

Background: The structures of infectious mammalian prions remain unclear.
Results: Based in part on NMR data, we developed models with single PrP molecules spanning the entire cross-section of prion fibrils.
Conclusion: These models are consistent with many empirical features of prion amyloids.
Significance: We provide a new basis for conceptualizing and experimentally evaluating the structures and propagation of infectious prions.
Keywords: Amyloid, Molecular Dynamics, Nuclear Magnetic Resonance (NMR), Prion, Protein Structure
Abstract
Structures of the infectious form of prion protein (e.g. PrPSc or PrP-Scrapie) remain poorly defined. The prevalent structural models of PrPSc retain most of the native α-helices of the normal, noninfectious prion protein, cellular prion protein (PrPC), but evidence is accumulating that these helices are absent in PrPSc amyloid. Moreover, recombinant PrPC can form amyloid fibrils in vitro that have parallel in-register intermolecular β-sheet architectures in the domains originally occupied by helices 2 and 3. Here, we provide solid-state NMR evidence that the latter is also true of initially prion-seeded recombinant PrP amyloids formed in the absence of denaturants. These results, in the context of a primarily β-sheet structure, led us to build detailed models of PrP amyloid based on parallel in-register architectures, fibrillar shapes and dimensions, and other available experimentally derived conformational constraints. Molecular dynamics simulations of PrP(90–231) octameric segments suggested that such linear fibrils, which are consistent with many features of PrPSc fibrils, can have stable parallel in-register β-sheet cores. These simulations revealed that the C-terminal residues ∼124–227 more readily adopt stable tightly packed structures than the N-terminal residues ∼90–123 in the absence of cofactors. Variations in the placement of turns and loops that link the β-sheets could give rise to distinct prion strains capable of faithful template-driven propagation. Moreover, our modeling suggests that single PrP monomers can comprise the entire cross-section of fibrils that have previously been assumed to be pairs of laterally associated protofilaments. Together, these insights provide a new basis for deciphering mammalian prion structures.
Introduction
The main event in the pathogenesis of prion diseases or transmissible spongiform encephalopathies is the conformational conversion of the normal monomeric prion protein, PrPC,4 to an ordered oligomeric/multimeric and infectious isoform, PrPSc (1–3). PrPC is a glycophosphatidylinositol (GPI)-anchored glycoprotein, the physiological and pathophysiological functions of which remain unclear (4, 5). The three-dimensional structures of many mammalian PrPC molecules are well understood and conserved evolutionarily, with a C-terminal domain (residues 125–231, hamster sequence) containing three α-helices (H1, residues 145–153; H2, 172–194; and H3, 200–226), a short anti-parallel β-sheet (B1, residues 129–133, and B2, 160–163), and a flexibly disordered N-terminal half (residues 23–124) (6). Relative to PrPC, PrPSc is usually relatively protease-resistant, and as such, PK-resistant species are often called PrPRes. The ultrastructures of PrPRes range from ill-defined, but apparently nonfibrillar or subfibrillar oligomers (7–12), to long linear unbranched amyloid fibrils (13, 14).
However, more detailed elucidation of PrPRes structure has been impeded by technical limitations of conformational analyses of insoluble, noncrystalline protein aggregates like PrPRes. Nevertheless, it has long been clear that PrPRes formation involves massive refolding of flexibly disordered segments of PrPC and at least some of the α-helices into a multimeric structure with a preponderance of β-sheets and -turns (15–17). After treatment with PK, an infectious PrPRes core remains, which varies with strain but typically spans residues ∼90–231 (i.e. PrPRes(90–231)) (18–21). Early circular dichroism (CD) analyses detected no α-helical content in PrPRes(90–231) (17), and initial Fourier transform infrared (FTIR) analyses suggested that less than half of the original α-helix remained (15, 16). More recently, improved purifications of PrPRes coupled with new FTIR spectroscopy and hydrogen/deuterium (H/D) exchange analyses have confirmed that it is highly unlikely that much, if any, of the native α-helices of PrPC remain in PrPRes amyloid, at least in rodent PrPRes isolates (22, 23).
In addition to studies performed directly on fully infectious brain-derived PrPRes, there have been numerous efforts to learn about PrP amyloid fibril formation by analyzing fibrils formed in vitro from recombinant PrP (rPrP) (3). Such fibrils tend to be either noninfectious or many orders of magnitude less infectious than natural tissue-derived PrPRes (24–26), but like bona fide PrPRes fibrils (13, 14), they are linear, unbranched, and of similar dimensions. The assembly of fibrils using rPrP molecules labeled with site-specific paramagnetic or isotopic probes has facilitated electron paramagnetic resonance (27) and solid-state (ss) NMR (28–31) analyses of the fibril architecture. Such studies have revealed that spontaneously formed rPrP fibrils are assembled via parallel in-register intermolecular β-sheets involving the core residues 173–224, including those formerly comprising helices 2 and 3 in PrPC (27, 29). Furthermore, PrP fragments comprising helices 2 and 3 can form high β-sheet amyloid fibrils under native-like conditions (32). These and other studies have clearly demonstrated that the majority of the helical secondary structure of rPrPC can refold into extended chains that are stacked in register to form fibrils. Studies with amyloid fibrils of recombinant PrP N-terminal residues 23–144 have also indicated parallel in-register β-sheet structures involving residues ∼113–125 and ∼130–140 (28, 30, 31). Moreover, comparisons of in vitro generated spontaneous rPrP fibrils to brain-derived PrPRes fibrils by FTIR spectroscopy and H/D exchange mass spectrometry indicate that major refolding of both the flexible N-terminal and helical C-terminal domains of PrPC occur in forming fully infectious PrPRes (22, 23). Finally, key precedents for parallel in-register intermolecular β-sheet structures in prions come from the yeast prions URE3, PSI+, and PIN+, which are self-propagating infectious amyloids of Ure2p, Sup35p, and Rnq1p domains, respectively (33, 34). Each of these yeast prions have such architectures (35–37), as is the case for amyloid of the Aβ peptide (34, 38, 39) using ssNMR (40, 41).
Here, we report the use of ssNMR to test whether, like noninfectious rPrP fibrils formed spontaneously under partially denaturing conditions (29), prion-seeded rPrP fibrils formed under more physiological conditions also have a parallel in-register β-sheet architecture in the C-terminal disulfide-linked loop formed from helices 2 and 3 in PrPC. We then considered the implications that such packing would have on the folding and intermolecular interactions of the more N-terminal residues that are tightly packed in fully infectious PrPRes (22). Combining these findings with a variety of other constraints on PrPRes structure, we have assembled the first detailed parallel in-register intermolecular β-sheet-based models of PrPRes fibrils and subjected them to energy minimizations and molecular dynamics (MD) simulations.
EXPERIMENTAL PROCEDURES
Protein Expression and Purification
DNA sequences encoding residues 90–231 of the Syrian hamster PrP sequence were cloned into the pET-23 vector as described previously (42). Rosetta BL21(DE3) Escherichia coli cells were transformed with this construct, and starter cultures were grown overnight at 37 °C in LB broth supplemented with chloramphenicol (34 μg/ml) and kanamycin (15 μg/ml). Cells were transferred to 1 liter of minimal medium that contained, per liter, 200 ml of salts (65 g/liter KH2PO4, 50 g/liter K2HPO4, 45 g/liter Na2PO4, 10 g/liter NH4Cl), 10 ml of trace elements (6 g/liter FeSO4, 6 g/liter CaCl2, 1.2 g/liter MnCl2, 0.8 g/liter CoCl2, 0.7 g/liter ZnSO4, 0.3 g/liter CuCl2, 0.25 g/liter (NH4)6Mo7O24, 5 g/liter EDTA), 10 ml of 1 m MgSO4, 12 ml of 40% dextrose, 100 ml of TAU (0.3 g/liter thiamine, 2 g/liter adenine sulfate, 2 g/liter uracil), 100 ml of amino acid mix (1 g/liter of each amino acid) and kanamycin. Cells were grown at 37 °C to A600 = 2, collected by centrifugation, and suspended in 1 liter of fresh minimal medium with the carbonyl 13C-labeled isoleucine, phenylalanine, or leucine in place of the corresponding unlabeled amino acid at 200 mg/liter. After 30 min of recovery, protein expression was induced by the addition of 1 mm isopropyl 1-thio-β-d-galactopyranoside and growth continued overnight. Cells were harvested by centrifugation and the protein was purified as described previously (42).
RT-QuIC of 263K-seeded rPrPC(90–231)
Purified rPrPC(90–231) labeled with either [1-13C]Ile, [1-13C]Phe, or [1-13C]Leu was converted into seeded aggregates using the RT-QuIC method as described previously (43). Stock solutions of seed fibrils were prepared by inoculating RT-QuIC reaction mixtures containing unlabeled rPrPC with brain homogenates from Syrian golden hamsters clinically ill with 263K hamster-adapted scrapie. A 1:1000 dilution of this reaction was then used to seed another round of rPrPC conversions. This process was repeated a total of five times to dilute out the amount of input infectious seed to levels below that capable of initiating infection. The final sample was generated using 13C-labeled rPrPC as the substrate. A total of 10 mg of converted labeled aggregates were produced for ssNMR studies. The reactions were pooled and aggregates harvested by centrifugation. The fibrils were then washed a total of three times with 1× PBS to remove unconverted rPrPC. Conversion was verified by PK digestion and immunoblotting.
PK Digestion and Immunoblotting
PK digestion and immunoblotting of the RT-QuIC reaction products were performed as described previously (42). Membranes were probed with R20 primary antiserum (44) diluted 1:15,000 and visualized with the Attophos AP fluorescent substrate system (Promega) according to the manufacturer's recommendations.
Solid-state NMR
Amyloid of PrP labeled with [1-13C]Ile, [1-13C]Phe, or [1-13C]Leu in RT-QuIC buffer was spun into thick-walled 3.2-mm Varian zirconium rotors. An InfinityPlus (Varian) spectrometer at 9.39 tesla (100.4 MHz 1H Larmor frequency) with a magic angle spinning probe (Varian) was used for all measurements. All measurements were done at room temperature and with magic angle spinning of 20 kHz. One-dimensional NMR spectra were obtained by 1H-13C cross-polarization and 1H decoupling at 110 kHz during the free induction decay. A 13C carrier frequency of 100.401 MHz (∼191 ppm) was used with 4096 scans for [1-13C]Ile- or [1-13C]Phe-labeled PrP amyloid and 65,536 scans for [1-13C]Leu-labeled PrP amyloid. Dipolar recoupling experiments used the PITHIRDS-CT method (41) with pulsed spin-lock detection for improved signal-to-noise ratio (45). PITHIRDS-CT data were corrected for the 1.1% natural abundance 13C as described previously (37).
Electron Microscopy
For negative staining of amyloid fibrils, Formvar and ultrathin carbon-coated grids (Ted Pella, Inc., Redding, CA) were subjected to glow discharge, quickly placed onto 10-μl droplets of fibril suspensions, and incubated for 30 min at room temperature. The grids were transferred sequentially for 5 min each onto three droplets of deionized water. The grids were then stained negatively with either methylamine tungstate (Nanoprobes, Inc., Yaphank, NY) or 1% uranyl acetate in water for 15 s, wicked of excess fluid, and air dried. Negatively stained grids were examined at 80 kV on a model H-7500 transmission electron microscope (Hitachi High Technologies, Dallas, TX) equipped with a model HR-100 digital camera system (Advanced Microscopy Techniques, Woburn, MA). Fibrils prepared for dark-field scanning transmission electron microscopy (STEM) were prepared as above without staining. The grids were examined under low temperatures using cryo-conditions by high angle dark field STEM at 300 kV with a model Titan Krios microscope (FEI, Inc., Hillsboro, OR), equipped with an annular dark field detector (Fischione Instruments, Export, PA).
Model Construction
For all models, a mouse (or hamster in the case of the ssNMR model) PrPRes monomer consisting of residues 90–231 was constructed using SYBYL Version 7.3 that incorporated constraints from experimental data (Fig. 5). Individual strands within the monomer were constructed and aligned in space based on the positioning of the artificial disulfide bonds (46). Once aligned, the segments were all connected, and a short energy minimization was performed in SYBYL using the AMBER99 force field with AMBER atom types and charges to remove any long bonds or bad geometries. The single molecule was then copied and stacked to generate multimers containing eight molecules. An energy minimization (200,000 steps using a conjugate gradient method) was performed on the octamer using the NAMD program Version 2.9 on the Biowulf Linux cluster. Models were solvated explicitly using the TIP3 water model along with counter-ions (sodium and chloride). The CHARMM27 force field was used with CHARMM atom types and charges and periodic boundary conditions were added along with the use of a Particle-Mesh Ewald summation to handle nonbonded interactions. After minimization, molecular dynamics was begun by slowly warming the system from ∼40 to 310 K in 10 K steps (with each step equilibrating for 5 ps) using a 2-fs time step. Data were gathered from a production run at a temperature of 310 K. Protein Data Bank coordinates for each energy minimized model are available in the supplemental material. This study utilized the high performance computational capabilities of the Biowulf Linux cluster at the National Institutes of Health, Bethesda (http://biowulf.nih.gov). Models were analyzed, and images were created using VMD and UCSF Chimera. Secondary structure was analyzed with the DSSP algorithm (47).
FIGURE 5.
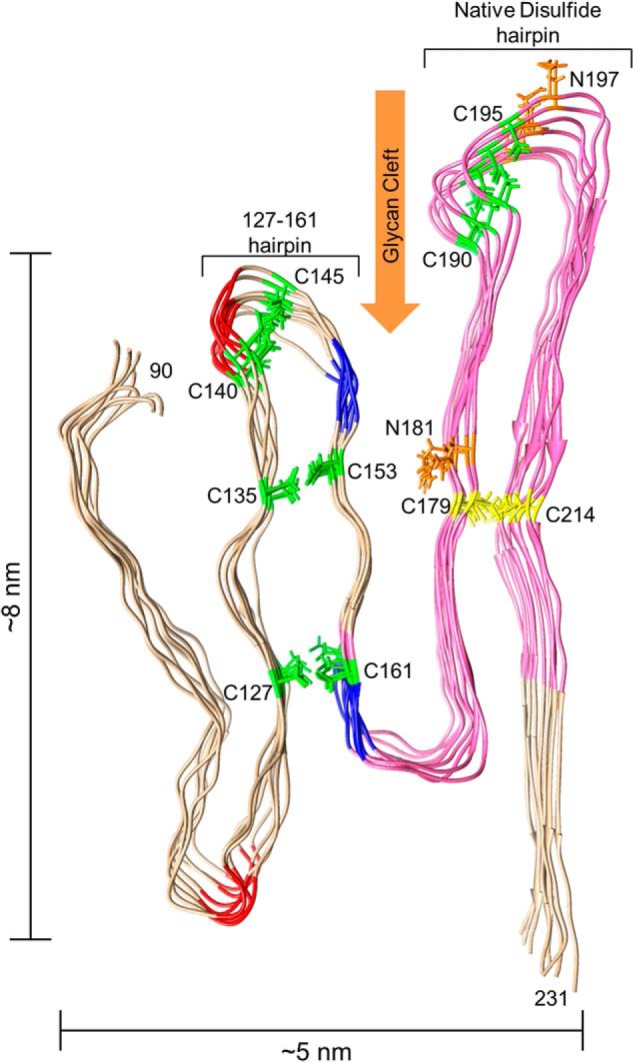
Model of an octameric PrPRes amyloid segment subject to the experimental constraints outlined in text. The view is looking down the axis of an energy-minimized octamer of mouse PrP(90–231) molecules stacked parallel in-register and with the main hairpins (denoted on the model) defined by either the natural disulfide or the artificial disulfides (yellow or green, respectively) that have been shown to be compatible with PrPSc formation (46). The segment that is known to form parallel in-register intermolecular β-sheet in unseeded rPrP amyloid fibrils (see text) is pink. Also labeled are potential YYR antibody epitopes (blue) and PK cleavage sites (red) that, at least under some conditions, are preferentially exposed in PrPSc. Asn-linked glycosylation sites (orange) and the glycan cleft (orange arrow) are also indicated. Size constraints are shown on the outside of the model.
RESULTS
Comparison of Prion-seeded Versus Denaturation-induced rPrP Amyloid Fibrils
To study the effects of prion templating on rPrP amyloid fibril formation, we generated amyloid fibrils from hamster rPrP(90–231) that were either seeded initially with scrapie (263K strain)-infected brain homogenate (263K BH) under nondenaturing conditions or allowed to arise spontaneously under destabilizing conditions (2 m guanidine HCl). The latter conditions were those used to make rPrP(90–231) fibrils for the previous PITHIRDS-CT ssNMR study (29). Consistent with our previous studies (43, 48, 49), the prion-seeded fibrils gave prominent PK-resistant bands of ∼11, 12, and 17 kDa, with the relative intensity of the latter being enhanced by the presence of Sarkosyl during the PK treatment (Fig. 1A, compare lanes 4 and 5). In contrast, spontaneously arising fibrils gave a predominant 11–12-kDa band even in the presence of Sarkosyl (Fig. 1A, lane 7). Additionally, under our nondenaturing conditions, partially PK-resistant amyloid fibril formation required the addition of scrapie-associated seeds (42), yielding fibrils that are distinct biochemically (Fig. 1A, compare lanes 6 and 7) and ultrastructurally (i.e. less twisted, Fig. 1B) from those formed spontaneously under destabilizing conditions (29).
FIGURE 1.

Characterization of prion-seeded [1-13C]Ile rPrP amyloid. A, purity of the [1-13C]Ile rPrPC substrate is shown by Coomassie Blue staining of an SDS-polyacrylamide gel in lane 1 with molecular mass standards labeled in kDa to the left. Immunoblots of 263K BH-seeded [1-13C]Ile rPrP RT-QuIC product is shown before and after PK digestion in the presence or absence of Sarkosyl in lanes 2–6. For comparison, the PK-treated product of rPrP amyloid fibrils formed under unseeded destabilizing conditions as used in Ref. 29 is shown in lane 7. C-terminal PrP antiserum R20 was used for immunoblotting. B, ultrastructure of negatively stained 263K BH-seeded RT-QuIC amyloid by transmission electron microscopy (bar, 20 nm).
ssNMR of Prion-seeded rPrP Aggregates
To probe the conformation of prion-seeded fibrils using ssNMR, we purified rPrP labeled with [1-13C]Ile, [1-13C]Phe, or [1-13C]Leu (Fig. 1A, lane 1) and seeded its aggregation with 263K BH (Fig. 1A, lanes 2–6). Similar to the observations with spontaneously formed rPrP fibrils (29), the one-dimensional spectrum of [1-13C]Ile -labeled material showed two peaks, both shifted to lower frequencies that are indicative of Ile being arranged in β-sheet conformations (Fig. 2A and Table 1) (50). To examine the spacing between labeled atoms, we used dipolar recoupling by the PITHIRDS-CT method (Fig. 2) (29). In this experiment, the rate of decay of magnetization on a given 13C atom is proportional to 1/r3, where r is the distance to the other closest 13C atom. The experiment can detect distances of between about 0.3 and 0.8 nm. The distance between β-strands of a β-sheet is ∼0.48 nm and thus for a given labeled residue will be the nearest neighbor distance in an intermolecular β-sheet structure that is parallel and in-register. The average decay rates of the four Ile residues at positions 182, 184, 203, and 205 were representative of intermolecular spacings of ∼0.5 nm (Fig. 2B, magenta triangles). As explained by Tycko et al. (29) in the context of their identically labeled spontaneous rPrP fibrils, this spacing is indicative of parallel in-register intermolecular β-sheet structures that have intermolecular spacings of ∼0.48 nm. We note that the distances between the 1-13C labels of the closely spaced isoleucine pairs, i.e. 182 and 184 and 203 and 205, are ∼0.5–0.6 nm within α-helices 2 and 3 within PrPC. However, the chemical shifts of these 13C labels indicated that they were in extended β-chains, where the intramolecular spacings along the polypeptide backbone would be ∼0.7 nm rather than ∼0.5 nm and thus would not be responsible for the rapid decay rates that we have observed. Moreover, as noted above, H/D exchange and FTIR spectroscopy data (22, 23, 49) make it highly unlikely that α-helices 2 and 3 exist in prion-seeded rPrP fibrils or in PrPRes. Thus, these results strongly suggest that upon prion-seeded conversion of rPrPC(90–231) to amyloid fibrils, Ile-182, -184, -203, and -205 refold from α-helices into parallel in-register intermolecular β-sheet structures.
FIGURE 2.

13C ssNMR analysis of 263K BH-seeded RT-QuIC products. A, one-dimensional NMR spectra of 263K BH-seeded amyloid of specifically labeled rPrP. Negative shifts are indicative of β-sheet structure. B, dipolar recoupling experiments using PITHIRDS-CT ssNMR. Solid lines indicate standard decay rates for the given intermolecular distances of 0.4–0.7 nm. Lines with markers indicate observed decay rate for the given labeled amino acid within RT-QuIC products. Inset shows the location of labeled amino acids (by color) within PrPC (Protein Data Bank code 1AG2).
TABLE 1.
Chemical shifts of [1-13C]Ile-, [1-13C]Phe-, and [1-13C]Leu-labeled PrP amyloid obtained from one-dimensional spectra
Random coil chemical shifts are from Wishart et al. (45). Peak fractions were estimated by deconvolution of the spectrum into Gaussian curves using Spinsight software.
| PrP sample | Peak width | Peak fraction | Chemical shift | Random coil |
|---|---|---|---|---|
| Hz | ppm | ppm | ||
| [1-13C]Phe | 68 | 0.48 | 173.53 | 174.1 |
| 122 | 0.52 | 172.62 | ||
| [1-13C]Ile | 94 | 0.5 | 173.94 | 174.7 |
| 92 | 0.5 | 171.9 | ||
| [1-13C]Leu | ∼500 | 173.76 | 175.9 |
With the [1-13C]Phe-labeled fibrils, the one-dimensional 13C NMR spectrum showed two peaks in the carbonyl region, with both chemical shifts indicative of β-sheet (Fig. 2A and Table 1). The average decay rate of the three Phe residues (Phe-140, -174, and -197), indicative of an average intermolecular distance of 0.5–0.6 nm, was slightly greater than was observed with the Ile residues. This result could be explained if two-thirds of all the Phe residues in the fibril were stacked in-register i.e. ∼0.48 nm apart, and the other third was not (Fig. 2B, black squares), which was also observed in spontaneously formed rPrP fibrils (29).
In contrast, the two leucines at the more N-terminal positions 125 and 130 gave an average decay rate consistent with a separation of >0.7 nm, which is incompatible with the adoption of uniform parallel in-register β-sheet structure (Fig. 2B, green triangles). Although the Leu residues are not in a uniform parallel in-register intermolecular β-sheet, the one-dimensional NMR spectrum of [1-13C]Leu-labeled material showed a single broad carbonyl peak (Fig. 2A and Table 1) indicative of still being in a β-sheet structure. This is consistent with observations that in vitro generated rPrP fibrils are more disordered in this N-terminal region than bona fide PrPSc amyloid (22, 49).
Building Parallel In-register β-Sheet Models
The indications that PrPSc-seeded rPrP amyloid fibrils have carboxy-proximal parallel in-register intermolecular β-sheets prompted us to build new detailed models of PrP(90–231) oligomers based on such structures to guide new insights and investigations into the structure of PrPRes. These models build on a schematic representation by Cobb et al. (27) of a limited amyloid core of PrP residues ∼160–220 in which each monomer stacks with their polypeptide backbones being perpendicular to the fibril axis. The basic layout for this domain is consistent with available data, including evidence that the native disulfide bond between residues 179 and 214 (murine sequence) is intact in PrPSc (51–53). This disulfide establishes a hairpin, hereafter designated the native disulfide (ND) hairpin (depicted in a representative ribbon diagram on the right sides of Fig. 3, A and B), containing both N-linked glycosylation sites (not depicted). Our use of the term hairpin here is not to be confused with “β-hairpin,” a specific structure in which backbone hydrogen bonding occurs intramolecularly across the hairpin rather than intermolecularly between stacked hairpins along the fibril axis as in a parallel in-register intermolecular β-sheet. Our modeling provides the first atomistic parallel in-register β-sheet-based models of PrP amyloid and expands on previously articulated ideas to include the entire span of residues that typically make up the tightly packed PK-resistant core of fully infectious brain-derived PrPRes, i.e. residues ∼90–231. Our efforts were based on the following constraints.
FIGURE 3.

Fibril linearity requires extended flattened structure. A, theoretical diagrammatic parallel in-register intermolecular β-sheet model for residues 90–231 PrPRes amyloid. B, destabilizing effects of combining a parallel in-register β-sheet architecture in the C-terminal ND hairpin (A and B, ND Hairpin) with two-tiered β-helices toward the N terminus (B, left side). The different thicknesses of these two architectures (β-helix versus parallel intermolecular β-sheet) within monomers would not appear to allow growth of stable linear fibrils, leaving major gaps along the fibril axis on the C-terminal side (B). The figure in A is adapted from Ref. 2.
Linearity of Fibrils
The parallel in-register β-sheet architecture that is evident for rPrP amyloid core residues ∼160–220 suggests that the more N-terminal residues that are also part of the PrPRes amyloid core assume a configuration in which they are not only tightly packed but also lie roughly on the same plane and, on average, contribute ∼0.5 nm of axial thickness along the fibril axis perpendicular to the β-strands (Fig. 3A). Otherwise, the fibril would not be linear because the N-terminal residues of each monomer would contribute greater thickness than the C-terminal residues, forcing the fibril to be unstable or discontinuous with respect to ND hairpin stacking (Fig. 3B, compare more N-terminal region, including a minimal β-helix, to ND hairpin in “fibril” schematic). This flattening of protein layers within amyloid fibrillar cores has been described more generally as a requirement imposed by in-register assembly via “amyloid spines” (54). This constraint argues against options such as the packing of α-helices (∼1.2 nm diameter) or β-helical or β-solenoid configurations in which the N-terminal chain (i.e. residues 90–159) loops back onto itself to form intramolecular β-sheets (at least 0.96 nm thick) perpendicular to the fibril axis (Fig. 3B).
Cross-sectional Dimensions
Each monomer must pack within the cross-sectional dimensions of the fibril and, as explained above, within an average axial thickness of ∼0.5 nm. Previous measurements of fully infectious PrPRes fibril cross-sections by negative stain transmission EM and atomic force microscopy have typically given ∼5–6 nm in the narrowest dimension and 9–20 nm in the wider dimension (13, 14). To augment these measurements by another approach that avoids potentially confounding effects of electron-dense stains (14), we collected dark field images using scanning transmission electron microscopy (STEM) of unstained, PK-treated GPI-anchorless PrPRes amyloid fibrils (Fig. 4A). Isolated fibrils (Fig. 4A, red arrowheads) were measured across their widths providing a distribution of 6–24 nm. Within this distribution, two apparent populations are evident, which are likely to represent single (Fig. 4A, single arrowhead) and double (Fig. 4A, double arrowhead) fibril populations with mean widths of ∼12 and ∼20 nm, respectively (Fig. 4B). Polypeptide β-strands are ∼0.3 nm long per residue, so a fully extended chain of residues 90–231 would be ∼42 nm (141 residues × 0.3 nm) long (Fig. 4C). Thus, the polypeptide backbone must fold back on itself multiple times in a “super-pleated sheet” (35, 55) to fit within the fibril cross-section (Fig. 4C). Distances between the backbone folds could vary depending upon the packing (or lack thereof) of side chains forming the faces of the β-sheets but is typically ∼1.0 nm in tightly packed amyloid fibrils containing steric zippers (54). As such, the ND hairpin as depicted in the Cobb et al. (27) model should be ∼5 × ∼2 nm incorporating ∼35 residues. A fibril cross-section of 5 × 9 nm could accommodate as many as ∼140 residues in four hairpins each with ∼35 residues (Fig. 4C), with 140 being the approximate number of residues in monomers of PK-treated PrPRes. Thus, we expect that multiple hairpins (in addition to that defined by the native disulfide) are present within PrPRes.
FIGURE 4.

Cross-sections of PrPRes amyloid fibrils. A, STEM imaging of PK-treated brain-derived GPI-anchorless fibrils (22L strain) (bar, 100 nm). Single and double red arrowheads indicate apparent single and double fibrils, respectively. B, fibril width distribution measured from STEM images and fit to a Sum of Two Gaussians linear regression to define two populations of apparent single and double fibrils. C, scale diagrams of the length of the PrP(90–231) polypeptide as an extended linear chain relative to potential folded architectures that would be required to fit such a polypeptide within the fibril widths given a coplanar parallel intermolecular β-sheet architecture. Yellow bars represent the native disulfide.
Glycans
The asparagine side chains to which glycans are often attached (Asn-181 and -197) should be oriented toward the outside of the ND hairpin because it is highly unlikely, if not impossible, that bulky hydrophilic glycans would fit inside the hairpin (Fig. 5; Asn glycosylation sites are colored orange). The location of these glycan sites, particularly Asn-181, provides an insight into where flexible loops, or “hinges,” within the structure may form (Fig. 5, glycan cleft). Additionally, knowing the side chain orientation of these residues helps establish the side chain orientations of nearby residues if they are in parallel in-register β-sheets because the side chains alternate from one face of a β-sheet to the other.
Artificial Disulfides
Hafner-Bratkovic et al. (46) described artificial disulfide cross-links within PrPC that are compatible not only with PrPC secondary structures but also with PrPRes formation in RML scrapie-infected cells. These cross-links were between the following pairs of residues: 127–161, 135–153, 140–145, and 190–195. Although these authors interpreted their results to mean that the native PrPC secondary structure domains containing these residues, including the α-helices, are retained in PrPSc, we note that the 190–195 bridge (Fig. 5, green) is also compatible with the ND hairpin described above (Fig. 5, yellow). More importantly, the 127–161, 135–153, and 140–143 bridges could be consistent with the formation of another hairpin in a parallel in-register intermolecular β-sheet configuration composed of at least residues 127–161 (Fig. 5, green), hereafter called the 127–161 hairpin. Furthermore, because the side chains of the cysteine residues that form the artificial disulfide bonds must also be oriented toward the interior of their respective hairpins, the same is likely to be true of the native residues that were replaced by cysteines in the mutants in the study by Hafner-Bratkovic et al. (46).
Epitopes and Proteolytic Cleavage Sites
With two hairpins and side chain orientations suggested by compatibility with the native and artificial disulfides and the N-glycosylation sites, we tentatively predicted further turn/loop positions based on known epitope exposures and sensitivities to proteolytic digestion. For example, one or both Tyr-Tyr-Arg epitopes (Fig. 5, blue) may be exposed within PrPRes but not PrPC (56). Furthermore, as the PrPRes structure is loosened by moderate guanidine HCl treatments, residues 117–119 and 140–143 (Fig. 5, red) become exposed to PK (57–59). Such epitopes and proteolytic sites are likely to be in more flexible turns or loops with exposure to the fibril surface, at least after partial unfolding and/or unbundling of fibrils.
Hairpin Orientation
Consistent with the idea of specific prion strains being based on distinct, self-propagating PrPRes conformers, the constraints detailed above are compatible with multiple configurations that are based on parallel in-register β-sheet packing. For instance, models can differ in the relative orientation of the 127–161 and ND hairpins and in the number of hairpins involving more extreme N- and C-terminal residues. One fundamental permutation of our first native disulfide model (PIRIBS-A, Fig. 6) is based on a 180° flipping of the 127–161 hairpin (hereafter the inverted hairpin) relative to the ND hairpin, resulting in reorientation of the loops near the N terminus (PIRIBS-B, Fig. 6). We also show the possibility of the C-terminal residues folding back against the ND hairpin (Fig. 6, PIRIBS-B and -C) as well as the potential for the extreme N terminus to adopt an additional hairpin or loop (PIRIBS-C, Fig. 6). The inverted hairpin configuration (PIRIBS-B and -C) also seems more compatible with observations that residues 141–176 can be deleted from PrPC expressed in transgenic mice and retain susceptibility to prion infection, allowing formation of PrPRes “miniprions” (60). This configuration brings the flanking residues 140 and 177 close together in space so that wild-type PrPRes might incorporate Δ141–176 PrP molecules if the latter simply bridged the gap between residues 140 and 177, bypassing the intervening hairpin on the wild-type template while retaining parallel-in-register alignment of corresponding residues (Fig. 6, PIRIBS-C, black). Alternative permutations of these models can be envisioned as the basis for the propagation of distinct prion strains from a single protein sequence.
FIGURE 6.

Energy-minimized states of three distinct models of an octameric segment of PrPRes amyloid based on permutations of parallel in-register β-sheet architectures. In each view the individual monomers are designated by a different color with the broader arrows designating predicted β-strands. The end-on view is looking down the fibril axis. Arrows indicate the rotation of the fibril for the alternative views. A, Parallel In-Register Intermolecular β-Sheet-A (PIRIBS-A) model. B, PIRIBS-B model in which the 127–161 hairpin constrained by the artificial disulfides are inverted relative to the natural disulfide hairpin. This model also depicts the possibility that the extended C-terminal domain depicted in A might instead be folded back against the natural disulfide hairpin. C, PIRIBS-B model, which has an additional N-terminal loop at residue 127 compared with PIRIBS-B. In the latter model, the deletion (Δ141–176) that has been shown to be compatible with the formation of PrPSc “miniprions” (60) is shown in black. For all models, the N and C termini are denoted, as are the hairpins, the residues that define the hairpins, and the glycan cleft (orange arrows).
Molecular Dynamics Simulations
We sought evidence that our parallel in-register intermolecular β-sheet-based structural prototypes were stable in silico without violating basic rules of structural biology. To refine and begin evaluating these models, we constructed assemblies of eight PrP molecules based on the aforementioned constraints and subjected them to energy minimization and molecular dynamics simulations. In its energy-minimized state PIRIBS-A (Fig. 6) contained 29% β-sheet structure and essentially no α-helical component (Table 2). The initial simulation was run using an octamer that still contained the artificial disulfide bonds (46) used to align the structures (Fig. 7, PIRIBS-A). The simulation was run for 63 ns to provide ample time to see side chain movement and secondary structure changes. Over the course of the simulation, parallel in-register β-sheet structures were largely conserved showing ∼7% loss in β-structure (Table 2). Consistent with the biochemical and biophysical data from initially prion-seeded rPrP(90–231) amyloid fibrils (27, 49), residues ∼124–227 maintained a more ordered and tightly packed core than residues ∼90–120 (Fig. 7, PIRIBS-A). Once we determined that the model was compatible with the disulfide bonds and the core was stable throughout the simulation, the mutant cysteines were reverted back to their wild-type residues, and the simulation was repeated (Fig. 8, PIRIBS-A). Again, even in the absence of the disulfides, the monomers remained tightly associated over the 63-ns simulation. In this simulation, a larger degree of disorder was observed for residues 90 to ∼145 (Fig. 8, PIRIBS-A) than was observed with the artificial disulfides intact (Fig. 7, PIRIBS-A). Flexibility in this region can often be seen empirically with rPrP(90–231) fibrils generated spontaneously in vitro (49). Most of the remainder of the molecule (residues 146–231) remained well packed and maintained much of its β-sheet content, again showing only ∼7% loss in β-structure (Table 2). Overall, our PIRIBS-A structure maintained a high degree of order in the C-terminal half (residues ∼145–231) with less order toward the N terminus (residues 90–144). Although compatible with the artificial disulfides, the PIRIBS-A structure retained a stable core even in the absence of the artificial disulfide cross-links.
TABLE 2.
Secondary structure analysis of PrPRes models
Values are represented for the energy-minimized state, the MD-simulated state with (MD-artificial disulfides) and without (MD-native disulfide) the artificial disulfides as % secondary structure.
| β-Sheet | α-Helix | Other | |
|---|---|---|---|
| PIRIBS-A model | |||
| Energy-minimized | 29 | 0.3 | 70.8 |
| MD-artificial sisulfides | 22.4 | 2.9 | 74.7 |
| MD-native disulfide | 22.6 | 2 | 75.4 |
| PIRIBS-B model | |||
| Energy-minimized | 36.1 | 0 | 63.9 |
| MD-artificial disulfides | 16.7 | 3.3 | 80 |
| MD-native disulfide | 15.2 | 2.7 | 82 |
| PIRIBS-C model | |||
| Energy-minimized | 42 | 0 | 58 |
| MD-artificial disulfides | 22 | 1.4 | 76.6 |
| MD-native disulfide | 17.1 | 2.2 | 80.7 |
FIGURE 7.

Molecular dynamics simulations of the models shown in Fig. 6, retaining the mutant cysteines and disulfide bonds. A, PIRIBS-A model after 63 ns. B, PIRIBS-B model after 100 ns. C, PIRIBS-C model after 100 ns.
FIGURE 8.

Molecular dynamics simulations of the models shown in Fig. 6 with the mutant cysteines reverted to their wild-type residues. A, PIRIBS-A model after 63 ns. B, PIRIBS-B model after 100 ns. C, PIRIBS-C model after 155 ns.
We then ran simulations on the inverted hairpin models (PIRIBS-B and -C), again using octamers containing the artificial disulfide bonds (Fig. 7, PIRIBS-B and -C). With the extra hairpins, a larger β-sheet content was observed initially (∼36 and ∼42% without and with the extra N-terminal fold at residue 127, respectively), with no α-helical component (Fig. 6, PIRIBS-B and -C, and Table 2). Simulations were run without (Fig. 7, PIRIBS-B) and with (Fig. 7, PIRIBS-C) the extra N-terminal fold for 155 and 100 ns, respectively. Both models demonstrated compatibility with the artificial disulfides and displayed stability nearly equivalent to the previous models for residues ∼126–227 without and with the N-terminal fold (Fig. 7, PIRIBS-B and -C). As in PIRIBS-A, the more N-terminal residues (90 to ∼120) were more disordered than the more C-terminal regions (160–231). In these simulations, the overall architecture was maintained, albeit with a loss of ∼10 or ∼20% β-sheet without or with the extra N-terminal fold, respectively (Fig. 7, PIRIBS-B and -C, and Table 2), attributed mainly to loss of structure in the N-terminal half (∼90–160). β-Sheet structure predominates from residues 166 to 231 with some short β-sheet segments as far N-terminally as residue 97 (Fig. 7, PIRIBS-B and -C). 100-ns simulations of these models with the mutant cysteines reverted back to their wild-type residues showed again that much of the original β-sheet structure (with an overall loss of ∼11% or 25% without or with the extra N-terminal fold, respectively, in the N-terminal half) was conserved within the core, which extended toward the N terminus over residues 166–231 (Fig. 8, PIRIBS-B and -C and Table 2) and in some subunits as far N-terminally as residues 152 (Fig. 8, PIRIBS-B) and 154 (Fig. 8, PIRIBS-C). Order was maintained, albeit to a lesser degree than in the C-terminal core, from residues ∼126 to 165 as has been seen consistently across all models. The largest degree of flexibility in the molecule was again observed in residues ∼90–125 (Fig. 8, PIRIBS-B and -C). Interestingly, the flexibility in the loops between the multiple hairpins allowed accordion-like expansion and contraction between widths of ∼6–11 nm, consistent with known fibril dimensions (14).
In summary, whereas the different parallel in-register β-sheet models showed varying degrees of order (or disorder) in these simulations, each structure was able to maintain substantial order in the C-terminal region (residues ∼124–231) with much of the original β-sheet structure conserved, especially in the C-terminal 160–231 region (Figs. 7 and 8, side). The flexibility in the N-terminal region (residues ∼90–125) was expected as it is consistent with that seen empirically in in vitro formed fibrils of pure rPrP without cofactors (27, 49).
Comparison of Parallel In-register β-Sheet Models to ssNMR Data
As explained above, the ∼0.5-nm intermolecular spacing of the four Ile residues indicated by our ssNMR analysis (Fig. 2B) is indicative of their being located in parallel in-register intermolecular β-sheets. Consistent with this, each of the Ile residues (Fig. 9, magenta) are within such sheets in the ND hairpin of our new model of PrPRes. One of the Phe residues (Fig. 9, residue 175, black) is also within a β-sheet, and the second (Fig. 9, residue 198, black) is located in a turn in or immediately adjacent to a β-sheet in the ND hairpin, and the third (Fig. 9, residue 141, black) lies at the edge of loop more proximal to the N terminus. This arrangement is consistent with an average intermolecular distance of 0.5–0.6 nm (Fig. 2B), which is slightly greater than that expected if all three Phe residues were in parallel in-register β-sheets (i.e. 0.48 nm). In contrast, the Leu at positions 125 and 130 (Fig. 9, green) are located in N-terminal domains that are less ordered in the models, especially after MD (Fig. 9B), consistent with their greater intermolecular distance (>0.7 nm) indicated by ssNMR (Fig. 2B). Thus our current models are consistent with the distances between these residues measured by ssNMR (Figs. 2 and 9). These data are also consistent with H/D exchange analyses of prion-seeded rPrP(90–231) fibrils (in the absence of cofactors), which indicated that the Ile and Phe residues are located in sequences that are more tightly packed and isolated from solvent than the Leu residues (49).
FIGURE 9.

Location of isotope-labeled residues within the new PIRIBS-A model of PrPRes. An energy-minimized model as depicted in Fig. 5 but using the hamster PrP sequence for comparison with the ssNMR data, showing β-sheets (blue) and the positions of the Ile (magenta), Phe (black), and Leu (green) residues that were 13C-labeled for the ssNMR analyses before (A) and after (B) 163-ns molecular dynamics simulation.
DISCUSSION
Although much remains to be done to establish the detailed atomistic accuracy of these models and permutations thereof, our development of parallel in-register intermolecular β-sheet-based models of PrPRes amyloid structure is grounded in multiple lines of empirical data and constraints. Layering of tightly packed molecules, in which a single PrP molecule forms the entire fibrillar cross-section, gives rise to well ordered, unbranched PrP fibrils that satisfy key features of PrPRes fibrils. Electron micrographs of scrapie-associated fibrils have long shown the accumulation of stain between two or more main axial elements (e.g. Fig. 10, red arrows). Most authors have assumed that these elements represent parallel protofilaments, each of which contain one or more PrP molecules comprising the protofilament cross-section (9, 13, 14, 61). Indeed, this assumption is reflected in most of the prevailing PrPSc models. Here, we propose alternatively that what appeared to be two or more individual protofilaments aligned in parallel is actually a single protofilament composed of hairpins or combinations of hairpins connected by a hinge(s), giving a “celery stalk” or half-pipe appearance (Fig. 10C, expansion).
FIGURE 10.

Celery stalk or half-pipe appearance of brain-derived GPI-anchorless (A, C (upper), and D) and wild-type PrPRes (B and C (lower)) amyloid (22L strain) by negative stain transmission electron microscopy. Flattened bends that have been observed in extended fibrils are marked with arrowheads. Red arrows designate the gap that was previously thought to be the spacing between individual protofilaments (14) but that we now think represents the expandable trough between the major hairpins (as diagrammed in C, expansion). Blue arrows flank the edges of what we perceive to be individual fibrils. A–C are adapted with permission from Ref. 14. All scale bars, 100 nm.
Previous descriptions have noted that the distance between “protofilaments,” proposed here as the trough between N- and C-proximal hairpins, ranged from “too narrow to measure” to 10.6 nm (14). Flexibility in the hinge loops between N- and C-proximal hairpins (Fig. 4C), particularly in the glycan cleft (Figs. 5–8), could explain this variability in fibril width. Indeed, it seems plausible that the spreading between the N- and C-proximal hairpins could be artificially influenced by interactions of the fibril with the EM or atomic force microscopy grids as diagrammed in Figs. 10C and 4C. Sim and Caughey (14) reported a flattening of fibril structures at bends as shown in Fig. 10, A and B. Such flattening would be predicted to occur with the architecture that we are proposing, much like the flattening that occurs if one bends any flexible half-pipe structure. With wild-type PrPRes fibrils, glycans attached to Asn-181 would be expected to protrude into the gap between the 127–161 and ND hairpins (Fig. 10B, red arrow), and it could account for a dynamic range of spreading between the N- and C-proximal hairpins, preventing their close apposition. Consistent with this possibility are observations that wild-type fibrils tend to be more widely spread than the much less glycosylated anchorless fibrils (14).
In the context of our models, one would expect that the appearance of PrPRes fibrils in electron micrographs would depend on fibril orientation on the grid. For instance, if the celery stalk is lying either on its “back” or face down as shown in Fig. 5, spreading between the N- and C-terminal hairpins (Fig. 10, blue arrows) might be promoted, and the trough in the middle (Fig. 5, glycan cleft, and Fig. 10, red arrows) could collect stain to emphasize a half-pipe appearance (Fig. 10C, expansion, red arrow). With more closely opposed hairpins, the trough would narrow giving the appearance of axial ribbing (Fig. 10D). Consistent with this possibility, both the celery stalk and the ribbed orientations can be observed (Fig. 10D). When the celery stalk or “half-pipe” is on its side, our models would predict that a smoother (without axial ribbing) surface might be exposed (Fig. 10, A and B, white arrowhead). Occasionally, fibril substructures have been observed that have been interpreted as individual protofilaments (14). However, we suspect that such structures arise from relatively rare proteolytic cleavages within the hinge between hairpins, allowing complete separation of the N- and C-proximal hairpins. Combined, these observations support a parallel in-register intermolecular β-sheet structure in which a single PrP molecule forms the entire fibrillar cross-section and the hairpins are separated by flexible hinge regions.
The parallel in-register β-sheet architecture in the C terminus requires that a portion of the protein backbone is perpendicular to the fibril axis. Although the same seems likely to be true of the remainder of the molecule, the 4.8 Å (0.48 nm) thickness might also be maintained if the N-terminal segments were tilted with respect to the C-terminal core, in which case they could not be in-register, but instead they are slightly out of register to maintain the linearity of the protofilament. As we have no explicit evidence for a parallel in-register alignment in the N-proximal residues within amyloids of PrP(23–231) or -(90–231), we cannot distinguish between these possibilities. However, ssNMR studies of rPrP(23–144) amyloid fibrils have established an overall parallel in-register alignment for this N-terminal construct (31), presumably within residues ∼113–140 (28). This provides a precedent for this architecture near the N terminus, in addition to the ND hairpin near the C terminus, of the protease-resistant core of PrPRes (residues ∼90–231).
The least constrained, and therefore least predictable region in our models, is near the N terminus of the PrP(90–231) PK-resistant core. This is reflected in both the disordered nature of these residues in the MD simulations and in the flexibility of this region in rPrP fibrils as has been revealed biochemically (49). A consistent indication from our MD simulations is that N-proximal residues (∼90–125) are less prone to forming stable stacked β-sheets on their own than most of the rest of the sequence (Fig. 8). This is in agreement with extensive H/D exchange and protease exposure analyses of spontaneously formed rPrP fibrils that have shown much greater flexibility and proteolytic exposure toward the N terminus (43, 48, 49). In contrast, such analyses indicate much greater protection from solvent and proteases of these residues in bona fide brain-derived PrPRes (48, 49). The reasons for this are unclear, but we suspect that this is likely due in part to the fact that there are two prolines (Pro-102 and -105) and four positively charged lysines within residues 101–110. Without some sort of charge compensation or delocalization, in-register stacking of these lysines would be disfavored by electrostatic repulsion. Indeed, polyanionic cofactors play an important role in forming infectious PrPRes both in vivo and in vitro (11, 62, 63), and we suspect that one of their key functions is to compensate the charges of these cationic lysine side chains, allowing them to pack tightly together as they would in a parallel in-register β-sheet architecture. Folding of this N-terminal region is of particular interest because it is this region that seems to distinguish infectious PrPRes from noninfectious rPrP fibrils and is most affected by the inclusion of cofactors that are critical in the formation of infectious PrPSc (11, 62, 63) but are absent in our in silico simulations.
Additional evidence consistent with a parallel in-register alignment in the N terminus of PrPRes is provided in a study by Onisko et al. (64) in which dimers and trimers cross-linked via Gly-90 with bis(sulfosuccinimidyl) suberate (BS3) were observed. A stacked in-register alignment of the N terminus would space Gly-90 at ∼4.8 or 9.6 Å between dimers or trimers, respectively. Such dimers and trimers would be susceptible to cross-linking by BS3, which has a maximum cross-linking distance of 11.4 Å. In contrast, four or more stacked monomers would be spaced at a distance greater than 14.4 Å and would therefore be outside the cross-linking range of BS3; accordingly, no cross-linked tetramers were observed (64). Significantly, these findings would also be incompatible with some of the current models in which the axial spacing between the same residues on adjacent monomers would be greater than 14.4 Å.
One attractive aspect of our modeling is that it seems straightforward to envision that variations in the relative orientations of β-sheets, hairpins, and loops could define distinct prion conformers or strains. Indeed, ssNMR studies have demonstrated that polymorphisms within the amyloid could result in shifting of the core β-sheets while maintaining the overall architectures and characteristics (28, 65). Such structures could propagate faithfully through a nucleated polymerization mechanism where incoming PrP molecules would align along the template established by the polypeptide backbone of the preceding molecule at the fibril end (65). The ability to propagate strain-specific conformational information through nucleated polymerization would seem to be more challenging in highly α-helical models in which much smaller portions of incoming PrP molecules would contact the fibrillar template.
In our models, polypeptide segments that protrude from the structure would be less stably bound and/or subject to proteolysis. Consistent with this idea is that during the course of the MD simulations the outermost molecules of the octamer display greater flexibility than the middle most molecules (Fig. 8, side). This instability at the edges of the growing fibril may provide a flexible docking point for incoming PrPC molecules while still conveying the underlying structure from the core of the fibril. As the fibril grows and more molecules are added to the ends, the previously exposed molecules may adopt a more stable structure reflecting the core of the fibril. Meanwhile, the outermost molecules might remain less constrained and flexible to assist in docking incoming PrP molecules. Indeed there is evidence for a two-step conversion process in which rapid binding of incoming PrP monomers is followed by a slower conformational transformation to the protease-resistant state (66, 67).
It is possible that an octamer, simulated in our studies, does not accurately reflect the stability of a true fibril core. A longer fibril with a longer “core” region may be more stable and efficient at propagating its structure. Indeed, we previously reported that oligomers of 5 or less units have virtually no infectivity, whereas particles ranging from 14- to 28-mers had the highest specific infectivity (10). This is also consistent with the lag time seen for nucleation in vitro, which may result from short fibrils needing to generate more stable cores through incorporation of additional PrP molecules before more rapid elongation of the fibril is promoted (68). The potential drawbacks of modeling a short fibril, notwithstanding the stability of our octameric models, suggest that parallel in-register intermolecular β-sheet structures can occur and provide a strong rationale for strain specificity via templated propagation of distinct core structures varying in their placement of loops and turns. Simulations are limited in that they include explicit water and enough counter-ions to neutralize the system, whereas the fibrils that form under more physiologically relevant conditions include different ionic strengths and cofactor molecules. However, we anticipate that the approaches and models that we have described here will provide a basis for the further elucidation of prion structure and propagation mechanisms.
Supplementary Material
Acknowledgments
We thank Dr. Robert Tycko (NIDDK, National Institutes of Health) for use of the NMR spectrometer and for helpful discussions. We also thank Drs. Bruce Chesebro, Suzette Priola, and Roger Moore for critical evaluation of this manuscript; Anita Mora and Heather Murphy for graphics assistance; and David Dorward for microscopy assistance.
This work was supported, in whole or in part, by National Institutes of Health Intramural Programs of the NIAID and the NIDDK.

The Protein Data Bank models for each energy-minimized model are available as supplemental material.
- PrPC
- cellular prion protein
- PrP
- prion protein
- PrPSc
- PrP-scrapie
- GPI
- glycophosphatidylinositol
- rPrP
- recombinant PrP
- BS3
- bis(sulfosuccinimidyl) suberate
- ssNMR
- solid-state NMR
- MD
- molecular dynamics
- PK
- proteinase K
- STEM
- scanning transmission electron microscopy
- ND
- native disulfide
- H/D
- hydrogen/deuterium
- BH
- brain homogenate.
REFERENCES
- 1. Prusiner S. B. (1998) Prions. Proc. Natl. Acad. Sci. U.S.A. 95, 13363–13383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Kraus A., Groveman B. R., Caughey B. (2013) Prions and the potential transmissibility of protein misfolding diseases. Annu. Rev. Microbiol. 67, 543–564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Caughey B., Baron G. S., Chesebro B., Jeffrey M. (2009) Getting a grip on prions: oligomers, amyloids, anchors and pathological membrane interactions. Annu. Rev. Biochem. 78, 177–204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Caughey B., Baron G. S. (2006) Prions and their partners in crime. Nature 443, 803–810 [DOI] [PubMed] [Google Scholar]
- 5. Linden R., Martins V. R., Prado M. A., Cammarota M., Izquierdo I., Brentani R. R. (2008) Physiology of the prion protein. Physiol. Rev. 88, 673–728 [DOI] [PubMed] [Google Scholar]
- 6. Wüthrich K., Riek R. (2001) Three-dimensional structures of prion proteins. Adv. Protein Chem. 57, 55–82 [DOI] [PubMed] [Google Scholar]
- 7. McKinley M. P., Taraboulos A., Kenaga L., Serban D., Stieber A., DeArmond S. J., Prusiner S. B., Gonatas N. (1991) Ultrastructural localization of scrapie prion proteins in cytoplasmic vesicles of infected cultured cells. Lab. Invest. 65, 622–630 [PubMed] [Google Scholar]
- 8. Gabizon R., McKinley M. P., Groth D. F., Kenaga L., Prusiner S. B. (1988) Properties of scrapie prion protein liposomes. J. Biol. Chem. 263, 4950–4955 [PubMed] [Google Scholar]
- 9. Govaerts C., Wille H., Prusiner S. B., Cohen F. E. (2004) Evidence for assembly of prions with left-handed β-helices into trimers. Proc. Natl. Acad. Sci. U.S.A. 101, 8342–8347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Silveira J. R., Raymond G. J., Hughson A. G., Race R. E., Sim V. L., Hayes S. F., Caughey B. (2005) The most infectious prion protein particles. Nature 437, 257–261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Deleault N. R., Walsh D. J., Piro J. R., Wang F., Wang X., Ma J., Rees J. R., Supattapone S. (2012) Cofactor molecules maintain infectious conformation and restrict strain properties in purified prions. Proc. Natl. Acad. Sci. U.S.A. 109, E1938–E1946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Piro J. R., Wang F., Walsh D. J., Rees J. R., Ma J., Supattapone S. (2011) Seeding specificity and ultrastructural characteristics of infectious recombinant prions. Biochemistry 50, 7111–7116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Merz P. A., Somerville R. A., Wisniewski H. M., Iqbal K. (1981) Abnormal fibrils from scrapie-infected brain. Acta Neuropathol. 54, 63–74 [DOI] [PubMed] [Google Scholar]
- 14. Sim V. L., Caughey B. (2009) Ultrastructures and strain comparison of under-glycosylated scrapie prion fibrils. Neurobiol. Aging 30, 2031–2042 [DOI] [PubMed] [Google Scholar]
- 15. Caughey B. W., Dong A., Bhat K. S., Ernst D., Hayes S. F., Caughey W. S. (1991) Secondary structure analysis of the scrapie-associated protein PrP 27–30 in water by infrared spectroscopy. Biochemistry 30, 7672–7680 [DOI] [PubMed] [Google Scholar]
- 16. Pan K.-M., Baldwin M., Nguyen J., Gasset M., Serban A., Groth D., Mehlhorn I., Huang Z., Fletterick R. J., Cohen F. E., Prusiner S. B. (1993) Conversion of α-helices into β-sheets features in the formation of the scrapie prion protein. Proc. Natl. Acad. Sci. U.S.A. 90, 10962–10966 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Safar J., Roller P. P., Gajdusek D. C., Gibbs C. J., Jr. (1993) Conformational transitions, dissociation, and unfolding of scrapie amyloid (prion) protein. J. Biol. Chem. 268, 20276–20284 [PubMed] [Google Scholar]
- 18. Oesch B., Westaway D., Wälchli M., McKinley M. P., Kent S. B., Aebersold R., Barry R. A., Tempst P., Teplow D. B., Hood L. E. (1985) A cellular gene encodes scrapie PrP 27–30 protein. Cell 40, 735–746 [DOI] [PubMed] [Google Scholar]
- 19. McKinley M. P., Bolton D. C., Prusiner S. B. (1983) A protease-resistant protein is a structural component of the scrapie prion. Cell 35, 57–62 [DOI] [PubMed] [Google Scholar]
- 20. Hope J., Multhaup G., Reekie L. J., Kimberlin R. H., Beyreuther K. (1988) Molecular pathology of scrapie-associated fibril protein (PrP) in mouse brain affected by the ME7 strain of scrapie. Eur. J. Biochem. 172, 271–277 [DOI] [PubMed] [Google Scholar]
- 21. Basler K., Oesch B., Scott M., Westaway D., Wälchli M., Groth D. F., McKinley M. P., Prusiner S. B., Weissmann C. (1986) Scrapie and cellular PrP isoforms are encoded by the same chromosomal gene. Cell 46, 417–428 [DOI] [PubMed] [Google Scholar]
- 22. Smirnovas V., Baron G. S., Offerdahl D. K., Raymond G. J., Caughey B., Surewicz W. K. (2011) Structural organization of brain-derived mammalian prions examined by hydrogen-deuterium exchange. Nat. Struct. Mol. Biol. 18, 504–506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Baron G. S., Hughson A. G., Raymond G. J., Offerdahl D. K., Barton K. A., Raymond L. D., Dorward D. W., Caughey B. (2011) Effect of glycans and the glycophosphatidylinositol anchor on strain dependent conformations of scrapie prion protein: improved purifications and infrared spectra. Biochemistry 50, 4479–4490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Wang F., Wang X., Yuan C. G., Ma J. (2010) Generating a prion with bacterially expressed recombinant prion protein. Science 327, 1132–1135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kim J. I., Cali I., Surewicz K., Kong Q., Raymond G. J., Atarashi R., Race B., Qing L., Gambetti P., Caughey B., Surewicz W. K. (2010) Mammalian prions generated from bacterially expressed prion protein in the absence of any mammalian cofactors. J. Biol. Chem. 285, 14083–14087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Makarava N., Kovacs G. G., Bocharova O., Savtchenko R., Alexeeva I., Budka H., Rohwer R. G., Baskakov I. V. (2010) Recombinant prion protein induces a new transmissible prion disease in wild-type animals. Acta Neuropathol. 119, 177–187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Cobb N. J., Sönnichsen F. D., McHaourab H., Surewicz W. K. (2007) Molecular architecture of human prion protein amyloid: a parallel, in-register β-structure. Proc. Natl. Acad. Sci. U.S.A. 104, 18946–18951 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Jones E. M., Wu B., Surewicz K., Nadaud P. S., Helmus J. J., Chen S., Jaroniec C. P., Surewicz W. K. (2011) Structural polymorphism in amyloids: new insights from studies with Y145Stop prion protein fibrils. J. Biol. Chem. 286, 42777–42784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Tycko R., Savtchenko R., Ostapchenko V. G., Makarava N., Baskakov I. V. (2010) The α-helical C-terminal domain of full-length recombinant PrP converts to an in-register parallel β-sheet structure in PrP fibrils: evidence from solid state nuclear magnetic resonance. Biochemistry 49, 9488–9497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Helmus J. J., Surewicz K., Nadaud P. S., Surewicz W. K., Jaroniec C. P. (2008) Molecular conformation and dynamics of the Y145Stop variant of human prion protein in amyloid fibrils. Proc. Natl. Acad. Sci. U.S.A. 105, 6284–6289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Helmus J. J., Surewicz K., Apostol M. I., Surewicz W. K., Jaroniec C. P. (2011) Intermolecular alignment in Y145Stop human prion protein amyloid fibrils probed by solid-state NMR spectroscopy. J. Am. Chem. Soc. 133, 13934–13937 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Adrover M., Pauwels K., Prigent S., de Chiara C., Xu Z., Chapuis C., Pastore A., Rezaei H. (2010) Prion fibrillization is mediated by a native structural element that comprises helices H2 and H3. J. Biol. Chem. 285, 21004–21012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Wickner R. B., Edskes H. K., Bateman D. A., Kelly A. C., Gorkovskiy A., Dayani Y., Zhou A. (2013) Amyloids and yeast prion biology. Biochemistry 52, 1514–1527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Toyama B. H., Weissman J. S. (2011) Amyloid structure: conformational diversity and consequences. Annu. Rev. Biochem. 80, 557–585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Shewmaker F., Wickner R. B., Tycko R. (2006) Amyloid of the prion domain of Sup35p has an in-register parallel β-sheet structure. Proc. Natl. Acad. Sci. U.S.A. 103, 19754–19759 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Baxa U., Wickner R. B., Steven A. C., Anderson D. E., Marekov L. N., Yau W. M., Tycko R. (2007) Characterization of β-sheet structure in Ure2p1–89 yeast prion fibrils by solid-state nuclear magnetic resonance. Biochemistry 46, 13149–13162 [DOI] [PubMed] [Google Scholar]
- 37. Wickner R. B., Dyda F., Tycko R. (2008) Amyloid of Rnq1p, the basis of the [PIN+] prion, has a parallel in-register β-sheet structure. Proc. Natl. Acad. Sci. U.S.A. 105, 2403–2408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Antzutkin O. N., Balbach J. J., Leapman R. D., Rizzo N. W., Reed J., Tycko R. (2000) Multiple quantum solid-state NMR indicates a parallel, not antiparallel, organization of β-sheets in Alzheimer's β-amyloid fibrils. Proc. Natl. Acad. Sci. U.S.A. 97, 13045–13050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Tycko R. (2011) Solid-state NMR studies of amyloid fibril structure. Annu. Rev. Phys. Chem. 62, 279–299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Benzinger T. L., Gregory D. M., Burkoth T. S., Miller-Auer H., Lynn D. G., Botto R. E., Meredith S. C. (1998) Propagating structure of Alzheimer's β-amyloid(10–35) is parallel β-sheet with residues in exact register. Proc. Natl. Acad. Sci. U.S.A. 95, 13407–13412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Tycko R. (2007) Symmetry-based constant-time homonuclear dipolar recoupling in solid state NMR. J. Chem. Phys. 126, 064506 [DOI] [PubMed] [Google Scholar]
- 42. Wilham J. M., Orrú C. D., Bessen R. A., Atarashi R., Sano K., Race B., Meade-White K. D., Taubner L. M., Timmes A., Caughey B. (2010) Rapid end-point quantitation of prion seeding activity with sensitivity comparable to bioassays. PLoS Pathog. 6, e1001217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Atarashi R., Wilham J. M., Christensen L., Hughson A. G., Moore R. A., Johnson L. M., Onwubiko H. A., Priola S. A., Caughey B. (2008) Simplified ultrasensitive prion detection by recombinant PrP conversion with shaking. Nat. Methods 5, 211–212 [DOI] [PubMed] [Google Scholar]
- 44. Caughey B., Raymond G. J., Ernst D., Race R. E. (1991) N-terminal truncation of the scrapie-associated form of PrP by lysosomal protease(s): implications regarding the site of conversion of PrP to the protease-resistant state. J. Virol. 65, 6597–6603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Petkova A. T., Tycko R. (2002) Sensitivity enhancement in structural measurements by solid state NMR through pulsed spin locking. J. Magn. Reson. 155, 293–299 [DOI] [PubMed] [Google Scholar]
- 46. Hafner-Bratkovic I., Bester R., Pristovsek P., Gaedtke L., Veranic P., Gaspersic J., Mancek-Keber M., Avbelj M., Polymenidou M., Julius C., Aguzzi A., Vorberg I., Jerala R. (2011) Globular domain of the prion protein needs to be unlocked by domain swapping to support prion protein conversion. J. Biol. Chem. 286, 12149–12156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Klose D. P., Wallace B. A., Janes R. W. (2010) 2Struc: the secondary structure server. Bioinformatics 26, 2624–2625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Atarashi R., Moore R. A., Sim V. L., Hughson A. G., Dorward D. W., Onwubiko H. A., Priola S. A., Caughey B. (2007) Ultrasensitive detection of scrapie prion protein using seeded conversion of recombinant prion protein. Nat. Methods 4, 645–650 [DOI] [PubMed] [Google Scholar]
- 49. Smirnovas V., Kim J. I., Lu X., Atarashi R., Caughey B., Surewicz W. K. (2009) Distinct structures of scrapie prion protein (PrPSc)-seeded versus spontaneous recombinant prion protein fibrils revealed by hydrogen/deuterium exchange. J. Biol. Chem. 284, 24233–24241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Wishart D. S., Sykes B. D., Richards F. M. (1991) Relationship between nuclear-magnetic-resonance chemical-shift and protein secondary structure. J. Mol. Biol. 222, 311–333 [DOI] [PubMed] [Google Scholar]
- 51. Welker E., Raymond L. D., Scheraga H. A., Caughey B. (2002) Intramolecular versus intermolecular disulfide bonds in prion proteins. J. Biol. Chem. 277, 33477–33481 [DOI] [PubMed] [Google Scholar]
- 52. Herrmann L. M., Caughey B. (1998) The importance of the disulfide bond in prion protein conversion. Neuroreport 9, 2457–2461 [DOI] [PubMed] [Google Scholar]
- 53. Turk E., Teplow D. B., Hood L. E., Prusiner S. B. (1988) Purification and properties of the cellular and scrapie hamster prion proteins. Eur. J. Biochem. 176, 21–30 [DOI] [PubMed] [Google Scholar]
- 54. Eisenberg D., Jucker M. (2012) The amyloid state of proteins in human diseases. Cell 148, 1188–1203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Shewmaker F., Ross E. D., Tycko R., Wickner R. B. (2008) Amyloids of shuffled prion domains that form prions have a parallel in-register β-sheet structure. Biochemistry 47, 4000–4007 [DOI] [PubMed] [Google Scholar]
- 56. Paramithiotis E., Pinard M., Lawton T., LaBoissiere S., Leathers V. L., Zou W. Q., Estey L. A., Lamontagne J., Lehto M. T., Kondejewski L. H., Francoeur G. P., Papadopoulos M., Haghighat A., Spatz S. J., Head M., Will R., Ironside J., O'Rourke K., Tonelli Q., Ledebur H. C., Chakrabartty A., Cashman N. R. (2003) A prion protein epitope selective for the pathologically misfolded conformation. Nat. Med. 9, 893–899 [DOI] [PubMed] [Google Scholar]
- 57. Kocisko D. A., Lansbury P. T., Jr., Caughey B. (1996) Partial unfolding and refolding of scrapie-associated prion protein: evidence for a critical 16-kDa C-terminal domain. Biochemistry 35, 13434–13442 [DOI] [PubMed] [Google Scholar]
- 58. Vázquez-Fernández E., Alonso J., Pastrana M. A., Ramos A., Stitz L., Vidal E., Dynin I., Petsch B., Silva C. J., Requena J. R. (2012) Structural organization of mammalian prions as probed by limited proteolysis. PLoS ONE 7, e50111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Sajnani G., Pastrana M. A., Dynin I., Onisko B., Requena J. R. (2008) Scrapie prion protein structural constraints obtained by limited proteolysis and mass spectrometry. J. Mol. Biol. 382, 88–98 [DOI] [PubMed] [Google Scholar]
- 60. Supattapone S., Bosque P., Muramoto T., Wille H., Aagaard C., Peretz D., Nguyen H. O., Heinrich C., Torchia M., Safar J., Cohen F. E., DeArmond S. J., Prusiner S. B., Scott M. (1999) Prion protein of 106 residues creates an artificial transmission barrier for prion replication in transgenic mice. Cell 96, 869–878 [DOI] [PubMed] [Google Scholar]
- 61. DeMarco M. L., Daggett V. (2004) From conversion to aggregation: protofibril formation of the prion protein. Proc. Natl. Acad. Sci. U.S.A. 101, 2293–2298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Deleault N. R., Harris B. T., Rees J. R., Supattapone S. (2007) Formation of native prions from minimal components in vitro. Proc. Natl. Acad. Sci. U.S.A. 104, 9741–9746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Deleault N. R., Geoghegan J. C., Nishina K., Kascsak R., Williamson R. A., Supattapone S. (2005) Protease-resistant prion protein amplification reconstituted with partially purified substrates and synthetic polyanions. J. Biol. Chem. 280, 26873–26879 [DOI] [PubMed] [Google Scholar]
- 64. Onisko B., Fernández E. G., Freire M. L., Schwarz A., Baier M., Camiña F., García J. R., Rodríguez-Segade Villamarín S., Requena J. R. (2005) Probing PrPSc structure using chemical cross-linking and mass spectrometry: evidence of the proximity of Gly-90 amino termini in the PrP 27–30 aggregate. Biochemistry 44, 10100–10109 [DOI] [PubMed] [Google Scholar]
- 65. Wickner R. B., Shewmaker F., Edskes H., Kryndushkin D., Nemecek J., McGlinchey R., Bateman D., Winchester C. L. (2010) Prion amyloid structure explains templating: how proteins can be genes. FEMS Yeast Res. 10, 980–991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. DebBurman S. K., Raymond G. J., Caughey B., Lindquist S. (1997) Chaperone-supervised conversion of prion protein to its protease-resistant form. Proc. Natl. Acad. Sci. U.S.A. 94, 13938–13943 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Horiuchi M., Priola S. A., Chabry J., Caughey B. (2000) Interactions between heterologous forms of prion protein: binding, inhibition of conversion, and species barriers. Proc. Natl. Acad. Sci. U.S.A. 97, 5836–5841 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Orrú C. D., Wilham J. M., Raymond L. D., Kuhn F., Schroeder B., Raeber A. J., Caughey B. (2011) Prion disease blood test using immunoprecipitation and improved quaking-induced conversion. MBio. 2, e00078–e00011 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.