

Abstract
We present an integrated approach that predicts and validates novel anti-cancer drug targets. We first built a classifier that integrates a variety of genomic and systematic datasets to prioritize drug targets specific for breast, pancreatic and ovarian cancer. We then devised strategies to inhibit these anti-cancer drug targets and selected a set of targets that are amenable to inhibition by small molecules, antibodies and synthetic peptides. We validated the predicted drug targets by showing strong anti-proliferative effects of both synthetic peptide and small molecule inhibitors against our predicted targets.
Electronic supplementary material
The online version of this article (doi:10.1186/s13073-014-0057-7) contains supplementary material, which is available to authorized users.
Background
Treatment options for a variety of deadly cancers remain limited and the productivity of existing drug development pipelines, despite years of biomedical research, has been steadily declining. This is partly because current drug discovery efforts are mainly focusing on previously validated 'druggable' protein families such as kinases [1]. This leaves a vast space of the protein universe unexploited by cancer drugs. Hence, there is an urgent need for the identification and validation of new cancer-relevant targets. Fortunately, the emergence of high-throughput techniques, such as short hairpin RNA (shRNA) screening [2], transcriptional profiling [3], DNA copy number detection [4] and deep sequencing [5], has led to substantial advances in our understanding of human cancer biology. While the wealth of information in these datasets presents an opportunity to leverage these for finding novel drug targets, it remains a challenge to systematically integrate all these highly heterogeneous sources of information to identify novel anti-cancer drug targets. Several previous studies have analyzed a few different biological aspects in cancers with the purpose of cancer gene identification. For instance, one group found that genes whose expression and DNA copy number are increased in cancer are involved in core cancer pathways [6,7], while another showed that cancer drivers tend to have correlations of somatic mutation frequency and expression level [8,9]. Moreover, past studies that combined large-scale datasets have mainly focused on the simple characterization of cancer-related genes without any venue to inhibit and validate these targets [10,11]. Therefore, it is essential to develop a novel computational approach that can effectively integrate all available large-scale datasets and prioritize potential anti-cancer drug targets. Furthermore, while such predictions are useful, it is of crucial importance to experimentally validate them. A straightforward way for validation is to generate inhibitors to such targets and test them in model systems.
Overall, there exist roughly three broad ways to generate an inhibitor (and lead compound for drug development) to a given target protein. First, small molecules comprise the major class of pharmaceutical drugs and can act either on intra- or extra-cellular targets blocking receptor signaling and interfering with downstream intracellular molecules. The classic approach to find a novel small molecule is to screen very large chemical libraries. An alternative route is to find new therapeutic indications of currently available drugs (drug repositioning). Several studies have assessed potential anti-cancer properties of existing drugs and natural compounds that are initially used for the treatment of non-cancer diseases [12]. Recently, system biology approaches have been intensively applied to discover novel effects for existing drugs by analyzing large data sets such as gene expression profiles [13], side-effect similarity [14] and disease-drug networks [15]. In particular, sequence and structural similarities among drug targets have been successfully utilized to find new clinical indications of existing drugs [16]. Second, antibodies that interfere with an extracellular target protein have shown great efficacy, such as altering growth signals and blood vessel formation of cancer cells. Recently developed technologies, such as hybridoma or phage-display, have led to the efficient generation of antibodies against given targets [17]. Finally, synthetic peptides are a promising class of drug candidates. Their properties lie between antibodies and small molecules, and there have been numerous efforts to create peptides that can affect intracellular targets [18,19]. As with antibodies, several approaches to systematically generate inhibitory peptides have been developed [20]. A successful approach for drug target prediction and validation needs to include both a method to generate a list of target candidates and a systematic approach to validate targets using one or more of the ways described above.
Here, we developed a computational framework that integrates various types of high-throughput data for genome-wide identification of therapeutic targets of cancers. We systematically analyzed these targets for possible inhibition strategies and validate a subset by generating and testing inhibitors. Specially, we identified novel targets that are specific for breast (BrCa), pancreatic (PaCa) and ovarian (OvCa) cancers, which are major sources of mortality throughout the world. By analyzing the relevance of sequence, functional and network topological features, we prioritized a set of proteins according to their probability of being suitable cancer drug targets. We also examined each target for potential inhibition strategies with small molecules, antibodies and synthetic peptides. For the case of small molecules, we further identified several compounds already approved as drugs for different clinical indications; these drugs are ideal candidates for trials as potential novel anti-cancer agents. To validate a subset of targets, we used phage display to generate high-affinity peptide inhibitors against our predicted targets and showed their biological effects in cancer cells. Furthermore, we validated additional targets using high-throughput chemical library screening, proving potential efficacy of small molecule inhibitors against our predicted targets.
Methods
Biological and network-topological signatures
We examined 13 biological and network-topological properties of cancer drug targets (Additional file 1). Gene essentiality data were obtained from large-scale shRNA screening against 29 BrCa, 28 PaCa and 15 OvCa cell lines [2]. To examine gene essentiality, we used average GARP (Gene Activity Ranking Profile) score across cell lines corresponding to their cancer type. We compiled mRNA expression data and DNA copy number profiles from the Cancer Cell Line Encyclopedia (CCLE) [21]. CCLE contains the information of 58 breast, 44 pancreatic and 50 ovarian cancer cell lines. Similar to gene essentiality, we examined average robust multi-array average (RMA)-generated gene expression values and average DNA copy number depending on cancer types. In addition, we measured how many times genomic properties of known cancer drug targets are altered in tested cancer samples (for example, significantly essentialized, over-expressed, and highly amplified). On average, all genomic signatures of known cancer drug targets were significantly altered in 20% of cancer samples; meanwhile, putative non-drug targets showed the alterations in less than 8% of samples (Additional file 2). We considered that a gene is altered if its genomic signature value (for example, essentiality, mRNA expression, and DNA copy number) is ranked within the top 10% of all genomic signature values in a given cancer sample.
Mutation data were downloaded from the COSMIC database [22]. We counted the number of all observed mutations in DNA sequence to decide mutation occurrence of each gene-product. Position enrichment is the maximum number of mutations that are observed at the specific position of one gene product. We measured the ratio of the number of non-synonymous mutations to the number of synonymous mutations (dN/dS ratio). Since each protein has a different size (coding sequence length) and different number of mutations, we normalized mutation occurrence (number of mutations in a gene) by coding sequence length for the fair comparison of the mutational property of proteins. Position enrichment is measured using this normalized mutation occurrence. To assess whether mutation information of the COSMIC database is biased to a set of specific genes that are frequently studied, we calculated mutation occurrence of each gene product using an independent set of whole-genome sequencing data [6,23–26]. We compared the number of mutations observed from the COSMIC database with those from whole-genome sequencing data and found that there is a positive correlation between two datasets, indicating that COSMIC data contain reliable mutation information (Additional file 3). In the case of BrCa, we considered all reported mutations in BrCa instead of considering subtype-specific mutations.
The human interactome was built on a network of integrated global protein-protein interactions [27]. All network-topological features were calculated by the Python package NetworkX [28]. Briefly, degree is defined as the number of links to node. Betweenness is the sum of the fraction of all the shortest paths that pass through the gene. Closeness centrality is a reciprocal of average distance to all other nodes from the gene. Clustering coefficient is the fraction of possible triangles that exist.
Predicting targets for cancer drugs
Generating datasets
Cancer drugs, including approved drugs and clinical trial drugs, were collected from NCI data files [29] and the Therapeutic Target Database, which is a richly annotated database of drugs, drug targets and their clinical indications [30]. We selected drugs that are used for the treatment of BrCa, PaCa, and OvCa. After collecting anti-BrCa, -PaCa and -OvCa drugs, their targets were identified from DrugBank [31] and the Therapeutic Target Database. In total, 62 known BrCa drug targets, 69 known PaCa targets and known 45 OvCa targets constituted the positive dataset. Putative non-drug targets (negative set) are defined as proteins that (1) are non-existent or absent from the DrugBank and Therapeutic Target Database, (2) are not annotated as cancer-associated proteins, (3) do not physically interact with known cancer drug targets, and (4) do not share Pfam functional domains [32] and sequence similarity (<30% of sequence identity) with known cancer drug targets. We removed the cancer-associated proteins from the negative set in order to obtain the best possible series of non-drug targets for cancer treatment. To collect cancer-associated proteins, we used a text-mining method and examined experimental applications of 15,663 human proteins to cancer studies based on the pre-existing literature on cancer pathogenesis. In total, 5,169 proteins were considered as putative non-drug targets (Additional file 4).
Machine learning and feature selection
Support vector machines (SVM) with radial basis function (RBF) kernels (software available at [33]) were used to classify proteins into two classes: cancer drug targets and non-cancer drug targets. SVMs are a widely used supervised learning algorithm with excellent performance on many applications in cancer biomarker identification [34], inferring gene-disease association [35] and drug target identification [36]. SVMs are particularly attractive in the application of genome-wide identification of anti-cancer drug targets since they can handle both large and noisy datasets and are robust to over-training. Studies that compared several multivariate methods showed that SVMs provide the most accurate model to the training set, allowing the reliable classification of data sets [37].
After collecting 13 biological and network-topological features (Additional file 1), we identified the most relevant features using the SVM-recursive feature elimination (SVM-REF) method [38]. Some features correlated with each other; for example, the number of interacting partners of a given node (degree) generally shows a positive correlation with the number of shortest paths that pass through a given node (betweenness) in protein-protein interaction (PPI) networks [39]. SVM-REF removes such redundant features generalizing performance and provides the ranking of each feature on all the training set. SVM-REF implements backward feature elimination and searches an optimal subset of features. First, SVM-REF trains the SVM on the data set with all tested features. Next, tested features are ranked according to the weight vector of the SVM and the least important feature is identified and removed according to a ranking criterion. The process repeats with the remaining features until SVM-REF achieves the highest accuracy of classification. Finally, SVM-REF provides the ranking of each feature. We selected the best five features that represent each biological and network-topological property. The highest scoring features are GARP score for gene essentiality, RMA intensity for mRNA expression, row chromosomal copy number for DNA copy number, mutation occurrence for somatic mutation pattern and closeness centrality for network-topological property. These selected relevant features are used to build the final optimized classifiers that distinguish cancer drug targets from other proteins.
To select the best prediction model after selecting the optimal set of features, 10-fold cross-validation was conducted on a training set composed of known BrCa, PaCa and OvCa drug targets as a positive set and putative non-drug targets as a negative set. We used a grid search to find the best combination of model parameters (C, γ and weights for cancer drug targets and putative non-drug targets) for the SVM-RBF kernel. Since the size of the positive dataset (known drug targets) is smaller compared to the size of the negative dataset (putative non-drug targets), the learning weight of the positive dataset is increased in order to create a balanced dataset. We introduced an error penalty parameter to ensure generalization of the classifier. The ratio of the error penalty for known drug targets:putative non-drug targets is set to 100:1 and applied in the SVM program using the weight parameter (Wi) for both classes (positive targets and negative non-targets). The benchmark study that compared several methods to deal with unbalanced data sets showed that assigning a penalty value (Wi) outperforms alternative methods, including generation of pseudo-positive samples and re-sampling (random selection of a few negative samples) [40]. Various pairs of parameters (C, γ and Wi for drug targets and non-drug targets) are evaluated and the one with the best cross-validation accuracy is picked. After selecting the best model parameters, the whole training set is trained again to generate the final classifier. The optimal parameters of cancer-specific classifiers and prediction models are shown in Additional file 5. Two procedures to optimize the final prediction model (SVM-REF and grid-search) have different roles in generating final predictors and are performed separately. Thus, there is no effect and influence of one procedure on the other though they show a synergistic effect when combined.
Target classification according to therapeutic classes
Identified drug targets are divided into three groups depending on their therapeutic class; antibodies, synthetic peptides or small molecules. To do this, we examined the structural properties and cellular localizations of predicted targets. UniProt subcellular localization annotations were used to assign cellular localization to the predicted targets [41]. For the identification of antibody targets, we selected secreted proteins and membrane proteins that have extracellular domains based on the notion that antibodies generally recognize protein fragments in the extracellular space [42]. To examine whether membrane proteins have extracellular domains, we used the topological information of membrane proteins that have been deposited in Uniprot. If membrane proteins have extracellular fragments longer than 40 amino acids, we considered them as extracellular domains. Targets for synthetic peptides were selected if proteins have known peptide-binding domains and localize at the cytoplasm and/or nucleus based on the notion that peptides can penetrate lipid bilayer. The PepX database was used to determine a list of known peptide-binding domains derived from protein fragments or protein domains in complex with peptides [43]. To characterize targets for small molecules, we selected targets for which small molecules have been characterized and used for clinical applications. We used the ChEMBL database (version 15) [44], from which we extracted compounds that are characterized as inhibitors or antagonists of our predicted targets as well as approved and experimental drugs. We considered targets if their STITCH score is more than 0.7. The STITCH database compiles chemical-protein interactions curated by text-mining of the literature and provides confidence scores that reflect the level of significance and certainty of interaction between small molecules and targets. A STITCH score of 0.7 is generally used as a cutoff to define high-confidence interactions [45,46]. To find more reliable targets of small molecules, we selected targets that are already known targets of approved and experimental drugs in the DrugBank and Therapeutic Target Database. These drugs and targets were considered as repositioned drugs and repositioned targets, respectively.
Computational evaluation of predicted drug targets
First, we assessed how many predicted targets are related to cancer pathogenesis. To do this, we collected cancer-related proteins whose functional alterations are causally implicated in oncogenesis (for example, oncogene products, tumor-suppressor gene products and proteins in core cancer pathways). We identified 1,367 cancer-related proteins, compiled from CancerGenes [47] using the queries 'oncogene' and 'tumor suppressor' [48] and two consensus studies [49,50]. To identify cancer disease gene products, we used the Online Mendelian Inheritance in Man (OMIM) database, which contains the most complete known disease-gene associations [51]; this revealed 27 BrCa-, 10 PaCa- and 14 OvCa-related genes. Second, we investigated the experimental applications of bioactive small molecules (inhibitors and antagonists) of predicted targets based on the assumption that if predicted targets are associated with cancer pathogenesis, their bioactive molecules would be applied to cancer research. Using text-mining, we examined whether predicted targets and 'cancer' are commonly observed as keywords in the title and/or abstract of literature deposited in NCBI's PubMed database. Third, we examined the shortest path length between predicted targets and cancer disease gene products. The shortest path length for integrated global protein-protein interactions [27] was calculated using the Python package networkX.
Experimental evaluation of predicted targets using peptide inhibitors
Target selection
To select targets of peptide inhibitors, we manually examined if predicted targets have peptide-binding domains that have relevance to cancer. We selected PPWD1 and NXF1 since they have well-characterized peptide domains (WD40 domain for PPWD1 and LRR domain for NXF1), and these domains have critical roles in cancer pathogenesis. The WD40 domain mediates signal transduction and transcriptional regulation during the cell cycle and apoptosis [52]. The LRR domain mediates specific binding to constitutive transport element (CTE) RNA and metabolism of various post-transcriptional mRNAs that regulate cancer cell proliferation and transformation [53,54]. Though PPWD1 and NXF1 have functional relevance in cancer pathogenesis and potential to be reliable drug targets, both proteins have not been used as targets for peptide and small molecule drug design. Based on these observations, we decided to generate peptide inhibitors of PPWD1 and NXF1.
Cloning and protein expression
For cloning target domains, the PPWD1-WD40 domain and NXF1-LRR domain boundaries were defined as the union of the domain regions identified by Pfam [32], SMART [55], and Gene3D [56]. DNA fragments encoding the identified domains were synthesized (GenScript Inc., Piscataway, NJ, USA) and cloned into a vector designed for the expression and purification of domains fused to the carboxyl terminus of glutathione S-transferase (GST), as described previously [57]. All plasmid constructs were verified by DNA sequencing.
Selection of peptide library
A random hexadecapeptide library (X16, where X is any amino acid) was fused to the N-terminus of the gene -8 major coat protein of M13 filamentous phage. The phage displayed peptide library (>1010 unique members) was used to select peptide binders for the collection of purified GST-target domain fusion proteins. High-throughput phage display selections were carried out as described previously [57]. In short, five rounds of phage selections, including absorption, washing, elution and amplification, were conducted to enrich the bound phage for each domain. We isolated individual clones from the phage pools to test for positive interactions with the cognate target domains by phage ELISA as described [58]. In total, 44 clones with an ELISA protein/GST signal above 3 were sequenced, resulting in the isolation of 4 unique peptide sequences for PPWD1-WD40 domain and 11 for NXF1-LRR domain, which were manually aligned by an expert. The sequence with the highest ELISA signal was selected for cell viability assays.
Lentiviral vector preparation
Sense and antisense oligonucleotides containing peptide sequences were obtained from Sigma (Oakville, Ontario, Canada). Oligonucleotide pairs were annealed at 55°C and extended by PCR using Accuprime pfx DNA polymerase (Invitrogen, Burlington, ON, Canada) at a concentration of 1 μM in a volume of 50 μl. PCR reactions were done in a thermal cycler (Biorad, Mississauga, Ontario, Canada). The peptide sequences in the PCR products were introduced in pLJM1 plasmid containing a green fluorescent protein (GFP) sequence at the N-terminus. The PCR fragments containing the peptide sequences and the pLJM1 plasmid (5 μg) were doubly digested by EcoRI (New England Biolabs, Whitby, Ontario, Canada) and XmaI (New England Biolabs) for 4 to 6 h. DNA fragments were then separated using a 1% agarose gel. The vector and insert bands were excised and DNA was extracted using Qiaquick gel extraction kit (QIAGEN, Valencia, CA, USA). DNA was eluted with TE buffer. Ligation was performed in 20 μl reaction volume using 400 units of T4 DNA ligase (New England Biolabs) with 2 μl of PCR fragments and 15 to 20 ng of prepped vector for overnight at 16°C. Ligation mixture (2 μl) was used to transform 25 μl Max Efficiency DH5™ T1 Phage-resistant Competent Cells (Invitrogen), following the manufacturer’s heat shock protocol. The transformed cells were recovered in 200 μl of super optimal broth with catabolite repression (SOC) for 1 h at 37°C and plated on agar plates.
Lentiviral production
Lentiviruses were made by transfecting packaging cells (293 T) with a three-plasmid system as previously described [59,60]. Transfections were performed in six-well plates at a density of 2.5 × 105 cells per well in 2 ml media (Dulbecco's modified Eagle's medium (DMEM)/10% inactivated fetal bovine serum (IFS)/no antibiotics) 24 h before transfection and grown at 37°C in 5% CO2. DNA for transfections was prepared by mixing 900 ng pCMV-dR8.74psPAX2 and 100 ng pMD2.G with 1 μg peptide expressing plasmids in each well. A mixture of 96 μl OptiMEM (Gibco, Burlington, ON, Canada) and 6 μl FUGENE (Roche, Mississauga, ON, Canada) was then added to the DNA and this mixture was incubated for 15 minutes before addition to the packaging cells. Cells were incubated for 24 h, and the medium was changed to remove remaining transfection reagent. Lentiviral supernatants were collected at 48 and 72 h post-transfection. Lentiviruses were frozen at -20°C or -80°C for long-term storage.
Cell infection and cell viability assay
RWP1 cells were seeded at a density of 5,000 cells per well in 96-well plates in a final volume of 100 μl of culture medium per well. Cells were infected with lentiviruses expressing the desired constructs at different multiplicity of infection (MOI) values. To determine the MOI values, we measured the proportion of cells that acquire resistance to puromycin treatment following infection, as described previously. Polybrene (5 μg/ml) was added to the cell medium. Cells were incubated overnight in a humidified incubator at 37°C in 5% CO2. On the next day, the medium was replaced with fresh medium. Cells were incubated at 37°C for a period of 72 h in a humidified incubator. To measure cell viability, 10 μl of AlamarBlue was added to each well of a 96-well plate. After a 2 h incubation (37°C), fluorescence intensity was measured using PHERAstar SpectraMax Plus384 microplate reader (BMG LABTECH, Cary, NC, USA) with an excitation filter of 535 nm and an emission filter of 590 nm.
Screening of chemical compounds against predicted drug targets
Cell lines and reagents
Panc0813 cells were maintained in RPM1 (ATCC) and cultured at 37°C in 5% CO2 in their recommended medium supplemented with 10% fetal bovine serum and 1% penicillin/streptomycin/glutamine. AlamarBlue reagent was purchased from Invitrogen.
High-throughput chemical compound screening
We performed high-throughput chemical library screening for the experimental evaluation of predicted drug targets. The TocRis chemical Library® (TocRis Chemical, Inc.) and Kinome Library, which is a collection of 1,580 biologically and pharmacologically established chemicals, including a variety of marketed drugs, was obtained from several vendors and purposely assembled by the SMART Laboratory and the Ontario Institute for Cancer Research to specifically target molecular entities associated with signaling pathways, cellular processes and disparate metabolic events. Next, these chemical libraries were applied to Panc0813 pancreatic cancer cells. Targets of 1,580 small molecules were determined using the STITCH [46] and ChEMBL databases [44]. We selected targets if they have a high STITCH confidence score (>0.7) against given small molecules and considered small molecules if they are reported as inhibitors or antagonists of given targets using the ChEMBL database. The Beckman BioMek FX and the Samuel Lunenfeld Research Institute High-Throughput Screening Robotics platform were applied for cell seeding, treatment and viability assessment. Panc0813 cells were seeded to 384-well culture plates (Corning, Corning, NY, USA) at a density of 600 cells/well and incubated for 24 h. Compounds were added to a concentration of 1 μM. Cells treated with 0.1% dimethyl sulfoxide (DMSO) vehicle alone were used as the negative controls, while media only was used to determine the assay noise. To measure cell viability, 10 μl of AlamarBlue was added to each well after a 72 h incubation with chemical compounds. After a 2 h incubation (37°C), fluorescence intensity was measured using PHERAstar microplate reader (BMG LABTECH) with an excitation filter of 535 nm and an emission filter of 590 nm. To get dose-dependent response curves of eight PaCa inhibitors and two non-PaCa inhibitors, stock solutions of kinase inhibitors (Dasatinib, BI-2536 and BMS-536924; 1 mg/ml) and other small molecule inhibitors (A-205804, ACDPP hydrochloride, rolipram, loperamide, yohimbine, D4476 and 2-hydroxysaclogen; 5 mg/ml) were prepared in DMSO and serially diluted two-fold in the same solvent. Aliquots (0.2 μl) from a series of dilutions (10 dilutions) were added to a plate. Cell viability was measured after a 72 h incubation with small molecule inhibitors. Each small molecule concentration was presented in duplicate.
Results
Systematic genomics analysis of existing drug targets
We devised a systematic pipeline that integrates various biological properties and identifies novel potential anti-cancer drug targets (Figure 1A). First, we collected genome-level datasets representing a wealth of information about human cancers, including gene essentiality, mRNA expression, DNA copy number alteration, somatic mutation patterns as well as PPI network data. We then systematically analyzed how much these genomic and systems properties can distinguish drug targets from other proteins. Next, we generated three cancer type-specific classifiers to characterize targets that have functional relevance in given cancers.
Figure 1.
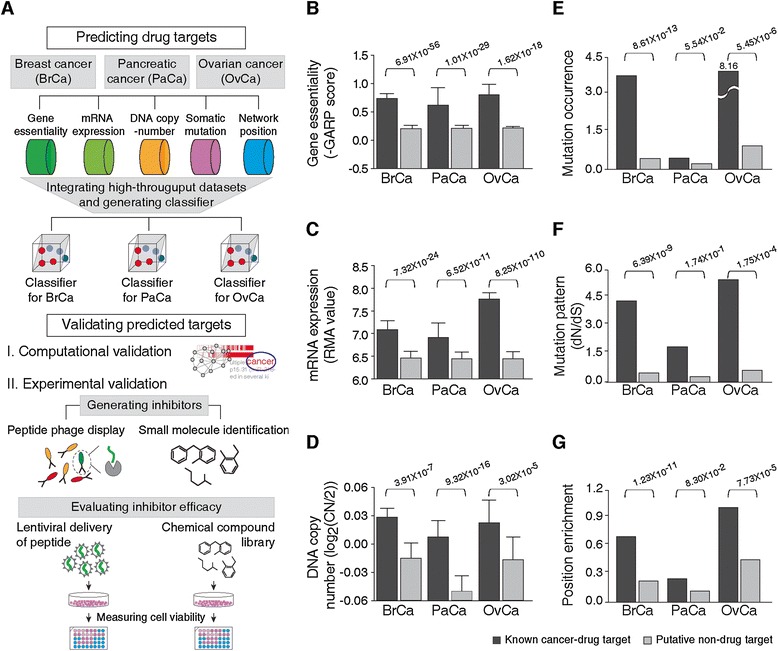
Biological properties of drug targets. Genomic signatures of drug targets for breast cancer (BrCa), pancreatic cancer (PaCa) and ovarian cancer (OvCa) are shown. (A) Overview of the systematic pipeline to identify and validate novel anti-cancer drug targets. (B) Gene essentiality, (C) mRNA expression, (D) DNA copy number, (E) mutation occurrence, (F) mutation pattern and (G) position enrichment of known cancer drug targets (black bars) and putative non-drug targets (gray bars) are compared. Error bars indicate standard deviation of drug targets.
To build a classifier, we first needed to generate a gold standard set. To this end, we generated a positive set (known targets of approved and clinically tested anti-cancer drugs, which are specific to cancer types) by extracting 62, 69 and 45 drug targets for BrCa, PaCa and OvCa. As a negative set, we generated a list of 5,169 putative non-drug targets by focusing on proteins that are not currently drug targets and that are not associated with cancer pathogenesis (for details see Methods and Additional file 4). We found that cancer drug targets possessed particular genomic signatures specific to the given cancer type. First, drug targets tend to be specifically essential in the given cancer types. The essentialities of BrCa, PaCa and OvCa drug targets (indicating lethality in response to shRNA-mediated knockdown in cell lines derived from this particular cancer type) are 3.88, 3.04 and 3.64 times higher, respectively, than those of putative non-drug targets in the given cancer types (average P-value = 1.62 × 10-18; Figure 1B). In mRNA expression studies in cancer cell lines, drug targets also show significantly higher expression levels in the given cancer type compared with putative non-drug targets (average P-value = 2.17 × 10-11; Figure 1C). Moreover, the likelihood of being a drug target increases consistently with increasing gene essentiality and mRNA expression (Additional file 6). Similar to gene essentiality and mRNA expression, drug targets have higher DNA copy number in given cancer types (average P-value = 1.08 × 10-5; Figure 1D) and are likely to be localized in the amplified region (copy number >2.5) in chromosomes compared to putative non-drug targets (Additional file 6).
We also found that cancer drug targets have unique somatic mutation patterns. Drug targets are mutated more frequently in the given cancer compared to putative non-drug targets (Figure 1E). In BrCa, on average, there were 3.32 mutations per BrCa drug target, while there were 0.37 mutations in a putative non-drug target. Likewise, mutation occurrences of PaCa and OvCa drug targets are 2.05 and 9.99 times higher than those of putative non-drug targets. Also, drug targets show signs of selection; we measured the ratio of the number of non-synonymous mutations to the number of synonymous mutations (dN/dS ratio) and found that drug targets showed significantly higher dN/dS ratios than putative non-drug targets in all cancer types (Figure 1F). Moreover, there is a significant clustering of mutations in specific amino acid positions in drug targets (Figure 1G), consistent with the fact that specific positions in drug targets act as drivers and play important roles in cancer pathogenesis [61].
In order to investigate cancer drug targets in the context of other proteins, we examined network positions of cancer drug targets in a global human PPI network. Drug targets are located at the center of the network; they show significantly higher degree, betweenness and closeness centrality (P-value < 10-7) and have lower clustering coefficients compared with the non-drug targets (average P-value = 1.16 × 10-3; Additional file 7). We observed similar network properties of drug targets when we used a different type of PPI network derived from the high-throughput yeast two-hybrid screens [62,63] (Additional file 8). Taken together, unique genomic and network topological properties of known cancer drug targets allow for distinguishing novel cancer drug targets from other proteins. We hence sought to integrate the differentiating power of these features to extract an optimized priority list of drug targets for each of the three cancer types.
Predicting novel cancer drug targets
We then adopted a machine learning algorithm to integrate the above features into a unified classifier that can distinguish potential drug targets specific to cancer type from other proteins. Based on the notion that choosing a relevant subset of the original features avoids overfitting and leads to better performance in machine learning [64], we evaluated these genomic and network topological properties as input features for machine learning and selected the most relevant features using a SVM-REF method [38] (see Methods and Additional file 1). These relevant features are gene essentiality score (GARP score), mRNA expression intensity (RMA score), DNA copy number, mutation occurrence and closeness centrality in the PPI network. Using these features and an SVM algorithm with a RBF kernel, we generated three classifiers that can predict potential drug targets specific to BrCa, PaCa and OvCa. From 10-fold cross-validation on the data sets (known BrCa, PaCa and OvCa drug targets as a positive set and putative non-drug targets as a negative set), we correctly assigned 55 of 62 BrCa drug targets (88.71% sensitivity), 43 of 69 PaCa drug targets (62.32% sensitivity) and 29 of 45 OvCa drug targets (64.44% sensitivity). Overall, the three classifiers showed an accuracy of 91.69% and a specificity of 91.91% (Table 1). Next, we evaluated individual features in terms of their discriminative power by measuring the area under the receiver operating characteristic curve (AUC) and found that our integrated approach far outperformed all single features (Figure 2A; Additional file 9). The average performance of our classifiers (AUC) is 0.78. Meanwhile, on average, single feature-based approaches achieve an AUC of 0.61. For a more comprehensive evaluation, we compared the precision-recall characteristics of our approach with those of single feature-based predictions. The areas under the precision-recall curves (AUCPRs) of our classifiers are 4 to 15 times higher than AUCPRs of single-based predictions (Additional file 9).
Table 1.
Performance evaluation of classifiers
| Classifier | ACC | Sensitivity | Specificity | BAC a | AUC |
|---|---|---|---|---|---|
| BrCa | 93.33 | 88.71 | 93.38 | 91.05 | 78.46 |
| PaCa | 89.65 | 62.32 | 90.02 | 76.17 | 77.47 |
| OvCa | 92.08 | 64.44 | 92.32 | 78.38 | 79.31 |
aBAC is balanced accuracy, which is defined as the arithmetic mean of sensitivity and specificity.
Figure 2.
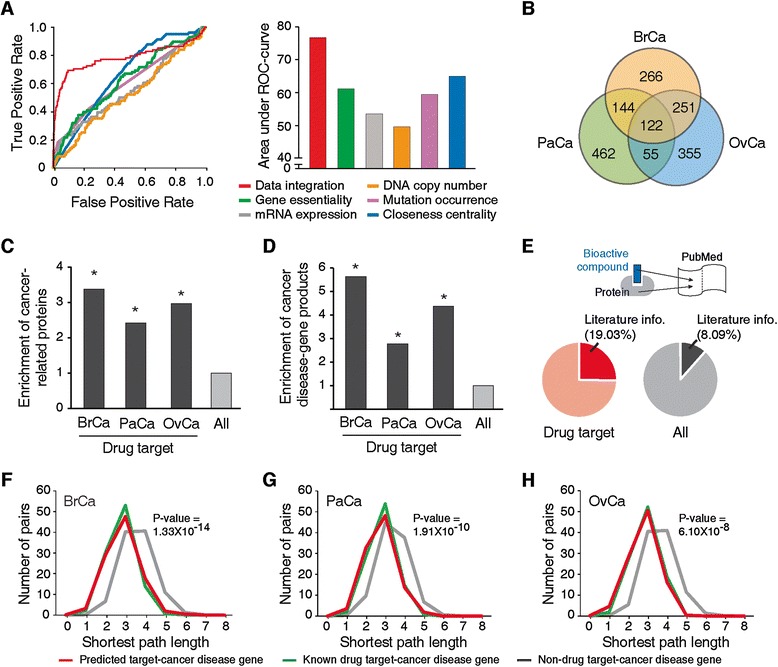
Performance evaluation of classifier and biological properties of predicted anti-cancer drug targets. (A) Receiver operating characteristic (ROC) curve (left) and area under the ROC curve (AUC; right) of integrated approach and single dataset-based approaches are compared. (B) Venn diagram of predicted drug targets for BrCa, PaCa and OvCa. (C,D) The possibility to find cancer-related proteins (C) and to find cancer disease genes (D) in cancer drug targets (black) are compared with those possibilities of all human proteins (All, gray). (E) Analysis of pre-existing literature dealing with cancer pathogenesis. The applications of bioactive compounds (inhibitors and antagonists) of cancer drug targets (red) and all human proteins (All, gray) are compared. (F-H) Distributions of shortest path lengths of BrCa (F), PaCa (G) and OvCa (H). Shortest path length between predicted cancer drug targets and cancer disease genes (red), between known cancer drug targets and cancer disease genes (green) and between non-drug targets and cancer disease genes (gray) in a PPI network are shown. *P-value < 1.00 × 10-5.
Finally, to predict cancer drug targets on a genome-wide scale, we applied our optimized classifiers to 15,663 human proteins and measured the probability of each to be a suitable drug target specific to cancer types. We considered all putative cancer drug targets that are within the top 5% of our probability scores. At this cutoff, predicted cancer drug targets showed low false-positive rates ranging from 1.41% to 2.20% depending on cancer types (Additional file 10). Of the predicted drug targets, 122 are global-cancer targets that are observed in all cancer types and 266, 462 and 355 are specific to BrCa, PaCa and OvCa, respectively (Figure 2B; Additional file 11). Our scores represent a prioritization of potential cancer drug targets, which is representative of their importance in cancer as measured by the various features we integrated. Of course, it is in itself not yet an identification of real drug targets, but more to be understood as a guideline. Whether there is a potential venue for inhibition (that is, some measure of 'druggability') is investigated below.
To evaluate the reliability of our prediction results, we compared our predictions with two other approaches that used different methods to identify anti-cancer drug targets. For the identification of drug targets, one approach modeled metabolic networks [65] and the other studies identified target candidates that have negative genetic interactions [66]. We found that a total of 22.1% of targets from the two approaches overlap with our predictions (Additional file 12A), while there is no overlap between the predictions of the two approaches. Also, we compiled a list of known drug targets (116 targets) of very well-studied anti-cancer drugs using a cancer drug resistance database (CancerDR) [67]. Our predictions overlap significantly with these known anti-cancer drug targets (P-value = 8.29 × 10-54; Additional file 12B). About 60% of known anti-cancer drug targets (69 targets) are predicted as drug targets. Meanwhile, only two predicted targets from the other approaches overlap with these known anti-cancer targets (P-value >0.5). Furthermore, when we relax the score cutoff, 95% of the known anti-cancer drug targets (110 targets) are ranked within the top 30% of probability scores, suggesting that our probability score is reliable to identify potential anti-cancer drug targets (Additional file 12C).
Properties of putative drug targets
With the list of putative targets at hand, we investigated their biological and cellular properties and evaluated their reliability using several genome- and network-wide analyses. First, we assessed whether predicted cancer drug targets are enriched with cancer-related proteins (for example, oncogene and tumor-suppressor gene products) and cancer disease gene products (their genetic defects are directly implicated in oncogenesis; see Methods). We observed that predicted targets are significantly related to cancer pathogenesis. As shown in Figure 2C, the chance to find cancer-related proteins in the predicted targets is about three times higher than in all human proteins (P-values of all cancer types <1.00 × 10-5). Also, the chances of predicted targets to be the products of cancer-disease genes are 5.63 (BrCa), 2.77 (PaCa) and 4.37 (OvCa) times higher than those of all human proteins (P-values of all cancer types <1.00 × 10-5; Figure 2D). Next, we validated the predicted targets based on the pre-existing literature on cancer pathogenesis. Using a text-mining method, we examined the experimental applications of bioactive compounds (inhibitors or antagonists) and found that compounds inhibiting our predicted targets are more frequently used for cancer research compared to the compounds of other human proteins (P-value = 1.90 × 10-3). Bioactive compounds associated with 19.03% of drug targets (315 targets) have been applied to cancer research, while 8.09% of human proteins and their compounds have been used in cancer research (Figure 2E).
Finally, we examined the relationship between predicted cancer drug targets and cancer disease gene products in the human PPI network. We hypothesized that suitable cancer drug targets are likely to be located close to disease gene products in the network [68]. Indeed, we observed that predicted drug targets are significantly closer to cancer disease gene products than other proteins (P-values of all cancer types <1.00 × 10-5; red lines in Figure 2F-H). In particular, the average shortest path length between predicted targets and cancer disease gene products is 2.84, which is similar to the shortest path length between known drug targets and cancer disease gene products (2.81). Meanwhile, the average shortest path length between non-drug targets and disease gene products is 3.55 (the average shortest path length of the entire network is 3.39). These results imply that our classifier correctly captures potential anti-cancer drug targets. Having a set of potential cancer drug target candidates at hand, we sought to devise strategies to find inhibitors to these molecules.
Analysis of druggability according to drug classes
We next investigated structural and cellular properties of our predicted cancer drug target candidates to obtain potential avenues for their inhibition (Figure 3A). We focused on three classes of potential cancer therapeutics, namely antibodies, peptide-based compounds and small molecules.
Figure 3.
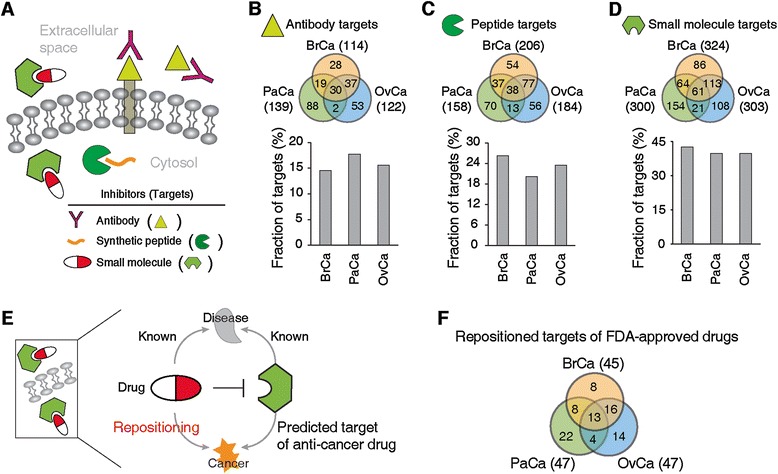
Classification of predicted targets depending on therapeutics classes. (A) Biochemical and cellular properties of targets depending on therapeutic classes. (B) Targets for antibodies. (C) Targets for synthetic peptides. (D) Targets for small molecules. Venn diagrams show the overlap of therapeutic class-specific targets depending on cancer type and gray bars represent fraction of therapeutic class-specific targets in all predicted anti-cancer targets. (E) Identification of repositioned drugs and their targets. (F) Overlap of repositioned targets of Food and Drug Administration (FDA)-approved drugs that have high specificity (number of targets is less than five proteins).
First, to define targets for antibodies, we searched for targets that have extracellular domains and are thus serum-accessible, as antibodies cannot traverse the cell membrane (Figure 3A). We found a total of 257 potential antibody targets (Figure 3B; Additional file 11). Among them, 30 are predicted to affect all cancer types, whereas 28, 88 and 53 are specific to BrCa, PaCa and OvCa, respectively. These antibody targets comprise about 16% of predicted targets in each cancer type. We found that antibodies against several of our predicted targets have been shown to have efficacy in pre-clinical settings. For example, antibodies against CD44 (BrCa drug target), FLT3 (PaCa drug target) and EPHB2 (OvCa drug target) reduce tumor cell invasion and engraftment in cancer leading to antibody-dependent cell-mediated cytotoxicity [69–71]. Also, immunotoxins against CD22 and CD19, which are global cancer targets, showed their effectiveness in eliminating acute lymphoblastic leukemia cells [72]. These identified proteins could be targeted with antibodies using established techniques such as hybridoma or phage display [17].
Second, we identified 345 potential peptide targets that have known peptide-binding domains and are thus targetable with synthetic peptides. As extracellular targets can be efficiently targeted using antibodies, we focused here on intracellular targets (assuming that peptides can be manipulated to cross the membrane [18,19]). On average, 23.33% of predicted targets have these features in each cancer type. Among them, 54, 70 and 56 are specific targets for BrCa, PaCa and OvCa (Figure 3C; Additional file 11). It has been shown that synthetic peptides binding to Zap70, Lck and Src (global cancer targets) block signaling downstream of these proteins and induce apoptosis in cancer cells [73,74]. For validations, we generated inhibitory peptides against a number of these targets as potential drug leads and evaluated their efficacy (see below).
Finally, for potential small molecule targets, we mined extensive databases of existing small molecule inhibitors and their targeting proteins. We selected all predicted targets for which small molecules have been characterized and used for experimental studies (inhibitors and antagonists) and clinical applications (approved and experimental drugs). We found that a total of 607 targets can be inhibited by small molecules in a given cancer (Figure 3D; Additional file 11). They comprise about 40% of predicted targets in each cancer type. Among them, 86 (BrCa), 154 (PaCa) and 108 (OvCa) are cancer type-specific targets. Indeed, we found that small molecule targets have functional relevance in specific cancer pathogenesis. For instance, the inhibitors of aurora kinase B (AURKB; a BrCa drug target) and serine/threonine protein kinase Chk1 (CHEK1; a PaCa drug target) have been shown to reduce cancer cell proliferation [75] and induce DNA damage [76]. The inhibition of pituitary tumor-transforming protein (PTTG1), an OvCa target, restricts sister chromatic separation and tumorigenesis, and thus has been studied as an important target for ovarian cancer chemotherapy [77].
Of particular interest are small molecules that are already approved as drugs. Associating alternative indications with approved drugs is a rapid way to find potential cancer drug therapies (repositioned drugs) and target molecules (repositioned targets). To explore the repositioned drugs and their targets, we searched for the subset of small molecules that are already approved drugs and inhibit our predicted cancer drug targets (Figure 3E). We found 85 repositioned targets that are inhibited by 224 US Food and Drug Administration (FDA) -approved drugs that have relatively high specificity (that is, that target less than five proteins). Among the repositioned targets, 13 are global cancer targets while 8, 22 and 14 are specific targets for BrCa, PaCa and OvCa, respectively (Figure 3F). Furthermore, we identified additional potential targets that are inhibited by approved compounds of lower specificity (see Additional file 13 for details). These targets would be prime candidates for validation as inhibitory small molecules are already available.
Generation of peptide inhibitors against predicted targets
We next sought to validate our methodology and assess the feasibility of our targets. To this end, we generated synthetic peptide inhibitors to two of our peptide binding PaCa targets: spliceosome-associated cyclophilin (PPWD1) and Nuclear RNA export factor 1 (NXF1). PPWD1 (WD40 domain) and NXF1 (LRR domain) have well-characterized peptide binding domains that play important roles in cancer pathogenesis (top and left panels in Figure 4A,B; see Methods for details). PPWD1 and NXF1 have similar biological properties of known drug targets in PaCa cell lines. They show high levels of mRNA expression, gene essentiality, DNA copy number, and closeness centrality in PPI networks resulting in high probability scores to be reliable PaCa targets (top and right panels in Figure 4A,B). Using peptide-phage display, we successfully obtained peptide binders against PPWD1-WD40 and NXF1-LRR (see Methods for details). After five rounds of panning against PPWD1-WD40 and NXF1-LRR, 44 clones were amplified and phage ELISA revealed 4 unique peptide binders for PPWD1-WD40 and 11 for NXF1-LRR, which we can represent as a position weight matrix (graphically as a Logo as in Figure 4A,B). Identified PPWD1-WD40-binding peptides commonly have the (G/A)P motif, while NXF1-LRR-binding peptides have the GFEXLR motif (bottom and left panels in Figure 4A,B). We selected the peptides showing the highest affinity signal (phage ELISA value) for further studies; KVYTAPNRQDNYVIQN for PPWD1-WD40 and GFETLWARHAQGQTQV for NXF1-LRR.
Figure 4.
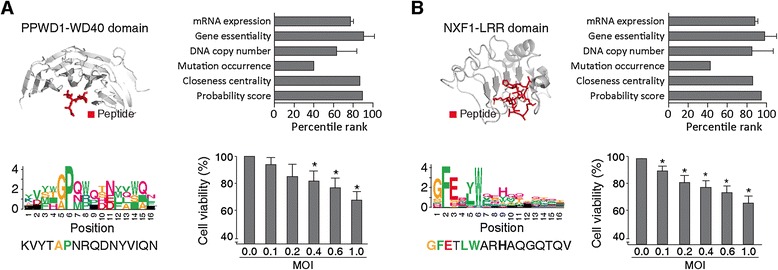
Generation and biological evaluation of peptide binders against predicted drug targets. (A,B) Peptide targets PPWD1-WD40 domain (A) and NXF1-LRR domain (B) are selected based on the known domain-peptide structures (top and left) and genomic/network topological properties and probability score (top and right). Structures of WD40 domain (Protein Data Bank (PDB) ID: 1NEX) and LRR domain (PDB ID: 3P72) are shown. High-affinity peptide binders against PPWD1-WD and NXF1-LRR and their sequence motifs are shown (bottom and left). Cell viabilities depending on the infection efficiency of lentiviruses are compared (bottom and right). Both peptides show decrease in cell viability in a dose-dependent manner as opposed to GFP control (Additional file 13). MOI represents multiplicity of infection. *P-value <0.1.
Next, we evaluated the biological effects of the two peptide binders in cells. To introduce peptide binders into cells, we used a lentiviral delivery system, which is a powerful tool to deliver protein or peptide of interest to cells with high transduction efficiency [78]. RWP1 PaCa cells, which exhibit high mRNA expression levels of PPWD1 and NXF1, were infected with peptide-expressing lentiviruses (with the peptide fused on a GFP scaffold; see Methods for details). We measured cell viabilities by changing the MOI of lentivirus and found that peptide binders have suppressive effects in PaCa cells. As shown in Figure 4 (bottom and right panels), lentiviral infection reduced cell viability in a dose-dependent manner, whereas a GFP control construct resulted in no significant changes in cell viability (Additional file 14). Lentivirus infection caused 30.11% (PPWD1-WD40) and 31.22% (NXF1-LRR) reductions of cell viability at the highest concentration (MOI = 1). These results suggest that drugs based on these peptides could be used as therapeutic agents for cancer therapy.
Validation of small molecule inhibitors to our drug targets
We next sought to validate the identified small molecule targets using high-throughput chemical library screens, by measuring their effects on the viability of PaCa cells. To this end, we selected two commercially available libraries that contain 137 inhibitors of 113 PaCa targets and 1,206 compounds that inhibit other proteins (Figure 5A; Additional file 15). To define reliable targets of inhibitors, we only considered targets that have strong interactions with inhibitors as derived from the STITCH database (STITCH score > 0.7) [46]. We then measured the viability of Panc0813 cells, a PaCa cell line, after treatment with these libraries. We selected the Panc0813 cell line as it shows relatively high expression levels of our targets, including the target of dasatinib, our positive control. While screening in a single cell line is not definite proof for the efficacy of the inhibitors of our targets, we do believe it goes a long way to emphasize the strength of our method. Our results showed that PaCa inhibitors exhibit strong anti-cancer activity as treatment leads to significant reduction of Panc0813 cell viability compared to treatment with other compounds (P-value = 2.64 × 10-3; Additional file 16). As shown in Figure 5B, inhibitors of predicted PaCa targets were almost twice as likely to show strong inhibition (reducing cell viability more than 50%, 17.52% versus 9.87%; left panel of Figure 5B). Meanwhile, treatment using the majority of other compounds (59.20%) resulted in unchanged cell viability (cell viability ≥70%; right panel of Figure 5B).
Figure 5.
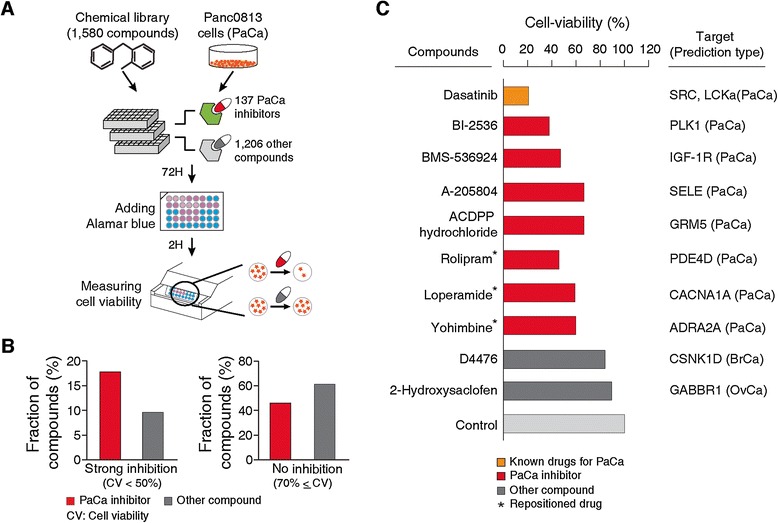
Screening of chemical compounds against predicted cancer drug targets. (A) Procedure to screen high-throughput chemical library against Panc0813 cells. (B) The fraction of compounds that lead to strong inhibition (cell viability <50%, left) and unchanged cell viability (cell viability ≥70%, right). Red bars indicate the inhibitors of PaCa targets; gray bars indicate other tested compounds that inhibit non-PaCa targets or non-drug targets. (C) Cell viabilities after treatment with positive control, dasatanib, which is an experimentally/clinically studied small molecule for pancreatic cancer treatment (orange), inhibitors of PaCa targets (red) and other compounds (dark gray) are compared. DMSO is considered as control (light gray). Asterisks indicate repositioned drugs.
We especially focused on eight PaCa inhibitors that have high specificities (number of binding proteins ≤5) and that have been shown to efficiently inhibit our predicted targets (half maximal inhibitory concentrations (IC50) ranging from 0.2 nM to 870 nM; Additional file 17). The targets of these eight PaCa inhibitors show high levels of mRNA expression, gene essentiality, DNA copy number, and closeness centrality in PPI networks, resulting in high probability scores to be reliable PaCa targets (Additional file 18). To evaluate the effect of PaCa inhibitors on cell viability, we compared their effect on cell viability with the effect of a negative set of other compounds. To address the issue that many compounds have multiple targets, we chose multiple sets with varying overlap in their targets with the targets of our PaCa inhibitors. We found that PaCa inhibitors reduced cell viability significantly stronger than the negative set. The compounds binding to none of the proteins that are bound by PaCa inhibitors did not affect PaCa cell viability (cell viability of 76.16%; the overall cell viability measured by all screened compounds was 76.72%). Also, compounds with limited overlapping sets of targets (share one to three targets with PaCa inhibitors) showed similar levels of cell viability (73.25% cell viability). Meanwhile, eight PaCa inhibitors resulted in about 53% cell viability (P-value <0.05; Additional file 19A). Furthermore, we measured the statistical significance of cell viability that is induced by a single PaCa inhibitor. All PaCa inhibitors reduced cell viability significantly (P-value <0.05; Additional file 19B).
We found several studies that show the potential efficacies of PaCa inhibitors. For instance, BI-2536 inhibits Polo-like kinase (PLK1; STITCH score = 0.973 and IC50 = 0.83 nM), a predicted PaCa target, and has shown anti-proliferative potency against pancreatic adenocarcinoma in both in vitro and in vivo studies [79]. BMS-536924, which is an inhibitor of insulin-like grown factor-1 receptor (IGF-1R; STITCH score = 0.987 and IC50 = 100 nM), blocks cancer cell growth and mediates apoptosis [80]. Indeed, treatment with BI-2536 and BMS-536924 showed significant loss of cell viability (cell viabilities of 37.77% and 47.15%, respectively; Figure 5C) in cell line screens. Meanwhile, treatment with other compounds (for example, D4476, a CSNK1D inhibitor with STITCH score = 0.084 and IC50 = 300 nM; and 2-hydroxysaclofen, a GABBR1 inhibitor with STITCH score = 0.77 and IC50 = 11 μM) does not affect PaCa cell viability (90% cell viability; dark gray bars in Figure 5C; Additional file 19B).
Importantly, we were able to validate three of the 'drug repositioning' targets mentioned above (Figure 5C). We observed reduced viability of Panc0813 cells in response to treatments with several approved and experimental drugs. For example, one FDA-approved anti-gastroenteritis drug, loperamide, shows loss of cell viability (59.11%), presumably by targeting the voltage-gated calcium channel subunit alpha-1A (CACNA1A; STITCH score = 0.82 and IC50 = 870 nM). Yohimbine, which is an approved inhibitor of adrenoceptor alpha 2A (ADRA2A; STITCH score = 0.997 and IC50 = 3.67 nM) and has been explored as a therapeutic for impotence and type II diabetes, also leads to loss of viability of PaCa cells (59.92%). Furthermore, rolipram, which is under phase II clinical trial as an anti-inflammatory drug, reduces cell viability (45.86%) by inhibiting phosphodiesterase 4D (PDE4D; STITCH score = 0.997 and IC50 = 31.6 nM). As these are approved drugs (loperamide and yohimbine) and in clinical trials (rolipram), they are prime candidates for further study as potential new pancreatic cancer drugs.
To evaluate the dosage dependence of the effect of our small molecule inhibitors (eight PaCa inhibitors and two non-PaCa inhibitors) on PaCa cell survival, we measured cell viability using different small molecule concentrations. We found that more than half of PaCa inhibitors reduced cell viability in the low micromolar range. Of eight PaCa inhibitors, five reduced cell viability in a dose-dependent manner (Additional file 20; dasatinib, BMS-536924, A-205804, rolipram and loperamide). Dasatinib, which has been studied as a drug for pancreatic cancer treatment [81], reduced cell viability to 50% at a concentration of 0.063 μM. Other PaCa inhibitors, BMS-536924, A-205804, rolipram and loperamide, reduced cell viability to 70% or less at concentrations ranging from 0.5 μM to 2.5 μM. In particular, we found that two repositioned drugs, rolipram and loperamide, reduced cell viability to 50% at a concentration of around 5 μM. Meanwhile, the negative controls, two non-PaCa inhibitors (D-4476 and 2-hydroxysaclofen) did not affect PaCa cell viability. On average, they resulted in cell viability of 98.07% regardless of their concentration. Taken together, these results suggest that a portion of our predicted targets (five of eight in these validations) show dose-dependent effects upon inhibition, thereby offering further experimental validation of our approach. As no prediction is perfect, three of our eight predicted PaCa small molecule inhibitors did not show a dose-dependent effect, although neither of the two negative controls did either.
As a complementary approach to validate our predicted small molecule targets, we analyzed available high-throughput drug screening data that were compiled in CancerDR [67]. We examined IC50 values against 1,054 various types of cancer cell lines (including 51 BrCa, 37 PaCa and 18 OvCa cell lines that have been used to test more than three inhibitors of our predicted targets) after treating with 148 small molecules, including 39 known inhibitors of our predicted targets. The inhibitors of our predicted targets showed inhibitory activities at lower concentrations compared to the inhibitors of non-targets in the given cancer types (Additional file 21). Inhibitors of BrCa targets showed lower IC50 values in 41 BrCa cell lines (80.39% of tested BrCa cell lines). Similarly, inhibitors of PaCa targets (30 out of 37 cell lines) and OvCa targets (18 out of 18 cell lines) showed better performance. Furthermore, we found that inhibition efficiencies of cancer target inhibitors are stronger in the given cancer compared with other cancer types (Additional file 22). For example, the IC50 value of PaCa target inhibitors is 25.45 μm in PaCa cell lines, 2.81 times stronger than in other cancer cells (71.48 μm). Inhibitors of BrCa targets and OvCa targets also showed stronger effects in given cancer cell lines. These results suggest that our predicted cancer drug targets have functional relevance in cancer pathogenesis and thus would be appropriate candidates for anti-cancer therapeutics design.
Discussion
In this study, we demonstrate the importance of large-scale data integration in identifying novel anti-cancer drug targets. While there have been previous attempts to predict drug targets, they have been limited due to a lack of diversity of their datasets. Our results emphasize the strong individual roles of gene essentiality, mRNA expression, somatic mutation, DNA copy number and network centrality to determine anti-cancer drug targets. Indeed, we found that potential cancer drug targets are likely to be essential, over-expressed, amplified and frequently mutated in the given cancer types and have crucial roles to maintain the PPI network. It suggests that effective integration of genomic and systemic uniqueness of drug targets captured dynamic regulation properties of cancer drug targets, leading to the improved prediction. Identification and validation of novel drug targets is of course a lengthy and difficult procedure; we believe that our work is helpful to give an initial prioritization of proteins.
In addition to five major biological properties that we used as features for cancer drug target identification, several biological properties that are related to gene expression/function regulation and genome evolution would be applied as features to identify potential drug targets. It has been shown that SNPs that affect rheumatoid arthritis-related pathways are enriched in drug targets that are known to be used for the treatment of rheumatoid arthritis [82]. Also, systematic mapping of tumor-specific transcriptional networks and identification of negative genetic interactions have been applied as a feature to identify therapeutic targets for cancer [66,83]. Though these features have been applied for drug target identification, their relatively low coverage, due to low-throughput screening and/or low coverage of the human genome, limits their usefulness for genome-wide identification of drug targets. In the future, when genome-wide data on those features are available, we expect that we can include them in our predictor and provide more accurate and reliable target information.
Inhibitory strategies that can ultimately lead to the development of new therapeutics are of crucial importance. We thus present an integrated approach that shows three different inhibitory strategies for the predicted cancer drug targets: using antibodies, synthetic peptides and small molecules (Figure 4). We thus show a direct route to validate these targets in further experiments. We did so in a few initial experiments to demonstrate the validity of our approach. To this end, we performed high-throughput chemical compound screening to evaluate the validity of our results under more physiological conditions and found several compounds that reduce cell viability by inhibiting our predicted targets (Figure 5; Additional file 16). Of course, these are only preliminary validations and more experiments are needed to establish our predictions as bona fide novel targets. As well as repositioned drugs, we suggest that the discussed small molecule inhibitors have potential applications for cancer therapeutics. For example, treatment with A-205804 (E-selectin (SELE) inhibitor) and ACDPP hydrochloride (metabotropic glutamate receptor 5 (GRM5) inhibitor) resulted in drastic reduction of the viability of Panc0813 cells (66% cell viability; Figure 5C). It has been suggested that down-regulation of SELE and GRM5 significantly reduces cancer metastasis [84] and cancer tumorigenesis [85]. Even if further chemical optimizations of A-205804 and ACDPP hydrochloride are required to improve efficacy and specificity, these results imply possible applications of these inhibitors for further development against pancreatic cancer. Furthermore, we identified 92 novel inhibitor candidates of PaCa targets that resulted in reduced cell viability (cell viability <70%; Additional file 23).
One particular promising venue to obtain novel therapies is by repositioning existing drugs, as many of the pitfalls of classical drug development can be sidestepped this way. Indeed, we compiled a list of existing approved drugs that inhibit some of our predicted targets. Interestingly, we found that some repositioned drugs have clinical indications related to cancer. For instance, yohimbine, which inhibits ADRA2A and has been explored as a treatment for type II diabetes, has been investigated for its ability to induce apoptosis and inhibit cell proliferation of pancreatic cancer cells [86]. It has been suggested that inhibition of ADRA2A alters the p21ras-mitogen-activated protein (MAP) kinase cascade via a Gi-mediated pathway and leads to apoptosis of cancer cells [87]. Interestingly, it has been shown that cancer development is correlated with the development of type II diabetes by the common alteration of the insulin-like growth factor 1 receptor signaling pathway, which is sensitive to insulin resistance and affects growth and differentiation of cancer cells [88]. Rolipram, which is an anti-inflammatory drug and inhibits PDE4D, has been shown to alter cell cycle progression, leading to apoptosis of leukemia cells [89]. It has been shown that inhibition of PDE4D enhances intracellular cAMP, which controls several inflammatory cell functions [90], and increased levels of cAMP induce apoptosis and cell cycle arrest in cancer cells [91]. Also, several studies have found that anti-inflammatory drugs exert their anti-inflammatory and anti-tumor effects through the inhibition of the cyclooxygenase-2 (COX-2) signaling pathway [92]. These results imply that treatments using repositioned drugs can modify metabolic flux and signaling pathways affecting the common pathophysiologic mechanisms underlying cancer and consequently alter cancer growth and proliferation.
Ultimately, our integrated approach generates a number of promising leads for novel cancer therapies, which are now straightforward to follow-up on. The obvious next steps are to perform similar analyses focusing on genetically (rather than histologically) defined cancer subtypes. Recently, several studies performed meta-analyses of cancer signatures (for example, somatic mutations and copy number changes) with thousands of tumors and suggested shared and cancer type-specific oncogenic properties [10,93]. Such oncogenic signatures could be incorporated into our predictor for the reliable prediction of cancer subtype-specific drug targets. In future studies, two or more of our predicted cancer drug targets, as well as existing drug targets, could be exploited using combinatorial drug therapy by blocking different signaling pathways and preventing cross-talk between pathways in cancer. Correctly predicting possible synergistic effects between two or more drugs will be an exciting venue for future studies.
Conclusion
An ongoing challenge of cancer research is to prioritize the selection of cancer drug targets, as is evident by the slow development of novel anti-cancer agents. We developed a computational model to identify and validate novel anti-cancer drug targets on a genome-wide scale. We generated peptide inhibitors to high-scoring targets using phage display and validate a subset of our novel drug targets by showing efficacy of their inhibitors in cancer cell lines. Furthermore, we carried out high-throughput chemical library screens showing novel effects of known inhibitory small molecule compounds. Beyond the three types of cancers we analyzed, there are many other types of diseases for which various genomic and systematic datasets are available. We believe that the application of our integrated approach has the potential to provide a list of drug target candidates for other human diseases.
Acknowledgements
We thank Roland Arnold and all lab members for technical assistance and valuable discussion. This work was supported by an operating grant of the Canadian Institute for Health Research (MOP-123526), Basic Science Research Program through the National Research Foundation of Korea funded by the Ministry of Education, Science and Technology (357-2011-1-C00143) and NSERC-CREATE Training Program (384338-10). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Abbreviations
- AUC
area under the receiver operating characteristic curve
- AUCPR
area under the precision-recall curve
- BrCa
breast cancer
- CCLE
Cancer Cell Line Encyclopedia
- DMSO
dimethyl sulfoxide
- ELISA
enzyme-linked immunosorbent assay
- FDA
Food and Drug Administration
- GARP
Gene Activity Ranking Profile
- GFP
green fluorescent protein
- GST
glutathione S-transferase
- MOI
multiplicity of infection
- OMIM
Online Mendelian Inheritance in Man
- OvCa
ovarian cancer
- PaCa
pancreatic cancer
- PCR
polymerase chain reaction
- PPI
protein-protein interaction
- RBF
radial basis function
- RMA
robust multi-array average
- ROC
receiver operating characteristic
- shRNA
short hairpin RNA
- SVM
support vector machine
- SVM-REF
support vector machine-recursive feature elimination
Additional files
Tested genomic and systemic properties of proteins.
Fraction of samples depending on different biological properties of known drug targets.
Number of mutations observed from COSMIC database and exome sequencing data. Mutations that are observed in (A) BrCa, (B) PaCa and (C) OvCa are compared.
Known anti-cancer drug targets and non-drug targets.
Optimized SVM parameters and prediction models.
Likelihood ratios of (A) gene essentiality, (B) mRNA expression and (C) DNA copy number.
Network topological properties.
Network topological properties of high-throughput yeast two-hybrid screens.
Performance evaluations of classifiers. ROC curves and AUCs of (A) PaCa drug target classifier and (B) OvCa drug target classifier. Precision-recall curves of (C) BrCa, (D) PaCa and (E) OvCa classifiers are presented. Performance of integrated approach and single data-based approaches are compared.
Optimization of probability score. False positive rates are calculated depending on the probability scores. Red bars indicate the false positive rate at the top 5% of probability scores.
Targets for small molecules, antibodies, and synthetic peptides.
Performance comparison. (A) Venn diagram of prediction results of our approach and another two approaches. (B) Enrichment of known anti-cancer drug targets in our prediction (top), a modeling-based approach (middle) and a genetic interaction-based approach (bottom). (C) Enrichment of known anti-cancer drug targets depending on probability score. Red dot indicates the number of known anti-cancer drug targets that are ranked within the top 5% of probability scores.
Repositioned targets and drugs.
Cell viabilities depending on the infection efficiency of lentiviruses. Cell viability is measured after infecting PaCa cells with GFP-expressing lentiviruses.
Inhibitors of predicted targets.
Cell viability distributions. The cell viability associated with PaCa inhibitors (red) and other compounds (gray) is shown.
Target specificity of PaCa inhibitors.
Percentile ranks of each biological property and overall prediction score of tested targets of small molecule inhibitors.
Statistical significance of PaCa inhibitor-induced cell viability. (A) Comparison of cell viabilities that are changed by PaCa inhibitors (red) and other compounds that have limited overlapping sets of PaCa inhibitor targets (gray). (B) Cell viability distribution of all screened compounds.
Dose-response curves of PaCa inhibitors. Eight PaCa inhibitors and two non-PaCa inhibitors at 10 different concentrations were used to treat PaCa cells. Observed cell viability is represented by gray circles. Red line represents the fitted dose-response curve.
Half maximal inhibitory concentration (IC50) of inhibitors of BrCa, PaCa and OvCa targets. IC50 values in (A) 51 BrCa cell lines, (B) 37 PaCa cell lines and (C) 18 OvCa cell lines are compared. These cell lines are used to test more than three inhibitors of our predicted targets.
Half maximal inhibitory concentration (IC50) of inhibitors in different cell types. (A) IC50 of BrCa inhibitors in BrCa cell lines, (B) IC50 of PaCa inhibitors in PaCa cell lines and (C) IC50 of OvCa inhibitors in OvCa cell lines are compared with IC50 values in other cell lines.
Novel PaCa target inhibitors.
Footnotes
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
JJ implemented the integrated method and analyzed data. SN and AD performed chemical compound library screening. JT performed the phage display screening and characterization of the peptides. SN performed the lentiviral delivery and growth experiments. SSS, JM, JW and PMK contributed to the conception of drug target identification and experimental validation. JJ and PMK drafted the manuscript. All authors read and approved the final manuscript.
Contributor Information
Jouhyun Jeon, Email: jouhyun.jeon@utoronto.ca.
Satra Nim, Email: satra.nim@kimlab.org.
Joan Teyra, Email: j.teyra@utoronto.ca.
Alessandro Datti, Email: datti@lunenfeld.ca.
Jeffrey L Wrana, Email: wrana@lunenfeld.ca.
Sachdev S Sidhu, Email: sachdev.sidhu@utoronto.ca.
Jason Moffat, Email: j.moffat@utoronto.ca.
Philip M Kim, Email: pi@kimlab.org.
References
- 1.Rask-Andersen M, Almen MS, Schioth HB. Trends in the exploitation of novel drug targets. Nat Rev Drug Discov. 2011;10:579–590. doi: 10.1038/nrd3478. [DOI] [PubMed] [Google Scholar]
- 2.Marcotte R, Brown KR, Suarez F, Sayad A, Karamboulas K, Krzyzanowski PM, Sircoulomb F, Medrano M, Fedyshyn Y, Koh JL, van Dyk D, Fedyshyn B, Luhova M, Brito GC, Vizeacoumar FJ, Vizeacoumar FS, Datti A, Kasimer D, Buzina A, Mero P, Misquitta C, Normand J, Haider M, Ketela T, Wrana JL, Rottapel R, Neel BG, Moffat J. Essential gene profiles in breast, pancreatic, and ovarian cancer cells. Cancer Discov. 2012;2:172–189. doi: 10.1158/2159-8290.CD-11-0224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Liotta L, Petricoin E. Molecular profiling of human cancer. Nat Rev Genet. 2000;1:48–56. doi: 10.1038/35049567. [DOI] [PubMed] [Google Scholar]
- 4.Beroukhim R, Mermel CH, Porter D, Wei G, Raychaudhuri S, Donovan J, Barretina J, Boehm JS, Dobson J, Urashima M, Mc Henry KT, Pinchback RM, Ligon AH, Cho YJ, Haery L, Greulich H, Reich M, Winckler W, Lawrence MS, Weir BA, Tanaka KE, Chiang DY, Bass AJ, Loo A, Hoffman C, Prensner J, Liefeld T, Gao Q, Yecies D, Signoretti S, et al. The landscape of somatic copy-number alteration across human cancers. Nature. 2010;463:899–905. doi: 10.1038/nature08822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mardis ER, Wilson RK. Cancer genome sequencing: a review. Hum Mol Genet. 2009;18:R163–R168. doi: 10.1093/hmg/ddp396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cancer Genome Atlas Research N Integrated genomic analyses of ovarian carcinoma. Nature. 2011;474:609–615. doi: 10.1038/nature10166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cancer Genome Atlas Research N Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature. 2008;455:1061–1068. doi: 10.1038/nature07385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Masica DL, Karchin R. Correlation of somatic mutation and expression identifies genes important in human glioblastoma progression and survival. Cancer Res. 2011;71:4550–4561. doi: 10.1158/0008-5472.CAN-11-0180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hodis E, Watson IR, Kryukov GV, Arold ST, Imielinski M, Theurillat JP, Nickerson E, Auclair D, Li L, Place C, Dicara D, Ramos AH, Lawrence MS, Cibulskis K, Sivachenko A, Voet D, Saksena G, Stransky N, Onofrio RC, Winckler W, Ardlie K, Wagle N, Wargo J, Chong K, Morton DL, Stemke-Hale K, Chen G, Noble M, Meyerson M, Ladbury JE, et al. A landscape of driver mutations in melanoma. Cell. 2012;150:251–263. doi: 10.1016/j.cell.2012.06.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ciriello G, Miller ML, Aksoy BA, Senbabaoglu Y, Schultz N, Sander C. Emerging landscape of oncogenic signatures across human cancers. Nat Genet. 2013;45:1127–1133. doi: 10.1038/ng.2762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Taylor BS, Schultz N, Hieronymus H, Gopalan A, Xiao Y, Carver BS, Arora VK, Kaushik P, Cerami E, Reva B, Antipin Y, Mitsiades N, Landers T, Dolgalev I, Major JE, Wilson M, Socci ND, Lash AE, Heguy A, Eastham JA, Scher HI, Reuter VE, Scardino PT, Sander C, Sawyers CL, Gerald WL. Integrative genomic profiling of human prostate cancer. Cancer Cell. 2010;18:11–22. doi: 10.1016/j.ccr.2010.05.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Telleria CM. Drug repurposing for cancer therapy. J Cancer Sci Ther. 2012;4:ix–xi. doi: 10.4172/1948-5956.1000e108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lamb J, Crawford ED, Peck D, Modell JW, Blat IC, Wrobel MJ, Lerner J, Brunet JP, Subramanian A, Ross KN, Reich M, Hieronymus H, Wei G, Armstrong SA, Haggarty SJ, Clemons PA, Wei R, Carr SA, Lander ES, Golub TR. The Connectivity Map: using gene-expression signatures to connect small molecules, genes, and disease. Science. 2006;313:1929–1935. doi: 10.1126/science.1132939. [DOI] [PubMed] [Google Scholar]
- 14.Campillos M, Kuhn M, Gavin AC, Jensen LJ, Bork P. Drug target identification using side-effect similarity. Science. 2008;321:263–266. doi: 10.1126/science.1158140. [DOI] [PubMed] [Google Scholar]
- 15.Hu G, Agarwal P. Human disease-drug network based on genomic expression profiles. PLoS One. 2009;4:e6536. doi: 10.1371/journal.pone.0006536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Keiser MJ, Setola V, Irwin JJ, Laggner C, Abbas AI, Hufeisen SJ, Jensen NH, Kuijer MB, Matos RC, Tran TB, Whaley R, Glennon RA, Hert J, Thomas KL, Edwards DD, Shoichet BK, Roth BL. Predicting new molecular targets for known drugs. Nature. 2009;462:175–181. doi: 10.1038/nature08506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bradbury AR, Sidhu S, Dubel S, McCafferty J. Beyond natural antibodies: the power of in vitro display technologies. Nat Biotechnol. 2011;29:245–254. doi: 10.1038/nbt.1791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Vallespi MG, Fernandez JR, Torrens I, Garcia I, Garay H, Mendoza O, Granadillo M, Falcon V, Acevedo B, Ubieta R, Guillen GE, Reyes O. Identification of a novel antitumor peptide based on the screening of an Ala-library derived from the LALF(32-51) region. J Pept Sci. 2010;16:40–47. doi: 10.1002/psc.1192. [DOI] [PubMed] [Google Scholar]
- 19.Ueyama H, Horibe T, Nakajima O, Ohara K, Kohno M, Kawakami K. Semaphorin 3A lytic hybrid peptide binding to neuropilin-1 as a novel anti-cancer agent in pancreatic cancer. Biochem Biophys Res Commun. 2011;414:60–66. doi: 10.1016/j.bbrc.2011.09.021. [DOI] [PubMed] [Google Scholar]
- 20.Aina OH, Sroka TC, Chen ML, Lam KS. Therapeutic cancer targeting peptides. Biopolymers. 2002;66:184–199. doi: 10.1002/bip.10257. [DOI] [PubMed] [Google Scholar]
- 21.Barretina J, Caponigro G, Stransky N, Venkatesan K, Margolin AA, Kim S, Wilson CJ, Lehar J, Kryukov GV, Sonkin D, Reddy A, Liu M, Murray L, Berger MF, Monahan JE, Morais P, Meltzer J, Korejwa A, Jané-Valbuena J, Mapa FA, Thibault J, Bric-Furlong E, Raman P, Shipway A, Engels IH, Cheng J, Yu GK, Yu J, Aspesi P, Jr, de Silva M, et al. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature. 2012;483:603–607. doi: 10.1038/nature11003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Forbes SA, Bindal N, Bamford S, Cole C, Kok CY, Beare D, Jia M, Shepherd R, Leung K, Menzies A, Teague JW, Campbell PJ, Stratton MR, Futreal PA. COSMIC: mining complete cancer genomes in the Catalogue of Somatic Mutations in Cancer. Nucleic Acids Res. 2011;39:D945–D950. doi: 10.1093/nar/gkq929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shah SP, Roth A, Goya R, Oloumi A, Ha G, Zhao Y, Turashvili G, Ding J, Tse K, Haffari G, Bashashati A, Prentice LM, Khattra J, Burleigh A, Yap D, Bernard V, McPherson A, Shumansky K, Crisan A, Giuliany R, Heravi-Moussavi A, Rosner J, Lai D, Birol I, Varhol R, Tam A, Dhalla N, Zeng T, Ma K, Chan SK, et al. The clonal and mutational evolution spectrum of primary triple-negative breast cancers. Nature. 2012;486:395–399. doi: 10.1038/nature10933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wood LD, Parsons DW, Jones S, Lin J, Sjoblom T, Leary RJ, Shen D, Boca SM, Barber T, Ptak J, Silliman N, Szabo S, Dezso Z, Ustyanksky V, Nikolskaya T, Nikolsky Y, Karchin R, Wilson PA, Kaminker JS, Zhang Z, Croshaw R, Willis J, Dawson D, Shipitsin M, Willson JK, Sukumar S, Polyak K, Park BH, Pethiyagoda CL, Pant PV, et al. The genomic landscapes of human breast and colorectal cancers. Science. 2007;318:1108–1113. doi: 10.1126/science.1145720. [DOI] [PubMed] [Google Scholar]
- 25.Jones S, Zhang X, Parsons DW, Lin JC, Leary RJ, Angenendt P, Mankoo P, Carter H, Kamiyama H, Jimeno A, Hong SM, Fu B, Lin MT, Calhoun ES, Kamiyama M, Walter K, Nikolskaya T, Nikolsky Y, Hartigan J, Smith DR, Hidalgo M, Leach SD, Klein AP, Jaffee EM, Goggins M, Maitra A, Iacobuzio-Donahue C, Eshleman JR, Kern SE, Hruban RH, et al. Core signaling pathways in human pancreatic cancers revealed by global genomic analyses. Science. 2008;321:1801–1806. doi: 10.1126/science.1164368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang L, Tsutsumi S, Kawaguchi T, Nagasaki K, Tatsuno K, Yamamoto S, Sang F, Sonoda K, Sugawara M, Saiura A, Hirono S, Yamaue H, Miki Y, Isomura M, Totoki Y, Nagae G, Isagawa T, Ueda H, Murayama-Hosokawa S, Shibata T, Sakamoto H, Kanai Y, Kaneda A, Noda T, Aburatani H. Whole-exome sequencing of human pancreatic cancers and characterization of genomic instability caused by MLH1 haploinsufficiency and complete deficiency. Genome Res. 2012;22:208–219. doi: 10.1101/gr.123109.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bossi A, Lehner B. Tissue specificity and the human protein interaction network. Mol Syst Biol. 2009;5:260. doi: 10.1038/msb.2009.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.ᅟ: NetworkX.ᅟ ᅟ, ᅟ:ᅟ [https://networkx.lanl.gov]
- 29.ᅟ: NCI Drug Dictionary.ᅟ ᅟ, ᅟ:ᅟ [http://www.cancer.gov/drugdictionary]
- 30.Chen X, Ji ZL, Chen YZ. TTD: Therapeutic Target Database. Nucleic Acids Res. 2002;30:412–415. doi: 10.1093/nar/30.1.412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Knox C, Law V, Jewison T, Liu P, Ly S, Frolkis A, Pon A, Banco K, Mak C, Neveu V, Djoumbou Y, Eisner R, Guo AC, Wishart DS. DrugBank 3.0: a comprehensive resource for 'omics' research on drugs. Nucleic Acids Res. 2011;39:D1035–D1041. doi: 10.1093/nar/gkq1126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Punta M, Coggill PC, Eberhardt RY, Mistry J, Tate J, Boursnell C, Pang N, Forslund K, Ceric G, Clements J, Heger A, Holm L, Sonnhammer EL, Eddy SR, Bateman A, Finn RD. The Pfam protein families database. Nucleic Acids Res. 2012;40:D290–D301. doi: 10.1093/nar/gkr1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.ᅟ: LIBSVM - A library for Support Vector Machines.ᅟ ᅟ, ᅟ:ᅟ [http://www.csie.ntu.edu.tw/~cjlin/libsvm]
- 34.Nam H, Chung BC, Kim Y, Lee K, Lee D. Combining tissue transcriptomics and urine metabolomics for breast cancer biomarker identification. Bioinformatics. 2009;25:3151–3157. doi: 10.1093/bioinformatics/btp558. [DOI] [PubMed] [Google Scholar]
- 35.Radivojac P, Peng K, Clark WT, Peters BJ, Mohan A, Boyle SM, Mooney SD. An integrated approach to inferring gene-disease associations in humans. Proteins. 2008;72:1030–1037. doi: 10.1002/prot.21989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Plaimas K, Eils R, Konig R. Identifying essential genes in bacterial metabolic networks with machine learning methods. BMC Syst Biol. 2010;4:56. doi: 10.1186/1752-0509-4-56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gertrudes JC, Maltarollo VG, Silva RA, Oliveira PR, Honorio KM, da Silva AB. Machine learning techniques and drug design. Curr Med Chem. 2012;19:4289–4297. doi: 10.2174/092986712802884259. [DOI] [PubMed] [Google Scholar]
- 38.Saeys Y, Inza I, Larranaga P. A review of feature selection techniques in bioinformatics. Bioinformatics. 2007;23:2507–2517. doi: 10.1093/bioinformatics/btm344. [DOI] [PubMed] [Google Scholar]
- 39.Valente TW, Coronges K, Lakon C, Costenbader E. How Correlated Are Network Centrality Measures? Connect (Tor) 2008;28:16–26. [PMC free article] [PubMed] [Google Scholar]
- 40.Tang Y, Zhang YQ, Chawla NV, Krasser S. SVMs modeling for highly imbalanced classification. IEEE Trans Syst Man Cybern B Cybern. 2009;39:281–288. doi: 10.1109/TSMCB.2008.2002909. [DOI] [PubMed] [Google Scholar]
- 41.Magrane M, Consortium U. UniProt Knowledgebase: a hub of integrated protein data. Database (Oxford) 2011;2011:bar009. doi: 10.1093/database/bar009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gerber DE. Targeted therapies: a new generation of cancer treatments. Am Fam Physician. 2008;77:311–319. [PubMed] [Google Scholar]
- 43.Vanhee P, Reumers J, Stricher F, Baeten L, Serrano L, Schymkowitz J, Rousseau F. PepX: a structural database of non-redundant protein-peptide complexes. Nucleic Acids Res. 2010;38:D545–D551. doi: 10.1093/nar/gkp893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gaulton A, Bellis LJ, Bento AP, Chambers J, Davies M, Hersey A, Light Y, McGlinchey S, Michalovich D, Al-Lazikani B, Overington JP. ChEMBL: a large-scale bioactivity database for drug discovery. Nucleic Acids Res. 2012;40:D1100–D1107. doi: 10.1093/nar/gkr777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhao XM, Iskar M, Zeller G, Kuhn M, van Noort V, Bork P. Prediction of drug combinations by integrating molecular and pharmacological data. PLoS Comput Biol. 2011;7:e1002323. doi: 10.1371/journal.pcbi.1002323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kuhn M, Szklarczyk D, Franceschini A, von Mering C, Jensen LJ, Bork P. STITCH 3: zooming in on protein-chemical interactions. Nucleic Acids Res. 2012;40:D876–D880. doi: 10.1093/nar/gkr1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.ᅟ: CancerGenes Database.ᅟ ᅟ, ᅟ:ᅟ [http://www.cbioportal.org/public-portal/index.do]
- 48.Higgins ME, Claremont M, Major JE, Sander C, Lash AE. CancerGenes: a gene selection resource for cancer genome projects. Nucleic Acids Res. 2007;35:D721–D726. doi: 10.1093/nar/gkl811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Futreal PA, Coin L, Marshall M, Down T, Hubbard T, Wooster R, Rahman N, Stratton MR. A census of human cancer genes. Nat Rev Cancer. 2004;4:177–183. doi: 10.1038/nrc1299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Vogelstein B, Kinzler KW. Cancer genes and the pathways they control. Nat Med. 2004;10:789–799. doi: 10.1038/nm1087. [DOI] [PubMed] [Google Scholar]
- 51.Hamosh A, Scott AF, Amberger JS, Bocchini CA, McKusick VA. Online Mendelian Inheritance in Man (OMIM), a knowledgebase of human genes and genetic disorders. Nucleic Acids Res. 2005;33:D514–D517. doi: 10.1093/nar/gki033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Stirnimann CU, Petsalaki E, Russell RB, Muller CW. WD40 proteins propel cellular networks. Trends Biochem Sci. 2010;35:565–574. doi: 10.1016/j.tibs.2010.04.003. [DOI] [PubMed] [Google Scholar]
- 53.Liker E, Fernandez E, Izaurralde E, Conti E. The structure of the mRNA export factor TAP reveals a cis arrangement of a non-canonical RNP domain and an LRR domain. EMBO J. 2000;19:5587–5598. doi: 10.1093/emboj/19.21.5587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Culjkovic-Kraljacic B, Borden KL. Aiding and abetting cancer: mRNA export and the nuclear pore. Trends Cell Biol. 2013;23:328–335. doi: 10.1016/j.tcb.2013.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Letunic I, Doerks T, Bork P. SMART 7: recent updates to the protein domain annotation resource. Nucleic Acids Res. 2012;40:D302–D305. doi: 10.1093/nar/gkr931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lees J, Yeats C, Perkins J, Sillitoe I, Rentzsch R, Dessailly BH, Orengo C. Gene3D: a domain-based resource for comparative genomics, functional annotation and protein network analysis. Nucleic Acids Res. 2012;40:D465–D471. doi: 10.1093/nar/gkr1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Huang H, Sidhu SS. Studying binding specificities of peptide recognition modules by high-throughput phage display selections. Methods Mol Biol. 2011;781:87–97. doi: 10.1007/978-1-61779-276-2_6. [DOI] [PubMed] [Google Scholar]
- 58.Tonikian R, Zhang Y, Boone C, Sidhu SS. Identifying specificity profiles for peptide recognition modules from phage-displayed peptide libraries. Nat Protoc. 2007;2:1368–1386. doi: 10.1038/nprot.2007.151. [DOI] [PubMed] [Google Scholar]
- 59.Naldini L, Blomer U, Gallay P, Ory D, Mulligan R, Gage FH, Verma IM, Trono D. In vivo gene delivery and stable transduction of nondividing cells by a lentiviral vector. Science. 1996;272:263–267. doi: 10.1126/science.272.5259.263. [DOI] [PubMed] [Google Scholar]
- 60.Zufferey R, Nagy D, Mandel RJ, Naldini L, Trono D. Multiply attenuated lentiviral vector achieves efficient gene delivery in vivo. Nat Biotechnol. 1997;15:871–875. doi: 10.1038/nbt0997-871. [DOI] [PubMed] [Google Scholar]
- 61.Greenman C, Stephens P, Smith R, Dalgliesh GL, Hunter C, Bignell G, Davies H, Teague J, Butler A, Stevens C, Edkins S, O'Meara S, Vastrik I, Schmidt EE, Avis T, Barthorpe S, Bhamra G, Buck G, Choudhury B, Clements J, Cole J, Dicks E, Forbes S, Gray K, Halliday K, Harrison R, Hills K, Hinton J, Jenkinson A, Jones D, et al. Patterns of somatic mutation in human cancer genomes. Nature. 2007;446:153–158. doi: 10.1038/nature05610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Rual JF, Venkatesan K, Hao T, Hirozane-Kishikawa T, Dricot A, Li N, Berriz GF, Gibbons FD, Dreze M, Ayivi-Guedehoussou N, Klitgord N, Simon C, Boxem M, Milstein S, Rosenberg J, Goldberg DS, Zhang LV, Wong SL, Franklin G, Li S, Albala JS, Lim J, Fraughton C, Llamosas E, Cevik S, Bex C, Lamesch P, Sikorski RS, Vandenhaute J, Zoghbi HY, et al. Towards a proteome-scale map of the human protein-protein interaction network. Nature. 2005;437:1173–1178. doi: 10.1038/nature04209. [DOI] [PubMed] [Google Scholar]
- 63.Stelzl U, Worm U, Lalowski M, Haenig C, Brembeck FH, Goehler H, Stroedicke M, Zenkner M, Schoenherr A, Koeppen S, Timm J, Mintzlaff S, Abraham C, Bock N, Kietzmann S, Goedde A, Toksöz E, Droege A, Krobitsch S, Korn B, Birchmeier W, Lehrach H, Wanker EE. A human protein-protein interaction network: a resource for annotating the proteome. Cell. 2005;122:957–968. doi: 10.1016/j.cell.2005.08.029. [DOI] [PubMed] [Google Scholar]
- 64.Dobson PD, Doig AJ. Distinguishing enzyme structures from non-enzymes without alignments. J Mol Biol. 2003;330:771–783. doi: 10.1016/S0022-2836(03)00628-4. [DOI] [PubMed] [Google Scholar]
- 65.Folger O, Jerby L, Frezza C, Gottlieb E, Ruppin E, Shlomi T. Predicting selective drug targets in cancer through metabolic networks. Mol Syst Biol. 2011;7:501. doi: 10.1038/msb.2011.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Lu X, Kensche PR, Huynen MA, Notebaart RA. Genome evolution predicts genetic interactions in protein complexes and reveals cancer drug targets. Nat Commun. 2013;4:2124. doi: 10.1038/ncomms3124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kumar R, Chaudhary K, Gupta S, Singh H, Kumar S, Gautam A, Kapoor P, Raghava GP. CancerDR: cancer drug resistance database. Sci Rep. 2013;3:1445. doi: 10.1038/srep01445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Yildirim MA, Goh KI, Cusick ME, Barabasi AL, Vidal M. Drug-target network. Nat Biotechnol. 2007;25:1119–1126. doi: 10.1038/nbt1338. [DOI] [PubMed] [Google Scholar]
- 69.Dohadwala M, Luo J, Zhu L, Lin Y, Dougherty GJ, Sharma S, Huang M, Pold M, Batra RK, Dubinett SM. Non-small cell lung cancer cyclooxygenase-2-dependent invasion is mediated by CD44. J Biol Chem. 2001;276:20809–20812. doi: 10.1074/jbc.C100140200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Small D. Targeting FLT3 for the treatment of leukemia. Semin Hematol. 2008;45:S17–S21. doi: 10.1053/j.seminhematol.2008.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Landen CN, Jr, Lu C, Han LY, Coffman KT, Bruckheimer E, Halder J, Mangala LS, Merritt WM, Lin YG, Gao C, Schmandt R, Kamat AA, Li Y, Thaker P, Gershenson DM, Parikh NU, Gallick GE, Kinch MS, Sood AK. Efficacy and antivascular effects of EphA2 reduction with an agonistic antibody in ovarian cancer. J Natl Cancer Inst. 2006;98:1558–1570. doi: 10.1093/jnci/djj414. [DOI] [PubMed] [Google Scholar]
- 72.Herrera L, Farah RA, Pellegrini VA, Aquino DB, Sandler ES, Buchanan GR, Vitetta ES. Immunotoxins against CD19 and CD22 are effective in killing precursor-B acute lymphoblastic leukemia cells in vitro. Leukemia. 2000;14:853–858. doi: 10.1038/sj.leu.2401779. [DOI] [PubMed] [Google Scholar]
- 73.Nishikawa K, Sawasdikosol S, Fruman DA, Lai J, Songyang Z, Burakoff SJ, Yaffe MB, Cantley LC. A peptide library approach identifies a specific inhibitor for the ZAP-70 protein tyrosine kinase. Mol Cell. 2000;6:969–974. doi: 10.1016/S1097-2765(05)00085-7. [DOI] [PubMed] [Google Scholar]
- 74.Eldar-Finkelman H, Eisenstein M. Peptide inhibitors targeting protein kinases. Curr Pharm Des. 2009;15:2463–2470. doi: 10.2174/138161209788682253. [DOI] [PubMed] [Google Scholar]
- 75.VanderPorten EC, Taverna P, Hogan JN, Ballinger MD, Flanagan WM, Fucini RV. The Aurora kinase inhibitor SNS-314 shows broad therapeutic potential with chemotherapeutics and synergy with microtubule-targeted agents in a colon carcinoma model. Mol Cancer Ther. 2009;8:930–939. doi: 10.1158/1535-7163.MCT-08-0754. [DOI] [PubMed] [Google Scholar]
- 76.Toledo LI, Murga M, Fernandez-Capetillo O. Targeting ATR and Chk1 kinases for cancer treatment: a new model for new (and old) drugs. Mol Oncol. 2011;5:368–373. doi: 10.1016/j.molonc.2011.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Panguluri SK, Yeakel C, Kakar SS. PTTG: an important target gene for ovarian cancer therapy. J Ovarian Res. 2008;1:6. doi: 10.1186/1757-2215-1-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Cockrell AS, Kafri T. Gene delivery by lentivirus vectors. Mol Biotechnol. 2007;36:184–204. doi: 10.1007/s12033-007-0010-8. [DOI] [PubMed] [Google Scholar]
- 79.Mross K, Dittrich C, Aulitzky WE, Strumberg D, Schutte J, Schmid RM, Hollerbach S, Merger M, Munzert G, Fleischer F, Scheulen ME. A randomised phase II trial of the Polo-like kinase inhibitor BI 2536 in chemo-naive patients with unresectable exocrine adenocarcinoma of the pancreas - a study within the Central European Society Anticancer Drug Research (CESAR) collaborative network. Br J Cancer. 2012;107:280–286. doi: 10.1038/bjc.2012.257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Hofmann F, Garcia-Echeverria C. Blocking the insulin-like growth factor-I receptor as a strategy for targeting cancer. Drug Discov Today. 2005;10:1041–1047. doi: 10.1016/S1359-6446(05)03512-9. [DOI] [PubMed] [Google Scholar]
- 81.Morton JP, Karim SA, Graham K, Timpson P, Jamieson N, Athineos D, Doyle B, McKay C, Heung MY, Oien KA, Frame MC, Evans TR, Sansom OJ, Brunton VG. Dasatinib inhibits the development of metastases in a mouse model of pancreatic ductal adenocarcinoma. Gastroenterology. 2010;139:292–303. doi: 10.1053/j.gastro.2010.03.034. [DOI] [PubMed] [Google Scholar]
- 82.Bakir-Gungor B, Sezerman OU. A new methodology to associate SNPs with human diseases according to their pathway related context. PLoS One. 2011;6:e26277. doi: 10.1371/journal.pone.0026277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Karamouzis MV, Papavassiliou AG. Transcription factor networks as targets for therapeutic intervention of cancer: the breast cancer paradigm. Mol Med. 2011;17:1133–1136. doi: 10.1007/s00894-010-0818-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Yoshimoto K, Tajima H, Ohta T, Okamoto K, Sakai S, Kinoshita J, Furukawa H, Makino I, Hayashi H, Nakamura K, Oyama K, Inokuchi M, Nakagawara H, Itoh H, Fujita H, Takamura H, Ninomiya I, Kitagawa H, Fushida S, Fujimura T, Wakayama T, Iseki S, Shimizu K. Increased E-selectin in hepatic ischemia-reperfusion injury mediates liver metastasis of pancreatic cancer. Oncol Rep. 2012;28:791–796. doi: 10.3892/or.2012.1896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Prickett TD, Samuels Y. Molecular pathways: dysregulated glutamatergic signaling pathways in cancer. Clin Cancer Res. 2012;18:4240–4246. doi: 10.1158/1078-0432.CCR-11-1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Shen SG, Zhang D, Hu HT, Li JH, Wang Z, Ma QY. Effects of alpha-adrenoreceptor antagonists on apoptosis and proliferation of pancreatic cancer cells in vitro. World J Gastroenterol. 2008;14:2358–2363. doi: 10.3748/wjg.14.2358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Alblas J, van Corven EJ, Hordijk PL, Milligan G, Moolenaar WH. Gi-mediated activation of the p21ras-mitogen-activated protein kinase pathway by alpha 2-adrenergic receptors expressed in fibroblasts. J Biol Chem. 1993;268:22235–22238. [PubMed] [Google Scholar]
- 88.Tognon CE, Sorensen PH. Targeting the insulin-like growth factor 1 receptor (IGF1R) signaling pathway for cancer therapy. Expert Opin Ther Targets. 2012;16:33–48. doi: 10.1517/14728222.2011.638626. [DOI] [PubMed] [Google Scholar]
- 89.Moon E, Lee R, Near R, Weintraub L, Wolda S, Lerner A. Inhibition of PDE3B augments PDE4 inhibitor-induced apoptosis in a subset of patients with chronic lymphocytic leukemia. Clin Cancer Res. 2002;8:589–595. [PubMed] [Google Scholar]
- 90.Hatzelmann A, Schudt C. Anti-inflammatory and immunomodulatory potential of the novel PDE4 inhibitor roflumilast in vitro. J Pharmacol Exp Ther. 2001;297:267–279. [PubMed] [Google Scholar]
- 91.Savai R, Pullamsetti SS, Banat GA, Weissmann N, Ghofrani HA, Grimminger F, Schermuly RT. Targeting cancer with phosphodiesterase inhibitors. Expert Opin Investig Drugs. 2010;19:117–131. doi: 10.1517/13543780903485642. [DOI] [PubMed] [Google Scholar]
- 92.Wang D, Dubois RN. The role of COX-2 in intestinal inflammation and colorectal cancer. Oncogene. 2010;29:781–788. doi: 10.1038/onc.2009.421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Kandoth C, McLellan MD, Vandin F, Ye K, Niu B, Lu C, Xie M, Zhang Q, McMichael JF, Wyczalkowski MA, Leiserson MD, Miller CA, Welch JS, Walter MJ, Wendl MC, Ley TJ, Wilson RK, Raphael BJ, Ding L. Mutational landscape and significance across 12 major cancer types. Nature. 2013;502:333–339. doi: 10.1038/nature12634. [DOI] [PMC free article] [PubMed] [Google Scholar]