

Abstract
The zinc transporter protein ZIP13 plays critical roles in bone, tooth, and connective tissue development, and its dysfunction is responsible for the spondylocheirodysplastic form of Ehlers-Danlos syndrome (SCD-EDS, OMIM 612350). Here, we report the molecular pathogenic mechanism of SCD-EDS caused by two different mutant ZIP13 proteins found in human patients: ZIP13G64D, in which Gly at amino acid position 64 is replaced by Asp, and ZIP13ΔFLA, which contains a deletion of Phe-Leu-Ala. We demonstrated that both the ZIP13G64D and ZIP13ΔFLA protein levels are decreased by degradation via the valosin-containing protein (VCP)-linked ubiquitin proteasome pathway. The inhibition of degradation pathways rescued the protein expression levels, resulting in improved intracellular Zn homeostasis. Our findings uncover the pathogenic mechanisms elicited by mutant ZIP13 proteins. Further elucidation of these degradation processes may lead to novel therapeutic targets for SCD-EDS.
Keywords: Proteasome, SCD-EDS, VCP, zinc transporter, ZIP13
Introduction
Zinc (Zn) transporters are pivotal for Zn homeostasis, which is important for human health (Fukada & Kambe, 2011). Zn contributes to a variety of cellular functions and physiological events (Fukada et al, 2014), and impaired Zn regulation can cause a variety of diseases (Prasad, 1995; MacDonald, 2000; Lichten & Cousins, 2009; Fukada et al, 2011b; Ryu et al, 2011). One such disease is acrodermatitis enteropathica (AE), a pediatric disorder resulting from Zn deficiency. Patients with autosomal recessive AE have mutations in the SLC39A4 gene (Wang et al, 2002; Dufner-Beattie et al, 2007), which encodes ZIP4, a membrane protein that mediates Zn influx across the cell membrane. A loss-of-function SLC39A4 gene mutation in humans results in growth retardation, dermatitis, and hair loss (Wang et al, 2002; Dufner-Beattie et al, 2007). ZIP4 may also affect pancreatic cancer pathogenesis and progression (Li et al, 2007; Zhang et al, 2013), and intestinal integrity (Geiser et al, 2012). ZIP6 is reported to control metastasis (Yamashita et al, 2004; Hogstrand et al, 2013), ZIP7 is involved in the progression and proliferation of breast cancer cells (Taylor et al, 2007), and ZIP8 plays a key role in osteoarthritis-related cartilage destruction (Kim et al, 2014). Transient neonatal Zn deficiency is a disease related to the SLC30A2 gene, which encodes the Zn efflux protein ZnT2. A heterozygous mutation in ZnT2 causes a low Zn concentration in mothers' milk, resulting in Zn deficiency in their breast-fed infants (Chowanadisai et al, 2006; Itsumura et al, 2013). ZnT8, which is expressed in pancreatic β cells, is essential for packaging insulin crystals (Bosco et al, 2010; Hardy et al, 2011), and variants in the SLC30A8/ZnT8 gene are associated with an increased risk for type 2 diabetes (Xu et al, 2012; Tamaki et al, 2013).
The spondylocheirodysplastic form of Ehlers-Danlos syndrome (SCD-EDS, OMIM 612350), a genetic disorder of connective tissues, bones, and teeth, is also related to Zn imbalance (Fukada et al, 2008; Giunta et al, 2008; Warman et al, 2011; Byers & Murray, 2012). SCD-EDS patients show short stature, skeletal dysplasia of the spine, and clinical abnormalities of the hands and teeth, in addition to the common features of EDS such as skin and joint looseness. A mouse model of SCD-EDS, the Slc39a13/Zip13-deficient (Zip13-KO) mouse, has features similar to those of human patients, that is, abnormal development of the skin, bone, teeth, and craniofacial structures. (Fukada et al, 2008, 2011a; Munemasa et al, 2014). Molecular analyses revealed that the mesenchymal-originated cells from Zip13-KO mice have impaired BMP/TGF-β signaling, indicating that ZIP13 is critical for the development of hard and connective tissues (Fukada et al, 2008). By homozygosity mapping of Portuguese patients with SCD-EDS, we identified a pathogenic mutation (c.221G>A, G74D) in the SLC39A13 gene (Fukada et al, 2008). The ectopic expression of the G74D ZIP13 mutant could not fully rescue Zip13-KO primary osteoblasts or dermal fibroblasts, indicating that G74D was a loss-of-function mutation (Fukada et al, 2008). This mutation was later renamed G64D, after identification of the de facto start codon 10 amino acids downstream from the conventional start codon, and its membrane topology was refined (Bin et al, 2011). Another mutant ZIP13 protein, in which phenylalanine–leucine–alanine (FLA) is deleted (ZIP13ΔFLA), was also reported in human SCD-EDS patients (Giunta et al, 2008). Characterization of the wild-type (WT) ZIP13 protein revealed that it is localized to the Golgi, possesses 8 putative transmembrane domains (TMs) with luminal N- and C-termini, and forms homo-dimers (Fukada et al, 2008; Bin et al, 2011), and its luminal loop was proposed to be responsible for Zn selection (Potocki et al, 2013). However, it remains unknown how the identified ZIP13 mutations lead to SCD-EDS.
Here, we demonstrate that both the ZIP13G64D and ZIP13ΔFLA proteins are rapidly degraded via the valosin-containing protein (VCP)-linked ubiquitin proteasome pathway, leading to an imbalance of intracellular Zn homeostasis. Furthermore, the protein expression levels and Zn homeostasis were recovered by inhibiting the proteasome machinery. This is the first demonstration of the mechanism by which these mutations cause the loss of ZIP13 function and SCD-EDS, and our findings may suggest potential therapies for treating this disease.
Results
The level of ZIP13G64D protein is decreased in cultured cells
To characterize the pathogenic ZIP13G64D protein, in which a glycine at amino acid position 64 (G64), located within TM1, is replaced by aspartic acid (Fig 1A), we first introduced ZIP13WT- and ZIP13G64D-expressing plasmids into 293T cells. While ZIP13WT increased the Metallothionein 1 (MT1) gene expression (Fig 1B) reflecting an increased intracellular Zn level (Supplementary Fig S1), ZIP13G64D did not, even though the ZIP13G64D and ZIP13WT transcript levels were equivalent (Fig 1C). In addition, the ZIP13 protein was barely detected by the anti-ZIP13 antibody ab-A1 (Fig 1D) in transiently ZIP13G64D-expressing 293T cells (Fig 1E). Similar results were obtained in HeLa cells stably expressing ZIP13G64D (Supplementary Fig S2A). These findings suggested that the ZIP13G64D protein was unstable, resulting in an imbalance of intracellular Zn homeostasis.
Figure 1. ZIP13 with the pathogenic G64D mutation shows a decreased protein expression level.
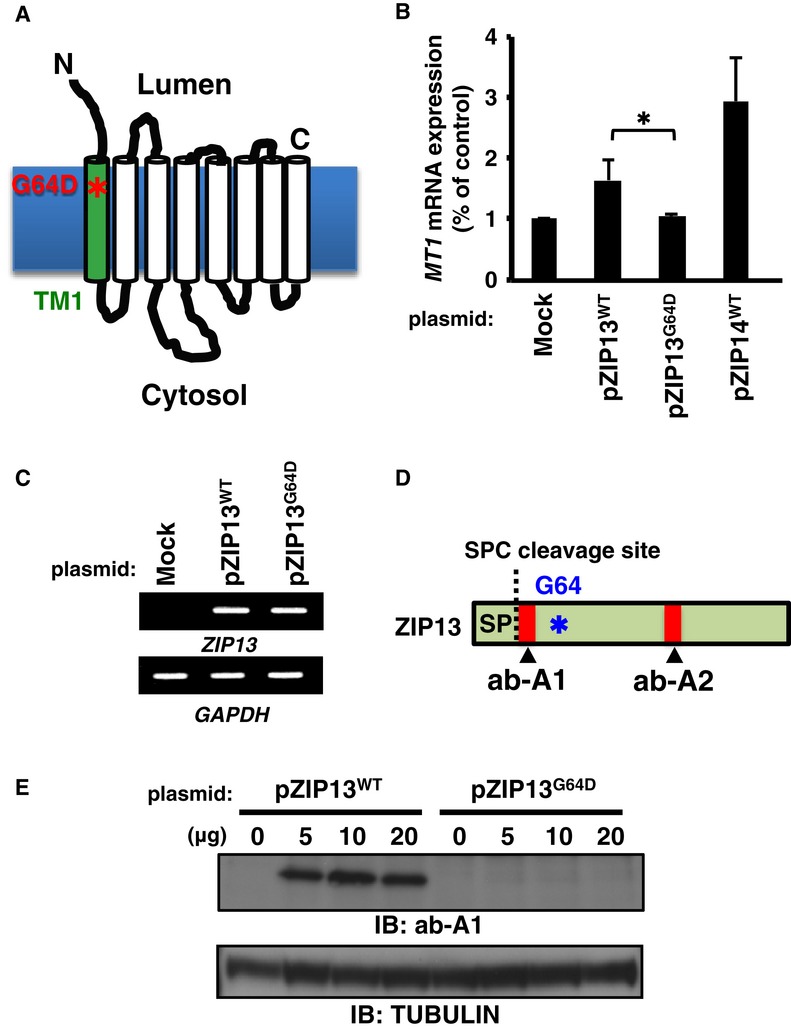
A Location of the G64D mutation in ZIP13. Asterisk (*) indicates the G64D mutation.
B Metallothionein 1 (MT1) expression. 293T cells transfected with the indicated DNA constructs were treated with 50 μM ZnSO4 for 6 h, and then, the MT1 mRNA expression level was analyzed by RT-qPCR. Data are representative of three experiments and shown as mean ± s.e.m. (*P = 0.037). ZIP14WT was included as a positive control.
C ZIP13 transcript levels in 293T cells expressing wild-type or G64D mutant ZIP13. 293T cells were transfected with plasmids for ZIP13WT or ZIP13G64D. Twenty-four hours later, RT–PCR was performed using primers for the indicated genes (Fukada et al, 2008).
D Schematic diagram showing the recognition sites of anti-ZIP13 antibodies. Asterisk (*) indicates the G64D mutation. SP, signal peptide; SPC, signal peptidase complex; ab-A1 and ab-A2 indicate anti-ZIP13 antibodies that recognize amino acids 23–35 of human ZIP13 and 184–201 of mouse ZIP13, respectively.
E ZIP13 protein levels in 293T cells expressing wild-type and G64D mutant ZIP13. Cell lysates were analyzed by Western blot (IB) using the ab-A1 antibody.
Source data are available online for this figure.
The G64D mutation affects the stability of the ZIP13 protein
We previously identified the signal peptide (SP) of the ZIP13 protein (Fig 1D) (Bin et al, 2011). SP is cleaved to yield the “mature” protein, that is, the functional protein with the correct intracellular distribution. To determine whether the G64D mutation affects the level of the mature ZIP13 or the SP-uncleaved “immature” protein, we generated two anti-ZIP13 antibodies: one against a synthetic peptide corresponding to an internal sequence (amino acids 23–35) in human ZIP13, proximal to the signal peptidase complex (SPC) cleavage site (ab-A1) and another against amino acids 184–201 of mouse ZIP13 (ab-A2) (Figs 1D and 2A). When the lysates of 293T cells expressing N-terminally 3xFLAG-tagged wild-type ZIP13 (Fig 2A) were immunoprecipitated using anti-FLAG antibody, separated by SDS–PAGE, and subjected to silver staining, two unique bands were observed with molecular weights between 29 and 47 kDa (band-A and band-B) (Fig 2B, left). In contrast, when cells expressing mutant ZIP13 (F-G64D) were treated similarly, band-B was severely decreased while band-A remained (Fig 2B, left). Western blot using an anti-FLAG antibody revealed that band-A contained FLAG and was therefore the SP-uncleaved, immature ZIP13 protein (Fig 2B, middle). Band-B was recognized in the F-WT sample by ab-A1 (Fig 2B, right), but not by the anti-FLAG antibody (Fig 2B, middle), indicating that it was the SP-cleaved, mature ZIP13WT protein. No bands were detected by the ab-A1 antibody in the F-G64D sample (Fig 2B, right), indicating that the SP-cleaved ZIP13G64D mature protein was specifically decreased in the cells. Western blot with the ab-A2 antibody revealed band-B at a lower position, most likely corresponding to the SP-cleaved, mature ZIP13 protein (Fig 2C, middle), and the amount of band-B yielded by the expression plasmid for F-G64D was markedly decreased (Fig 2C, middle). Furthermore, when the lysates from cells expressing a C-terminally V5 epitope-tagged ZIP13 (ZIP13-V5) (Fig 2D) were subjected to Western blot with an anti-V5 antibody, the V5-tagged mutant (G64D-V5) levels were lower (Fig 2E and Supplementary Fig S2A), similar to the results with F-G64D (Fig 2B). While immunoprecipitation analysis showed the same two bands in both the wild-type (WT-V5) and G64D-V5 samples (Fig 2E, band-A and band-B), the G64D-V5-expressing cells contained a reduced amount of band-B, indicating that the expression of SP-cleaved G64D mature protein was greatly reduced in these cells.
Figure 2. The pathogenic G64D mutation affects the stability of the SP-cleaved mature ZIP13 protein.
A Schematic diagram of the N-terminally 3xFLAG-tagged ZIP13 protein (F-ZIP13). Asterisk (*) indicates the G64D mutation. SP, signal peptide; SPC, signal peptidase complex; ab-A1 and ab-A2: anti-ZIP13 antibodies.
B Protein expression of F-ZIP13 in 293T cells. N-terminally 3xFLAG-tagged wild-type (F-WT) and G64D mutant (F-G64D) ZIP13 proteins were immunoprecipitated (IP) with an anti-FLAG antibody, and then, the immunoprecipitates were analyzed by silver staining and Western blot using an anti-FLAG or anti-ZIP13 (ab-A1) antibody. IgH, heavy chain of IgG; IgL, light chain of IgG; A: SP-uncleaved immature ZIP13 protein; B: SP-cleaved mature ZIP13 protein.
C SP-cleaved mature ZIP13 protein was detected by ab-A2. A: SP-uncleaved immature ZIP13 protein; B: SP-cleaved mature ZIP13 protein.
D Schematic diagram of the C-terminally V5 epitope-tagged ZIP13 protein (ZIP13-V5).
E Protein expression of ZIP13-V5 in 293T cells. V5 epitope-tagged wild-type or G64D mutant ZIP13 protein (WT-V5 or G64D-V5) was immunoprecipitated using an anti-V5 antibody, and then, the immunoprecipitate was analyzed by Western blot using an anti-V5 antibody. A: SP-uncleaved immature ZIP13 protein; B: SP-cleaved mature ZIP13 protein.
F Dimer formation assay. The dimer formation of ZIP13 was analyzed by blue native-PAGE using the lysates of 293T cells expressing F-WT or F-G64D.
F Monomer–monomer interaction assay. 293T cells were co-transfected with expression plasmids for F-G64D and G64D-V5 ZIP13, followed by immunoprecipitation with the indicated antibodies. Western blotting analysis was performed with either an anti-V5 or anti-FLAG antibody.
Source data are available online for this figure.
Since ZIP13 protein forms a homo-dimer (Bin et al, 2011) and the G87R mutation in the zinc transporter ZnT2 is reported to cause neonatal zinc deficiency due to a dominant-negative effect on its homo-dimerization (Lasry et al, 2012), we next examined whether the G64D mutation affects the oligomeric state of the ZIP13 protein. Blue native-PAGE analysis of lysates from F-ZIP13-expressing 293T cells showed a lower expression of F-G64D than F-WT, but the F-G64D apparently still formed dimers similar to F-WT (Fig 2F). We further evaluated the monomer–monomer interaction between ZIP13G64D proteins in 293T cells that were co-transfected with plasmids encoding F-G64D and G64D-V5, followed by immunoprecipitation with anti-FLAG or anti-V5 antibodies. Western blotting analysis clearly showed that F-G64D and G64D-V5 formed a complex (Fig 2G). Taken together, these results indicated that the loss of function of the G64D mutation was mainly attributable to a large reduction in the quantity of the mature ZIP13 protein, rather than to a disruption in ZIP13's ability to form a complex due to a change in its biochemical characteristics.
Proteasome-dependent pathways are involved in the degradation of ZIP13G64D protein
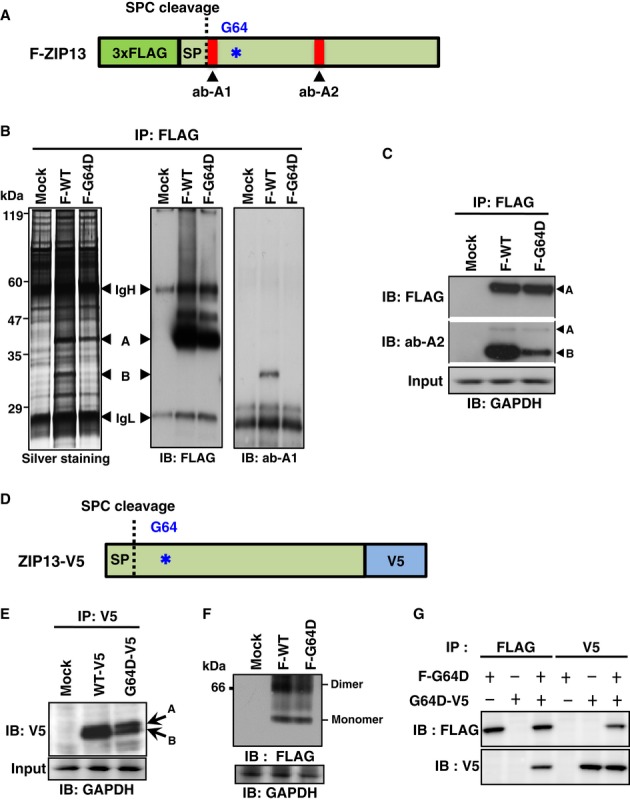
Given that the expression level of ZIP13G64D protein but not its mRNA was reduced, it was likely that a protein degradation pathway was involved. To address this possibility, we expressed ZIP13-V5 (Fig 2D) in 293T cells, followed by treatment with MG132, an inhibitor of proteasome-dependent degradation pathways, or bafilomycin, an inhibitor of lysosome-dependent degradation pathways (Lee & Goldberg, 1998; Ishidoh & Kominami, 2002). The cells were then lysed by a detergent-containing buffer, and the lysates were separated into soluble and insoluble fractions by brief centrifugation and subjected to Western blotting analysis. The protein level of G64D-V5 in the NP40-detergent-soluble fraction was lower than that of WT-V5 (Fig 3A, left), similar to the result using non-tagged ZIP13s (Fig 1E). While bafilomycin had no apparent effect on the protein expression patterns, MG132 preferentially increased the amount of WT-V5 and G64D-V5 protein in the NP40-detergent-insoluble fraction, which contained numerous ubiquitinated proteins, and in which the level of G64D-V5 was greater than that of WT-V5 (Fig 3A, right). These findings indicated that ZIP13 is normally degraded by a proteasome-dependent pathway and that the G64D mutation alters the protein's properties so that more of it accumulates in the detergent-insoluble fraction.
Figure 3. ZIP13G64D protein is readily degraded by a proteasome-dependent mechanism.

A Proteasome inhibitor treatments. 293T cells were transfected with WT-V5 or G64D-V5 ZIP13 and treated with 10 μM MG132 or 1 μM bafilomycin for 6 h. Cells were lysed in 1% NP-40 and then separated into soluble and insoluble fractions. Western blotting analysis was performed with an anti-V5 or anti-ubiquitin antibody.
B HeLa cells expressing WT-V5 or G64D-V5 (Supplementary Fig S2A) were treated with 10 μM MG132 for the indicated periods. (Upper) Total cell lysates were analyzed by Western blot using an anti-V5 antibody. (Lower) The hCD8 levels indicate the amount of transfected plasmid DNA (pMX-WT-IRES-hCD8 or pMX-G64D-IRES-hCD8). Cells were analyzed by flow cytometry using APC-conjugated anti-hCD8 antibody. Histograms were gated on hCD8-positive cells.
C Confocal images of ZIP13. HeLa cells stably expressing the indicated proteins were treated with or without MG132. Nuclei (blue), ZIP13 (green), Golgi (red), and actin (magenta) were stained with DAPI, anti-V5 antibody, anti-GM130 antibody, and Phalloidin, respectively.
D HeLa cells stably expressing the indicated proteins were treated with proteasome inhibitors 10 μM MG132 or 1 μM lactacystin for 6 h, followed by Western blot of whole-cell lysates using an anti-V5 antibody.
E Location of pathogenic mutations in TM1. Amino acid alignment of the TM1 of human ZIP family members. Red: hydrophobic amino acids; blue: acidic amino acids; magenta: basic amino acids; green: hydrophilic amino acids. AE (G340D): amino acid substitution in ZIP4 of AE patients; SCD-EDS (G64D): amino acid substitution in ZIP13 of SCD-EDS patients.
F The 64th amino acid influences ZIP13 protein stability. C-terminally V5-tagged ZIP13 expression plasmids with a mutation at position 64 were transfected into 293T cells and analyzed by Western blot using an anti-V5 antibody.
G Mutant ZIP13 constructs with an acidic amino acid at position 64. 293T cells were transfected with C-terminally V5-tagged ZIP13 expression plasmids, treated with MG132, lysed in NP-40, separated into soluble and insoluble fractions, and analyzed using an anti-V5 antibody.
H Mutant ZIP13 constructs in which glycine 64 was replaced with asparagine (G64N) or glutamine (G64Q). Total cell lysates were analyzed by Western blot using an anti-V5 antibody.
Source data are available online for this figure.
To confirm that the ZIP13G64D protein was degraded via a proteasome-dependent pathway, we repeated the experiment using a different cell line. HeLa cell lines stably expressing WT-V5 or G64D-V5 were established by blasticidin selection. Clones containing similar amounts of transfected cDNA were selected by monitoring their internal ribosome entry site (IRES)-driven human CD8 expression (Fig 3B, lower, and Supplementary Fig S2C), then treated with the proteasome inhibitor MG132. Western blotting analysis showed that MG132 treatment led to an increase in the G64D-V5 protein over time (Fig 3B, upper), accumulating it most likely in the Golgi (Fig 3C), where ZIP13 is normally localized (Fukada et al, 2008). Furthermore, treatment with lactacystin, another proteasome inhibitor, upregulated the G64D-V5 protein expression (Fig 3D). The ZIP13 homodimers were also increased when MG132 was applied (Supplementary Fig S3). These findings suggested that the G64D protein enters a proteasome-dependent degradation pathway.
Amino acid alignment showed that ZIP family members share a small and neutral amino acid at the site corresponding to the 64th position of ZIP13 (Fig 3E). To determine how the amino acid composition at this position affects protein stability, we next substituted different amino acids at the 64th position, using a variety of approaches. Replacement of G64 with an amino acid containing a small side chain, such as alanine (G64A), cysteine (G64C), or serine (G64S), caused little change in the protein expression level from that of wild-type ZIP13 (Fig 3F). However, the replacement with an amino acid containing a large side chain, isoleucine (G64I) or leucine (G64L), or with the basic amino acid arginine (G64R) significantly reduced the protein level, although not to the same extent as with aspartic acid, an acidic amino acid (G64D) (Fig 3F). We thus hypothesized that the acidic side chain in G64D interferes with the stability of the ZIP13 protein. To address this possibility, we replaced G64 with another acidic amino acid, glutamic acid (G64E), and observed a severe decrease in the ZIP13G64E protein level, comparable to ZIP13G64D (Fig 3F and G). Notably, the transcript levels of these mutants were all comparable to that of wild type (Supplementary Fig S4A), and MG132 treatment caused ZIP13G64E protein to be recovered in the insoluble fraction, similar to ZIP13G64D protein (Fig 3G). The replacement of G64 with asparagine (G64N) or glutamine (G64Q) also reduced the protein level, but to a lesser extent than G64D (Fig 3H), while the transcription level was similar to wild-type cells (Supplementary Fig S4B). Based on these findings, we concluded that a small and neutral amino acid at the 64th position is critical for the stability of the ZIP13 protein. The replacement of G64 with an amino acid having a large or basic side chain caused its protein level to decrease, and acidity at the 64th position was fatal to the ZIP13 protein, leading to its clearance by the proteasome-dependent (20S proteasome-independent: Supplementary Fig S5) degradation pathway.
Pathogenic ZIP13 proteins are degraded by the ubiquitination-dependent pathway
To determine whether the ZIP13G64D protein was ubiquitinated, 6 × histidine-tagged mono-ubiquitin was co-expressed with ZIP13WT-V5 or ZIP13G64D-V5 in 293T cells; then, the ubiquitin-containing proteins were purified using Ni-NTA agarose under denaturing conditions. Ubiquitinated ZIP13WT or ZIP13G64D protein was elevated in the MG132-treated samples (Supplementary Fig S6). Consistent with this finding, cotreatment with PYR-41 (a ubiquitin-activating enzyme E1 inhibitor) and the protein synthesis inhibitor cyclohexamide (CHX) suppressed the decrease in mutant ZIP13 protein expression in HeLa cells (Fig 4A). In addition, we noted an increase in the slowly migrating ubiquitinated wild-type ZIP13 protein after MG132 treatment (Fig 4B, left) and that the ubiquitinated/non-ubiquitinated G64D protein ratio was significantly higher than that of wild type (Fig 4B, right). These findings suggested that the wild-type ZIP13 protein is turned over by the ubiquitin proteasome pathway, but the G64D mutant is more extensively degraded by this pathway.
Figure 4. ZIP13G64D protein is degraded by a ubiquitination-dependent pathway.
A Treatment with PYR-41, a ubiquitin E1 inhibitor, suppressed the downregulation of ZIP13G64D protein in the presence of cycloheximide (CHX). HeLa cells stably expressing WT-V5 or G64D-V5 were treated with 10 μM MG132 or 10 μM PYR-41 together with CHX for the indicated times. Total cell lysates were subjected to Western blotting analysis with an anti-V5 antibody. Right panel shows the relative expression levels of ZIP13 proteins. Data are representative of two independent experiments.
B HeLa cells stably expressing WT-V5 or G64D-V5 (Supplementary Fig S2A) were treated with 10 μM MG132 for 6 h. The cell lysates were analyzed by Western blot using an anti-V5 antibody. The ubiquitinated/non-ubiquitinated G64D protein ratio was upregulated compared to that of wild type (right panel). Data are shown as mean ± s.e.m. (*P = 0.036).
C Single cycle kinetic analysis of ZIP13 protein binding to the amine-coupled antibody 35B11 on a Biacore sensor tip. Solution-phase ZIP13-35B11 binding was measured by surface plasmon resonance (BIAcore). A representative BIAcore sensorgram shows the response over time (resonance units [RU]) during the binding of purified recombinant human ZIP13 protein to immobilized 35B11 antibodies. Purified human ZIP13 protein at concentrations of 25, 50, 100, 200, and 400 nM was added at 0, 190, 380, 570, and 760 s, respectively. The graph is representative of four independent experiments.
D Intracellular flow cytometric analysis of the endogenous ZIP13 expression in a healthy female donor or female SCD-EDS patient. Cultured primary human fibroblasts were treated with DMSO or 10 μM MG132 for 6 h. After fixation and permeabilization, the cells were stained with the monoclonal antibody 35B11, followed by goat anti-mouse Alexa 488. Data are representative of two independent experiments. Similar results were obtained in a healthy male donor and male SCD-EDS patient.
Source data are available online for this figure.
Next, we investigated whether these results were applicable to cells from SCD-EDS patients. We first generated the monoclonal anti-human ZIP13 antibody 35B11 clone using the “liposome immunization” system and the three-step screening method (Hino et al, 2013). This method is useful for producing antibodies that recognize the tertiary structure of a membrane protein with high affinity (Hino et al, 2013). The 35B11 clone was confirmed to bind the purified ZIP13 protein, assessed by surface plasmon resonance (SPR) experiments (Fig 4C). Sensorgrams fitted to a 1:1 binding model using the Biacore T200 Evaluation Software yielded the following average kinetic constants: ka, 1.34 ± 0.04 × 104 M−1 s−1; kd, 2.59 ± 0.3 × 10−4 s−1; KD, 19.3 ± 2.7 nM. Flow cytometric analyses using 35B11 demonstrated that the level of ZIP13G64D protein was significantly reduced compared to ZIP13WT protein in HeLa stable lines (Supplementary Fig S7), confirming that this antibody was also useful for detecting the cellular ZIP13 proteins. We next prepared primary cultured fibroblasts from the biopsies of healthy donors and SCD-EDS patients who expressed the ZIP13G64D protein and compared the ZIP13 protein levels. Consistent with the results in cell lines, the expression level of ZIP13 protein was decreased in the cells from patients compared to those from healthy donors (Fig 4D, blue line versus dotted line). Importantly, MG132 treatment of the SCD-EDS patient cells increased the total ZIP13G64D protein expression to the level of healthy donors (Fig 4D, red line versus dotted line), indicating that the pathogenic G64D mutation of ZIP13 in SCD-EDS patients causes degradation of the functional protein by the proteasome-dependent pathway.
We also studied the effect on protein levels of another ZIP13 mutation (Giunta et al, 2008), in which three amino acids (phenylalanine–leucine–alanine: FLA) in TM3 are deleted as the result of a frame shift (ZIP13ΔFLA, Fig 5A and B). The ZIP13ΔFLA protein expression was also reduced although it was more unstable than the ZIP13ΔG64D protein, and failed to increase the intracellular Zn level in 293T cells and in HeLa cells stably introduced with its expression plasmid (Fig 5C–G, Supplementary Figs S1 and S2). Moreover, ZIP13ΔFLA protein was readily restored after MG132 treatment (Fig 5F), indicating that it was degraded by the proteasome-dependent pathway as well as the ZIP13G64D protein.
Figure 5. ZIP13ΔFLA protein is degraded by a proteasome-dependent pathway.

A Location of the ΔFLA mutation (deletion of phenylalanine–leucine–alanine in TM3) in ZIP13.
B Amino acid alignment of the TM3 of human ZIP family members. Amino acids conserved in all of the indicated zinc transporters (*), conserved substitutions (:), semi-conserved substitutions (.). Red: hydrophobic amino acids; blue: acidic amino acids; magenta: basic amino acids; green: hydrophilic amino acids.
C Protein expression level of G64D-V5 in 293T stable lines. The cell lysates of two representative clones stably expressing WT-V5 or the G64D-V5 mutant were analyzed by Western blot using an anti-V5 antibody.
D Protein expression level of ΔFLA-V5 in 293T stable lines. The cell lysates of two representative clones stably expressing WT-V5 or the ΔFLA-V5 mutant were analyzed by Western blot using an anti-V5 antibody.
E The hCD8 expression levels in 293T stable lines, as an indicator of the amount of transfected plasmid DNA (pMX-WT-IRES-hCD8, pMX-G64D-IRES-hCD8, or pMX-ΔFLA-IRES-hCD8). Two representative clones stably expressing WT-V5 or the G64D-V5 or ΔFLA-V5 mutant were analyzed by flow cytometry using an APC-conjugated anti-hCD8 antibody. Histograms were gated on hCD8-positive cells.
F Recovery of mutant ZIP13 protein expression by MG132 treatment. Representative 293T clones stably expressing WT-V5 (#1), G64D-V5 (#1), or ΔFLA-V5 (#2), were treated with 10 μM MG132 for the indicated times, followed by Western blotting analysis with an anti-V5 antibody.
G Posttranslational degradation of mutant ZIP13 proteins. HeLa clones stably expressing WT-V5, G64D-V5, or ΔFLA-V5 were treated with 10 μM CHX for the indicated times. Total cell lysates were analyzed by Western blot using an anti-V5 antibody (upper). Right graph shows the relative expression level of ZIP13 proteins over time. Data are representative of three independent experiments.
H Protein expression level of the SCD-EDS pathogenic mutants in human fibroblasts in the presence of bortezomib. Human dermal fibroblasts transiently expressing ZIP13 mutants were treated with 10 nM bortezomib for 6 h, followed by Western blotting analysis using an anti-V5 antibody.
Source data are available online for this figure.
Bortezomib is a therapeutic proteasome inhibitor that acts by reversibly binding to the catalytic site of the 26S proteasome (Teicher et al, 1999; Lightcap et al, 2000). Using the human dermal fibroblast and 293T cells, we found that bortezomib restored the ZIP13G64D and ZIP13ΔFLA mutant protein levels (Fig 5H and Supplementary Fig S8A and B), accompanied by normalization of the intracellular Zn level (Supplementary Fig S8C) as the MG132 treatment does (Supplementary Fig S9). These observations suggested that 26S proteasome inhibitors could restore the impaired intracellular Zn homeostasis by the ZIP13 mutants; thus, the manipulation of 26S proteasome activity by inhibitory compounds might be a therapeutic approach for SCD-EDS caused by pathogenic mutant ZIP13 proteins.
VCP is involved in the degradation of the mutant ZIP13 proteins
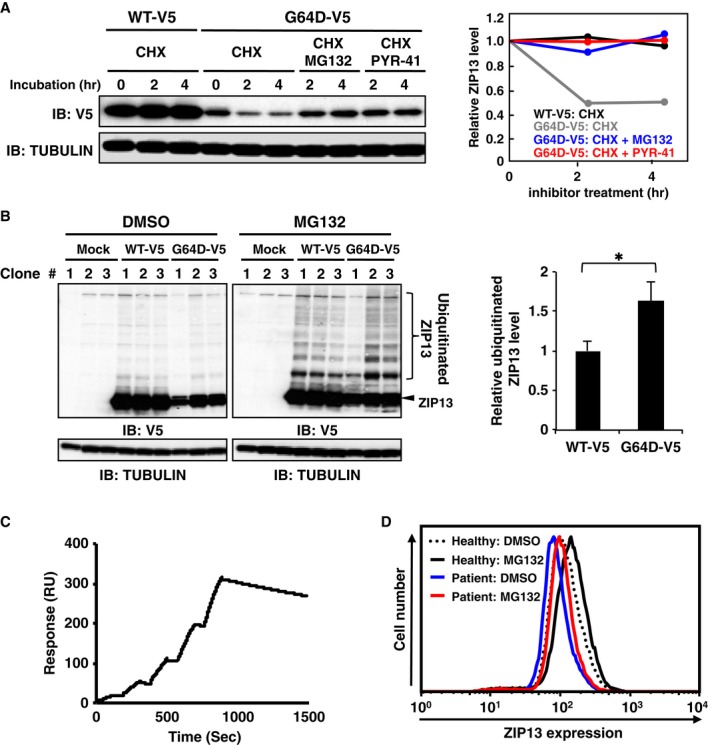
To further elucidate the molecular mechanisms involved in normal and pathogenic ZIP13 homeostasis, we isolated ZIP13-associated molecules by immunoprecipitation. Of these, we identified VCP/Cdc48/p97 by mass spectrometric analysis (Fig 6A). VCP belongs to the AAA superfamily, which mediates multiple functions, including the ubiquitination-dependent proteasome system (Ye et al, 2001, 2004; Richly et al, 2005). In addition to ZIP13WT, VCP bound to and co-localized with the mutant ZIP13G64D protein (Fig 6A–C). Intriguingly, more VCP was associated with ZIP13G64D than with ZIP13WT (Fig 6B, lower), indicating that the VCP protein might preferentially interact with the pathogenic ZIP13G64D protein. To understand VCP's role in the degradation of the mutant ZIP13 protein, we knocked down VCP by siRNAs or suppressed its function by expressing a dominant-negative form of VCP. VCP siRNAs reduced the protein level of the endogenous VCP (Fig 6D, middle) and restored the protein level of ZIP13G64D (Fig 6D, upper). Furthermore, the ectopic expression of dominant-negative VCP, F-VCPE305Q/E578Q, restored the protein level of ZIP13G64D (Fig 6E). In addition, a VCP inhibitor DBeQ (Chou et al, 2011) could suppress the decay of the ZIP13G64D protein (Fig 6F). These findings suggested that the VCP-linked proteasome-dependent pathway is involved in the normal steady-state turnover of wild-type ZIP13 and is critical for the clearance of the pathogenic mutant ZIP13 protein.
Figure 6. The mutant ZIP13 protein is degraded through a VCP-dependent mechanism.
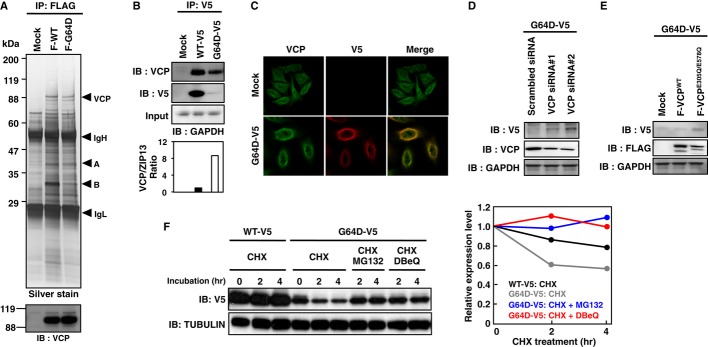
A Identification of VCP/Cdc48/p97 as a ZIP13-associating protein. Whole-cell lysates from 293T cells transfected with FLAG-tagged ZIP13 were immunoprecipitated with an anti-FLAG antibody, followed by SDS–PAGE and silver staining. Unique bands were cut out and analyzed by TOF/MASS to identify the proteins. A protein band near 88 kDa was determined to be VCP/Cdc48/p97. VCP was also detected by Western blot using an anti-VCP antibody (lower). IgH: heavy chain of IgG; IgL: light chain of IgG; A: SP-uncleaved immature ZIP13 protein; B: SP-cleaved mature ZIP13 protein.
B VCP binds to ZIP13. Whole-cell lysates from 293T cells transfected with expression plasmids for V5-tagged ZIP13 proteins were immunoprecipitated with an anti-V5 antibody, followed by SDS–PAGE. VCP and ZIP13 proteins were detected by Western blot using anti-VCP and anti-V5 antibodies, respectively. The VCP/ZIP13 ratio was analyzed using ImageJ software (http://rsbweb.nih.gov/ij/download.html) (bottom).
C Confocal images of VCP in HeLa cells stably expressing G64D-V5. VCP (green) and G64D-V5 (red) were stained with anti-V5 and anti-VCP antibodies, respectively.
D Effect of VCP siRNA on the protein expression of G64D-V5 in HeLa cells. VCP siRNA was transfected into HeLa cells stably expressing G64D-V5. Seventy-two hours posttransfection, the cells were harvested and subjected to Western blotting analysis using anti-V5 or anti-VCP antibodies.
E Effect of a dominant-negative form of VCP on the protein expression of G64D-V5 in HeLa cells. 3xFLAG-tagged wild-type VCPWT and dominant-negative VCPE305Q/E578Q were transfected into HeLa cells stably expressing G64D-V5. Twenty-four hours later, the cells were lysed and then subjected to Western blotting analysis with anti-V5 or anti-FLAG antibodies.
F Effect of a VCP inhibitor, DBeQ on the protein expression of G64D-V5 in HeLa cells. HeLa cells stably expressing WT-V5 or G64D-V5 were treated with 10 μM MG132 or 10 μM DBeQ together with CHX for the indicated times. The cell lysates were subjected to Western blotting analysis with an anti-V5 antibody. Right graph shows the relative expression level of ZIP13 proteins. Data are representative of two independent experiments.
Source data are available online for this figure.
Discussion
In the present study, we investigated the molecular pathogenic basis of the mutant ZIP13 proteins ZIP13G64D and ZIP13ΔFLA, which are responsible for SCD-EDS, to determine how these mutations lead to the loss of ZIP13 function. We demonstrated that the degradation of functional ZIP13 proteins by the VCP-linked ubiquitin proteasome pathway is the major pathogenic consequence of these mutations and that the resultant disturbance of intracellular Zn homeostasis can cause SCD-EDS (Fig 7).
Figure 7. Pathogenic mutations in ZIP13 result in its rapid reduction and zinc imbalance, leading to SCD-EDS.
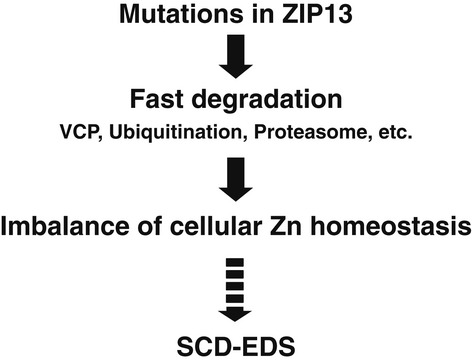
Pathogenic mutations cause the mutant ZIP13 proteins to enter the VCP-linked ubiquitin proteasome degradation pathway, resulting in reduced protein expression levels and imbalance of cellular Zn homeostasis.
In both the ZIP13G64D and ZIP13ΔFLA proteins, the pathogenic mutation occurs in a TM domain (Fukada et al, 2008; Giunta et al, 2008). TM domains are generally composed of hydrophobic amino acids, which interact with lipids and often form a helix (Singer & Nicolson, 1972). The Gly-X-X-Gly motif, a well-known motif found in helices, plays a critical role in helix-helix packing (Dohan & Carrasco, 2003; Kim et al, 2004; Munter et al, 2010). In this motif, the first and last glycine can be replaced by another amino acid with a small side chain (alanine, serine, or cysteine) (Dohan & Carrasco, 2003; Kim et al, 2004; Munter et al, 2010). In the case of ZIP13G64D, we demonstrated that replacing glycine 64, which is within a Ser-X-X-Gly motif, with a bulky amino acid with a large side chain (leucine, isoleucine, glutamic acid, or arginine) reduced the protein expression level, but replacement with alanine, serine, or cysteine did not (Fig 3F), revealing that an amino acid with a small side chain at position 64 is important for ZIP13's protein stability. In the proton-coupled folate transporter (PCFT), a Gly-X-X-Gly motif is proposed to provide conformational flexibility due to the lack of a side chain and was shown to be involved in PCFT's stability (Zhao et al, 2012). In our study, only the substitution of glycine 64 with an acidic amino acid, glutamic acid (G64E mutation), reduced the mutant ZIP13 protein level as severely as the G64D mutation, indicating that not only the size of the side chain, but also its negative charge may be important for the loss of G64D function. Reports on another Zn-imbalance disorder, AE, reveal a variety of mutations in the human ZIP4 gene from these patients (Andrews, 2008). These mutations include G340D, G384R, G643R, and L382P in Gly-X-X-Gly motif-like and leucine zipper-like regions; of these, G384R, G643R, and L382P reduce the protein level, although the mechanism underlying this decrease is not fully known (Wang et al, 2002). Intriguingly, the improper posttranslational modification of ZIP4's N-terminal ectodomain is observed in some cases (Kambe & Andrews, 2009). When Zn is deficient, the N-terminal ectodomain of the mouse ZIP4 protein is cleaved, and the resulting protein accumulates on the plasma membrane to up-regulate Zn import. The G340D, G384R, and G643R mutants of ZIP4 show decreased ectodomain cleavage in response to Zn deficiency. In contrast to ZIP4, ZIP13 does not possess an ectodomain cleavage site at its N-terminus (Kambe & Andrews, 2009; Bin et al, 2011), implying that a mutation in ZIP13's Gly-X-X-Gly motif induces loss of function by a mechanism distinct from that elicited by ZIP4 mutations. The G340D ZIP4 mutation in AE patients occurs in a Gly-X-X-Gly motif in TM1, comparable to the G64 position in ZIP13 (Fig 3E), consistent with the importance of this motif in ZIP family members. Our finding that the FLA deletion in TM3 caused the rapid proteasome-dependent degradation of ZIP13 (Fig 5 and Supplementary Fig S2) suggests that SCD-EDS by the FLA deletion is also initially caused by a reduction in functional ZIP13 protein (Jeong et al, 2012).
Our biochemical analyses demonstrated that the pathogenic mutations caused the ZIP13 protein to be unstable and enter a proteasome-dependent degradation pathway (Figs 3, 4, 5, 6 and 7). In the case of ZIP4, elevated Zn promotes the endocytosis and degradation of the ZIP4 protein. In this process, lysines near the histidine-rich cluster between TM3 and TM4 of ZIP4 are modified by ubiquitination (Mao et al, 2007). We detected ubiquitinated ZIP13 protein (Fig 4B), although ZIP13 does not contain a typical histidine-rich cluster between TM3 and TM4, nor any other histidine clusters (Bin et al, 2011). We also found that VCP associates with either wild-type or mutant ZIP13 proteins, even though it preferentially interacts with the mutant ZIP13, suggesting that the VCP–ZIP13 interaction is important for both the normal steady-state turnover of wild-type ZIP13 and the clearance of ZIP13 proteins containing critical mutations (Fig 6). VCP was originally identified as a valosin-containing protein in pigs (Koller & Brownstein, 1987) and has roles in nucleus reformation, membrane fusion, protein quality control, autophagy, and other cellular processes (Latterich et al, 1995; Bukau et al, 2006; Ramadan et al, 2007; Buchan et al, 2013). VCP may mediate the retro-translocation of ZIP13 from the membrane into the cytosol before or after ZIP13's ubiquitination, along with various chaperones and ubiquitin-binding proteins that help deliver it to the proteasome for degradation (Ye et al, 2001, 2004; Richly et al, 2005). In addition to VCP, heat-shock proteins may be involved, because we found that the treatment of 17AAG, an HSP90 inhibitor, also restored the expression level of ZIP13G64D protein (Supplementary Fig S10), supporting the idea that various molecules take part in ZIP13's degradation. The precise mechanism for ZIP13's degradation awaits future studies, but clues may lie in the identification of proteins that bind the extra/intracellular loops of ZIP13. Although mutated proteins sometimes induce ER stress before being degraded (Vidal et al, 2011), the expression level of ER-stress-responsive molecules was comparable between the cells expressing ZIP13WT and the pathogenic mutants (Supplementary Fig S11), indicating that ER stress might not significantly participate in the pathogenic process of mutant ZIP13 proteins.
Importantly, our results lend credence to the potential use of proteasome inhibitors in clinical investigations of SCD-EDS and its therapeutics (Figs 3, 4, 5, and Supplementary Figs S8 and S9). We also found that VCP inhibitor improved the protein level of the pathogenic ZIP13 mutants (Fig 6F), further supporting the therapeutic potential of compounds targeted to proteasome pathways. Cystic fibrosis is a genetic disease caused by mutations in the cystic fibrosis transmembrane conductance regulator (CFTR). Ninety percent of the patients have a ΔF508 mutation, which prevents proper folding and processing of the CFTR protein; as a result, little of the mutant protein reaches the cell surface (Rommens et al, 1988; Riordan et al, 1989; Ward et al, 1995). Much research has focused on elucidating the folding, trafficking, and degradation properties of CFTR pathogenic mutants, and on developing drugs that are either “potentiators” of CFTR itself or “correctors” of its degradation pathway (Wang et al, 2008; Becq, 2010; Gee et al, 2011). VX-809 is the latest CFTR drug. It was obtained from a screen as a compound that reduces degradation of the ΔF508 mutant protein and increases CFTR accumulation on the cell surface and is currently in clinical trials (Van Goor et al, 2011). Another mutation, G551D, which accounts for about 5% of the cystic fibrosis patients, does not affect the protein's trafficking, but prohibits proper channel gating. Kalydeco (VX-770) was developed to treat cystic fibrosis patients carrying the G551D mutation (Van Goor et al, 2009; Accurso et al, 2010). It acts as a “potentiator” to open the gate of CFTR for proper chloride transport (Rowe & Verkman, 2013). In the case of SCD-EDS patients, therapeutic strategies analogous to those used to treat cystic fibrosis, as either molecular “potentiators” or “correctors”, may be effective depending on the functional consequences of the mutation. Moreover, we cannot exclude the possible involvement of another degradation pathway or translational defects of the ZIP13 mutants as a consequence of the mutation, given that the ZIP13ΔFLA protein level recovered much more than the ZIP13G64D protein level after MG132 treatment (Fig 5F and H) although the ZIP13ΔFLA protein was more unstable than the ZIP13G64D protein (Fig 5G). Future investigations of the molecular details underlying the degradation of G64D and ΔFLA mutants, and of the protein structure and homeostasis of ZIP13, will provide a framework to develop potential treatments for SCD-EDS and for the related metabolic diseases since ZIP13 is also implicated in adipose and muscle tissues homeostasis (Fukada et al, 2008). In this regard, mutant ZIP13 gene knock-in mice could be useful animal models to develop therapeutics for SCD-EDS, and the development of Zn transport assay system using proteoliposomes with purified ZIP13 proteins may also facilitate further understandings of the physio-pathogenesis of ZIP13.
Taken together, we have gained insight into the mechanism underlying the loss of function of ZIP13 mutants in SCD-EDS patients (Fig 7). This mechanism involves the disruption of Zn regulation through a reduction of the ZIP13 protein level via the VCP-linked ubiquitin and proteasome-dependent degradation pathway. We found that conserved amino acid(s) in TMs are critical for the stability of ZIP13 protein, and compounds that inhibit protein degradation are potential therapeutics for SCD-EDS. Further exploration of the pathogenic mechanism of SCD-EDS will reveal new avenues for clinical interventions.
Materials and Methods
Cell culture and compounds
293T, HeLa, HT1080, and the human dermal fibroblast (Lonza) were maintained in DMEM+GlutaMAX medium (Gibco) with 10% FBS and antibiotics at 37°C. To construct stable cell lines, plasmids were transfected using Lipofectamine 2000 (Invitrogen), and cells were selected with 100 μg/mL HygroGold (Invivogen) for 293T cells and 10–50 μg/mL blasticidin (Invivogen) for HeLa cells. To monitor the amount of transfected plasmid, the cDNAs of ZIP13 and its mutants were subcloned into pMX-IRES-hCD8 (Yamasaki et al, 2006). Bafilomycin (Sigma), MG132 (Sigma), lactacystin (Enzo Life Sciences), PYR-41 (Sigma), DBeQ (Sigma), bortezomib (Cell Signaling), and cycloheximide (Sigma) were dissolved in DMSO.
Plasmid constructs
FLAG-tagged ZIP13 and V5-tagged ZIP13 were constructed as previously described (Fukada et al, 2008; Bin et al, 2011). Plasmids used for the ubiquitination analysis were kind gifts from Drs. Takashi Tanaka and Chin Ha Jung. The plasmid encoding a dominant-negative form of VCP (E305Q/E578Q) (Shirogane et al, 1999) was reconstructed into p3xFLAG-Myc-CMV-26 (Sigma). The various G64 mutants were constructed using the EZchange™ Site-directed Mutagenesis kit (Enzynomics) with designated primers (Supplementary Table S1) as described by the manufacturer. The reporter vector pGL4.12-MT-264/+42 contained the mouse MT-1 promoter was a gift from Dr. Tomoki Kimura (Kimura et al, 2008).
Western blotting analysis
Cells were collected in 1% NP-40 containing 0.05 M Tris–HCl, pH 7.5, 0.15 M NaCl, and 0.01 M MgCl2. After centrifugation at 15,000 × g for 5 min, the supernatant was collected and analyzed as the soluble fraction. The pellet was re-suspended in 1% SDS containing 0.05 M Tris–HCl, pH 7.5, 0.15 M NaCl, and 0.01 M MgCl2 and analyzed as the insoluble fraction. Those fractions were boiled for 5 min in SDS–PAGE sample buffer containing 0.125 M Tris–HCl, pH 6.8, 20% glycerol, 4% SDS, 10% 2-mercaptoethanol, and 0.004% bromophenol blue and loaded onto a 5–20% or 10–20% polyacrylamide gradient gel. The ER stress antibody sampler kit was obtained from Cell Signaling Technology. Blue native-PAGE was performed as previously described (Bin et al, 2011). Anti-V5 (Invitrogen), anti-tubulin (Santa Cruz), anti-ubiquitinated proteins (Biomol), anti-FLAG (Sigma), and anti-VCP (Abcam) antibodies, and an anti-ER stress antibody sampler kit (Cell Signaling) were used for protein detection.
Quantitative Real-time PCR
cDNA was synthesized using ReverTra Ace (Toyobo). The mRNA levels of ZIP13 were analyzed as previously reported (Bin et al, 2011). The mRNA levels of CHOP and BIP were analyzed using the TaqMan® Gene Expression Assay following the manufacturer's instructions (Applied Biosystems).
Generation of anti-ZIP13 antibodies
The ab-A1 and ab-A2 anti-ZIP13 antibodies were generated in rabbits against synthetic peptides corresponding to amino acids 23–35 of human ZIP13 for ab-A1, and 184–201 of mouse ZIP13 for ab-A2 (Fukada et al, 2008). The monoclonal antibody 35B11 was produced using the method of Hino and others (Hino et al, 2012, 2013). Briefly, purified ZIP13 (Bin et al, 2011) was reconstituted into phospholipid vesicles consisting of egg L-α-phosphatidylcholine and Lipid A (Sigma) as an adjuvant. The antibodies were screened by ELISA and dot blot analyses, as described previously (Hino et al, 2013).
Biacore
The binding affinity of 35B11 (IgG2a) for ZIP13 was tested by SPR spectroscopy using a Biacore T200 analyzer (GE Healthcare). A monoclonal anti-mouse Fcγ fragment-specific antibody was immobilized on a sensor chip (CM5), and the culture media after hybridoma cell cultivation were then loaded. Antibodies in the supernatant were tightly trapped by the anti-Fc antibody. The running buffer was 0.02 M HEPES (pH 7.4), 0.15 M NaCl, and 0.04% dodecyl-β-D-maltopyranoside (DDM). Purified ZIP13 protein in 0.04% DDM was then passed over the surface. Analyte concentrations were calculated using the absorbance at 280 nm with the theoretical extinction coefficients.
Confocal microscopy
Cells were seeded onto glass coverslips in 35-mm glass dishes (Iwaki) overnight and were treated with or without 10 μM MG132 for 6 h. The cells were then fixed with 4% paraformaldehyde in PBS. For immunostaining, the cells were made permeable with BD Perm/Wash buffer containing antibodies and 1% BSA. Fluorescence was detected with an inverted spectral Confocal Scanning system, TCS SP2 AOBS (Leica), with an oil immersion 63× objective. Images were processed with Adobe Photoshop CS3 version 10.0. DAPI (Molecular Probes), anti-V5 antibody (Invitrogen), anti-GM130 antibody (clone35, BD Transduction Laboratories), and Alexa Fluor®635 phalloidin (Molecular Probes) were used to visualize nuclei, ZIP13, Golgi, and actin, respectively. Alexa Fluor®546 goat anti-mouse IgG F(ab')2 fragment was used for the secondary staining of GM130.
Flow cytometric analysis
Cells were fixed and permeabilized with cytofix/cytoperm reagent (BD Biosciences) for 15 min at room temperature. After washing with Perm/Wash buffer, the cells were blocked with 0.5% BSA containing Perm/Wash buffer for 30 min at room temperature. The cells were then stained with 20 μg/ml anti-ZIP13 antibody (clone 35B11) in 0.5% BSA containing perm/wash buffer for 1 h at room temperature, washed extensively with Perm/Wash buffer, and then further incubated with goat anti-mouse Alexa 488 (Molecular Probes) for 1 h at room temperature. After more extensive washing with Perm/Wash buffer, the cells were subjected to flow cytometric analysis.
Immunoprecipitation and mass spectrometry
Cells were disrupted in 1% NP-40 lysis buffer containing 0.05 M Tris–HCl, pH 7.5, 0.15 M NaCl, and 0.01 M MgCl2, and the cell debris was removed by centrifugation at 15,000 × g for 5 min. After incubation with an anti-V5 or anti-FLAG antibody for 3–4 h, the immune complexes were pulled down with protein G (GE Healthcare) for 2–3 h and then washed with 0.05% NP-40 lysis buffer. The complexes were dissociated in 1% SDS–PAGE sample buffer and subjected to SDS–PAGE and silver staining. Single bands were cut out and analyzed by mass spectrometry, and VCP (NP_009057.1) was identified.
Ni-NTA purification
For Ni-NTA purification, cells were harvested into a denaturing lysis buffer (0.05 M Tris–HCl and 6 M GuHCl, adjusted to pH 8.0 using NaOH). The cell debris was disrupted by sonication, and Ni-NTA agarose was added. The mixture was then incubated for over 2 h. The Ni-NTA agarose was washed with 0.05 M Tris–HCl and 8 M urea, pH 6.3, and the proteins were eluted into 0.05 M Tris–HCl and 8 M urea, pH 4.5.
siRNA transfection
Cells were transiently transfected with 100 pM siRNA (Genolution) using Lipofectamine RNAimax (Invitrogen), according to the manufacturer's instructions. VCP-targeting siRNAs were constructed using the human VCP mRNA sequence at nucleotides 599–619 (TGTAGGGTATGATGACATTG) or 480–500 (TAACCTTCGTGTACGCCTA). PA28-targeting siRNAs were constructed using the published human PA28 mRNA sequence (GAAUCAAUAUGUCACUCUAUU) or (UCUGAAGGAACCAAUCUUAUU) (Chen et al, 2007).
Measurement of Zn level
Zn fluorescence staining was performed with slight modification (Taniguchi et al, 2013). 293T cells were treated with 10 nM bortezomib for 6 h. Afterwards, they were incubated with 1 μM FluoZin-3 for 30 min, and then with 10 μM Zn pyrithione for 10 min. The cells were washed with PBS and fixed with 4% paraformaldehyde in PBS. Fluorescence was detected with an inverted fluorescence imaging system, EVOS f1 (AMG). To quantify the cellular Zn level, 1 × 106–107 cells were subjected to a modified acid deproteinizing method (Nomoto, 1987) and then analyzed by inductively coupled plasma-atomic emission spectrometry (ICP-AES).
Patient cells
Written informed consents were obtained from the subjects. The study was approved by ethics committees of participating institutions.
Statistical analysis
The two-tailed Student's t-test was used to analyze the difference between two groups.
The paper explained
Problem
The spondylocheirodysplastic form of Ehlers-Danlos syndrome (SCD-EDS, OMIM 612350), a genetic disorder of connective tissues, bones, and teeth, is related to an imbalance in the cellular handling of zinc caused by mutation in the zinc transporter ZIP13; however, the pathogenic mechanism of the mutation is poorly understood.
Results
We found that pathogenic ZIP13 proteins are degraded by the VCP-linked ubiquitin proteasome pathway. Interrupting this pathway restored the ZIP13 expression levels, resulting in improvement of the intracellular Zn homeostasis.
Impact
Our data revealed the pathogenic mechanism of mutant ZIP13 proteins and lend credence to the therapeutic potential of inhibitors for proteasome-dependent pathways. Further studies may lead to new therapeutic intervention strategies for SCD-EDS.
Acknowledgments
We are grateful for support from Drs. Toshio Hirano, Shigeo Koyasu, Masaru Taniguchi, and Kenji Mishima, helpful assistance from Mr. Masami Kawamura and Ms Norie Takeuchi, and for fruitful discussion with Dr. Ik Soo Kim. We also thank Dr. So Iwata and his colleagues for instruction and monoclonal antibody generation, and Mr. Masaru Emura, Dr. Shinji Shimoda, and Dr. Takeaki Nagamine, Genostaff and Shino-Test for technical advises. The pMX-IRES-hCD8 vector was a kind gift from Dr. Sho Yamasaki. This work was supported by KAKENHI Grant (TF) of Japan Society for the Promotion of Science (JSPS), Life Science Foundation of Japan (TF), Japan Osteoporosis Foundation (TF), the Targeted Proteins Research Program (SY), RIKEN Junior Research Associate Program (TM), and the Platform for Drug Discovery, Informatics, and Structural Life Science (SY) from the Ministry of Education, Culture, Sports, Science, and Technology (MEXT).
Author contributions
BHB, SH, TH, and TF conceived and designed the experiments. TH, MI, TKS, MS, and SY generated and analyzed the monoclonal anti-ZIP13 antibody (35B11). BHB, SH, JB, HK, TM, KF, TK, JS, KHK, DHC, YJN, and WO performed the rest of the experiments. BHB, SH, EGC, TRL, JB, DH, and TF analyzed the data. BHB, SH, TH, AF, YF, ASF, SI, TRL, and TF wrote and reviewed the manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting Information
for this article is available online: http://embomolmed.embopress.org
References
- Accurso FJ, Rowe SM, Clancy JP, Boyle MP, Dunitz JM, Durie PR, Sagel SD, Hornick DB, Konstan MW, Donaldson SH, et al. Effect of VX-770 in persons with cystic fibrosis and the G551D-CFTR mutation. N Engl J Med. 2010;363:1991–2003. doi: 10.1056/NEJMoa0909825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrews GK. Regulation and function of Zip4, the acrodermatitis enteropathica gene. Biochem Soc Trans. 2008;36:1242–1246. doi: 10.1042/BST0361242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becq F. Cystic fibrosis transmembrane conductance regulator modulators for personalized drug treatment of cystic fibrosis: progress to date. Drugs. 2010;70:241–259. doi: 10.2165/11316160-000000000-00000. [DOI] [PubMed] [Google Scholar]
- Bin BH, Fukada T, Hosaka T, Yamasaki S, Ohashi W, Hojyo S, Miyai T, Nishida K, Yokoyama S, Hirano T. Biochemical characterization of human ZIP13 protein: a homo-dimerized zinc transporter involved in the Spondylocheiro dysplastic Ehlers-Danlos syndrome. J Biol Chem. 2011;286:40255–40265. doi: 10.1074/jbc.M111.256784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosco MD, Mohanasundaram DM, Drogemuller CJ, Lang CJ, Zalewski PD, Coates PT. Zinc and zinc transporter regulation in pancreatic islets and the potential role of zinc in islet transplantation. Rev Diabet Stud. 2010;7:263–274. doi: 10.1900/RDS.2010.7.263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchan JR, Kolaitis RM, Taylor JP, Parker R. Eukaryotic Stress Granules Are Cleared by Autophagy and Cdc48/VCP Function. Cell. 2013;153:1461–1474. doi: 10.1016/j.cell.2013.05.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bukau B, Weissman J, Horwich A. Molecular chaperones and protein quality control. Cell. 2006;125:443–451. doi: 10.1016/j.cell.2006.04.014. [DOI] [PubMed] [Google Scholar]
- Byers PH, Murray ML. Heritable collagen disorders: the paradigm of the ehlers-danlos syndrome. J Invest Dermatol. 2012;132:E6–E11. doi: 10.1038/skinbio.2012.3. [DOI] [PubMed] [Google Scholar]
- Chen X, Barton LF, Chi Y, Clurman BE, Roberts JM. Ubiquitin-independent degradation of cell-cycle inhibitors by the REGgamma proteasome. Mol Cell. 2007;26:843–852. doi: 10.1016/j.molcel.2007.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou TF, Brown SJ, Minond D, Nordin BE, Li K, Jones AC, Chase P, Porubsky PR, Stoltz BM, Schoenen FJ, et al. Reversible inhibitor of p97, DBeQ, impairs both ubiquitin-dependent and autophagic protein clearance pathways. Proc Natl Acad Sci USA. 2011;108:4834–4839. doi: 10.1073/pnas.1015312108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chowanadisai W, Lonnerdal B, Kelleher SL. Identification of a mutation in SLC30A2 (ZnT-2) in women with low milk zinc concentration that results in transient neonatal zinc deficiency. J Biol Chem. 2006;281:39699–39707. doi: 10.1074/jbc.M605821200. [DOI] [PubMed] [Google Scholar]
- Dohan O, Carrasco N. Advances in Na(+)/I(-) symporter (NIS) research in the thyroid and beyond. Mol Cell Endocrinol. 2003;213:59–70. doi: 10.1016/j.mce.2003.10.059. [DOI] [PubMed] [Google Scholar]
- Dufner-Beattie J, Weaver BP, Geiser J, Bilgen M, Larson M, Xu W, Andrews GK. The mouse acrodermatitis enteropathica gene Slc39a4 (Zip4) is essential for early development and heterozygosity causes hypersensitivity to zinc deficiency. Hum Mol Genet. 2007;16:1391–1399. doi: 10.1093/hmg/ddm088. [DOI] [PubMed] [Google Scholar]
- Fukada T, Asada Y, Mishima K, Shimoda S, Saito I. Slc39a13/Zip13: a crucial zinc transporter involved in tooth development and inherited disorders. J Oral Biosci. 2011a;53:1–12. [Google Scholar]
- Fukada T, Civic N, Furuichi T, Shimoda S, Mishima K, Higashiyama H, Idaira Y, Asada Y, Kitamura H, Yamasaki S, et al. The zinc transporter SLC39A13/ZIP13 is required for connective tissue development; its involvement in BMP/TGF-beta signaling pathways. PLoS ONE. 2008;3:e3642. doi: 10.1371/journal.pone.0003642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukada T, Hojyo S, Bin B. Zinc signal in growth control and bone diseases. In: Fukada T, Kambe T, editors. Zinc Signals in Cellular Functions and Disorders. Tokyo: Springer; 2014. , in press. [Google Scholar]
- Fukada T, Kambe T. Molecular and genetic features of zinc transporters in physiology and pathogenesis. Metallomics. 2011;3:662–674. doi: 10.1039/c1mt00011j. [DOI] [PubMed] [Google Scholar]
- Fukada T, Yamasaki S, Nishida K, Murakami M, Hirano T. Zinc homeostasis and signaling in health and diseases: Zinc signaling. J Biol Inorg Chem. 2011b;16:1123–1134. doi: 10.1007/s00775-011-0797-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gee HY, Noh SH, Tang BL, Kim KH, Lee MG. Rescue of DeltaF508-CFTR trafficking via a GRASP-dependent unconventional secretion pathway. Cell. 2011;146:746–760. doi: 10.1016/j.cell.2011.07.021. [DOI] [PubMed] [Google Scholar]
- Geiser J, Venken KJ, De Lisle RC, Andrews GK. A mouse model of acrodermatitis enteropathica: loss of intestine zinc transporter ZIP4 (Slc39a4) disrupts the stem cell niche and intestine integrity. PLoS Genet. 2012;8:e1002766. doi: 10.1371/journal.pgen.1002766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giunta C, Elcioglu NH, Albrecht B, Eich G, Chambaz C, Janecke AR, Yeowell H, Weis M, Eyre DR, Kraenzlin M, et al. Spondylocheiro dysplastic form of the Ehlers-Danlos syndrome–an autosomal-recessive entity caused by mutations in the zinc transporter gene SLC39A13. Am J Hum Genet. 2008;82:1290–1305. doi: 10.1016/j.ajhg.2008.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardy AB, Serino AS, Wijesekara N, Chimienti F, Wheeler MB. Regulation of glucagon secretion by zinc: lessons from the beta cell-specific Znt8 knockout mouse model. Diabetes Obes Metab. 2011;13(Suppl 1):112–117. doi: 10.1111/j.1463-1326.2011.01451.x. [DOI] [PubMed] [Google Scholar]
- Hino T, Arakawa T, Iwanari H, Yurugi-Kobayashi T, Ikeda-Suno C, Nakada-Nakura Y, Kusano-Arai O, Weyand S, Shimamura T, Nomura N, et al. G-protein-coupled receptor inactivation by an allosteric inverse-agonist antibody. Nature. 2012;482:237–240. doi: 10.1038/nature10750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hino T, Iwata S, Murata T. Generation of functional antibodies for mammalian membrane protein crystallography. Curr Opin Struct Biol. 2013;23:563–568. doi: 10.1016/j.sbi.2013.04.007. [DOI] [PubMed] [Google Scholar]
- Hogstrand C, Kille P, Ackland ML, Hiscox S, Taylor KM. A mechanism for epithelial-mesenchymal transition and anoikis resistance in breast cancer triggered by zinc channel ZIP6 and STAT3 (signal transducer and activator of transcription 3) Biochem J. 2013;455:229–237. doi: 10.1042/BJ20130483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishidoh K, Kominami E. Processing and activation of lysosomal proteinases. Biol Chem. 2002;383:1827–1831. doi: 10.1515/BC.2002.206. [DOI] [PubMed] [Google Scholar]
- Itsumura N, Inamo Y, Okazaki F, Teranishi F, Narita H, Kambe T, Kodama H. Compound Heterozygous Mutations in SLC30A2/ZnT2 Results in Low Milk Zinc Concentrations: a Novel Mechanism for Zinc Deficiency in a Breast-Fed Infant. PLoS ONE. 2013;8:e64045. doi: 10.1371/journal.pone.0064045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeong J, Walker JM, Wang F, Park JG, Palmer AE, Giunta C, Rohrbach M, Steinmann B, Eide DJ. Promotion of vesicular zinc efflux by ZIP13 and its implications for spondylocheiro dysplastic Ehlers-Danlos syndrome. Proc Natl Acad Sci USA. 2012;109:E3530–E3538. doi: 10.1073/pnas.1211775110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kambe T, Andrews GK. Novel proteolytic processing of the ectodomain of the zinc transporter ZIP4 (SLC39A4) during zinc deficiency is inhibited by acrodermatitis enteropathica mutations. Mol Cell Biol. 2009;29:129–139. doi: 10.1128/MCB.00963-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JH, Jeon J, Shin M, Won Y, Lee M, Kwak JS, Lee K, Rhee J, Ryu JH, Chun CH, et al. Regulation of the Catabolic Cascade in Osteoarthritis by the Zinc-ZIP8-MTF1 Axis. Cell. 2014;156:730–743. doi: 10.1016/j.cell.2014.01.007. [DOI] [PubMed] [Google Scholar]
- Kim S, Chamberlain AK, Bowie JU. Membrane channel structure of Helicobacter pylori vacuolating toxin: role of multiple GXXXG motifs in cylindrical channels. Proc Natl Acad Sci USA. 2004;101:5988–5991. doi: 10.1073/pnas.0308694101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura T, Li Y, Okumura F, Itoh N, Nakanishi T, Sone T, Isobe M, Andrews GK. Chromium(VI) inhibits mouse metallothionein-I gene transcription by preventing the zinc-dependent formation of an MTF-1-p300 complex. Biochem J. 2008;415:477–482. doi: 10.1042/BJ20081025. [DOI] [PubMed] [Google Scholar]
- Koller KJ, Brownstein MJ. Use of a cDNA clone to identify a supposed precursor protein containing valosin. Nature. 1987;325:542–545. doi: 10.1038/325542a0. [DOI] [PubMed] [Google Scholar]
- Lasry I, Seo YA, Ityel H, Shalva N, Pode-Shakked B, Glaser F, Berman B, Berezovsky I, Goncearenco A, Klar A, et al. A Dominant Negative Heterozygous G87R Mutation in the Zinc Transporter, ZnT-2 (SLC30A2), Results in Transient Neonatal Zinc Deficiency. J Biol Chem. 2012;287:29348–29361. doi: 10.1074/jbc.M112.368159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Latterich M, Frohlich KU, Schekman R. Membrane fusion and the cell cycle: Cdc48p participates in the fusion of ER membranes. Cell. 1995;82:885–893. doi: 10.1016/0092-8674(95)90268-6. [DOI] [PubMed] [Google Scholar]
- Lee DH, Goldberg AL. Proteasome inhibitors: valuable new tools for cell biologists. Trends Cell Biol. 1998;8:397–403. doi: 10.1016/s0962-8924(98)01346-4. [DOI] [PubMed] [Google Scholar]
- Li M, Zhang Y, Liu Z, Bharadwaj U, Wang H, Wang X, Zhang S, Liuzzi JP, Chang SM, Cousins RJ, et al. Aberrant expression of zinc transporter ZIP4 (SLC39A4) significantly contributes to human pancreatic cancer pathogenesis and progression. Proc Natl Acad Sci USA. 2007;104:18636–18641. doi: 10.1073/pnas.0709307104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lichten LA, Cousins RJ. Mammalian zinc transporters: nutritional and physiologic regulation. Annu Rev Nutr. 2009;29:153–176. doi: 10.1146/annurev-nutr-033009-083312. [DOI] [PubMed] [Google Scholar]
- Lightcap ES, McCormack TA, Pien CS, Chau V, Adams J, Elliott PJ. Proteasome inhibition measurements: clinical application. Clin Chem. 2000;46:673–683. [PubMed] [Google Scholar]
- MacDonald RS. The role of zinc in growth and cell proliferation. J Nutr. 2000;130:1500S–1508S. doi: 10.1093/jn/130.5.1500S. [DOI] [PubMed] [Google Scholar]
- Mao X, Kim BE, Wang F, Eide DJ, Petris MJ. A histidine-rich cluster mediates the ubiquitination and degradation of the human zinc transporter, hZIP4, and protects against zinc cytotoxicity. J Biol Chem. 2007;282:6992–7000. doi: 10.1074/jbc.M610552200. [DOI] [PubMed] [Google Scholar]
- Munemasa T, Idaira Y, Fukada T, Shimoda S, Asada Y. Histological Analysis of Dentinogenesis Imperfecta in Slc39a13/Zip13 Knockout Mice. J Hard Tissue Biol. 2014;23:163–170. [Google Scholar]
- Munter LM, Botev A, Richter L, Hildebrand PW, Althoff V, Weise C, Kaden D, Multhaup G. Aberrant amyloid precursor protein (APP) processing in hereditary forms of Alzheimer disease caused by APP familial Alzheimer disease mutations can be rescued by mutations in the APP GxxxG motif. J Biol Chem. 2010;285:21636–21643. doi: 10.1074/jbc.M109.088005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nomoto S. A simplified method for trace metal analysis of biological materials. J UOEH. 1987;9(Suppl):111–122. [PubMed] [Google Scholar]
- Potocki S, Valensin D, Camponeschi F, Kozlowski H. The extracellular loop of IRT1 ZIP protein–the chosen one for zinc? J Inorg Biochem. 2013;127:246–252. doi: 10.1016/j.jinorgbio.2013.05.003. [DOI] [PubMed] [Google Scholar]
- Prasad AS. Zinc: an overview. Nutrition. 1995;11:93–99. [PubMed] [Google Scholar]
- Ramadan K, Bruderer R, Spiga FM, Popp O, Baur T, Gotta M, Meyer HH. Cdc48/p97 promotes reformation of the nucleus by extracting the kinase Aurora B from chromatin. Nature. 2007;450:1258–1262. doi: 10.1038/nature06388. [DOI] [PubMed] [Google Scholar]
- Richly H, Rape M, Braun S, Rumpf S, Hoege C, Jentsch S. A series of ubiquitin binding factors connects CDC48/p97 to substrate multiubiquitylation and proteasomal targeting. Cell. 2005;120:73–84. doi: 10.1016/j.cell.2004.11.013. [DOI] [PubMed] [Google Scholar]
- Riordan JR, Rommens JM, Kerem B, Alon N, Rozmahel R, Grzelczak Z, Zielenski J, Lok S, Plavsic N, Chou JL, et al. Identification of the cystic fibrosis gene: cloning and characterization of complementary DNA. Science. 1989;245:1066–1073. doi: 10.1126/science.2475911. [DOI] [PubMed] [Google Scholar]
- Rommens JM, Zengerling S, Burns J, Melmer G, Kerem BS, Plavsic N, Zsiga M, Kennedy D, Markiewicz D, Rozmahel R, et al. Identification and regional localization of DNA markers on chromosome 7 for the cloning of the cystic fibrosis gene. Am J Hum Genet. 1988;43:645–663. [PMC free article] [PubMed] [Google Scholar]
- Rowe SM, Verkman AS. 2013. Cystic fibrosis transmembrane regulator correctors and potentiators. Cold Spring Harbor perspectives in medicine 3.
- Ryu MS, Langkamp-Henken B, Chang SM, Shankar MN, Cousins RJ. Genomic analysis, cytokine expression, and microRNA profiling reveal biomarkers of human dietary zinc depletion and homeostasis. Proc Natl Acad Sci USA. 2011;108:20970–20975. doi: 10.1073/pnas.1117207108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shirogane T, Fukada T, Muller JM, Shima DT, Hibi M, Hirano T. Synergistic roles for Pim-1 and c-Myc in STAT3-mediated cell cycle progression and antiapoptosis. Immunity. 1999;11:709–719. doi: 10.1016/s1074-7613(00)80145-4. [DOI] [PubMed] [Google Scholar]
- Singer SJ, Nicolson GL. The fluid mosaic model of the structure of cell membranes. Science. 1972;175:720–731. doi: 10.1126/science.175.4023.720. [DOI] [PubMed] [Google Scholar]
- Tamaki M, Fujitani Y, Hara A, Uchida T, Tamura Y, Takeno K, Kawaguchi M, Watanabe T, Ogihara T, Fukunaka A, et al. The diabetes-susceptible gene SLC30A8/ZnT8 regulates hepatic insulin clearance. J Clin Invest. 2013;123:4513–4524. doi: 10.1172/JCI68807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taniguchi M, Fukunaka A, Hagihara M, Watanabe K, Kamino S, Kambe T, Enomoto S, Hiromura M. Essential role of the zinc transporter ZIP9/SLC39A9 in regulating the activations of Akt and Erk in B-cell receptor signaling pathway in DT40 cells. PLoS ONE. 2013;8:e58022. doi: 10.1371/journal.pone.0058022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor KM, Morgan HE, Smart K, Zahari NM, Pumford S, Ellis IO, Robertson JF, Nicholson RI. The emerging role of the LIV-1 subfamily of zinc transporters in breast cancer. Mol Med. 2007;13:396–406. doi: 10.2119/2007-00040.Taylor. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teicher BA, Ara G, Herbst R, Palombella VJ, Adams J. The proteasome inhibitor PS-341 in cancer therapy. Clin Cancer Res. 1999;5:2638–2645. [PubMed] [Google Scholar]
- Van Goor F, Hadida S, Grootenhuis PD, Burton B, Cao D, Neuberger T, Turnbull A, Singh A, Joubran J, Hazlewood A, et al. Rescue of CF airway epithelial cell function in vitro by a CFTR potentiator, VX-770. Proc Natl Acad Sci USA. 2009;106:18825–18830. doi: 10.1073/pnas.0904709106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Goor F, Hadida S, Grootenhuis PD, Burton B, Stack JH, Straley KS, Decker CJ, Miller M, McCartney J, Olson ER, et al. Correction of the F508del-CFTR protein processing defect in vitro by the investigational drug VX-809. Proc Natl Acad Sci USA. 2011;108:18843–18848. doi: 10.1073/pnas.1105787108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vidal R, Caballero B, Couve A, Hetz C. Converging pathways in the occurrence of endoplasmic reticulum (ER) stress in Huntington's disease. Curr Mol Med. 2011;11:1–12. doi: 10.2174/156652411794474419. [DOI] [PubMed] [Google Scholar]
- Wang B, Heath-Engel H, Zhang D, Nguyen N, Thomas DY, Hanrahan JW, Shore GC. BAP31 interacts with Sec61 translocons and promotes retrotranslocation of CFTRDeltaF508 via the derlin-1 complex. Cell. 2008;133:1080–1092. doi: 10.1016/j.cell.2008.04.042. [DOI] [PubMed] [Google Scholar]
- Wang K, Zhou B, Kuo YM, Zemansky J, Gitschier J. A novel member of a zinc transporter family is defective in acrodermatitis enteropathica. Am J Hum Genet. 2002;71:66–73. doi: 10.1086/341125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward CL, Omura S, Kopito RR. Degradation of CFTR by the ubiquitin-proteasome pathway. Cell. 1995;83:121–127. doi: 10.1016/0092-8674(95)90240-6. [DOI] [PubMed] [Google Scholar]
- Warman ML, Cormier-Daire V, Hall C, Krakow D, Lachman R, LeMerrer M, Mortier G, Mundlos S, Nishimura G, Rimoin DL, et al. Nosology and classification of genetic skeletal disorders: 2010 revision. Am J Med Genet A. 2011;155A:943–968. doi: 10.1002/ajmg.a.33909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J, Wang J, Chen B. SLC30A8 (ZnT8) variations and type 2 diabetes in the Chinese Han population. Genet Mol Res. 2012;11:1592–1598. doi: 10.4238/2012.May.24.1. [DOI] [PubMed] [Google Scholar]
- Yamasaki S, Ishikawa E, Sakuma M, Ogata K, Sakata-Sogawa K, Hiroshima M, Wiest DL, Tokunaga M, Saito T. Mechanistic basis of pre-T cell receptor-mediated autonomous signaling critical for thymocyte development. Nat Immunol. 2006;7:67–75. doi: 10.1038/ni1290. [DOI] [PubMed] [Google Scholar]
- Yamashita S, Miyagi C, Fukada T, Kagara N, Che YS, Hirano T. Zinc transporter LIVI controls epithelial-mesenchymal transition in zebrafish gastrula organizer. Nature. 2004;429:298–302. doi: 10.1038/nature02545. [DOI] [PubMed] [Google Scholar]
- Ye Y, Meyer HH, Rapoport TA. The AAA ATPase Cdc48/p97 and its partners transport proteins from the ER into the cytosol. Nature. 2001;414:652–656. doi: 10.1038/414652a. [DOI] [PubMed] [Google Scholar]
- Ye Y, Shibata Y, Yun C, Ron D, Rapoport TA. A membrane protein complex mediates retro-translocation from the ER lumen into the cytosol. Nature. 2004;429:841–847. doi: 10.1038/nature02656. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Yang J, Cui X, Chen Y, Zhu VF, Hagan JP, Wang H, Yu X, Hodges SE, Fang J, et al. A novel epigenetic CREB-miR-373 axis mediates ZIP4-induced pancreatic cancer growth. EMBO Mol Med. 2013;5:1322–1334. doi: 10.1002/emmm.201302507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao R, Shin DS, Fiser A, Goldman ID. Identification of a functionally critical GXXG motif and its relationship to the folate binding site of the proton-coupled folate transporter (PCFT-SLC46A1) Am J Physiol Cell Physiol. 2012;303:C673–C681. doi: 10.1152/ajpcell.00123.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.