

Abstract
Recent studies of protein post-translational modifications revealed that various types of lysine acylation occur in eukaryotic and bacterial proteins. Lysine propionylation, a newly discovered type of acylation, occurs in several proteins, including some histones. In this study, we identified 361 propionylation sites in 183 mid-exponential phase and late stationary phase proteins from Thermus thermophilus HB8, an extremely thermophilic eubacterium. Functional classification of the propionylproteins revealed that the number of propionylation sites in metabolic enzymes increased in late stationary phase, irrespective of protein abundance. The propionylation sites on proteins expressed in mid-exponential and late stationary phases partially overlapped. Furthermore, amino acid frequencies in the vicinity of propionylation sites differed, not only between the two growth phases but also relative to acetylation sites. In addition, 33.8% of mid-exponential phase–specific and 80.0% of late stationary phase–specific propionylations (n ≥ 2) implied that specific mechanisms regulate propionylation in the cell. Moreover, the limited degree of overlap between lysine propionylation (36.8%) and acetylation (49.2%) sites in 67 proteins that were both acetylated and propionylated strongly suggested that the two acylation reactions are regulated separately by specific enzymes and may serve different functions. Finally, we also found that eight propionylation sites overlapped with acetylation sites critical for protein functions such as Schiff-base formation and ligand binding.
Protein structure and function are deeply influenced by post-translational modifications (PTMs).1 Recent reports have described more than 300 types of PTMs, such as phosphorylation, methylation, glycosylation, and acylation (1, 2). The combination of enrichment techniques for post-translationally modified peptides and mass-spectrometric approaches for the identification of PTMs has been responsible for the discovery of many diverse protein modifications (3). Among these, acylation at lysine residues is one of the most prevalent PTMs (4). In particular, acetylation, succinylation, and malonylation occur at thousands of modification sites (5–17). Lysine acylation was first reported on histones in the 1960s (18, 19). For many decades, our knowledge of the roles of this modification in cells was limited to observations made on histones and a few transcription-associated proteins in eukaryotes. However, recent studies revealed thousands of acetylation sites on both eukaryotic and bacterial proteins (8, 9, 11), and a large number of studies of the functions of protein acetylation have demonstrated that this modification is biologically significant (20, 21). In a previous study, we performed structural mapping of acetylation sites on proteins; based on the results, we proposed potential roles for acetylation in protein–nucleic acid interactions, inter- and intra-subunit interactions, Schiff-base formation, and ligand binding (22). Lysine succinylation and malonylation were discovered more recently (5, 6, 10, 12, 13). Like acetylation, these modifications occur in a diverse range of proteins. Thus, proteome-wide analyses have expanded our knowledge of lysine acylation and suggested that it exerts influence on a wide range of biological functions.
Other types of lysine acylations have been described recently, such as formylation (23, 24), propionylation (25), butyrylation (25), and crotonylation (26) in histones and other types of proteins. Lysine propionylation was initially reported in histones (25), but subsequently three non-histone proteins (p53, p300, and CREB-binding protein) were shown to be propionylated (27–30). More recently, Fritz et al. reported several candidate propionylproteins in mouse liver (31). A few studies have investigated the significance of lysine propionylation. First, lysine propionylation was proposed to act as a histone mark. It is well known that alterations in histone modifications affect gene expression and bring about epigenetic regulation (28). For example, the propionylation level on Lys-23 of histone H3 in myeloid precursor leukemia cells decreases during monocytic differentiation (30). Second, propionylation was shown to be induced in mouse liver mitochondria under chronic ethanol ingestion, suggesting that it plays a role in cellular stress responses (31). Third, lysine propionylation was detected not only in eukaryotes, but also in bacteria (32). For example, propionylation at Lys-592 of propionyl-CoA synthetase of Salmonella enterica inhibits its enzymatic activity (32). Although some of the functions of this modification have begun to be revealed, the cellular distribution of lysine propionylation in eukaryotes and bacteria remains poorly characterized relative to the distributions of acetylation and succinylation.
Recently, we reported the results of phosphoproteome and acetylome analyses performed during a proteomic characterization of Thermus thermophilus HB8, an extremely thermophilic Gram-negative eubacterium (22, 33). The 2.2-Mb genome of T. thermophilus HB8 contains 2238 open reading frames, about half the number in Escherichia coli. The simplicity of this genome is greatly advantageous in attempts to achieve a comprehensive understanding of the bacterium's physiology. We have studied T. thermophilus as a model organism with the goal of understanding biological phenomena in bacteria, using “-omics” approaches such as structural genomics (34–36), transcriptomics (37), metabolomics (38), whole-cell proteomics (39), and PTM proteomics (22, 33).
In this study, we enriched propionylated peptides using an anti-propionyllysine polyclonal antibody and then identified the modification using hybrid quadrupole TOF-MS combined with nano-scale liquid chromatography (nano-LC). Using this approach, we demonstrated for the first time that lysine propionylation is a prevalent PTM; specifically, we identified 361 propionylation sites in 183 proteins expressed in the mid-exponential and late stationary phases of T. thermophilus. We performed comparative analyses of lysine propionylation in the two growth phases based on the modification sites, functional classification, and local sequence context around propionylation sites. We found that the number of propionylation sites increased about 3-fold in stationary phase, and that this increase in propionylation was not related to protein expression level. Moreover, the low degree of overlap between propionylation sites in mid-exponential and late stationary phases suggested that propionylation and depropionylation are controlled by distinct mechanisms in vivo.
EXPERIMENTAL PROCEDURES
Media and Culture Conditions
Tryptone and yeast extract were purchased from Nihon Seiyaku (Tokyo, Japan). Phytagel for solid cultivation was obtained from Sigma-Aldrich (St. Louis, MO). Formic acid and acetonitrile for liquid chromatography and other chemical compounds were purchased from Wako (Tokyo, Japan). A single colony of T. thermophilus HB8 (ATCC27634) on a TT plate (medium composition: 0.4% tryptone, 0.2% yeast extract, 0.1% NaCl, 1.5 mm MgCl2, 1.5 mm CaCl2, and 1.5% phytagel) was inoculated into TT broth (medium composition: 0.4% tryptone, 0.2% yeast extract, 0.1% NaCl, 0.4 mm MgCl2 and 0.4 mm CaCl2) and cultivated overnight at 70¦°C with vigorous shaking. A 50-μl aliquot of the T. thermophilus HB8 culture was inoculated into 5 ml of TT broth for a seed culture. The cells were then cultured at 70¦°C until mid-exponential phase (A600 = 0.8), and then 1.5 ml of the culture was inoculated into 150 ml of TT broth for the main culture. Cultivated cells that had reached either mid-exponential phase (A600 = 0.8, ∼4 to 5 h of cultivation) or late stationary phase (A600 = 2.5, 24-h cultivation) were harvested via centrifugation at 7000 × g for 5 min. Cell pellets were washed twice with cold phosphate-buffered saline (PBS) and then disrupted immediately. Mid-exponential and late stationary phase cell pellets were each obtained from three independent biological replicates.
Preparation of Crude Extracts and Tryptic Digests
A cell pellet harvested at either mid-exponential or late stationary phase was suspended in 5 ml of lysis buffer (50 mm Tris-HCl containing 5 mm EDTA and 1 mm phenylmethylsulfonyl fluoride, pH 8.0) and then disrupted using an ultrasonic disruptor (UD-201, TOMY, Tokyo, Japan) at 4¦°C. To minimize the effect of non-enzymatic modifications during the cell lysis procedure, the supernatant was immediately mixed with four volumes of cold acetone after the removal of cell debris by centrifugation at 7000 × g for 5 min and then kept at −30¦°C overnight. Using an aliquot of lysate, the concentration of total proteins was determined via the Bradford method (Bio-Rad, Hercules, CA). The remaining mixture stored at −30¦°C was centrifuged at 10,000 × g for 30 min; the resultant pellet was washed in cold acetone, and then the precipitate was dried to remove remaining acetone. For protein denaturation, the pellet was dissolved in 6 m guanidine hydrochloride and then heated at 95¦°C for 10 min. After dilution with 50 mm ammonium bicarbonate buffer (pH 8.0) to a final guanidine hydrochloride concentration of 1.5 m, the denatured lysate was digested with TPCK trypsin (Thermo Scientific, Rockford, IL) at 37¦°C for 16 h using an enzyme/substrate ratio of 1/100 (w/w). The trypsinized lysate was further digested with TPCK trypsin (1/100 w/w) at 37¦°C overnight to achieve complete digestion, and then cysteines were alkylated as follows: The fully trypsinized lysate was reduced with 5 mm dithiothreitol (DTT) at 50¦°C for 30 min and then alkylated with 10 mm iodoacetamide at room temperature in the dark for 30 min. The reaction was stopped with 10 mm DTT. Finally, the resultant solution was passed through a Sep-Pak Plus C18 cartridge (Waters, Milford, MA), and the eluent was lyophilized using a Freezone 4.5 (Labconco, Kansas City, MO) overnight.
Propionylpeptide Enrichment
We generated an antibody against propionylated bovine serum albumin (BSA), as previously described (40). Briefly, BSA was chemically propionylated with propionic anhydride; the chemical propionylation of primary amines was monitored by means of the ninhydrin reaction until about 90% of the amino groups had been propionylated. The generation of anti-propionyllysine antiserum from a rabbit (homemade) was carried out by Kitayama Labs Co. (Nagano, Japan).
A 50-μl aliquot of the anti-propionyllysine antibody was conjugated to protein A-agarose (Santa Cruz Biotechnology, Dallas, TX) in 500 μl of PBS (pH 7.5) by gentle shaking at 4¦°C for 4 h. The conjugated beads were washed three times with 1.0 ml of NETN buffer (50 mm Tris-HCl, pH 8.0, containing 100 mm NaCl, 1.0 mm EDTA, and 0.5% Nonidet P40). A 5-mg tryptic digest suspended in 500 μl of NETN buffer was mixed with the antibody-conjugated beads and then gently shaken using an RT-30 mini (TAITEC, Tokyo, Japan) at 4¦°C for 6 h. The beads were washed with 1.0 ml of NETN buffer and then washed twice with 1.0 ml of ETN buffer (50 mm Tris-HCl containing 100 mm NaCl and 1.0 mm EDTA, pH 8.0). The enriched propionylpeptides were eluted three times with 200 μl of 0.1% trifluoroacetic acid, and the eluates were pooled. These immunoprecipitation steps were repeated twice per biological replicate. The eluted peptides were further desalted using a Sep-Pak Plus C18 cartridge and then lyophilized in a Freezone 4.5 for 3 h. Prior to mass spectrometry, the dried sample was dissolved in 20 μl of 0.1% formic acid. Two immunoprecipitation samples were obtained for each biological replicate. In total, six immunoprecipitations (i.e. two samples of each of three biological replicates) from each growth phase were used for propionylpeptide enrichment (Fig. 1B).
Fig. 1.

A, chemical structures of acetylated and propionylated lysine. B, schematic overview of identification of propionylation sites. Analysis of each biological sample involved three biological replicates (S1, S2, and S3), two anti-propionyllysine immunoprecipitations (IP1 and IP2) for each biological replicate, and two LC-MS/MS runs (Run1 and Run2) for each affinity-enriched sample. C, propionylation sites in mid-exponential and late stationary phase proteins identified in T. thermophilus; numbers adjacent to the columns indicate the number of biological replicates in which the sites were detected. ME, mid-exponential phase; LS, late stationary phase.
Nano-LC and MS/MS Analyses
Half (10 μl) of each enriched sample was injected via the autosampler of a Proxeon EASY-nLC (Bruker Daltonics, Billerica, MA) and subsequently washed with solvent A (0.1% formic acid in water) on an NS-MP-10 BioSphere trap column (C18, 5 μm, 120 Å, 100-μm inner diameter, 20-mm length; Nanoseparations, Nieuwkoop, The Netherlands) at a flow rate of 8 μl/min for 3 min. The peptides were separated at a flow rate of 200 nL/min on an NS-AC-11-C18 BioSphere analytical column (C18, 5 μm, 120 Å, 75-μm inner diameter, 150-mm length; Nanoseparations) with the following gradient: 5% solvent B (0.1% formic acid and acetonitrile) for 5 min; 5% to 40% solvent B for 120 min; 40% to 90% solvent B for 5 min; and 90% solvent B for 40 min. The eluted peptides were introduced into a micrOTOF-Q II mass spectrometer (Bruker Daltonics) via electrospray ionization, typically at a capillary voltage of −1.5 kV and a drying gas temperature of 150¦°C. MicrOTOF control software version 3.0 controlled the MS and MS/MS analyses by automatically switching between MS scanning and MS/MS fragmentation for the three most abundant ions within the m/z range from 300 to 3000. MS/MS fragmentation of precursor ions by means of collision-induced dissociation with Ar gas was performed at 20 to 40 eV, depending on their charge states and m/z values. Each enriched sample was examined twice via nano-LC and hybrid quadrupole TOF-MS for identification of peptide propionylation sites.
Data Processing and Mascot Analysis
MS and MS/MS spectral data obtained in 24 nano-LC-MS/MS runs were analyzed using DataAnalysis 4.0 SP5 software (Bruker Daltonics). Peak lists, including the m/z and intensity values of precursor ions, along with those of their product ions, were generated by means of the Compound-Auto MS(n) option of the DataAnalysis 4.0 SP5 software. Charge-assigned peaks after deconvolution and the 50 most abundant non-deconvoluted peaks above the intensity threshold of 150 from each MS/MS spectrum were exported to peak list files. The exported peaks were queried against a T. thermophilus HB8 database (compiled in-house) containing 2238 protein sequence entries from GenBank accession numbers AP008226, AP008227, and AP008228 of the complete genome sequence; searches were performed using the Mascot search engine (version 2.4; Matrix Science, London, UK). Propionylated peptides were identified using a mass tolerance of ±0.05 Da for precursor and product ions, and a maximum of two miscleavage sites were allowed for trypsin. Carbamidomethylation of cysteine residues was selected as a fixed modification, and propionylation of lysine residues was selected as a variable modification. Only peptides with Mascot ion scores in the 99% confidence range (p value < 0.01) were considered to have been identified. Finally, MS/MS results that simultaneously matched to amino acid sequences of tryptic peptides from potentially contaminating rabbit serum and yeast extract were eliminated. The identification results and MS/MS spectral data (ProteomeXchange accession PXD000544; PRIDE accession number 31942) were deposited in the ProteomeXchange Consortium (http://proteomecentral.proteomexchange.org) and PRIDE (www.ebi.ac.uk/pride/) databases. Because propionylated lysine is not cleaved by trypsin, putative proprionylation of C-terminal lysines was filtered out. The false discovery rate was determined by means of a Mascot decoy database search (supplemental Table S2). To improve confidence in the results, misidentified propionylpeptides were excluded via manual inspection.
2-DE Preparation and Image Analysis
Each 300-μg aliquot of protein sample was precipitated with three volumes of ice-cold acetone, and then the pellets were washed with three volumes of ice-cold acetone. After drying, the protein pellets were suspended in 250 μl of rehydration buffer (7 m urea, 2 m thiourea, 30% glycerol, 2% CHAPS, 40 mm DTT, 0.5% IPG buffer, and a trace of bromphenol blue) for IEF. The first electro-focusing process was carried out using 13-cm Immobiline DryStrips (linear, pH 4–7 and pH 3–10 NL; GE Healthcare, Piscataway, NJ) on an IPGPhor II apparatus (GE Healthcare Biosciences). Proteins were adsorbed onto the dry strips, and the strip gels were rehydrated overnight at 20¦°C. The voltage set was as follows: 0.2 kV (6 h), 0.5 kV (1 h), 1.0 kV (1 h), 8.0 kV (gradient 1 h), and 8.0 kV (up to 20,000 Vh). Prior to the second SDS-PAGE, the focused IPG strips were equilibrated for 15 min in equilibrating solution (7 m urea, 2.0% SDS, 30% glycerol, and 50 mm Tris-HCl, pH 6.8) containing 1.0% DTT and the same buffer containing 2.5% iodoacetamide. Second-dimension PAGE was carried out on a 12.5% polyacrylamide linear gradient gel (13 cm × 15 cm × 1 mm) at a constant voltage of 100 V until the dye had eluted from the bottom of the gel. Protein spots on the gel were detected by means of CBB G-250 staining. The ImageMaster 2-D Platinum software package (GE Healthcare Biosciences) and an ImageScanner (GE Healthcare Biosciences) were employed to digitalize gel images and detect protein spots. The 2-DE images were acquired at 300 dpi resolution in Melanie format, and the background of each image was then subtracted.
Validation of Identifications Using Synthetic Propionylpeptides
Two propionylpeptides, ALFAEKprDGR from TTHA1742 and LLFKprDEVR from TTHA1552, were selected for verification of propionylation sites that we identified in this study. These peptides were synthesized by Hokkaido System Science Co. (Sapporo, Japan). A 2-μl aliquot of 100 pm synthetic propionylpeptide was injected with an autosampler and then subjected to nano-LC and micrOTOF-QII using the same method and procedure described above. The MS/MS patterns and elution times of the two chemically synthesized propionylpeptides were compared with those of the corresponding propionylpeptides identified in this study.
In Silico Analysis
Functional annotation was based on the Clusters of Orthologous Groups of Proteins database (www.ncbi.nlm.nih.gov/COG/) (41), with compensation for “function unknown” and “general function prediction only” using Gene Ontology annotation (42) and KEGG Orthology (43). Ultimately, definitions of protein functions were updated based on results from all three of these annotations.
The normalized amino acid sequence frequency around propionylated lysine residues was analyzed using the iceLogo software (44). Ten amino acids on the N-terminal and C-terminal sides of propionylated lysine residues were subjected to amino acid sequence context analysis, and the results were plotted as the percent difference (p value < 0.05).
RESULTS
Identification of Propionylation Sites in T. thermophilus HB8
To identify lysine propionylation sites in T. thermophilus, we used an anti-propionyllysine polyclonal antibody to enrich propionylated peptides. We designed the experiment as follows: three biological replicates were generated for each growth stage; two affinity enrichments of propionylpeptides were performed for each replicate, and two nano-LC-MS/MS runs were performed for each affinity-enriched sample (Fig. 1B; detailed description in “Experimental Procedures”). From the data obtained, we identified hundreds of propionylated peptides from three biological replicates of T. thermophilus HB8 cells in mid-exponential and late stationary phases (p value < 0.01) (Fig. 1C). To evaluate the enrichment efficiency, we also performed mass spectrometric analyses of non-enriched peptides (data not shown) and then compared the identified unique propionylpeptides with the affinity-enriched ones. In late stationary phase, no propionylpeptides were detected among 651 unique peptides in the non-enriched samples. By contrast, 366 propionylated and 123 non-propionylated peptides were uniquely identified in the enriched samples. In mid-exponential phase, we identified no propionylated peptides among 404 unique peptides in the non-enriched samples, whereas 127 propionylated and 186 non-propionylated peptides were identified in the affinity-enriched samples. These results indicated that the enrichment procedure using the anti-propionyllysine antibody was highly efficient. After elimination of overlapping propionylation sites in each growth phase, we manually removed unclear and misidentified results. Overall, we identified a total of 361 unique propionylation sites in 183 proteins in the mid-exponential and late stationary phases (Table I and supplemental Table S1). In mid-exponential phase, we detected 121 propionylation sites in 80 proteins, and in late stationary phrase, we identified 323 lysine propionylations in 163 proteins (Fig. 1C). In mid-exponential phase, 36.4% (44/121) of the lysine propionylations were detected in all three biological replicates, and 58.7% (71/121) of the propionylations were identified at least twice (Fig. 1C). In the late stationary phase, 53.9% (174/323) of the lysine propionylations were identified in the three replicates, and 74.0% (239/323) of the propionylation sites were identified at least twice. The MS/MS results and false discovery rates are summarized in supplemental Fig. S1 and supplemental Table S2, respectively.
Table I. List of identified propionylproteins in T. thermophilus HB8.
| ORF | Definition | Propionylation sites |
|---|---|---|
| Metabolism | ||
| Energy production and conversion | ||
| TTHA0027 | Probable potassium channel, β subunit (oxidoreductase) | K31, K252 |
| TTHA0089 | NADH-quinone oxidoreductase chain 1 | K58 |
| TTHA0185 | Pyruvate dehydrogenase complex, E1 component | K323, K429 |
| TTHA0206 | Nicotinamide nucleotide transhydrogenase, α subunit 1 | K235 |
| TTHA0229 | 2-oxoisovalerate dehydrogenase, E1 α subunit | K294 |
| TTHA0230 | 2-oxoisovalerate dehydrogenase, E1 β subunit | K33, K181 |
| TTHA0232 | Pyruvate dehydrogenase complex, E2 component | K265, K274 |
| TTHA0233 | Pyruvate dehydrogenase complex, E3 component | K76, K82, K121, K170, K232, K240, K280, K284 |
| TTHA0278 | ATP-dependent phosphoenolpyruvate carboxykinase | K512 |
| TTHA0287 | 2-oxoglutarate dehydrogenase E3 component | K71, K220, K239 |
| TTHA0288 | 2-oxoglutarate dehydrogenase E2 component | K131, K219, K227, K393 |
| TTHA0466 | Alcohol dehydrogenase | K225 |
| TTHA0506 | Malate synthase | K144, K329 |
| TTHA0520 | NAD-dependent malic enzyme (malate dehydrogenase) | K10 |
| TTHA0537 | Succinyl-CoA synthetase α chain | K42, K112, K143 |
| TTHA0538 | Succinyl-CoA synthetase β chain | K45, K73, K84, K298, K311, K334, K362 |
| TTHA0558 | Fumarate hydratase class II (EC 4.2.1.2) | K121, K420 |
| TTHA0968 | Phenylacetic acid degradation protein PaaZ | K132, K305 |
| TTHA0996 | Succinate-semialdehyde dehydrogenase | K82 |
| TTHA1146 | Electron transfer flavoprotein, α subunit | K17 |
| TTHA1272 | V-type ATP synthase subunit B | K86 |
| TTHA1273 | V-type ATP synthase subunit A | K97 |
| TTHA1275 | V-type ATP synthase subunit | K271 |
| TTHA1279 | V-type ATP synthase, subunit (VAPC-THERM) | K31, K67, K97 |
| TTHA1343 | Citrate synthase | K273, K300 |
| TTHA1378 | Homoisocitrate dehydrogenase | K76 |
| TTHA1454 | Succinate dehydrogenase, flavoprotein subunit | K125, K557, K563, K573 |
| TTHA1535 | Isocitrate dehydrogenase | K46, K97, K105, K185, K196, K203, K343, K357, K368, K421, K457 |
| TTHA1578 | 1-pyrroline-5-carboxylate dehydrogenase | K73 |
| TTHA1836 | Isocitrate lyase | K190, K324, K387 |
| TTHA1965 | Inorganic pyrophosphatase | K160 |
| TTHB240 | 5-carboxy-2-hydroxymuconate semialdehyde dehydrogenase | K8 |
| Amino acid transport and metabolism | ||
| TTHA0046 | Aspartate aminotransferase | K250 |
| TTHA0216 | Alanine dehydrogenase | K73 |
| TTHA0457 | 3-phosphoshikimate 1-carboxyvinyltransferase | K22 |
| TTHA0545 | Aspartate-semialdehyde dehydrogenase | K274 |
| TTHA0582 | Aspartate aminotransferase, subgroup IV | K299, K317 |
| TTHA0722 | Histidinol dehydrogenase | K59 |
| TTHA1210 | 2-isopropylmalate synthase (LeuA) | K115, K357, K415 |
| TTHA1211 | Probable ketol-acid reductoisomerase (IlvC) | K55, K120 |
| TTHA1212 | Acetolactate synthase, small subunit (ilvN) | K62 |
| TTHA1496 | Arginase | K176 |
| TTHA1524 | Serine hydroxymethyltransferase | K245, K326 |
| TTHA1698 | Carboxypeptidase G2 | K102 |
| TTHA1755 | Acetylornithine/acetyl-lysine aminotransferase | K346, K356 |
| TTHA1781 | GTPase ObgEa | K30, K147 |
| TTHA1928 | γ-Glutamyl phosphate reductase | K75, K232, K317 |
| Carbohydrate transport and metabolism | ||
| TTHA0002 | Enolase (2-phosphoglycerate dehydratase) | K61 |
| TTHA0003 | Pyruvate kinase | K74 |
| TTHA0106 | Ribulose-phosphate 3-epimerase | K72 |
| TTHA0108 | Transketolase | K25, K347 |
| TTHA0116 | Phosphonopyruvate decarboxylase | K192, K217, K300 |
| TTHA0299 | Glucokinase | K21, K33 |
| TTHA0481 | Oligo-1,6-glucosidase | K435 |
| TTHA0650 | Putative phosphoglucomutase/phosphomannomutase | K127 |
| TTHA0905 | Glyceraldehyde 3-phosphate dehydrogenase | K75, K112, K190, K221, K246, K267, K294, K297 |
| TTHA0906 | Phosphoglycerate kinase | K10, K122, K244 |
| TTHA0980 | Bifunctional fructose 1,6-bisphosphate aldolase/phosphatasea | K84, K233 |
| TTHA1299 | Ribose 5-phosphate isomerase | K67 |
| TTHA1446 | Fructose-1,6-bisphosphatase, class II | K31 |
| TTHA1773 | Fructose-1,6-bisphosphate aldolase | K148, K240, K241, K273, K279 |
| Coenzyme transport and metabolism | ||
| TTHA0341 | Molybdopterin biosynthesis enzyme, MoaB | K133 |
| TTHA0345 | Porphobilinogen deaminase | K26 |
| TTHA0617 | Nicotinate phosphoribosyltransferase | K428 |
| TTHA0634 | Magnesium chelatase related protein | K110 |
| TTHA0966 | Phenylacetyl-CoA ligase | K439 |
| TTHA1120 | Methylene-THF dehydrogenase (cyclohydrolase) | K169 |
| TTHA1642 | S-adenosylmethionine synthetase | K37 |
| TTHA1738 | 3-oxoadipate enol-lactonasea | K100, K135 |
| TTHA1789 | Molybdenum cofactor biosynthesis protein C | K19 |
| TTHB053 | Precorrin-6Y C5,15-methyltransferase [decarboxylating] | K85, K111, K186, K231 |
| TTHB054 | Precorrin-2 methylase | K170 |
| TTHB055 | Precorrin-4 C11-methyltransferase | K57 |
| TTHB057 | Cobalamin biosynthesis protein CbiG | K91, K274, K336 |
| Lipid transport and metabolism | ||
| TTHA0413 | 3-oxoacyl-[acyl carrier protein] synthase II | K245 |
| TTHA0415 | 3-oxoacyl-[acyl carrier protein] reductase | K38 |
| TTHA0559 | Acetyl coenzyme A acetyltransferase (thiolase) | K16, K188, K212 |
| TTHA0750 | 3-oxoacyl-[acyl carrier protein] reductase | K95, K196 |
| TTHA0890 | Putative 3-hydroxyacyl-CoA dehydrogenase | K304, K467 |
| TTHA0892 | Acyl-CoA dehydrogenase | K221, K266 |
| TTHA0987 | β-Ketoadipyl CoA thiolase | K211, K214 |
| TTHA1124 | Acetyl-CoA carboxylase, biotin carboxyl carrier protein | K111, K143 |
| TTHA1248 | Acetyl-coenzyme A synthetase | K368, K612, K629 |
| TTHA1262 | 3-hydroxybutyryl-CoA dehydrogenase | K58, K285 |
| TTHA1434 | 3-hydroxybutyryl-CoA dehydratase | K81, K87, K142 |
| TTHA1768 | Acetyl-CoA carboxylase carboxyl transferase, β subunit | K87 |
| TTHB019 | MaoC-related acyl dehydratase | K114 |
| Nucleotide transport and metabolism | ||
| TTHA0075 | Ribonucleoside-diphosphate reductase | K51, K200, K2024, K2060 |
| TTHA0188 | Nucleoside diphosphate kinase | K55, K94 |
| TTHA0432 | IMP dehydrogenase/GMP reductase | K411, K417 |
| TTHA1466 | CTP synthase | K462 |
| TTHA1552 | GMP synthase | K102, K359 |
| TTHA1614 | Adenine phosphoribosyltransferase | K176 |
| TTHA1671 | Adenylate kinase | K150 |
| TTHA1742 | Orotate phosphoribosyltransferase | K88, K98 |
| TTHB209 | Ribonucleoside-diphosphate reductase, α subunit | K38 |
| Inorganic ion transport and metabolism | ||
| TTHA0045 | Probable potassium uptake protein TrkA | K372 |
| TTHA0122 | Manganese-containing pseudocatalase | K217 |
| TTHA0557 | Superoxide dismutase [Mn] | K6, K104, K105, K118, K120 |
| TTHA0706 | Cation-transporting ATPase | K70, K197 |
| TTHA1028 | Thiosulfate sulfurtransferase | K207, K216 |
| TTHA1477 | TrkA domain proteina | K92 |
| Secondary metabolite biosynthesis, transport, and catabolism | ||
| TTHA0223 | Type 11 methyltransferasea | K176 |
| TTHA0483 | Probable transcriptional regulator, ArsR family | K4 |
| TTHA0809 | Probable 2-hydroxyhepta-2,4-diene-1,7-dioate isomerase | K154 |
| General function prediction only | ||
| TTHA0474 | Acetoin utilization protein AcuB (acetoin dehydrogenase) | K62 |
| TTHA0829 | Putative acetoin utilization protein, acetoin dehydrogenase | K50 |
| TTHA0972 | Phenylacetic acid degradation protein PaaA | K230 |
| Translation | ||
| TTHA0142 | Peptide chain release factor 2 | K335 |
| TTHA0161 | Leucyl-tRNA synthetase | K20, K205, K215, K468, K653 |
| TTHA0162 | 30S ribosomal protein S1 | K105 |
| TTHA0210 | 50S ribosomal protein L12 | K65, K87, K90, K95, K105, K112 |
| TTHA0246 | 50S ribosomal protein L1 | K85 |
| TTHA0250 | 50S ribosomal protein L33 | K17, K38 |
| TTHA0251 | Translation elongation factor EF-Tu.B | K3 |
| TTHA0366 | Aspartyl/glutamyl-tRNA amidotransferase subunit B | K256 |
| TTHA0543 | Glycyl-tRNA synthetase | K322, K443, K475 |
| TTHA0551 | Translation initiation factor 3 (IF-3) | K65 |
| TTHA0712 | Histidyl-tRNA synthetase | K10 |
| TTHA0858 | Ribosome recycling factor (Rrf) | K4, K112, K145 |
| TTHA0860 | Elongation factor Ts (EF-Ts) | K22, K120 |
| TTHA0875 | Seryl-tRNA synthetase | K66 |
| TTHA1067 | Isoleucyl-tRNA synthetase (IleRS) | K878 |
| TTHA1125 | Elongation factor P (EF-P) | K42 |
| TTHA1139 | Polynucleotide phosphorylase | K532 |
| TTHA1169 | Valyl-tRNA synthetase (ValRS) | K97, K845 |
| TTHA1294 | Ribosomal subunit interface protein | K122, K165 |
| TTHA1298 | Methionyl-tRNA synthetase | K429 |
| TTHA1397 | 30S ribosomal protein S20 | K48, K54 |
| TTHA1399 | Tyrosyl-tRNA synthetase (TyrRS) | K328 |
| TTHA1485 | Peptide chain release factor 1a | K4, K107 |
| TTHA1663 | 50S ribosomal protein L17 | K78 |
| TTHA1667 | 30S ribosomal protein S13 | K46, K79 |
| TTHA1675 | 30S ribosomal protein S5 | K145 |
| TTHA1678 | 30S ribosomal protein S8 | K46 |
| TTHA1680 | 50S ribosomal protein L5 | K47 |
| TTHA1684 | 30S ribosomal protein L29 | K8 |
| TTHA1695 | Elongation factor G (EF-G) | K10, K381, K513 |
| TTHA1696 | 30S ribosomal protein S7 | K53 |
| TTHA1697 | 30S ribosomal protein S12 | K111 |
| TTHA1875 | Threonyl-tRNA synthetase | K62, K249 |
| Post-translational modification, protein turnover, chaperones | ||
| Protein refolding | ||
| TTHA0271 | 60 kDa chaperonin, GroEL | K41, K42, K50, K116, K121, K129, K131, K169, K325, K334, K336, K337, K349, K362, K388, K429, K430, K440, K529 |
| TTHA0272 | 10 kDa chaperonin, GroES | K26, K28, K45, K66 |
| TTHA0614 | Trigger factor | K67 |
| TTHA1480 | Small heat shock protein, HSP20 family | K15, K123 |
| TTHA1484 | Small heat shock protein, HSP20 family | K8 |
| TTHA1487 | ATP-dependent Clp protease, ClpB | K424, K433, K448, K499, K510, K558, K630 |
| TTHA1491 | Chaperone protein DnaK (heat shock protein 70) | K84, K126, K245, K514 |
| Stress response | ||
| TTHA0370 | Thioredoxin reductase related protein | K108 |
| TTHA0593 | Probable thiol-disulfide isomerase/thioredoxin | K140 |
| TTHA1300 | Putative bacterioferritin comigratory protein/thiol peroxidase | K114 |
| TTHA1482 | Bacterioferritin | K69 |
| TTHA1625 | Osmotically inducible protein OsmC | K90, K120 |
| TTHA1635 | Iron-sulfur cluster biosynthesis protein IscA | K115 |
| TTHA1714 | Heme peroxidasea | K149 |
| TTHA1838 | SufC protein (ATP-binding protein) | K124, K149, K243 |
| TTHA1840 | SufD protein (membrane protein) | K14, K25, K28, K41, K42, K109, K336, K415 |
| TTHB162 | Crispr-associated ramp cmr1 familya | K359 |
| TTHB190 | Crispr-associated protein cse4a | K303, K318, K360 |
| Transcription | ||
| TTHA0008 | Phage shock protein A | K74 |
| TTHA0101 | Transcriptional repressor, TetR family | K185 |
| TTHA0248 | Transcription antitermination protein NusG | K27 |
| TTHA0701 | N utilization substance protein A (NusA) | K143, K182 |
| TTHA1664 | DNA-directed RNA polymerase α chain | K7, K270 |
| TTHA1812 | DNA-directed RNA polymerase β′ chain (RpoC) | K428 |
| TTHA1813 | DNA-directed RNA polymerase β chain (RpoB) | K86 |
| Replication, recombination, and repair | ||
| TTHA0180 | DNA polymerase III α subunit | K505 |
| TTHA1349 | DNA-binding protein HU | K23, K28, K68, K72, K76 |
| TTHA1583 | Site-specific DNA-methyltransferase | K338 |
| Intracellular trafficking and secretion | ||
| TTHA0365 | Type IV pilus assembly protein, PilT | K347 |
| TTHA0443 | Putative membrane protein | K298 |
| TTHA1251 | Preprotein translocase SecA subunit | K649 |
| Cell cycle control, mitosis, and meiosis | ||
| TTHA0564 | Cell division initiation protein DivIVA | K112 |
| TTHA1816 | Rod shape-determining protein MreB | K224 |
| Cell wall/membrane biogenesis | ||
| TTHA0850 | SpoVS related protein | K26 |
| General function prediction only | ||
| TTHA0051 | Conserved hypothetical protein | K8 |
| TTHA0420 | Putative flavoprotein | K84 |
| TTHA0810 | Metallo-β-lactamase related protein | K239, K291 |
| TTHA0961 | Flavin reductase component (HpaC)a | K6 |
| TTHA1483 | Conserved putative protein | K45, K147, K221 |
| TTHA1662 | Conserved hypothetical protein | K85 |
| Function unknown | ||
| TTHA1048 | Conserved hypothetical protein | K303 |
| TTHA1554 | Conserved hypothetical protein | K26 |
| TTHB059 | Hypothetical protein | K42 |
a The definition of protein function was updated using Gene Ontology annotation and KEGG Orthology.
Validation of Propionylation Sites with Chemically Synthesized Peptides
Because each peptide exhibited a composition-specific MS/MS spectrum and elution time on C18 reverse phase nano-LC, comparison of the MS/MS spectral patterns and elution times of identified propionylpeptides and the corresponding synthetic peptides allowed us to validate the newly identified PTMs (5, 10, 25). Thus, we attempted to validate the lysine propionylations (+56.026215-Da mass shift) using chemically synthesized propionylpeptides. Fig. 2 shows the similarities in elution times and MS/MS spectra between a propionylpeptide identified in this study, ALFAEKprDGR (m/z 531.782, +2) from TTHA1742, and the corresponding synthetic peptide. This finding strongly supports our identification of this lysine residue of TTHA1742 as a propionylation site. Additionally, we verified the propionylation of another peptide, LLFKprDEVR (m/z 538.311, +2) from TTHA1552, by comparing its nano-LC elution time and MS/MS spectral pattern with those of the corresponding synthetic peptide (supplemental Fig. S2).
Fig. 2.

Verification of propionylation of the enriched peptide (ALFAEKprDGR) in vivo by comparison of elution times and MS/MS spectra of the synthetic peptide. Extracted ion current chromatograms (A) and MS/MS spectra (B) of m/z 531.782 for the affinity-enriched propionylpeptide and the synthetic propionylpeptide.
Functional Classification of Propionylated Proteins
To determine which cellular functions were influenced by lysine propionylations, we assessed the functions of the proteins in which we had identified propionylation sites. We assigned a functional group to each propionylprotein using information from Clusters of Orthologous Groups, Gene Ontology annotation, and KEGG Orthology. From this analysis, we identified 10 distinct functional classes (Fig. 3A and Table I). Our functional classification revealed that 58.7% of propionylproteins belonged to the “metabolism” class. Further subcategorization of the “metabolism” class into nine subclasses revealed that the “energy production and conversion” subclass was the largest group (32 proteins), followed by the “coenzyme,” “carbohydrate,” “amino acid,” and “lipid transport and metabolism” subclasses (15, 14, 14, and 13 proteins, respectively) (Fig. 3B). The “energy production and conversion” class included highly propionylated enzymes such as isocitrate dehydrogenase (11 sites), pyruvate dehydrogenase complex (15 sites), 2-oxoisovalerate dehydrogenase complex (10 sites), and succinyl-CoA synthetase complex (10 sites). The “carbohydrate transport and metabolism” subclass also included a highly propionylated enzyme, glyceraldehyde 3-phosphate dehydrogenase (eight sites). These multiply propionylated proteins are involved in central metabolism. Overall, our analyses revealed that a total of 24 enzymes in the central metabolism pathways (glycolysis/gluconeogenesis and citric acid cycle) were propionylated (supplemental Fig. S3).
Fig. 3.

Functional classification of propionylproteins and the amino acid sequence context around propionylated lysine. A, distributions of the functional classes of 184 propionylproteins. B, distributions of functional subclasses of 108 propionylproteins in the “metabolism” class. C, the frequency of amino acid residues around propionylated lysines (p value = 0.05).
The “translation” class was the second largest group, including a total of 33 proteins: 13 ribosomal proteins, 9 aminoacyl-tRNA synthetases, 4 translation elongation factors, 2 peptide release factors, 1 ribosome recycling factor, and 1 translation initiation factor. The third largest class, “post-translational modification, protein turnover, chaperones,” contained 18 proteins, which were further categorized into two subclasses, “protein refolding” (7 proteins) and “stress response” (11 proteins). These classes included highly propionylated proteins such as GroEL, which has the largest number of propionylation sites (19 sites); ClpB (7 sites); and the SufCD proteins (total of 11 sites).
It was possible that the large numbers of propionylproteins belonging to the “metabolism” class, as well as the relatively large numbers of propionylation sites in individual members of this class, might have been a consequence of protein abundance. To test this idea, we investigated the abundance of the propionylated proteins using 2-DE reference maps of protein abundance in T. thermophilus, compiled in a previous study by our group (39). This analysis revealed, however, that 16 of the 41 most abundant proteins were not propionylated (supplemental Table S5), suggesting that protein abundance does not contribute to propionylation.
Local Sequence Context around Propionylation Sites
To determine whether lysine propionylation tends to occur near specific amino acids, we investigated the amino acid sequence frequencies of the 10 residues on either side of propionylated lysines (Fig. 3C). Negatively charged amino acids, mainly glutamic acid, tended to be distributed near propionylated lysines. In particular, the +1 position was preferentially occupied by either glutamic acid or aspartic acid, whereas the −1 position exhibited no such preference. This is reminiscent of our previous observation that acidic amino acids frequently occur near acetylation sites in T. thermophilus (22). Basic amino acids, typically lysine, were relatively more abundant in the general vicinity of propionylation sites (i.e. from the −10 to +10 positions) than acidic amino acids. In addition, propionylation was less common near hydrophobic amino acids, particularly leucine. However, this preference was sensitive to position: the +1 position robustly rejected leucine, whereas the −1 position actually favored leucine.
Comparison of Lysine Propionylation between the Mid-exponential and Late Stationary Phases
We observed a significant increase in propionylation in late stationary phase (Fig. 1C). Therefore, we investigated whether the number of propionylation sites differed between the mid-exponential and late stationary phases, as well as how many propionylation sites overlapped between the two phases (Fig. 4). As shown in supplemental Fig. S4, Western blot analysis using anti-propionyllysine polyclonal antibody demonstrated that the level of propionylation increased in late stationary phase. For further comparative analysis of the two phases, we focused on propionylation sites identified at least twice in three different biological samples (Fig. 1B). Of these, 71 unique propionylation sites in 52 proteins were identified in mid-exponential phase, and 239 propionylation sites in 130 proteins were detected in late stationary phase (Fig. 4). Thus, the numbers of propionylation sites and propionylated proteins increased 3.3-fold and 2.5-fold, respectively, in late stationary phase relative to mid-exponential phase. Comparison of propionylation sites between the two growth phases revealed that 24 sites in 12 proteins were specific to mid-exponential phase, and 192 sites in 90 proteins were specific to late stationary phase. Thus, 47 propionylation sites in 40 proteins overlapped between the two growth phases (Fig. 4).
Fig. 4.

Comparison of propionylation sites between mid-exponential and late stationary phases. The numbers of identified propionylproteins (n ≥ 2) in each growth phase are shown in the upper Venn diagram, and the numbers of propionylation sites (n ≥ 2) are shown in the left Venn diagram. The numbers of propionylation sites on 40 proteins that were propionylated in both growth phases are shown in the right Venn diagram.
To rule out the possibility that elevated protein expression resulted in the greater number of propionylation sites in stationary phase, we compared the expression levels of propionylated proteins between mid-exponential and late stationary phases via 2-DE analysis of whole-cell lysates. For many propionylated proteins that were frequently propionylated in stationary phase, the expression levels did not significantly differ in exponential phase (Fig. 5 and supplemental Fig. S5). For instance, two propionylation sites in TTHA1028, a thiosulfurtransferase, were detected only in mid-exponential phase, and not in late stationary phase, even though the expression of TTHA1028 was increased about 2-fold in stationary phase. Propionylation of TTHA0593, a probable thiol-disulfide/thioredoxin, was not elevated in late stationary phase, even though the expression level increased dramatically (over 2.5-fold) (supplemental Fig. S5). Moreover, we compared propionylation sites in 40 proteins that were identified in both mid-exponential and late stationary phases (supplemental Table S1). A total of 122 propionylation sites in these 40 proteins were detected in both phases (Fig. 4); among these, 47 (38.5%) overlapped between the two phases. Remarkably, 65 propionylation sites were detected uniquely in late stationary phase, whereas 10 propionylation sites were unique to mid-exponential phase. Thus, the expression level of a protein might not contribute to its propionylation. The growth-phase-dependent differences in propionylation sites suggest that propionylation plays a biologically significant role in the growth of T. thermophilus.
Fig. 5.

Comparative analyses of the expression levels of representative propionylated proteins between exponential and stationary phase. Two replicates for each growth phase were prepared. A reference gel image of mid-exponential phase of T. thermophilus, which reported spot numbers and identification results from our previous study (36), was used for comparative analysis as one of the exponential phase images in this study. Full images of 2-DE gels (pI 4 to 7, 13 cm) and each spot number are shown in supplemental Fig. S5. The left side shows the three-dimensional spot image, and the right side shows the magnified two-dimensional spot image of each propionylated protein on 2-DE gels. Red arrows indicate spots corresponding to target proteins. The number of identified propionylation sites in each propionylprotein is given in parentheses. The black number indicates the total number of identified propionylation sites (n ≥ 1) in both growth phases. The blue and red numbers indicate propionylation sites identified in mid-exponential and late stationary phase, respectively. The changes of theoretical pI values are presented in the upper 2-DE images. ME and LS indicate mid-exponential and late stationary phase, respectively. The spot numbers of specific proteins on the 2-DE reference gel are as follows: TTHA0161, Spot No. 3; TTHA0543, Spot No. 46; TTHA1028, Spot Nos. 180 and 181; TTHA1535, Spot No. 47; TTHA1838, Spot No. 150; TTHA1839, Spot Nos. 24 and 25; TTHA1840, Spot Nos. 60 and 61.
Comparative Analysis of Functional Classes of Propionylated Proteins between Two Growth Phases
Because we observed differences in protein propionylation between mid-exponential and late stationary phases, we sought to determine which biological processes are influenced by propionylation in a growth-phase-dependent manner (Fig. 6). In this analysis, we categorized the propionylproteins identified in each phase into 10 functional classes, and further categorized the “metabolism” class into nine subclasses, in the manner described above. Even though the percentage of proteins in the “translation” class dramatically decreased in late stationary phase, the absolute number did not change meaningfully (12 proteins in the mid-exponential phase versus 14 proteins in the late stationary phase) (Fig. 6A). By contrast, both the number and the percentage of identified proteins in the “metabolism” class significantly increased in late stationary phase (87 proteins, 69.6%) relative to mid-exponential phase (29 proteins, 55.7%) (Fig. 6A). Additionally, in the “metabolism” subclasses, the level of propionylation in late stationary phase increased globally (Fig. 6A). However, the proportions of most subclasses did not change between the two growth phases, with two exceptions: “amino acid transport and metabolism,” which increased from 10.3% to 17.2% between mid-exponential and late stationary phase, and “lipid transport and metabolism,” which decreased from 20.7% to 12.6%. (Fig. 6B). The subclass “post-translational modification, protein turnover, chaperones” was also larger in late stationary phase (15 proteins) than in mid-exponential phase (7 proteins) (Fig. 6A). The number of propionylation sites in proteins of the “metabolism” and “post-translational modification, protein turnover, chaperones” classes also increased dramatically in late stationary phase (supplemental Table S3). By contrast, the number of propionylation sites in the “translation” class was hardly affected by growth phase.
Fig. 6.

Functional classification of propionylproteins and the amino acid sequence context around propionylated lysines in mid-exponential and late stationary phases. Propionylation sites that were detected at least twice in biological triplicates were counted for these analyses. A, distributions of functional classes of propionylproteins. B, distributions of functional subclasses of propionylproteins in the “metabolism” class. C, the frequency of amino acid residues around propionylated lysines (p value = 0.05).
Comparative analysis of the amino acid sequence context near propionylated lysine residues between the two growth phases revealed that the relative frequency of lysine in the vicinity of propionylation sites (i.e. from the −10 to the +10 position) was elevated in stationary phase, whereas the frequency of acidic amino acids did not change significantly (Fig. 6C). Additionally, hydrophobic amino acids, especially leucine, were relatively less abundant near propionylation sites in late stationary phase (Fig. 6C).
DISCUSSION
Lysine propionylation, a post-translational acylation, has been previously observed on several proteins, including propionyl-CoA synthetase in bacteria and histones in eukaryotes (27–32). In an earlier study, we analyzed the acetylome of T. thermophilus using an immunoprecipitation method and identified 197 lysine acetylation sites in 127 proteins (22). Given the similarity of the chemical structures of acetylated and propionylated lysine residues, it is reasonable to assume that the same strategy used to identify acetylpeptides could be applied to propionylpeptides (Fig. 1A). Through Western blot analysis using an anti-propionyllysine antibody developed by our group, we found that propionylation occurred in T. thermophilus, and that the levels of this modification increased in stationary phase (supplemental Fig. S4). By means of affinity enrichment using this antibody, we identified 361 propionylation sites in 183 proteins, indicating that lysine propionylation is one of the most prevalent PTMs in T. thermophilus, comparable to acetylation and phosphorylation (33, 45).
Next, we showed that lysine propionylation mainly occurred on proteins associated with the “metabolism” functional class (Fig. 3A). In this class, “energy production and conversion” was the largest subclass (Fig. 3B). These results suggest that lysine propionylation may regulate metabolism. A deep relationship between lysine acetylation and metabolism has been proposed in various eukaryotes and bacteria (6, 20, 21, 46, 47). The similarity between lysine acetylation and propionylation suggests that these two types of acylation may control the activities of metabolic enzymes; however, the specific role of each acylation remains unclear.
In our analyses, we carefully considered the possibility that protein abundance might influence the rate of propionylation of many metabolic enzymes. Indeed, many of the propionylproteins in the “metabolism” class are included in the protein abundance list presented in our previous report (39). However, 16 of the 41 most abundant proteins were not propionylated, supporting the idea that specific mechanisms regulate the propionylation levels of metabolic enzymes (supplemental Table S5). Furthermore, Fig. 5 and supplemental Fig. S5 show that the greater number of propionylation sites in late stationary phase relative to mid-exponential phase was not associated with an increase in protein expression (i.e. the rate of propionylation was not correlated with protein levels).
Among the 361 propionylation sites we identified in 183 proteins, 121 sites in 80 proteins were detected in mid-exponential phase, and 323 sites in 163 proteins were detected in late stationary phase (Fig. 1B). To elucidate the significance of the dependence of propionylation on the growth phase, we performed a comparative analysis of propionylproteins between mid-exponential and late stationary phases (Fig. 6). In stationary phase, metabolism-related proteins were highly propionylated, but the number of translation-related proteins did not significantly change (Fig. 6A). The finding that 61.5% of propionylations were growth-stage-specific also excludes the possibility that the greater number of propionylation sites on metabolic enzymes in stationary phase was a consequence of protein abundance. These results provide further support for the idea that lysine propionylation is regulated by specific mechanisms that depend on growth stage. Additionally, amino acid sequence context analysis revealed growth-phase-specific differences, such as the relative enrichment of basic amino acids and the relative depletion of leucine, in late stationary versus mid-exponential phase (Fig. 6C). Because the amino acid sequence context around modified sites reflects the substrate specificity of the enzymes that catalyze the modifications, these differences in sequence context between the two growth phases suggest that propionylation is regulated by enzymes that are expressed in a growth-phase-dependent manner.
Recent studies have proposed that non-enzymatic acylation may occur as a PTM in E. coli, yeast, mouse liver, and several human cell lines (11, 12, 48). In particular, these reports argued that non-enzymatic acylation by acyl donors such as acetylphosphate, acetyl-CoA, and succinyl-CoA occurs frequently in cellular proteins, and that acylation levels depend on the concentrations of these metabolites. Because the properties of these donors are similar to those of acetyl-CoA and acetylphosphate, propionylation is also predicted to occur non-enzymatically; if the same principle holds true, propionyl-CoA and propionylphosphate would be candidate acyl donors. A previous study on non-enzymatic acylation on proteins revealed that over 50% of succinylation sites are also acetylated in E. coli, yeast, and mouse liver cells, suggesting that most of the accessible lysine residues might be targeted by succinylation (12).
In this study, we compared the propionylation sites with the previously reported acetylation sites of T. thermophilus (22). Specifically, we compared 197 acetylation sites in 127 proteins identified in late stationary phase with 323 propionylation sites in 163 proteins in the same growth phase. The relatively high frequency of acidic amino acids near propionylation sites was similar to the distribution of these residues near acetylation sites. However, whereas glutamic acid was often present at the −1 position of acetylation sites, this position was much more likely to be occupied by leucine and lysine adjacent to a propionylation site. Moreover, the distribution of basic amino acids, especially lysine, near propionylation sites was distinct from the distribution near acetylation sites. The differences in amino acid frequencies in the vicinities of propionylation and acetylation sites supports the idea that these modifications are regulated by distinct machineries and play different biological roles in T. thermophilus. We also found that only 19.5% (63/323) of the propionylation sites and 35% (63/197) of the acetylation sites overlapped (Fig. 7 and supplemental Table S4). The overlap between propionylation and acetylation in T. thermophilus is much smaller than that between succinylation and acetylation in E. coli, yeast, and mouse liver cells (12). Furthermore, we investigated 67 proteins that were both acetylated and propionylated (acetylpropionylproteins) and found that 63% (108/171) of the propionylation sites and 51% (65/128) of the acetylation sites from these acetylpropionylproteins did not overlap (Fig. 7). Therefore, it is likely that the majority of these two types of acylations are regulated separately, consistent with the idea that lysine propionylation and acetylation mediate different functions.
Fig. 7.
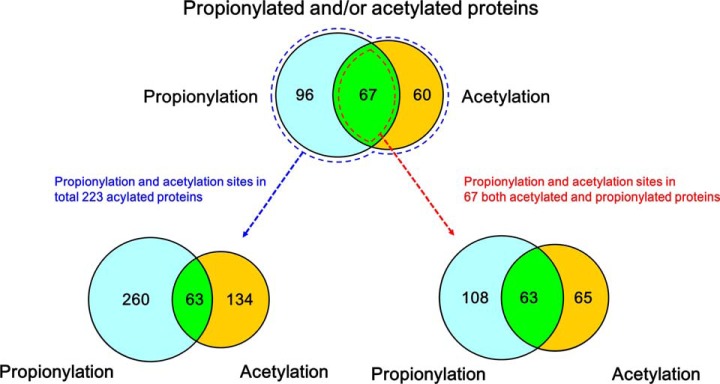
Comparison of propionylation and acetylation sites in T. thermophilus. The numbers of identified propionylproteins and acetylproteins are shown in the upper Venn diagram, and the numbers of modification sites are shown the left Venn diagram. The numbers of propionylation and acetylation sites in 47 acetylproprionylproteins are shown in the right Venn diagram.
We propose two possible mechanisms for the distinct regulation of these modifications. First, lysine propionylation and acetylation may be catalyzed by different acyltransferases. Second, even though acylations can occur non-enzymatically, the modification levels of propionylation and acetylation may be individually regulated by distinct sets of deacylases. Recently, Cheng et al. reported that Tip60, a histone acetyltransferase, exhibits different activities toward acetyl-CoA and propionyl-CoA when using these compounds as acyl donors to modify p53 (28). Feldman and colleagues reported differences in the catalytic activities of sirtuins (SIRT1 through SIRT6), NAD+-dependent protein deacetylases, toward 13 types of acyllysine residues (38). These studies support our hypothesis that distinct sets of acyltransferases and/or deacylases regulate acetylation and propionylation. In fact, we found three possible deacylases (TTHA1392, Sirt2 family; TTHA0475, AcuC; TTHA0052, histone deacetylase homolog) in the complete genome sequence of T. thermophilus HB8. We speculate that these three deacylases have different substrate specificities. Therefore, we hypothesize that the regulation of different types of deacylation via distinct mechanisms results in the prevalence of propionylation over acetylation in T. thermophilus.
Although the overlap between propionylation and acetylation sites is consistent with the idea of a non-enzymatic reaction, we should note that some of these modifications could be catalyzed by the same enzymes. Notably, we found that eight propionylation sites overlapped with acetylation sites that were previously reported to be critical for protein functions such as Schiff-base formation, ligand binding, and nucleic-acid binding (supplemental Table S4). Given that some of the enzymes that catalyze reactions involving acyl groups, such as acyl-CoA synthetases (49) and acetate kinase (50), recognize different lengths or types of acyl chains, it is conceivable that acyltransferases and deacylases with broad substrate specificities catalyze the two types of acylation and thereby regulate enzymatic activities.
In the past few years, several new types of lysine acylation have been reported. However, except for acetylation and succinylation, the distributions of other types of acylation remain poorly characterized. In this study, we showed that propionylation is a prevalent PTM and that it occurs in a large set of metabolic enzymes in a growth-phase-dependent manner, indicating that propionylation may control metabolism. The existence of growth-phase-specific propionylation sites, as well as the low degree of overlap between acetylation and propionylation sites, suggests that specific mechanisms regulate propionylation and acetylation and that the majority of the two modifications are catalyzed separately. Moreover, the small number of propionylation and acetylation sites that do overlap (19.5%) includes eight sites critical for protein functions, indicating that acetylation and propionylation play important roles in cells and may be regulated by enzymes with broad substrate specificities.
Supplementary Material
Acknowledgments
The mass spectrometry proteomics data have been deposited with the ProteomeXchange Consortium (http://proteomecentral.proteomexchange.org) via the PRIDE partner repository with the dataset identifier PXD000544 and DOI 10.6019/PXD000544.
Footnotes
Author contributions: H.O., K.K., R.M., and S.K. designed research; H.O. and K.K. performed research; H.O. and K.K. contributed new reagents or analytic tools; H.O. and K.K. analyzed data; H.O., K.K., R.M., and S.K. wrote the paper.
 This article contains supplemental material.
This article contains supplemental material.
1 The abbreviations used are:
- PTM
- post-translational modification
- nano-LC
- nano-scale liquid chromatography
- 2-DE
- two-dimensional gel electrophoresis
- IPG
- immobilized pH gradient
- IEF
- isoelectric focusing.
REFERENCES
- 1. Witze E. S., Old W. M., Resing K. A., Ahn N. G. (2007) Mapping protein post-translational modifications with mass spectrometry. Nat. Methods 4, 798–806 [DOI] [PubMed] [Google Scholar]
- 2. Walsh C. T. (2006) Posttranslational Modification of Proteins: Expanding Nature's Inventory, Roberts & Co., Englewood, CO [Google Scholar]
- 3. Zhao Y., Jensen O. N. (2009) Modification-specific proteomics: strategies for characterization of post-translational modifications using enrichment techniques. Proteomics 9, 4632–4641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Lin H., Su X., He B. (2012) Protein lysine acylation and cysteine succination by intermediates of energy metabolism. ACS Chem. Biol. 7, 947–960 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Zhang Z., Tan M., Xie Z., Dai L., Chen Y., Zhao Y. (2011) Identification of lysine succinylation as a new post-translational modification. Nat. Chem. Biol. 7, 58–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Park J., Chen Y., Tishkoff D. X., Peng C., Tan M., Dai L., Xie Z., Zhang Y., Zwaans B. M., Skinner M. E., Lombard D. B., Zhao Y. (2013) SIRT5-mediated lysine desuccinylation impacts diverse metabolic pathways. Mol. Cell 50, 919–930 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kim S. C., Sprung R., Chen Y., Xu Y., Ball H., Pei J., Cheng T., Kho Y., Xiao H., Xiao L., Grishin N. V., White M., Yang X. J., Zhao Y. (2006) Substrate and functional diversity of lysine acetylation revealed by a proteomics survey. Mol. Cell 23, 607–618 [DOI] [PubMed] [Google Scholar]
- 8. Choudhary C., Kumar C., Gnad F., Nielsen M. L., Rehman M., Walther T. C., Olsen J. V., Mann M. (2009) Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science 325, 834–840 [DOI] [PubMed] [Google Scholar]
- 9. Zhang K., Zheng S., Yang J. S., Chen Y., Cheng Z. (2013) Comprehensive profiling of protein lysine acetylation in Escherichia coli. J. Proteome Res. 12, 844–851 [DOI] [PubMed] [Google Scholar]
- 10. Peng C., Lu Z., Xie Z., Cheng Z., Chen Y., Tan M., Luo H., Zhang Y., He W., Yang K., Zwaans B. M., Tishkoff D., Ho L., Lombard D., He T. C., Dai J., Verdin E., Ye Y., Zhao Y. (2011) The first identification of lysine malonylation substrates and its regulatory enzyme. Mol. Cell. Proteomics 10, M111.012658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Weinert B. T., Iesmantavicius V., Wagner S. A., Scholz C., Gummesson B., Beli P., Nystrom T., Choudhary C. (2013) Acetyl-phosphate is a critical determinant of lysine acetylation in E. coli. Mol. Cell 51, 265–272 [DOI] [PubMed] [Google Scholar]
- 12. Weinert B. T., Scholz C., Wagner S. A., Iesmantavicius V., Su D., Daniel J. A., Choudhary C. (2013) Lysine succinylation is a frequently occurring modification in prokaryotes and eukaryotes and extensively overlaps with acetylation. Cell Rep. 4, 842–851 [DOI] [PubMed] [Google Scholar]
- 13. Du J., Zhou Y., Su X., Yu J. J., Khan S., Jiang H., Kim J., Woo J., Kim J. H., Choi B. H., He B., Chen W., Zhang S., Cerione R. A., Auwerx J., Hao Q., Lin H. (2011) Sirt5 is a NAD-dependent protein lysine demalonylase and desuccinylase. Science 334, 806–809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Yu B. J., Kim J. A., Moon J. H., Ryu S. E., Pan J. G. (2008) The diversity of lysine-acetylated proteins in Escherichia coli. J. Microbiol. Biotechnol. 18, 1529–1536 [PubMed] [Google Scholar]
- 15. Zhang J., Sprung R., Pei J., Tan X., Kim S., Zhu H., Liu C. F., Grishin N. V., Zhao Y. (2009) Lysine acetylation is a highly abundant and evolutionarily conserved modification in Escherichia coli. Mol. Cell. Proteomics 8, 215–225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Wang Q., Zhang Y., Yang C., Xiong H., Lin Y., Yao J., Li H., Xie L., Zhao W., Yao Y., Ning Z. B., Zeng R., Xiong Y., Guan K. L., Zhao S., Zhao G. P. (2010) Acetylation of metabolic enzymes coordinates carbon source utilization and metabolic flux. Science 327, 1004–1007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Zhao S., Xu W., Jiang W., Yu W., Lin Y., Zhang T., Yao J., Zhou L., Zeng Y., Li H., Li Y., Shi J., An W., Hancock S. M., He F., Qin L., Chin J., Yang P., Chen X., Lei Q., Xiong Y., Guan K. L. (2010) Regulation of cellular metabolism by protein lysine acetylation. Science 327, 1000–1004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Phillips D. M. (1963) The presence of acetyl groups of histones. Biochem. J. 87, 258–263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Allfrey V. G., Faulkner R., Mirsky A. E. (1964) Acetylation and methylation of histones and their possible role in the regulation of RNA synthesis. Proc. Natl. Acad. Sci. U.S.A. 51, 786–794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Patel J., Pathak R. R., Mujtaba S. (2011) The biology of lysine acetylation integrates transcriptional programming and metabolism. Nutr. Metab. 8, 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Thao S., Escalante-Semerena J. C. (2011) Control of protein function by reversible Nε-lysine acetylation in bacteria. Curr. Opin. Microbiol. 14, 200–204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Okanishi H., Kim K., Masui R., Kuramitsu S. (2013) Acetylome with structural mapping reveals the significance of lysine acetylation in Thermus thermophilus. J. Proteome Res. 12, 3952–3968 [DOI] [PubMed] [Google Scholar]
- 23. Jiang T., Zhou X., Taghizadeh K., Dong M., Dedon P. C. (2007) N-formylation of lysine in histone proteins as a secondary modification arising from oxidative DNA damage. Proc. Natl. Acad. Sci. U.S.A. 104, 60–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Wisniewski J. R., Zougman A., Mann M. (2008) Nε-formylation of lysine is a widespread post-translational modification of nuclear proteins occurring at residues involved in regulation of chromatin function. Nucleic Acids Res. 36, 570–577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Chen Y., Sprung R., Tang Y., Ball H., Sangras B., Kim S. C., Falck J. R., Peng J., Gu W., Zhao Y. (2007) Lysine propionylation and butyrylation are novel post-translational modifications in histones. Mol. Cell. Proteomics 6, 812–819 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Tan M., Luo H., Lee S., Jin F., Yang J. S., Montellier E., Buchou T., Cheng Z., Rousseaux S., Rajagopal N., Lu Z., Ye Z., Zhu Q., Wysocka J., Ye Y., Khochbin S., Ren B., Zhao Y. (2011) Identification of 67 histone marks and histone lysine crotonylation as a new type of histone modification. Cell 146, 1016–1028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Zhang K., Chen Y., Zhang Z., Zhao Y. (2009) Identification and verification of lysine propionylation and butyrylation in yeast core histones using PTMap software. J. Proteome Res. 8, 900–906 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Cheng Z., Tang Y., Chen Y., Kim S., Liu H., Li S. S., Gu W., Zhao Y. (2009) Molecular characterization of propionyllysines in non-histone proteins. Mol. Cell. Proteomics 8, 45–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Tweedie-Cullen R. Y., Brunner A. M., Grossmann J., Mohanna S., Sichau D., Nanni P., Panse C., Mansuy I. M. (2012) Identification of combinatorial patterns of post-translational modifications on individual histones in the mouse brain. PLoS One 7, e36980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Liu B., Lin Y., Darwanto A., Song X., Xu G., Zhang K. (2009) Identification and characterization of propionylation at histone H3 lysine 23 in mammalian cells. J. Biol. Chem. 284, 32288–32295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Fritz K. S., Green M. F., Petersen D. R., Hirschey M. D. (2013) Ethanol metabolism modifies hepatic protein acylation in mice. PLoS One 8, e75868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Garrity J., Gardner J. G., Hawse W., Wolberger C., Escalante-Semerena J. C. (2007) N-lysine propionylation controls the activity of propionyl-CoA synthetase. J. Biol. Chem. 282, 30239–30245 [DOI] [PubMed] [Google Scholar]
- 33. Takahata Y., Inoue M., Kim K., Iio Y., Miyamoto M., Masui R., Ishihama Y., Kuramitsu S. (2012) Close proximity of phosphorylation sites to ligand in the phosphoproteome of the extreme thermophile Thermus thermophilus HB8. Proteomics 12, 1414–1430 [DOI] [PubMed] [Google Scholar]
- 34. Iino H., Naitow H., Nakamura Y., Nakagawa N., Agari Y., Kanagawa M., Ebihara A., Shinkai A., Sugahara M., Miyano M., Kamiya N., Yokoyama S., Hirotsu K., Kuramitsu S. (2008) Crystallization screening test for the whole-cell project on Thermus thermophilus HB8. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 64, 487–491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Yokoyama S., Hirota H., Kigawa T., Yabuki T., Shirouzu M., Terada T., Ito Y., Matsuo Y., Kuroda Y., Nishimura Y., Kyogoku Y., Miki K., Masui R., Kuramitsu S. (2000) Structural genomics projects in Japan. Nat. Struct. Biol. 7, 943–945 [DOI] [PubMed] [Google Scholar]
- 36. Cava F., Hidalgo A., Berenguer J. (2009) Thermus thermophilus as biological model. Extremophiles 13, 213–231 [DOI] [PubMed] [Google Scholar]
- 37. Agari Y., Kashihara A., Yokoyama S., Kuramitsu S., Shinkai A. (2008) Global gene expression mediated by Thermus thermophilus SdrP, a CRP/FNR family transcriptional regulator. Mol. Microbiol. 70, 60–75 [DOI] [PubMed] [Google Scholar]
- 38. Ooga T., Ohashi Y., Kuramitsu S., Koyama Y., Tomita M., Soga T., Masui R. (2009) Degradation of ppGpp by nudix pyrophosphatase modulates the transition of growth phase in the bacterium Thermus thermophilus. J. Biol. Chem. 284, 15549–15556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Kim K., Okanishi H., Masui R., Harada A., Ueyama N., Kuramitsu S. (2012) Whole-cell proteome reference maps of an extreme thermophile, Thermus thermophilus HB8. Proteomics 12, 3063–3068 [DOI] [PubMed] [Google Scholar]
- 40. Guan K. L., Yu W., Lin Y., Xiong Y., Zhao S. (2010) Generation of acetyllysine antibodies and affinity enrichment of acetylated peptides. Nat. Protoc. 5, 1583–1595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Tatusov R. L., Fedorova N. D., Jackson J. D., Jacobs A. R., Kiryutin B., Koonin E. V., Krylov D. M., Mazumder R., Mekhedov S. L., Nikolskaya A. N., Rao B. S., Smirnov S., Sverdlov A. V., Vasudevan S., Wolf Y. I., Yin J. J., Natale D. A. (2003) The COG database: an updated version includes eukaryotes. BMC Bioinformatics 4, 41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Conesa A., Gotz S., Garcia-Gomez J. M., Terol J., Talon M., Robles M. (2005) Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 21, 3674–3676 [DOI] [PubMed] [Google Scholar]
- 43. Kanehisa M., Goto S., Kawashima S., Okuno Y., Hattori M. (2004) The KEGG resource for deciphering the genome. Nucleic Acids Res. 32, D277–D280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Colaert N., Helsens K., Martens L., Vandekerckhove J., Gevaert K. (2009) Improved visualization of protein consensus sequences by iceLogo. Nat. Methods 6, 786–787 [DOI] [PubMed] [Google Scholar]
- 45. Wu W. L., Liao J. H., Lin G. H., Lin M. H., Chang Y. C., Liang S. Y., Yang F. L., Khoo K. H., Wu S. H. (2013) Phosphoproteomic analysis reveals the effects of PilF phosphorylation on type IV pilus and biofilm formation in Thermus thermophilus HB27. Mol. Cell. Proteomics 12, 2701–2713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Moellering R. E., Cravatt B. F. (2013) Functional lysine modification by an intrinsically reactive primary glycolytic metabolite. Science 341, 549–553 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Osborne B., Cooney G. J., Turner N. (2014) Are sirtuin deacylase enzymes important modulators of mitochondrial energy metabolism? Biochim. Biophys. Acta 1840, 1295–1302 [DOI] [PubMed] [Google Scholar]
- 48. Wagner G. R., Payne R. M. (2013) Widespread and enzyme-independent Nε-acetylation and Nε-succinylation in the chemical conditions of the mitochondrial matrix. J. Biol. Chem. 288, 29036–29045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Crosby H. A., Pelletier D. A., Hurst G. B., Escalante-Semerena J. C. (2012) System-wide studies of N-lysine acetylation in Rhodopseudomonas palustris reveal substrate specificity of protein acetyltransferases. J. Biol. Chem. 287, 15590–15601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Rose I. A., Grunberg-Manago M., Korey R. S., Ochoa S. (1954) Enzymatic phosphorylation of acetate. J. Biol. Chem. 211, 737–756 [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.