 50 mm Tris/HCl buffer, pH 7.5, containing 0.2 m NaCl and 1 mm CaCl2. The sample was denatured for 5 min at 45 °C and refolded at 25 °C by switching between two water baths. All curves were averaged by at least three independent measurements. The curve in the presence of enzyme was subtracted from the curve of enzyme itself.
Peptidyl-Prolyl Cis-Trans Isomerase Assays Using Peptide Substrates
Measurements of the catalytic efficiency (kcat/Km) for the isomerization reaction were performed as described (56), based on the α-chymotrypsin assay (57) with the following modifications. Stock solutions of substrates were prepared in DMSO, and the final DMSO concentration in the assay was 0.352% for kcat/Km measurements. Kinetic measurements were made at 5 °C to minimize the non-enzymatic isomerization reaction in 35 mm HEPES buffer, pH 7.8, containing 1 mm CaCl2. Final substrates of Suc-Ala-Ala/Leu-Pro-Phe-AMC and chymotrypsin concentrations were 8.8 and 12.8 μm, respectively. Fluorescence changes were monitored at 380 nm with a HiTech stopped-flow spectrophotometer (TgK Scientific Ltd., Bradford-on-Avon, UK). The assay was started by the mixing of chymotrypsin and substrate. Progression curves were analyzed by fitting to a second-order exponential decay function with Origin (OriginLab Corp., Northampton, MA). Values for kcat/Km were calculated according to kcat/Km = (kobs − ku)/[E], where ku is rate constant for the unanalyzed isomerization reaction, and kobs is the rate constant for the catalyzed reaction in the presence of enzymes at a given concentration of [E]. k values were calculated using Origin. Suc-Ala-Ala-Pro-Phe-AMC and Suc-Ala-Leu-Pro-Phe-AMC peptide were obtained from Peptide Institute, Inc. (Osaka, Japan) and Bachem (Bubendorf, Switzerland), respectively. Cyclophilin B was used as a positive control, and FKBP22 was added up to 0.8 μm to obtain detectable enzyme activity to calculate kcat/Km.
Refolding of the Carboxyl-terminal Quarter Fragment of Type III Collagen with and without Prolyl 4-Hydroxylation in the Presence and Absence of FKBP22
Refolding was monitored by circular dichroism measurements at 220 nm. Collagens were denatured for 5 min at 45 °C and then added into precooled reaction buffer (50 mm Tris/HCl, pH 7.5, containing 0.2 m NaCl and 1 mm CaCl2) at 10 °C in the presence and absence of FKBP22. Refolding was monitored for 2700 and 6000 s for the prolyl 4- and non-hydroxylated carboxyl-terminal quarter fragment of type III collagen, respectively. The initial folding kinetics were calculated using data points of 60–350 and 60–2000 s for the prolyl 4- and non-hydroxylated carboxyl-terminal quarter fragment of type III collagen, respectively. Protein concentrations are 2 and 6 μm for both carboxyl-terminal quarter fragments of type III collagens and FKBP22, respectively. All curves are the average of at least three independent measurements.
Interaction between FKBP22 and FK506
Binding studies were performed on a SLM8000C instrument modified by ISS and operated with the Vinci software (ISS, Champaign, IL). The excitation wavelength was 280 nm, and the emission signals were measured over the range of 300–400 nm. The measurements were run at room temperature, and samples were kept stirring during measurements. A stock solution of FK506 was prepared in DMSO and further diluted with TBS buffer containing 1 mm CaCl2. The assays were started after preincubation of various concentrations of FK506 with FKBP22 (final concentration 15 nm) at room temperature into a reaction cell for 5 min. Free FKBP22 was calculated using the fluorescence intensities in the absence and presence of saturating amounts of FK506. The absorption spectra of 300 nm FK506 were measured in a Cary4 spectrophotometer over the range of 250–350 nm.
Circular Dichroism Measurements in the Presence and Absence of Calcium
FKBP22 was dialyzed into 1 mm Tris/HCl buffer with Chelex 100 resin, analytical grade (Bio-Rad), pH 7.5, at 4 °C for the measurements of the effect of calcium on the structure after the measurement in presence of calcium. The measurement condition is the same as above.
Refolding of Full-length Type III Collagen Measured by Circular Dichroism
Refolding of full-length native type III collagen used essentially the same procedures as refolding of the quarter fragment of type III collagen in the presence and absence of FKBP22 (see above). Time of measurement and refolding temperature were modified to 4500 s and 25 °C, respectively. Protein concentrations were 0.2 and 2.0 μm for full-length type III collagen and FKBP22, respectively. The wavelength was 226 nm for the measurements in presence and absence of FK506 due to absorption of DMSO. FKBP22 was preincubated with 10 μm FK506 for 5 min at room temperature. The final DMSO concentration was 0.6%. The reaction buffer and FKBP22 were treated with Chelex 100 resin analytical grade (Bio-Rad) for the measurements in the presence and absence of calcium. All curves are the average of at least three independent measurements.
Citrate Synthase Thermal Aggregation Assay
The aggregation of citrate synthase upon thermal denaturation was measured by the method of Shao et al. (58). Citrate synthase (Sigma-Aldrich) was diluted 200-fold to a final concentration of 0.15 μm into prewarmed 40 mm Hepes buffer, pH 7.4, containing 1 mm CaCl2 at 43 °C. The aggregation of citrate synthase was monitored by absorbance at 500 nm in a Cary4000 spectrophotometer (Varian Inc., Palo Alto, CA). All enzyme concentrations were determined by amino acid analysis.
Chemically Denatured Rhodanese and Citrate Synthase Refolding and Aggregation Assay
Another frequently used assay for chaperone activity is the inhibition of aggregation of chemically denatured rhodanese (59, 60). Bovine rhodanese (Sigma-Aldrich) was denatured in 30 mm Tris/HCl buffer, pH 7.4, containing 6 m guanidine hydrochloride and 1 mm dithiothreitol at 25 °C for 1 h and then diluted 100-fold to a final concentration of 0.2 μm in 30 mm Tris/HCl buffer, pH 7.2, containing 50 mm NaCl and 1 mm CaCl2. The aggregation of denatured rhodanese was monitored at 320 nm with a Cary4000 spectrophotometer. Chemically denatured citrate synthase was prepared by the same procedures as rhodanese and used as an additional model substrate. The final concentration of denatured citrate synthase was 0.15 μm. All protein concentrations were determined by amino acid analysis.
Surface Plasmon Resonance Analysis
Surface plasmon resonance experiments were carried out using a BIACore X instrument (GE Healthcare). Purified collagens were immobilized on a CM5 sensor chip by amide coupling. The approximate coupled collagen concentrations were 6.0 ng/mm2 (6000 RU) of type I collagen, 1.2 ng/mm2 (1200 RU) of type II collagen, 3.0 ng/mm2 (3000 RU) of type III collagen, 2.0 ng/mm2 (2000 RU) of type V collagen, 4.5 ng/mm2 (4500 RU) of type VI collagen and 3.0 ng/mm2 (3000 RU) of type X collagen. The experiments were carried out at a flow rate of 10 μl/min and 20 °C in HBS-P buffer (10 mm Hepes buffer, pH 7.4, containing 150 mm NaCl and 0.005% Surfactant P20) containing 1 mm CaCl2. Hsp47 was injected as a positive control. Various concentrations of FKBP22 were injected to six different types of collagens on chips. All curves are the average of at least three independent measurements. For the analysis of binding of FKBP22 to type VI and X collagen, the steady state affinity model was used, and for all other interactions, the data were fitted with the Langmuir binding model (BIAevaluation software, GE Healthcare).
RESULTS
Biochemical Characterization of Human Recombinant FKBP22
We expressed human FKBP22 in E. coli to test interactions with collagens. Fig. 1A shows an SDS-polyacrylamide gel of purified human FKBP22 after removal of the His tag in the presence and absence of reducing agent. The purified protein under non reducing conditions migrates slightly faster than under reducing condition. The two cysteine residues in the sequence form a disulfide bridge (38), resulting in a different migration under reducing and non-reducing conditions. Fig. 2B and Table 1 show the CD spectra and the content of secondary structures predicted by CD, respectively. FKBP12 and FKBP65 were included for a comparison. A high content of α-helix is observed in FKBP22 compared with the other FKBPs. This is due to the presence of two EF-hand motifs, which have an α-helical structure (38). FKBP65 also contains two EF-hand motifs like FKBP22. However, the contribution of α-helix to the overall structure is smaller with three additional FKBP domains in FKBP65. Bacterial FKBP22 forms a dimer in solution (42). Recombinant human FKBP22 showed a mixed population of monomers and dimers in solution with a shift toward the dimer upon increasing protein concentrations. Strikingly, increased temperature also facilitated the formation of dimers (38).
FIGURE 1.
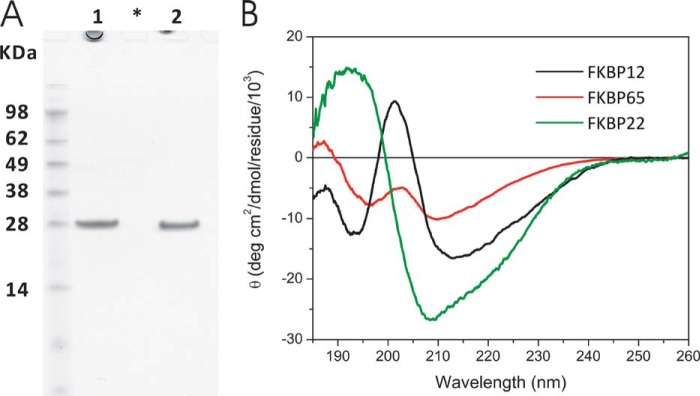
Characterization of purified human FKBP22. A, SDS-PAGE analysis of purified recombinant human FKBP22. Human FKBP22 was purified from an E. coli expression system, and the figure shows the final purified material in the presence and absence of dithiothreitol for lanes 1 and 2, respectively. The purified FKBP22 was run on NuPAGE Novex BisTris 12% gel (Invitrogen) and stained with GelCode Blue Stain Reagent. *, blank lane. B, circular dichroism spectra of FKBPs. The circular dichroism spectrum was measured at 4 °C in 1 mm Tris/HCl, pH 7.5, containing 0.05 mm CaCl2. The concentration of human FKBP22 (green) was 0.05 mg/ml. The spectra for human FKBP12 (black) and chick FKBP65 (red) are included for comparison.
FIGURE 2.

Interaction of FKBP22 with collagens. Fibril formation of type I (A) and type III (B) collagen in the presence and absence of Hsp47 or FKBP22. A stock solution of type I and type III collagen in 50 mm acetic acid was diluted to a final concentration of 0.1 and 0.2 μm, respectively. The measurements were performed in 0.1 m sodium bicarbonate buffer, pH 7.8, containing 0.15 m NaCl and 1 mm CaCl2 at 34 °C. Hsp47 was used as a positive control. A, the curves indicate the absence (black) and presence of 0.05 μm Hsp47 (red) and 0.2 μm FKBP22 (green). B, the curves indicate the absence (black) and presence of 0.05 μm Hsp47 (red) and 0.05 μm (green) and 0.1 μm (blue) FKBP22. Shown are thermal melting curves of type I collagen (C) and full-length type III collagen (D) in the presence (blue) and absence (red) of FKBP22. The final protein concentrations were 0.2 and 0.6 μm for collagens and FKBP22, respectively. The melting curves in the presence of FKBP22 are shown after subtraction of the FKBP22-only melting curve.
TABLE 1.
Comparison of the secondary structure content between chick FKBP65, human FKBP12, and human FKBP22
| FKBP65 | FKBP12 | FKBP22 | |
|---|---|---|---|
| % | % | % | |
| α-Helix | 8.3 | 7.4 | 28 |
| β-Sheets | 26.6 | 28.8 | 11.7 |
| Turns | 22.9 | 17.4 | 24.2 |
| Other structures | 41.9 | 43.4 | 40.5 |
Interaction of FKBP22 with Folded Type I and Type III Collagen
To test whether FKBP22 is involved in collagen quality control, we tested the effect on the thermal stability of the triple helix and collagen fibril formation in presence and absence of FKBP22. First we measured the effect on collagen fibril formation. Hsp47, a collagen chaperone, was shown to inhibit collagen fibril formation in vitro (61, 62). Fibril formation of both type I and III collagen was inhibited by Hsp47 at a 0.5- and 0.25-fold molar excess to type I and III collagen, respectively (Fig. 2, A and B). FKBP22 did not show any inhibition of fibril formation of type I collagen even at a 2- or 4-fold molar excess to type I collagen or Hsp47, respectively (Fig. 2A). On the other hand, a delay in fibril formation was observed for type III collagen (Fig. 2B). 0.05 μm Hsp47 and 0.1 μm FKBP22 showed comparable inhibition. FKBP22 stabilized neither type I collagen (Fig. 2C) nor type III (Fig. 2D). The melting curves for type I and type III collagen did not change in the presence or absence of a 3-fold molar excess of FKBP22.
PPIase Activity of FKBP22
FKBP22 was active as a PPIase when type III collagen was used as the substrate. Fig. 3 shows that the rate of refolding of denatured type III collagen is faster in the presence of FKBP22. Moreover, the presence of FKBP22 enhanced the final amount of refolded type III collagen. To calculate the catalytic efficiency of FKBP22, we used classical model peptides as substrates. Surprisingly, we only detected very low activities toward these peptides (Table 2). Considering these results, this phenomenon could be attributed to two possible hypotheses, substrate preference for 4-hydroxylproline instead of proline and/or length dependence of proline-containing polypeptides. To evaluate the first hypothesis, we used the carboxyl-terminal quarter fragment of type III collagen with and without prolyl 4-hydroxylation prepared from fetal bovine calf skin and expressed in bacteria, respectively (54, 55). FKBP22 more effectively catalyzed triple helix formation of prolyl 4-hydroxylated carboxyl-terminal quarter fragment than that of non-hydroxylated quarter fragment (Fig. 4 and Table 3). The amount of folded prolyl 4-hydroxylated carboxyl-terminal quarter fragment was increased as well as the amount of full-length type III collagen (Fig. 4A and Table 3). On the other hand, there was no significant difference for non-hydroxylated quarter fragment (Fig. 4C and Table 3). These results suggest that FKBP22 preferentially recognizes the X–4Hyp bond in collagen sequences.
FIGURE 3.

Refolding of full-length type III collagen in the presence and absence of FKBP22. Type III collagen was denatured for 5 min at 45 °C and then refolded at 25 °C for 90 min. Refolding was monitored by optical rotatory dispersion at 365 nm. The increased slope of the linear refolding phase indicates catalysis of the cis-trans isomerization. The final concentrations of type III collagen and FKBP22 were 0.075 and 0.75 μm, respectively.
TABLE 2.
The PPIase activities of FKBP22 and cyclophilin B with Suc-AXPF-AMC peptides as substrates
|
kcat/Km |
||
|---|---|---|
| Suc-AAPF-AMC | Suc-ALPF-AMC | |
| mm−1 s−1 | ||
| FKBP22 | 1.3 ± 0.8 | 30.7 ± 12.5 |
| Cyclophilin B | 23,270 ± 5000 | 7900 ± 400 |
FIGURE 4.

Refolding of the quarter fragment of type III collagen with and without prolyl 4-hydroxylation in the presence and absence of FKBP22. Refolding was monitored by a circular dichroism spectrum at 220 nm. Protein concentrations were 2 and 6 μm for the quarter fragments of type III collagens and FKBP22, respectively. A, refolding of the prolyl 4-hydroxylated quarter fragment of type III collagen in the presence (blue) and absence (red) of FKBP22. The black curve represents FKBP22 by itself. B, determination of the initial folding rate of prolyl 4-hydroxylated quarter fragment of type III collagen in the presence (blue) and absence (red) of FKBP22. Open circles and solid straight lines, raw data points and calculated initial folding rate from A, respectively. The slope of the straight lines reflected the initial rate of folding of prolyl 4-hydroxylated quarter fragment of type III collagen. Open black circles, raw data points of FKBP22 alone. C, refolding of non-4-hydroxylated quarter fragment of type III collagen in the presence (blue) and absence (red) of FKBP22. D, the determination of initial folding kinetics of the non-4-hydroxylated quarter fragment of type III collagen in presence (blue) and absence (red) of FKBP22. Open circles and solid straight lines, raw data points and calculated initial folding rate from C, respectively. The slope of the straight line reflected the initial rate of folding of the non-hydroxylated quarter fragment of type III collagen.
TABLE 3.
The rate of folding and final folded amount of 4-hydroxylated and non-4-hydroxylated quarter fragment of type III collagen in the presence and absence of FKBP22
| 4-Hydroxylated quarter fragment |
Non-4-hydroxylated quarter fragment |
|||
|---|---|---|---|---|
| Without FKBP22 | With FKBP22 | Without FKBP22 | With FKBP22 | |
| k (millidegrees/s) × 10−4 | 52.8 ± 3.18a | 60.8 ± 0.54a | 18.3 ± 0.22 | 19.7 ± 0.94 |
| Fraction folded | 0.74 ± 0.10b | 0.86 ± 0.19b | 0.81 ± 0.20 | 0.85 ± 0.09 |
a p < 0.01.
b p < 0.3.
The Effect of FK506 and Calcium Ion on the PPIase Activity of FKBP22
Two experiments were performed for further characterization of the PPIase activity of FKBP22. The first study is the inhibition of the PPIase activity with FK506, which generally blocks the active site of FKBP domains. To test the binding ability of FK506 to the FKBP domain of FKBP22, the fluorescence spectrum of hydrophobic amino acids located near the active site (e.g. Tyr-33, Trp-69, and Phe-108) was monitored by excitation at 280 nm and monitoring emission from 300 to 400 nm. The spectrum of free FKBP22 is shown by a solid line in Fig. 5A. The spectrum was shifted because of fluorescence derived from these hydrophobic residues covered by FK506 (dashed line in Fig. 5A). This quenching showed a concentration-dependent manner of FK506 (Fig. 5B). This binding affected the function during full-length type III collagen refolding (Fig. 5C). The rate of folding became slower in presence of FK506, and the increase of the folded amount of type III collagen was somewhat reduced (Fig. 5C). The amino-terminal region of FKBP22 is located on the opposite side of the catalytic site of the FKBP domain, and this region shows major structural differences compared with FKBP12, which did not act as a molecular chaperone in vitro (12). Several side chains of hydrophobic amino acid residue are exposed to the surface (38); therefore, this region may be responsible for the function as a molecular chaperone even in presence of FK506. The influence of the EF-hand motifs on the PPIase activity of FKBP22 was studied in the absence and presence of calcium. The calcium ion is required to coordinate the structure of the EF hands in FKBP22 (38). The α-helical structures of the EF-hand motif disappears after removing calcium (Fig. 6A). However, FKBP22 showed PPIase activity to the full-length type III collagen (Fig. 6B). This indicates that the EF-hand motifs do not contribute to the PPIase activity or the chaperone activity.
FIGURE 5.
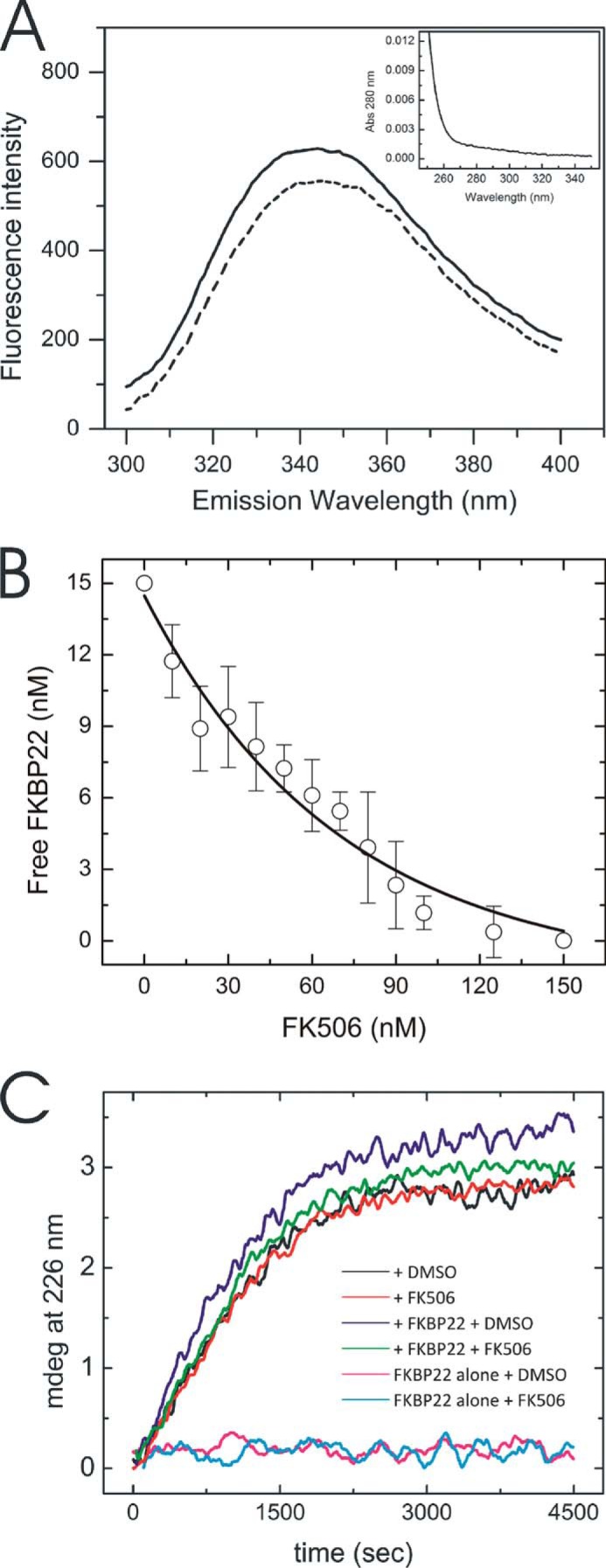
Influence of FK506 on the structure and PPIase activity of FKBP22. A, fluorescence spectra of 15 nm FKBP22 in presence (dotted line) and absence (solid line) of 150 nm FK506 resulting from tryptophan fluorescence at 280-nm excitation. Inset, background absorbance of 300 nm FK506 at 280 nm. B, titration curve of free FKBP22 in the presence of various concentrations of FK506. Free FKBP22 was calculated using the fluorescence signal at 340 nm. The concentration of FKBP22 was 15 nm. C, effect of FK506 on the refolding of full-length type III collagen monitored by CD at 220 nm. The protein concentrations were 0.2 and 2.0 μm for full-length type III collagen and FKBP22, respectively. FKBP22 was preincubated with 10 μm FK506 for 5 min at room temperature. Refolding of type III collagen with DMSO is shown in the presence (blue) and absence (black) of FKBP22 or with FK506 in the presence (green) and absence (red) of FKBP22. FKBP22 alone with DMSO (magenta) and with FK506 (cyan) are also shown.
FIGURE 6.
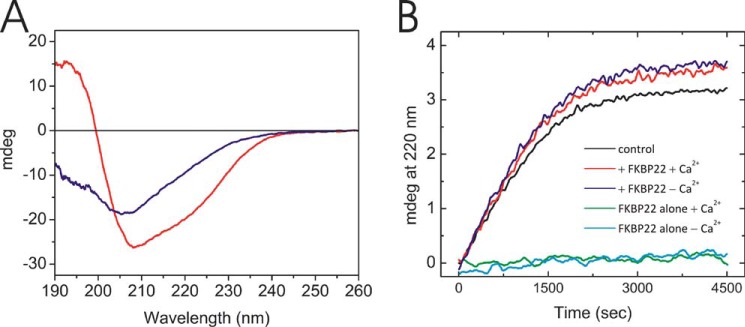
The effect of calcium on the FKBP22 structure and activity. A, circular dichroism spectra of FKBP22 in the presence (red) and absence (blue) of calcium are shown. The circular dichroism spectra were measured at 4 °C in 1 mm Tris/HCl, pH 7.5, treated with Chelex 100 resin, analytical grade. The concentration of both Chelex-treated and untreated FKBP22 was 8.4 μm. B, effect of calcium on the refolding of full-length type III collagen monitored by CD at 220 nm. The protein concentrations were 0.2 and 2.0 μm for full-length type III collagen and FKBP22, respectively. Refolding of type III collagen in the presence of FKBP22 with (red) and without (blue) calcium is shown. Type III collagen alone (black) or FKBP22 alone with (green) and without (cyan) calcium is also shown.
Classical Molecular Chaperone Assays for FKBP22 Using Model Substrates
FKBP22 inhibits fibril formation of type III collagen. This inhibition could indicate a molecular chaperone activity. We have previously characterized classical molecular chaperone activities against model substrates by procollagen molecular chaperones FKBP65 and prolyl 3-hydroxylase 1·cartilage-associated protein·CypB complex, which is composed of three procollagen-related rER proteins (12, 61). Here we tested three different enzyme assay systems using FKBP22, a thermal aggregation assay of citrate synthase (Fig. 7A), and a refolding and aggregation assay of chemical denatured citrate synthase (Fig. 7B) and rhodanese (Fig. 7C). FKBP22 did not show molecular chaperone activity against any of these substrates unlike protein-disulfide isomerase, which was used as positive control.
FIGURE 7.
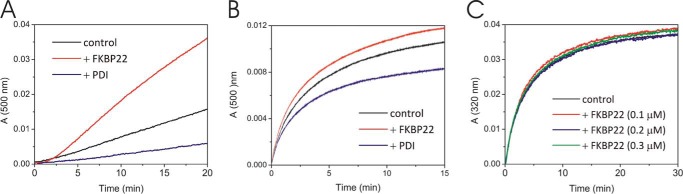
Classical chaperone activity assays using model substrates. A, the thermal aggregation of citrate synthase was monitored at 500 nm. A 30 μm citrate synthase solution was diluted 200-fold into prewarmed 40 mm Hepes buffer, pH 7.5, at 43 °C. The curves present the absence (black) and presence of 0.1 μm protein-disulfide isomerase (blue) and 0.5 μm FKBP22 (red). B, chemically denatured citrate synthase was diluted 100-fold (0.15 μm final concentration) into 30 mm Tris/HCl buffer, pH 7.2, containing 50 mm NaCl. Absorbance (light scattering) was monitored at 500 nm. The curves present the absence (black) and presence of 0.15 μm protein-disulfide isomerase (blue) and 0.25 μm FKBP22 (red). C, chemically denatured rhodanese was diluted 100-fold (0.2 μm final concentration) into 30 mm Tris/HCl buffer, pH 7.2, containing 50 mm NaCl. Absorbance (light scattering) was monitored at 320 nm. The curves present the absence (black) and presence of 0.1 μm (red), 0.2 μm (blue), and 0.3 μm (green) FKBP22.
Quantitation of Direct Binding of the FKBP22 to Various Types of Collagens
To test whether FKBP22 interacts with other types of collagen, surface plasmon resonance experiments were carried out using a BIACore X instrument. Hsp47 was previously shown to bind to native type I, II, III, and V collagen by this method (63) and was used as a positive control. Interestingly, FKBP22 did not show a direct binding response to type I, type II, and type V collagen. Binding was detected to type III, type VI, and type X collagen (Fig. 8, A–F). Binding to these collagens occurred in a concentration-dependent manner (Fig. 8, G–I). The equilibrium dissociation constants of the interaction between FKBP22 and type III, type VI, and type X collagen were ∼30 and 300 times weaker than the interaction of Hsp47 for type X and type III and VI collagen (Table 4). These values indicate a more transient interaction compared with Hsp47, which was used as a control.
FIGURE 8.

Direct binding kinetics of FKBP22 to collagens. Direct binding kinetics were monitored by surface plasmon resonance analysis using a BIAcore X instrument. A, 40 μm FKBP22 (red) and 0.05 μm Hsp47 (blue) as positive control were injected over a CM5 chip with immobilized bovine type I collagen. B, 25 μm FKBP22 (red) and 0.1 μm Hsp47 (blue) as positive control were injected over a CM5 chip with immobilized bovine type II collagen. C, 60 μm FKBP22 (red) and 0.05 μm Hsp47 (blue) as positive control were injected over a CM5 chip with immobilized bovine type III collagen. D, 30 μm FKBP22 (red) and 0.3 μm Hsp47 (blue) as positive control were injected over a CM5 chip with immobilized bovine type V collagen. E, 30 μm FKBP22 (red) and 0.3 μm Hsp47 (blue) as positive control were injected over a CM5 chip with immobilized human type VI collagen. F, 24 μm FKBP22 (red) and 0.4 μm Hsp47 (blue) as positive control were injected over CM5 chip-immobilized human type X collagen. G, various concentrations of FKBP22 were run over the type III collagen chip. The following binding curves are shown: 60 μm (black), 40 μm (red), 30 μm (blue), and 20 μm (green) FKBP22. H, various concentrations of FKBP22 were run over the type VI collagen chip. The following binding curves are shown: 30 μm (black), 24 μm (red), 18 μm (blue), and 12 μm (green) FKBP22. I, various concentrations of FKBP22 were run over the type X collagen chip. The following binding curves are shown: 24 μm (black), 16 μm (red), 12 μm (blue), and 8 μm (green) FKBP22.
TABLE 4.
Binding of Hsp47 and FKBP22 to various types of collagen
| ka | kd | Kd | χ2 value | ||
|---|---|---|---|---|---|
| ms−1 | s−1 | μm | |||
| Type I collagen | Hsp47a | 1.4 ± 1.24·104 | 3.1 ± 2.6·10−2 | 4.08 ± 2.8 | 12.8 ± 6.5 |
| Hsp47 (63) | 2.08·104 | 2.36·10−2 | 1.1 | ||
| FKBP22 | NOb | ||||
| Type II collagen | Hsp47a | 18.6 ± 12·104 | 5.3 ± 0.38·10−2 | 0.406 ± 0.24 | 8.2 ± 1.7 |
| Hsp47 (63) | 2.38·104 | 1.71·10−2 | 0.71 | ||
| FKBP22 | NO | ||||
| Type III collagen | Hsp47a | 11.9 ± 1.0·104 | 2.8 ± 0.38·10−2 | 0.24 ± 0.1 | 14.9 ± 2.4 |
| Hsp47 (63) | 2.18·104 | 1.55·10−2 | 0.71 | ||
| FKBP22a | 4.9 ± 0.7·104 | 3.4 ± 0.5·10−2 | 69 ± 1.7 | 12.5 ± 6.3 | |
| Type Vcollagen | Hsp47a | 10.0 ± 3.11·104 | 2.4 ± 0.05·10−2 | 0.24 ± 0.08 | 4.7 ± 1.5 |
| Hsp47 (63) | 1.89·104 | 1.65·10−2 | 0.87 | ||
| FKBP22 | NO | ||||
| Type VI collagen | Hsp47a | 13.9 ± 9.01·104 | 2.4 ± 0.01·10−2 | 0.21 ± 0.14 | 9.7 ± 0.7 |
| FKBP22c | 62 ± 46 | 10.4 ± 4.3 | |||
| Type X collagen | Hsp47a | 11.9 ± 11·104 | 9.3 ± 1.9·10−2 | 1.6 ± 1.2 | 6.3 ± 1.0 |
| FKBP22c | 43 ± 33 | 4.6 ± 3.7 |
a Data were calculated by a global fit of the concentration-dependent measurements using the Langmuir model.
b NO, not observed.
c Data were calculated by a steady state fit from the concentration-dependent measurements.
DISCUSSION
We have purified recombinantly expressed human FKBP22 and tested whether FKBP22 might have a role in collagen biosynthesis by analyzing its interactions in vitro. Our data show that FKBP22 can act as a PPIase in the formation of the triple helix and that it interacts with triple helical type III, type VI, and type X collagen. The interaction of FKBP22 with folded collagens points to a molecular chaperone activity. Indeed, increasing amounts of refolded type III collagen are found in the presence of FKBP22. Direct binding studies show that higher concentrations of FKBP22 are required for binding than that of Hsp47, indicating a transient interaction, as expected for chaperone activity (Table 4 and Fig. 8). This weak interaction is not sufficient to stabilize the triple helix (Fig. 2D) but could be strong enough to prevent the premature interactions of collagen molecules in the rER (Fig. 2B) because enlarged rER was observed in patient cells that lack FKBP22 (44). Similar results are observed between the prolyl 3-hydroxylase 1 complex and type I collagen. This complex acts as a procollagen-related multifunctional complex (61, 64). This complex also enhances the amount of folded type III collagen (61) (Fig. 3). Consistent with these results, FKBP22 is probably involved in the folding and quality control of type III, type VI, and type X procollagen as a molecular chaperone in the rER. However, FKBP22 did not show general molecular chaperone activity against model substrates (Fig. 7). One possible explanation is that FKBP22 has unique substrate recognition for certain types of collagen. Further studies are required to understand this phenomenon.
FKBPs generally show PPIase activity to proline-containing model peptides as PPIases (35, 65–66). FKBP22 acts as PPIase for the folding of type III collagen but did not catalyze small peptides (Fig. 3 and Table 2). In contrast, FKBP65 did not accelerate the rate of folding of type III collagen to a significant degree but had activity for model peptides (35). FKBP22 catalyzes the folding of the 4-hydroxylated quarter fragment of type III collagen but not the non-4-hydroxylated form, and it does not catalyze Pro-containing peptides. Therefore, we speculate that FKBP22 preferentially recognizes 4-hydroxyproline. The crystal structure of human FKBP22 suggests that the active site in the FKBP domain has a larger pocket for the ligand, and this space may better accommodate modified versions of proline (3- and 4-hydroxyproline) (38).
Type III collagen is the most practical model substrate for a collagen refolding experiment in vitro because disulfide bonds at the carboxyl-terminal end prevent three individual α chains from dissociating under denaturing conditions. Given that all types of collagen contain 4-hydroxyproline, it is likely that FKBP22 is globally involved in triple helix formation. However, the lack of a change in electrophoretic mobility in type I collagen of patients without FKBP22 (44) suggests that this PPIase does not play a major role in the folding of type I collagen.
Human mutations in FKBP14 cause Ehlers-Danlos syndrome with a wider array of clinical features (44). We hypothesized that this broader spectrum is caused by a distinct substrate preference of FKBP22. We observed that FKBP22 interacts with type III collagen but not type I collagen. This result provides a reasonable explanation for why human mutations in FKBP14 do not lead to an OI phenotype. Mutations in type III collagen result in a vascular type of Ehlers-Danlos syndrome (48, 49). One patient with an FKBP14 mutation was reported to have vascular abnormalities (44). Only this clinical feature correlates with the interaction of type III collagen and FKBP22. FKBP22 seems to be part of the molecular ensemble for type III, type VI, and type X collagen maturation. This may lead to the other observed phenotypes of Ehlers-Danlos syndrome due to the lack of FKBP22. Type VI collagen was disrupted in patient fibroblasts (44), and we observe that FKBP22 does interact with type VI collagen. These patients also showed muscle problems consistent with the observation that type VI collagen is abundant in muscles. FKBP22 binds to type X collagen but not type II collagen. This result suggests that FKBP22 is involved in the growth of the hypertrophic zone in cartilage, abundant in type X collagen, but not in development of articular cartilage, abundant in type II collagen. Joint hypermobility is observed in these patients, and it may result from these substrate preferences. We tested six different types of collagen out of 29 types. We summarize our findings in Fig. 9. It is possible that FKBP22 interacts with other minor types of collagen and extracellular matrix proteins, such as fibrillin, fibronectin, COMP, etc.
FIGURE 9.

Schematic diagram of the functions of FKBP22 during collagen biosynthesis in the rER. The PPIase and molecular chaperone activity are shown in the collagen biosynthesis steps with potential substrates.
Neurospora crassa FKBP22 was reported to form a complex with BiP and other chaperones (41, 67). However, this FKBP22 and the mammalian FKBP22 are structurally distinct except for the FKBP domain. The biological correlation between them is not clear. Various molecular chaperones and folding enzymes form the complex during procollagen biosynthesis (4). There is no report of other binding partners of human FKBP22. The finding of novel substrates and interaction partners of FKBP22 may provide clues to understanding the mechanism of this variant of Ehlers-Danlos syndrome.
Acknowledgments
We thank Dr. Takako Sasaki for providing type VI collagen and the HEK 293 clone stably transfected with human type X collagen and prolyl 4-hydroxylase α and β subunits. We also thank the Analytical Core Facility of Shriners Hospitals for Children in Portland for mass spectrometry analysis and amino acid analysis.
This work was supported by Grants SHC 85500 and SHC 85100 from Shriners Hospitals for Children (to H. P. B.).
- rER
- rough endoplasmic reticulum
- FKBP
- FK506-binding protein
- CypB
- cyclophilin B
- OI
- osteogenesis imperfecta
- PPIase
- peptidyl-prolyl cis-trans isomerase
- BisTris
- 2-[bis(2-hydroxyethyl)amino]-2-(hydroxymethyl)propane-1,3-diol
- AMC
- 7-amino-4-methylcoumarin
- RU
- resonance units.
REFERENCES
- 1. Bächinger H. P., Mizuno K., Vranka J., Boudko S. (2010) Collagen Formation and Structure. in Comprehensive Natural Products II: Chemistry and Biology (Mander L., Liu H.-W., eds) pp. 469–530, Elsevier, Oxford [Google Scholar]
- 2. Bateman J. F., Boot-Handford R. P., Lamandé S. R. (2009) Genetic diseases of connective tissues: cellular and extracellular effects of ECM mutations. Nat. Rev. Genet. 10, 173–183 [DOI] [PubMed] [Google Scholar]
- 3. Makareeva E., Aviles N. A., Leikin S. (2011) Chaperoning osteogenesis: new protein-folding disease paradigms. Trends Cell Biol. 21, 168–176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Ishikawa Y., Bächinger H. P. (2013) A molecular ensemble in the rER for procollagen maturation. Biochim. Biophys. Acta 1833, 2479–2491 [DOI] [PubMed] [Google Scholar]
- 5. Eyre D. R., Weis M. A. (2013) Bone collagen: new clues to its mineralization mechanism from recessive osteogenesis imperfecta. Calcif. Tissue Int. 93, 338–347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bächinger H. P., Bruckner P., Timpl R., Engel J. (1978) The role of cis-trans isomerization of peptide bonds in the coil leads to and comes from triple helix conversion of collagen. Eur. J. Biochem. 90, 605–613 [DOI] [PubMed] [Google Scholar]
- 7. Bächinger H. P., Morris N. P., Davis J. M. (1993) Thermal stability and folding of the collagen triple helix and the effects of mutations in osteogenesis imperfecta on the triple helix of type I collagen. Am. J. Med. Genet. 45, 152–162 [DOI] [PubMed] [Google Scholar]
- 8. Bruckner P., Eikenberry E. F. (1984) Formation of the triple helix of type I procollagen in cellulo. Temperature-dependent kinetics support a model based on cis in equilibrium trans isomerization of peptide bonds. Eur. J. Biochem. 140, 391–395 [DOI] [PubMed] [Google Scholar]
- 9. Steinmann B., Bruckner P., Superti-Furga A. (1991) Cyclosporin A slows collagen triple-helix formation in vivo: indirect evidence for a physiologic role of peptidyl-prolyl cis-trans-isomerase. J. Biol. Chem. 266, 1299–1303 [PubMed] [Google Scholar]
- 10. Rulten S. L., Kinloch R. A., Tateossian H., Robinson C., Gettins L., Kay J. E. (2006) The human FK506-binding proteins: characterization of human FKBP19. Mamm. Genome 17, 322–331 [DOI] [PubMed] [Google Scholar]
- 11. Yao Q., Li M., Yang H., Chai H., Fisher W., Chen C. (2005) Roles of cyclophilins in cancers and other organ systems. World J. Surg. 29, 276–280 [DOI] [PubMed] [Google Scholar]
- 12. Ishikawa Y., Vranka J., Wirz J., Nagata K., Bächinger H. P. (2008) The rough endoplasmic reticulum-resident FK506-binding protein FKBP65 is a molecular chaperone that interacts with collagens. J. Biol. Chem. 283, 31584–31590 [DOI] [PubMed] [Google Scholar]
- 13. Ishikawa Y., Vranka J. A., Boudko S. P., Pokidysheva E., Mizuno K., Zientek K., Keene D. R., Rashmir-Raven A. M., Nagata K., Winand N. J., Bächinger H. P. (2012) Mutation in cyclophilin B that causes hyperelastosis cutis in American Quarter Horse does not affect peptidylprolyl cis-trans isomerase activity but shows altered cyclophilin B-protein interactions and affects collagen folding. J. Biol. Chem. 287, 22253–22265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Alanay Y., Avaygan H., Camacho N., Utine G. E., Boduroglu K., Aktas D., Alikasifoglu M., Tuncbilek E., Orhan D., Bakar F. T., Zabel B., Superti-Furga A., Bruckner-Tuderman L., Curry C. J., Pyott S., Byers P. H., Eyre D. R., Baldridge D., Lee B., Merrill A. E., Davis E. C., Cohn D. H., Akarsu N., Krakow D. (2010) Mutations in the gene encoding the RER protein FKBP65 cause autosomal-recessive osteogenesis imperfecta. Am. J. Hum. Genet. 86, 551–559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Barnes A. M., Cabral W. A., Weis M., Makareeva E., Mertz E. L., Leikin S., Eyre D., Trujillo C., Marini J. C. (2012) Absence of FKBP10 in recessive type XI osteogenesis imperfecta leads to diminished collagen cross-linking and reduced collagen deposition in extracellular matrix. Hum. Mutat. 33, 1589–1598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Barnes A. M., Carter E. M., Cabral W. A., Weis M., Chang W., Makareeva E., Leikin S., Rotimi C. N., Eyre D. R., Raggio C. L., Marini J. C. (2010) Lack of cyclophilin B in osteogenesis imperfecta with normal collagen folding. N. Engl. J. Med. 362, 521–528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Barnes A. M., Duncan G., Weis M., Paton W., Cabral W. A., Mertz E. L., Makareeva E., Gambello M. J., Lacbawan F. L., Leikin S., Fertala A., Eyre D. R., Bale S. J., Marini J. C. (2013) Kuskokwim syndrome, a recessive congenital contracture disorder, extends the phenotype of FKBP10 mutations. Hum. Mutat. 34, 1279–1288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kelley B. P., Malfait F., Bonafe L., Baldridge D., Homan E., Symoens S., Willaert A., Elcioglu N., Van Maldergem L., Verellen-Dumoulin C., Gillerot Y., Napierala D., Krakow D., Beighton P., Superti-Furga A., De Paepe A., Lee B. (2011) Mutations in FKBP10 cause recessive osteogenesis imperfecta and Bruck syndrome. J. Bone Miner. Res. 26, 666–672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Pyott S. M., Schwarze U., Christiansen H. E., Pepin M. G., Leistritz D. F., Dineen R., Harris C., Burton B. K., Angle B., Kim K., Sussman M. D., Weis M., Eyre D. R., Russell D. W., McCarthy K. J., Steiner R. D., Byers P. H. (2011) Mutations in PPIB (cyclophilin B) delay type I procollagen chain association and result in perinatal lethal to moderate osteogenesis imperfecta phenotypes. Hum. Mol. Genet. 20, 1595–1609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Schwarze U., Cundy T., Pyott S. M., Christiansen H. E., Hegde M. R., Bank R. A., Pals G., Ankala A., Conneely K., Seaver L., Yandow S. M., Raney E., Babovic-Vuksanovic D., Stoler J., Ben-Neriah Z., Segel R., Lieberman S., Siderius L., Al-Aqeel A., Hannibal M., Hudgins L., McPherson E., Clemens M., Sussman M. D., Steiner R. D., Mahan J., Smith R., Anyane-Yeboa K., Wynn J., Chong K., Uster T., Aftimos S., Sutton V. R., Davis E. C., Kim L. S., Weis M. A., Eyre D., Byers P. H. (2013) Mutations in FKBP10, which result in Bruck syndrome and recessive forms of osteogenesis imperfecta, inhibit the hydroxylation of telopeptide lysines in bone collagen. Hum. Mol. Genet. 22, 1–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Setijowati E. D., van Dijk F. S., Cobben J. M., van Rijn R. R., Sistermans E. A., Faradz S. M., Kawiyana S., Pals G. (2012) A novel homozygous 5 bp deletion in FKBP10 causes clinically Bruck syndrome in an Indonesian patient. Eur. J. Med. Genet. 55, 17–21 [DOI] [PubMed] [Google Scholar]
- 22. Shaheen R., Al-Owain M., Faqeih E., Al-Hashmi N., Awaji A., Al-Zayed Z., Alkuraya F. S. (2011) Mutations in FKBP10 cause both Bruck syndrome and isolated osteogenesis imperfecta in humans. Am. J. Med. Genet. A 155A, 1448–1452 [DOI] [PubMed] [Google Scholar]
- 23. Shaheen R., Al-Owain M., Sakati N., Alzayed Z. S., Alkuraya F. S. (2010) FKBP10 and Bruck syndrome: phenotypic heterogeneity or call for reclassification? Am. J. Hum. Genet. 87, 306–307; author reply 308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Steinlein O. K., Aichinger E., Trucks H., Sander T. (2011) Mutations in FKBP10 can cause a severe form of isolated Osteogenesis imperfecta. BMC Med. Genet. 12, 152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. van Dijk F. S., Nesbitt I. M., Zwikstra E. H., Nikkels P. G., Piersma S. R., Fratantoni S. A., Jimenez C. R., Huizer M., Morsman A. C., Cobben J. M., van Roij M. H., Elting M. W., Verbeke J. I., Wijnaendts L. C., Shaw N. J., Högler W., McKeown C., Sistermans E. A., Dalton A., Meijers-Heijboer H., Pals G. (2009) PPIB mutations cause severe osteogenesis imperfecta. Am. J. Hum. Genet. 85, 521–527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Venturi G., Monti E., Dalle Carbonare L., Corradi M., Gandini A., Valenti M. T., Boner A., Antoniazzi F. (2012) A novel splicing mutation in FKBP10 causing osteogenesis imperfecta with a possible mineralization defect. Bone 50, 343–349 [DOI] [PubMed] [Google Scholar]
- 27. Mikol V., Kallen J., Walkinshaw M. D. (1994) X-ray structure of a cyclophilin B/cyclosporin complex: comparison with cyclophilin A and delineation of its calcineurin-binding domain. Proc. Natl. Acad. Sci. U.S.A. 91, 5183–5186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Horibe T., Yosho C., Okada S., Tsukamoto M., Nagai H., Hagiwara Y., Tujimoto Y., Kikuchi M. (2002) The chaperone activity of protein disulfide isomerase is affected by cyclophilin B and cyclosporin A in vitro. J. Biochem. 132, 401–407 [DOI] [PubMed] [Google Scholar]
- 29. Kozlov G., Bastos-Aristizabal S., Määttänen P., Rosenauer A., Zheng F., Killikelly A., Trempe J. F., Thomas D. Y., Gehring K. (2010) Structural basis of cyclophilin B binding by the calnexin/calreticulin P-domain. J. Biol. Chem. 285, 35551–35557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Smith T., Ferreira L. R., Hebert C., Norris K., Sauk J. J. (1995) Hsp47 and cyclophilin B traverse the endoplasmic reticulum with procollagen into pre-Golgi intermediate vesicles. A role for Hsp47 and cyclophilin B in the export of procollagen from the endoplasmic reticulum. J. Biol. Chem. 270, 18323–18328 [DOI] [PubMed] [Google Scholar]
- 31. Vranka J. A., Sakai L. Y., Bächinger H. P. (2004) Prolyl 3-hydroxylase 1, enzyme characterization and identification of a novel family of enzymes. J. Biol. Chem. 279, 23615–23621 [DOI] [PubMed] [Google Scholar]
- 32. Boudko S. P., Ishikawa Y., Lerch T. F., Nix J., Chapman M. S., Bächinger H. P. (2012) Crystal structures of wild-type and mutated cyclophilin B that causes hyperelastosis cutis in the American Quarter Horse. BMC Res. Notes 5, 626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Choi J. W., Sutor S. L., Lindquist L., Evans G. L., Madden B. J., Bergen H. R., 3rd, Hefferan T. E., Yaszemski M. J., Bram R. J. (2009) Severe osteogenesis imperfecta in cyclophilin B-deficient mice. PLoS Genet. 5, e1000750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Patterson C. E., Schaub T., Coleman E. J., Davis E. C. (2000) Developmental regulation of FKBP65. An ER-localized extracellular matrix binding-protein. Mol. Biol. Cell 11, 3925–3935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Zeng B., MacDonald J. R., Bann J. G., Beck K., Gambee J. E., Boswell B. A., Bächinger H. P. (1998) Chicken FK506-binding protein, FKBP65, a member of the FKBP family of peptidylprolyl cis-trans isomerases, is only partially inhibited by FK506. Biochem. J. 330, 109–114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Grabarek Z. (2011) Insights into modulation of calcium signaling by magnesium in calmodulin, troponin C and related EF-hand proteins. Biochim. Biophys. Acta 1813, 913–921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Dong H., Li X., Lou Z., Xu X., Su D., Zhou X., Zhou W., Bartlam M., Rao Z. (2008) Crystal-structure and biochemical characterization of recombinant human calcyphosine delineates a novel EF-hand-containing protein family. J. Mol. Biol. 383, 455–464 [DOI] [PubMed] [Google Scholar]
- 38. Boudko S. P., Ishikawa Y., Nix J., Chapman M. S., Bächinger H. P. (2014) Structure of human peptidyl-prolyl cis-trans isomerase FKBP22 containing two EF-hand motifs. Protein Sci. 23, 67–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Budiman C., Koga Y., Takano K., Kanaya S. (2011) FK506-binding protein 22 from a psychrophilic bacterium, a cold shock-inducible peptidyl prolyl isomerase with the ability to assist in protein folding. Int. J. Mol. Sci. 12, 5261–5284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Jana B., Sau S. (2012) The helix located between the two domains of a Mip-like peptidyl-prolyl cis-trans isomerase is crucial for its structure, stability, and protein folding ability. Biochemistry 51, 7930–7939 [DOI] [PubMed] [Google Scholar]
- 41. Tremmel D., Duarte M., Videira A., Tropschug M. (2007) FKBP22 is part of chaperone/folding catalyst complexes in the endoplasmic reticulum of Neurospora crassa. FEBS Lett. 581, 2036–2040 [DOI] [PubMed] [Google Scholar]
- 42. Budiman C., Angkawidjaja C., Motoike H., Koga Y., Takano K., Kanaya S. (2011) Crystal structure of N-domain of FKBP22 from Shewanella sp. SIB1: dimer dissociation by disruption of Val-Leu knot. Protein Sci. 20, 1755–1764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. van de Hoef D. L., Bonner J. M., Boulianne G. L. (2013) FKBP14 is an essential gene that regulates Presenilin protein levels and Notch signaling in Drosophila. Development 140, 810–819 [DOI] [PubMed] [Google Scholar]
- 44. Baumann M., Giunta C., Krabichler B., Rüschendorf F., Zoppi N., Colombi M., Bittner R. E., Quijano-Roy S., Muntoni F., Cirak S., Schreiber G., Zou Y., Hu Y., Romero N. B., Carlier R. Y., Amberger A., Deutschmann A., Straub V., Rohrbach M., Steinmann B., Rostásy K., Karall D., Bönnemann C. G., Zschocke J., Fauth C. (2012) Mutations in FKBP14 cause a variant of Ehlers-Danlos syndrome with progressive kyphoscoliosis, myopathy, and hearing loss. Am. J. Hum. Genet. 90, 201–216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Steinmann B., Eyre D. R., Shao P. (1995) Urinary pyridinoline cross-links in Ehlers-Danlos syndrome type VI. Am. J. Hum. Genet. 57, 1505–1508 [PMC free article] [PubMed] [Google Scholar]
- 46. Yeowell H. N., Walker L. C. (2000) Mutations in the lysyl hydroxylase 1 gene that result in enzyme deficiency and the clinical phenotype of Ehlers-Danlos syndrome type VI. Mol. Genet. Metab. 71, 212–224 [DOI] [PubMed] [Google Scholar]
- 47. Giunta C., Randolph A., Al-Gazali L. I., Brunner H. G., Kraenzlin M. E., Steinmann B. (2005) Nevo syndrome is allelic to the kyphoscoliotic type of the Ehlers-Danlos syndrome (EDS VIA). Am. J. Med. Genet. A 133A, 158–164 [DOI] [PubMed] [Google Scholar]
- 48. Steinmann B., Royce P. M., Superti-Furga A. (2003) in Connective Tissue and Its Heritable Disorders (Royce P. M., Steinman B., eds) pp. 431–523, John Wiley & Sons, Inc., New York [Google Scholar]
- 49. Mao J.-R., Bristow J. (2001) The Ehlers-Danlos syndrome: on beyond collagens. J. Clin. Invest. 107, 1063–1069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Böhm G., Muhr R., Jaenicke R. (1992) Quantitative analysis of protein far UV circular dichroism spectra by neural networks. Protein Eng. 5, 191–195 [DOI] [PubMed] [Google Scholar]
- 51. Pokidysheva E., Zientek K. D., Ishikawa Y., Mizuno K., Vranka J. A., Montgomery N. T., Keene D. R., Kawaguchi T., Okuyama K., Bächinger H. P. (2013) Posttranslational modifications in type I collagen from different tissues extracted from wild type and prolyl 3-hydroxylase 1 null mice. J. Biol. Chem. 288, 24742–24752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Mizuno K., Bächinger H. P., Imamura Y., Hayashi T., Adachi E. (2013) Fragility of reconstituted type V collagen fibrils with the chain composition of α1(V)α2(V)α3(V) respective of the D-periodic banding pattern. Connect Tissue Res. 54, 41–48 [DOI] [PubMed] [Google Scholar]
- 53. Wagner K., Pöschl E., Turnay J., Baik J., Pihlajaniemi T., Frischholz S., von der Mark K. (2000) Coexpression of α and β subunits of prolyl 4-hydroxylase stabilizes the triple helix of recombinant human type X collagen. Biochem. J. 352, 907–911 [PMC free article] [PubMed] [Google Scholar]
- 54. Davis J. M., Bächinger H. P. (1993) Hysteresis in the triple helix-coil transition of type III collagen. J. Biol. Chem. 268, 25965–25972 [PubMed] [Google Scholar]
- 55. Mizuno K., Boudko S., Engel J., Bächinger H. P. (2013) Vascular Ehlers-Danlos syndrome mutations in type III collagen differently stall the triple helical folding. J. Biol. Chem. 288, 19166–19176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Harrison R. K., Stein R. L. (1990) Mechanistic studies of peptidyl prolyl cis-trans isomerase: evidence for catalysis by distortion. Biochemistry 29, 1684–1689 [DOI] [PubMed] [Google Scholar]
- 57. Fischer G., Bang H., Mech C. (1984) Determination of enzymatic catalysis for the cis-trans-isomerization of peptide binding in proline-containing peptides. Biomed. Biochim. Acta 43, 1101–1111 [PubMed] [Google Scholar]
- 58. Shao F., Bader M. W., Jakob U., Bardwell J. C. (2000) DsbG, a protein disulfide isomerase with chaperone activity. J. Biol. Chem. 275, 13349–13352 [DOI] [PubMed] [Google Scholar]
- 59. Martin J., Langer T., Boteva R., Schramel A., Horwich A. L., Hartl F. U. (1991) Chaperonin-mediated protein folding at the surface of groEL through a “molten globule”-like intermediate. Nature 352, 36–42 [DOI] [PubMed] [Google Scholar]
- 60. Mendoza J. A., Rogers E., Lorimer G. H., Horowitz P. M. (1991) Chaperonins facilitate the in vitro folding of monomeric mitochondrial rhodanese. J. Biol. Chem. 266, 13044–13049 [PubMed] [Google Scholar]
- 61. Ishikawa Y., Wirz J., Vranka J. A., Nagata K., Bächinger H. P. (2009) Biochemical characterization of the prolyl 3-hydroxylase 1·cartilage-associated protein·cyclophilin B complex. J. Biol. Chem. 284, 17641–17647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Thomson C. A., Ananthanarayanan V. S. (2000) Structure-function studies on hsp47: pH-dependent inhibition of collagen fibril formation in vitro. Biochem. J. 349, 877–883 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Natsume T., Koide T., Yokota S., Hirayoshi K., Nagata K. (1994) Interactions between collagen-binding stress protein HSP47 and collagen. Analysis of kinetic parameters by surface plasmon resonance biosensor. J. Biol. Chem. 269, 31224–31228 [PubMed] [Google Scholar]
- 64. Ishikawa Y., Bächinger H. P. (2013) An additional function of the rough endoplasmic reticulum protein complex prolyl 3-hydroxylase 1·cartilage-associated protein·cyclophilin B: the CXXXC motif reveals disulfide isomerase activity in vitro. J. Biol. Chem. 288, 31437–31446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Feng M., Gu C., Ma S., Wang Y., Liu H., Han R., Gao J., Long Y., Mi H. (2011) Mouse FKBP23 mediates conformer-specific functions of BiP by catalyzing Pro117 cis/trans isomerization. Biochem. Biophys. Res. Commun. 408, 537–540 [DOI] [PubMed] [Google Scholar]
- 66. Nielsen J. B., Foor F., Siekierka J. J., Hsu M. J., Ramadan N., Morin N., Shafiee A., Dahl A. M., Brizuela L., Chrebet G. (1992) Yeast FKBP-13 is a membrane-associated FK506-binding protein encoded by the nonessential gene FKB2. Proc. Natl. Acad. Sci. U.S.A. 89, 7471–7475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Tremmel D., Tropschug M. (2007) Neurospora crassa FKBP22 is a novel ER chaperone and functionally cooperates with BiP. J. Mol. Biol. 369, 55–68 [DOI] [PubMed] [Google Scholar]