

Abstract
This review discusses mechanisms that link allelic variants of MHC class II molecules (MHCII) to immune pathology. We focus on HLA-DQ (DQ) alleles associated with celiac disease (CD) and type 1 diabetes (T1D) and the role of the murine DQ-like allele, H2-Ag7 (Ag7), in murine T1D. MHCII molecules bind peptides, and alleles vary in their peptide-binding specificity. Disease-associated alleles permit binding of disease-inducing peptides, such as gluten-derived, Glu-/Pro-rich gliadin peptides in CD and peptides from islet autoantigens, including insulin, in T1D. In addition, the CD-associated DQ2.5 and DQ8 alleles are unusual in their interactions with factors that regulate their peptide loading, invariant chain (Ii) and HLA-DM (DM). The same alleles, as well as other T1D DQ risk alleles (and Ag7) share nonpolar residues in place of Asp at β57 and prefer peptides that place acidic side chains in a pocket in the MHCII groove (P9). Antigen-presenting cells from T1D-susceptible mice and humans retain CLIP due to poor DM editing, although underlying mechanisms differ between species. We propose that these effects on peptide presentation make key contributions to CD and T1D pathogenesis.
Background
MHCII alleles are associated with immune-related diseases
Major histocompatibility complex (MHC) genes were the first genes found to be associated with diseases that involve T cell-mediated pathology [reviewed in (1)]. These strong associations account for a substantial proportion of genetic risk. The MHC is a multi-locus region of the genome, which comprises several sub-regions containing clustered genes involved in immune function, as illustrated for the HLA (human leukocyte antigen) region, the human MHC, in Fig. 1A (2). The class II sub-region, which carries many of the strongest disease associations, encodes the α and β chains of several heterodimeric MHC class II (MHCII) glycoproteins, called HLA-DR, -DP, and DQ in humans. MHCII proteins are expressed constitutively on B cells, dendritic cells (DCs), thymic epithelial cells (TECs), and monocytes/macrophages (collectively, antigen-presenting cells [APCs]) and are inducible on other cell types (3).
Fig. 1. MHCII-linked susceptibility to CD and T1D.

(A) Schematic map of the human MHC (HLA) on human chromosome 6q23; polymorphic “classical” class I and II genes are shown as black boxes, with DQ genes indicated by red “*”. (B) Polymorphisms discussed in the text, which are implicated in the differential associations of HLA-DQ and H2-A subtypes with CD and T1D. These alleles differ in other respects, but the key polymorphisms are identified by comparison to non-susceptibility alleles (not shown) and shown in single-letter code. δ, deletion. Residue numbering for DQ α chain sequences in this Figure, as throughout this manuscript, follows that for H2-A and HLA-DR (90) to facilitate identification of homologous residues; native residue numbers for DQα would be higher by 3 (α25, α56, α79). (C) Anchor motifs for peptide binding to these risk alleles. Single letter code is used. Charged residues are red (+) or blue (-). Groupings: ϕ = hydrophobic; n = neutral aliphatic, p = polar, ± = charged. Preferences discussed in the text are highlighted in bold.
MHCII genes exhibit extensive structural polymorphism, with many alleles existing in human populations at most loci (except for the α chain of DR), all of which differ from one another at multiple positions. Most individuals are heterozygous at most MHCII loci and express both alleles (termed co-dominance). In the international HLA gene nomenclature (4), each allele is identified by its locus name followed by two pairs of digits, which identify structural variants (e.g., DRB1*04:01 for one of the structural variants of the gene coding for the HLA-DR β chain). Different MHCII alleles are overrepresented in patients with different immune-related disorders, particularly in autoimmune diseases with a significant contribution of T cells to pathogenesis. In murine models of these conditions, genetic studies also indicate a marked effect on disease susceptibility of genotype at the two MHCII loci (called H2-A and H2-E, with alleles designated by superscripts, e.g., H2-Ed).
Structure and normal function of MHCII proteins
The normal function of MHCII glycoproteins is to capture peptides in endocytic compartments of APCs and to present them at the surface of these cells, where they can engage clonally variable antigen receptors (T cell receptors, TCR) on T lymphocytes expressing the CD4 molecule (CD4+ T cells), a co-receptor for MHCII (5) (Fig. 2A). Crystal structures of MHCII proteins reveal a single, groove-shaped peptide binding site, which accommodates peptides with a 9-amino acid binding “core”, usually with N- and C-terminal overhangs of varying length [reviewed in (6)]. Certain amino acid side chains are preferentially found at critical positions within the core (“anchor residues” at relative positions 1, 4, 6, 7, and 9), where they make complementary contacts with specificity pockets lining the groove. The complex also is stabilized by multiple hydrogen bonds between the peptide backbone and the groove. These mechanisms enable MHCII molecules to bind tens of thousands of peptides that satisfy the binding preferences at anchor residues (7). There is no discrimination between peptides derived from endosomal proteolysis of self proteins or from invading pathogens. Much MHCII polymorphism involves short, recombined sequence cassettes that diversify the peptide binding groove with remarkably little impact on the overall tertiary structure (examples are shown in Fig. 2B, with relevant polymorphisms listed in Fig. 1B). Thus, each MHCII allelic variant presents a distinct peptide repertoire (8), albeit with some overlap (e.g., Fig. 1C).
Fig. 2. Structural basis for MHCII allele-specific peptide binding.
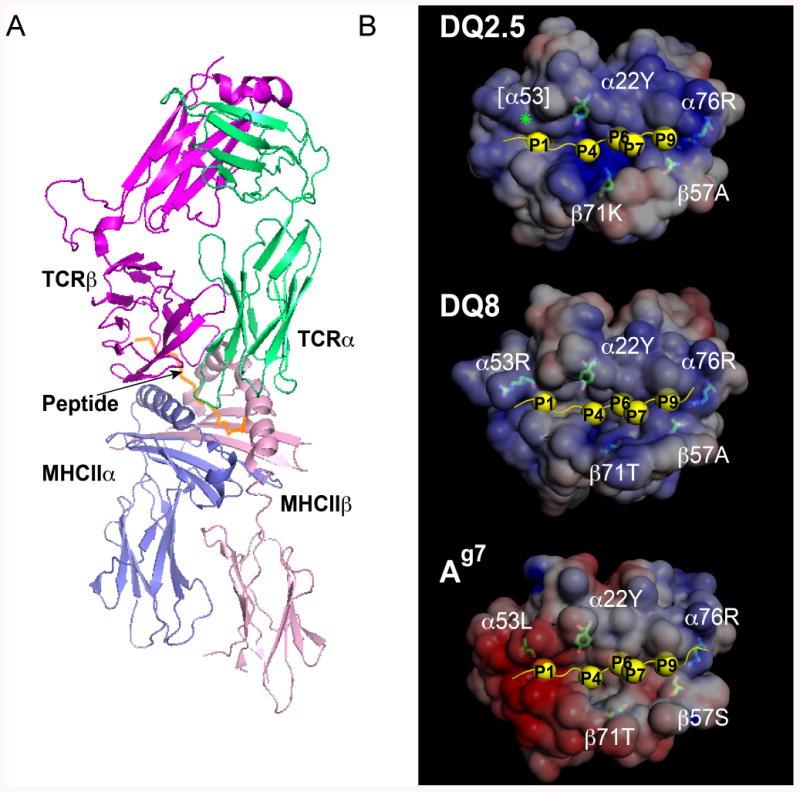
(A) Ribbon diagram of a crystal structure showing TCR engagement by peptide/MHCII complex on APCs. The 21.30 TCR (magenta and green) is shown engaging the murine MHCII molecule I-Ag7 (blue and pink), with the HEL11-27 peptide (orange) bound to it. Image was generated from Protein Data Bank (PDB) coordinates 3MBE (146) using PyMol software (154). (B) Mapping of key polymorphic residues in Fig. 1B onto the binding grooves of the disease-associated human MHCII alleles DQ2.5, DQ8 and murine alleles Ag7. Crystal structures are shown of DQ2.5 complex with P6-deamidated αI-gliadin peptide (residues 57-68) [PDB code: 1S9V (70); top], DQ8 complex with α2-gliadin peptide (residues 223-240) deamidated at P1 and P9 [PDB code: 2NNA (73); middle], and Ag7/GAD peptide (residues 207-220) [PDB code: 1ES0 (124); bottom]. Each MHCII molecule is shown in a space-filling representation with surfaces color-coded according to electrostatic potential (red, negative; blue, positive). Side chains of key polymorphic residues discussed in the text are shown in a stick representation. The bound peptide is shown as a yellow ribbon with anchor residues as yellow spheres. Images were generated using Pymol along with the APBS tool and PDB2PQR web service and software package (154-157).
CD4+ T cells normally remain tolerant to self peptides, as well as to peptides derived from innocuous foreign material (commensal organisms, food proteins, dust), whilst mounting protective responses against pathogens. This is possible because the functional consequences of antigen recognition depend both on the strength of the TCR interaction with peptide/MHCII and the biological context. In the thymus, developing thymocytes require weak TCR ligation by self peptide/MHC complexes on cortical TECs to survive (positive selection). Subsequently, strong TCR ligation by peptide/MHC on thymic medullary APCs (TECs or DCs) results in killing of thymocytes with dangerous levels of self-reactivity (negative selection). This key mechanism of central tolerance is supported by the expression of diverse peripheral self antigens in medullary TECs under the control of the autoimmune regulator (AIRE) transcription factor (9). Thymic positive and negative selection depends on the level of MHCII expression and on recognition of specific peptides (10). The percentage of surviving CD4+ thymocytes is small, and these cells enter the circulation either as short-lived regulatory CD4+ T cells (Tregs) (11-12), which dampen effector T cell responses, or as long-lived naïve T helper cell precursors. Weak ligation by self peptide/MHCII molecules on peripheral APCs reinforces self tolerance (10) and promotes the survival of functional CD4+ T cells (13). Thus, all peripheral CD4+ T cells are self-reactive, but normally maintain self tolerance. In contrast, during infection, CD4+ T cells initiate adaptive immune responses to non-self. DCs in infected tissues phagocytose or endocytose material from pathogens, which is fragmented into peptides, some of which bind to MHCII molecules. Triggered by proinflammatory signals, the DCs migrate to draining lymph nodes and present the pathogen-derived, MHCII-bound peptides to naïve and memory CD4+ T cells, triggering their activation, proliferation, and differentiation (14). Antigen presentation to the activated effector CD4+ T cells by other APCs (e.g., B cells, tissue macrophages) subsequently maintains antigenic specificity during the effector arm of the immune response.
Challenges in explaining MHCII/disease associations
Autoimmune diseases result from the activation of pathogenic effector T helper cells by self antigens presented by MHCII molecules [for example, (15)]. Non-specific inflammation or infection appears to be required to break T-cell tolerance. Thereafter, the chronic activation of infiltrating pathogenic T cells by local APCs contributes to the destruction of affected organs, alongside the pathogenic effects of autoantibodies made with T-cell help. Similarly, allergic diseases result from aberrant activation of T helper cells specific for normal, otherwise innocuous non-self peptides, usually at mucosal surfaces. In contrast to autoimmune disease, the thymus is not thought to contribute to tolerance here, but mucosal tolerance mechanisms must be overcome.
Different MHCII alleles associated with one autoimmune disease often share polymorphisms in the groove (e.g., Fig. 1B and Fig. 2B). This suggests that the disease-associated polymorphisms determine the binding of self peptides (e.g., Fig. 1C) and their presentation to autoaggressive T cells. Indeed, such T cells preferentially recognise autoantigen-derived peptides presented by the disease-associated MHCII alleles, rather than by other alleles that are co-expressed (see below). Such observations do not explain, however, why the presentation of self peptides by the same MHCII alleles in the thymus and periphery is unable to maintain tolerance. Neither does it explain why other MHCII allelic variants, which usually can present other peptides from the same self antigens, confer no risk. Thus, additional mechanisms must account for the association of particular MHCII alleles with loss of T cell tolerance. Analogous considerations apply to MHCII-associated allergic diseases.
Complex regulation of peptide binding to MHCII molecules in vivo
Antigen presentation by MHCII molecules is shaped by many factors besides their specificity for peptides. Importantly, peptide binding to MHCII molecules is regulated by other proteins during MHCII maturation and trafficking (Fig. 3)[reviewed in (5-16-18)]. Newly synthesised MHCII α and β chains assemble in the endoplasmic reticulum (ER) as αβ heterodimers and rapidly associate with the invariant chain (Ii). A disordered region of Ii, including residues 81-104, folds into the peptide binding groove of the newly assembled MHCII molecules (19) and prevents its premature occupation by other ligands in the ER (20). Directed by the cytoplasmic tail of Ii [reviewed in (16)], the nascent αβIi complexes accumulate transiently in late endosomal multivesicular or multilamellar compartments. There, Ii cleavage by endosomal proteases yields MHCII molecules with their grooves still occupied by Ii remnant peptides, spanning residues 81-104 of Ii (now termed CLIP, for “class II-associated Ii peptides”). CLIP must dissociate to allow binding of self and non-self peptides resulting from endosomal protein catabolism.
Fig. 3. Regulation of MHCII peptide loading by Ii and DM.
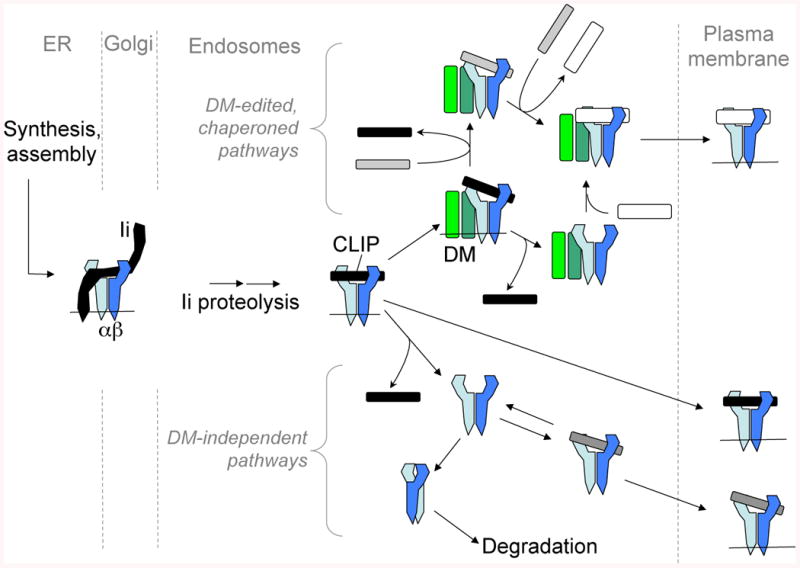
MHCII chains are shown in shades of blue, Ii in black, and DM in green. Events in different subcellular compartments are separated by gray dashed lines. Peptides derived from endosomal proteolysis are shown in gray or white, depending on their stability of association with MHCII (weak: gray; strong: white). Lipid membranes (thin horizontal lines) are sometimes omitted for clarity. HLA-DQ susceptibility alleles for CD and T1D, as well as the cellular context in NOD APCs, appear to favor DM-independent peptide loading pathways.
CLIP release may occur spontaneously (21), but is accelerated by HLA-DM (H2-DM or H2-M in mice) (22-24). DM structurally resembles the peptide-binding MHCII molecules, but lacks a peptide binding groove (25). Its lateral interaction with MHCII/CLIP complexes in endosomal membranes (26-27) catalyses CLIP release, stabilises empty MHCII molecules (28-29), accelerates exchange of endosomal peptides other than CLIP (22-24) and, as a result of this editing process, favours the binding of peptides with slow rates of dissociation (Fig. 3, top right). Peptide-loaded MHCII molecules are finally deposited on the APC surface for inspection by CD4+ T cells.
This elegant mechanism, defined in cultured APC lines, only hints at the complexity of physiological antigen presentation. APC types differ in their content of endosomal proteases, which affects processing of Ii and protein antigens (30-32). Delivery of exogenous antigens into the MHCII pathway may involve phagocytosis (in macrophages and DCs) or receptor-mediated endocytosis, using surface immunoglobulin in B cells or lectin-type receptors in DCs [reviewed in (5)]. Endogenous antigens may enter from the secretory pathway or, via autophagy, from the cytosol [reviewed in (5)]. In subsets of B cells and DCs, DM activity is regulated by differential expression of HLA-DO (H2-O or –DO in mice), another distant MHCII homologue (33-34). DO binds to DM and is thought to inhibit its interaction with MHCII molecules. APC types may differ in the relative abundance and trafficking of empty, CLIP-loaded, and peptide-loaded MHCII molecules. For example, cortical TECs express relatively high levels of CLIP/MHCII (35), whereas immature DCs express DM and empty MHCII molecules, available for peptide loading, at the cell surface (36). Internalization and turnover of MHCII molecules in immature DCs is regulated by β-chain ubiquitination; activation of DCs by proinflammatory stimuli shuts down this pathway, allowing deposition of long-lived, cell surface peptide/MHCII complexes (37). Moreover, peptide loading pathways within one cell are heterogeneous: many peptides require Ii and DM for presentation to T cells, but other peptides are presented independently of these molecules, or may be inhibited by them (38-39). Thus, peptide loading of MHCII molecules is modified by numerous cellular variables.
Are disease-associated MHCII molecules biochemically unusual?
These complexities are relevant to the question of how particular MHCII alleles predispose towards autoimmune disease, because MHCII alleles vary in their post-translational fate [reviewed in (40)]. Some MHCII alleles release CLIP spontaneously, without DM, while others form very stable CLIP complexes, requiring DM for CLIP release and loading with antigenic peptides (41-42). The surface expression of many allelic variants with low CLIP affinity is reduced in the absence of DM, and their turnover is increased (43-44). This is due to the loss of empty MHCII molecules after spontaneous CLIP release, or due to poor rescue by low-affinity peptide binding, in the absence of DM chaperoning and editing (Fig. 3, bottom right).
We have noted previously that many (but not all) MHCII alleles associated with autoimmune diseases form relatively unstable complexes with CLIP and discussed possible implications for autoimmune pathogenesis (40). Here, we review our current understanding of the overlapping MHCII associations with CD, a gastrointestinal hypersensitivity to dietary glutens, and with T1D, an idiopathic autoimmune disease of pancreatic islets. We describe how structural polymorphism shapes both the inherent specificity for peptides and the post-translational fate of MHCII molecules, and discuss how these effects may promote immune pathology.
Celiac disease
Clinical features and association with HLA-DQ
CD is a chronic inflammatory enteropathy with diarrhea, steatorrhea, malabsorption, weight loss and, in children, growth retardation. It affects 1% of the population in developed countries, with a female bias and onset typically either in early childhood or following menopause (45). CD is triggered by dietary exposure to wheat glutens and their homologues in rye and barley. Heterogeneous proteins in these allergens (glutenins and gliadins, with subtypes designated by Greek letters and Roman numerals) trigger cellular and humoral hypersensitivity. Symptoms improve when the dietary triggers are eliminated, but compliance can be difficult and symptoms return upon re-exposure. Histology of affected small intestinal tissue shows villous atrophy, crypt hyperplasia, and infiltration by memory and effector T helper cells into the lamina propria (46).
CD has a clearly defined genetic component, shown by high concordance rates in monozygotic twins (47). MHCII associations account for 35-40% of genetic risk (48); many additional genetic risk factors have recently been identified, primarily by genome-wide association studies (GWAS) (48). The two MHCII alleles associated with CD are DQA1*05:01/DQB1*02:01 and DQA1*03/DQB1*03:02, coding for the DQ2.5 and DQ8 proteins, respectively (49-51); the older protein nomenclature will be used subsequently. DQ2.5 is found in 90-95% of patients, vs. 20-30% of controls; almost all DQ2.5-negative CD patients carry DQ8 (52). DQ2.5 homozygosity confers a fivefold increased risk of CD, compared to heterozygotes, with intermediate risk for DQ2.5/DQ8 compound heterozygotes. The closely related alleles, DQ2.2 (DQA*0201/DQB*02) and DQ0602 (DQA1*0102/DQB1*0602), carry a very low risk for CD (53). Interestingly, latent CD (i.e., positive serology without clinical disease) tends to be associated with low-risk DQ genotypes (54).
T-cell hyper-responsiveness to modified gluten proteins
The unique immune-stimulatory properties of gluten proteins result from a combination of their inherent properties and their processing and presentation by host APCs. The high Pro content of certain gliadin peptides renders them unusually resistant to intestinal proteolysis (55-56). Gliadin peptides may have direct pro-inflammatory effects (57-58), but their pathogenicity depends on their specific recognition by CD4+ T cells in the lamina propria. Such T cells have been isolated from lesional tissue and blood of patients with CD after gluten challenge (59-60). They are diverse in their TCR gene usage, but predominantly require peptide presentation by disease-associated DQ molecules (59-60). APCs from individuals who are homozygous for the DQ susceptibility alleles present gliadin-derived peptides more efficiently than heterozygotes, correlating with disease risk (61).
Surprisingly, peptides derived from native gliadin sequences are not stimulatory; they bind poorly to DQ2.5 or DQ8 and consequently fail to elicit DQ-restricted CD4+ T cells. However, specific glutamine residues (Gln) within these peptides are substrates for deamidation to glutamate (Glu) by tissue transglutaminase (TG2), an enzyme in the gut epithelium (62), which specifically targets Glu-X-Pro or Glu-X-X-ϕ sequence motifs (ϕ = bulky hydrophobic residue) (63-64). Deamidation can be mimicked by chemical synthesis of Gln→Glu-substituted peptides or promoted non-specifically by heat treatment. Deamidation greatly enhances the ability of gliadin peptides to stimulate DQ2.5- or DQ8-restricted CD4+ T cells (62-65-66). The positioning of native Gln residues and of TG2-catalysed conversion to Glu in gliadin peptides is critical for T-cell recognition (66-67). The presentation of glutenin peptides by DQ8, however, is less dependent on TG2 (68).
Disease-associated DQ alleles bind modified gluten peptides
Pro-rich peptides, such as gliadin epitopes in CD, are poor binding substrates for most MHCII alleles, because in the core MHCII-binding region, the imidazole ring of the Pro side chain prevents participation in hydrogen bonds that tether peptides in the groove. Unusually, however, DQ2.5, the strongest CD risk allele, accepts Pro at P1, where a conserved hydrogen bond usually forms with the nitrogen atom of the peptide bond (69-70)(Fig. 1C and 2B, top). This is due to a deletion at position 53 of the DQ2.5 α chain, which renders this hydrogen bond dispensable (Fig. 1B; Fig. 1 legend shows DQα residue numbering conventions). At P6, DQ2.5 also can tolerate Pro, due to the unique presence of Ser30β. Disruption of hydrogen bonding normally disfavours Pro even at non-anchor positions in the binding core, but DQ2.5 allows Pro at P3 and P8 (69). Indeed, many gliadin epitopes have Pro residues at one or more of these positions.
DQ2.5 also is unusual in its ability to accommodate negatively charged peptide residues (Glu or Asp) at any position within the binding core (69) (Fig. 1C), due to positively charged residues (notably Arg76α and Lys71β) and extensive hydrogen bonding interactions in the groove (70). Except at P1, Gln is markedly less well tolerated at anchor residues than Glu, which explains the requirement for TG2 modification. The relative positions of Pro and Glu residues also matter. With Pro at P8 or a hydrophobic residue at P9 of a gliadin peptide, Gln at P6 becomes a substrate for TG2 (similarly for Pro at P6 with Gln deamidation at P4), improving binding to DQ2.5. The side chains of the remaining, unmodified Gln residues of gliadin peptides may assist binding by complementing defects in the main-chain hydrogen bond network due to the Pro residues (70-71).
Peptide binding specificity also explains some of the variation in CD risk associated with DQ subtypes. DQ8, which confers less risk than DQ2.5, presents fewer Pro-rich α-gliadin peptides (56). One contributing factor is the presence of Arg at α53, which prohibits interaction with Pro at P1. Furthermore, whereas for HLA-DQ2.5 a single deamidation step at P4, P6 or P7 of a gluten peptide is sufficient to evoke a CD4+ T-cell response, for HLA-DQ8, deamidation at two positions (P1 and P9) is preferred. This may further limit the efficiency of generating DQ8-binding gliadin peptides (72-73). Both factors may contribute to the lower disease risk associated with DQ8 (74). Unexpectedly, the non-associated DQ2.2 variant has a peptide binding motif that closely resembles that of DQ2.5, but presents gluten peptides less well. This may relate to difficulties in accommodating Pro at the P3 position in DQ2.2 (61-75-76), although recent work (77) contests this and favours another explanation, discussed below.
Unusual mode of peptide acquisition by disease-associated DQ alleles
In addition to its unique peptide binding specificity, DQ2.5 also interacts abnormally with Ii and DM. Most MHCII alleles bind CLIP in a single binding register (“CLIP1”), shown in the crystal structure of CLIP bound to DR3 and using Met91 (human Ii numbering) to occupy the P1 position of the groove (19). DQ2.5, however, is able to bind in a non-canonical “CLIP2” binding register, with Pro96 in P1 (78-79). In vitro, the rate of spontaneous CLIP release, in either register, from recombinant soluble DQ2.5 molecules is slower than CLIP release from DR3, and even slower than that of some gliadin peptides. Quantitative peptide extraction experiments indicate that CLIP naturally occupies the majority of DQ2.5 molecules, similar to the high occupancy of DR3 with CLIP in cells lacking DM (77-78).
This phenotype is exaggerated by a poor interaction of DQ2.5 with DM (80). CLIP release from DQ2.5 is slow even in DM-sufficient cells. In vitro, at least 10-fold more recombinant DM is required to release CLIP (either register) from DQ2.5 molecules than from DR3. CLIP complexes with DQ1 (DQA1*01:01, DQB1*05:01) exhibit a normal degree of DM susceptibility. The deletion of residue α53 in DQ2.5 (as in all DQ alleles with DQA1*04 and *05 chains) is critical, because insertion of Gly, the residue at α53 in DQ1, restores sensitivity to DM (80). Interestingly, the insertion of Arg at this position (found in DQA1*03, and thus in DQ8) fails to restore sensitivity to DM, suggesting that DQ8 also interacts poorly with DM.
Extreme CLIP retention would prevent antigen presentation (81). However, DQ2.5 allows sufficient CLIP release for gliadin peptide presentation. Interestingly, recent studies show that DQ2.5 resistance to DM editing actually promotes the presentation of certain gliadin peptides (80). Model APCs expressing wild-type DQ2.5 molecules present an immunodominant αII-gliadin (residues 62-70) peptide to T cells, with or without DM present. Expression of the DM-sensitive DQ mutant (α53Gly insertion) markedly suppresses presentation of this peptide, whereas presentation in the absence of DM is maintained. The DQ2.5 DM interaction defect could enable the presentation of other gluten peptides in CD, which would otherwise be edited by DM.
For most other MHCII alleles, lack of DM interaction results in the presentation of peptides that form “unedited”, kinetically unstable complexes. However, one factor contributing to the prolonged retention of CLIP and several gliadin peptides is the presence of a Tyr at position α22 in DQ2.5, which participates in hydrogen bonding networks with the peptide main chain (77). This feature is shared with most DQ alleles, including DQ8. A few subtypes, including DQ2.2, have Phe at this position. Introduction of this change into DQ2.5 results in faster release of CLIP and gliadin peptides and reduces the persistence with which antigens are presented (77). This may explain why DQ2.2 does not confer substantial risk of CD, even though its anchor motif closely resembles that of the DQ2.5 high-risk allele. The Tyr α22 effect is DM-independent: introduction of Phe at α22 into DQ2.5 does not restore DM-dependent peptide exchange (80).
A Series of Unfortunate Events
In summary, the genetic association of CD with HLA-DQ2.5 can be explained by a “series of unfortunate events” (Fig. 4). The model explains why the DQ risk alleles are necessary for CD, but does not explain why, despite inescapable exposure to glutens in normal Western diets, most individuals with DQ risk alleles do not develop CD. Other genetic factors or environmental triggers, or both, must be important. The model endorses the development, now underway, of gliadin-degrading enzymes to destroy immune-stimulatory peptides in the gut, of TG2 inhibitors to prevent their immunogenic modification, of varieties of grain that lack relevant T-cell epitopes, and of allele-specific, non-immunogenic MHCII-blocking peptides. A novel possibility is to mimic DM editing of DQ2.5 and DQ8 to reduce the presentation of gliadin peptides. Small-molecule “chemical chaperones” that promote peptide exchange by MHCII have been developed (82), It would be interesting to know whether these agents act on DQ2.5 molecules despite their DM resistance. One caveat is that self peptide exchange by such agents might create neo-epitopes that could trigger autoimmunity.
Fig. 4. Role of DQ2.5 polymorphisms in the presentation of gliadin peptides to trigger CD.

Peptides derived from dietary glutens, rendered uniquely resistant to proteolysis by their high Pro content, survive digestion in the small intestine, only to be deamidated at critical Gln residues by epithelial TG2, enabling them to bind DQ2.5, by virtue of its unusual peptide binding properties. The presentation of at least some peptides is boosted by inefficient DM editing of the peptide repertoire bound to DQ2.5, in combination with prolonged, sequence-independent, retention of peptides. These factors promote activation of gluten-specific, disease-causing CD4+ helper T cells. This constellation of properties appears to be shared, to varying degrees, by the risk alleles, but not by the non-susceptibility alleles. The less strongly associated DQ8 allele shares many of the key features of DQ2.5, but has a different peptide binding motif that is less well matched to the intestinal processing and modification patterns of gliadins. The low-risk allele, DQ2.2, has a permissive peptide binding motif but does not retain gliadin peptides as persistently; other alleles lack suitable binding motifs, and some interact better with DM.
The existence of several HLA-DQ-transgenic mouse models, on various genetic backgrounds, will be helpful in exploring these therapeutic possibilities. These mice mount strong mucosal CD4+ T cell responses to gliadins, although in many studies the prevailing cytokine secretion patterns are anti-inflammatory; clinically significant enteropathy develops only on a genetic background with particular autoimmune susceptibility [review in (83)].
Type 1 diabetes
Clinical features and association with DQ
In T1D, insulin-secreting β-cells in the pancreatic islets of Langerhans are destroyed, causing insulin deficiency and leading to life-threatening hyperglycemia and ketoacidosis [reviewed in (84)]. The disease may occur at any age but usually has juvenile onset. Insulin replacement therapy prevents acute T1D and must be maintained for life by insulin injections and glucose monitoring. Even so, life expectancy is reduced due to chronic complications from imperfect glucose control.
Human T1D has a well-recognised genetic component. MHC genes are the most important susceptibility region (85), accounting for ≈ 50% of genetic risk (86). In the MHCII region, the strongest association signals are found at HLA-DQB1, even though the attribution of risk to this locus is complicated by linkage disequilibrium with adjacent DQA1 and DR loci, creating conserved haplotypes. With few exceptions, a sequence feature shared between DQ risk alleles is the presence of an uncharged residue (Val, Ser, or Ala) at position 57 of the β chain. Non-susceptibility alleles have a negatively charged aspartic acid (Asp) residue at this position. The most strongly associated alleles in Caucasians are DQ8, followed by DQ2 [(87) and review in (88); Fig. 1B] – the same loci implicated in CD, but in reverse order. Interestingly, DR3/DR4 heterozygotes are at particularly high risk of T1D. This is thought to be due to the generation of mixed-haplotype DQ αβ heterodimers “in trans” and suggests that α-chain pairing is also important. Consistent with this, one study has suggested that Arg49α is a risk-associated polymorphism (89). Another complication is that heterozygosity for some Asp57β alleles confers dominant protection from disease. Other MHC polymorphisms confer risk independently of DQ. In the class II region, most DR4 subtypes confer increased risk, but DRB1*04:03 does not; in the class I region, associations with HLA-A and –B are found in large family studies (90-91). Polymorphisms at the insulin gene and several genes implicated in T-cell regulation (IL2 and its receptor, CTLA4, PTPN22) and many less well-defined loci show weak associations with T1D in GWAS (92).
The histopathology of T1D confirms immune involvement, characterised by lymphocytic infiltration into pancreatic islets; overt T1D ensues once a majority of β cells are lost (93). Autoantibodies to islet-specific autoantigens, including insulin and glutamic acid decarboxylase (GAD), and CD4+ effector T cells specific for peptides from these autoantigens, are found in the blood of T1D patients. These autoreactivities may develop long before clinical disease onset (94).
H2-Ag7-associated T1D in nonobese diabetic mice
The role of MHCII molecules in T1D pathogenesis has also been studied in the nonobese diabetic (NOD) mouse, a diabetes-prone mutant strain (95), which exhibits remarkable similarities with human T1D. NOD mice develop spontaneous insulitis with T-cell infiltration and autoantibodies to islet autoantigens. T1D develops around 3 months of age, with greater penetrance in females than males. Both CD4+ and CD8+ T cells are important, based on studies of T cell-deficient mutant mice (96) and adoptive transfer experiments (97).
MHCII genotype is critical for T1D in NOD mice. Their H2-Aα chain gene is identical to that of Ad (found in non-susceptible strains), but the β chain, designated Aβg7, is unique amongst inbred mouse strains, differing from Aβd at 17 positions (98). Antibody blocking of Ag7 prevents T1D (99). Like the T1D-associated DQ alleles, Ag7 has a non-Asp residue (Ser) in position β57; the adjacent position His56 is not shared with DQ (98). Transgenic expression of mutant Aβg7 chains, with Proβ56 or Aspβ57, results in competition of mutant molecules with endogenous wild-type Aβg7 for pairing with Aα and greatly reduces T1D, indicating a critical role for these polymorphisms (100-101). Thus, the key disease-related MHCII polymorphism in autoimmune T1D is the same in mice and humans.
Homozygous expression of I-Ag7 is required for disease in NOD mice (102). This is reminiscent of the increased risk of T1D in humans homozygous for non-Asp57β alleles of DQ and of dominant protective effects of some Asp57β alleles. NOD mice lack expression of H2-Eα, the orthologue of HLA-DRα; thus, H2-A is the sole expressed MHCII protein, except possibly for low-level “mixed-isotype” pairing of Aα with Eβ. Transgenic expression of Eα confers protection of NOD mice (101); this may be analogous to protective effects of some DR alleles in humans. Mapping of other disease genes outside the H2 region supports the idea that NOD mice recapitulate much of the pathogenesis of human T1D (91).
In NOD mice, pancreatic islet development may be abnormal at weaning (103). Myeloid APCs exhibit early developmental defects (104), and may contribute to abnormal islet function and homeostasis (105). B-cell development is also abnormal in NOD spleen, with altered representation of transitional and marginal zone B cells (106). Progression from insulitis to overt diabetes is accompanied by expansion of marginal zone B cells and their invasion into pancreatic islets, suggesting that antigen presentation by this subset may contribute to pathogenesis (107). Indeed, B cell-deficient NOD mice do not develop diabetes (108). Antigen presentation by pancreatic DC has also been implicated in T1D pathogenesis (109). Together, these findings suggest that antigen presentation evolves over time during T1D pathogenesis and involves several APC types.
MHCII association with T1D: a role for covalently modified self?
T-cell epitopes from islet cell autoantigens, including GAD, preproinsulin, and insulin B chain, have been identified in mouse and man and are generally presented by the disease-associated MHCII alleles (15). It should be noted, however, that the earliest infiltrating CD4+ T cells show evidence of clonal expansion but respond to none of the defined islet autoantigens (110-111), suggesting that the known specificities arise from epitope spreading. The peptide binding motifs of the disease-associated DQ8 and DQ2 alleles, and of Ag7 in mice, are unusual, but the ability of islet autoepitopes to satisfy these requirements is not, by itself, sufficient to explain why T1D risk is increased by these particular risk alleles, rather than any others.
Studies of particular islet autoantigens suggest some explanations for loss of self tolerance to specific peptides. One immunodominant peptide from insulin A chain requires a non-native disulfide bond to stimulate T cells (112). Here, disulfide bond reshuffling may break self tolerance by generating neo-self determinants. In a different mechanism for generating neo-determinants, NOD CD4+ T cells respond to an insulin peptide bound to Ag7 in a register that is not efficiently generated by normal processing of whole insulin; in contrast, T cells to an overlapping, well-processed peptide are deleted (113). Incomplete central tolerance to insulin could also arise from the discrepancy between low level expression of insulin in the thymus and extremely high local concentrations of insulin in pancreatic islets. Indeed, insulin promoter polymorphisms that reduce thymic expression of insulin increase risk of T1D [reviewed in (91)]. However, a link to particular DQ alleles or Ag7 has yet to emerge: similar mechanisms might operate in the context of other MHCII alleles that do not confer T1D risk.
Quantitative defects in antigen presentation and tolerance
Although islet autoepitopes bind the disease-associated MHCII variants, a high binding affinity does not appear necessary. At least one immunodominant insulin peptide in NOD mice does not form highly stable complexes with Ag7 (114); an autoepitope from chromogranin A appears to have an unusual mode of binding with partial occupancy of the groove (115). Consistent with this, some crystal structures of T1D-associated MHCII alleles show suboptimal P9 side chain/pocket interactions (70). The relative instability of some of these complexes resembles findings in other autoimmune diseases (116), but contrasts with the good correlation between stable MHCII binding and immunodominance in responses to foreign antigens (117). Thymic presentation of moderate affinity peptides may be inefficient, leading to escape from deletion of the T cells recognising them.
Presentation defects in NOD mice may reflect a generalised propensity towards Ag7 -restricted autoreactivity. Immunisation of NOD mice with foreign protein in adjuvant leads to the activation of T cells that react with self antigens on NOD splenocytes (118). This autoproliferation requires Ag7 homozygosity and is suppressed by expression of other H2-A alleles, or of H2-E, matching the genetic regulation of T1D (118). Autoreactive T cells in mouse strains carrying I-Ag7 (NOD and the H2-congenic B6.g7 strain) are more frequent compared to mice of H-2b haplotype (NOD.H-2b and B6)(119). Thus, expression of Ag7 as the sole MHCII allele confers a defect in T-cell tolerance, which is not islet-specific. Deletion of self-reactive transgenic TCRs is highly efficient in other mouse strains, but autoreactive T cells escape into the periphery in NOD mice. Depending on the TCR, autoimmunity may affect pancreatic β cells (e.g., BDC2.5 TCR) or other target organs (120). These transgenic systems require Ag7 for peptide presentation to pathogenic T cells, but a single copy of Ag7 is sufficient and other class II alleles fail to protect. Recent work suggests that BDC2.5 NOD mice generate an excess of αβ (vs. γδ) T cell precursors, compared to H2-identical B6.g7 mice, which may alter their subsequent thymic selection independently of Ag7 (121), but these findings do not contradict the notion that Ag7 is inefficient at enforcing self tolerance. Collectively, these findings argue that T1D in NOD mice arises in part due to defective thymic negative selection of diabetogenic T cells; Ag7 also may be limited in its ability to maintain peripheral tolerance and Tregs (122).
Is Ag7 a promiscuous peptide binder?
The peptide binding specificity of the Ag7 groove may promote this generalized autoreactivity. Its peptide binding motif, shown in Fig. 1C and based on quantification of amino acids systematically over-represented in naturally processed Ag7-binding peptides, does not seem unusual. However, in vitro assays (123-124) show that many other side chains are permitted at P1; the small side chains preferred at P4 and P6 provide little scope for stabilizing interactions. P9 shows a clear statistical preference for acidic residues, but many binding peptides have small neutral amino acids at P9. These unusually permissive side chain preferences of Ag7 allow promiscuous binding of diverse peptides from self antigens, compared to greater selectivity of other murine MHCII alleles (123-124). Structural studies of Ag7 are consistent with these findings (124). Moreover, Ag7 is thought to exhibit a defect in the retention of bound peptides, (125). Thus, Ag7 may have an intrinsic tendency to display many peptides at lower abundance and more fleetingly than other alleles, increasing the risk of errors during T cell negative selection. This tendency of Ag7 differs from the non-specific peptide retention by DQ2.5 and DQ8, which confer high risk of T1D, but resembles the poor peptide retention by DQ2.2, which is not a high risk allele despite having a non-Asp residue at β57 (90). Paradoxically, Ag7 shares Tyr22α, the residue implicated in peptide retention by DQ2.5 and DQ8. Thus, the role of polymorphism at α22 in peptide retention, and thus its potential contribution to T1D pathogenesis, may differ between mice and humans.
The peptide binding defect of Ag7 is not universal. Peptides from autoantigens, as well as from uninvolved self proteins, with a length of at least 13 amino acids and an acidic residue at the C-terminus, exhibit stable binding to Ag7 (126). Similarly, acidic P9 residues are found frequently in naturally processed peptides from APCs and pancreatic islet β-cells (127-129). Crystal structures of other H2-A alleles show an inter-chain salt bridge between Aspβ57 and Arg in position α76. The structure of Ag7 bound to a GAD65 peptide shows, as expected, that Ser at β57 lacks this salt bridge, but the negatively charged P9 residue (Glu217) of the bound peptide compensates (124). Thus, the binding characteristics of Ag7-bound self peptides with negative charges at P9 may be more typical of those bound to other MHCII alleles, whereas self peptides with neutral residues at P9 may bind more loosely. The preference for peptides with acidic P9 residues is shared with non-Asp57β alleles of DQ.
Does Ag7 exhibit defective maturation and peptide loading?
The Ser57β polymorphism also is thought to affect the post-translational fate of Ag7. Pulse/chase labelling studies show no major defects in assembly, early maturation, and ER to Golgi export of Ag7 (130), but defects are detectable in other assays. In Ii-deficient cells, substitution of Asp57β to Ser in Ad (as in Ag7) impairs surface expression (131). Thus, lack of inter-chain salt bridging may compromise αβ heterodimer assembly in non-Asp57β alleles. However, as murine APC populations in vivo typically express Ii, and Ii transfection overcomes the expression defect of Ad (Aspβ57→Ser) (131), assembly defects are unlikely to contribute much to immune defects associated with lack of Aspβ57 in vivo. Moreover, Ag7 αβ heterodimers expressed as recombinant soluble molecules in insect cells have been used in peptide binding and crystallization studies without evidence of instability (132).
Following Ii proteolysis, Ag7 may be unusual in its further maturation post-Golgi. At endosomal pH, CLIP is released rapidly in vitro from recombinant soluble Ag7 (132). The rates suggest that spontaneous CLIP release (i.e., without DM) should readily occur during endosomal trafficking of nascent MHCII molecules. This may afford opportunities for loading of peptides without participation of the editing function of DM (40), exacerbating promiscuous peptide binding in vivo. The low stability of Ag7/CLIP is partly attributable to the P9 pocket preferences of this allele, because stabilisation can be achieved by replacing Met98 of CLIP (murine Ii numbering) at P9 with a negatively charged residue (44).
The α51-53 region, which modulates DM interactions of DQ alleles, is polymorphic amongst H2-A alleles, including Ag7 (90). However, there is no evidence that Ag7 fails to interact with DM as a result. In transfected cells, expression of DM can aid CLIP release from Ag7 (133). Moreover, Ag7 expression is reduced in mutant APC lacking H2-DM, due to accelerated turnover (44). The surprising observation that NOD splenocytes nonetheless have increased levels of CLIP at the surface therefore suggests a CLIP exchange defect (134). The cellular regulation of peptide exchange also may be abnormal in PBMCs from T1D patients, which express elevated CLIP levels (135). This observation is consistent with the inherent resistance of the human DQ2 and DQ8 risk alleles to DM-catalysed CLIP release (see above), but other studies suggest an abnormal environment for peptide loading. In B cells from twins discordant for T1D, expression of the p35 isoform of Ii is lower in T1D and associated with a greater ability to load MHCII molecules with exogenous peptides (136). Moreover, a DR4-restricted GAD autoepitope was found to be presented less efficiently in cells expressing high levels of DM, suggesting that poor editing may reveal this epitope in vivo (137). Thus, poor DM-mediated peptide editing and loading with suboptimal peptides may be shared features of human and murine T1D antigen presentation, although the mechanisms differ and only the human DQ alleles exhibit a pronounced inherent DM interaction defect (cf. Figs. 5 and 6).
Fig. 5. Model for effects of Ag7 polymorphisms in murine T1D pathogenesis.

Both peptide binding specificity and use of un-edited peptide loading pathways are affected by β57 polymorphism and further modified by other polymorphisms and cellular context. Features shared with DQ8 (red text, also see Fig. 6) include the P9 preference for negatively charged residues, SDS instability, and poor DM editing. Note that Ag7, unlike DQ8, has not been shown to exhibit a DM interaction defect in transfected cells, suggesting that cellular variables impair DM editing in NOD mice. The exact causal relationships between the biochemical defects and effector T-cell selection/activation remain speculative (indicated by gray arrows).
Fig. 6. Model for effects of DQ8 polymorphisms in human T1D pathogenesis.

See legend to Fig. 5 and text for details.
Is Ag7 unstable in NOD mice?
Another phenotype associated with the Ag7 in NOD mice is accelerated protein turnover, for example in cultured NOD splenocytes (125). The MHCII molecules in these experiments derive mostly from B cells, but Ag7 expression by NOD bone marrow-derived DCs is also diminished (104). The in vivo instability of Ag7 could contribute to inefficient thymic selection and peripheral tolerance. Little is known about how the disposal of internalized MHCII molecules is regulated by peptide binding, DM chaperoning, and proteolysis, and how these variables are affected by MHCII polymorphism. Important variables that affect measured Ag7 turnover in NOD splenocytes remain to be addressed. Allele-specific monoclonal antibodies used for immunoprecipitation may differ in their binding to MHCII conformations with greater or lesser stability in vivo, confounding the effects of structural polymorphism (138). The relative proportion of functional subsets of splenic B cells differs between NOD and control mice. Similarly, the maturation and function of DCs is abnormal in the NOD strain (104-139).
Most MHCII alleles are stabilised by the binding of high-affinity peptides so that, at room temperature, they resist chain dissociation by the detergent, sodium dodecyl sulphate (SDS). In contrast, complexes of most alleles with Ii and CLIP are unstable in SDS and acquisition of SDS stability is normally dependent on DM-dependent endosomal peptide loading (140-141). H2-Ag7 is a notable exception, dissociating readily during SDS-PAGE analysis of un-boiled samples (125). The β56/57 polymorphisms are a key determinant of SDS instability. Human DQ alleles also vary in their SDS stability; lower SDS stability correlates closely with increased risk for T1D conferred by DQ2 and DQ8 subtypes (142). The biological significance of this correlation is difficult to assess, and SDS stability may simply be a biochemical correlate of the genetic polymorphism. When compared with other H2-A alleles, Ag7 falls at the lower end of a spectrum of stability, rather than being an extreme outlier, but this does not rule out a contribution of Ag7 instability to the autoimmunity of NOD mice.
Peptide loading regulators and T1D pathogenesis in mice
Studies of mutant NOD mice lacking Ii confirm the importance of peptide loading regulators in T1D pathogenesis. These mice do not develop T1D and development of insulitis is impaired, even though, surprisingly, Ii-null NOD APCs present islet autoantigens well (143). Protection from T1D in the absence of Ii is associated with a reduced ratio of effector to regulatory CD4+ T cells. In contrast, B-cell development, which is impaired by Ii knockout in other mouse strains, is unaffected in NOD mice. In the absence of Ii, Ag7 molecules form heterogeneous, SDS-stable complexes, probably with ER polypeptides; moreover, H2-DM expression is reduced in Ii-deficient mice (144). A role for peptide loading cofactors in pathogenesis is further supported by an elegant study, in which DM-catalysed peptide exchange was modulated in NOD by expression of human DO in myeloid DCs under the control of the CD11c promoter (145). Strikingly, this intervention protected NOD mice from diabetes, implying a critical role for DM in regulating the presentation of tolerogenic vs. pathogenic self peptides, although these studies have not yet been able to address directly the functional relevance of un-edited peptide loading or MHCII instability in pathogenesis.
Does Ag7 select unusual TCRs?
Lastly, recent work suggests that the lack of Asp57β in Ag7 also affects interactions with the TCR. This idea is consistent with an enrichment for negatively charged residues observed in TCR β-chain CDR3 sequences of CD4+ T cells from NOD mice, regardless of peptide binding specificity (146). Structural studies suggest that this is due to long-range electrostatic interactions with Arg76α. Conceivably, a gain of interactions of CDR3 regions with MHCII at the expense of interactions with the peptide may compensate for the diminished abundance of some peptides, yet increase the likelihood of errors in self/non-self discrimination, and thus of autoimmunity.
In summary, a functional link between poor editing of peptides presented by the T1D-associated MHCII alleles and autoimmune propensity is plausible, but remains hypothetical (Figs. 5 and 6). Islet-specific effector CD4+ Th1 cells may escape thymic deletion and peripheral tolerance mechanisms due to the inefficient baseline presentation of poorly edited, low-abundance self peptides presented by the MHCII risk alleles; higher autoantigen doses in injured islets and pancreatic lymph nodes may overcome this defect during the initiation of autoimmunity. Moreover, inefficient self antigen presentation in the context of T1D risk alleles may impair the generation and maintenance of Tregs that dampen autoimmunity.
Outstanding research questions
Broader conclusions emerge from these studies. The first and foremost link between the MHCII alleles and immune pathology is the fact that disease-related polymorphisms enable the presentation of peptides that are derived from otherwise innocuous self or non-self antigens (gliadins in CD, islet proteins in T1D) and activate pathogenic effector T cells. This alone does not, however, provide a mechanism for the MHCII association with disease, because it leaves unexplained why tolerance to these self peptides failed, and why other MHCII alleles are not permissive for presentation of disease-related peptides. In CD, at least, the link of risk-associated polymorphisms with pathogenesis is much stronger: specific covalent modifications of gluten-derived peptides are required to enable selective binding to precisely those risk alleles. The modified peptides are unlikely to be presented consistently or efficiently in the thymus or in peripheral APCs at baseline, providing a mechanism for the lack of T-cell tolerance. A very similar paradigm has emerged recently in rheumatoid arthritis, where it is thought that modification of Arg to citrulline by peptidyl arginine deiminases at sites of inflammation may generate neo-self determinants with greatly enhanced ability to bind to the disease-associated HLA-DR alleles [reviewed in (147)]. Much remains to be learned, however, about the events that lead to the pathogenic convergence of antigen modification and antigen presentation in vivo.
A second principle is that altered interactions with peptide loading cofactors are relevant to the presentation of neo-self determinants by MHCII risk alleles. In the CD example, DQ2.5 is permissive for gliadin peptide presentation due to its capacity for some spontaneous CLIP release, poor editing of the peptide repertoire by DM, and its ability to retain un-edited peptides via hydrogen bond networks. Importantly, these features can be attributed to specific polymorphisms that distinguish DQ2.5 from non-susceptibility alleles with similar peptide binding specificity. The role of peptide editing merits further study in other autoimmune diseases. For example, in rheumatoid arthritis, some genetic evidence suggests contributions of presentation co-factors DM and its inhibitor DO to pathogenesis (148-150).
In T1D, there is now a good understanding of the general rules for determinant selection by the risk alleles. In addition, several interrelated biochemical defects of the disease-associated DQ alleles and their murine Ag7 homologue have been identified. Nonetheless, our understanding of mechanisms and causation remains incomplete. We do not know whether, in T1D, the MHCII risk alleles are unique in their ability to present islet autoantigens, rather than merely being permissive for their presentation. Meanwhile, there is evidence in mice for increased self-reactivity of CD4+ T cells, which is associated with homozygous expression of Ag7, and for escape of pathogenic, self-reactive TCRs from negative selection. We propose that biochemical features of Ag7 and DQ risk alleles contribute to generalised perturbations in the thymic development and peripheral homeostasis/regulation of CD4+ T cells, which raise the propensity towards autoimmunity.
Several broad questions remain. Which of the several interrelated biochemical defects of the T1D risk alleles are present in vivo, shared between species, and relevant to pathogenesis and how might their contributions to T-cell dysregulation be disentangled? Which of several possible homeostatic or regulatory mechanisms in T cells are disrupted by Ag7? How do other MHCII alleles confer dominant protection in this system? Future studies will capitalise on our increasing ability to manipulate interactions of non-Aspβ57 alleles with peptide loading cofactors. Advances in our ability to quantify homeostatic abnormalities in vivo, through the measurement of protein turnover and cellular homeostasis by stable isotope techniques (151-153), will also be helpful.
MHCII associations with immune pathology were first recognised over 30 years ago, but this has not led to successful therapies. However, recent work, reviewed here, has shown that MHCII risk alleles may present neo-determinants to effector CD4+ T cells and contribute to T-cell dysregulation, both of which may be amenable to manipulation in established, chronic disease. This has revived prospects that further insights into these mechanisms will not only fill our considerable gaps in understanding of genotype/phenotype relationships in complex autoimmune diseases, but could, at long last, lead to novel therapies.
Acknowledgments
E.D.M. and R.B. thank the current and past members of their laboratories for the work that contributed to the ideas synthesised in this review. R.B. and A.D.R. are funded by a Senior Research Fellowship from Arthritis Research UK (ref. 18543, to R.B.). T.H., W.J. and E.D.M are funded by the National Institutes of Health (AI089080, AR061297, AI095813), by the Stanford NIH/NCRR CTSA award number UL1 RR025744, and by the Lucile Packard Foundation for Children's Health.
References
- 1.Carpenter CB. Autoimmunity and HLA. J Clin Immunol. 1982;2(3):157–165. doi: 10.1007/BF00915217. [DOI] [PubMed] [Google Scholar]
- 2.Klein J. Natural History of the Major Histocompatibility Complex. John Wiley & Sons; New York: 1986. [Google Scholar]
- 3.Ting JP, Trowsdale J. Genetic control of MHC class II expression. Cell. 2002;109(Suppl):S21–33. doi: 10.1016/s0092-8674(02)00696-7. [DOI] [PubMed] [Google Scholar]
- 4.Marsh SG, et al. Nomenclature for factors of the HLA system, 2010. Tissue Antigens. 2010;75(4):291–455. doi: 10.1111/j.1399-0039.2010.01466.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Neefjes J, et al. Towards a systems understanding of MHC class I and MHC class II antigen presentation. Nat Rev Immunol. 2011;11(12):823–836. doi: 10.1038/nri3084. [DOI] [PubMed] [Google Scholar]
- 6.Suri A, Lovitch SB, Unanue ER. The wide diversity and complexity of peptides bound to class II MHC molecules. Current opinion in immunology. 2006;18(1):70–77. doi: 10.1016/j.coi.2005.11.002. [DOI] [PubMed] [Google Scholar]
- 7.Velazquez C, DiPaolo R, Unanue ER. Quantitation of lysozyme peptides bound to class II MHC molecules indicates very large differences in levels of presentation. J Immunol. 2001;166(9):5488–5494. doi: 10.4049/jimmunol.166.9.5488. [DOI] [PubMed] [Google Scholar]
- 8.Rammensee HG, Friede T, Stevanoviic S. MHC ligands and peptide motifs: first listing. Immunogenetics. 1995;41(4):178–228. doi: 10.1007/BF00172063. [DOI] [PubMed] [Google Scholar]
- 9.Anderson MS, et al. Projection of an immunological self shadow within the thymus by the aire protein. Science. 2002;298(5597):1395–1401. doi: 10.1126/science.1075958. [DOI] [PubMed] [Google Scholar]
- 10.Morris GP, Allen PM. How the TCR balances sensitivity and specificity for the recognition of self and pathogens. Nat Immunol. 2012;13(2):121–128. doi: 10.1038/ni.2190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Liston A, Rudensky AY. Thymic development and peripheral homeostasis of regulatory T cells. Current opinion in immunology. 2007;19(2):176–185. doi: 10.1016/j.coi.2007.02.005. [DOI] [PubMed] [Google Scholar]
- 12.Vukmanovic-Stejic M, et al. Measurement of proliferation and disappearance of regulatory T cells in human studies using deuterium-labeled glucose. Methods Mol Biol. 2011:707243–261. doi: 10.1007/978-1-61737-979-6_16. [DOI] [PubMed] [Google Scholar]
- 13.De Riva A, et al. Noncognate interaction with MHC class II molecules is essential for maintenance of T cell metabolism to establish optimal memory CD4 T cell function. J Immunol. 2007;178(9):5488–5495. doi: 10.4049/jimmunol.178.9.5488. [DOI] [PubMed] [Google Scholar]
- 14.Zygmunt B, Veldhoen M. T helper cell differentiation: more than just cytokines. Adv Immunol. 2011:109159–196. doi: 10.1016/B978-0-12-387664-5.00005-4. [DOI] [PubMed] [Google Scholar]
- 15.Di Lorenzo TP, Peakman M, Roep BO. Translational mini-review series on type 1 diabetes: Systematic analysis of T cell epitopes in autoimmune diabetes. Clin Exp Immunol. 2007;148(1):1–16. doi: 10.1111/j.1365-2249.2006.03244.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Busch R, Mellins ED. Developing and shedding inhibitions: how MHC class II molecules reach maturity. Current opinion in immunology. 1996;8(1):51–58. doi: 10.1016/s0952-7915(96)80105-1. [DOI] [PubMed] [Google Scholar]
- 17.Busch R, et al. Accessory molecules for MHC class II peptide loading. Current opinion in immunology. 2000;12(1):99–106. doi: 10.1016/s0952-7915(99)00057-6. [DOI] [PubMed] [Google Scholar]
- 18.Cresswell P. Assembly, transport, and function of MHC class II molecules. Annu Rev Immunol. 1994:12259–293. doi: 10.1146/annurev.iy.12.040194.001355. [DOI] [PubMed] [Google Scholar]
- 19.Ghosh P, et al. The structure of an intermediate in class II MHC maturation: CLIP bound to HLA-DR3. Nature. 1995;378(6556):457–462. doi: 10.1038/378457a0. [DOI] [PubMed] [Google Scholar]
- 20.Busch R, et al. Invariant chain protects class II histocompatibility antigens from binding intact polypeptides in the endoplasmic reticulum. EMBO J. 1996;15(2):418–428. [PMC free article] [PubMed] [Google Scholar]
- 21.Kropshofer H, Vogt AB, Hämmerling GJ. Structural features of the invariant chain fragment CLIP controlling rapid release from HLA-DR molecules and inhibition of peptide binding. Proc Natl Acad Sci U S A. 1995;92(18):8313–8317. doi: 10.1073/pnas.92.18.8313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sloan VS, et al. Mediation by HLA-DM of dissociation of peptides from HLA-DR. Nature. 1995;375(6534):802–806. doi: 10.1038/375802a0. [DOI] [PubMed] [Google Scholar]
- 23.Denzin LK, Cresswell P. HLA-DM induces CLIP dissociation from MHC class II alpha beta dimers and facilitates peptide loading. Cell. 1995;82(1):155–165. doi: 10.1016/0092-8674(95)90061-6. [DOI] [PubMed] [Google Scholar]
- 24.Sherman MA, Weber DA, Jensen PE. DM enhances peptide binding to class II MHC by release of invariant chain-derived peptide. Immunity. 1995;3(2):197–205. doi: 10.1016/1074-7613(95)90089-6. [DOI] [PubMed] [Google Scholar]
- 25.Fremont DH, et al. Crystal structure of mouse H2-M. Immunity. 1998;9(3):385–393. doi: 10.1016/s1074-7613(00)80621-4. [DOI] [PubMed] [Google Scholar]
- 26.Busch R, et al. Secondary structure composition and pH-dependent conformational changes of soluble recombinant HLA-DM. J Biol Chem. 1998;273(42):27557–27564. doi: 10.1074/jbc.273.42.27557. [DOI] [PubMed] [Google Scholar]
- 27.Pashine A, et al. Interaction of HLA-DR with an acidic face of HLA-DM disrupts sequence-dependent interactions with peptides. Immunity. 2003;19(2):183–192. doi: 10.1016/s1074-7613(03)00200-0. [DOI] [PubMed] [Google Scholar]
- 28.Vogt AB, et al. HLA-DM stabilizes empty HLA-DR molecules in a chaperone-like fashion. Immunol Lett. 1997;57(1-3):209–211. doi: 10.1016/s0165-2478(97)00061-8. [DOI] [PubMed] [Google Scholar]
- 29.Denzin LK, Hammond C, Cresswell P. HLA-DM interactions with intermediates in HLA-DR maturation and a role for HLA-DM in stabilizing empty HLA-DR molecules. J Exp Med. 1996;184(6):2153–2165. doi: 10.1084/jem.184.6.2153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hsing LC, Rudensky AY. The lysosomal cysteine proteases in MHC class II antigen presentation. Immunol Rev. 2005:207229–241. doi: 10.1111/j.0105-2896.2005.00310.x. [DOI] [PubMed] [Google Scholar]
- 31.Costantino CM, et al. Lysosomal cysteine and aspartic proteases are heterogeneously expressed and act redundantly to initiate human invariant chain degradation. J Immunol. 2008;180(5):2876–2885. doi: 10.4049/jimmunol.180.5.2876. [DOI] [PubMed] [Google Scholar]
- 32.Reich M, et al. Invariant chain processing is independent of cathepsin variation between primary human B cells/dendritic cells and B-lymphoblastoid cells. Cell Immunol. 2011;269(2):96–103. doi: 10.1016/j.cellimm.2011.03.012. [DOI] [PubMed] [Google Scholar]
- 33.Denzin LK, et al. Negative regulation by HLA-DO of MHC class II-restricted antigen processing. Science. 1997;278(5335):106–109. doi: 10.1126/science.278.5335.106. [DOI] [PubMed] [Google Scholar]
- 34.van Ham SM, et al. HLA-DO is a negative modulator of HLA-DM-mediated MHC class II peptide loading. Curr Biol. 1997;7(12):950–957. doi: 10.1016/s0960-9822(06)00414-3. [DOI] [PubMed] [Google Scholar]
- 35.Kasai M, et al. CLIP-derived self peptides bound to MHC class II molecules of medullary thymic epithelial cells differ from those of cortical thymic epithelial cells in their diversity, length, and C-terminal processing. Eur J Immunol. 2000;30(12):3542–3551. doi: 10.1002/1521-4141(200012)30:12<3542::AID-IMMU3542>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- 36.Santambrogio L, et al. Extracellular antigen processing and presentation by immature dendritic cells. Proc Natl Acad Sci U S A. 1999;96(26):15056–15061. doi: 10.1073/pnas.96.26.15056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Shin JS, et al. Surface expression of MHC class II in dendritic cells is controlled by regulated ubiquitination. Nature. 2006;444(7115):115–118. doi: 10.1038/nature05261. [DOI] [PubMed] [Google Scholar]
- 38.Lightstone L, et al. In the absence of the invariant chain, HLA-DR molecules display a distinct array of peptides which is influenced by the presence or absence of HLA-DM. Proc Natl Acad Sci U S A. 1997;94(11):5772–5777. doi: 10.1073/pnas.94.11.5772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Katz JF, et al. Invariant chain and DM edit self-peptide presentation by major histocompatibility complex (MHC) class II molecules. J Exp Med. 1996;184(5):1747–1753. doi: 10.1084/jem.184.5.1747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Busch R, et al. Achieving stability through editing and chaperoning: regulation of MHC class II peptide binding and expression. Immunol Rev. 2005:207242–260. doi: 10.1111/j.0105-2896.2005.00306.x. [DOI] [PubMed] [Google Scholar]
- 41.Sette A, et al. Binding of major histocompatibility complex class II to the invariant chain-derived peptide, CLIP, is regulated by allelic polymorphism in class II. J Exp Med. 1995;181(2):677–683. doi: 10.1084/jem.181.2.677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Koonce CH, et al. DM loss in k haplotype mice reveals isotype-specific chaperone requirements. J Immunol. 2003;170(7):3751–3761. doi: 10.4049/jimmunol.170.7.3751. [DOI] [PubMed] [Google Scholar]
- 43.Rinderknecht CH, et al. Posttranslational regulation of I-Ed by affinity for CLIP. J Immunol. 2007;179(9):5907–5915. doi: 10.4049/jimmunol.179.9.5907. [DOI] [PubMed] [Google Scholar]
- 44.Rinderknecht CH, et al. I-Ag7 is subject to post-translational chaperoning by CLIP. Int Immunol. 2010;22(8):705–716. doi: 10.1093/intimm/dxq056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Green PH, Cellier C. Celiac disease. N Engl J Med. 2007;357(17):1731–1743. doi: 10.1056/NEJMra071600. [DOI] [PubMed] [Google Scholar]
- 46.Halstensen TS, Brandtzaeg P. Activated T lymphocytes in the celiac lesion: non-proliferative activation (CD25) of CD4+ alpha/beta cells in the lamina propria but proliferation (Ki-67) of alpha/beta and gamma/delta cells in the epithelium. Eur J Immunol. 1993;23(2):505–510. doi: 10.1002/eji.1830230231. [DOI] [PubMed] [Google Scholar]
- 47.Houlston RS, Ford D. Genetics of coeliac disease. QJM. 1996;89(10):737–743. doi: 10.1093/qjmed/89.10.737. [DOI] [PubMed] [Google Scholar]
- 48.Dubois PC, et al. Multiple common variants for celiac disease influencing immune gene expression. Nat Genet. 2010;42(4):295–302. doi: 10.1038/ng.543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sollid LM, et al. Evidence for a primary association of celiac disease to a particular HLA-DQ alpha/beta heterodimer. J Exp Med. 1989;169(1):345–350. doi: 10.1084/jem.169.1.345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Spurkland A, et al. Susceptibility to develop celiac disease is primarily associated with HLA-DQ alleles. Hum Immunol. 1990;29(3):157–165. doi: 10.1016/0198-8859(90)90111-2. [DOI] [PubMed] [Google Scholar]
- 51.Spurkland A, et al. Dermatitis herpetiformis and celiac disease are both primarily associated with the HLA-DQ (alpha 1*0501, beta 1*02) or the HLA-DQ (alpha 1*03, beta 1*0302) heterodimers. Tissue Antigens. 1997;49(1):29–34. doi: 10.1111/j.1399-0039.1997.tb02706.x. [DOI] [PubMed] [Google Scholar]
- 52.Johnson TC, et al. Relationship of HLA-DQ8 and severity of celiac disease: comparison of New York and Parisian cohorts. Clin Gastroenterol Hepatol. 2004;2(10):888–894. doi: 10.1016/s1542-3565(04)00390-8. [DOI] [PubMed] [Google Scholar]
- 53.Harmon GS, Lebeck LK, Weidner N. Gluten-dependent enteropathy and atypical human leukocyte antigen alleles. Hum Pathol. 2011;42(8):1112–1116. doi: 10.1016/j.humpath.2010.10.010. [DOI] [PubMed] [Google Scholar]
- 54.Schirru E, et al. High frequency of low-risk human leukocyte antigen class II genotypes in latent celiac disease. Hum Immunol. 2011;72(2):179–182. doi: 10.1016/j.humimm.2010.11.007. [DOI] [PubMed] [Google Scholar]
- 55.Hausch F, et al. Intestinal digestive resistance of immunodominant gliadin peptides. Am J Physiol Gastrointest Liver Physiol. 2002;283(4):G996–G1003. doi: 10.1152/ajpgi.00136.2002. [DOI] [PubMed] [Google Scholar]
- 56.Shan L, et al. Structural basis for gluten intolerance in celiac sprue. Science. 2002;297(5590):2275–2279. doi: 10.1126/science.1074129. [DOI] [PubMed] [Google Scholar]
- 57.Hue S, et al. A direct role for NKG2D/MICA interaction in villous atrophy during celiac disease. Immunity. 2004;21(3):367–377. doi: 10.1016/j.immuni.2004.06.018. [DOI] [PubMed] [Google Scholar]
- 58.Di Sabatino A, et al. Epithelium derived interleukin 15 regulates intraepithelial lymphocyte Th1 cytokine production, cytotoxicity, and survival in coeliac disease. Gut. 2006;55(4):469–477. doi: 10.1136/gut.2005.068684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Molberg O, et al. Gliadin specific, HLA DQ2-restricted T cells are commonly found in small intestinal biopsies from coeliac disease patients, but not from controls. Scand J Immunol. 1997;46(3):103–109. doi: 10.1046/j.1365-3083.1997.d01-93.x. [DOI] [PubMed] [Google Scholar]
- 60.Gjertsen HA, et al. T cells from the peripheral blood of coeliac disease patients recognize gluten antigens when presented by HLA-DR, -DQ, or -DP molecules. Scand J Immunol. 1994;39(6):567–574. doi: 10.1111/j.1365-3083.1994.tb03414.x. [DOI] [PubMed] [Google Scholar]
- 61.Vader W, et al. The HLA-DQ2 gene dose effect in celiac disease is directly related to the magnitude and breadth of gluten-specific T cell responses. Proc Natl Acad Sci U S A. 2003;100(21):12390–12395. doi: 10.1073/pnas.2135229100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Molberg O, et al. Tissue transglutaminase selectively modifies gliadin peptides that are recognized by gut-derived T cells in celiac disease. Nat Med. 1998;4(6):713–717. doi: 10.1038/nm0698-713. [DOI] [PubMed] [Google Scholar]
- 63.Vader LW, et al. Specificity of tissue transglutaminase explains cereal toxicity in celiac disease. J Exp Med. 2002;195(5):643–649. doi: 10.1084/jem.20012028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Fleckenstein B, et al. Gliadin T cell epitope selection by tissue transglutaminase in celiac disease. Role of enzyme specificity and pH influence on the transamidation versus deamidation process. J Biol Chem. 2002;277(37):34109–34116. doi: 10.1074/jbc.M204521200. [DOI] [PubMed] [Google Scholar]
- 65.van de Wal Y, et al. Selective deamidation by tissue transglutaminase strongly enhances gliadin-specific T cell reactivity. J Immunol. 1998;161(4):1585–1588. [PubMed] [Google Scholar]
- 66.Quarsten H, et al. HLA binding and T cell recognition of a tissue transglutaminase-modified gliadin epitope. Eur J Immunol. 1999;29(8):2506–2514. doi: 10.1002/(SICI)1521-4141(199908)29:08<2506::AID-IMMU2506>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- 67.Arentz-Hansen H, et al. The intestinal T cell response to alpha-gliadin in adult celiac disease is focused on a single deamidated glutamine targeted by tissue transglutaminase. J Exp Med. 2000;191(4):603–612. doi: 10.1084/jem.191.4.603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.van de Wal Y, et al. Glutenin is involved in the gluten-driven mucosal T cell response. Eur J Immunol. 1999;29(10):3133–3139. doi: 10.1002/(SICI)1521-4141(199910)29:10<3133::AID-IMMU3133>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- 69.Stepniak D, et al. Large-scale characterization of natural ligands explains the unique gluten-binding properties of HLA-DQ2. J Immunol. 2008;180(5):3268–3278. doi: 10.4049/jimmunol.180.5.3268. [DOI] [PubMed] [Google Scholar]
- 70.Kim CY, et al. Structural basis for HLA-DQ2-mediated presentation of gluten epitopes in celiac disease. Proc Natl Acad Sci U S A. 2004;101(12):4175–4179. doi: 10.1073/pnas.0306885101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Bergseng E, et al. Main chain hydrogen bond interactions in the binding of proline-rich gluten peptides to the celiac disease-associated HLA-DQ2 molecule. J Biol Chem. 2005;280(23):21791–21796. doi: 10.1074/jbc.M501558200. [DOI] [PubMed] [Google Scholar]
- 72.Tollefsen S, et al. HLA-DQ2 and -DQ8 signatures of gluten T cell epitopes in celiac disease. J Clin Invest. 2006;116(8):2226–2236. doi: 10.1172/JCI27620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Henderson KN, et al. A structural and immunological basis for the role of human leukocyte antigen DQ8 in celiac disease. Immunity. 2007;27(1):23–34. doi: 10.1016/j.immuni.2007.05.015. [DOI] [PubMed] [Google Scholar]
- 74.Margaritte-Jeannin P, et al. HLA-DQ relative risks for coeliac disease in European populations: a study of the European Genetics Cluster on Coeliac Disease. Tissue Antigens. 2004;63(6):562–567. doi: 10.1111/j.0001-2815.2004.00237.x. [DOI] [PubMed] [Google Scholar]
- 75.Qiao SW, et al. Refining the rules of gliadin T cell epitope binding to the disease-associated DQ2 molecule in celiac disease: importance of proline spacing and glutamine deamidation. J Immunol. 2005;175(1):254–261. doi: 10.4049/jimmunol.175.1.254. [DOI] [PubMed] [Google Scholar]
- 76.van de Wal Y, et al. Unique peptide binding characteristics of the disease-associated DQ(alpha 1*0501, beta 1*0201) vs the non-disease-associated DQ(alpha 1*0201, beta 1*0202) molecule. Immunogenetics. 1997;46(6):484–492. doi: 10.1007/s002510050309. [DOI] [PubMed] [Google Scholar]
- 77.Fallang LE, et al. Differences in the risk of celiac disease associated with HLA-DQ2.5 or HLA-DQ2.2 are related to sustained gluten antigen presentation. Nat Immunol. 2009;10(10):1096–1101. doi: 10.1038/ni.1780. [DOI] [PubMed] [Google Scholar]
- 78.Fallang LE, et al. Complexes of two cohorts of CLIP peptides and HLA-DQ2 of the autoimmune DR3-DQ2 haplotype are poor substrates for HLA-DM. J Immunol. 2008;181(8):5451–5461. doi: 10.4049/jimmunol.181.8.5451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Wiesner M, et al. Dominance of an alternative CLIP sequence in the celiac disease associated HLA-DQ2 molecule. Immunogenetics. 2008;60(9):551–555. doi: 10.1007/s00251-008-0310-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Hou T, et al. An insertion mutant in DQA1*0501 restores susceptibility to HLA-DM: implications for disease associations. J Immunol. 2011;187(5):2442–2452. doi: 10.4049/jimmunol.1100255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Miyazaki T, et al. Mice lacking H2-M complexes, enigmatic elements of the MHC class II peptide-loading pathway. Cell. 1996;84(4):531–541. doi: 10.1016/s0092-8674(00)81029-6. [DOI] [PubMed] [Google Scholar]
- 82.Falk K, et al. Ligand exchange of major histocompatibility complex class II proteins is triggered by H-bond donor groups of small molecules. J Biol Chem. 2002;277(4):2709–2715. doi: 10.1074/jbc.M109098200. [DOI] [PubMed] [Google Scholar]
- 83.Marietta EV, David CS, Murray JA. Important lessons derived from animal models of celiac disease. Int Rev Immunol. 2011;30(4):197–206. doi: 10.3109/08830185.2011.598978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Daneman D. Type 1 diabetes. Lancet. 2006:367847–858. doi: 10.1016/S0140-6736(06)68341-4. [DOI] [PubMed] [Google Scholar]
- 85.Davies JL, et al. A genome-wide search for human type 1 diabetes susceptibility genes. Nature. 1994;371(6493):130–136. doi: 10.1038/371130a0. [DOI] [PubMed] [Google Scholar]
- 86.Tisch R, McDevitt H. Insulin-dependent diabetes mellitus. Cell. 1996;85(3):291–297. doi: 10.1016/s0092-8674(00)81106-x. [DOI] [PubMed] [Google Scholar]
- 87.Todd JA, Bell JI, McDevitt HO. HLA-DQ beta gene contributes to susceptibility and resistance to insulin-dependent diabetes mellitus. Nature. 1987;329(6140):599–604. doi: 10.1038/329599a0. [DOI] [PubMed] [Google Scholar]
- 88.Wucherpfennig KW. MHC-linked susceptibility to type 1 diabetes: a structural perspective. Ann N Y Acad Sci. 2003:1005119–127. doi: 10.1196/annals.1288.013. [DOI] [PubMed] [Google Scholar]
- 89.Khalil I, et al. A combination of HLA-DQ beta Asp57-negative and HLA DQ alpha Arg52 confers susceptibility to insulin-dependent diabetes mellitus. J Clin Invest. 1990;85(4):1315–1319. doi: 10.1172/JCI114569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Cucca F, et al. A correlation between the relative predisposition of MHC class II alleles to type 1 diabetes and the structure of their proteins. Human molecular genetics. 2001;10(19):2025–2037. doi: 10.1093/hmg/10.19.2025. [DOI] [PubMed] [Google Scholar]
- 91.Polychronakos C, Li Q. Understanding type 1 diabetes through genetics: advances and prospects. Nat Rev Genet. 2011;12(11):781–792. doi: 10.1038/nrg3069. [DOI] [PubMed] [Google Scholar]
- 92.Barrett JC, et al. Genome-wide association study and meta-analysis find that over 40 loci affect risk of type 1 diabetes. Nat Genet. 2009;41(6):703–707. doi: 10.1038/ng.381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Nichols J, Cooke A. Overcoming self-destruction in the pancreas. Curr Opin Biotechnol. 2009;20(5):511–515. doi: 10.1016/j.copbio.2009.09.009. [DOI] [PubMed] [Google Scholar]
- 94.Mallone R, Brezar V, Boitard C. T cell recognition of autoantigens in human type 1 diabetes: clinical perspectives. Clin Dev Immunol. 2011;2011:513210. doi: 10.1155/2011/513210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Makino S, et al. Breeding of a non-obese, diabetic strain of mice. Jikken Dobutsu. 1980;29(1):1–13. doi: 10.1538/expanim1978.29.1_1. [DOI] [PubMed] [Google Scholar]
- 96.Makino S, et al. Absence of insulitis and overt diabetes in athymic nude mice with NOD genetic background. Jikken Dobutsu. 1986;35(4):495–498. doi: 10.1538/expanim1978.35.4_495. [DOI] [PubMed] [Google Scholar]
- 97.Matsumoto M, et al. Transfer of autoimmune diabetes from diabetic NOD mice to NOD athymic nude mice: the roles of T cell subsets in the pathogenesis. Cell Immunol. 1993;148(1):189–197. doi: 10.1006/cimm.1993.1101. [DOI] [PubMed] [Google Scholar]
- 98.Acha-Orbea H, McDevitt HO. The first external domain of the nonobese diabetic mouse class II I-A beta chain is unique. Proc Natl Acad Sci U S A. 1987;84(8):2435–2439. doi: 10.1073/pnas.84.8.2435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Boitard C, et al. Prevention of diabetes in nonobese diabetic mice by anti-I-A monoclonal antibodies: transfer of protection by splenic T cells. Proc Natl Acad Sci U S A. 1988;85(24):9719–9723. doi: 10.1073/pnas.85.24.9719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Quartey-Papafio R, et al. Aspartate at position 57 of nonobese diabetic I-Ag7 beta-chain diminishes the spontaneous incidence of insulin-dependent diabetes mellitus. J Immunol. 1995;154(10):5567–5575. [PubMed] [Google Scholar]
- 101.Lund T, et al. Prevention of insulin-dependent diabetes mellitus in non-obese diabetic mice by transgenes encoding modified I-A beta-chain or normal I-E alpha-chain. Nature. 1990;345(6277):727–729. doi: 10.1038/345727a0. [DOI] [PubMed] [Google Scholar]
- 102.Hattori M, et al. The NOD mouse: recessive diabetogenic gene in the major histocompatibility complex. Science. 1986;231(4739):733–735. doi: 10.1126/science.3003909. [DOI] [PubMed] [Google Scholar]
- 103.Pelegri C, et al. Islet endocrine-cell behavior from birth onward in mice with the nonobese diabetic genetic background. Mol Med. 2001;7(5):311–319. [PMC free article] [PubMed] [Google Scholar]
- 104.Strid J, et al. A defect in bone marrow derived dendritic cell maturation in the nonobese diabetic mouse. Clin Exp Immunol. 2001;123(3):375–381. doi: 10.1046/j.1365-2249.2001.01473.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Durant S, et al. Nonobese diabetic (NOD) mouse dendritic cells stimulate insulin secretion by prediabetic islets. Autoimmunity. 2002;35(7):449–455. doi: 10.1080/0891693021000040575. [DOI] [PubMed] [Google Scholar]
- 106.Rolf J, et al. The enlarged population of marginal zone/CD1d(high) B lymphocytes in nonobese diabetic mice maps to diabetes susceptibility region Idd11. J Immunol. 2005;174(8):4821–4827. doi: 10.4049/jimmunol.174.8.4821. [DOI] [PubMed] [Google Scholar]
- 107.Marino E, et al. Marginal-zone B-cells of nonobese diabetic mice expand with diabetes onset, invade the pancreatic lymph nodes, and present autoantigen to diabetogenic T-cells. Diabetes. 2008;57(2):395–404. doi: 10.2337/db07-0589. [DOI] [PubMed] [Google Scholar]
- 108.Serreze DV, et al. B lymphocytes are essential for the initiation of T cell-mediated autoimmune diabetes: analysis of a new “speed congenic” stock of NOD.Ig mu null mice. J Exp Med. 1996;184(5):2049–2053. doi: 10.1084/jem.184.5.2049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Mohan JF, et al. Unique autoreactive T cells recognize insulin peptides generated within the islets of Langerhans in autoimmune diabetes. Nat Immunol. 2010;11(4):350–354. doi: 10.1038/ni.1850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Baker FJ, et al. Restricted islet-cell reactive T cell repertoire of early pancreatic islet infiltrates in NOD mice. Proc Natl Acad Sci U S A. 2002;99(14):9374–9379. doi: 10.1073/pnas.142284899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Gelber C, et al. Isolation of nonobese diabetic mouse T-cells that recognize novel autoantigens involved in the early events of diabetes. Diabetes. 1994;43(1):33–39. doi: 10.2337/diab.43.1.33. [DOI] [PubMed] [Google Scholar]
- 112.Mannering SI, et al. The insulin A-chain epitope recognized by human T cells is posttranslationally modified. J Exp Med. 2005;202(9):1191–1197. doi: 10.1084/jem.20051251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Mohan JF, Petzold SJ, Unanue ER. Register shifting of an insulin peptide-MHC complex allows diabetogenic T cells to escape thymic deletion. J Exp Med. 2011;208(12):2375–2383. doi: 10.1084/jem.20111502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Suri A, Levisetti MG, Unanue ER. Do the peptide-binding properties of diabetogenic class II molecules explain autoreactivity? Curr Opin Immunol. 2008;20(1):105–110. doi: 10.1016/j.coi.2007.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Stadinski BD, et al. Chromogranin A is an autoantigen in type 1 diabetes. Nat Immunol. 2010;11(3):225–231. doi: 10.1038/ni.1844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Fairchild PJ, et al. An autoantigenic T cell epitope forms unstable complexes with class II MHC: a novel route for escape from tolerance induction. Int Immunol. 1993;5(9):1151–1158. doi: 10.1093/intimm/5.9.1151. [DOI] [PubMed] [Google Scholar]
- 117.Sant AJ, et al. The relationship between immunodominance, DM editing, and the kinetic stability of MHC class II:peptide complexes. Immunol Rev. 2005:207261–278. doi: 10.1111/j.0105-2896.2005.00307.x. [DOI] [PubMed] [Google Scholar]
- 118.Ridgway WM, et al. Analysis of the role of variation of major histocompatibility complex class II expression on nonobese diabetic (NOD) peripheral T cell response. J Exp Med. 1998;188(12):2267–2275. doi: 10.1084/jem.188.12.2267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Kanagawa O, et al. Autoreactivity of T cells from nonobese diabetic mice: an I-Ag7-dependent reaction. Proc Natl Acad Sci U S A. 1998;95(4):1721–1724. doi: 10.1073/pnas.95.4.1721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Mangialaio S, et al. The arthritogenic T cell receptor and its ligand in a model of spontaneous arthritis. Arthritis Rheum. 1999;42(12):2517–2523. doi: 10.1002/1529-0131(199912)42:12<2517::AID-ANR3>3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]
- 121.Mingueneau M, et al. Thymic negative selection is functional in NOD mice. J Exp Med. 2012;209(3):623–637. doi: 10.1084/jem.20112593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Mellanby RJ, et al. Both central and peripheral tolerance mechanisms play roles in diabetes prevention in NOD-E transgenic mice. Autoimmunity. 2008;41(5):383–394. doi: 10.1080/08916930801991021. [DOI] [PubMed] [Google Scholar]
- 123.Stratmann T, et al. The I-Ag7 MHC class II molecule linked to murine diabetes is a promiscuous peptide binder. J Immunol. 2000;165(6):3214–3225. doi: 10.4049/jimmunol.165.6.3214. [DOI] [PubMed] [Google Scholar]
- 124.Corper AL, et al. A structural framework for deciphering the link between I-Ag7 and autoimmune diabetes. Science. 2000;288(5465):505–511. doi: 10.1126/science.288.5465.505. [DOI] [PubMed] [Google Scholar]
- 125.Carrasco-Marin E, et al. The class II MHC I-Ag7 molecules from non-obese diabetic mice are poor peptide binders. J Immunol. 1996;156(2):450–458. [PubMed] [Google Scholar]
- 126.Reich EP, et al. Self peptides isolated from MHC glycoproteins of non-obese diabetic mice. J Immunol. 1994;152(5):2279–2288. [PubMed] [Google Scholar]
- 127.Suri A, et al. Natural peptides selected by diabetogenic DQ8 and murine I-A(g7) molecules show common sequence specificity. J Clin Invest. 2005;115(8):2268–2276. doi: 10.1172/JCI25350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Suri A, et al. Specificity of peptide selection by antigen-presenting cells homozygous or heterozygous for expression of class II MHC molecules: The lack of competition. Proc Natl Acad Sci U S A. 2003;100(9):5330–5335. doi: 10.1073/pnas.0330859100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Suri A, et al. First signature of islet beta-cell-derived naturally processed peptides selected by diabetogenic class II MHC molecules. J Immunol. 2008;180(6):3849–3856. doi: 10.4049/jimmunol.180.6.3849. [DOI] [PubMed] [Google Scholar]
- 130.Reizis B, et al. Molecular characterization of the diabetes-associated mouse MHC class II protein, I-Ag7. Int Immunol. 1997;9(1):43–51. doi: 10.1093/intimm/9.1.43. [DOI] [PubMed] [Google Scholar]
- 131.Nalefski EA, Shaw KT, Rao A. An ion pair in class II major histocompatibility complex heterodimers critical for surface expression and peptide presentation. J Biol Chem. 1995;270(38):22351–22360. doi: 10.1074/jbc.270.38.22351. [DOI] [PubMed] [Google Scholar]
- 132.Hausmann DH, et al. pH-dependent peptide binding properties of the type I diabetes-associated I-Ag7 molecule: rapid release of CLIP at an endosomal pH. J Exp Med. 1999;189(11):1723–1734. doi: 10.1084/jem.189.11.1723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Peterson M, Sant AJ. The inability of the nonobese diabetic class II molecule to form stable peptide complexes does not reflect a failure to interact productively with DM. J Immunol. 1998;161(6):2961–2967. [PubMed] [Google Scholar]
- 134.Bhatnagar A, et al. Nonobese diabetic mice display elevated levels of class II-associated invariant chain peptide associated with I-Ag7 on the cell surface. J Immunol. 2001;166(7):4490–4497. doi: 10.4049/jimmunol.166.7.4490. [DOI] [PubMed] [Google Scholar]
- 135.Silva DG, et al. Elevated lymphocyte expression of CLIP is associated with type 1 diabetes and may be a useful marker of autoimmune susceptibility. Ann N Y Acad Sci. 2004:103765–68. doi: 10.1196/annals.1337.009. [DOI] [PubMed] [Google Scholar]
- 136.Yan G, et al. Impaired processing and presentation by MHC class II proteins in human diabetic cells. J Immunol. 2003;170(1):620–627. doi: 10.4049/jimmunol.170.1.620. [DOI] [PubMed] [Google Scholar]
- 137.Lich JD, et al. Editing of an immunodominant epitope of glutamate decarboxylase by HLA-DM. J Immunol. 2003;171(2):853–859. doi: 10.4049/jimmunol.171.2.853. [DOI] [PubMed] [Google Scholar]
- 138.Arneson LS, Peterson M, Sant AJ. The MHC class II molecule I-Ag7 exists in alternate conformations that are peptide dependent. J Immunol. 2000;165(4):2059–2067. doi: 10.4049/jimmunol.165.4.2059. [DOI] [PubMed] [Google Scholar]
- 139.Marleau AM, Summers KL, Singh B. Differential contributions of APC subsets to T cell activation in nonobese diabetic mice. J Immunol. 2008;180(8):5235–5249. doi: 10.4049/jimmunol.180.8.5235. [DOI] [PubMed] [Google Scholar]
- 140.Morris P, et al. An essential role for HLA-DM in antigen presentation by class II major histocompatibility molecules. Nature. 1994;368(6471):551–554. doi: 10.1038/368551a0. [DOI] [PubMed] [Google Scholar]
- 141.Germain RN, Rinker AG., Jr Peptide binding inhibits protein aggregation of invariant-chain free class II dimers and promotes surface expression of occupied molecules. Nature. 1993;363(6431):725–728. doi: 10.1038/363725a0. [DOI] [PubMed] [Google Scholar]
- 142.Ettinger RA, et al. Exceptional stability of the HLA-DQA1*0102/DQB1*0602 alpha beta protein dimer, the class II MHC molecule associated with protection from insulin-dependent diabetes mellitus. J Immunol. 1998;161(11):6439–6445. [PubMed] [Google Scholar]
- 143.Mellanby RJ, et al. Loss of invariant chain protects nonobese diabetic mice against type 1 diabetes. J Immunol. 2006;177(11):7588–7598. doi: 10.4049/jimmunol.177.11.7588. [DOI] [PubMed] [Google Scholar]
- 144.Koonce CH, Bikoff EK. Dissecting MHC class II export, B cell maturation, and DM stability defects in invariant chain mutant mice. J Immunol. 2004;173(5):3271–3280. doi: 10.4049/jimmunol.173.5.3271. [DOI] [PubMed] [Google Scholar]
- 145.Yi W, et al. Targeted regulation of self-peptide presentation prevents type I diabetes in mice without disrupting general immunocompetence. J Clin Invest. 2010;120(4):1324–1336. doi: 10.1172/JCI40220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Yoshida K, et al. The diabetogenic mouse MHC class II molecule I-Ag7 is endowed with a switch that modulates TCR affinity. J Clin Invest. 2010;120(5):1578–1590. doi: 10.1172/JCI41502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Klareskog L, et al. Genes, environment and immunity in the development of rheumatoid arthritis. Curr Opin Immunol. 2006;18(6):650–655. doi: 10.1016/j.coi.2006.06.004. [DOI] [PubMed] [Google Scholar]
- 148.Lee HS, et al. Several regions in the major histocompatibility complex confer risk for anti-CCP-antibody positive rheumatoid arthritis, independent of the DRB1 locus. Mol Med. 2008;14(5-6):293–300. doi: 10.2119/2007-00123.Lee. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Louis-Plence P, et al. The down-regulation of HLA-DM gene expression in rheumatoid arthritis is not related to their promoter polymorphism. J Immunol. 2000;165(9):4861–4869. doi: 10.4049/jimmunol.165.9.4861. [DOI] [PubMed] [Google Scholar]
- 150.Toussirot E, et al. The association of HLA-DM with rheumatoid arthritis in eastern France. Hum Immunol. 2000:61303–308. doi: 10.1016/s0198-8859(99)00126-3. [DOI] [PubMed] [Google Scholar]
- 151.De Riva A, et al. Measurement of protein synthesis using heavy water labeling and peptide mass spectrometry: Discrimination between major histocompatibility complex allotypes. Anal Biochem. 2010;403(1-2):1–12. doi: 10.1016/j.ab.2010.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Busch R, et al. Measurement of protein turnover rates by heavy water labeling of nonessential amino acids. Biochim Biophys Acta. 2006;1760(5):730–744. doi: 10.1016/j.bbagen.2005.12.023. [DOI] [PubMed] [Google Scholar]
- 153.Busch R, et al. Measurement of cell proliferation by heavy water labeling. Nat Protoc. 2007;2(12):3045–3057. doi: 10.1038/nprot.2007.420. [DOI] [PubMed] [Google Scholar]
- 154.Schrodinger L. The PyMOL Molecular Graphics System, Version 1.3r1 2010 [Google Scholar]
- 155.Baker NA, et al. Electrostatics of nanosystems: application to microtubules and the ribosome. Proc Natl Acad Sci U S A. 2001;98(18):10037–10041. doi: 10.1073/pnas.181342398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Dolinsky TJ, et al. PDB2PQR: expanding and upgrading automated preparation of biomolecular structures for molecular simulations. Nucleic Acids Res. 2007;35(Web Server issue):W522–525. doi: 10.1093/nar/gkm276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Dolinsky TJ, et al. PDB2PQR: an automated pipeline for the setup of Poisson-Boltzmann electrostatics calculations. Nucleic Acids Res. 2004;32(Web Server issue):W665–667. doi: 10.1093/nar/gkh381. [DOI] [PMC free article] [PubMed] [Google Scholar]