

Abstract
When applied extracellularly, myo-inositol hexakisphosphate (InsP6) and myo-inositol pentakisphosphate (InsP5) can inhibit the growth and proliferation of tumour cells. There is debate about whether these effects result from interactions of InsP6 and InsP5 with intracellular or extracellular targets. We synthesised FAM-InsP5, a fluorescent conjugate of InsP5 that allows direct visualisation of its interaction with cells. FAM-InsP5 was internalised by H1229 tumour cells, a finding that supports earlier reports that externally applied inositol phosphates can—perhaps surprisingly—enter into cells. Close examination of the process of FAM-InsP5 uptake suggests a mechanism of non-receptor-mediated endocytosis, which is blocked at 4 °C and probably involves interaction of the ligand with the glycocalyx. However, our results are difficult to reconcile with antiproliferative mechanisms that require direct interactions of externally applied InsP5 or InsP6 with cytosolic proteins, because internalised FAM-InsP5 appears in lysosomes and apparently does not enter the cytoplasm. Studies using FAM-InsP5 are less difficult and time-consuming than experiments using InsP5 or InsP6, a factor that allowed us to analyse cellular uptake across a range of human cell types, identifying strong cell-specific differences.
Keywords: cyclitols, fluorescent probes, inositol, natural products, phytic acid
Introduction
myo-Inositol hexakisphosphate (InsP6, 1, Scheme 1) is the most abundant inositol phosphate in mammalian cells.[1] Despite the widespread occurrence of InsP6 in plants,[2] current evidence supports the idea that animal cells synthesise all their own InsP6 from Ins(1,4,5)P3[3] rather than acquiring it from dietary sources. No significant amounts of InsP6 could be detected in analyses of human serum, platelet-free plasma and urine.[3] Several studies (reviewed in ref. [4]) have demonstrated that high concentrations (≥100 μm) of InsP6 applied extracellularly inhibit proliferation of tumour cells. myo-Inositol 1,3,4,5,6-pentakisphosphate (InsP5, 2) has also been reported to have similar antiproliferative effects and to be active at lower concentrations (<50 μm).[5]
Scheme 1.
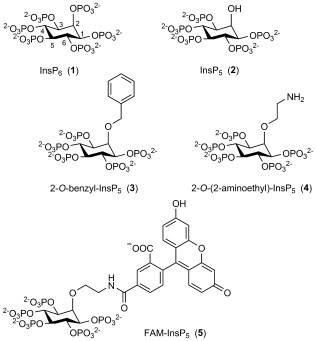
Structures of myo-inositol hexakisphosphate (InsP6, 1), myo-inositol 1,3,4,5,6-pentakisphosphate (InsP5, 2) and InsP5 analogues 3, 4 and 5.
The mechanisms underlying the effects of applying InsP6 or InsP5 to cells (reviewed in ref. [4]) are not clear. Some workers have postulated that the antiproliferative effects of InsP6 (and, by implication, InsP5) originate in extracellular interactions, such as chelation of metal cations or binding to cell-surface receptors or growth factors. Others have proposed that direct interactions of InsP6, InsP5 or their metabolites with intracellular proteins involved in the control of proliferation and apoptosis might be involved. This would require externally applied inositol phosphates to be able to enter cells and gain access to these proteins in the cytosol, something that many consider unlikely for such highly charged polyanionic molecules. Nevertheless, there have been reports that extracellularly added radiolabelled InsP6[6] and InsP5[7] can be taken up and metabolised by tumour cells. A biphasic effect of externally applied InsP6 on lung tumour (H1299) cells was recently reported:[8] low concentrations of InsP6 (≤50 μm) stimulate cell growth whereas high concentrations (≥100 μm) inhibit growth. In these experiments, uptake and metabolism of InsP6 in H1299 cells was confirmed by use both of radiolabelled InsP6 and of unlabelled InsP6 with analysis by metal dye detection (MDD) HPLC. It was shown that, prior to and after cellular uptake, InsP6 was dephosphorylated to inositol by multiple inositol polyphosphate phosphatase 1 (MINPP1). The authors propose that MINPP1 protects cells from InsP6-mediated depletion of cations from the medium and that dephosphorylation of internalised InsP6 provides an additional source of micronutrients.
The idea that animal cells might be able to take up inositol phosphates such as InsP5 and InsP6 from their environment is still not widely accepted, and methods of studying uptake with radiolabelled InsPn and/or MDD-HPLC can be difficult and time-consuming. Inositol phosphates readily associate with various surfaces, including culture dishes and cell surfaces,[8] for example, and great care to distinguish cell-surface binding from true internalisation must therefore be taken. We thus reasoned that a suitable fluorescent derivative of InsP6 or InsP5 might be used to visualise its internalisation by cells directly in real time by confocal microscopy. Furthermore, by using a fluorescent InsPn it should be straightforward to track the distribution of the internalised conjugate and to measure rates and extents of internalisation and distribution.
Here we report the design, synthesis and use of a fluorescent conjugate of InsP5 that allows visualisation of its uptake by cells. We describe the time course of internalisation of the fluorescent InsP5 conjugate by H1299 cells and its intracellular distribution. We then go on to compare the efficiency of internalisation of fluorescent InsP5 across a range of cell types.
Results and Discussion
Design of FAM-InsP5, a fluorescent InsP5 conjugate
Previously, we synthesised 2-O-benzyl-Ins(1,3,4,5,6)P5 (2-O-Bn-InsP5, 3, Scheme 1), in which a hydrophobic benzyl group is attached to the axial 2-O atom of the myo-inositol ring. Compound 3 was found to have more potent antiproliferative effects than InsP5 itself in sensitive cells, and was also active against cell lines resistant to InsP5.[9] In the present work, we reasoned that synthetic conjugates of InsP5 based on the structure of 3 might be used to probe the molecular mechanisms underlying the effects of InsP5, InsP6 and 3 on cells. We therefore identified 2-O-(2-aminoethyl)-InsP5 (4) as a suitable starting point for the construction of such conjugates.
In 4, the benzyl group of 3 is replaced with a short (two-carbon) chain to a reactive primary amine group, allowing conjugation with a range of fluorophores and other amine-reactive probes. The robust ether linkage directly to the axial 2-O atom of the myo-inositol ring, rather than through a phosphate group, was chosen to maximise the stability of conjugates. Moreover, because conjugates of 4 can retain the meso symmetry of InsP5 and InsP6, the problem of enantiomeric pairs is avoided and synthesis is thereby simplified. The linker is kept as short as possible to limit the size of conjugates and to minimise the number of rotatable bonds. The latter consideration is important if fluorescent conjugates are to be used for fluorescence polarisation (FP) studies of binding to proteins.[10] We chose 5-carboxyfluorescein as the fluorophore for conjugation with 4 to give FAM-InsP5 (5) because it is bright, inexpensive and available in single-isomer form and gives a stable amide linkage in conjugates with amines.
Synthesis of FAM-InsP5
We synthesised 4 (Scheme 2) from the known diol 6, which is available directly from myo-inositol in multi-gram quantities.[11, 12] The required 2-aminoethyl linker was installed through introduction of a cyanomethyl ether[13] at the axial 2-O atom of the myo-inositol ring. Although regioselective 2-O-alkylation of 6 is possible,[12] we could not achieve satisfactory results by direct cyanomethylenation of 6 due to insolubility of this diol in the required solvent (acetonitrile). We therefore chose to protect the equatorial 5-OH group of 6 temporarily as a benzoate ester, by regioselective acylation with benzoic anhydride. Alkylation of 7 with sodium hydride and bromoacetonitrile in acetonitrile then proceeded smoothly to give the 2-O-cyanomethyl ether 8. Earlier attempts at 2-O-alkylation using the 5-O-acetate ester of 6 gave a mixture of mono- and dialkylated material due to partial loss of the acetate group, but the 5-O-benzoate ester was unaffected under the same conditions. Treatment of 8 with LiAlH4 simultaneously removed the benzoate ester and gave the 2-O-(2-aminoethyl) ether, which was temporarily protected in situ by treatment with ethyl trifluoroacetate to give alcohol 9. The butane-2,3-diacetal (BDA) protecting groups were now cleanly removed by use of aqueous TFA, and phosphitylation of the pentaol product, followed by oxidation of phosphites, then gave fully protected 10. After the benzyl protecting groups on the phosphates had been removed by catalytic hydrogenolysis, the N-trifluoroacetyl protecting group was cleaved by alkaline hydrolysis with either triethylamine (TEA) or N,N-diisopropylethylamine (DIPEA) in water at 60 °C, giving the TEA or DIPEA salts of 4, respectively.
Scheme 2.
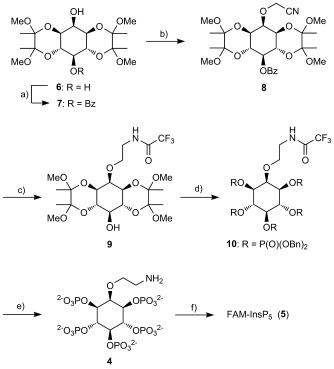
Synthesis of FAM-InsP5 (5). a) Benzoic anhydride, pyridine, DMAP, 0 °C to RT, 84 %; b) NaH, BrCH2CN, CH3CN, −30 °C to RT, 85 %; c) i: LiAlH4, THF, 0 °C; ii: ethyl trifluoroacetate, THF, 66 %; d) i: TFA, CH2Cl2, H2O; ii: (BnO)2PNPri2, 5-phenyltetrazole, CH2Cl2; iii: mCPBA, CH2Cl2, −78 °C to RT, 88 %; e) i: Pd(OH)2/C, MeOH, H2O, H2, 1 atm; ii: DIPEA, H2O, 60 °C; quantitative; f) i: 5-carboxyfluorescein succinimidyl ester, propan-2-ol, DIPEA, 60 °C; ii: Q Sepharose Fast Flow, eluting with aqueous 0 to 2.0 m TEAB; iii: LichroPrep RP-18, eluting with 0 to 30 % CH3CN in aqueous 0.05 m TEAB, 79 %. Bn, benzyl; Bz, benzoyl; TEAB, triethylammonium bicarbonate.
Attempts to achieve reaction between 4 and the succinimidyl esters (SEs) or pentafluorophenyl esters of 5-carboxyfluorescein[14] in aqueous buffers or methanol were unsuccessful, giving little or no product as judged by analytical reversed-phase HPLC. Clearly, the reactivity of the amine group in 4 is reduced by the nearby phosphate groups, and competing hydrolysis of the reactive ester of 5-carboxyfluorescein occurs more rapidly than the conjugation reaction. When anhydrous DMSO was used as the solvent, the target conjugate 5 was slowly formed, but 31P NMR spectroscopy and analytical RP-HPLC showed that small amounts of cyclic pyrophosphate esters of InsP5 also accumulated. The low solubility of the triethylammonium salt of 4 in other organic solvents precluded their use, but the DIPEA salt had good solubility in several organic solvents, including dichloromethane, acetonitrile and propan-2-ol. Conjugation reactions with the DIPEA salt of 4 in all three of these solvents were successful although slow, taking several days to complete. Eventually, it was found that propan-2-ol at elevated temperatures (60 °C) and 5-carboxyfluorescein SE gave the best results. The reaction was complete within 24 h, with no sign of pyrophosphate formation, and under these conditions there was little reaction between propan-2-ol and the SE.
For the intended experiments with cells, isolation of pure 5 was essential. Purification of the crude reaction product by anion-exchange chromatography on Q Sepharose Fast Flow resin, with elution with a gradient of aqueous triethylammonium bicarbonate (TEAB), removed excess dye and traces of unreacted 4. High concentrations of TEAB (2.0 m) were necessary to elute 5 because of the five phosphate groups and because fluorescein is itself strongly retained on Q Sepharose. This procedure gave a product that was pure by 31P NMR spectroscopy and RP HPLC, but 1H and 13C NMR spectroscopy showed signals corresponding to cationic debris from the Q Sepharose column, which we have often observed when high concentrations of TEAB are used. A second purification step by ion-pair chromatography on Lichroprep RP-18 was effective in removing these contaminants, giving pure 5 as the triethylammonium salt.
FAM-InsP5 is internalised by H1299 cells
In a recent study,[8] some of us demonstrated that H1299 cells are able to take up InsP6. In order to show whether 5 is taken up, H1299 cells were incubated with 5 overnight. As a control, cells were also incubated with fluorescein. Uptake of 5 or fluorescein was analysed by performing z-stacks using light and fluorescence micrographs. Detection of fluorescence in the middle cell layers was defined as cellular uptake (see Figure S1 in the Supporting Information for details of the analysis). We did not detect any fluorescence signals in fluorescein-treated cells (data not shown), but cells treated with FAM-InsP5 show an accumulation of small fluorescent dots inside the cells. Figure 1 A shows focus stackings of bright and fluorescence light overlays at high magnification. The fluorescent dots inside the cells represent large aggregates of 5, and it could be seen that, after internalisation, the cell had accumulated five to fifty of these aggregates. Interestingly, the ability of H1299 cells to internalise 5 seems to depend on cellular confluence, because the ability of cells grown to low and middle (20 to 50 %) confluence was about four times higher than that of cells grown to high (80 %) confluence (Figures 1 B and S2).
Figure 1.
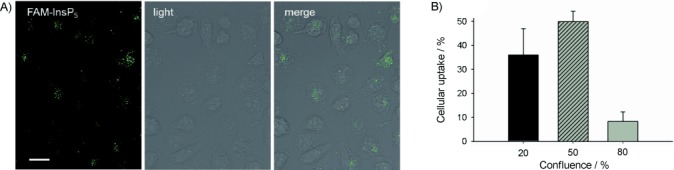
Uptake of FAM-InsP5 (5) in H1299 cells. A) H1299 cells were grown to 50 % confluence and treated with 5 (20 μm). After 16 h, the cells were washed, fixed with paraformaldehyde and embedded in Fluoromount-G medium. Shown are focus stackings (deep focus fusion) of bright and fluorescence light overlays of one representative micrograph at 600-fold magnification. Scale bar: 10 μm. B) H1299 cells were grown to 20, 50 or 80 % confluence, treated with 5 (20 μm) for 16 h and fixed and embedded as described above. XZ-stacks of bright and fluorescence light micrographs were performed (see Figures S1 and S2) and used to analyse internalisation of 5, and the percentage of cells that had internalised 5 was calculated.
Time and temperature dependence of FAM-InsP5 uptake
To establish whether uptake of 5 into H1299 cells is specific or nonspecific, the time dependence of uptake was analysed. Specific receptor-mediated uptake of extracellular substances is very fast (over a timescale of about 1 to 2 min) and does not further increase with time, whereas uptake by nonspecific endocytosis is much slower.[15] Using z-stack analyses, we found that, after two minutes of incubation, aggregates of 5 were only detected at the cell surface (Figure S3), and after ten minutes the first aggregates were visible inside the cell body. From this time point on, the number of cells that had internalised 5 continuously increased and was highest after 16 h of incubation (Figures 2 A and S3). Because uptake of 5 first started between 2 and 10 min and increased over time, we conclude that 5 is endocytosed by a nonspecific mechanism.
Figure 2.
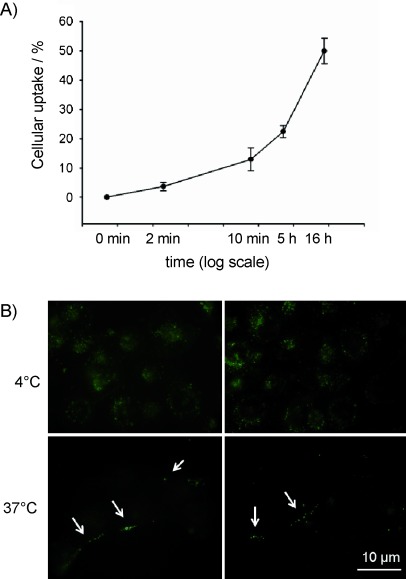
FAM-InsP5 (5) is internalised by endocytosis. A) The same experiment as described in (Figure 1 A) was performed with cells that were grown to 50 % confluence, and internalisation of 5 was analysed (see Figure S3) after the time points indicated in the figure. Values shown are mean±SEM. B) H1299 cells grown to 50 % confluence were incubated with 5 for 50 min at 4 °C (upper panels) or 37 °C (lower panels). Uptake of 5 was analysed as described in Figure 1. At 4 °C, endocytosis is inhibited, and z-stack analysis (not shown) revealed that under these conditions no internalisation but only cell surface-localisation was visible (upper panel). In control cells incubated at 37 °C, z-stack analysis revealed that around 10 % of the cells had internalised 5. Representative samples of these cells are shown in the second row, with small aggregates of internalised 5 marked by arrows.
In order to verify that 5 is taken up by endocytosis, H1299 cells were incubated with 5 for one hour either at 37 °C or at 4 °C, a temperature at which endocytosis is blocked. When cells were incubated at 4 °C, large aggregates of 5 were visible at the cell surface (Figure 2 B) but we did not detect any cells that had internalised 5. However, for cells incubated at 37 °C, z-stack analysis revealed that about 10 % of the cells had internalised 5, which in the xy-orientation is seen as small aggregates (Figure 2 B, marked by arrows). These results strongly indicate that 5 is taken up by endocytosis.
Localisation of internalised FAM-InsP5
Next, we examined the distribution of internalised 5 in H1229 cells by using indirect double immunofluorescence. Cells preincubated with 5 for 20 h were treated with antibodies against EEA1 (early endosome antigen 1) and LAMP-2 (lysosome-associated membrane protein-2), which are marker proteins for endosomes and lysosomes, respectively. We detected colocalisation of 5 with LAMP-2 (lysosomes) but not with EEA1 (early endosomes; Figure 3 A). These results are consistent because, after long incubation times, 5-containing early endosomes should have become fused with, or matured to become, lysosomes. Our finding that 5 accumulated in lysosomes shows that its fate after cellular uptake is similar to that of InsP6, which also resides in lysosomes after internalisation.[8]
Figure 3.
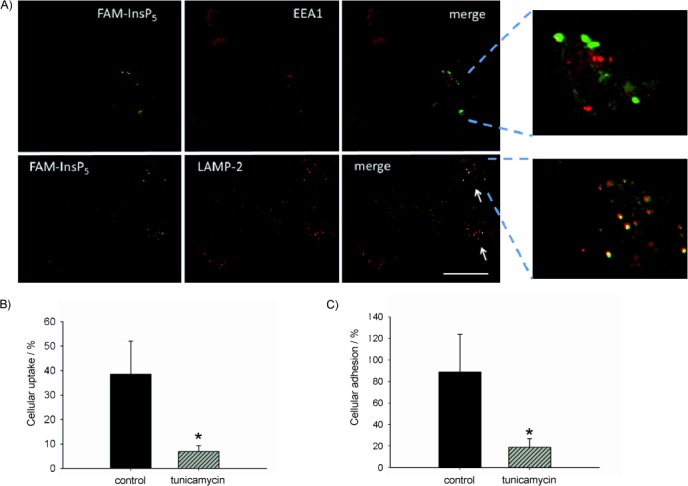
Uptake of FAM-InsP5 (5) into H1299 cells. A) H1299 cells were grown to 50 % confluence and treated with 5 (20 μm). After 16 h the cells were washed, fixed with paraformaldehyde and treated with primary antibodies against marker proteins for early endosomes (EEA1) and LAMP-2 (lysosomes), which were then detected with the aid of secondary antibodies labelled with Alexa Fluor 568. The yellow colour in the merge micrographs indicates colocalisation between 5 and the corresponding marker proteins (see arrows). Scale bar: 10 μm. B) Inhibition of FAM-InsP5 (5) uptake by tunicamycin. H1299 cells grown to 50 % confluence were either treated with tunicamycin (25 μm) for 6 h or remained untreated (control). After washing of the cells twice with PBS, they were treated with 5 (20 μm) and 16 h later they were washed and fixed with paraformaldehyde. Internalisation of 5 was analysed as described in Figure 1. C) Cells were treated as described in B. Then, the washed cells were treated with 5 (20 μm) for 50 min at 4 °C and the fluorescence at the cell surface was analysed in the XY-plane. Percentage cellular adhesion of 5 was calculated from ten micrographs, and the results are depicted in the histogram (mean values±SEM) * p<0.0001.
Disruption of the cellular glycocalyx reduces uptake of FAM-InsP5
We previously showed[8] that the internalisation of InsP6 by H1299 cells is reduced by treating the cells with tunicamycin, a nucleoside antibiotic that inhibits N-linked glycosylation of proteins, leading to partial degradation of the cellular glycocalyx.[16] We suggested that cationic complexes of InsP6 with metal cations might bind to the negatively charged glycocalyx prior to internalisation and that disruption of the glycocalyx by tunicamycin reduces the affinity of InsP6-metal complexes for the cell surface, thus leading to reduced uptake of InsP6. We therefore examined the effect of tunicamycin treatment on internalisation of 5. As shown in Figure 3 B, treatment with tunicamycin strongly inhibited uptake of 5 by H1299 cells. Internalisation of 5 was reduced by (82±39) % (p<0.0001) relative to control cells not treated with tunicamycin. This result suggests that, as in the case of InsP6, uptake of 5 depends on interactions with molecules of the glycocalyx.
To verify that 5 interacts with the glycocalyx, we examined adhesion of 5 to the cell surface in the presence or absence of tunicamycin. To prevent cellular uptake, the cells were incubated at 4 °C after addition of 5. Analysis revealed that in the absence of tunicamycin 90 % of the cells showed adhesion of 5 to the cell surface, whereas in the presence of tunicamycin only 20 % of the cells were associated with 5 (Figure 3 C). Tunicamycin thus strongly inhibits adhesion of 5 to the cell surface.
Cell lines differ in their ability to internalise FAM-InsP5
In the next step, we examined whether uptake of 5 differs between cell lines. We compared uptake of 5 between malignant [HCT-116 (colon 1), MDA-MDB-231 (breast 1), Mevo (melanoma), LN2343 (lung 1)] and well differentiated [CaCo-2 (colon 2), MCF-7 (breast 2), fibroblasts, PT4323 (lung 2)] cell lines from different entities (see the Experimental Section). As shown in Figure 4, the abilities of malignant HCT-116 and MDA-MDB-231 cells to take up 5 were 3.5 and 17 times higher, respectively, than those of the well differentiated CaCo-2 and MCF-7 cells from the corresponding tissues. Healthy skin fibroblasts, however, had an ability to take up 5 ten times higher than their ability to take up malignant melanoma cells (Mevo), and uptake of 5 in lung tumour cells derived from a primary tumour (PT4323) was four times higher than in lung tumour cells derived from a lymph node metastasis (LN2343). There are thus large differences between the abilities of cell lines to take up 5, but these abilities do not seem to depend on cellular differentiation status.
Figure 4.
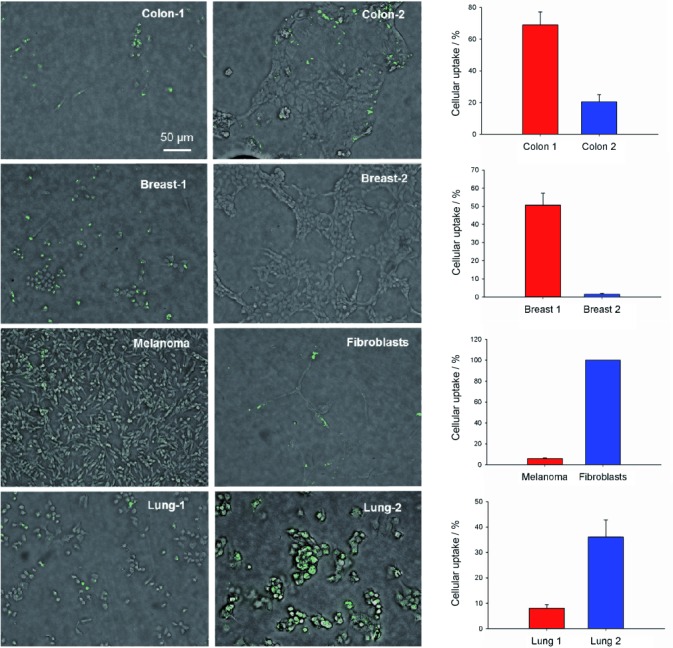
Uptake of FAM-InsP5 (5) is different between cell lines. Each cell line was grown to 50 % and to 80 % confluence. The cells were treated with 5 (20 μm) for 16 h, fixed with paraformaldehyde and embedded in Fluoromount. Shown are focus stackings of bright and fluorescence light overlays of one representative micrograph at 200-fold magnification. Uptake of 5 was determined (see above) from cells grown to 50 and to 80 % confluence (from five micrographs each) and the mean values±SEM were calculated. Histograms show uptake for well differentiated (blue) and more malignant (red) cell lines.
Effect of cell size and contacts on uptake of FAM-InsP5
The finding that uptake of 5 depends on a functional glycocalyx (Figure 3 B and C), which covers the cell surface, suggests that uptake of 5 could be related to the cell size. To examine this hypothesis, we analysed a potential correlation between uptake (particles per cell and percentage cellular uptake) of 5 and cell size (Figure 5 A and B). We also examined the relationship between uptake of 5 and the extent of cell–cell contacts, which also influences cell size (Figure 5 C and D). For the MCF-7 line, the cells are small and grow in clusters (therefore 100 % cell–cell contacts), and uptake of 5 as measured by particles per cell or by percentage cellular uptake was very low. At the other extreme, fibroblasts are large and do not form cell–cell contacts. In this case, 100 % of the cells took up 5 and there were large numbers of particles per cell. However, for several cell lines of intermediate size and/or cell–cell contacts, there was no clear correlation with uptake of 5. LN2343 and HCT-116 cells, for example, had similar sizes and cell contacts, but they differed widely in uptake of 5. From these data we conclude that cellular uptake of 5 is not always determined by the cell size or the extent of cell–cell contacts, but might also depend on the abilities of different cell types to phago- or endocytose.
Figure 5.
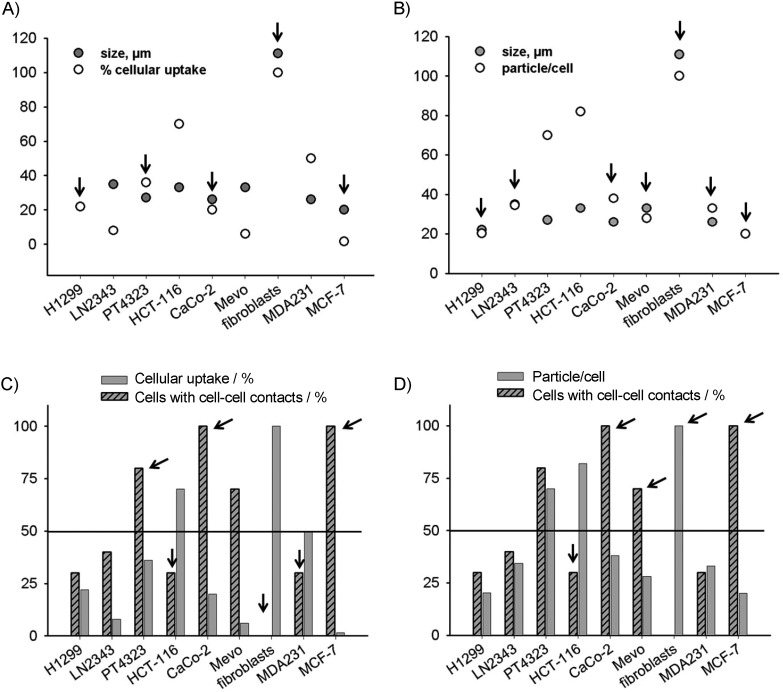
Effect of cell size and contacts on uptake of FAM-InsP5 (5). A), B) Cell size was determined, and the number of aggregates (particles) of 5 per cell was counted or percentage cellular uptake of 5 was determined as described in Figure 1. Cells lines for which cell size and uptake of 5 were correlated are marked with arrows. C), D) Percentage of cell–cell contacts was determined and plotted either against percentage uptake of 5 or against the number of particles of 5 per cell. Cells with few contacts and high uptake of 5 and cells with many contacts and low uptake of 5 are marked with arrows.
Extents of FAM-InsP5 and InsP6 uptake compared
The data above indicate that uptake of 5 occurs by a similar mechanism to uptake of InsP6. Both InsP6[8] and 5 bind to the glycocalyx, are endocytosed by a nonspecific mechanism and are stored in lysosomes. To compare the extents of uptake between 5 and InsP6, cells were treated in parallel with 5 or InsP6 for 3 h, and the InsPs were then extracted and their concentrations analysed. The concentration of 5 was easily measured by fluorescence detection (see the Experimental Section for details), whereas InsP6 was analysed by MDD-HPLC as previously reported.[8] As shown in Figure 6 A, about 1 % of the originally applied 5 and 3 % of the applied InsP6 could be extracted from the extensively washed cells. These results indicate that uptake of InsP6 is more efficient than uptake of 5. The fluorescein group might therefore inhibit internalisation of 5. It is also possible that the differences result from the different extraction methods. However, analysis of the washing fractions (Figure 6 B) revealed nearly the same pattern for 5 as previously observed for InsP6.[8] The majority of 5 was washed from the cells in fractions 1 and 2, and lower amounts were detected in fraction 3. In fractions 4 and 5 only background fluorescence was measured. This result again confirms that 5 adheres to the cell surface in similar manner to that observed for InsP6.
Figure 6.
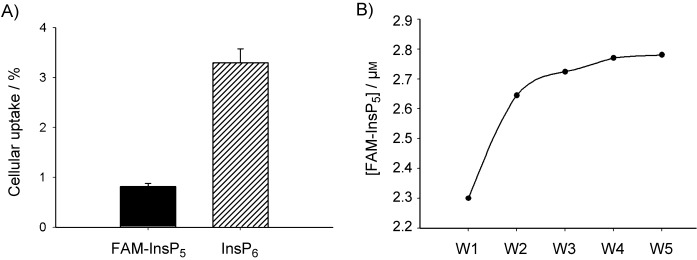
Comparison of uptake of FAM-InsP5 (5) and InsP6. A) H1299 cells were incubated with 5 or InsP6 (20 μm) for 3 h at 37 °C. After washing of the cells five times with PBS, InsP6 was extracted as previously described[8] and analysed by MDD-HPLC. For analysis of the concentration of 5, washed cells were extracted as described in the Experimental Section, and the concentration of 5 was analysed with a fluorescence reader, on the basis of a standard curve for 5 at excitation 490 nm and emission 535 nm. B) Analysis of fluorescence of the washing fractions of cells treated with 5; see (A).
Conclusions
We have developed an InsP5-based fluorescent probe—FAM-InsP5 (5)—that allows direct observation of its internalisation by cells. When applied extracellularly to H1299 tumour cells, 5 is internalised and appears in the lysosomes. Internalisation of 5 is slow, and does not occur at 4 °C; this provides strong evidence that the mechanism of uptake is by nonspecific endocytosis. Uptake of 5 is markedly reduced in cells treated with tunicamycin, a reagent that degrades the cellular glycocalyx, and this suggests that 5 initially binds to the glycocalyx, perhaps as a complex with multivalent metal ions. These results are consistent with the findings of a recent study[8] that showed that InsP6 was internalised by nonspecific endocytosis in H1299 cells.
The concordance of results between this study with 5 and the previous study[8] with InsP6 (similar timescale, distribution and tunicamycin dependence of uptake) suggests that, at least in H1229 cells, 5 is internalised in a similar way to InsP6. However, in contrast to methods based on radiolabelled inositol phosphates or metal dye detection, analyses of internalisation with 5 can be carried out relatively quickly and easily, making it possible to screen multiple cell lines and sets of conditions in parallel. This has allowed us to compare uptake of 5 by a range of cell lines and to show that the efficiency of internalisation differs markedly between cell types. These differences in internalisation might arise from differences in the ability of cells to phagocytose extracellular particles.[17] They might also reflect variation in the composition of the glycocalyx, in which case probes such as 5 could find use as markers for cell surface composition.
Our results with 5 are consistent with the findings of earlier reports[6, 7] that provided evidence for internalisation of InsP6 and InsP5 by various tumour cell lines. These studies also found antiproliferative effects for InsP6, InsP5 and 2-O-benzyl-InsP5 (3). However, at least for the cell lines and conditions used in the present study, our findings are difficult to reconcile with the idea that these antiproliferative effects can be attributed to direct interactions with cytosolic enzymes; it appears that internalised 5 remains in the lysosomes and does not reach the cytosol. Further studies with 5 and related probes will explore these aspects and will aim to elucidate the detailed mechanism of cell-specific uptake of highly phosphorylated inositols by human cells.
Experimental Section
Synthesis of FAM-InsP5
General chemistry experimental: All chemicals and solvents were supplied by Sigma–Aldrich and Alfa–Aesar. Unless otherwise stated, HPLC-grade solvents were used, and commercial reagents were used without further purification. Thin-layer chromatography (TLC) was performed on precoated plates (Merck TLC aluminium sheets silica 60 F254) with detection by UV light or with phosphomolybdic acid in methanol or alkaline aqueous KMnO4, followed by heating. Flash chromatography was performed on an ISCO CombiFlash Rf automated flash chromatography system with RediSep Rf disposable flash columns. Ion-exchange chromatography was carried out on Q Sepharose Fast Flow with a Pharmacia Biotech Gradifrac system and a P-1 pump, with elution at 5 mL min−1 with gradients of aqueous triethylammonium bicarbonate (TEAB) buffer. Low-pressure reversed-phase chromatography was performed on Lichroprep RP-18 (Merck) with use of the Gradifrac system and elution at 5 mL min−1 with gradients of acetonitrile in TEAB buffer (0.05 m). All water used in the purification of water-soluble polyphosphates was of MilliQ quality. During the manipulation of fluorescent compounds, light was excluded by covering reaction vessels, columns etc. with aluminium foil. RP-HPLC analyses of FAM-InsP5 (5) were performed with a Waters 2695 Alliance module fitted with a Waters 2996 photodiode array detector (210–600 nm). The chromatographic system consisted of a Phenomenex Security Guard cartridge system for HPLC and a Phenomenex Gemini 5 μm C18 10 Å column (150×4.60 mm), with elution at 1 mL min−1 with a gradient (5 % to 70 %) of acetonitrile in aqueous triethylammonium acetate (0.1 m) over 10 min, with detection at 254 nm. Proton 1H NMR spectra were recorded with a Bruker Avance III (400 MHz) spectrometer. Proton chemical shifts are reported in ppm (δ) relative to internal tetramethylsilane (TMS, 0.0 ppm) or with the solvent reference relative to TMS employed as the internal standard ([D6]DMSO: 2.50 ppm; D2O: 4.79 ppm). 13C and DEPT spectra were recorded with a Bruker Avance III (100 MHz) spectrometer with complete proton decoupling. Carbon chemical shifts are reported in ppm (δ) relative to internal tetramethylsilane (TMS: 0.0 ppm) or with the solvent resonance relative to TMS employed as the internal standard ([D6]DMSO: 39.51 ppm). 31P NMR spectra were recorded with Bruker Avance III (109 MHz and 162 MHz) spectrometers with complete proton decoupling. Phosphorus chemical shifts are reported in ppm (δ) relative to an H3PO4 (85 %) external standard (H3PO4: 0.0 ppm). Melting points were determined with a Reichert–Jung Thermo Galen Kofler block or a Stanford Research Systems Optimelt MPA100 automated melting point system and are uncorrected. Microanalysis was carried out at the University of Bath microanalysis service. Mass spectra were recorded at the SERC Mass Spectrometry Service Centre, Swansea, and at the University of Bath on VG Autospec or MicroTOF instruments.
1,6:3,4-Bis-[O-(2,3-dimethoxybutane-2,3-diyl)]-myo-inositol (6): Trimethyl orthoformate (100 mL), butanedione (25 mL, 285 mmol) and (±)-10-camphorsulphonic acid (0.5 g) were added to a stirred suspension of myo-inositol (25.0 g, 139 mmol) in MeOH (250 mL). The mixture was heated under N2 at reflux for 96 h and then allowed to cool, giving a cherry-red suspension. The precipitate was filtered off, washed with MeOH (200 mL) and allowed to dry, giving crude diol 6 as a white solid (22.4 g, 85 % pure by 1H NMR). This material was crystallised from boiling CHCl3/MeOH (1:1, v/v, 650 mL) and dried under vacuum at 60 °C to give 6 as colourless crystals (15.4 g, 37.7 mmol, 27 %); m.p. >300 °C with sublimation and decomposition; Rf=0.24 (EtOAc); Rf=0.29 (CHCl3/acetone 2:1); 1H NMR (400 MHz, [D6]DMSO): δ=1.17 (s, 6 H; CH3), 1.18 (s, 6 H; CH3), 3.13 (s, 6 H; OCH3), 3.15 (s, 6 H; OCH3), 3.25 (dt, 3J=5.6, 9.3 Hz, 1 H; H-5), 3.35 (dd, 3J=10.2, 2.4 Hz, 2 H; H-1, H-3), 3.67 (dd, 3J=9.8, 9.8 Hz, 2 H; H-4, H-6), 3.76 (dt, 3J=4.6, 2.4 Hz, 1 H; H-2), 4.99 (d, 3J=4.8 Hz, 1 H; OH-2), 5.05 ppm (d, 3J=5.6 Hz, 1 H; OH-5); 13C NMR (100 MHz, [D6]DMSO): δ=17.64 (CH3), 47.12 (OCH3), 47.42 (OCH3), 67.57 (C-2), 68.37 (C-1, C-3), 69.13 (C-5), 69.33 (C-4, C-6), 98.41 (BDA quaternary C), 98.97 ppm (BDA quaternary C); HRMS: m/z calcd for C18H32O10: 431.1888 [M+Na]+; found: 431.1880; elemental analysis calcd (%) for C18H32O10: C 52.93, H 7.90; found: C 52.6, H 7.92.
5-O-Benzoyl-1,6:3,4-bis-[O-(2,3-dimethoxybutane-2,3-diyl)]-myo-inositol (7): A solution of diol 6 (2.04 g, 5.00 mmol) in anhydrous pyridine (20 mL) was stirred at 0 °C under N2. Benzoic anhydride (1.24 g, 5.5 mmol) was added, followed by a catalytic amount of DMAP (60 mg, 0.50 mmol). The cooling bath was removed, and the solution was allowed to reach room temperature. After 18 h, the solution was diluted with CH2Cl2 (200 mL), washed well with HCl (1.0 m, 2×250 mL) and saturated NaHCO3 solution (100 mL) and then dried (MgSO4) and concentrated under reduced pressure. The residue was purified by flash chromatography on silica (EtOAc in CH2Cl2, 0 to 40 %) to give the 5-O-benzoate ester 7 as a white solid (2.15 g, 4.19 mmol, 84 %); crystals from boiling EtOH, m.p. 292.5–295.5 °C; Rf=0.24 (CH2Cl2/EtOAc 5:1); 1H NMR (400 MHz, CDCl3): δ=1.19 (s, 6 H; CH3), 1.32 (s, 6 H; CH3), 2.44 (br s, 1 H; 5-OH), 3.13 (s, 6 H; OCH3), 3.25 (s, 6 H; OCH3), 3.72 (dd, 3J=10.2, 2.6 Hz, 2 H; H-1, H-3), 4.10 (t, 3J=2.6 Hz, 1 H; H-2), 4.26 (dd, 3J=10.1, 9.9 Hz, 2 H; H-4, H-6), 5.39 (t, 3J=9.9 Hz, 1 H; H-5), 7.41–7.45 (m, 2 H; meta-H of Bz), 7.55 (tt, 3J=7.4, 1.3 Hz, 1 H; para-H of Bz), 8.05–8.08 ppm (m, 2 H, ortho-H of Bz); 13C NMR (100 MHz, CDCl3): δ=17.61 (CH3), 17.64 (CH3), 47.58 (OCH3), 48.00 (OCH3), 67.28 (C-4, C-6), 68.63 (C-1, C-3), 68.95 (C-2), 70.96 (C-5), 99.33 (BDA quaternary C), 100.13 (BDA quaternary C), 128.37 (Bz meta-C), 129.59 (Bz ortho-C), 130.40 (Bz ipso-C), 132.76 (Bz para-C), 165.16 ppm (Bz C=O); HRMS: m/z calcd for C25H36O11: 535.2150 [M+Na]+; found: 535.2153; elemental analysis calcd for C25H36O11: C 58.58, H 7.08; found: C 58.5, H 7.14.
5-O-Benzoyl-2-O-cyanomethyl-1,6:3,4-bis-[O-(2,3-dimethoxybutane-2,3-diyl)]-myo-inositol (8): Sodium hydride (468 mg of a 60 % suspension in mineral oil, 11.7 mmol) was added under N2 to a suspension of 7 (2.00 g, 3.90 mmol) in dry acetonitrile (20 mL). The suspension was stirred at room temperature for 30 min. The suspension first became almost clear, and then thickened as evolution of gas ceased. The suspension was then cooled to −30 °C (acetonitrile/solid CO2 bath), and bromoacetonitrile (1.5 mL, 23 mmol) was added dropwise over 2 min. The suspension was stirred at −20 °C for 3.5 h and then allowed to reach room temperature overnight. The resulting brown suspension was concentrated under reduced pressure to give a solid residue, which was dispersed in dichloromethane (100 mL in portions with use of an ultrasound bath) and filtered through Celite, leaving a pale yellow solution. The solution was concentrated, and the residue was purified by flash chromatography (EtOAc in CH2Cl2, 0 to 50 %) to give 8 as a white solid (1.83 g, 3.32 mmol, 85 %); m.p. 264–266 °C (from EtOH); Rf=0.54 (CH2Cl2/EtOAc 5:1); 1H NMR (400 MHz, CDCl3): δ=1.17 (s, 6 H; CH3), 1.30 (s, 6 H; CH3), 3.13 (s, 6 H; OCH3), 3.25 (s, 6 H; OCH3), 3.76 (dd, 3J=10.3, 2.4 Hz, 2 H; H-1, H-3), 4.01 (t, 3J=2.4 Hz, 1 H; H-2), 4.14 (dd, 3J=10.2, 10.0 Hz, 2 H; H-4, H-6), 4.69 (s, 2 H; CH2CN), 5.37 (t, 3J=9.8 Hz, 1 H; H-5), 7.42–7.45 (m, 2 H; meta-H of Bz), 7.55 (tt, 3J=7.2, 1.2 Hz, 1 H; para-H of Bz), 8.05–8.08 ppm (m, 2 H; ortho-H of Bz); 13C NMR (100 MHz, CDCl3): δ=17.57 (CH3), 17.58 (CH3), 47.64 (OCH3), 48.09 (OCH3), 56.77 (OCH2CN), 67.43 (C-4, C-6), 68.68 (C-1, C-3), 71.03 (C-5), 76.22 (C-2), 99.27 (BDA quaternary C), 99.97 (BDA quaternary C), 116.28 (OCH2CN), 128.39 (Bz meta-C), 129.62 (Bz ortho-C), 130.29 (Bz ipso-C), 132.83 (Bz para-C), 165.16 ppm (Bz C=O); HRMS: m/z calcd for C27H37NO11: 574.2259 [M+Na]+; found: 574.2269; elemental analysis calcd for C27H37NO11: C 58.79, H 6.76, N 2.54; found: C 58.4, H 6.85, N 2.48.
1,6:3,4-Bis-[O-(2,3-dimethoxybutane-2,3-diyl)]-2-O-[2-(2,2,2-trifluoroacetylamino)ethyl]-myo-inositol (9): A solution of 8 (1.78 g, 3.23 mmol) in dry THF (25 mL) was added dropwise over 30 min, under N2 at 0 °C, to a solution of LiAlH4 in THF (10 mL of a 1.0 m solution, 10 mmol). The mixture was stirred at room temperature for a further 1 h and then quenched by careful addition of a saturated solution of potassium sodium tartrate (75 mL). Ether (75 mL) was then added, and the mixture was stirred vigorously for 30 min until two distinct layers formed. The ether layer was separated, and the aqueous layer was re-extracted with ether (2×100 mL). The combined organic extracts were dried (MgSO4) and concentrated to give the crude amine (1.8 g, Rf=0.16 in CH2Cl2/MeOH/NH3, 200:20:1) as a foam, which was taken up in dry THF (10 mL) and stirred with ethyl trifluoroacetate (2 mL, 8.4 mmol) at room temperature. After 2 h, the solution was concentrated, and the residue was purified by flash chromatography (ethyl acetate in petroleum ether, 0 to 100 %) to give 9 as a white solid (1.16 g, 2.12 mmol, 66 %); m.p. 206–208 °C (from EtOAc/petroleum ether); Rf=0.25, (CH2Cl2/EtOAc 1:1); 1H NMR (400 MHz, CDCl3): δ=1.29 (s, 6 H; CH3), 1.32 (s, 6 H; CH3), 2.62 (br s, 1 H; 5-OH), 3.24 (s, 6 H; OCH3), 3.28 (s, 6 H; OCH3), 3.54 (dd, 3J=10.2, 2.6 Hz, 2 H; H-1, H-3), 3.54–3.57 (m, 2 H; OCH2CH2N), 3.62 (t, 3J=2.6 Hz, 1 H; H-2), 3.65 (td, 3J=9.5, 2.1 Hz, 1 H; H-5), 3.81 (dd, 3J=5.1, 4.7 Hz, 2 H; OCH2CH2N), 3.94 (dd, 3J=10.0, 9.8 Hz, 2 H; H-4, H-6), 7.32 ppm (br t, 1 H; NHC(O)CF3); 13C NMR (100 MHz, CDCl3): δ=17.50 (CH3), 17.69 (CH3), 39.52 (OCH2CH2N), 47.96 (OCH3), 48.06 (OCH3), 68.47 (C-1, C-3), 69.26 (C-4, C-6), 70.11 (C-5), 70.80 (OCH2CH2N), 77.46 (C-2), 99.27 (BDA quaternary C), 99.89 (BDA quaternary C), 115.99 (q, 2JH,F=288 Hz; CF3), 157.24 ppm (q, 3JH,F=37 Hz; C(O)CF3); HRMS: m/z calcd for C22H36F3NO11: 570.2133 [M+Na]+; found: 570.2151; elemental analysis calcd for C22H36F3NO11: C 48.26, H 6.63, N 2.56; found: C 48.1, H 6.52, N 2.41.
2-O-[2-(2,2,2-Trifluoroacetylamino)ethyl]-myo-inositol 1,3,4,5,6-pentakis(dibenzylphosphate) (10): Compound 9 (450 mg, 0.822 mmol) was dissolved in aqueous TFA (95 %, 10 mL). The solution was stirred at room temperature for 20 min and then concentrated by evaporation under reduced pressure. Ethanol was added and evaporated several times to remove traces of TFA. The solid pentaol product (Rf=0.20, CH2Cl2/MeOH 3:1) was dried under vacuum for 16 h and then suspended in dry dichloromethane (10 mL). The suspension was stirred at room temperature under N2, and 5-phenyltetrazole (900 mg, 6.16 mmol) was added, followed by bis(benzyloxy)diisopropylaminophosphine (1.7 mL, 4.9 mmol). The mixture was stirred at room temperature for 2 h, after which time a clear solution remained. The solution was cooled to −78 °C, and 3-chloroperoxybenzoic acid (57 %, 2.5 g, 8.2 mmol) was added in portions over 1 min. A precipitate formed during the oxidation reaction. The resulting suspension was allowed to warm to room temperature and diluted with EtOAc (50 mL), giving a clear solution. The solution was washed with aqueous sodium sulfite solution (10 %, 2×50 mL), dried over MgSO4 and concentrated, leaving an oily residue, which was purified by flash chromatography with elution with acetone in dichloromethane (0 to 50 %) to give 10 (1.17 g, 0.722 mmol, 88 %) as a colourless oil; Rf=0.20 (CH2Cl2/acetone 5:1); 1H NMR (400 MHz, CDCl3): δ=3.31–3.34 (m, 2 H; OCH2CH2N), 3.69 (t, 3J=4.6 Hz, 2 H; OCH2CH2N), 4.25 (ddd, 3JH,H=9.5, 2.2 Hz, 3JH,P=9.5 Hz, 2 H; H-1, H-3), 4.40–4.49 (m, 2 H; H-2, H-5), 4.92–5.03 (m, 22 H; CH2Ph, H-4, H-6), 7.15–7.29 (m, 50 H; Ph), 8.06 ppm (broad, 1 H; amide NH); 13C NMR (100 MHz, CDCl3): δ=40.09 (OCH2CH2N), 69.59–69.96 (with 3JC,P couplings; OPOCH2Ph), 71.04 (OCH2CH2N), 74.67, 75.24, 75.49 and 75.86 (broad signals with JC,P couplings; inositol ring CH), 115.95 (1JC,F=288 Hz; CF3), 127.96–128.71 (CH of Ph), 135.21–135.80 (ipso-C of POCH2Ph), 157.62 ppm (2JC,F=36.8 Hz; C(O)CF3); 19F NMR (376 MHz, CDCl3): δ=−75.14 ppm; 31P NMR (162 MHz, CDCl3): δ=−2.05 (2 P), −1.45 (1 P), −1.43 ppm (2 P); HRMS: m/z calcd for C80H81F3NO22P5: 1618.3818 [M]−; found: 1618.3788.
2-O-(2-Aminoethyl)-myo-inositol 1,3,4,5,6-pentakisphosphate (4): Palladium hydroxide on activated charcoal (Fluka, 20 %, 50 % water, 100 mg) was added to a solution of 10 (380 mg, 0.235 mmol) in MeOH (30 mL) and deionised water (8 mL). The suspension was stirred vigorously under hydrogen (balloon) for 16 h. The catalyst was removed by filtration through a PTFE syringe filter (0.2 μm) to give a colourless solution, which was neutralised by addition of N,N-diisopropylethylamine (DIPEA, 0.5 mL). The solvents were then removed by evaporation under reduced pressure. A 1H NMR spectrum of the product in D2O at this stage showed no residual aromatic signals, thus indicating that hydrogenolysis was complete. The product was redissolved in deionised water (1 mL), excess DIPEA (1 mL) was added, and the solution was heated under N2 at 60 °C for 20 h. The solution was concentrated, and the residue was redissolved in deionised water and lyophilised to give the diisopropylethylammonium salt of 4 as a fawn solid (325 mg); 1H NMR (400 MHz, D2O): δ=1.18–1.22 (m, 75 H; DIPEA CH3), 3.07 (q, 3J=7.4 Hz,10 H; DIPEA CH2), 3.13 (t, 3J=5.1 Hz, 2 H; OCH2CH2NH3+), 3.58 (heptet, 3J=6.7 Hz,10 H; DIPEA CH), 3.96 (t, 3J=5.1 Hz, 2 H; OCH2CH2NH3+), 4.02–4.08 (m, 3 H; H-1, H-3, H-5), 4.11 (t, 3J=2.5 Hz, 1 H; H-2), 4.38 ppm (q, 3J=9.7 Hz, 2 H; H-4, H-6); 13C NMR (100 MHz, D2O): δ=12.11 (DIPEA CH2CH3), 16.22 (DIPEA CH(CH3)2), 17.69 (DIPEA CH(CH3)2), 39.52 (OCH2CH2NH3+), 42.51 (DIPEA CH2CH3), 54.32 (DIPEA CH(CH3)2), 69.37 (OCH2CH2NH3+), 73.89 and 76.13 (C-1, C-3, C-4, C-6), 77.27 (C-5), 79.17 ppm (C-2); 31P NMR (162 MHz, D2O): δ=−0.39 (2 P), 0.63 (2 P), 0.80 ppm (1 P; P-5); HRMS: m/z calcd for C8H22NO21P5: 621.9300 [M]−; found: 621.9311.
This material was used in subsequent conjugation reactions, but for biological evaluations of 4, a portion was purified by ionexchange chromatography on Q Sepharose Fast Flow resin with elution with TEAB (0 to 2.0 m) to give the triethylammonium salt of 4, which was accurately quantified by total phosphate assay.
2-O-[2-(5-Fluoresceinylcarboxy)aminoethyl]-myo-inositol 1,3,4,5,6-pentakisphosphate (5): Dry DIPEA (10 μL) was added to a suspension of 4 (20 mg DIPEA salt, 14 μmol) in dry propan-2-ol. Solid 5-carboxyfluorescein NHS ester[14] (13 mg, 28 μmol) was added to the resulting clear solution, followed by further dry DIPEA (60 μL). The flask was covered in foil to exclude light, and the reaction mixture was stirred under N2 at 60 °C for 24 h and was then allowed to cool and concentrated under reduced pressure. The residue was dissolved in TEAB (0.05 m, pH approx. 7.5, 5 mL) and applied to a column of Q Sepharose Fast Flow resin (bicarbonate form, 70 mm×20 mm). The column was washed well with milliQ water, followed by TEAB (0.8 m, pH approx. 7.8) until the eluent ran colourless. This required approximately 400 mL of buffer. The column was then eluted with a gradient of TEAB (0.8 to 2.0 m, over 300 mL), with collection of 10 mL fractions. A fluorescent product eluted at high buffer concentration (>1.6 m TEAB). Fractions containing this product were combined and concentrated to give an orange solid, which was re-dissolved in TEAB (0.05 m, pH approx. 7.5, 5 mL) and applied to a small column (100 mm×10 mm) of Lichroprep RP-18. The column was eluted with a gradient of acetonitrile (0 to 30 % in 0.05 m TEAB over 300 mL), with collection of 10 mL fractions. Fluorescent fractions were combined and concentrated to leave a solid residue, which was re-dissolved in milliQ water and lyophilised to give the pure triethylammonium salt of 5 (containing 4.5 Et3NH+ per equiv of 5) as a fluffy orange solid (16 mg, 11 μmol, 79 %); 1H NMR (400 MHz, D2O): δ=1.12 [t, 3J=7.5 Hz, approx. 40 H; (CH3CH2)3NH+], 3.03 [q, 3J=7.5 Hz, approx. 27 H; (CH3CH2)3NH+], 3.61 (t, 3J=5.1 Hz, 2 H; OCH2CH2NH), 3.97 (t, 3J=5.1 Hz, 2 H; OCH2CH2NH), 4.04–4.14 (m, 3 H; H-1, H-3, H-5), 4.19 (br s, 1 H; H-2), 4.41 (dt, 3JH,P=9.4 Hz, 3JH,H=9.4 Hz, 2 H; H-4, H-6), 6.65–6.68 (m, 4 H; fluorescein H-2′, H-4′, H-5′, H-7′), 6.99 (d, 3J=9.7 Hz, 2 H; fluorescein H-1′, H-8′), 7.43 (d, 3J=8.2 Hz, 1 H; fluorescein H-7), 8.13 (dd, 3J=8.2 Hz, 4J=1.9 Hz, 1 H; fluorescein H-6), 8.31 ppm (d, 4J=1.9 Hz, 1 H; fluorescein H-4); 13C NMR (100 MHz, D2O): δ=8.14 [(CH3CH2)3NH+], 40.34 (OCH2CH2NH), 46.53 [(CH3CH2)3NH+], 71.67 (OCH2CH2NH), 74.10 and 76.29 (C-1, C-3, C-4, C-6), 77.49 (C-5), 78.62 (C-2), 102.64, 112.66, 116.55, 126.60, 127.77, 130.97, 131.45, 132.48, 135.97, 144.62, 155.23, 165.24, 169.21 (C=O), 170.98 ppm (C=O); 31P NMR (109 MHz, CD3OD): δ=0.97 (2 P), 1.89 (2 P), 2.21 ppm (1 P; P-5); HRMS: m/z calcd for C29H31NO27P5−: 979.9777 [M]−; found: 979.9741; analytical RP HPLC: tR=4.40 min (see General Chemistry Experimental).
Cell lines and cell culture: NCI-H1299 (H1299), HCT-116, CaCo-2, MDA-MDB-231, MCF-7 and Mevo cells were purchased from ATCC. Skin fibroblasts cells were a gift from Ulla Kasten-Pisula (Hamburg, Germany), and PT4323 (primary tumour) and LN2343 (the corresponding lymph node metastasis) were both freshly isolated from primary lung adenocarcinomas (for details see ref. [8]). The well-established cell line H1299 derives from a lymph node metastasis of lung adenocarcinoma cells. HCT-116 and CaCo-2 are colon cancer cells, MDA-MDB-231 and MCF-7 are breast cancer cells, and Mevo are melanoma cells. The cell lines MDA-MDB-231, NCI-H1299 and HCT-116 were cultured in Dulbecco's modified Eagle's medium (DMEM), Mevo and fibroblasts were grown in RPMI, PT4323 and LN2343 in RPMI with supplements (see ref. [8]) and CaCo-2 in MEM alpha medium. All media were supplemented with foetal calf serum (FCS, 10 %, v/v), l-glutamine (4 mm), streptomycin (100 μg mL−1) and penicillin (100 U mL−1) and were purchased from Invitrogen.
Analysis of cellular uptake of FAM-InsP5 (5) in intact cells by fluorescence microscopy: Cells were grown on poly-l-lysine-covered cover slips to 20, 50 or 80 % confluence, and 5 or fluorescein (TEA+ salt in each case, 20 μm) as control (both diluted in deionised water) was added. After incubation for different times, the cells were washed three times with phosphate saline (PBS), fixed with paraformaldehyde (3 %), washed again three times with PBS and finally embedded in Fluoromount-G (Southern Biotech, Birmingham, Alabama. USA). Uptake of 5 was analysed by performing z-stacks (about 50 stacks per image of 1 to 1.5 μm in size) of bright and fluorescence light with a BZ-9000 E microscope from Keyence (Neu Isenburg, Germany). After overlay of these z-stacks, a 3D analysis was performed (software: BZ-H1RE), and the position of 5 was determined in xz- and in xy-layers of single stacks. Positioning of 5 in the middle cell layers was defined as cellular uptake, whereas accumulation in the first layers was defined as cell surface localisation of 5. The micrographs shown here represent focus stackings of bright and fluorescence light overlays by using software (BZ-H1RE) that selects the sharpest areas from multiple frames. Every experiment was performed in triplicate, and at least 100 cells were analysed per experiment.
Analysis of cellular uptake of FAM-InsP5 (5) in lysed cells by fluorescence photometry: H1299 cells grown to about 80 % confluence in 10 cm dishes were treated with 5 (25 μm) for 3 h. The cells were then washed five times with PBS, MPER buffer (1.1 mL) was added, the cells were scraped, and the suspension was frozen in N2 and thawed twice. The resulting suspension was diluted 1:1 with PBS, and the fluorescence was analysed with a Tecan Infinitive M100 fluorescence reader at excitation 490 nm and emission 535 nm. To prepare a standard curve, different concentrations of 5 were added to untreated lysed cells and, after cautious mixing, the fluorescence of the standards was measured in parallel to the samples as well as to the washing fractions. To analyse bleaching, a solution of 5 was added to cell suspension or to PBS, and fluorescence was analysed as above over a time period of 3 h. Under both sets of conditions (cell suspension and PBS) we measured 20 % loss by bleaching.
Immunofluorescence: Cells preincubated with 5 for 16 h in chamber slides were washed twice with PBS, fixed with paraformaldehyde (3 %) for 10 min, washed three times with PBS and treated with Triton-100 (0.3 %) for 5 min at RT. After washing of the cells three times with PBS they were blocked in BSA/PBS (Sigma—Aldrich, 2.5 %) for 20 min, incubated with antibodies against early endosome antigen 1 (EEA1; Abcam, ab2900, Cambridge, UK) or lysosome-associated membrane protein-2 (LAMP-2; Santa Cruz #sc-5571), respectively, at a dilution of 1:200 in BSA/PBS (0.7 %, w/v) for 16 h at 8 °C. After having been washed again three times with PBS, the cells were finally treated with anti-rabbit secondary antibodies coupled to Alexa Fluor 568 at a dilution of 1:1000 for 1 h at 22 °C. After the cells had been washed with PBS, images were captured on a fluorescence microscope (Keyence BZ-9000).
Acknowledgments
We thank the Wellcome Trust for support (Programme Grant 082837 to A.M.R. and B.V.L.P.).
Supporting Information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re-organized for online delivery, but are not copy-edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
miscellaneous_information
References
- 1.Shears SB. Cell. Signalling. 2001;13:151–158. doi: 10.1016/s0898-6568(01)00129-2. [DOI] [PubMed] [Google Scholar]
- 2.Raboy V. Phytochemistry. 2003;64:1033–1043. doi: 10.1016/s0031-9422(03)00446-1. [DOI] [PubMed] [Google Scholar]
- 3.Letcher AJ, Schell MJ, Irvine RF. Biochem. J. 2008;416:263–270. doi: 10.1042/BJ20081417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Vucenik I, Shamsuddin AM. Nutr. Cancer. 2006;55:109–125. doi: 10.1207/s15327914nc5502_1. [DOI] [PubMed] [Google Scholar]
- 5.Piccolo E, Vignati S, Maffucci T, Innominato PF, Riley AM, Potter BVL, Pandolfi PP, Broggini M, Iacobelli S, Innocenti P, Falasca M. Oncogene. 2004;23:1754–1765. doi: 10.1038/sj.onc.1207296. [DOI] [PubMed] [Google Scholar]
- 6a.Vucenik I, Shamsuddin AM. J. Nutr. 1994;124:861–868. doi: 10.1093/jn/124.6.861. [DOI] [PubMed] [Google Scholar]
- 6b.Ferry S, Matsuda M, Yoshida H, Hirata M. Carcinogenesis. 2002;23:2031–2041. doi: 10.1093/carcin/23.12.2031. [DOI] [PubMed] [Google Scholar]
- 7.Maffucci T, Piccolo E, Cumashi A, Iezzi M, Riley AM, Saiardi A, Godage HY, Rossi C, Broggini M, Iacobelli S, Potter BVL, Innocenti P, Falasca M. Cancer Res. 2005;65:8339–8349. doi: 10.1158/0008-5472.CAN-05-0121. [DOI] [PubMed] [Google Scholar]
- 8.Windhorst S, Lin H, Blechner C, Fanick W, Brandt L, Brehm MA, Mayr GW. Biochem. J. 2013;450:115–125. doi: 10.1042/BJ20121524. [DOI] [PubMed] [Google Scholar]
- 9.Falasca M, Chiozzotto D, Godage HY, Mazzoletti M, Riley AM, Previdi S, Potter BVL, Broggini M, Maffucci T. Br. J. Cancer. 2010;102:104–114. doi: 10.1038/sj.bjc.6605408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ding Z, Rossi AM, Riley AM, Rahman T, Potter BVL, Taylor CW. Mol. Pharmacol. 2010;77:995–1004. doi: 10.1124/mol.109.062596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11a.Montchamp JL, Tian F, Hart ME, Frost JW. J. Org. Chem. 1996;61:3897–3899. doi: 10.1021/jo960170n. [DOI] [PubMed] [Google Scholar]
- 11b.Riley AM, Jenkins DJ, Potter BVL. Carbohydr. Res. 1998;314:277–281. [Google Scholar]
- 11c.Baeschlin DK, Chaperon AR, Green LG, Hahn MG, Ince SJ, Ley SV. Chem. Eur. J. 2000;6:172–186. doi: 10.1002/(sici)1521-3765(20000103)6:1<172::aid-chem172>3.0.co;2-5. [DOI] [PubMed] [Google Scholar]
- 12.Riley AM, Wang HC, Weaver JD, Shears SB, Potter BVL. Chem. Commun. 2012;48:11292–11294. doi: 10.1039/c2cc36044f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Malet C, Hindsgaul O. J. Org. Chem. 1996;61:4649–4654. doi: 10.1021/jo960284z. [DOI] [PubMed] [Google Scholar]
- 14.Adamczyk M, Fishpaugh JR, Heuser KJ. Bioconjugate Chem. 1997;8:253–255. doi: 10.1021/bc9600877. [DOI] [PubMed] [Google Scholar]
- 15.Canton I, Battaglia G. Chem. Soc. Rev. 2012;41:2718–2739. doi: 10.1039/c2cs15309b. [DOI] [PubMed] [Google Scholar]
- 16a.Brandish PE, Kimura K, Inukai M, Southgate R, Lonsdale JT, Bugg TDH. Antimicrob. Agents Chemother. 1996;40:1640–1644. doi: 10.1128/aac.40.7.1640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16b.Crean SM, Meneski JP, Hullinger TG, Reilly MJ, DeBoever EH, Taichman RS. Br. J. Haematol. 2004;124:534–546. doi: 10.1046/j.1365-2141.2003.04786.x. [DOI] [PubMed] [Google Scholar]
- 17.Fais S. Cancer Lett. 2007;258:155–164. doi: 10.1016/j.canlet.2007.09.014. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
miscellaneous_information